
Thermal evolution of solid solution of silica-embedded AgPt alloy NPs in the large miscibility gap†
Hemant
Jatav
 ab,
Anusmita
Chakravorty
a,
Ambuj
Mishra
a,
Matthias
Schwartzkopf
c,
Andrei
Chumakov
c,
Stephan V.
Roth
cd and
Debdulal
Kabiraj
*a
ab,
Anusmita
Chakravorty
a,
Ambuj
Mishra
a,
Matthias
Schwartzkopf
c,
Andrei
Chumakov
c,
Stephan V.
Roth
cd and
Debdulal
Kabiraj
*a
aMaterials Science Department, Inter-University Accelerator Centre, New Delhi, India. E-mail: d.kabiraj@gmail.com; hemantphy95@gmail.com
bSchool of Physics, Devi Ahilya Vishwavidhyalaya, Indore, India
cDeutsches Elektronen-Synchrotron (DESY), Notkestraße 85, D-22607 Hamburg, Germany
dDepartment of Fibre and Polymer Technology, KTH Royal Institute of Technology, Teknikringen 56-58, SE-100 44 Stockholm, Sweden
First published on 4th February 2025
Abstract
Understanding the phase behavior of immiscible elements in bimetallic nanomaterials is essential for controlling their structure and properties. At the nanoscale, the miscibility of these immiscible elements often deviates from their behavior in bulk materials. Despite its significance, comprehensive and quantitative experimental insights into the dynamics of the immiscible-to-miscible transition, and vice versa, remain limited. In this study, we investigate the nucleation and growth kinetics of silica-embedded AgPt nanoparticles (NPs) across a wide range of annealing temperatures (25 °C to 900 °C) to elucidate temperature-dependent nanoalloy phase transitions and NP size distribution. Our findings reveal that the alloy phase persists up to 400 °C, with a corresponding average NP size of ∼2 nm. Beyond this temperature, phase instability begins to occur. We propose a three-stage process of nucleation and growth: (1) initial AgPt nanoalloy formation during deposition, (2) growth via thermal energy-assisted diffusion up to 400 °C, and (3) Ag atom emission from the nanoalloy above 500 °C, indicating Ag diffusion towards the surface, followed by partial sublimation of Ag atoms at 900 °C. These results provide crucial insights into the thermal limits for the dealloying of NPs, growth kinetics, and phase stability or instability under varying thermal conditions.
New conceptsAgPt bimetallic alloy nanoparticles (NPs) have gained significant attention due to their enhanced catalytic performance and cost-effectiveness compared to platinum-based nanostructures. However, the AgPt system exhibits limited miscibility at the nanoscale, making alloy formation and stabilization within the miscibility gap challenging. The phase behavior of AgPt nanoalloys is influenced by the composition, morphology, and surface atomic arrangement of the constituent metals. In this study, we investigated the temperature-dependent phase behavior of silica-embedded AgPt NPs up to 900 °C, using transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), in situ grazing incidence small-angle X-ray scattering (GISAXS), grazing incidence wide-angle X-ray scattering (GIWAXS), X-ray photoelectron spectroscopy (XPS), and ultraviolet-visible (UV-Vis) absorption spectroscopy. Our findings reveal that AgPt NPs exhibit phase instability beyond 400 °C. Based on our comprehensive analysis, we propose a three-stage growth mechanism that accounts for surface energy, intermixing, cohesive energy, and strain energy during the alloying and dealloying processes. We conclude that embedding ultrasmall metallic NPs in a dielectric matrix significantly enhances their thermal stability in terms of both size and alloy phase. |
1 Introduction
Pt-based nanostructures have demonstrated promising catalytic performance among all other noble metal-based nanostructures.1–4 However, the high cost of Pt limits its applicability and compels researchers to seek cost-effective alternative metals. Alloying Pt with less costly metals addresses the issue of cost-effectiveness, improves the overall physiochemical properties and thermal stability, making it suitable for practical applications.5–13 Recently, AgPt bimetallic alloy nanoparticles (NPs) have gained a lot of attention owing to the cost-effectiveness of Pt alloy-based nanostructures and their higher catalytic performance.14–16 AgPt is a fascinating system not only from an application standpoint but also for gaining a fundamental scientific understanding of thermodynamic alloy phase stability. AgPt exhibits a large bulk miscibility gap; however, at the nanoscale, it can exist as alloy NPs when the composition is approximately Ag50Pt50 at%.17,18 The formation and stabilization of the nanoalloy phase within the miscibility gap is challenging and strongly depend on the composition, morphology (size and shape), and surface atomic arrangement of the constituent metals.19–22Highlighting the impact of size and alloy phase, ultrasmall AgPt NPs (diameter <10 nm) in the alloy phase exhibit excellent electrocatalytic activities for the ethylene glycol oxidation reaction (EGOR) and glycerol oxidation reaction (GOR) compared to larger NPs with different segregation geometries.5,13 These ultrasmall bimetallic NPs not only improve the chances of alloy/intermetallic formation,23,24 but are also efficient for applications due to their significant surface-to-volume ratio, enhanced catalytic activity, and selectivity compared to their larger counterparts. Recent studies have indicated that the complete miscibility of bulk-immiscible elements, such as Au and Rh, is highly size-dependent. NPs of these two immiscible elements undergo a phase-separation-to-alloy transition as their size decreases, achieving complete miscibility in the sub-2 nm particle size range across the entire compositional range.25 Pirart et al. similarly reported that a stable alloy phase of AgPt NPs is maintained only up to a particle size of 2 nm. In conclusion, the miscibility of bulk-immiscible elements in nanoalloys decreases with the increasing size of the NPs.26–28 Temperature-dependent studies on AgPt NPs have been performed on thin films grown on substrates or liquid supports. Existing reports suggest that AgPt NPs segregate into different structural geometries, such as core–shell or random mixture29–31 and completely dealloyed at high annealing temperatures (ATs) of (∼300 °C).32–35 With an increase in AT, the size of the supported AgPt NPs increases initially, followed by thermodynamically driven surface segregation, alloy phase degradation, and complete sublimation of Ag concentration at approximately 500–600 °C.34,35 Therefore, thermal coarsening is a significant challenge with metal NPs. Particularly, annealing at high temperatures increases the size of NPs, which may affect the thermodynamic phase stability of nanoalloys. Therefore, a systematic temperature-dependent growth study is required to understand the thermal stability of AgPt bimetallic NPs, their growth kinetics, and to determine the temperature range within which both the ultrasmall size and nanoalloy phase are retained.
Generally, chemically prepared NPs are coated with a capping layer to preserve their physicochemical properties. When such NPs are subjected to high temperatures (above ∼400 °C), the protective capping layers are unable to prevent the loss of active surface area due to coarsening.36–39 Therefore, we proposed the preparation of nanocomposite (NC) thin films containing metallic NPs surrounded by a silica dielectric matrix. Embedding metallic NPs in a silica matrix stabilizes the NPs by preventing coalescence, migration, and oxidation, thereby enhancing thermal stability. The SiO2 matrix promotes confinement by stabilizing the structure and preventing diffusion and phase segregation of NPs. It may also support in maintaining the phase stability of two immiscible metals, particularly at elevated temperatures. We recently demonstrated the importance of embedding NPs in a silica matrix by investigating the size and structural stability of AuAg/SiO2 NCs and comparing their properties with those of Au/SiO2 and Ag/SiO2 NCs.40–42 (Here, the abbreviation XY/SiO2 represent the system of XY NPs embedded in silica matrix.) We showed that Au and Ag NPs maintain an ultrasmall size (<10 nm) up to 800 °C. The miscible system of bimetallic AuAg/SiO2 NC also preserves homogeneity of the alloy phase up to an AT of 900 °C while maintaining the NP size (<10 nm). In conclusion, embedding ultrasmall metallic NPs in a dielectric host matrix ensures their thermal stability and reduces thermal coarsening by limiting atomic diffusivities.
Notably, in the as-synthesized form, the catalytic performance of the atom beam co-sputtering (ABS)-synthesized silica-embedded NPs may not be as effective as that of chemically synthesized NPs. However, post-growth treatment processes can be employed to improve the catalytic efficacy of the ABS-synthesized silica-embedded NPs. Some notable post-growth treatment methods are as follows: (1) wet chemical etching, where chemical etchants, such as hydrofluoric acid (HF) or ammonia, are used to selectively dissolve the silica matrix, exposing the embedded NPs. This method is particularly effective for silica-based matrices.43–45 (2) Laser ablation, where short-pulse lasers ablate the matrix material, allowing for rapid exposure of the embedded NPs over large areas.46 (3) Formation of Porous Silica, where controlled etching or thermal treatment can transform the silica matrix into a controlled porous structure.47,48 These processes enhance NP accessibility while preserving the structural integrity of the matrix, making the NPs more accessible for catalytic reactions.
Till date, research on the temperature-dependent nucleation and growth kinetics of AgPt NPs is limited, with only a few studies addressing NP-embedded NCs. A comprehensive understanding of the thermodynamic evolution of the AgPt alloy phase, based on atomic diffusion, segregation, growth, and optical properties is still lacking. In this work, we utilize atom beam co-sputtering of silica and Ag and Pt metal foils to prepare thin films of metal–silica NCs. AgPt NPs are studied up to an AT of 900 °C and their growth kinetics, including the morphological and structural evolution, are compared with those of their Ag and Pt monometallic counterparts under the same annealing conditions. The experimental results show that, unlike the constituent Ag and Pt metals, the bulk-immiscible AgPt nanoalloys do not follow the classical theory of nucleation and growth. Finally, we propose a three-stage process for the nucleation and growth of silica-embedded AgPt NPs.
2 Results and discussion
2.1 Formation of AgPt ultrasmall alloy nanoparticles
The thickness, chemical composition, and elemental stoichiometry of the prepared NC films obtained after simulating the Rutherford backscattering spectrometry (RBS) spectra, are listed in Table 1 (simulated RBS spectra are shown in Fig. S1, ESI†). The NC thin films are made up of AgPt NPs (∼Ag60Pt40 at%) embedded inside silica matrix (∼Si1O2). The high-resolution transmission electron microscope (HRTEM) image, presented in Fig. 1(a), shows that the matrix-embedded NPs are spherical in shape, with an average diameter of ∼1.6 nm.| Nanocomposites (Si:O) | Film thickness (nm) | Composition (%) | Stoichiometry |
|---|---|---|---|
| AgPt/SiO2 | 295 ± 15 | Si: 31.0 ± 2 | Si1O2 |
| O: 61.4 ± 3 | |||
| Ag: 4.5 ± 0.2 | |||
| Pt: 3.1 ± 0.2 |
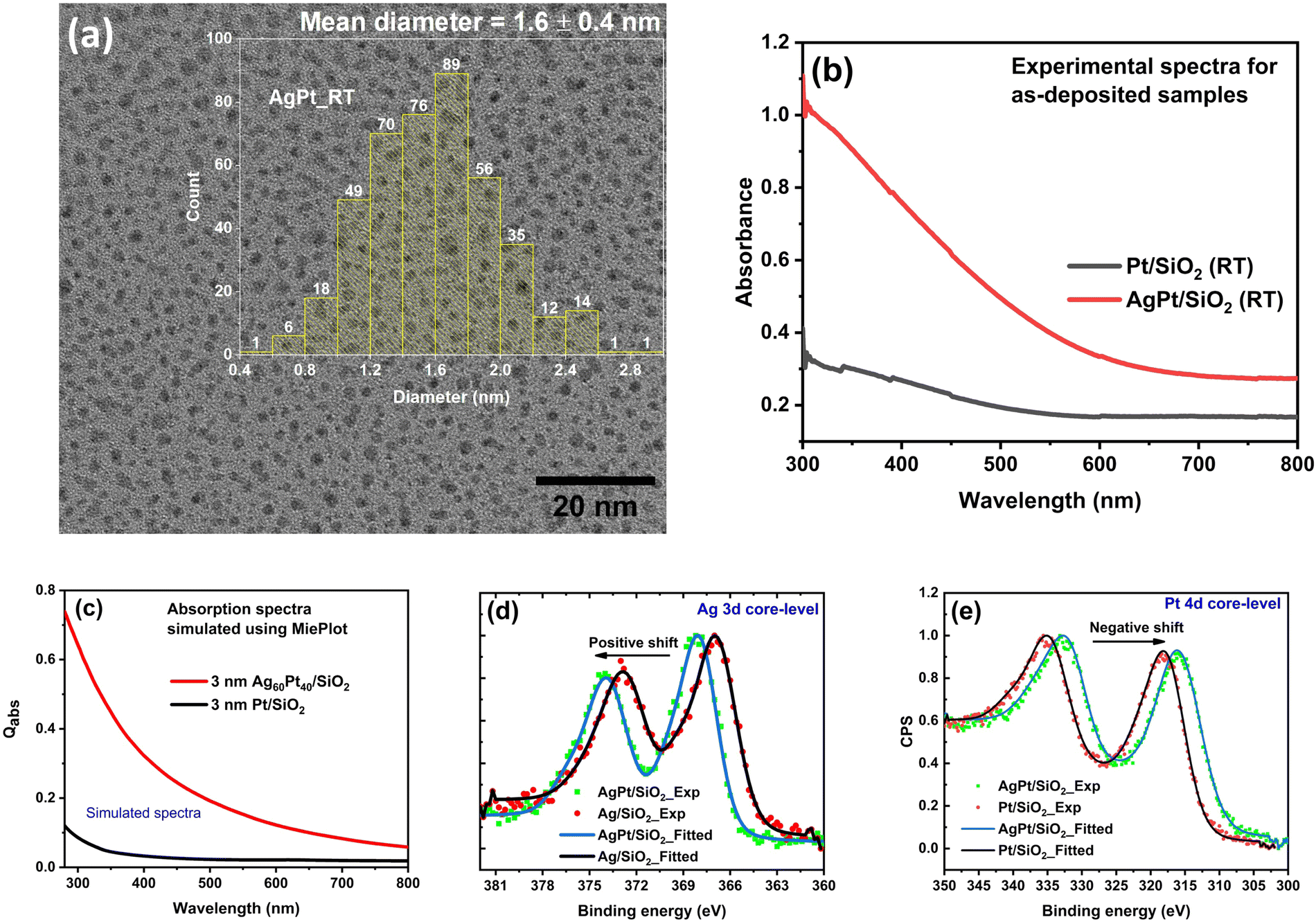 | ||
| Fig. 1 (a) HRTEM image of as-deposited AgPt/SiO2 NC along with the size distribution histogram, (b) The experimental absorption spectra of as-deposited AgPt/SiO2 and Pt/SiO2 NCs, (c) The simulated absorption spectra of as-deposited AgPt/SiO2 and Pt/SiO2 obtained using MiePlot. XPS spectra for core levels (d) Ag 3d and (e) Pt 4d components of Ag/SiO2, Pt/SiO2, and AgPt/SiO2 NCs. The experimentally observed spectra are represented by scattered points, while fitted curves are shown by continuous lines. | ||
The formation of AgPt nanoalloys is confirmed by X-ray photoelectron spectroscopy (XPS). XPS spectrum corresponding to the Ag 3d core-level of AgPt/SiO2 NC (Fig. 1(d)) exhibits a shift of 1.2 eV toward higher binding energy (B.E.) relative to that of Ag/SiO2 NC. In contrast, the Pt 4d core-level (Fig. 1(e)) of AgPt/SiO2 NC exhibits a negative shift of 0.2 eV toward lower B.E. relative to that of Pt/SiO2 NC. The observation of these shifts in the B.E. values indicates the presence of AgPt alloy NPs in the as-deposited sample.49 These observed shifts are caused by electrons transferring from Ag to Pt atoms owing to the electronegativity difference between them or by charges transferring from Ag to the surrounding silica matrix. Furthermore, XPS analysis confirms the presence of oxidation-free Ag and Pt NPs, as no additional tailing toward higher binding energies is observed in Ag 3d and Pt 4d core-level spectra.21 A detailed analysis and fitting of the core-level XPS spectra of Pt and Ag components are provided in the ESI† (Fig. S2).
The UV-visible (UV-vis) absorption analysis also supports the formation of AgPt alloy NPs embedded inside silica matrix, as no localized surface plasmon resonance (LSPR)-related peaks are observed in the absorption spectrum of the as-deposited sample (Fig. 1(b)). If the as-deposited sample contained a mixture of Ag and Pt NPs instead of AgPt alloy NPs, an LSPR peak corresponding to Ag NPs in the visible region would have been observed.50 To further validate our claim based on absorption analysis, the optical absorption cross-section for AgPt/SiO2 NC is simulated using MiePlot software-an electrodynamic simulation based on Mie theory.51 The dielectric function (DF) required as an input parameter for the MiePlot simulation is calculated using Rahm's model52 for Ag60Pt40 at% alloy NPs. In all of our simulations, the NPs are considered to be spherical, and the refractive index of the amorphous silica matrix is set as 1.45.53 The simulated absorption spectra of Ag60Pt40 at% alloy and Pt NPs (shown in Fig. 1(c)) exhibit an excellent agreement with the experimentally obtained absorption spectra (Fig. 1(b)). Our findings are consistent with a recent study which found that the two immiscible elements (Au and Rh)25 and (Ag and Pt)28 form alloy NPs only when their sizes are smaller than 2 nm.
2.2 Temperature-dependent particle growth and nanoalloy phase stability
The nanoalloy phase stability of AgPt NPs has been previously reported to decrease as the size of NPs increase.26,27The UV-Vis absorption spectra of AgPt/SiO2 NC after heating at different ATs is shown in Fig. 2(a). For AgPt/SiO2 NC, no LSPR peak is observed in the visible region for the as-deposited and 400 °C annealed samples. The absence of an LSPR peak below 400 °C indicates the presence of NPs in alloy form.50 After annealing at 500 °C, a single broad hump-like feature appears at ∼400 nm. We assign this feature as Peak 1. At 600 °C, two clearly resolvable peaks appear at ∼418 nm (peak 1) and ∼350 nm (peak 2). The two peaks obtained after annealing at a certain temperature can be linked to the quadrupole and multipole resonance due to the presence of larger NPs.54 Further annealing at 700 °C, peak 1 is observed at ∼426 nm and peak 2 is observed at ∼354 nm. At this temperature, both peaks become sharp, with peak 1 exhibiting a higher absorbance than peak 2. At 800 °C, the positions of peak 1 (∼437 nm) and peak 2 (∼358 nm) further shift to higher wavelengths, with peak 2 exhibiting a higher absorbance. The position of peak 1 and peak 2 as a function of AT is shown in Fig. 2(b). Both peaks shift to higher wavelengths with ATs. Interestingly, at an AT of 900 °C, both peaks disappear. This is possibly due to the presence of Pt NPs alone. At a high AT of 900 °C, it can be anticipated that all Ag atoms have diffused toward the outer edges or completely diffused out from the silica matrix.34
 | ||
| Fig. 2 UV-visible absorption spectra of (a) AgPt/SiO2 NCs, along with the corresponding (b) shifts in the mean position of peak 1 (right-side peak) and peak 2 (left-side peak) after annealing for 30 min at indicated temperatures. (c) Simulated absorption spectra for different sized Ag NPs (inset depicts the enlarged view of peak 1 region), along with the corresponding (d) shifts in the position of peak 1 (right-side peak) and peak 2 (left-side peak). | ||
To determine the origin of the peaks observed in the absorption spectra after annealing of AgPt/SiO2 NC at temperatures above 500 °C, simulations were performed. Simulated absorption spectra for Ag NPs of varying sizes embedded in a silica matrix are presented in Fig. 2(c). A remarkable similarity can be observed between the simulated spectra for silica-embedded Ag NPs and the experimentally obtained spectra for silica-embedded AgPt NPs annealed above 500 °C. The mean positions of peaks 1 and 2 observed in Fig. 2(c) are plotted as a function of NP size in Fig. 2(d). Similar to experimental observations (Fig. 2(b)), peaks 1 and 2 shift toward higher wavelengths as NP size increases (Fig. 2(d)). Thus, the peaks observed after 500 °C annealing can be assigned to originate from large Ag NPs that are formed as a consequence of annealing. In addition to Ag NPs, small AgPt NPs also continue to exist in the silica matrix after various anneals.
HRTEM images of AgPt/SiO2 NC after annealing at various temperatures are shown in Fig. 3(a)–(f), along with the corresponding histograms in Fig. 3(g)–(l) displaying the NP size distribution. The TEM images reveal that the majority of NPs are spherical, and their size distribution (diameter) depends on the AT. To obtain the size distribution of NPs, the diameters of over 300 NPs were measured. The mean NP size in the as-deposited sample (Fig. 3(a)) is estimated as 1.6 ± 0.4 nm. As shown in Fig. 3(b) and (h), a symmetric size distribution of the NPs, with an average size of 2.1 ± 0.5 nm, can be observed after annealing at 400 °C. After 500 °C annealing, the NP size distribution of exhibits a bimodal character, as can be seen in Fig. 3(c) and (i). At this AT, NPs with an average size of 2.0 ± 0.4 nm dominate, coexisting with a few larger NPs having a diameter of about 30 nm. After 600 °C annealing, the average NP size is determined as 2.9 ± 1.0 nm. As compared to the 500 °C annealed sample, average-sized NPs coexist with a greater proportion of large-sized NPs in the 600 °C annealed sample, as shown in Fig. 3(d). The average size of NPs increases to 5 ± 3 nm and 8 ± 6 nm after annealing at 800 °C (Fig. 3(e)) and 900 °C (Fig. 3(f)), respectively. After annealing at 800 °C, approximately 92% of the NPs are smaller than 10 nm, 6% are between 10 and 16 nm, and 2% are larger than 16 nm, with the largest NP measuring 28 nm. As the AT increases to 900 °C, the asymmetry becomes more pronounced, with some NPs as large as 40 nm. After annealing at 900 °C, approximately 78% of the NPs are less than 10 nm in size, 17% are between 10 and 20 nm in size, and the remaining 5% are larger than 20 nm. Overall, the average size of the NPs increases, and the symmetric size distribution observed at 400 °C becomes asymmetric with increasing AT. In addition, HRTEM analysis was performed to determine the structural stability of ultrasmall NPs at different ATs (Fig. S6, ESI†). For the majority of NPs, a single domain crystal structure is observed, along with a nearly constant lattice spacing of ∼0.23 nm across the entire range of ATs, up to 800 °C. From HRTEM images, it is not possible to uniquely identify the lattices of the metals present in the NC films, as the lattice spacings of Ag, Pt, and AgPt nanoalloy are nearly identical. However, a sharp change in crystallinity at the interface between Ag and amorphous silica in the HRTEM images (Fig. S6, ESI†) confirms the absence of a silver oxide layer.55
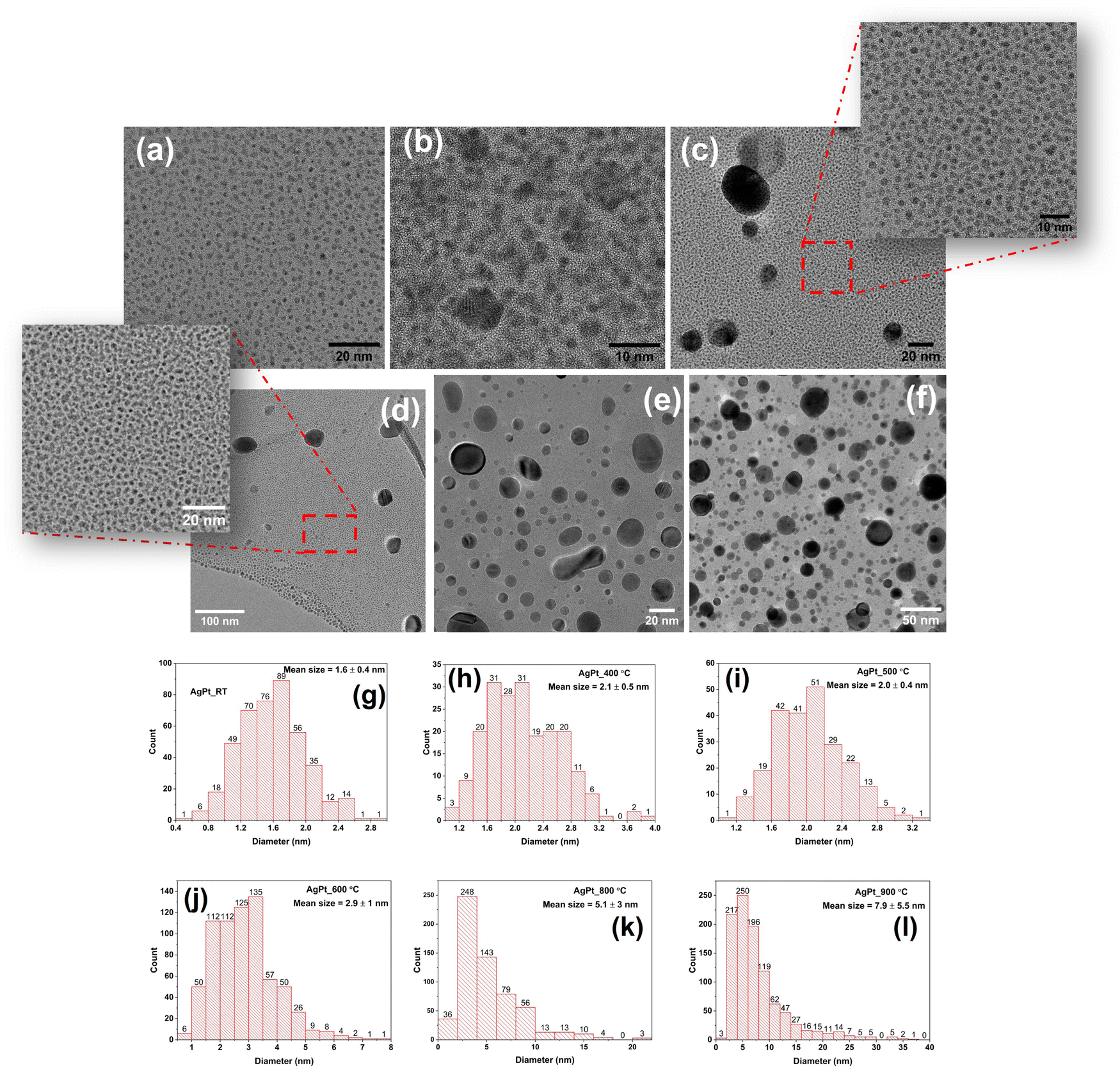 | ||
| Fig. 3 High-resolution TEM images of silica-embedded AgPt NPs for (a) as-deposited, (b) 400 °C, (c) 500 °C, (d) 600 °C, (e) 800 °C and (f) 900 °C samples and their corresponding histograms (g)–(l). Here, histograms show the NP size distribution after annealing at different temperatures. | ||
High-angle annular dark-field-scanning transmission electron microscopy (HAADF-STEM) and energy-dispersive X-ray spectroscopy (EDS) elemental mapping are used to determine the compositional distribution of elements in the NC films, particularly, the composition of larger NPs after 500 °C annealing. The observed Z-contrast in HAADF-STEM images (Fig. 4) of AgPt/SiO2 NC reveals the presence of spherical NPs, containing higher Z elements (Ag and Pt), that are distributed in the matrix of lighter Z elements (Si and O). After annealing at 500 °C (Fig. 4(a)), the Ag and Pt atoms uniformly distribute in the SiO2 matrix. EDS maps show that the larger NPs are primarily composed of Ag atoms. To further examine the influence of thermal annealing on the formation of larger NPs, STEM-EDS elemental maps and spectra were acquired after annealing the AgPt/SiO2 NC at different ATs; the results are shown in Fig. S5 (ESI†). Similar to the observations made on the sample after annealing at 500 °C, Ag and Pt atoms are uniformly distributed in the silica matrix with an almost constant ratio between Ag and Pt (Fig. S5(c)–(e), ESI†), even after annealing at 600 °C and 700 °C; however, the number density of larger Ag NPs increases as a function of AT. After annealing at 900 °C, the obtained EDS maps reveal that the overall NP size considerably increases, with Pt atoms comprising the majority (95 at%) of NPs, while Ag atoms constitute only 5 at% (Fig. S5(f) and (g), ESI†). The decrease in Ag concentration at 900 °C is a result of elevated diffusion of Ag atoms toward the NP edges or evaporation from the silica matrix.34 To further support this claim, RBS analysis of the samples annealed at 700 °C and 900 °C was performed. The RBS data, presented in Fig. S1d (ESI†), confirms that not only Ag atoms diffuse toward the surface of the NC thin film but the total concentration of Ag in the silica matrix also decreases at higher ATs. The obtained RBS results are in line with the observations made and conclusions drawn from UV-vis absorption and STEM-EDS analyses.
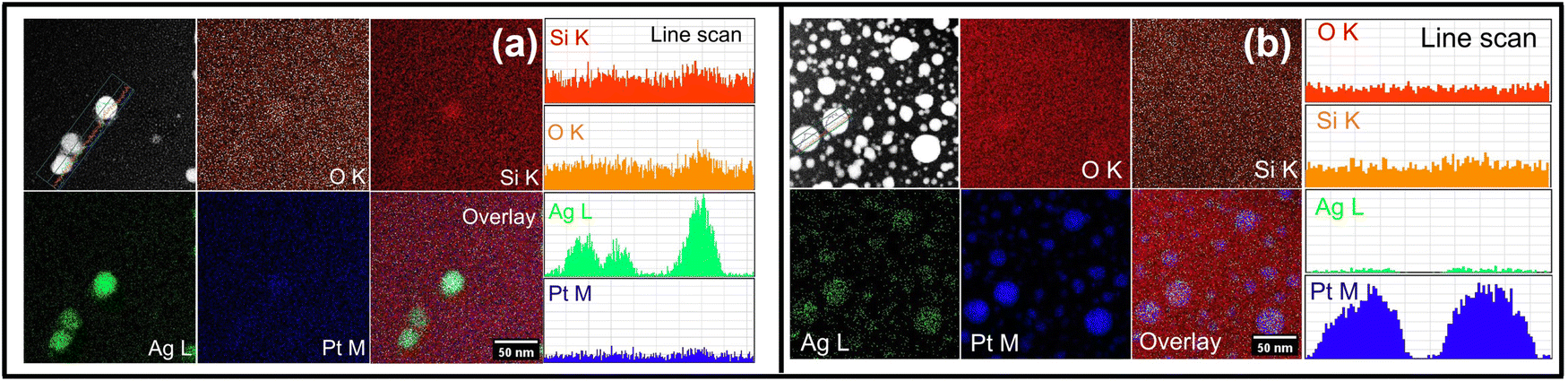 | ||
| Fig. 4 HAADF-STEM images of AgPt NPs embedded inside silica matrix after annealing at (a) 500 °C and (b) 900 °C. Corresponding elemental maps are shown to the right of (a) and (b). | ||
The temperature-dependent growth of silica-embedded AgPt NPs was also investigated using in situ grazing-incidence small-angle X-ray scattering (GISAXS). Details regarding GISAXS analysis can be found in ref. 56. To acquire signals from a wide range of scattering angles or q-values (from 0 to 6 nm−1), in situ annealing GISAXS signals are collected at two different sample-to-detector distances (SDD). Grazing-incidence ultra-small-angle X-ray scattering (GIUSAXS) signals collected at the SDD of 9475 mm provide information regarding the thermal evolution of larger NPs, while grazing-incidence medium-angle X-ray scattering (GIMAXS) signals obtained by placing the detector at the distance of 206 mm provide information about the thermal evolution of ultrasmall NPs (Fig. S3, ESI†). Fig. 5(a)–(i) present the two-dimensional (2D) GIMAXS and GIUSAXS maps, along with the corresponding horizontal and vertical line cuts. These line cuts are extracted at the critical angle (αc = 0.164°) using the method described in the ESI.†
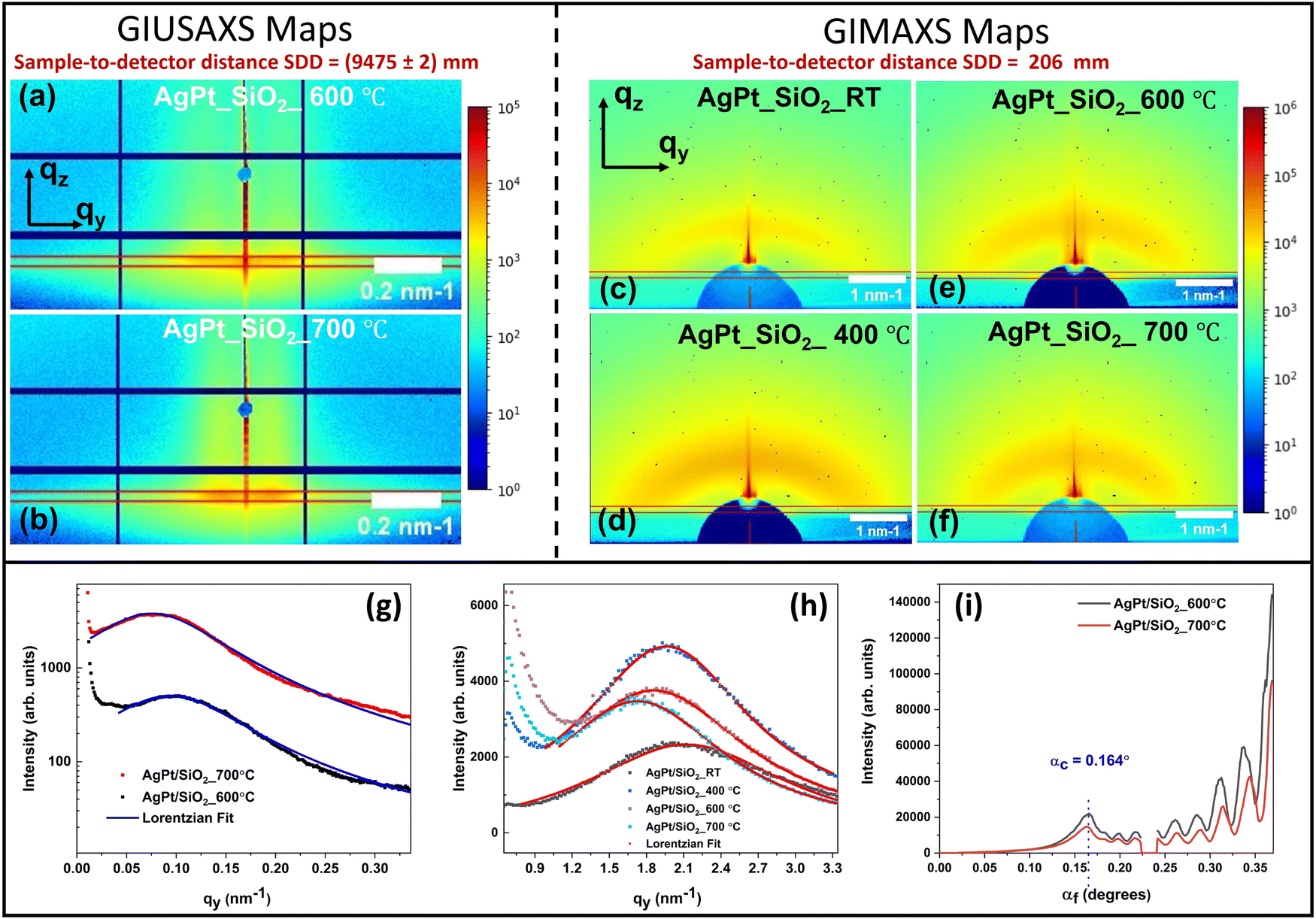 | ||
| Fig. 5 2D GIUSAXS maps for AgPt/SiO2 NCs after annealing at (a) at 600 °C and (b) at 700 °C, and (c)–(f) 2D GIMAXS maps extracted from GIWAXS maps for AgPt/SiO2 NCs after annealing at various annealing temperatures. (g) Shows the horizontal line cuts of maps (a), (b) and (h) show the horizontal line cuts of maps (c), (f), indicated by a red rectangular box. (i) Vertical line cuts of the 2D GIUSAXS maps, showing the critical angle from which the horizontal line cuts were taken. | ||
A geometrical model of cluster growth57 was used to extract the NP sizes and distribution from the GISAXS maps. The results are provided in Tables S1 and S2 (in ESI†). The details of the model and analysis method are also included therein. The NPs remain below <2 nm up to 400 °C. After annealing at 500 °C, a bimodal size distribution is observed, indicating the coexistence of both ultrasmall AgPt and large Ag NPs (consistent with STEM-EDS results shown in Fig. 4). The size of the ultrasmall NPs increases from ∼1.48 nm to ∼1.82 nm as the AT increases from RT to 700 °C. The size of larger Ag NPs is determined as ∼38 nm and ∼46 nm after annealing at 600 °C and 700 °C, respectively. Structural analysis using the GIWAXS data (Fig. S4, ESI†) confirms the existence of the crystalline Ag and AgPt clusters in the NC thin films. High intensity peaks at 2 values of 2θ = 25.96 ± 0.02 and 2θ = 29.94 ± 0.02 are observed, corresponding to the (111) and (200) planes of Ag and AgPt respectively, at an incidence wavelength of λ = 0.1048 nm.58
Obtained results confirm that AgPt alloy NPs are formed during the deposition process and remain as an alloy up to 400 °C. Above 400 °C, a few large Ag NPs appear in the silica matrix, indicating dealloying of AgPt NPs. As the temperature increases, the dealloying process of AgPt accelerates, leading to a further increase in the size of Ag NPs. At 900 °C, significant dealloying occurs, with primarily Pt NPs remaining in the silica matrix (Fig. S5(f) and (g), ESI†). A similar alloy phase degradation of substrate supported AgPt thin films with increasing ATs has been observed in previous studies.32–35 However, for substrate supported NPs, dealloying has been reported to begin at a comparatively lower AT of 250 °C, leading to the complete sublimation of Ag in the temperature range of 500–600 °C. In our study, the improved thermal stability of silica-embedded NPs can be attributed to the following factors: (a) formation of nanoalloys (particle size <5 nm) with lower defects: HRTEM images reveal a singular domain structure, indicating that embedded NPs smaller than 5 nm do not undergo significant scattering losses at the domain and core–shell interfaces;28,59 (b) influence of the surrounding silica matrix: in addition to the synergistic effect between Pt and Ag metals, the presence of the surrounding silica dielectric matrix shields the NPs from surface oxidation, lowers their surface energy, and reduces atomic and nanocluster diffusion. Consequently, thermally stable and surfactant-free ultrasmall NPs are formed.40,60
2.3 Nucleation and growth kinetics
Above 500 °C, the observation of large-sized Ag NPs in the AgPt/SiO2 NC system is not in line with the observation made in our previous study of miscible AuAg/SiO2 NC system.40 This indicates that AgPt NCs do not follow the classical theory of nucleation and growth, whereas their constituent metals, Ag and Pt, follow the classical theory of nucleation and growth.57,61–63 The nucleation process involved in the formation of silica-embedded Ag, Au and AgAu NPs occurs primarily during sputter deposition.40,41 Below 600 °C, the thermal energy-assisted diffusion process is responsible for the growth of these NPs. Above 600 °C, the growth is governed by a competitive process between coalescence and Ostwald ripening. At this stage, smaller NPs with large surface energy coalesce into larger NPs, minimizing the overall Gibbs’ free energy via particle migration. At 900 °C, the growth of silica-embedded NPs is dominated by the Ostwald ripening process. In conclusion, mono-metallic and miscible bimetallic AuAg alloy NPs, embedded inside silica matrix, follow the classical theory of nucleation and growth while maintaining their sizes in the ultrasmall regime. Additionally, the silica-embedded AuAg NPs retain their alloy phase up to 800 °C.The growth mechanism of AgPt NC cannot be explained solely by the classical theory of nucleation and growth. Based on the experimental results, the growth mechanism of AgPt/SiO2 can be described via a three-stage process as follows:
Stage 1: UV-vis absorption, TEM, and XPS results confirm the formation of AgPt NPs during the initial deposition process. The alloy nanocluster nucleation initiates in the gaseous phase prior to deposition.64 Another possibility is that nucleation occurs on the substrate during the initial phases of deposition initiated by a partial electron transfer from Ag to Pt atoms or to the surrounding silica.65,66 In the as-deposited sample, we speculate that in addition to the AgPt NPs (∼1.6 nm), a few unstable clusters of Ag and Pt with a radius smaller than the critical radius are also present in the silica matrix; however, these clusters cannot be detected due to the resolution limit of the characterization techniques.
Stage 2: the thermal evolution of AgPt/SiO2 NC between RT and 400 °C can be understood as an independent and simultaneous growth of bimetallic AgPt NPs and monometallic clusters (of Ag and Pt) via thermal energy-assisted diffusion. By independent, we mean that the growth of AgPt NPs has no effect on the growth of Ag and Pt clusters, and vice versa. Thus, the average size of AgPt, Ag, and Pt clusters increases in this region; however, the size of unstable clusters of Ag and Pt remains below the critical radius and has no effect on the structural and optical absorption properties. Below 400 °C, AgPt NPs remain as an alloy due to the combined effect of defect-free single domain structure, efficient charge exchange, and surrounding silica matrix.
Stage 3: the AgPt/SiO2 NC between 500 °C and 900 °C exhibits a unique and concurrent process of dealloying and selective growth. This stage combines various concurrent processes, including thermal energy-assisted diffusion, particle coalescence, and the attachment/detachment of mono-elemental clusters/atoms (Ag and Pt) from and on the surface of the AgPt alloy. At this stage, the emergence of larger Ag NPs becomes apparent alongside AgPt NPs. For better clarity, we present a separate discussion on the atomic level rearrangement within the AgPt NPs, size evolution of large Ag NPs and ultrasmall AgPt NPs, as follows:
I. Atomic rearrangement within AgPt NPs (selective surface migration of Ag atoms over Pt atoms): the selective diffusion of Ag atoms over Pt atoms in AgPt alloy NPs at elevated temperatures (>500 °C) is governed by a complex interplay of thermodynamic and kinetic parameters. A key factor driving this selective diffusion is the difference in their melting points. In bulk form, the melting points of Ag and Pt are approximately 961.8 °C and 1768.3 °C, respectively. NPs exhibit lower melting points than their bulk counterparts due to their higher surface-to-volume ratio.67,68 This effect is more pronounced for Ag than for Pt as particle size decreases to the nanoscale.68 The reduction in the melting point of Ag significantly enhances its atomic mobility and diffusivity relative to Pt at elevated temperatures.68 Additionally, the lower atomic mass of Ag compared to Pt further contributes to higher atomic mobility and diffusivity of Ag atoms.
The selective diffusion of Ag over Pt in AgPt alloy NPs is also influenced by differences in cohesive energy, bonding strength, and surface energy. Pt has a higher cohesive energy (∼−795 kJ mol−1) compared to Ag (∼−249 kJ mol−1), resulting in stronger intra-metallic bonds that restrict the movement of Pt atoms.69 This difference in cohesive energy affects the diffusion coefficients, with the activation energy for Ag diffusion (∼10–20 kJ mol−1)70 being significantly lower than that for Pt diffusion (∼187–218 kJ mol−1) in a silica matrix.71 As a result, Ag atoms exhibit a higher diffusion coefficient than Pt atoms in the SiO2 matrix, enabling them to overcome energy barriers for migration more easily.
Surface energy also plays a crucial role in driving the selective diffusion of Ag atoms. Ag has a lower surface energy (∼1.25 J m−2) compared to Pt (∼2.48 J m−2), making the migration of Ag atoms to the surface thermodynamically favorable for minimizing the overall Gibbs free energy.22,72,73 This thermodynamically driven surface rearrangement of Ag atoms is further supported by kinetic factors. Since Ag atoms have a lower activation energy for diffusion compared to Pt atoms, they exhibit faster migration kinetics.
The combined influence of these factors—melting point depression, cohesive energy, diffusion coefficients, surface energy, Gibbs free energy, and kinetic energy barriers—creates a dynamic system where Ag atoms preferentially diffuse toward the surface, while Pt atoms remain confined within the core of the AgPt alloy nanoparticles.
II. Growth of bigger Ag NPs: at sufficiently high ATs, Ag atoms in AgPt alloy NPs diffuse more rapidly toward the NP surface than Pt atoms due to the various aforementioned factors. The faster diffusion of Ag toward the NP surface results in the surface segregation of Ag atoms or the formation of a core–shell structure.74 Further increases in temperature lead to the emission of surface Ag atoms due to their lower melting point at the nanoscale.
Concurrently, above 500 °C, the smaller NPs may begin to melt from the surface to the interior, resulting in the liquid-state diffusion of constituent atoms inside the NP and a reduced binding energy of surface atoms.75 Annealing at temperatures above 500 °C provides sufficient thermal energy for Ag atoms migrating toward or originally on the surface of the AgPt NPs to overcome the surface binding energy. Subsequently, the surface Ag atoms are emitted from the AgPt NPs and diffuse into the silica matrix.
In addition, the large difference in the bulk modulus of constituent metals at the nanoscale significantly affects the phase stability of bimetallic NPs at higher ATs. Sluiter et al.18 carried out extensive studies on AgPt and AuPd NCs to determine why AgPt is immiscible. They concluded that AgPt is immiscible because of the strain energy component of mixing enthalpy, which results in a positive mixing enthalpy for AgPt and a negative mixing enthalpy for AuPd. The strain energy component of mixing enthalpy is directly related to the bulk modulus. It is found that the difference in bulk modulus between Ag (104 GPa) and Pt (283 GPa) is significant whereas the difference in bulk modulus between between Au (173 GPa) and Pd (193 GPa) is comparatively negligible. Particularly, at higher temperatures (above 400 °C), this the difference in bulk modulus affects the strain energy component of mixing enthalpy, inter-atomic force constant, and vibrational entropy, leading to the a weaker bond strength between Ag and Pt atoms. As a result, Ag atoms more easily break the bond with Pt and escape the AgPt NPs. Pirart et al. studied the size effect on AgPt NPs and observed that as the NP size increases, the internal stress changes. This change in internal stress alters the bulk modulus, leading to instability in the AgPt L11 phase.18,28
TEM and GISAXS analyses confirm the formation of larger Ag NPs above 500 °C. This drastic increase in the size of Ag NPs can be attributed to the dealloying of AgPt NPs. The dealloying process is well supported by EDS, RBS, and UV-vis absorption results. The Ag atoms emitted from AgPt NPs diffuse within the silica matrix, facilitated by their high diffusivity, and subsequently attach to neighbouring stable Ag NPs. Thus, in addition to the coalescence of smaller Ag clusters, the dealloying of Ag atoms from the AgPt NPs beyond the AT of 500 °C is responsible for the unexpected growth of larger Ag NPs.
III. Evolution of ultrasmall AgPt NPs: after 500 °C, resulting from the dealloying of AgPt NPs and subsequent emission of Ag atoms from AgPt NPs, the presence of Pt-rich AgPt NPs with reduced sizes is expected. EDS analysis performed on the samples annealed at 700 °C and 900 °C confirms the presence of Pt-rich AgPt NPs and a reduction in the Ag concentration in the NPs. Additionally, RBS analysis indicates an overall decrease in Ag concentration and diffusion of Ag atoms toward the thin film surface. In contrast to the expected NP size reduction, both GISAXS and TEM analyses reveal an increase in the average NP size. The observed NP size increase can be explained by the attachment of Pt atoms or clusters to Pt-rich AgPt NPs. The preferential attachment of Pt atoms or clusters to Pt-rich AgPt NPs is possibly driven by the superior stability of the Pt–Pt bond when compared to Ag–Pt, Ag–Ag, and AgP–AgPt bonds.22 The attachment of Pt atoms or clusters to Pt-rich AgPt NPs effectively compensates for the size reduction resulting from the emission of Ag atoms. Annealing to 900 °C appears to provide sufficient energy for most of the Ag atoms to leave the AgPt NPs and evaporate from the surface of the NC thin film, leaving behind only Pt NPs (∼8 nm) in the silica matrix.
To summarize, after annealing at 500 °C, the evolution of NPs within the AgPt/SiO2 NCs involves a complex and concurrent mechanism. The pictorial representation of this temperature-dependent evolution of NPs within the AgPt/SiO2 NCs is illustrated in Fig. 6. As shown in Fig. 6, the growth of Ag NPs results from the coalescence of smaller Ag clusters and the attachment of Ag atoms emitted from the AgPt alloy surface. Concurrently, the evolution of AgPt NPs involves the detachment of Ag atoms and the attachment of Pt clusters. This leads to a slight increase in the overall size of AgPt NPs, accompanied by a decrease in Ag concentration. At higher ATs, the emission of Ag atoms from the alloy surface contributes to the instability of the nanoalloy phase. The complex growth kinetics are influenced by differences in surface energies, interatomic diffusivities, atomic reconstruction, melting point depression, and strain energy. Notably, melting point depression and strain energy part of the mixing enthalpy between Ag and Pt, are the primary factors driving dealloying at elevated temperatures. This study finds that at 900 °C, there is a partial reduction in Ag concentration, demonstrating greater thermal stability of these embedded NCs compared to AgPt NPs on a substrate or in a liquid, where Ag atoms completely sublimated between 500 and 650 °C.34,35
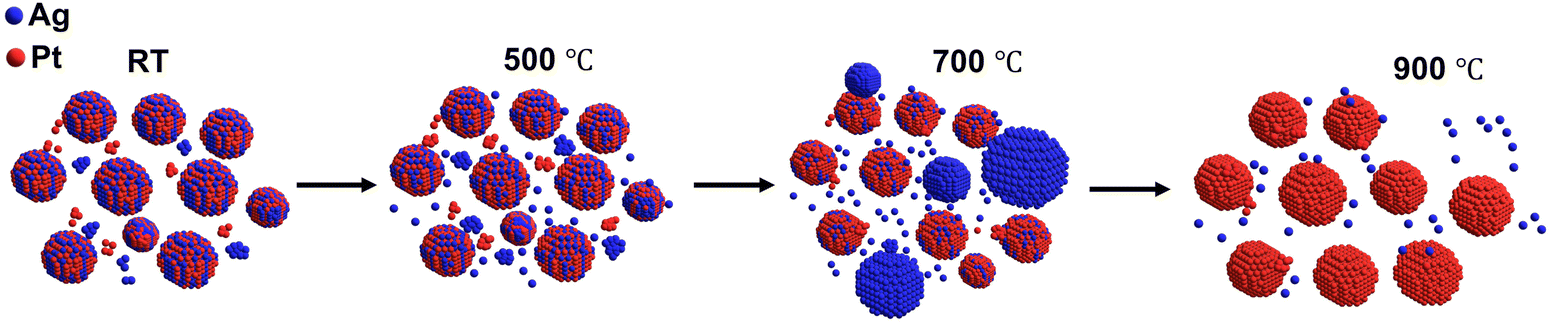 | ||
| Fig. 6 Pictorial representation of the growth kinetics of AgPt NPs at various temperatures. Initially, at room temperature (RT), AgPt nanoparticles exist alongside nanoclusters composed of Ag and Pt. At 500 °C, in addition to monometallic nanoclusters, Ag atoms emitted from the surface of AgPt nanoparticles also present. When the temperature is raised to 700 °C, the process of coalescence of nanoclusters and the emission of Ag atoms intensify. This is evidenced by the declining concentration of Ag atoms in AgPt nanoparticles, attributed to the ample thermal energy present, resulting in the formation of larger Ag nanoparticles. Concurrently, nanoclusters of Pt are attached to the Pt-rich surface of AgPt nanoparticles. Finally, at 900 °C, all Ag atoms are completely emitted from the AgPt nanoparticles, leaving only Pt nanoparticles behind. | ||
3 Conclusion
In this study, the temperature-dependent evolution of silica-embedded AgPt alloy NPs is investigated, with special emphasis on the ultrasmall size of NPs and nanoalloy phase stability. Complete miscibility of AgPt alloy NPs is sustained up to the annealing temperature of 400 °C, while the size of NPs remains in the ultrasmall region (changing from 1.6 nm to 2.1 nm). The AgPt NPs in this size regime often show higher thermal stability because of the oxidation-, defect- and stress-free surface. After 500 °C, a partial dealloying of AgPt NPs is observed, which is followed by an almost complete dealloying of AgPt NPs at 900 °C. A comprehensive interplay between different energies such as surface, inter-mixing, cohesive and strain energies are discussed to understand the growth and the process of alloying/dealloying. Based on our observations, a three-stage process is proposed: (1) AgPt nanoalloy formation during deposition (2) growth of alloy NPs via thermal energy-assisted diffusion up to 400 °C, (3) initiation of Ag atom emission from the AgPt nanoalloy surface above 500 °C, followed by nearly complete diffusion of Ag atoms out of the silica matrix at 900 °C. Our results demonstrate the importance of the silica matrix in maintaining the size of ultrasmall NPs and stabilizing nanoalloy phases at higher ATs. Our study shows that bulk phase diagrams are not fully applicable to nanoscale materials, highlighting the need to investigate the temperature-dependent growth dynamics of other immiscible bimetallic NPs and their size-dependent thermodynamic phase behaviour. Understanding their thermodynamics will enable precise engineering of bimetallic NP structures for practical applications.4 Experimental section
The atom beam co-sputtering technique was utilized for fabricating AgPt/SiO2 NC thin films. For depositing the desired NC films on quartz and silicon substrates (size: 1 × 1 cm2), four Ag and four Pt metal foils were pasted over a 3 inch diameter quartz target. Subsequently, these metal foils, along with Si and O from quartz target were co-sputtered under a high vacuum of 5 × 10−6 mbar to produce embedded NC thin films. The surface area of each metal foil was 5 × 5 mm2. The procedure for synthesis is described in detail in ref. 41 The substrates were kept at ∼25 °C during the co-sputtering process. The films deposited on optically transparent quartz substrates were used for UV-vis absorption spectroscopy measurements, whereas all other characterizations were conducted on the films deposited on silicon substrates. Post-deposition annealing was performed at 400 °C, 500 °C, 600 °C, 700 °C, 800 °C, and 900 °C in a tubular furnace under flowing Ar gas. A constant ramp rate of 10 °C min−1 was used for reaching different ATs. After reaching the desired AT, the temperature was maintained for 30 min, followed by natural cooling. UV-visible absorption spectroscopy was performed in the wavelength range of 300 nm to 800 nm using a Hitachi U-3300 spectrometer. Rutherford backscattering spectrometry (RBS) was performed using 2 MeV He+ ions at the pelletron accelerator RBS-AMS systems (PARAS) facility of the Inter-University Accelerator Centre (IUAC), New Delhi. The RBS spectra of the NCs are presented in Fig. S1 (ESI†). The thickness of the silica layer and concentrations of the embedded elements were determined by simulating the RBS spectra using the Rutherford Universal Manipulation Program (RUMP). To examine the structural and morphological modifications in the NCs, the JEOL JEM-F200 transmission electron microscope, equipped with a thermal field emission electron gun, was operated in TEM and HRTEM modes at 200 keV electron energy. Additionally, the TEM machine was operated in the scanning transmission electron microscope (STEM) mode to enhance image analysis through Z-contrast. For elemental mapping and quantification, energy-dispersive X-ray spectroscopy (EDS) measurements were performed in the STEM mode. The core level XPS measurements were performed at Indus 1, Raja Ramanna Centre for Advanced Technology (RRCAT), Indore, India, using an Al Kα (1486.6 eV) source and an Omicron energy analyzer (EA-125, Germany). XPS is a highly sensitive technique that enables precise detection of alloy-induced fluctuations in the electronic structure of bimetallic NCs. Before performing the XPS measurements, the samples were affixed to a holder inside a high vacuum chamber that was pumped to reach a base pressure of 10−10 Torr. The samples were then cleaned using surface sputtering with 3 keV Ar ions. For calibrating the XPS data, the binding energy of the C 1s peak at 284.8 eV was used. For analysis, the 4d and 3d core levels of Pt and Ag NCs were selected due to their intense and narrow XPS-related peaks. Finally, in situ grazing-incidence small-angle X-ray scattering (GISAXS) and grazing-incidence wide-angle X-ray scattering (GIWAXS) measurements were performed on the NCs after annealing at various temperatures (RT to 800 °C) to monitor the growth of embedded NPs within the silica matrix. The in situ measurements rule out any sample-dependent variations in initial conditions. The data was collected using a micro-focused X-ray beam with an energy of 11.83 keV at the P03 beamline at PETRA III, DESY, Germany.56 The spot size of the X-ray beam was 27 μm × 25 μm (horizontal × vertical). After reaching the desired AT, the temperature was maintained for 30 minutes, followed by natural cooling. In situ annealing was performed using a DHS 1100 (Anton Paar) heating stage set up with a carbon dome in an N2 gas environment. The incident angle of the X-ray beam was set at 0.4 degrees. Using a PILATUS 2M (Dectris) detector positioned at a distance of 9475 ± 2 mm from the sample, GISAXS signals were recorded. To collect the GIWAXS data, a LAMBDA 9M (X-spectrum) detector was positioned at a distance of 206 mm from the sample. In addition to the GISAXS data collected using the PILATUS 2M detector, GISAXS and GIWAXS data were also collected simultaneously using the LAMBDA 9M. A schematic of the setup is shown in ref. 41. After each measurement, the position of the X-ray beam on the sample was shifted by 2 mm to minimize radiation damage.Author contributions
Hemant Jatav: conceptualization, data curation, formal analysis, resources, and writing original draft. Anusmita Chakravorty: reviewing, and editing. Ambuj Mishra: formal TEM analysis. Matthias Schwartzkopf: setup GISAXS experiment, data analysis and reviewing. Andrei Chumakov: setup GISAXS experiment, data analysis and reviewing. Stephan V. Roth: formal discussions and reviewing. D. Kabiraj: resources, supervision, and writing, review, and editing.Data availability
All data generated or analyzed during this study are included in the published article and its ESI† and source data files.Conflicts of interest
The authors declare no competing financial interest or conflicts of interest.Acknowledgements
HJ acknowledges the Council of Scientific and Industrial Research (CSIR), India, for financial support through a PhD fellowship. The authors are thankful to Dr Ambuj Tripathi for providing the UV-Vis spectroscopy facility at IUAC, New Delhi. The authors are also thankful to Mr G. R. Umapathy for performing the RBS measurement at IUAC, New Delhi. The authors are thankful to Dr Ram Janay Choudhary for carrying out XPS measurements. In situ GISAXS/GIWAXS measurements of this research work were carried out at the light source PETRA III of DESY, a member of Helmholtz Association (HGF). Financial support from the Department of Science and Technology (Government of India) provided within the framework of the India@DESY collaboration is gratefully acknowledged. We thank the Department of Science and Technology for providing partial funding for the development of the Atom Beam Sputtering setup.Notes and references
- Z. Peng and H. Yang, Nano Today, 2009, 4, 143–164 CrossRef CAS.
- S. Mostafa, F. Behafarid, J. R. Croy, L. K. Ono, L. Li, J. C. Yang, A. I. Frenkel and B. R. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS PubMed.
- X. Zhou, Y. Gan, J. Du, D. Tian, R. Zhang, C. Yang and Z. Dai, J. Power Sources, 2013, 232, 310–322 CrossRef CAS.
- F. Grillo, H. Van Bui, D. La Zara, A. A. Aarnink, A. Y. Kovalgin, P. Kooyman, M. T. Kreutzer and J. R. van Ommen, Small, 2018, 14, 1800765 CrossRef PubMed.
- C. Yang, B. H. Ko, S. Hwang, Z. Liu, Y. Yao, W. Luc, M. Cui, A. S. Malkani, T. Li and X. Wang, et al. , Sci. Adv., 2020, 6, eaaz6844 CrossRef PubMed.
- C. Xie, C. Chen, Y. Yu, J. Su, Y. Li, G. A. Somorjai and P. Yang, Nano Lett., 2017, 17, 3798–3802 CrossRef CAS PubMed.
- B.-W. Zhang, Z.-C. Zhang, H.-G. Liao, Y. Gong, L. Gu, X.-M. Qu, L.-X. You, S. Liu, L. Huang and X.-C. Tian, et al. , Nano Energy, 2016, 19, 198–209 CrossRef CAS.
- P. Shi, R. Li, Y. Li, Y. Wen, Y. Zhong, W. Ren, Z. Shen, T. Zheng, J. Peng and X. Liang, et al. , Science, 2021, 373, 912–918 CrossRef CAS PubMed.
- Q. Chen, Z. Cao, G. Du, Q. Kuang, J. Huang, Z. Xie and L. Zheng, Nano Energy, 2017, 39, 582–589 CrossRef CAS.
- G. Zhang, Z. Yang, W. Zhang, H. Hu, C. Wang, C. Huang and Y. Wang, Nanoscale, 2016, 8, 3075–3084 RSC.
- G.-T. Fu, B.-Y. Xia, R.-G. Ma, Y. Chen, Y.-W. Tang and J.-M. Lee, Nano Energy, 2015, 12, 824–832 CrossRef CAS.
- A. Mahmood, N. Xie, M. A. U. Din, F. Saleem, H. Lin and X. Wang, Chem. Sci., 2017, 8, 4292–4298 RSC.
- P. Song, H. Xu, B. Yan, J. Wang, F. Gao, Y. Zhang, Y. Shiraishi and Y. Du, Inorg. Chem. Front., 2018, 5, 1174–1179 RSC.
- T. Fu, J. Fang, C. Wang and J. Zhao, J. Mater. Chem. A, 2016, 4, 8803–8811 RSC.
- H. You, Z. Peng, J. Wu and H. Yang, Chem. Commun., 2011, 47, 12595–12597 RSC.
- Z. Li, Y. Li, C. He and P. K. Shen, J. Mater. Chem. A, 2017, 5, 23158–23169 RSC.
- G. L. Hart, L. J. Nelson, R. R. Vanfleet, B. J. Campbell, M. H. Sluiter, J. H. Neethling, E. J. Olivier, S. Allies, C. I. Lang and B. Meredig, et al. , Acta Mater., 2017, 124, 325–332 CrossRef CAS.
- M. H. Sluiter, C. Colinet and A. Pasturel, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 174204 CrossRef.
- S. Fu, C. Zhu, J. Song, P. Zhang, M. H. Engelhard, H. Xia, D. Du and Y. Lin, Nanoscale, 2017, 9, 1279–1284 RSC.
- D. Qazzazie, O. Yurchenko, S. Urban, J. Kieninger and G. Urban, Nanoscale, 2017, 9, 6436–6447 RSC.
- M. Zhu, M. T. Nguyen, Y.-T. R. Chau, L. Deng and T. Yonezawa, Langmuir, 2021, 37, 6096–6105 CrossRef CAS PubMed.
- Z. Peng, H. You and H. Yang, ACS Nano, 2010, 4, 1501–1510 CrossRef CAS PubMed.
- Y.-T. Pan, Y. Yan, Y.-T. Shao, J.-M. Zuo and H. Yang, Nano Lett., 2016, 16, 6599–6603 CrossRef CAS PubMed.
- M. Zhou, C. Li and J. Fang, Chem. Rev., 2020, 121, 736–795 CrossRef PubMed.
- P.-C. Chen, M. Gao, C. A. McCandler, C. Song, J. Jin, Y. Yang, A. L. Maulana, K. A. Persson and P. Yang, Nat. Nanotechnol., 2024, 1–7 Search PubMed.
- S. Xiong, W. Qi, B. Huang and M. Wang, ChemPhysChem, 2011, 12, 1317–1324 CrossRef CAS PubMed.
- L. Vega, H. A. Aleksandrov, R. Farris, A. Bruix, F. Viñes and K. M. Neyman, Mater. Adv., 2021, 2, 6589–6602 RSC.
- J. Pirart, A. Front, D. Rapetti, C. Andreazza-Vignolle, P. Andreazza, C. Mottet and R. Ferrando, Nat. Commun., 2019, 10, 1982 CrossRef CAS PubMed.
- U. Aslam and S. Linic, Chem. Mater., 2016, 28, 8289–8295 CrossRef CAS.
- Z. Peng, H. You and H. Yang, Adv. Funct. Mater., 2010, 20, 3734–3741 CrossRef CAS.
- F. Mares-Briones, O. Barragán-Mares, J. López-Miranda, R. Esparza and G. Rosas, Mater. Res. Express, 2019, 6, 0850h8 CrossRef CAS.
- J. Onsgaard, P. J. Godowski and Z. Shen Li, J. Vac. Sci. Technol., A, 2012, 30, 01A111 CrossRef.
- S. Li, F. Tang, H. Wang, J. Feng and Z. Jin, RSC Adv., 2018, 8, 10237–10245 RSC.
- A. Y. Fedorov, A. Bukhtiyarov, M. Panafidin, I. Prosvirin, Y. Zubavichus and V. Bukhtiyarov, Appl. Surf. Sci., 2023, 157872 CrossRef CAS.
- P. Pandey, S. Kunwar and J. Lee, J. Alloys Compd., 2020, 813, 152193 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- C. Gao, Y. Hu, M. Wang, M. Chi and Y. Yin, J. Am. Chem. Soc., 2014, 136, 7474–7479 CrossRef CAS PubMed.
- C. Xue, X. Chen, S. J. Hurst and C. A. Mirkin, Adv. Mater., 2007, 19, 4071–4074 CrossRef CAS.
- M. Schwartzkopf, A. Rothkirch, N. Carstens, Q. Chen, T. Strunskus, F. C. Löhrer, S. Xia, C. Rosemann, L. Bießmann and V. Korstgens, et al. , ACS Appl. Nano Mater., 2022, 5, 3832–3842 CrossRef CAS.
- H. Jatav, M. Mičetic, A. Chakravorty, A. Mishra, M. Schwartzkopf, A. Chumakov, S. V. Roth and D. Kabiraj, Nanoscale, 2023, 15, 12025–12037 RSC.
- H. Jatav, M. Shabaninezhad, M. Micetic, A. Chakravorty, A. Mishra, M. Schwartzkopf, A. Chumakov, S. V. Roth and D. Kabiraj, Langmuir, 2022, 38, 11983–11993 CrossRef CAS PubMed.
- H. Jatav, A. Mishra and D. Kabiraj, Mater. Today: Proc., 2022, 57, 234–238 CAS.
- J. K. Kang and C. B. Musgrave, J. Chem. Phys., 2002, 116, 275–280 CrossRef CAS.
- A. Grau-Carbonell, S. Sadighikia, T. A. Welling, R. J. van Dijk-Moes, R. Kotni, M. Bransen, A. Van Blaaderen and M. A. Van Huis, ACS Appl. Nano Mater., 2021, 4, 1136–1148 CrossRef CAS PubMed.
- U. Jeong, J. B. Joo and Y. Kim, RSC Adv., 2015, 5, 55608–55618 RSC.
- B. Agrawalla, B. Dai and S. Allen, J. Vac. Sci. Technol., B: Microelectron. Process. Phenom., 1987, 5, 601–605 CrossRef CAS.
- J. Cheng, S. J. Rathi, P. Stradins, G. L. Frey, R. T. Collins and S. K. R. Williams, RSC Adv., 2014, 4, 7627–7633 RSC.
- S. Alberti, S. Schmidt, S. Hageneder, P. C. Angelomé, G. J. Soler-lllia, P. Vana, J. Dostalek, O. Azzaroni and W. Knoll, Mater. Chem. Front., 2023, 7, 4142–4151 RSC.
- N. Sui, K. Wang, X. Shan, Q. Bai, L. Wang, H. Xiao, M. Liu, V. L. Colvin and W. Y. William, Dalton Trans., 2017, 46, 15541–15548 RSC.
- F. Mares-Briones, A. Higareda, J. L. Lopez-Miranda, R. Mendoza-Cruz and R. Esparza, Nanomaterials, 2023, 13, 1396 CrossRef CAS PubMed.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli and P. Müller-Buschbaum, et al. , J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- J. M. Rahm and P. Erhart, J. Phys. Chem. C, 2018, 122, 28439–28445 CrossRef CAS.
- I. H. Malitson, J. Opt. Soc. Am., 1965, 55, 1205–1209 CrossRef CAS.
- M. Sui, S. Kunwar, P. Pandey and J. Lee, Sci. Rep., 2019, 9, 16582 CrossRef PubMed.
- A. L. Gould, S. Kadkhodazadeh, J. B. Wagner, C. R. A. Catlow, A. J. Logsdail and M. Di Vece, J. Phys. Chem. C, 2015, 119, 23767–23773 CrossRef CAS.
- A. Buffet, A. Rothkirch, R. Döhrmann, V. Körstgens, M. M. Abul Kashem, J. Perlich, G. Herzog, M. Schwartzkopf, R. Gehrke and P. Müller-Buschbaum, et al. , J. Synchrotron Radiat., 2012, 19, 647–653 CrossRef CAS PubMed.
- M. Schwartzkopf, A. Buffet, V. Körstgens, E. Metwalli, K. Schlage, G. Benecke, J. Perlich, M. Rawolle, A. Rothkirch and B. Heidmann, et al. , Nanoscale, 2013, 5, 5053–5062 RSC.
- H. Ananda Murthy, T. Desalegn Zeleke, C. Ravikumar, M. Anil Kumar and H. Nagaswarupa, Mater. Res. Express, 2020, 7, 055016 CrossRef.
- R. Ferrando, Nanoalloys, Elsevier, 2020, pp. 115–149 Search PubMed.
- C. Gao, F. Lyu and Y. Yin, Chem. Rev., 2020, 121, 834–881 CrossRef PubMed.
- M. Ohring, Materials Science of Thin Films: Deposition and Structure, Elsevier, 2001 Search PubMed.
- G. Santoro, S. Yu, M. Schwartzkopf, P. Zhang, S. Koyiloth Vayalil, J. F. Risch, M. A. Rübhausen, M. Hernández, C. Domingo and S. V. Roth, Appl. Phys. Lett., 2014, 104, 243107 CrossRef.
- N. T. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2014, 114, 7610–7630 CrossRef CAS PubMed.
- T.-W. Liao, A. Yadav, K.-J. Hu, J. van der Tol, S. Cosentino, F. D'Acapito, R. E. Palmer, C. Lenardi, R. Ferrando and D. Grandjean, et al. , Nanoscale, 2018, 10, 6684–6694 RSC.
- R. Watson, J. Hudis and M. Perlman, Phy. Rev. B, 1971, 4, 4139 CrossRef.
- W. Dachraoui, M. I. Bodnarchuk and R. Erni, ACS Nano, 2022, 16, 14198–14209 CrossRef CAS PubMed.
- D. Oucheriah, C. Mottet and H. Amara, et al. , Faraday Discuss., 2023, 242, 144–159 RSC.
- E. Toulkeridou, J. Kioseoglou and P. Grammatikopoulos, Nanoscale Adv., 2022, 4, 4819–4828 RSC.
- M. Turchanin and P. Agraval, Powder Metall. Met. Ceram., 2008, 47, 26–39 CrossRef CAS.
- I. Tanahashi, M. Yoshida, Y. Manabe and T. Tohda, J. Mater. Res., 1995, 10, 362–365 CrossRef CAS.
- G. Liu, K. Yang, J. Li, W. Tang, J. Xu, H. Liu, R. Yue and Y. Chen, J. Phys. Chem. C, 2014, 118, 22719–22729 CrossRef CAS.
- H. Liu, U. Pal and J. Ascencio, J. Phys. Chem. C, 2008, 112, 19173–19177 CrossRef CAS.
- C. Mottet, et al. , J. Phys.: Condens. Matter, 2021, 33, 154006 CrossRef PubMed.
- M. Chi, C. Wang, Y. Lei, G. Wang, D. Li, K. L. More, A. Lupini, L. F. Allard, N. M. Markovic and V. R. Stamenkovic, Nat. Commun., 2015, 6, 8925 CrossRef CAS PubMed.
- O. A. Yeshchenko, I. M. Dmitruk, A. A. Alexeenko and A. M. Dmytruk, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 085434 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nh00509k |
| This journal is © The Royal Society of Chemistry 2025 |