
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural study of Lindqvist polyoxovanadate–peptide conjugates†‡
H.
Yu
a,
C.
Honisch
ab,
M.
Frigo
a,
N.
Balice
a,
S.
Mantovani
a,
F.
Cammelli
 a,
G.
Saielli
ac,
V.
Cocetta
d,
A.
Sacilotto
d,
P.
Ruzza
*b and
M.
Carraro
*ac
a,
G.
Saielli
ac,
V.
Cocetta
d,
A.
Sacilotto
d,
P.
Ruzza
*b and
M.
Carraro
*ac
aDepartment of Chemical Science, University of Padova, Via F. Marzolo 1, Padova, Italy
bInstitute of Biomolecular Chemistry of CNR (ICB-CNR), Padova Unit, Via F. Marzolo 1, Padova, Italy. E-mail: paolo.ruzza@unipd.it
cInstitute on Membrane Technology of CNR (ITM-CNR), Padova Unit, Via F. Marzolo 1, Padova, Italy
dDepartment of Pharmaceutical and Pharmacological Sciences, University of Padova, Via Marzolo 5, Padova, Italy. E-mail: mauro.carraro@unipd.it
First published on 27th January 2025
Abstract
Polyoxometalates (POMs) are inorganic molecular clusters that exhibit significant biological activity against various pathologies, including cancer. Despite their therapeutic potential, the clinical application of POMs is limited by challenges such as low selectivity and toxicity. To overcome these limitations, conjugation with targeting moieties such as biomolecules and peptides has emerged as a promising strategy to enhance their selectivity. Within this context, the use of spacers can reduce intramolecular interactions between the POM surface and targeting molecules, thereby maintaining their accessibility for biological recognition. In this study, the Lindqvist hexavanadate cluster is functionalized with the antagonist peptide demobesin-1 (DB), with or without spacers, to evaluate the impact of this modification on peptide conformation and biological activity. Our findings provide valuable insights into the optimization of POM-based therapeutic agents with improved selectivity and reduced toxicity.
1 Introduction
Polyoxometalates (POMs) are metal oxide clusters formed by transition metals like molybdenum (Mo), tungsten (W), or vanadium (V), coordinated by oxygen.1,2 Their structural diversity3 makes them appealing for catalytic applications4 and for the creation of new materials and nanostructures5 with interesting electronic6 and magnetic properties.7Additionally, POMs exhibit biological activity in various therapeutic contexts:8 as anticancer,9,10 antiviral,11 and antibacterial12 agents. Their remarkable properties, including acidity, charge and redox activity, facilitate the interactions with biological macromolecules such as proteins, enzymes, and peptides. This interplay often results in degradation, hydrolysis, or inactivation of the targeted macromolecules.13–15 However, the low selectivity and stability of POMs limit their broader application. Strategies such as conjugation with organic moieties1 or encapsulation may improve bioavailability and stability, opening doors for more effective therapeutic uses.9,16 Hybrid POMs for targeting cancer cells are particularly promising, although examples remain limited.17
The Anderson–Evans polyoxomolybdate (POMo), with the inorganic core {MnMo6O24}, was previously functionalized with demobesin-1 (DB).18 DB (with sequence H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Leu-NHEt) is a peptide derived from bombesin. DB, as bombesin, is an antagonist for gastrin-releasing peptide receptors (GRPR), which are frequently overexpressed in cancers such as breast, prostate, lungs and pancreas.19 As a matter of fact, bombesin-related peptides have shown significant potential in cancer imaging and therapy. Specifically, inhibiting the interaction of gastrin-releasing peptide with GRPR may disrupt cancer growth, offering a promising therapeutic strategy.20
However, the POMo–DB conjugate did not exhibit enhanced efficacy compared to peptide-free POM. This limitation is likely due to peptide folding and the hybrid's propensity to aggregate into nanoparticles.18 Spacers such as Ttds (4,7,10-trioxa-1,13-tridecanediamine) and Glu-Glu-Glu-Glu-β-Ala (EEEEβA) showed promise in addressing these challenges by reducing aggregation and minimizing interference between active domains.21,22 Despite these enhancements, the anticancer activity observed in vitro remains insufficient for practical applications.
In this work, we have explored a different approach, by replacing Anderson–Evans polyoxomolybdate with the Lindqvist-type {V6O19} polyoxovanadate (POV). Both POMo and POV share the same number of addenda metals (Mo or V) and functionalization strategies, such as the introduction of triols as the tris(hydroxymethyl)aminomethane (TRIS),1,22,23 but they are characterized by different shape and charge.
POVs have demonstrated significant potential in biology and medicine.9,24 For instance, Na4[Co(H2O)6V10O28]·18H2O ({CoV10}) exhibited a remarkable activity against the human cancer cell lines SMMC-7721 (liver cancer) and SK-OV-3 (ovary cancer).25 K12[V18O42(H2O)]·6H2O ({V18}) was found to inhibit the proliferation of breast cancer cell lines, in a dose-dependent manner, even better than the positive control drug 5-fluorouracil (5-FU).26 Similarly, the decavanadate complex Na4{(HOCH2CH2)3NH2[V10O28]}·6H2O exhibited antitumor activity against several tumor cells, including human laryngeal carcinoma epithelial cell line (Hep-2), human breast cancer cell line (MDA-MB-231) and hepatocellular liver carcinoma cell line (HepG2), which was better than 5-FU.27 A self-assembled nanocage (termed VMOP-31), and resulting from the assembly of six {V5O9Cl} units was described as a promising candidate for chemotherapy of cancer with high efficacy and low toxicity.28 VMOP-31 was highly effective in inhibiting the cell growth of different solid tumors. Furthermore, compared with cis-platin, VMOP-31 had negligible side effects. POVs were also covalently bonded to amino acid esters as alanine29 or glycine,30 demonstrating higher inhibitory activity against the proliferation of human laryngeal carcinoma epithelial (Hep-2), rhabdomyosarcoma (RD), and breast adenocarcinoma (MCF-7) cell lines, compared to standard treatments. In particular, the Lindqvist POV functionalized with β-alanine ethyl ester, [Bu4N]2[V6O13{(OCH2)3CCH2OOCCH2CH2CONHCH2CH2COOCH2CH3}2] exhibited the strongest inhibitory effects against Hep-2 and MCF-7 cells, with IC50 values of 11.40 and 53.01 μM respectively.29
Herein, we describe the fabrication of POV–DB conjugates that exploit our previously reported spacer strategy.21,22 Specifically, to evaluate how different POM scaffolds may influence DB's secondary structure and accessibility, the Lindqvist hexavanadate was bis-functionalized with DB units featuring distinct spacers (Scheme 1). The novel hybrids were characterized using 2D NMR and CD spectroscopy to investigate the behavior of the peptides when grafted on the title POV. Our findings aim to provide further insights into the design of more selective POM–peptide hybrid nanodrugs.31
 | ||
| Scheme 1 Reaction scheme to obtain the hybrid derivatives starting from V6-OH (TBA2[V6O13{(OCH2)3CH2OH}2]). For clarity reasons, counter cations (TBA) are omitted. | ||
2 Results and discussion
2.1 Differences between the hexametalated polyoxoanions
With respect to Anderson–Evans molybdates, Lindqvist vanadates display different topology and charge. These POVs are, indeed, characterized by compact architecture with most metals exposed on the surface, high symmetry, low charge and high stability.32,33 Owing to such properties, they may be suitable compounds for a comparative analysis, in order to evaluate the impact of POM structure and charge on the peptide structure. Fig. 1 depicts the electrostatic potential, mapped on an electronic isosurface of the POM precursors (obtained from relativistic density functional theory calculations, see the experimental details Section), i.e. the two hexametalates bis-functionalized with TRIS molecules, and highlights the notable differences in terms of surface electron density. The lower surface charge of the POV is witnessed by the overall lower negative charge (−2), which is compensated by only two cations, while the POMo bears three counterions. Additional differences arise from the lack of heteroatoms’ electrons (Mn3+) in the POV. | ||
| Fig. 1 DFT calculated electrostatic potential mapped on an electronic isosurface of 0.01 density for the two POM precursors: (a) [V6O13{(OCH2)3CNH2}2]2− and (b) [MnMo6O18{(OCH2)3CNH2}2]3−. Red and blue regions indicate negative and positive potential, respectively. | ||
All these features may have an impact on the ability of the surface to interact with the appended, flexible, peptide chains, particularly in terms of electrostatic interactions and steric hindrance of the counterions.
2.2 Synthesis of the POV bio-conjugates and characterization
The POV–peptide hybrids prepared in this work are presented in Scheme 1. The EEEEβA sequence, consisting of four glutamate residues was used to provide repulsive interactions with the negatively charged POM surface.21,22 The flexible hydrophilic linker Ttds was introduced in the system to increase water solubility and bioavailability.Peptide synthesis was performed by manual solid-phase peptide synthesis (SPPS, see ESI,‡ Fig. S1–S12), combining previously reported procedures.18,21,22 In this way, DB (Pep-1) was converted to Pep-2 (with Ttds only), Pep-3 (with EEEEβA only) and Pep-4 (with both spacing units). To prepare the hybrid POMs, the initial step was the synthesis of the (OH)-functionalized polyoxovanadate (V6-OH, Scheme 1 and Fig. S13, S14, ESI‡), obtained starting from NaVO3, reacted with a stoichiometric amount of HCl and pentaerythritol (HOCH2)3CCH2OH, in the presence of nBu4NBr (tetrabutyl ammonium, TBA). With respect to TRIS, the use of pentaerythritol is convenient to avoid the formation of zwitterionic species resulting from amine protonation. The following steps allowed the functionalization of the free hydroxymethylene groups with succinic anhydride, to generate the corresponding by-succinate derivative (V6-succ, Scheme 1 and Fig. S15, S16, ESI‡), while the reaction with N-hydroxysuccinimide (NHS), in the presence of dicyclohexylcarbodiimide (DCC), yielded the activated species V6-NHS (Scheme 1 and Fig. S17–S19, ESI‡).29,30 A L-phenylalanine functionalized POV (V6-Phe) was initially prepared to test and optimize the derivatization of V6-NHS, and to get a further hybrid to compare with the POV–peptide conjugates.
This last reaction was performed with L-PheOMe·HCl and diisopropylethylamine (DIPEA), to give, after precipitation with diethyl ether, the desired product with 85% yield (Fig. S20–S23, ESI‡).
Afterward, the peptides with or without spacers, were combined with the NHS-activated POVs to yield POV-1, 2, 3, 4 (Scheme 1). Each synthetic step was monitored by FT-IR, ESI-MS, 1H- and 51V-NMR (Fig. S24–S45, ESI‡). 51V-NMR confirms the integrity of the inorganic core, showing only one broad signal at ∼−495 ppm for all compounds, in agreement with the occurrence of highly symmetrical, anti-Lindqvist hybrid isomers. ESI-MS(−) analysis was used to assess the successful grafting of two peptide chains on the POV. While the POV-1, 2, 3 mostly showed the di-charged anions, POV-4 displayed an extensive deprotonation, leading to a complex with more negative charges.
As depicted in Fig. 2, the FT-IR (KBr) spectra of POV–NHS and POV-spacer(s)-peptide exhibit no structural alterations in the region characteristic of the polyoxometalate framework (between 1000 cm−1 and 500 cm−1). Moreover, in POV-1, 2, 3, 4, the presence of a moderately intense band at 1534 cm−1 suggests the presence of –N–H bending, while signals related to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group of the –NHS (at 1783 and 1814 cm−1) are replaced by signals indicative of peptide bond stretching, around 1640 cm−1, confirming the successful addition of the peptide to the POV.
O group of the –NHS (at 1783 and 1814 cm−1) are replaced by signals indicative of peptide bond stretching, around 1640 cm−1, confirming the successful addition of the peptide to the POV.
 | ||
| Fig. 2 FTIR (KBr) spectra of V6-NHS (black), POV-1 (red), POV-2 (blue), POV-3 (green), POV-4 (purple), the orange dash-dot lines indicate the position of the characteristic peaks at 1814 cm−1, 1783 cm−1, 1640 cm−1 and 1534 cm−1 (see text). | ||
2.3 Antitumor activity
To assess whether the conjugates retain their cytotoxic activity, the compounds were tested on the A2780 ovarian cancer cell line, a widely accepted and standardized cell line in cancer research. The precursor V6-OH and the hybrid V6-Phe were used as reference compounds.Cell viability was measured using the Crystal Violet assay, following 24 hours of treatment with increasing concentrations of the compounds (ranging from 1 to 100 μM).
The results, presented in Fig. 3 and Table 1, which collects the IC50 values, demonstrate that the newly synthesized compounds exhibit activity in the same order of magnitude of previously reported POVs (with IC50 mostly in the range 1–50 μM for 24 h incubation),26,29,34–37 and very similar to that of the reference POVs V6-OH and V6-Phe.
 | ||
| Fig. 3 Viability tests on A2780 ovarian cancer cell line observed after 24 h. **p < 0.01, ****p < 0.0001 treatment vs. control. | ||
| # | Compound | IC50 (μM) |
|---|---|---|
| 1 | V6-OH | 42.3 ± 9.4 |
| 2 | V6-Phe | 37.1 ± 1.6 |
| 3 | POV-1 | 36.2 ± 16.9 |
| 4 | POV-2 | 43.2 ± 4.5 |
| 5 | POV-3 | 43.3 ± 5.5 |
| 6 | POV-4 | 55.7 ± 7.8 |
However, despite a favorable general retention of activity, no improvement was observed when the peptides were grafted on the POM and, surprisingly, the POV-4 was the less effective.
Due to these results, no additional cytotoxicity test was performed. The lack of peptide recognition by cancer cells indicates, indeed, that further structural refinement is necessary before conducting a comprehensive screening.38
The missing expected improvement has previously been attributed to undesired peptide folding. In the Anderson–Evans POM–peptide conjugates,18,39 indeed, the POM was responsible for promoting an evolution of the secondary structure towards α-helix, rendering it unavailable for receptor recognition.
In the following paragraphs, we will explore the structural changes of the peptide chains when conjugated to the Lindqvist POV, aiming to unveil a general POM-dependent behavior concerning peptide folding.
2.4 2D NMR characterization
The impact of the spacers on the POV–peptide interactions was evaluated by examining the secondary structure of the peptide constructs using 2D 1H NMR and circular dichroism (CD).NMR spectra (COSY, TOCSY and ROESY) were acquired in d6-DMSO. Through 2D homonuclear NMR experiments, the bis-functionalization with C2 symmetry and the sequential resonance assignment of both peptides and POV conjugates were assessed. For example, it is noticeable the presence of signals ascribed to β-Ala, NH-Ttds (amide bond between Ttds and DB) and NH-POV (amide bond between V6-succ and the peptides), together with the intense peak of the terminal NHEt, for POV-4 (see ESI‡).
In order to detect meaningful changes of peptide environment, the overlap of COSY NMR spectra in the NH amide and CHα resonance region for compounds Pep-1, POV-1, and POV-4 is reported in Fig. 4. Notably, the cross-peaks of POV-1 experience a more significant upfield shift, especially for Gln, Val and Ala residues, while the shift of POV-4 signals is less pronounced. As a comparison, COSY spectra of POV-2 and POV-3 are displayed in Fig. S46 (ESI‡), demonstrating intermediate behavior for the single-spacer hybrids.
 | ||
| Fig. 4 Overlaid COSY spectra (in d6-DMSO) for NH-CHα cross peaks belonging to free and V6-grafted DB (Pep-1 (blue), POV-1 (green) and POV-4 (red)). The spectra were acquired on a Bruker DMX-600 instrument operating at 599.90 MHz for 1H at 298 K. | ||
The COSY analysis confirms the expected behavior of the spacers in mitigating the effect of POV on altering the peptide environment. With a focus on the peptides, secondary chemical shift values were calculated as differences in the CHα chemical shift between DB and the three corresponding spacers-DB (Δδ in Fig. 5(A)).
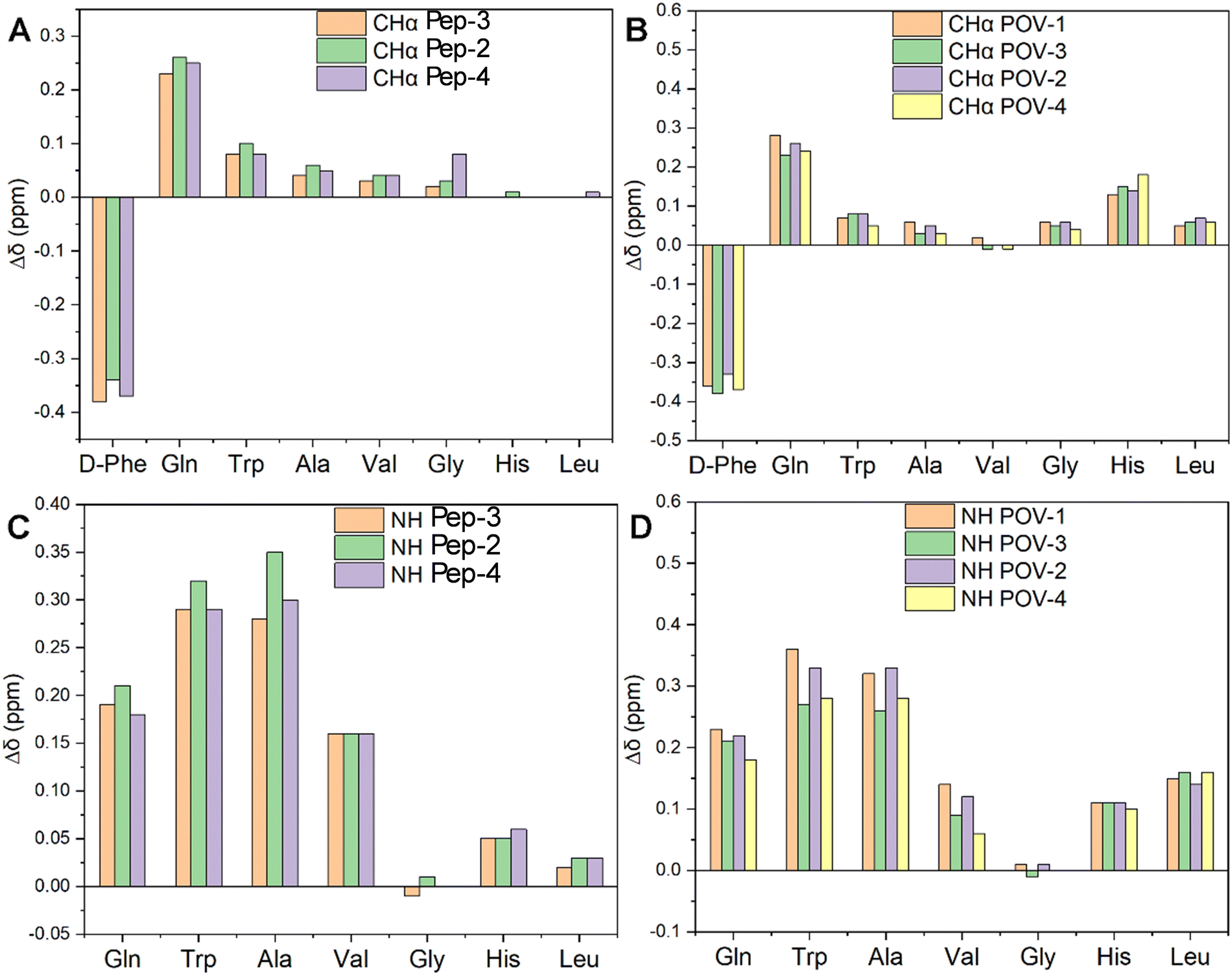 | ||
| Fig. 5 Influence of the POV-spacers on chemical shift of peptide. Δδ values were determined by subtracting the resonances of DB-spacer or POV-spacer-DB hybrids to the resonances of DB. (A) and (C) are Δδ of DB-spacers, (B) and (D) are Δδ of POV-spacers-DB. | ||
The resonance of CHα in the D-Phe residue displays a downfield shift due to the presence of the bond with the spacer, yielding a new amide group near CHα in Pep-2, Pep-3, Pep-4. Moreover, all spacers foster shielding effects on the other residues, becoming more evident in the N-terminal region of the peptides, i.e. closer to the spacers, with a gradual decrease towards the free C-terminal region.
When considering the corresponding hybrid POVs, CHα resonances of all amino acids (including terminal Leu and His) undergo an upfield shift (Fig. 5(B)). This behavior may suggest a rearrangement of the peptide, likely positioning the terminal amino acid in closer contact to the POV core, owing to a bending of the middle region of the peptide.
In Fig. 5(C), the NH resonance values of spacers-DB constructs (Pep-2, 3 and 4) were subtracted from those of DB (Pep-1). The resonance of NH in those residues are also upfield shifted, with a minimum effect on the Gly residue. The impact of POV in increasing the chemical shift difference (Δδ) with respect to the free peptide is indeed visible for all residues, except Gly (NH, Fig. 5(D)). POV-3 and POV-4 (both with the negatively charged spacer) exhibit a moderately lower shift for the residues before Gly (N-terminal region). Noteworthy, diagnostic CHα, N (i > i + 1) cross peaks between CHα (δ = 1.2 ppm) for Ala and NH (δ = 8.2 ppm for Gly, 8.3 ppm for Phe, separately) suggest the presence of an α-helix conformation for POV-1 (see TOCSY in the Fig. S27, ESI‡). Cross peaks also were observed between CH3 (δ = 0.86 ppm) for Leu and NH (δ = 8.2 ppm) for Gly.
2.5 CD spectroscopy
CD spectroscopy was used to evaluate the secondary structure of both peptides and POV hybrids (Fig. S47 and S48, ESI‡).The dichroic signals were monitored in the presence of a variable amount of 2,2,2-trifluoroethanol (TFE) in water, from 10% to 80 or 100% (v/v). The relatively lower polarity of TFE allows to mimic the behavior of the peptide in a less polar environment, so to provide useful information on the possible conformations of the peptide when interacting with the cell membrane. Fig. 6, in particular, shows two representative TFE concentrations (10 and 80% v/v).
 | ||
| Fig. 6 Far-UV CD spectra of Pep-4 (12.5 μM, dot-dashed lines) and POV-4 (12.5 μM, solid lines) at different TFE percentage. | ||
Herein, POV-4 was compared with its parent peptide Pep-4. While the bands of the peptide are preserved in POV-4, an increased intensity is generally observed, as a result of the double amount of peptide in the hybrid and of chirality induction by the peptide on the POV chromophore.40 Moreover, at 80%, the signal for POV-4 becomes much stronger, showing a great tendency of the appended peptide to rearrange its secondary structure. For a more thorough comprehension of the distinctions in circular dichroism (CD) spectra, additional analysis was conducted using CD Apps software.41 The histograms presented in Fig. 7 delineate the distribution of secondary structures of the investigated compounds across varying percentages of TFE. As its concentration increases, the structural tendency leans towards an α-helix, whereas in an aqueous environment, the peptide demonstrates a higher inclination for a β-sheet structure. Notably, POV-4 exhibits the highest sensitivity to TFE, already between 10% and 20% TFE, where a strong tendency towards the formation of α-helix folding can be observed, as a result of the combined effects of POM and TFE, both acting as stabilizing agents for the α-helical conformation.
 | ||
| Fig. 7 Percentage of α-helix (blue), β-strand (orange), turns (grey) and random coil (yellow) for peptides and POV-hybrids in 10%, 20%, 40%, 60%, 80% and 100% TFE solution. | ||
2.6 Transmission electron microscopy (TEM)
The molecular conformation is also expected to affect intermolecular interactions, leading to self-assembly and aggregation of the hybrid POVs. Transmission electron microscopy (TEM) has been used to monitor the morphology of the aggregates. Specifically, Fig. 8(A) illustrates that the POV-1 forms amorphous clusters. Similarly, Fig. 8(B) depicts the clustering of POV-3, with high polydispersity of aggregates (both small particles and large aggregates). Fig. 8(C) displays that POV-2 forms entangled fibers, likely controlled by the assembly of longer and solvated Ttds chains. Eventually, as shown in Fig. 8(D), POV-4 forms nano-sized particles, whose size is around 2–3 nm and which are uniformly distributed, without any further agglomeration into bigger structures or fibers. V6-OH is also scarcely aggregated, while V6-Phe forms flakes with length up to 200 nm (Fig. S49, ESI‡). | ||
| Fig. 8 TEM micrographs, 10−4 M solutions of (A) compound POV-1, (B) POV-3 (C) POV-2 (D) POV-4 in DMSO/water. | ||
3 Experimental
3.1 Material and methods
Fmoc-amino acids, hexafluorophosphate azabenzotriazole tetramethyl uronium (HATU), hydroxybenzotriazole (HOBt), hexafluorophosphate benzotriazole tetramethyl uronium (HBTU), trifluoroacetic acid, Fmoc-Ttds-OH, piperidine, Fmoc-ethyl-indole AM resin, H-Asp(OtBu)-2-Cl-Trt resin and N,N-diisopropylethylamine (DIPEA) were obtained from Iris Biotech. succinic anhydride, N,N′-dicyclohexylcarbodiimide (DCC), tris(hydroxymethyl)aminomethane (TRIS), N-hydroxysuccinimide (NHS), ethanol, acetonitrile (ACN), diethyl ether (Et2O), trifluoroacetic acid (TFA), piperidine, dimethylformamide (DMF), dichloromethane (DCM), triisopropylsilane (TIPS) were purchased from Sigma-Aldrich and used without any further treatment.HPLC-grade water was obtained through filtration using an Elga Veolia PURELAB system. The scales employed included a Mettler AE 240 scale and a Mettler ΔT21 Comparator scale.
3.2 Instruments
Circular Dichroism measurements were performed at 25 °C using a nitrogen flushed Jasco J-1500 spectropolarimeter (Jasco, Easton, MD, USA) equipped with a thermostated cell holder. The spectral window was 260–190 nm in a 0.5 cm quartz cuvette, with 0.2 nm data pitch and 16 accumulations at 50 nm min−1 scanning speed. The solutions used were 25 μM (peptide), 12.5 μM (POM-hybrids), separately, at 25 °C. Analysis of CD spectra were carried out using Spectra Manager software supplied by Jasco. The secondary structure estimation was performed using the CD Apps software41 with the CONTINLL algorithm and SP43 database (190–240 nm), estimating α-helix type 1 and α-helix type 2, β-strand type 1 and β-strand type 2, turns, unordered structures.NMR spectra were recorded on a Bruker Avance DMX 600 MHz (Bruker Corp., Billerica, MA, USA), operating at 599.90 MHz for 1H, at 298 K. Bruker 300 MHz UltraShield was used for monodimensional 1H NMR and 51V NMR (with the instrument operating at 78.9 MHz). Deuterated DMSO and acetonitrile were used as solvents for analysis. Analysis of NMR spectra was performed on MestReNova and TopSpin softwares.
FT-IR measurements were performed on a Jasco FT/IR-4100 spectrometer, by preparing KBr pellets. The spectra were analyzed by the Jasco Spectra Manager 2.00.07 software.
UV-vis measurements were carried out using a Varian Cary® 50 (Agilent) with a 1 cm path quartz cuvette. The data were processed by the Cary WinUV Application Software.
Preparative HPLC system for peptide purification (Shimadzu, Tokyo, Japan) was equipped with LC-8A pumps, SCL-8A controller and SPD-6A spectrophotometric detector, using a linear gradient (eluent A: 0.05% TFA in H2O; eluent B: 0.05% TFA in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v CH3CN–H2O; 20–45% B in 35 minutes, with 12 mL min−1 flow), and a Vydac C18 column (300 Å, 10 μm, 250 × 22 mm).
:1 v/v CH3CN–H2O; 20–45% B in 35 minutes, with 12 mL min−1 flow), and a Vydac C18 column (300 Å, 10 μm, 250 × 22 mm).
TEM measurements were obtained with a FEI Tecnai G2 transmission electron microscope (Thermo Fisher Scientific, Waltham, MA, USA), operating at an excitation voltage of 100 kV.
Mass spectrometry measurements were carried out using a LC/MSD Trap SL instrument equipped with an electrospray (ESI) source, supplied by Agilent Technologies. The hybrids, dissolved in acetonitrile, were injected by a HPLC 1100 series pump, and analyzed in negative or positive ion mode. LC-ESI-MS(+) analyses of the peptides were performed using an Agilent Technologies 1260 Infinity II system equipped with a 6130 Quadrupole LC/MS analyzer and an ESI ionization system. The column was a Kinetex 3.5 μm XB-C18 100 Å eluted with a linear binary gradient 5–95% B in 30 min (solvent A was water with 10% CH3CN + 0.1% TFA, and B was 90% CH3CN, 10% H2O + 0.1% TFA).
3.3 Cytotoxicity
Cytotoxicity tests were performed on A2780 human ovarian carcinoma cell line (Sigma-Aldrich) grown in RPMI-1640 (Corning) supplemented with 2 mM L-glutamine and 10% fetal bovine serum (Gibco). Cells were seeded in appropriate number in 96-well plates and, following overnight incubation, treated with compounds at 1–5–10–25–50–100 μM for 24 hours. After treatments, cells were fixed with 4% paraformaldehyde solution, rinsed with PBS and then stained with Crystal Violet solution 0.1%. After wash with water, cells were resuspended in acetic acid 1% in water. The absorbance was measured at 590 nm using a Victor3X multilabel plate counter (Wallac Instruments, Turku, Finland). The IC50 (half maximal inhibitory concentration) was calculated by ED50plus V1.0.3.4 The synthesis of peptides
Peptides 1, 2, 3, and 4 were synthesized using 200 mg of Fmoc-ethyl-indole AM resin (0.71 mmol g−1, 0.14 mmol) for each, employing solid-phase peptide synthesis (SPPS) following a traditional procedure.42 The initial amino acid, Leu, was introduced via double coupling using HATU (3 equiv.) as an activator, with subsequent coupling reactions utilizing HOBt (3 equiv.) and HBTU (3 equiv.). At each step, 6 equiv. of DIPEA were employed to deprotonate the carboxylic group. Peptide detachment from the resin occurred through treatment with TFA, followed by C-18 RP-HPLC purification using a linear binary gradient from 20% to 45% of eluent B over 35 minutes (eluent A: 0.05% TFA in H2O; eluent B: 0.05% TFA in 9:1 v/v CH3CN–H2O).
3.5 The synthesis of hybrids
Hybrids POV-1, POV-2, POV-3 and POV-4 were synthesized, starting from POV–NHS. 1 equiv. of POV–NHS (20 mg, 0.0089 mmol) and 2.1–2.3 equiv. of peptides (Pep-1, Pep-2, Pep-3 or Pep-4) were dissolved in 1.2 mL of DMF in presence of 15 equiv. of DIPEA (18 mg, 24 μL, 0.13 mmol). The solution was stirred for 24 hours at room temperature. ESI-MS(−) analysis confirmed the presence of hybrids. Crystallization was achieved under Et2O atmosphere overnight, and the obtained solid was dried under vacuum.3.6 DFT calculations
Relativistic DFT calculations were run using the software package ADF 2019.43 The level of theory used for the single point calculations was relativistic ZORA scalar unrestricted PBE0/TZ2P (all-electron). Geometry was taken from the X-ray structures of analogue compounds.44,45 The electrostatic potential was mapped on to the isoelectronic density using the software ADFview.46 For the MnMo cluster we considered two possible multiplicities due to the presence of an Mn3+ ion having electronic configuration [Ar]3d4, that is a high spin system with 4 unpaired electrons and a low-spin system with 2 unpaired electrons. The triplet system resulted 9.8 kcal mol−1 higher in energy than the quintet. Therefore, we have used the quintet electronic configuration for the calculation of the electrostatic potential in Fig. 1. We noted, however, that the electrostatic potential at the outer surface of the MnMo cluster is essentially unaffected by the different spin-density distributions at the internal Mn atom.4 Conclusions
The demobesin-1 peptide was used to decorate the hexavanadate by applying a slightly modified literature procedure, previously used for Anderson–Evans polyoxometalates. Since DB can be useful for cancer cell targeting, it is interesting to monitor its availability when conjugated with the POMs. To this aim, different spacers have been introduced to separate the polyanion and the peptide, and their effects in terms of peptide folding and aggregation have been evaluated. Our results show that the combined use of Ttds and Glu-Glu-Glu-Glu-β-Ala spacers spring in a synergistic ability to keep the peptide in a state like that of the starting peptide, as observed from 2D NMR, and in a less aggregated form, as seen from TEM. Concerning the secondary structure, however, the evolution to α-helix is still strongly favored, especially in a less polar medium, for all POV derivatives, with a response even more evident than that observed for the corresponding hybrids based on Anderson–Evans molybdates. As a result, the peptide becomes unavailable for recognition and the biological activity is the same as the peptide-free POV.Reducing the overall charge of the POM appears thus not relevant for decreasing the impact of the POM on the peptide. In going from {MnMo6O24} to {V6O19}, indeed, the available POM surface increases, accompanied by a concurrent reduction in the number of hindered counterions. This enhances the likelihood of interactions with the POM surface, favoring the conformational evolution, even in the presence of both spacers.
In conclusion, novel hybrid POVs have been prepared with straightforward methodologies and characterized. All compounds display significant cytotoxicity against selected cancer cells, demonstrating that the hybridization of the Lindqvist type polyoxovanadate does not decrease their overall activity. Despite the lack of selectivity, these data may drive the synthesis of a broader family of bio-hybrid vanadates. Future experiments should consider, for example, the use of rigid peptides, unable to fold towards the POM surface.
Author contributions
Conceptualization: P. R., M. C.; data curation: H. Y.; N. B., M. F., F. C.; formal analysis: H. Y.; N. B., M. F., S. M., F. C., C. H.; funding acquisition, project administration: P. R., M. C.; investigation: H. Y.; N. B., M. F., S. M., F. C., C. H., G. S., V. C., A. S.; methodology: G. S., P. R., M. C.; software: H. Y., M. F., N. B., G. S., C. H.; supervising: C. H., P. R., M. C.; validation, C. H., P. R., M. C.; visualization: H. Y., G. S.; writing – original draft preparation: H. Y., C. H., G. S., V. C; writing – review and editing: H. Y., C. H., P. R., M. C. All authors have read and agreed to the published version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by Next Generation EU-PRIN 2022-M4.C2.1.1, project: 2022CAS9ZT (HYMBAD). DFT calculations were run on the Linux clusters of the C3P community of the Department of Chemical Sciences of the University of Padova. H. Y. acknowledges the China Scholarship Council for supporting her PhD at Padova University. V. C. grant was supported by Fondazione Umberto Veronesi. We thank Laura Zanetti Polzi for useful discussion on data validation.References
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- A. S. Cherevan, S. P. Nandan, I. Roger, R. Liu, C. Streb and D. Eder, Adv. Sci., 2020, 7, 1903511 CrossRef CAS PubMed.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105–121 RSC.
- M. Samaniyan, M. Mirzaei, R. Khajavian, H. Eshtiagh-Hosseini and C. Streb, ACS Catal., 2019, 9, 10174–10191 CrossRef CAS.
- R. Sivakumar, J. Thomas and M. Yoon, J. Photochem. Photobiol., C, 2012, 13, 277–298 CrossRef CAS.
- X. Lopez, J. M. Maestre, C. Bo and J. M. Poblet, J. Am. Chem. Soc., 2001, 123, 9571–9576 CrossRef CAS PubMed.
- E. Coronado and C. J. Gomezgarcia, Comments Inorg. Chem., 1995, 17, 255–281 CrossRef CAS.
- M. B. Colovic, M. Lackovic, J. Lalatovic, A. S. Mougharbel, U. Kortz and D. Z. Krsti, Curr. Med. Chem., 2020, 27, 362–379 CrossRef CAS PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Angew. Chem., Int. Ed., 2019, 58, 2980–2999 CrossRef CAS PubMed.
- H. Yanagie, A. Ogata, S. Mitsui, T. Hisa, T. Yamase and M. Eriguchi, Biomed. Pharmacother., 2006, 60, 349–352 CrossRef CAS PubMed.
- B. Hasenknopf, Front. Biosci., 2005, 10, 275–287 CrossRef CAS PubMed.
- A. Bijelic, M. Aureliano and A. Rompel, Chem. Commun., 2018, 54, 1153–1169 RSC.
- M. Arefian, M. Mirzaei, H. Eshtiagh-Hosseini and A. Frontera, Dalton Trans., 2017, 46, 6812–6829 RSC.
- S. Lentink, D. E. Salazar Marcano, M. A. Moussawi and T. N. Parac-Vogt, Angew. Chem., Int. Ed., 2023, 62, e202303817 CrossRef PubMed.
- S. Fabbian, G. Giachin, M. Bellanda, C. Borgo, M. Ruzzene, G. Spuri, A. Campofelice, L. Veneziano, M. Bonchio, M. Carraro and R. Battistutta, Front. Mol. Biosci., 2022, 9, 906390 CrossRef CAS PubMed.
- D. E. Salazar Marcano and T. N. Parac-Vogt, Coord. Chem. Rev., 2024, 518, 216086 CrossRef CAS.
- H.-K. Yang, Y.-X. Cheng, M.-M. Su, Y. Xiao, M.-B. Hu, W. Wang and Q. Wang, Bioorg. Med. Chem. Lett., 2013, 23, 1462–1466 CrossRef CAS PubMed.
- D. Ventura, A. Calderan, C. Honisch, S. Krol, S. Serratì, M. Bonchio, M. Carraro and P. Ruzza, Pept. Sci., 2018, 110, e24047 CrossRef.
- D. Pooja, A. Gunukula, N. Gupta, D. J. Adams and H. Kulhari, Int. J. Biochem. Cell Biol., 2019, 114, 105567 CrossRef CAS PubMed.
- B. P. Rurarz, M. Bukowczyk, N. Gibka, A. W. Piastowska-Ciesielska, U. Karczmarczyk and P. Ulański, Int. J. Mol. Sci., 2023, 24, 3455 CrossRef CAS PubMed.
- V. Tagliavini, C. Honisch, S. Serrati, A. Azzariti, M. Bonchio, P. Ruzza and M. Carraro, RSC Adv., 2021, 11, 4952–4957 RSC.
- H. Yu, C. Honisch, M. Frigo, N. Balice, V. Tagliavini, X. Zhao, E. Stramiglio, A. Campofelice, S. Serratì, A. Azzariti, L. Porcelli, L. Zanetti Polzi, S. Corni, P. Ruzza and M. Carraro, Front. Chem. Biol., 2024, 3, 1377357 CrossRef.
- Z. Xiao, K. Chen, B. Wu, W. Li, P. Wu and Y. Wei, Eur. J. Inorg. Chem., 2016, 808–811 CrossRef CAS.
- F. Carvalho and M. Aureliano, Int. J. Mol. Sci., 2023, 24, 5043 CrossRef CAS PubMed.
- F. Zhai, X. Wang, D. Li, H. Zhang, R. Li and L. Song, Biomed. Pharmacother., 2009, 63, 51–55 CrossRef CAS PubMed.
- W. Qi, B. Zhang, Y. Qi, S. Guo, R. Tian, J. Sun and M. Zhao, Molecules, 2017, 22, 1535 CrossRef PubMed.
- M. Cheng, N. Li, N. Wang, K. Hu, Z. Xiao, P. Wu and Y. Wei, Polyhedron, 2018, 155, 313–319 CrossRef CAS.
- Y. Zheng, H. Gan, Y. Zhao, W. Li, Y. Wu, X. Yan, Y. Wang, J. Li, J. Li and X. Wang, Chem. – Eur. J., 2019, 25, 15326–15332 CrossRef CAS PubMed.
- X. Hu, H. Wang, B. Huang, N. Li, K. Hu, B. Wu, Z. Xiao, Y. Wei and P. Wu, J. Inorg. Biochem., 2019, 193, 130–132 CrossRef CAS PubMed.
- Y. Wang, F. Wang, D. Wang, A. Li, G. Chen, H. Xiong, Y. Wei, P. Wu and Z. Xiao, J. Mol. Struct., 2020, 1201, 127138 CrossRef CAS.
- H. Soria-Carrera, E. Atrián-Blasco, R. Martín-Rapún and S. G. Mitchell, Chem. Sci., 2023, 14, 10–28 RSC.
- Q. Chen, D. P. Goshorn, C. P. Scholes, X. L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS.
- A. A. Fertig, S. G. Rabbani, M. D. Koch, W. W. Brennessel, P. Miró and E. M. Matson, Nanoscale, 2021, 13, 6162–6173 RSC.
- M. Aureliano, N. I. Gumerova, G. Sciortino, E. Garribba, A. Rompel and D. C. Crans, Coord. Chem. Rev., 2021, 447, 214143 CrossRef CAS.
- M. d M. Barbosa, L. M. A. d Lima, W. A. d S. Alves, E. K. B. d Lima, L. A. d Silva, T. D. d Silva, K. Postal, M. Ramadan, K. Kostenkova, D. A. Gomes, G. G. Nunes, M. C. Pereira, W. E. d Silva, M. F. Belian, D. C. Crans and E. C. Lira, Pharmaceuticals, 2023, 16, 1232 CrossRef CAS PubMed.
- A. L. De Sousa-Coelho, M. Aureliano, G. Fraqueza, G. Serrão, J. Gonçalves, I. Sánchez-Lombardo, W. Link and B. I. Ferreira, J. Inorg. Biochem., 2022, 235, 111915 CrossRef CAS PubMed.
- A. Galani, V. Tsitsias, D. Stellas, V. Psycharis, C. P. Raptopoulou and A. Karaliota, J. Inorg. Biochem., 2015, 142, 109–117 CrossRef CAS PubMed.
- L. Chen, Z. Zhao, R. O. Diarimalala, Z. Chen, Y. Wang, T. Zhan, Y. Zhao, C. Ma, X. Wang, C. Zhao, Z. Xiao, K. Hu and P. Wu, Chem. Biodiversity, 2024, 21, e202301898 CrossRef CAS PubMed.
- V. Kulikov, N. A. Johnson, A. J. Surman, M. Hutin, S. M. Kelly, M. Hezwani, D. L. Long, G. Meyer and L. Cronin, Angew. Chem., Int. Ed., 2017, 56, 1141–1145 CrossRef CAS PubMed.
- M. Carraro, A. Sartorel, G. Scorrano, C. Maccato, M. H. Dickman, U. Kortz and M. Bonchio, Angew. Chem., Int. Ed., 2008, 120, 7385–7389 CrossRef.
- R. Hussain, K. Benning, T. Jávorfi, E. Longo, T. R. Rudd, B. Pulford and G. Siligardi, J. Synchrotron Radiat., 2015, 22, 465–468 CrossRef CAS PubMed.
- M. Amblard, J.-A. Fehrentz, J. Martinez and G. Subra, Mol. Biotechnol., 2006, 33, 239–254 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. F. Guerra, S. J. A. v Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef.
- E. Al-Sayed, A. Blazevic, A. Roller and A. Rompel, Chem. – Eur. J., 2015, 21, 17800–17807 CrossRef CAS PubMed.
- Z. Xiao, K. Chen, B. Wu, W. Li, P. Wu and Y. Wei, Eur. J. Inorg. Chem., 2016, 808–811 CrossRef CAS.
- X. Sun, T. M. Soini, J. Poater, T. A. Hamlin and F. M. Bickelhaupt, J. Comput. Chem., 2019, 40, 2227–2233 CrossRef CAS PubMed.
Footnotes |
| † Special issue dedicated to 13th International Vanadium Symposium. |
| ‡ Electronic supplementary information (ESI) available: Synthesis and characterization of peptides, POMs and hybrids. See DOI: https://doi.org/10.1039/d4nj02851a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |