
The well-defined three-dimensional matrix of a micro-sized silicon/carbon composite promoting lithium-ion transportation†
Denghui
Wang‡
ac,
Minghao
Ma‡
ab,
Wenqiang
Xu
ab,
Yingjie
Ma
 *a,
Lidong
Li
*b and
Xianglong
Li
*ac
*a,
Lidong
Li
*b and
Xianglong
Li
*ac
aCAS Key Laboratory of Nanosystem and Hierarchical Fabrication, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing, 100190, China. E-mail: lixl@nanoctr.cn; mayj@nanoctr.cn
bState Key Laboratory for Advanced Metals and Materials, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing, 100083, China. E-mail: lidong@mater.ustb.edu.cn
cUniversity of Chinese Academy of Sciences, Beijing, 100049, China
First published on 29th October 2024
Abstract
Micro-sized silicon is a promising anode material due to its high theoretical capacity and low cost. However, its bulk particle size poses a challenge during electrochemical cycling, and the long ion/electron transport paths within it limit the rate capability. Herein, we propose a structural engineering approach for establishing a well-defined three-dimensional (3D) micro-sized silicon/carbon matrix to achieve efficient omnidirectional ionic and electronic conductivity within micro-sized silicon and effectively mitigate the volume changes. The prepared materials, comprising ordered two-dimensional porous silicon nanosheets, offer direct two-dimensional electrolyte transport channels aligned parallel to the layer plane and porous channels oriented perpendicular to the layer plane. These well-defined omnidirectional pathways enable more efficient electrolyte mass transport than the disordered paths within the traditional 3D porous silicon anodes. A robust carbon shell, securely bonded to silicon through dual covalent bonding, effectively shields these pathways, buffering the volume changes and offering an electronically conductive 3D carbon network.
New conceptsThree-dimensional (3D) porous micro-sized Si–C hybrid materials, which integrate the benefits of micro-sized and nano-sized Si–C composites, have been developed to address the challenges of micro-sized silicon due to its considerable bulk particle size, such as inevitable particle fragmentation and elongated ion and electron transport pathways. However, disordered channels within these materials act as constraints like “country roads” and thus impede efficient electrolyte mass transport, hindering their electrochemical performance. We propose a novel structural engineering approach to achieve omnidirectional superior ionic and electronic conductivity within micro-sized silicon to overcome this limitation. This innovative technique precisely establishes 3D-defined pathways for superior lithium-ion conductivity, rather than relying on disorganized routes, offering an alternative similar to “motorways” instead of “country roads”. The enhanced ion transport kinetics with our material design ensures the full utilization of silicon, enabling the anode to realise higher reversible capacity along with rate performance. These 3D pathways are reinforced by a robust carbon shell, buffering the volume changes, minimizing the undesirable side reactions caused by silicon fractures, and offering an electronically conductive 3D carbon network. Our approach is appealing for electrode slurry technology in producing energy storage devices, as it requires anodes with highly efficient omnidirectional lithium-ion transport. |
1. Introduction
Currently, much smaller and more powerful high-performance lithium-ion batteries (LIBs) are needed to meet the increasing market demand for power sources.1,2 Unfortunately, commercial LIBs use graphite as the anode active material and thus cannot meet the market demand due to the low theoretical capacity of graphite. In recent years, silicon (Si) has emerged as the most attractive alternative to graphite because of its high theoretical capacity (4200 mA h g−1), low lithium intercalation potential (<0.4 V vs. Li/Li+), and abundant resources.3 However, the Si anode suffers from intrinsic low ionic and electronic conductivity and a significant volume expansion (>300%) during Li+ intercalation.Therefore, silicon–carbon (Si–C) nanocomposites, such as core–shell,4 nanowires,5,6 and yolk–shell,7,8 have been developed to overcome these issues. In Si–C nanocomposites, conductive carbon reinforcements are introduced as supports or coating layers to increase the electron conductivity of silicon anodes and buffer the volume changes of silicon during electrochemical cycling; the nano-sized silicon architecture can alleviate silicon mechanical fracture during volume changes and facilitate lithium-ion transport.9 Nevertheless, Si–C nanocomposites suffer from a low tap density and costly, low-yield production due to their complex synthesis processes. Instead, micro-sized Si–C anode materials are more favorable for practical applications due to their lower cost and higher tap density.10,11 However, micro-sized silicon particles much more easily undergo mechanical fracture upon volume changes, and they feature longer ionic and electronic transport pathways during charging/discharging processes, impeding their rate capabilities.12
As a result, micro-sized Si–C anode materials with embedded nanostructures, combining the advantages of micro-sized and nano-sized Si–C composites, have been fabricated to improve electrochemical performance.12–14 The typical materials are 3D porous micro-sized Si–C hybrid materials with a micrometer particle size and nanopores throughout the bulk particle.15–19 Their porous structure can offer sufficient ionic transport channels and mitigate the volume expansion without sacrificing the high tap density.20,21 Most importantly, their 3D porous architectures can meet conventional electrode slurry technology requirements, commonly used in energy storage device production, which needs anodes with efficient omnidirectional lithium-ion transport.22 Particularly, the pores densely distributed in the 3D porous materials make a considerable contribution to electrochemical performance, accommodating the volume expansion and thereby maintaining the structural integrity and forming channels to facilitate electrolyte transport.23 However, 3D porous Si–C materials typically have a large space and large surface area, resulting in a low tap density with a low volumetric capacity and inducing a low initial Coulombic efficiency (ICE).24 Consequently, a well-defined architecture is crucial to 3D porous Si–C materials. Besides, most of the previous 3D porous micro-sized Si–C anode materials have disordered channels inside, which, like the “country roads”, limit the electrolyte mass transport.12,25 Thus, it is necessary to develop new 3D porous micro-sized Si–C anode materials to supply much more efficient channels for lithium-ion transport.
Herein, we propose a structural engineering strategy for establishing a well-defined 3D matrix of a micro-sized silicon/carbon composite (3DS@C) to promote lithium-ion transportation through a dual lithium-ion intercalation/deintercalation process combined with carbon encapsulation (Fig. 1). The prepared 3DS@C consists of ordered layers of two-dimensional porous silicon nanosheets, thus providing direct two-dimensional channels parallel to the layer plane and porous channels perpendicular to the layer plane for fast lithium-ion transport. The resulting 3D hierarchical nanoarchitecture affords a well-defined pathway for omnidirectional superionic conductivity. Additionally, the 3D channels are reinforced by a robust carbon shell firmly attached to silicon through dual covalent bonding. This strategic reinforcement effectively minimizes the irreversible consumption of lithium ions and establishes a 3D carbon network throughout the active material for electron conductivity. Unlike the traditional 3D porous silicon anodes with disordered channels that provide “country roads” for lithium-ion transport,12 3DS@C offers highly efficient omnidirectional ionic and electronic conductivity “motorways”, achieving superior rate capability besides outstanding cycling stability.
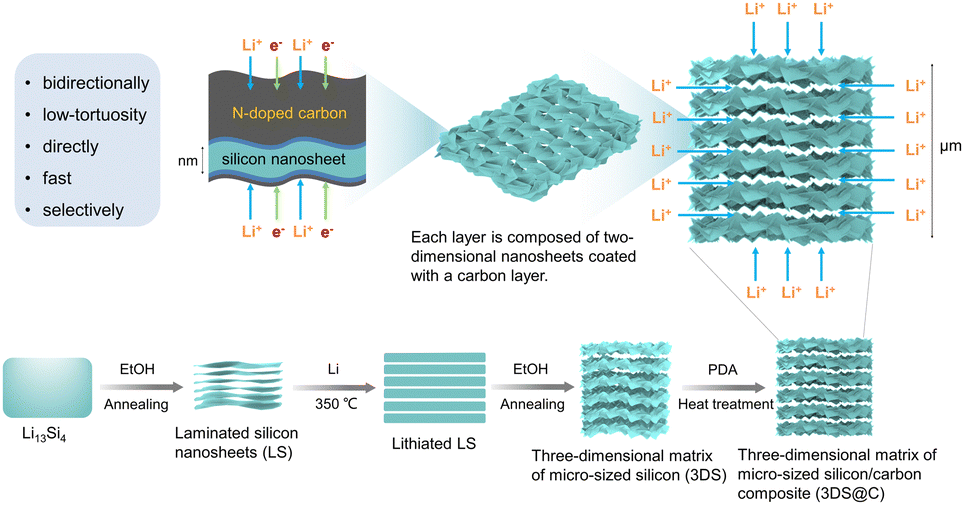 | ||
| Fig. 1 Schematic showing the fabrication process and structural advantages of 3DS@C and its corresponding characteristics for lithium storage. | ||
2. Results and discussion
Fig. 1 schematically illustrates the fabrication procedure and the structure of 3DS@C. 3DS@C was prepared using a dual lithium-ion intercalation/deintercalation process followed by carbon encapsulation. After the first lithium-ion deintercalation, the commercial macro-sized Li13Si4 was transformed into the intermediate product, laminated silicon nanosheets (LS). As shown in Fig. 2a, LS, consisting of ordered layers of 2D silicon nanosheets, features a hierarchically layered micro–nanostructure with particle sizes ranging from 5 to 10 μm. This vertically layered structure of LS emerges from the spontaneous separation of Li13Si4 during the initial delithiation process, succeeded by annealing, giving rise to stacked crystalline silicon nanosheets.26 After secondary lithiation, delithiation, and annealing, the three-dimensional matrix of micro-sized silicon (3DS) is acquired, inheriting the layered configuration of LS. Nevertheless, the crystalline nanosheets in 3DS possess a substantial number of nanopores formed by etching the lithiated LS. These pores establish conduits for the conduction of lithium ions in the vertical direction (Fig. 2b). Based on our previous work,22 the crystalline nanosheets in LS were partially lithiated by regulating the ratio of lithium metal to silicon, yielding the lithiated LS. In the subsequent procedures, nanopores were generated on the nanosheets through delithiation by etching with ethanol, followed by secondary annealing to produce 3DS. As a result, 3DS has two-dimensional channels parallel to the layer plane that offer a direct transport pathway for lithium ions and, in the meantime, possess lithium-ion transport channels perpendicular to the layer plane. Therefore, 3DS possesses a three-dimensional matrix structure that serves as a pathway for omnidirectional lithium-ion transport. The internal space within the matrix structure of 3DS should effectively buffer the volume expansion of silicon during cycling. Via in situ polymerization and carbonization of dopamine, these omnidirectional lithium-ion transport channels were reinforced to prevent them from collapsing and endow them with a stable interface against the electrolyte, giving 3DS@C. Fig. 2c shows that 3DS@C perfectly inherits the structure of 3DS, although it has an additional carbon layer compared to 3DS. | ||
| Fig. 2 Morphological and structural characterization of LS, 3DS, and 3DS@C. SEM images at different magnifications of (a) LS, (b) 3DS, and (c) 3DS@C. | ||
The microstructures of LS, 3DS, and 3DS@C were analyzed via transmission electron microscopy (TEM). As shown in Fig. 3a, the nanosheets within LS are flat two-dimensional layers, and no pores are observed on their surface. In contrast, the nanosheets in 3DS are characterized as thick 2D layers, while conspicuous nanopores are observed on their surface, attributable to the secondary lithiation and delithiation (Fig. 3b). As a result of the nanosheet stacking, these nanopores give rise to lithium-ion transport pathways that are oriented perpendicular to the plane of the layers, substantiated by the SEM experimental findings (Fig. 2b). 3DS@C acquired the structural traits of 3DS, as nanopores are also present on the nanosheets constituting 3DS@C, albeit with carbon encapsulation (Fig. 3c). The high-resolution TEM reveals the silicon–carbon interface in 3DS@C, and the carbon layer is doped by nitrogen (Fig. 3c).
 | ||
| Fig. 3 Morphological, structural, and componential characterization of LS, 3DS, and 3DS@C. TEM images at different magnifications of (a) LS, (b) 3DS, and (c) 3DS@C. (d) STEM and elemental mapping images of 3DS@C. The white dashed lines indicate the boundaries of silicon layers (a and b) and the interface between silicon and carbon layers (c). | ||
Nevertheless, Brunauer–Emmett–Teller (BET) experiments show that 3DS@C has a distinct pore structure compared to LS and 3DS (Fig. S1b, ESI†). LS and 3DS contain micro-pores (1.2–1.6 nm), mesopores (pore width: 2.5–50 nm), and macropores (pore width: 50–110 nm). 3DS has a significantly larger pore volume than LS, thus exhibiting a higher specific surface area (Fig. S1a and Table S1, ESI†). In contrast, 3DS@C contains micro-pores (1.2–1.6 nm), mesopores within the 6–50 nm range, and macropores within the 50–110 nm range while virtually lacking mesopores within the 2–5 nm range, which is probably attributed to the carbon coating blocking the micro-pores and mesopores within the 2–5 nm range. The observed substantial decrease in the specific surface area (18.96 vs. 29.06 m2 g−1) and pore volume (0.425 vs. 0.580 m3 g−1), and increase in the average pore size (8.96 vs. 7.89 nm) of 3DS@C relative to 3DS (Table S1, ESI†) provide additional evidence in support of this hypothesis. Notably, compared to LS, 3DS@C still has a larger pore volume (0.425 vs. 0.405 m3 g−1), demonstrating that 3DS@C has more abundant pores than LS. The mesopores and macropores constitute the primary pore types in 3DS@C, while the micropores are relatively scarce.
The polydopamine-derived carbon layer within 3DS@C uniformly encapsulates the silicon framework, confirmed by the even distribution of Si, C, O, and N in the STEM (Fig. 3d). The Raman spectrum of 3DS@C further corroborates the presence of carbon encapsulation, as evidenced by the distinct D-band (1350 cm−1) and G-band (1590 cm−1) characteristic of graphene-like carbon materials (Fig. 4a). The ID/IG value (a relative intensity ratio of the D-band to G-band) of 3DS@C is approximately 0.9, reflecting a flawed and disordered structure within the introduced carbon of 3DS@C. Thermogravimetric analysis (TGA) shows that 3DS@C has a low carbon content of around 1.9 wt%, implying a significant silicon content (98.1 wt%) contributing to its high capacity (Fig. 4b).
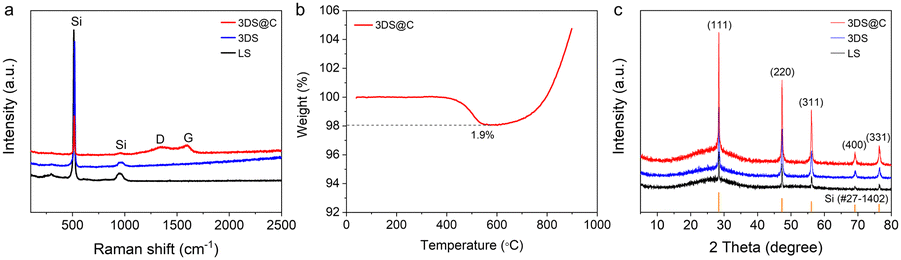 | ||
| Fig. 4 Structural and componential characterization of LS, 3DS, and 3DS@C. (a) Raman spectra, (b) TGA curves, and (c) XRD patterns of 3DS@C. | ||
As characterized by X-ray photoelectron spectroscopy (XPS), the silicon–carbon interface of 3DS@C consists of Si, C, N, and O (Fig. S2a, ESI†). The N 1s XPS measurement implies that the interface contains four types of nitrogen-doped carbon, including pyridine N (398.5 eV), pyrrole N (399.7 eV), and graphite N (401.1 eV), which would enhance electron and Li+ transport (Fig. S2b, ESI†). Moreover, the Si–N bond (397.6 eV), Si–N bond (102.6 eV), and C–N bond (285.8 eV) obtained from N 1s, Si 2p, and C 1s XPS spectra, respectively, disclose that the carbon layer is covalently bonded to the silicon framework (C–N–Si and C–O–Si; Fig. S2b–d, ESI†). The covalently bonded silicon–carbon interface should be able to guarantee that the Li+ transport channels of 3DS@C possess high mechanical flexibility and chemical stability to stabilize the solid electrolyte interface (SEI) layer. In addition, owing to the nitrogen-doped sites, this interface should facilitate efficient modulation and selective permeation of ions, promoting rapid lithium-ion transport at the Si/C interface.
Evidently, 3DS@C exhibits a distinct combination of a 3D matrix structure and a covalently bonded carbon layer, providing a pathway for omnidirectional ionic conductivity and establishing a robust silicon–carbon interface. The electrochemical performance of lithium storage and kinetic properties of LS, 3DS, and 3DS@C were assessed within the coin-type half-cell range to investigate the advantageous structural features of 3DS@C. The cyclic voltammogram patterns of the initial three cycles for each sample exhibit similar reduction and oxidation peaks, consistent with the lithiation/delithiation behaviors of crystalline and amorphous silicon (Fig. S3, ESI†). However, the 3DS electrode exhibits an initial reversible capacity of 2930 mA h g−1 with a Coulombic efficiency (CE) of 86.85% at 0.2 A g−1, much higher than that of the LS electrode (2716 mA h g−1 and 80.84%; Fig. 5a). This result suggests that, due to its matrix structure, the lithiation/delithiation process within the 3DS electrode involves a higher level of silicon participation, and there is a more stable solid electrolyte interphase (SEI) inside the electrode. After introducing the carbon layer, the initial reversible specific capacity can be enhanced to 3184 mA h g−1, accompanied by a higher Coulombic efficiency (89.24%). This improvement is attributed to the carbon layer of 3DS@C, which acts as a protective barrier between the silicon and the electrolyte, thereby preventing undesired side reactions caused by silicon fragmentation during the charging/discharging process. The superior capacity and Coulombic efficiency (CE) of 3DS@C, compared to LS and 3DS, highlight the structural advantages of 3DS@C, including its 3D matrix structure and carbon coating.
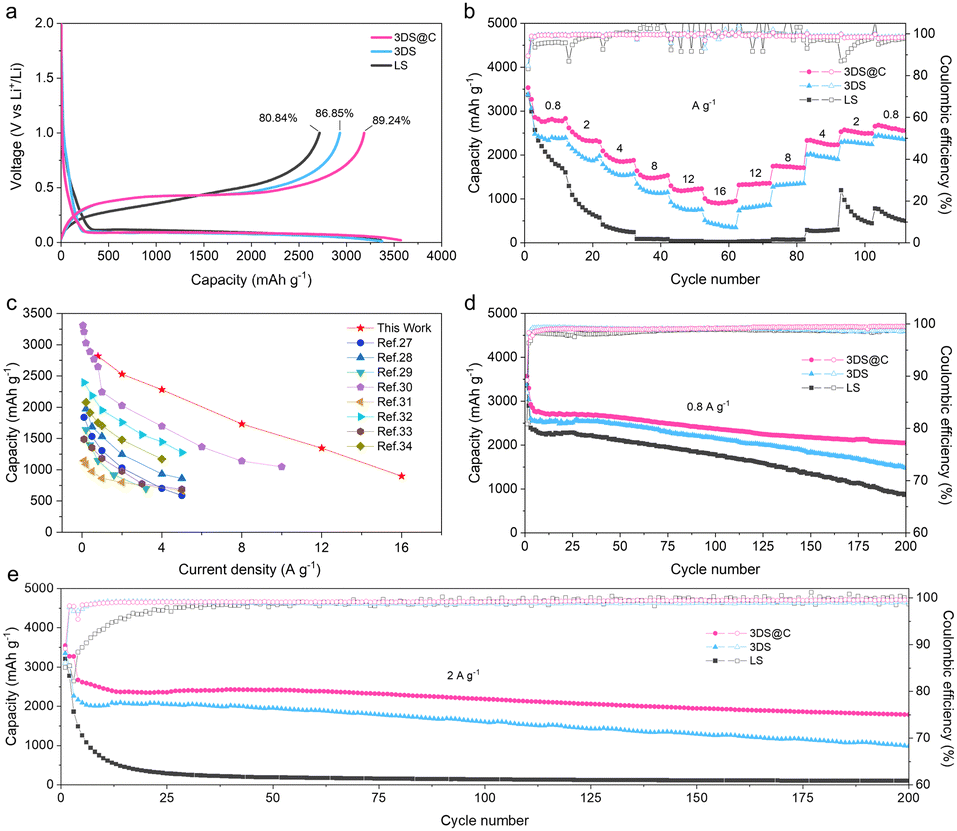 | ||
| Fig. 5 Electrochemical performance characterization of LS, 3DS, and 3DS@C. (a) Galvanostatic charge–discharge voltage profiles for the first cycle at 0.2 A g−1. (b) Capacities of LS, 3DS, and 3DS@C at different rates from 0.8 to 16 A g−1 (ten cycles for each rate). (c) Rate capability comparison of 3DS@C and Si/C anode materials in the previous work. (d) and (e) Cycling performance of LS, 3DS, and 3DS@C over 200 cycles tested at (d) 0.8 A g−1 and (e) 2 A g−1 (cycled at 0.2 A g−1 for the initial two cycles). | ||
Due to its advanced structural features, the 3DS@C electrode exhibits predominant rate capability compared to LS and 3DS electrodes, as illustrated in Fig. 5b. The LS electrode displays limited rate capability, as evidenced by a decrease in specific capacity with increasing current rate. Significantly, the specific capacity of the LS electrode approaches zero when the current density reaches 8 A g−1, and its recovery becomes challenging under extremely high current densities of up to 16 A g−1. In contrast, the 3DS electrode exhibits a notable enhancement in specific capacity compared to LS. Specifically, at current densities of 0.8, 2, 4, 8, 12, and 16 A g−1, the 3DS electrode demonstrates average specific capacities of approximately 2340, 2016, 1640, 1203, 844, and 460 mA h g−1, respectively. This improved rate capability of the 3DS electrode can be attributed to its 3D matrix structure, which facilitates efficient transport of Li+ and provides a more porous space to accommodate the volumetric changes associated with silicon. Notably, leveraging the synergistic effects of the 3D matrix structure and carbon protective layer, the 3DS@C electrode exhibits significantly higher reversible capacities than 3DS. Specifically, at identical current rates, the 3DS@C electrode shows higher reversible capacities of 2796, 2402, 1940, 1502, 1220, and 942 mA h g−1, surpassing the corresponding values of 3DS by 19.5%, 19.1%, 18.3%, 24.9%, 44.5%, and 104.8%, respectively. Moreover, even after cycling at current densities from 16 A g−1 to 0.8 A g−1, the 3DS@C electrode retains a specific capacity of 2660 mA h g−1, indicating near-complete restoration of its initial capacity. The 3DS@C electrode demonstrates a significantly superior rate capability to previously reported silicon/carbon anodes, as illustrated in Fig. 5c and Table S2 (ESI†),27–34 owing to the omnidirectional “motorways” for ionic and electronic conductivity within its structure. Although the reported micro-sized Si–C anode materials with porous structures can also offer sufficient ionic transport channels, most of them have disordered channels inside, which, like the “country roads”, limit the electrolyte mass transport.
The exceptional rate capability and remarkable reversibility observed in 3DS@C come from its unique structural characteristics. The 3D matrix structure of 3DS@C facilitates the efficient transport of Li+ and effectively accommodates the volumetric changes during cycling. Additionally, its carbon layer shields silicon from the electrolyte and provides enhanced electronic conductivity.
Electrochemical impedance spectroscopy (EIS) and the constant current intermittent titration technique (GITT) were employed to elucidate the enhanced lithium storage performance of the 3DS@C anode. As shown in Fig. S4 (ESI†), the Nyquist plots of the LS, 3DS, and 3DS@C electrodes exhibit two semicircles corresponding to the SEI impedance (RSEI) and the charge-transfer impedance (Rct) at the electrode/electrolyte interface alongside straight lines in the low-frequency region indicative of the diffusion resistance of lithium ions within the electrode material. These impedance characteristics were evaluated under a current density of 2 A g−1 (0.5C) for 1, 10, and 100 cycles, respectively. The LS, 3DS, and 3DS@C electrodes exhibit varying levels of ohmic impedance (RS), SEI impedance (RSEI), and charge-transfer impedance (Rct), with the LS electrode displaying the highest values, followed by the 3DS electrode and the 3DS@C electrode demonstrating the lowest values. This trend highlights the advantages of the structural characteristics of 3DS@C. The 3D matrix structure facilitates the efficient transport of lithium ions, while the carbon layer enhances electron transport. As a result, the 3DS@C electrode exhibits excellent ionic and electronic conductivity. Additionally, the RS and RSEI of the 3DS and 3DS@C electrodes remain relatively stable throughout cycling, unlike the LS electrode, which experiences a progressive increase in these impedances. This observation suggests that the 3D matrix structure effectively mitigates silicon crushing during cycling, leading to improved stability and performance of the 3DS@C electrode. The validity of this finding was further substantiated through a comparative analysis of the Li+ diffusion coefficients during charging and discharging states of the LS, 3DS, and 3DS@C electrodes, as determined using the GITT (Fig. S5, ESI†). The Li+ diffusion coefficients during charging and discharging states of the 3DS and 3DS@C electrodes are more than one order of magnitude higher than those of the LS electrode. This observation underscores the crucial role played by the 3D matrix structures of 3DS and 3DS@C electrodes in enhancing the transport of Li+. The high ionic conductivity of 3DS@C stems from constructing a three-dimensional lithium-ion transport network mainly by mesopores (6–50 nm) and macropores (50–110 nm), supported by the BET analysis.
Furthermore, the 3DS@C electrode exhibits a higher Li+ diffusion coefficient than the 3DS electrode, indicating the notable impact of the protective carbon layer of the 3DS@C electrode in enhancing the diffusion of Li+. The carbon layer endows 3DS@C with superior electrical conductivity, as evidenced by comparing the electrical resistivity of LS, 3DS, and 3DS@C powders (Fig. S6, ESI†). The powder electrical resistivity of LS, 3DS, and 3DS@C markedly decreases with the increase in pressure, and 3DS@C powder exhibits the lowest electrical resistivity under various pressures. Significantly, under different pressures, the resistivity of 3DS@C is only two- to three-thousandths of that of 3DS, demonstrating the superior electrical conductivity of 3DS@C. Notably, the electrical resistivity of 3DS is considerably higher than that of LS under different pressures. This phenomenon might be attributed to the densely distributed nanopores on the nanosheets in 3DS disrupting the originally continuous structure of the nanosheets and impeding the transmission of electrons in the lamellae. In LS, the nanosheet layers are intact, thereby causing the resistivity of 3DS to be much higher than that of LS.
The nitrogen-doped carbon layer of 3DS@C (Fig. S2b, ESI†) likely strengthens the rapid transport of Li+ at the silicon/carbon interface. It has been demonstrated that the contents of pyridinic N and pyrrolic N induce defects, coordinating with lithium ions (Li+) to generate active sites for Li+ storage and enhancing specific capacity.35 Additionally, owing to the synergistic effect of electron-deficient pyridinic N, pyrrolic N, and electron-rich graphitic N, the nitrogen-doped carbon layer is bestowed with favorable electron acceptor–donor properties, further facilitating electronic conductivity. In addition, the nitrogen-doped carbon layer is covalently bonded to the silicon layer through dual covalent bonds (C–N–Si and C–O–Si; as shown in Fig. S2b–d, ESI†), forming a stable silicon–carbon interface. This interface prevents direct contact between Si and the electrolyte, promotes selective permeation of lithium ions, and stabilizes the structure of 3DS@C.
Besides the excellent rate capability, the structural advantages inherent in 3DS@C impart exceptional cycling performance. Fig. 5d and e compare the cycling stability of LS, 3DS, and 3DS@C at a current density of 0.8 A g−1 (0.2C) and 2 A g−1 (0.5C), respectively. At current densities of 0.8 and 2 A g−1, the 3DS@C electrode demonstrates a remarkable reversible capacity of 2928 and 2615 mA h g−1, respectively, while maintaining good capacity retention (2045 and 1788 mA h g−1, respectively) for over 200 cycles. In contrast, the 3DS electrode exhibits a significantly lower reversible capacity of 2608 and 2094 mA h g−1 at current densities of 0.8 and 2 A g−1, respectively, with a further rapid decline observed during cycling. The LS electrode shows the lowest reversible capacity of 2394 and 1259 mA h g−1 at current densities of 0.8 and 2 A g−1, respectively. During cycling at 2 A g−1, the LS electrode displays a conspicuous occurrence of an abrupt capacity decay to zero within the initial 20 cycles.
The significant differences in cycling performance among LS, 3DS, and 3DS@C can be ascribed to their distinct structural characteristics. The LS electrode possesses channels solely oriented along the parallel direction of the silicon nanosheet plane, restricting the electrolyte's movement exclusively along the parallel direction rather than in the perpendicular direction to the nanosheet plane. Due to the random orientation of each active material particle during the fabrication process of the LS anode, there is no assurance that the plane direction of these particles aligns perpendicular to the electrode collector. Consequently, this misalignment impedes electrolyte diffusion, limiting the utilization of silicon in LS particles within the electrode and resulting in poor capacity. Besides, LS lacks sufficient inner space to accommodate the volumetric changes of silicon during cycling, resulting in silicon crushing and subsequently diminished cycling stability.
In contrast, 3DS and 3DS@C exhibit a 3D matrix structure that facilitates electrolyte transport in all directions, maximizing the utilization of silicon within both 3DS and 3DS@C. Thus, they demonstrate improved capacity performance compared to LS. Furthermore, the 3D matrix structures of 3DS and 3DS@C provide ample space to mitigate the volume changes of silicon, thereby giving rise to enhanced cycling stability. Moreover, it is significant that a nitrogen-doped protective carbon layer was covalently bonded onto the silicon interface in 3DS@C. This carbon layer enhances the electronic conductivity of the silicon, facilitates the penetration of lithium ions from the electrolyte into the silicon, and provides a protective shield for the silicon against the electrolyte. As a result, 3DS@C exhibits enhanced capacity and improved cycling stability compared to 3DS.
The elemental analysis of the SEI of LS, 3DS, and 3DS@C after cycling via XPS (Fig. S7, ESI†) further clarified the reasons for the superior cycling performance of 3DS@C. In the SEI of LS and 3DS, the concentrations of F and Li are low, and a large portion of element F is organic fluorine (Fig. S7 a, c, and d, ESI†). In contrast, in the SEI of 3DS@C, the concentration of C is significantly decreased, while the concentrations of Li and F gradually increase. The maximum content of F and Li elements in 3DS@C indicates that it possesses more LiF, which accounts for its outstanding stability in the cycle.
To further confirm the critical role of the covalently bonded silicon–carbon interface in 3DS@C during cycling, we compared the morphology and structure of the 3DS electrode and 3DS@C electrode before and after cycling (Fig. S8, ESI†). The thickness of the 3DS@C electrode exhibited a minimal increase of only ∼5.8% after undergoing 50 cycles at 2 A g−1. In contrast, the thickness of the 3DS electrode increased significantly by 19.7% after the same number of cycles at 2 A g−1. This observation highlights the significance of the covalently bonded carbon layer on silicon, which effectively buffers the volume changes experienced by silicon and shields it from the electrolyte, endowing 3DS@C with outstanding cycling stability.
3. Conclusions
In conclusion, we present a structural engineering strategy to achieve efficient omnidirectional lithium-ion and electron transport within micro-sized silicon anodes. Our approach surpasses conventional methods for fabricating 3D porous silicon by creating well-defined lithium-ion conductivity pathways instead of disordered paths, establishing superior ionic conductivity “motorways” instead of “country roads” throughout the bulk volume. This feature fulfills the conventional electrode slurry technology requirements in energy storage device fabrication, necessitating anodes exhibiting efficient omnidirectional lithium-ion transport. Moreover, the enhanced ion transport kinetics with our material design ensures full utilization of silicon, enabling the silicon anode to release higher reversible capacity besides rate performance. Additionally, the established lithium-ion conductivity paths are reinforced by a mechanical carbon shell, which buffers the volume changes of silicon particles, sets up a 3D electron transport network within the silicon particles, and forms a stable SEI. Our approach is a powerful technique to address the issues of micro-sized silicon, and it can be expanded to other electrode materials that require fast ion transport kinetics for maximized utilization.Author contributions
X. L., Y. M., and L. L. conceived and designed the project. D. W. and M. M. carried out materials synthesis and electrochemical measurements. W. X. contributed to the materials characterization. X. L., Y. M., and M. M. co-wrote the paper. All authors discussed the results and commented on the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (grant no. 22375053 and 52173250) and the Zhejiang Provincial Market Supervision and Administration Bureau (grant no. CY2023321).References
- F. Wu, J. Maier and Y. Yu, Chem. Soc. Rev., 2020, 49, 1569–1614 RSC.
- W. Cai, Y.-X. Yao, G.-L. Zhu, C. Yan, L.-L. Jiang, C. He, J.-Q. Huang and Q. Zhang, Chem. Soc. Rev., 2020, 49, 3806–3833 RSC.
- M. Je, D.-Y. Han, J. Ryu and S. Park, Acc. Chem. Res., 2023, 56, 2213–2224 CrossRef CAS PubMed.
- S. Chen, L. Shen, P. A. Van Aken, J. Maier and Y. Yu, Adv. Mater., 2017, 29, 1605650 CrossRef PubMed.
- S. Imtiaz, I. S. Amiinu, D. Storan, N. Kapuria, H. Geaney, T. Kennedy and K. M. Ryan, Adv. Mater., 2021, 33, 2105917 CrossRef CAS PubMed.
- G. Zhou, L. Xu, G. Hu, L. Mai and Y. Cui, Chem. Rev., 2019, 119, 11042–11109 CrossRef CAS PubMed.
- H. Su, A. A. Barragan, L. Geng, D. Long, L. Ling, K. N. Bozhilov, L. Mangolini and J. Guo, Angew. Chem., Int. Ed., 2017, 56, 10780–10785 CrossRef CAS.
- L. Zhang, C. Wang, Y. Dou, N. Cheng, D. Cui, Y. Du, P. Liu, M. Al-Mamun, S. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2019, 58, 8824–8828 CrossRef CAS.
- Z. Yan, J. Liu, Y. Lin, Z. Deng, X. He, J. Ren, P. He, C. Pang, C. Xiao, D. Yang, H. Yu and N. Du, Electrochim. Acta, 2021, 390, 138814 CrossRef CAS.
- D. H. S. Tan, Y.-T. Chen, H. Yang, W. Bao, B. Sreenarayanan, J.-M. Doux, W. Li, B. Lu, S.-Y. Ham, B. Sayahpour, J. Scharf, E. A. Wu, G. Deysher, H. E. Han, H. J. Hah, H. Jeong, J. B. Lee, Z. Chen and Y. S. Meng, Science, 2021, 373, 1494–1499 CrossRef CAS PubMed.
- Y. Li, K. Yan, H.-W. Lee, Z. Lu, N. Liu and Y. Cui, Nat. Energy, 2016, 1, 15029 CrossRef CAS.
- L. Sun, Y. Liu, L. Wang and Z. Jin, Adv. Funct. Mater., 2024, 2403032 CrossRef CAS.
- R. Jain, A. S. Lakhnot, K. Bhimani, S. Sharma, V. Mahajani, R. A. Panchal, M. Kamble, F. Han, C. Wang and N. Koratkar, Nat. Rev. Mater., 2022, 7, 736–746 CrossRef CAS.
- X. Zhang, D. Kong, X. Li and L. Zhi, Adv. Funct. Mater., 2019, 29, 1806061 CrossRef.
- R. Yi, F. Dai, M. L. Gordin, S. Chen and D. Wang, Adv. Energy Mater., 2013, 3, 295–300 CrossRef CAS.
- G. Huang, J. Han, Z. Lu, D. Wei, H. Kashani, K. Watanabe and M. Chen, ACS Nano, 2020, 14, 4374–4382 CrossRef CAS PubMed.
- H. Jia, X. Li, J. Song, X. Zhang, L. Luo, Y. He, B. Li, Y. Cai, S. Hu, X. Xiao, C. Wang, K. M. Rosso, R. Yi, R. Patel and J.-G. Zhang, Nat. Commun., 2020, 11, 1474 CrossRef CAS PubMed.
- W. An, B. Gao, S. Mei, B. Xiang, J. Fu, L. Wang, Q. Zhang, P. K. Chu and K. Huo, Nat. Commun., 2019, 10, 1447 CrossRef PubMed.
- Z. Yang, C. Wu, S. Li, L. Qiu, Z. Yang, Y. Zhong, B. Zhong, Y. Song, G. Wang, Y. Liu, Z. Wu and X. Guo, Adv. Funct. Mater., 2022, 32, 2107897 CrossRef CAS.
- H. Sun, J. Zhu, D. Baumann, L. Peng, Y. Xu, I. Shakir, Y. Huang and X. Duan, Nat. Rev. Mater., 2018, 4, 45–60 CrossRef.
- X. Li, Z. Chen, X. Liu, L. Guo, A. Li, X. Chen and H. Song, Adv. Funct. Mater., 2024, 2401686 CrossRef CAS.
- D. Wang, Y. Ma, W. Xu, S. Zhang, B. Wang, L. Zhi and X. Li, Adv. Mater., 2023, 35, 2212157 CrossRef CAS PubMed.
- P. Li, H. Kim, S.-T. Myung and Y.-K. Sun, Energy Storage Mater., 2021, 35, 550–576 CrossRef.
- Z.-L. Xu, X. Liu, Y. Luo, L. Zhou and J.-K. Kim, Prog. Mater. Sci., 2017, 90, 1–44 CrossRef CAS.
- Z. Cheng, H. Jiang, X. Zhang, F. Cheng, M. Wu and H. Zhang, Adv. Funct. Mater., 2023, 33, 2301109 CrossRef CAS.
- J. Lang, B. Ding, S. Zhang, H. Su, B. Ge, L. Qi, H. Gao, X. Li, Q. Li and H. Wu, Adv. Mater., 2017, 29, 1701777 CrossRef PubMed.
- Z. Zhao, J. Han, F. Chen, J. Xiao, Y. Zhao, Y. Zhang, D. Kong, Z. Weng, S. Wu and Q. Yang, Adv. Energy Mater., 2022, 12, 2103565 CrossRef CAS.
- Z. Li, Z. Zhao, S. Pan, Y. Wang, S. Chi, X. Yi, J. Han, D. Kong, J. Xiao, W. Wei, S. Wu and Q. Yang, Adv. Energy Mater., 2023, 13, 2300874 CrossRef CAS.
- R. Zhu, Z. Wang, X. Hu, X. Liu and H. Wang, Adv. Funct. Mater., 2021, 31, 2101487 CrossRef CAS.
- Y. Mu, M. Han, B. Wu, Y. Wang, Z. Li, J. Li, Z. Li, S. Wang, J. Wan and L. Zeng, Adv. Sci., 2022, 9, 2104685 CrossRef CAS PubMed.
- Y. Zhang, Z. Wang, K. Hu, J. Ren, N. Yu, X. Liu, G. Wu and N. Liu, Energy Storage Mater., 2021, 34, 311–319 CrossRef.
- Y. An, Y. Tian, H. Wei, B. Xi, S. Xiong, J. Feng and Y. Qian, Adv. Funct. Mater., 2020, 30, 1908721 CrossRef CAS.
- N. Liu, J. Liu, D. Jia, Y. Huang, J. Luo, X. Mamat, Y. Yu, Y. Dong and G. Hu, Energy Storage Mater., 2019, 18, 165–173 CrossRef.
- R. Gao, J. Tang, K. Zhang, K. Ozawa and L.-C. Qin, Nano Energy, 2020, 78, 105341 CrossRef CAS.
- Y. Yu, C. Yang, Y. Jiang, Z. Shang, J. Zhu, J. Zhang and M. Jiang, Adv. Energy Mater., 2024, 2403086 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00349g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |