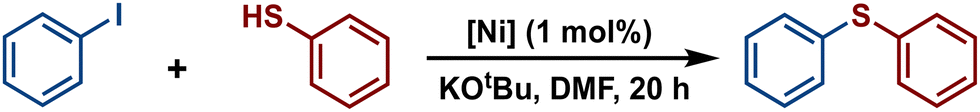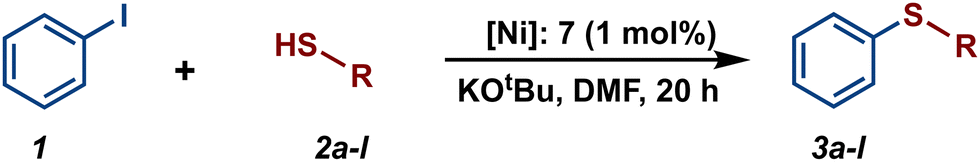DOI:
10.1039/D4NJ03776F
(Paper)
New J. Chem., 2025, Advance Article
Naphthyl and biphenyl para-substituted POCOP-Ni(II) pincer complexes as efficient catalysts in C–S cross-coupling reactions†
Received
27th August 2024
, Accepted 31st October 2024
First published on 31st October 2024
Abstract
A series of six novel para-poly-aromatic acyl-functionalized nickel POCOP-pincer complexes, [NiCl{C6H2-4-Y-2,6-(OPR2)2}] (Y = [1,1′-biphenyl]-4-carboxylate, 2-naphthoate, R = iPr, tBu, Ph), have been synthesized in a straightforward manner in high yields. All new complexes have been fully characterized by standard techniques and three of them unequivocally confirmed by single crystal X-ray diffraction analysis. The catalytic activity of the complexes was examined in C–S cross coupling reactions of iodobenzene with both aryl and alkyl thiols showing high conversions with low catalyst loadings.
Introduction
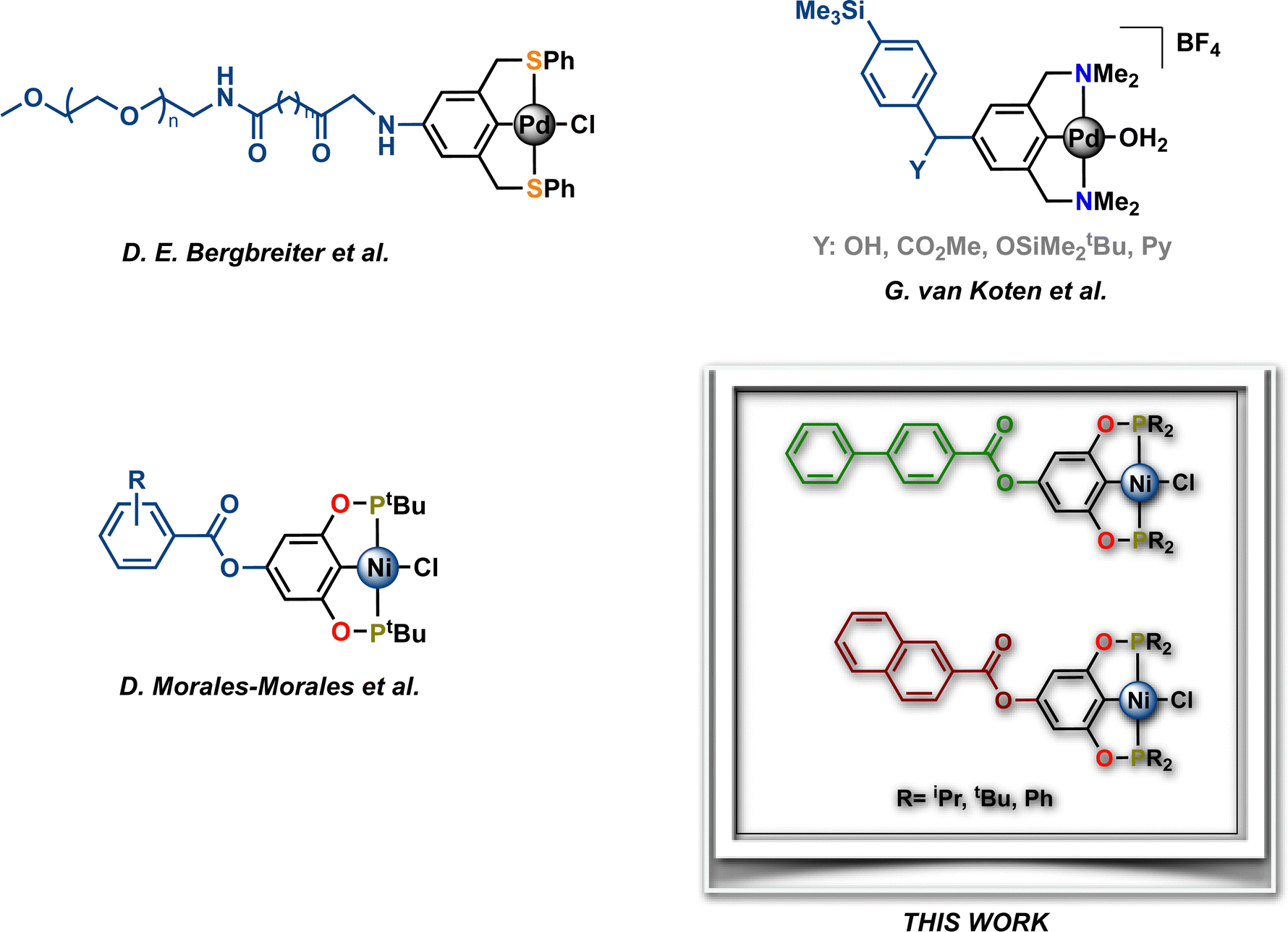
Pincer metal complexes provide robust architectures that continue to find notable applications in organometallic chemistry and homogenous catalysis.1–15 In particular, mer-tridentate ligands bearing aromatic backbones enable efficient fine-tuning of their electronic properties by substitution on the aryl moiety with either electron-donating or electron-withdrawing groups. Besides, the functionalization of the main framework with suitable groups allows the introduction of molecular recognition sites and the immobilization of these pincer complexes to solid supports. In this sense, some pincer complexes bearing anchoring groups (i.e. connectors) such as –OH, –NR2, –OSiR3 or halogens have been reported as precursors for preparing hybrid materials, such as metallodendrimers, and heterogeneous catalysts via their immobilization on e.g., silica or polymers.16–18 Hence, these hybrid materials have been successfully used as catalytic materials in both homogeneous and heterogeneous catalysis and materials science.19–31 In this context, Bergbreiter et al. prepared a para-NH2 functionalized SCS-Pd pincer complex, which was subsequently attached to a soluble polymer support, polyethylene glycol, affording a highly stable and recyclable catalyst for the Heck reaction (Chart 1).32 Other interesting examples have been described by van Koten et al., who synthesized a series of cationic para-substituted NCN-Pd pincer complexes using a strategically substituted group bearing two anchoring points, which allowed both the immobilization (–SMe3) and introduction of an additional functional group (Y). These functionalized NCN-Pd complexes were successfully used as catalysts in the aldol reaction between methyl isocyanoacetate and various substituted benzaldehydes, achieving moderate conversions (Chart 1).33 Noteworthily, the above-described species were prepared by the initial para-functionalization of the free ligands, followed by their metalation in multistep synthetic routes that produced pincer complexes, albeit in low yields.
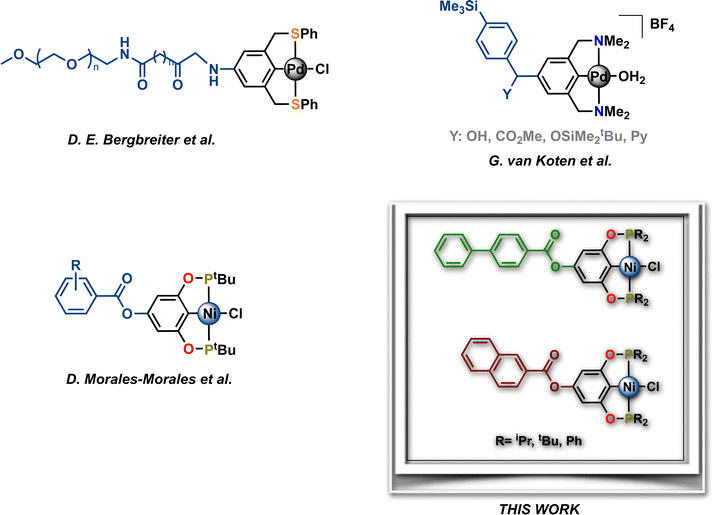 |
| | Chart 1 Selected examples of functionalized metallic pincer complexes. | |
Recently, we have become interested in the chemistry of para-substituted aryl phosphine-based pincers as an extension of our related work with POCOP-based variants. This interest has been motivated by the potential to exploit additional reaction control for the unique steric profile of these complexes through the functionalization of the main framework. In 2015, we reported the synthesis of a series of para-ester substituted Ni-POCOP pincer complexes (Chart 1).34 We have extended this work to novel pincer platforms to explore their organometallic chemistry, thus herein, we present our findings in the chemistry of nickel POCOP-pincer complexes, of the type [NiCl{C6H2-4-OY-2,6-(OPR2)2}] (R = iPr, tBu, Ph) with poly-aromatic acyl scaffolds (Y). These compounds were generated by the para-OH esterification from readily available starting materials and through a versatile and straightforward method in high yields. Poly-aromatic substituents were selected due to their high thermal stability and resistance to redox processes. The catalytic performance of these new functionalized nickel POCOP-pincer complexes in C–S couplings of iodobenzene with various aryl and alkyl thiol derivatives is also presented.
Results and discussion
Synthesis and functionalization of the para-hydroxy substituted POCOP nickel pincer complexes
Using a procedure previously reported by our group, para-hydroxy Ni-POCOP pincer compounds (1 = iPr, 2 = tBu and 3 = Ph) were prepared via a simple reaction of phloroglucinol and the corresponding chlorophosphine, followed by direct metalation with NiCl2. All the nickel complexes were characterized by NMR (1H, 13C and 31P) and structurally confirmed by direct comparison with previously reported data.34,35 Afterwards, the functionalization of the –OH group was carried out following a slightly modified procedure previously reported by our research group to afford its corresponding ester derivatives.34 This synthetic route is shown in Scheme 1.
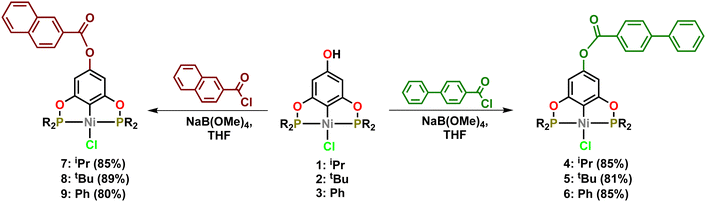 |
| | Scheme 1 Synthesis of para-functionalized POCOP-Ni(II) pincer complexes. | |
In the first step the –OH group was deprotonated by treatment with a base (NaB(OMe)4) in dry THF at room temperature for 2 h to give the phenoxide salts [{4-NaO-C6H2-2,6-(OPR2)}NiCl], then in situ the aromatic acyl chloride was added to the reaction mixture at room temperature to give, after 12 h of stirring, the air-stable complexes: 4, 5 and 6 [1,1′-biphenyl-4-carbonyl chloride] derivatives and the complexes 7, 8 and 9 produced by the treatment with 2-naphthoyl chloride. All functionalized Ni(II) pincer complexes (4–9) were purified by column chromatography obtaining analytically pure materials as yellow crystalline solids in good isolated yields (80–89%). All these new complexes were fully characterized by NMR spectroscopy (1H, 13C and 31P), FTIR, mass spectrometry and elemental analyses.
NMR analysis of complexes 4–9 suggests that they all have C2v symmetry in solution. In the 31P{1H} NMR spectra of the pincer compounds 4–9 a singlet was observed, consistent with the symmetric nature of the pincer complexes (4: δ 187.21 ppm, 5: δ 189.7 ppm, 6: δ 142.8 ppm, 7: δ 187.2 ppm, 8: δ 189.7 ppm, and 9: δ 142.7 ppm). Analysis by 1H NMR afforded spectra that displayed typical signals in the region for aromatic protons, which were assigned to the pincer backbone and either naphthyl or biphenyl protons between δ 8.75 and 6.37 ppm, along with the signals corresponding to the phosphine substituents (R = iPr, tBu, Ph). The 13C{1H} NMR spectra provided further structural information exhibiting signals of the metalated C–Ni carbon as apparent triplets due to their coupling with the mutually trans-phosphorous nuclei at 123.2–121.4 ppm with values of 2JCP between 21–24 Hz. In addition, the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O from the ester group was evidenced by its characteristic signal between δ 165.2–164.9 ppm. The FT-IR spectra of the series of complexes exhibited a key band between 1735 and 1722 cm−1 assigned to vCO. Mass spectra also confirmed the formation of the complexes, the resulting spectra exhibiting the molecular ions [M]+ at 741, 605, 661, 766, 631 and 688 m/z for complexes 4, 5, 6, 7, 8 and 9, respectively. The results obtained from elemental analysis of all the compounds are also in agreement with the proposed structural formulations.
O from the ester group was evidenced by its characteristic signal between δ 165.2–164.9 ppm. The FT-IR spectra of the series of complexes exhibited a key band between 1735 and 1722 cm−1 assigned to vCO. Mass spectra also confirmed the formation of the complexes, the resulting spectra exhibiting the molecular ions [M]+ at 741, 605, 661, 766, 631 and 688 m/z for complexes 4, 5, 6, 7, 8 and 9, respectively. The results obtained from elemental analysis of all the compounds are also in agreement with the proposed structural formulations.
X-Ray diffraction analysis
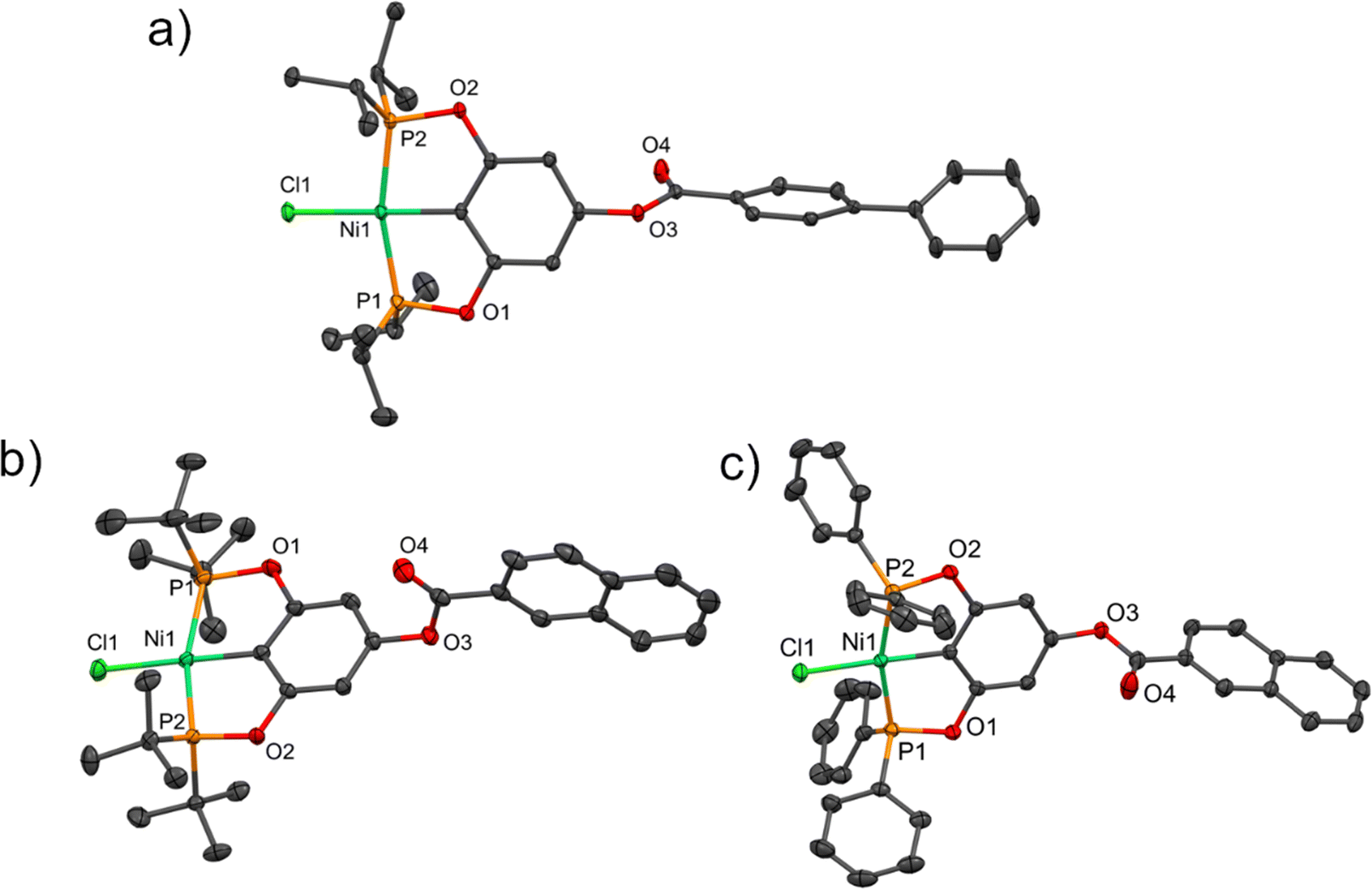
It was possible to obtain three crystalline structures of the complexes 4, 8 and 9. Crystals of complex 4 were obtained by slow evaporation from a saturated dichloromethane solution, while those of complexes 8 and 9 were produced from their corresponding concentrated toluene solutions at room temperature. All compounds crystallized in a monoclinic system. The asymmetric unit of crystal 4 consists of two molecules of the pincer compound and two molecules of dichloromethane. Complex 8 crystallized with a molecule of toluene, while complex 9 crystallized without dissolvent. The crystallographic data of the compounds are summarized in Table S1 (ESI†) and the selected bond distances are listed in Table S2 (ESI†). The molecular structures of the compounds are shown in Fig. 1.
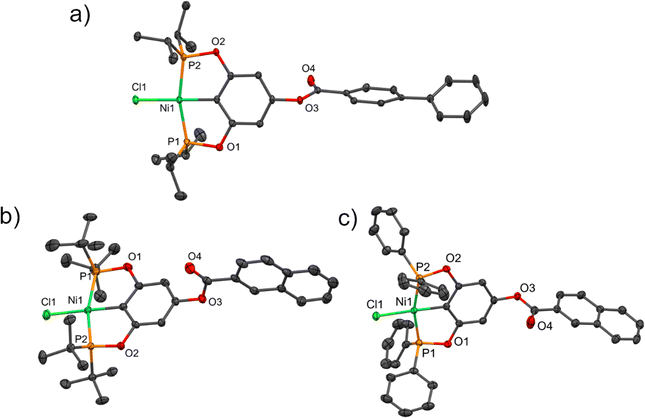 |
| | Fig. 1 Molecular structures of compounds (a) 4, (b) 8, and (c) 9. Thermal ellipsoids are drawn at a 30% probability level, the disordered part (compound 8), hydrogen atoms and solvent molecules are omitted for clarity. | |
The carbon–nickel (C–Ni) bond distances were found to be around 1.882 Å, while the Ni–Cl bond lengths ranged from 2.1829 to 2.2049 Å, consistent with similar compounds.36 On the other hand, the P–Ni bond lengths varied from 2.1533 to 2.1934 Å depending on the substituent of the phosphine group: ditertbutyl groups resulted in longer bonds and diisopropyl or diphenyl produced shorter bonds. The angles between the two atoms in the trans positions around nickel (∠C–Ni–Cl and ∠P–Ni–P) were close to a linear angle of 180° (Table S2, ESI†).
Supramolecular analysis of non-covalent interactions
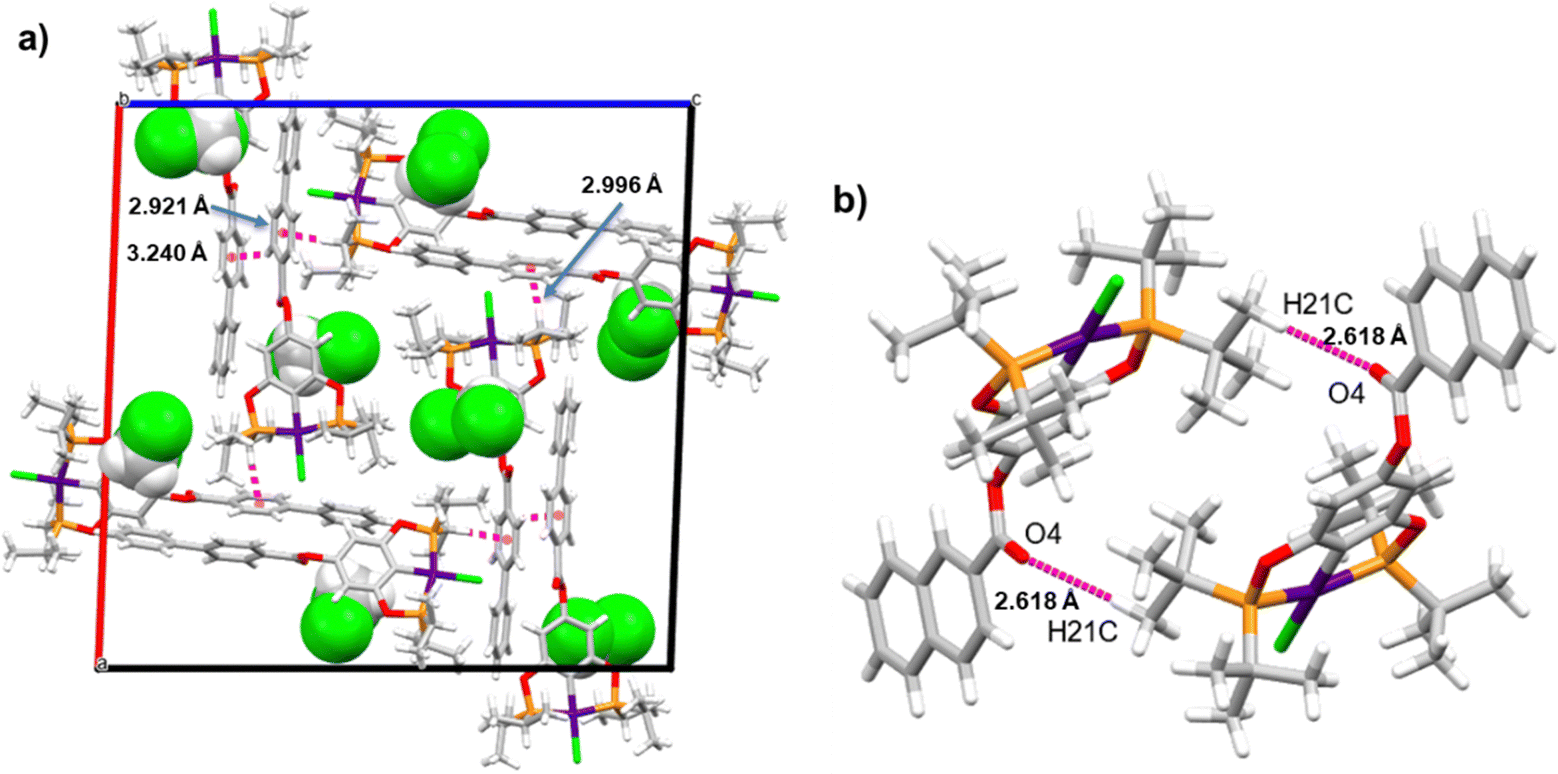
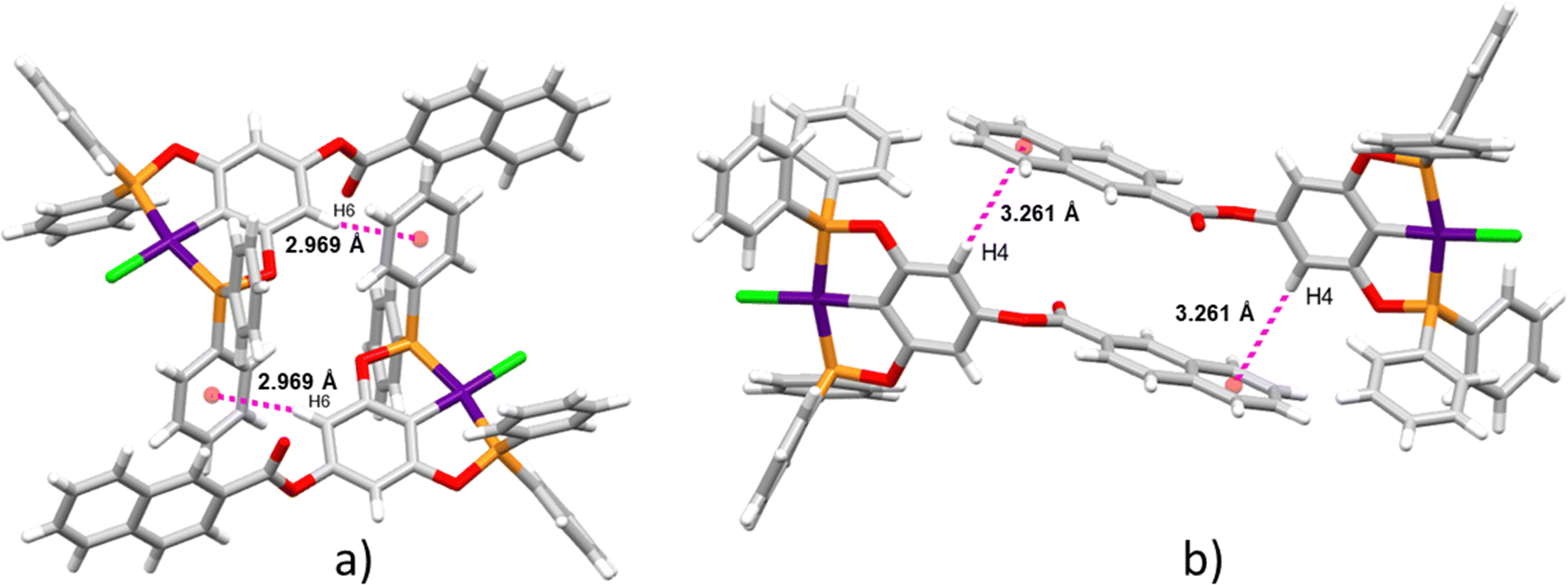
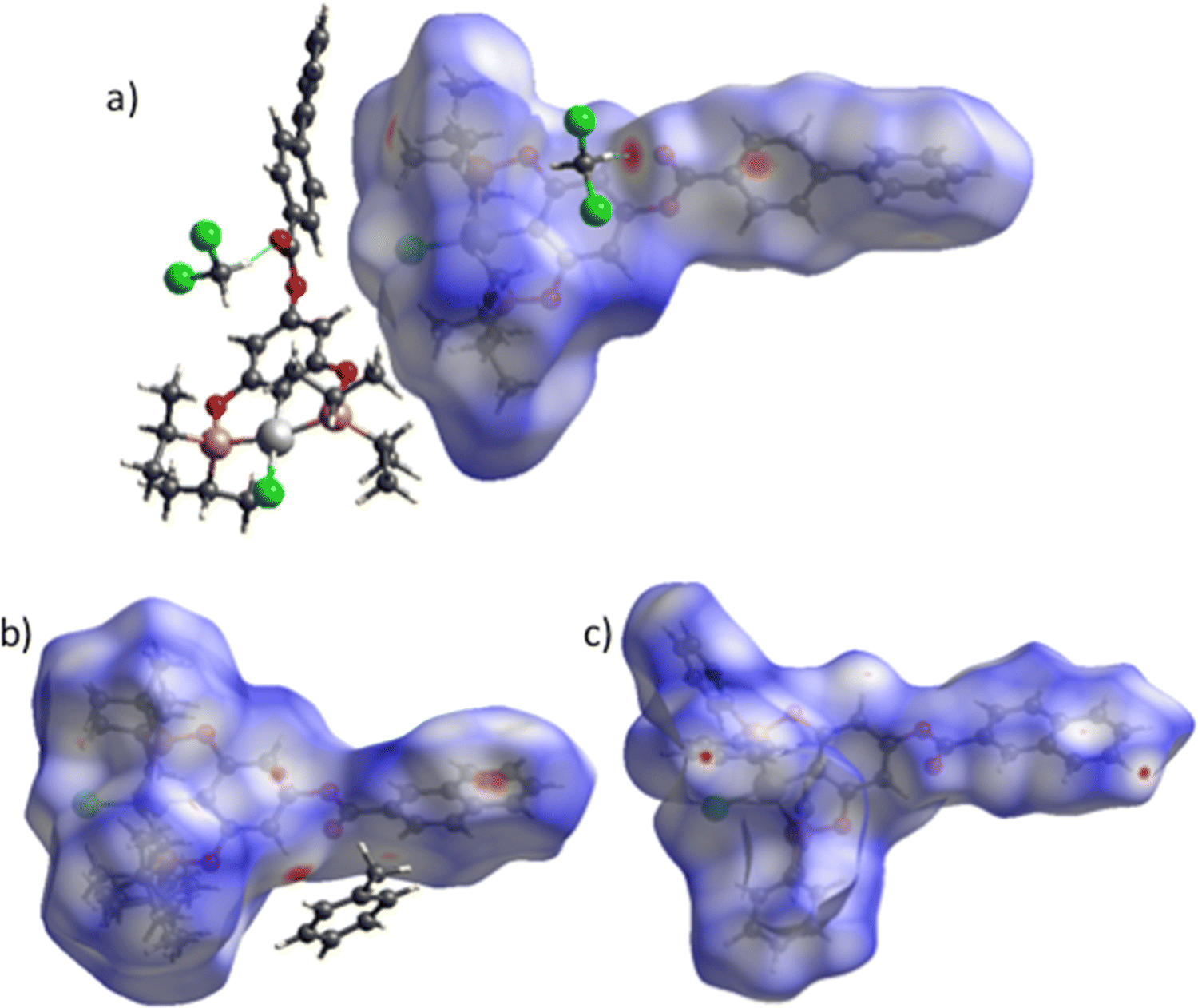
Supramolecular structures are supported via CH⋯π, O⋯H, and CH⋯Cl non-covalent interactions (Table S3, ESI†). Three kinds of CH–π interactions support the crystal packing of complex 4, in addition, the molecules of solvent (DCM) are in alternate positions, interacting via a CH⋯O interaction with the oxygen of the ester (Fig. 2a). A dimeric interaction supported by two reciprocal O⋯H interactions by the ester bridge was observed for the most stable conformation of complex 8 (Fig. 2b). Similarly, two dimeric interactions were observed in complex 9 this time both supported by CH–π interactions (Fig. 3a and b). Despite the three complexes containing many aromatic fragments, no π–π stacking was observed.
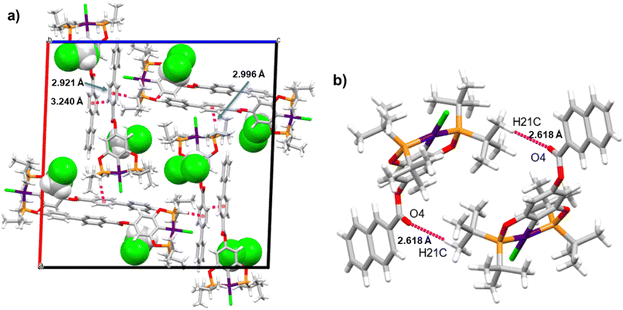 |
| | Fig. 2 (a) Crystal packing of 4 and (b) the dimeric structure interaction of compound 8. | |
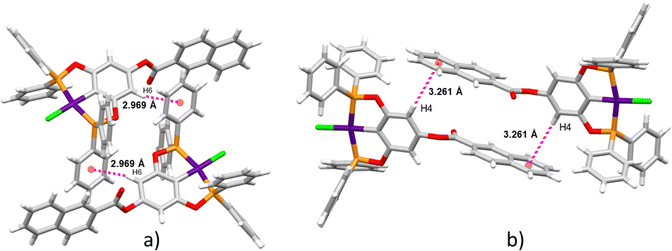 |
| | Fig. 3 Two dimeric interactions for complex 9via CH–π. | |
Hirshfeld surface analysis
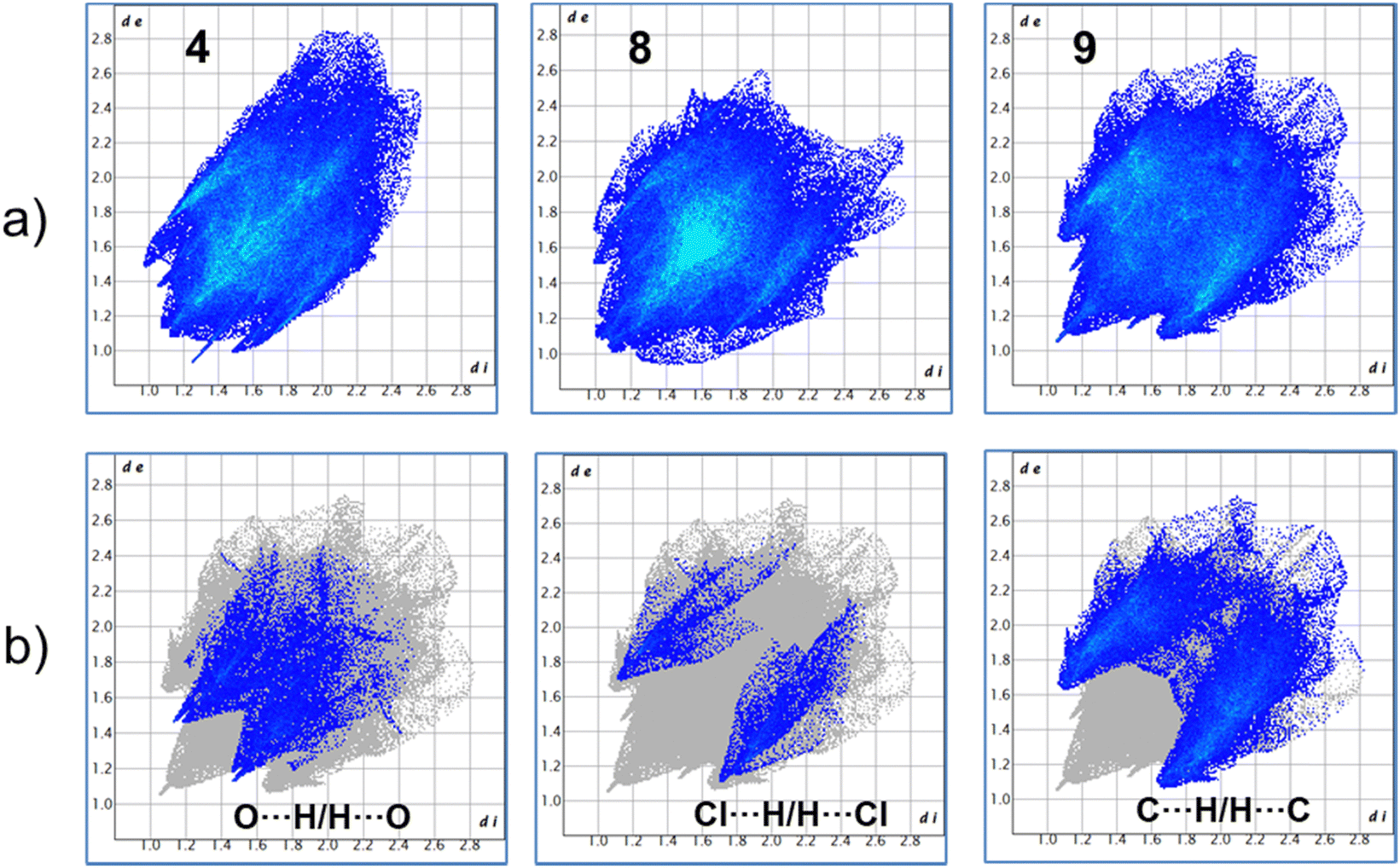
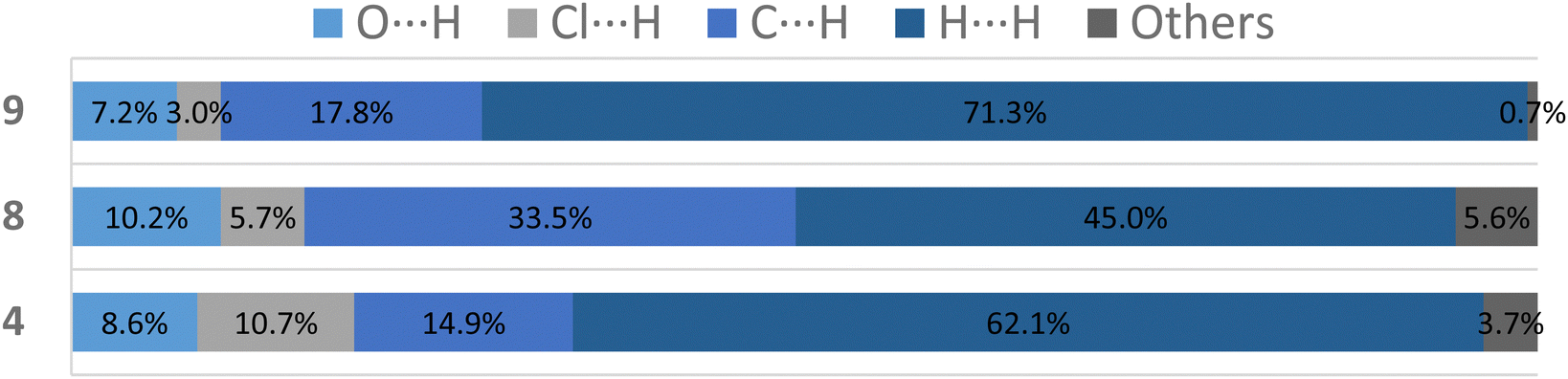
For more clarity, we examined the nature of intermolecular interactions of the molecular structure using CrystalExplorer37 software. Hirshfeld surface analysis and two-dimensional fingerprint plots38 for the three compounds (Fig. 5a) were computed. The principal interactions (red regions) on the surface (close contacts) were due to O⋯H/H⋯O, and other (H⋯H, H⋯C/C⋯H) interactions are highlighted by conventional mapping of dnorm on molecular Hirshfeld surfaces for the three complexes in Fig. 4. For compound 4, the surface was created for a single molecule of the pincer unity, while for 8 all orientations of the disordered molecule with their partial occupancies were included. Reciprocal O⋯H/H⋯O, C⋯H/H⋯C, and Cl⋯H/H⋯Cl contacts were represented in complex 9 as two symmetrical wings in the fingerprint decomposed plots (Fig. 5b). The most predominant interactions in all the complexes were H⋯H contacts (see Table 1 and Fig. 6).
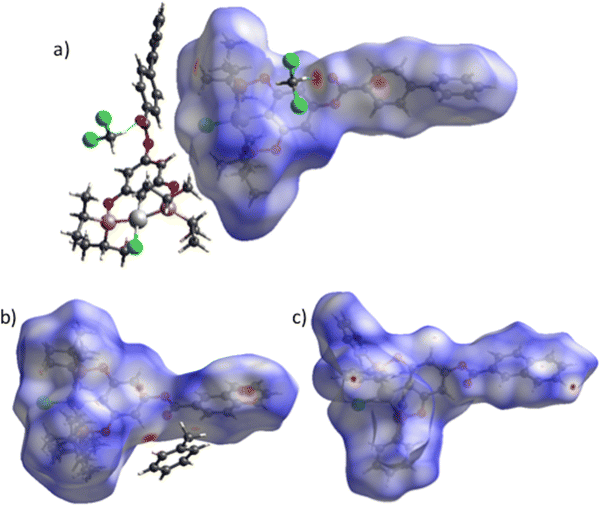 |
| | Fig. 4 Hirshfeld surface mapped over dnorm for compounds (a) 4 (b) 8 and (c) 9. | |
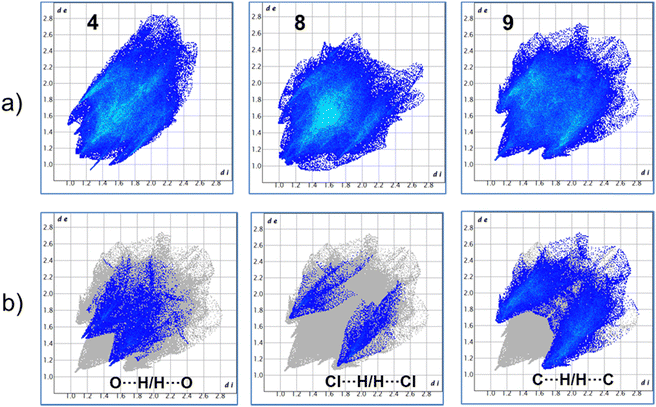 |
| | Fig. 5 (a) Fingerprints of complexes 4, 8 and 9. (b) Representative decomposed fingerprint plots for compound 9. | |
Table 1 Principal contacts in compounds 4, 8 and 9
| |
O⋯H (%) |
Cl⋯H (%) |
C⋯H (%) |
H⋯H (%) |
Others (%) |
|
4
|
8.6 |
10.7 |
14.9 |
62.1 |
3.7 |
|
8
|
10.2 |
5.7 |
33.5 |
45.0 |
5.6 |
|
9
|
7.2 |
3.0 |
17.8 |
71.3 |
0.7 |
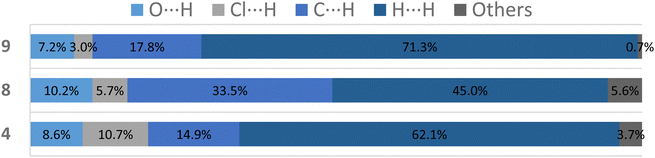 |
| | Fig. 6 Plot of percentages of contacts observed in complexes 4, 8 and 9. | |
Catalytic activity
Among cross-coupling reactions catalyzed by transition metals, C–S couplings have a significant importance to the pharmaceutical industry, enabling the production of a wide variety of important bioactive sulfur compounds such as truxal, metixene and nelfinavir, for instance.39–44 Because of this, the study of C–S bonds formation has attracted increasing interest, leading to the development of efficient methodologies for the production of these compounds utilizing transition metal catalysts. Particularly, catalytic cross-couplings of aryl halides and aryl thiols offer a simple and efficient route for the synthesis of diaryl sulfides and they have been developed using various metal-based catalysts such as Co,45,46 Cu,47,48 Pd,49,50 and Ni.51,52 Hence, considering that some Ni(II)-POCOP pincer complexes have been successfully employed as catalysts for catalytic C–S coupling reactions53–56 we decided to explore the scope of a series of novel para-poly-aromatic nickel POCOP-pincer complexes 4–9 in the thiolation of iodobenzene under the optimized reaction conditions previously established in our laboratory using analogous catalytic systems.
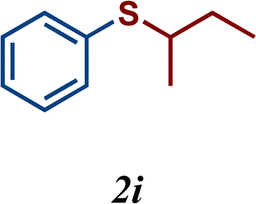
The catalytic activity of complexes 1–9 was evaluated in the C–S cross-coupling reaction. All nickel complexes were screened in the coupling reaction of iodobenzene with thiophenol (selected as the model reaction) using KOtBu as base in DMF at 110 °C for 20 h. Blank tests showed no significant activity without the Ni-POCOP catalyst (<0.5%). The results are summarized in Table 2. In general, all compounds, with the exception of complexes 4 and 2, demonstrated to be efficient catalysts in this process affording the coupled product (thioether) in high yields (87–100%). Regarding the functionalized complexes, the highest conversions were achieved with complexes 7 and 8. Interestingly, a comparison between complex 7 (99% yield) and its related non-substituted Ni-POCOP pincer complex 1 (100% yield) under the optimized reactions conditions afforded a very similar catalytic performance. Furthermore, the catalytic activity of complex 2 was significantly increased from 12% yield to 97% (5) and 99% (8) yield after functionalization with biphenyl and naphthyl fragments.
Table 2 Cross-coupling reaction of iodobenzene and thiophenol catalysed by nickel complexes 1–9a
|
|
| Entry |
[Ni] |
GC yieldb (%) |
|
Reaction conditions: 0.25 mmol of iodobenzene, 0.25 mmol of thiophenol, 0.25 mmol of KOtBu, 5 mL of DMF, 1 mol% of catalyst, 110 °C for 20 h.
Conversions were obtained by GC-MS and are based on residual iodobenzene and are the average of two runs.
|
| 1 |
1
|
100 |
| 2 |
2
|
12 |
| 3 |
3
|
100 |
| 4 |
4
|
45 |
| 5 |
5
|
97 |
| 6 |
6
|
98 |
| 7 |
7
|
99 |
| 8 |
8
|
99 |
| 9 |
9
|
87 |
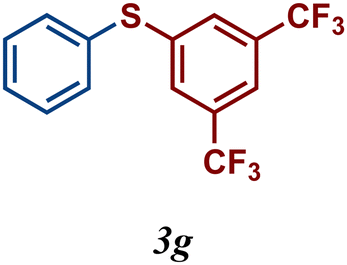
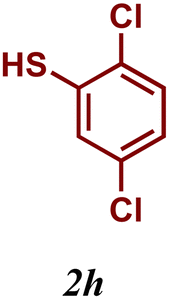
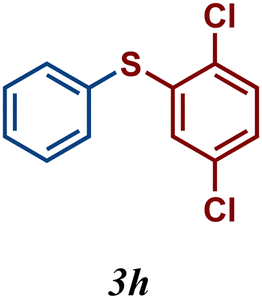
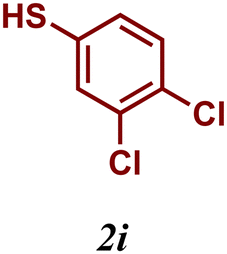
Encouraged by these results, we turned our attention to extend the scope of this reaction using a series of aromatic and aliphatic thiol derivatives and complex 7 as a catalyst (Table 3).
Table 3 C–S cross-coupling reaction catalyzed by complex 7a
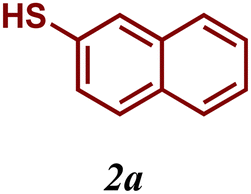
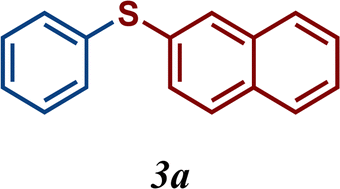
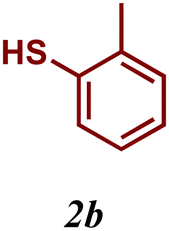
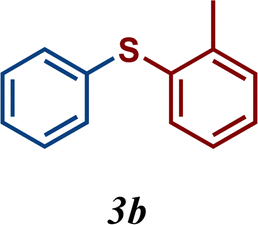
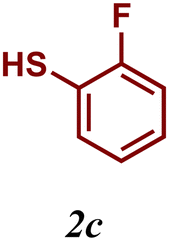
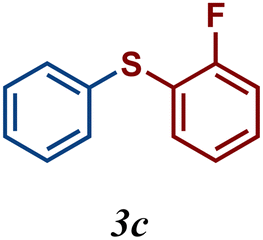
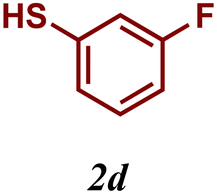
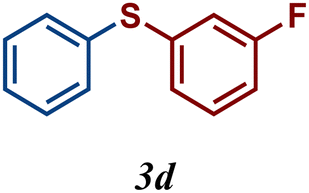
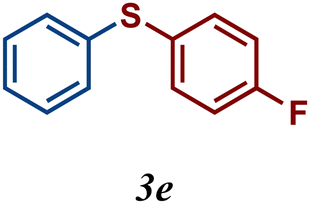
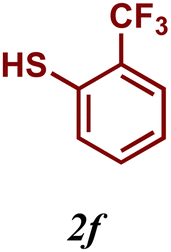
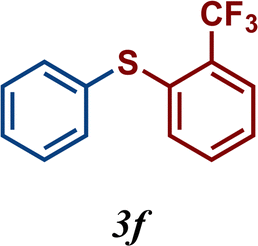
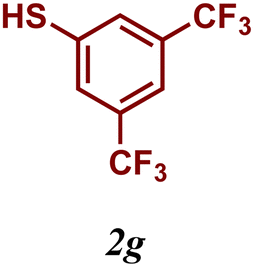
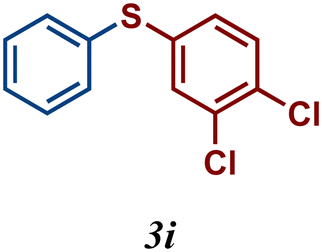
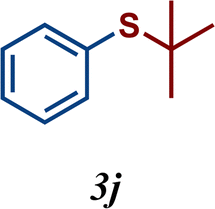
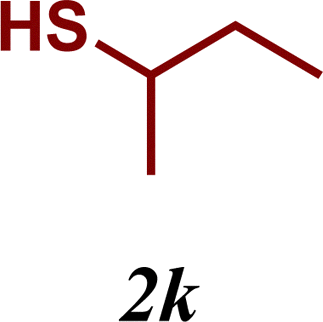
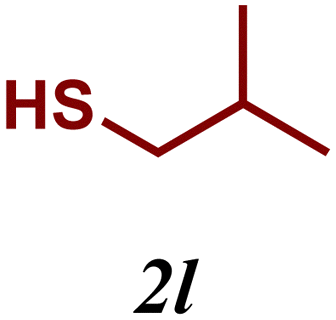
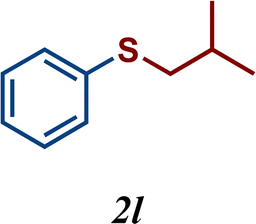
The variation of the electronic and steric nature of the aryl thiol had an impact on the reaction efficiency and as a result, aromatic thiols with both electron-donating and electron-withdrawing substituents at the ortho-position, such as 2b and 2c afforded the coupling product in a high yield (Table 3, entries 2 and 3), while those bearing an electron-withdrawing and sterically hindered substituent in the same position provided a low yield (48%, entry 6). On the other hand, simple aromatic thiols such as 2-mercapto naphthalene (2a) and the para-substituted (2e) reacted extremely well with iodobenzene, forming the appropriate products in high yields (Table 3, entries 1 and 5). Di-substituted aryl thiols are readily tolerated, and the desired products were obtained in good and excellent yields (entries 7, 8 and 9). In addition, to extend the study of C–S couplings, alkyl thiols were used. They were shown to be less effective than aromatic one, affording the coupling products in low yields (Table 3, entries 10–12). However, it is worth mentioning that the most sterically hindered alkyl thiol offered the best conversion (34%) among these substrates. These results clearly show the efficiency of C–S couplings to be strongly dependent on the sterics of the thiol precursor.
Conclusions
In summary, we report the facile synthesis of six novel para-poly-aromatic acyl-functionalized nickel POCOP-pincer complexes, [NiCl{C6H2-4-OH-2,6-(OPR2)2}] (R = iPr, tBu, Ph) in good yields. All the para-functionalized nickel pincer complexes are air- and moisture-stable and were fully characterized by NMR spectroscopy, mass spectrometry and elemental analysis. In addition, the molecular structures of complexes 4, 8 and 9 were unequivocally determined by single crystal X-ray diffraction studies, confirming the tridentate coordination of the pincer ligands. The catalytic activity of all the nickel complexes was evaluated in C–S couplings, exhibiting good performances, except for complex 4. Thus, in general the C–S couplings proceeded well with aryl thiols bearing either electron-withdrawing or electron-donating substituents, while the reactions using alkyl thiols only yielded low conversions. The successful use of these Ni(II) species suggests their potential in other chemical transformations. Thus we are further exploring their catalytic activity in other cross coupling reactions and the potential of the reactivity of the aromatic moieties at the para position of the pincer compounds. These results will be disclosed in due course.
Experimental section
All reactions were carried out under nitrogen using standard Schlenk techniques unless otherwise specified. All chemical compounds were obtained from Aldrich Chemical Co. and used as received without further purification. para-Hydroxy Ni(II)-POCOP pincer complexes 1, 2 and 3 were synthesized according to the reported procedures and structurally confirmed by direct comparison with literature data. Melting points were recorded on a Mel-Temp II apparatus and are reported without correction. 1H and 13C{1H} NMR spectra were recorded on a Bruker Ascend 500 spectrometer or on a JEOL GX300 spectrometer. Chemical shifts are reported in ppm down field of TMS using the residual signals in the solvent as the internal standard. Elemental analyses were performed on a PerkinElmer 240 analyzer. MS-Electrospray spectra were recorded on a Bruker Daltonics-Esquire 3000 plus electrospray mass spectrometer. Mass measurements in FAB+ were performed at a resolution of 3000 using magnetic field scans and the matrix ions as the reference material or, alternatively, by electric field scans with the sample peak bracketed by two (polyethylene glycol or cesium iodide) reference ions.
General esterification procedure for nickel complexes
A Schlenk flask was charged with the corresponding nickel complex (100 mg) along with NaB(OCH3)4 (1.5 eq.) in dry THF (30 mL). The resulting solution was stirred for 2 h at room temperature with an observed color change from yellow to red. Then, the corresponding acyl chloride (1 eq.) was added and the solution returned to its initial color. The reaction mixture was subsequently stirred for 12 h at room temperature. After this time, the solution was evaporated under vacuum and the solid residue was purified by column chromatography using dichloromethane as the eluent. Further removal of the solvent under vacuum yielded the pincer complexes as yellow microcrystalline powders.
Synthesis of 4
Yield: 118 mg (85%). M.p. 262 °C. 1H NMR (500 MHz, CDCl3) δ 8.22 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 7.72 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 7.65 (d, 3JH–H = 7.1 Hz, 2H, CHAr), 7.49 (t, 3JH–H = 7.5 Hz, 2H, CHAr), 7.42 (t, 3JH–H = 7.4 Hz, 1H, CHAr), 6.37 (s, 2H, CHAr), 2.43 (q, 3JH–H = 9 Hz, 2H, CHAr), 2.43 (sept, 3JH–H = 9 Hz, 4H, CH(CH3)2), 1.45 (q, 3JH–H = 17.3 Hz, 12H, CH3), 1.36 (q, 3JH–H = 14.5 Hz, 6H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 168.4 (t, 2JC–P = 10 Hz, C–OP), 164.9 (s, OC–O), 151.8 (s, C–O), 146.4 (s, Cipso), 139.7 (s, Cipso), 130.7 (s, CHAr), 129.1 (s, CHAr), 128.4 (s, CHAr), 128.3 (s, CHAr), 127.4 (s, CHA), 127.3 (t, CHAr), 121.9 (t, 2JC–P = 21 Hz, C–Ni), 99.6 (pt, CHAr), 27.9 (t, 1JC–P = 11 Hz, –CH–), 17.6 (s, CH3), 16.8 (s, CH3). 31P{1H} NMR (202 MHz, CDCl3): δ 187.21. MS (DART +): 631 m/z [M]+. IR (ATR, cm−1): 2962 (m), 2869 (w), 1728 (s), 1259 (m), 485 (m). Elem. anal. calcd for C31H39ClNiO4P2: C, 58.94; H, 6.22. Found: C, 58.92; H, 6.18.
Synthesis of 5
Yield: 120 mg (89%). M.p. 273–274 °C. 1H NMR (500 MHz, CDCl3) δ 8.22 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 7.72 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 7.66 (d, 3JH–H = 7 Hz, 2H, CHAr), 7.50–7.47 (m, 2H, CHAr), 7.43–7.40 (m, 2H, CHAr), 6.38 (s, 2H, CHAr), 1.51 (vt, 36H, –CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 169.0 (t, 2JC–P = 9 Hz, C–OP), 164.9 (s, OC–O), 151.5 (s, C–O), 146.4 (s, Cipso), 139.9 (s, Cipso), 130.7 (s, CHAr), 129.1 (s, CHAr), 128.4 (s, CHAr), 127.4 (s, CHAr), 127.3 (s, CHAr), 121.4 (t, 2JC–P = 21 Hz, C–Ni), 99.2 (t, 3JC–P = 6 Hz, CHAr), 39.4 (s, 1JC–P = 6 Hz, C(CH3)3), 28.1 (s, CH3). 31P{1H} NMR (202 MHz, CDCl3): δ 189.7. MS (DART +): 688 m/z [M]+. IR (ATR, cm−1): 2951 (m), 2855 (w), 1726 (s), 1255 (m), 473 (m). Elem. anal. calcd for C35H47ClNiO4P2: C, 61.12; H, 6.89. Found: C, 61.14; H, 6.92.
Synthesis of 6
Yield: 104 mg (80%). M.p. 269–270 °C. 1H NMR (500 MHz, CDCl3) δ 8.23 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 8.03–7.99 (m, 8H, CHAr), 7.73 (d, 3JH–H = 8.5 Hz, 2H, CHAr), 7.66 (d, 3JH–H = 7.1 Hz, 2H, CHAr), 7.55–7.47 (m, 14H, CHAr), 7.42 (t, 3JH–H = 7.3 Hz, 1H, CHAr), 6.58 (s, 2H, CHAr). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9 (t, 2JC–P = 11.8 Hz, C–OP), 164.9 (s, OC–O), 152.5 (s, C–O), 146.5 (s, Cipso), 139.9 (s, Cipso), 132.0 (s, CHAr), 129.1 (s, CHAr), 128.9 (s, CHAr), 128.4 (s, CHAr), 128.1 (s, CHAr), 127.4 (s, CHAr), 127.3 (s, CHAr), 123.2 (t, 2JC–P = 24 Hz, C–Ni), 101.1 (t, 3JC–P = 6.7 Hz, CHAr). 31P{1H} NMR (202 MHz, CDCl3): δ 142.8. MS (ESI+): 766 m/z [M]+. IR (ATR, cm−1): 2922 (m), 2853 (w), 1735 (s), 1577 (m), 1250 (m), 480 (m). Elem. anal. calcd for C43H31ClNiO4P2: C, 67.27; H, 4.07. Found: C, 67.25; H, 4.09.
Synthesis of 7
Yield: 107 mg (85%). M.p. 279–280 °C. 1H NMR (500 MHz, CDCl3) δ 8.73 (s, 1H, CHAr), 8.15 (dd, 3JH–H = 8.6 Hz, 4JH–H = 1.7 Hz, 1H, CHAr), 7.98 (d, 3JH–H = 8.0 Hz, 1H, CHAr), 7.93 (d, 3JH–H = 8.7 Hz, 1H, CHAr), 7.91 (d, 3JH–H = 8.2 Hz, 1H, CHAr), 7.65–7.61 (m, 1H, CHAr), 7.60–7.55 (m, 1H, CHAr), 6.60 (m, 2H, CHAr). 2.44 (sept, 3JH–H = 9 Hz, 4H, CH(CH3)2), 1.45 (q, 3JH–H = 17.3 Hz, 12H, CH3), 1.36 (q, 3JH–H = 14.5 Hz, 12H, CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 168.5 (t, 2JC–P = 9 Hz, C–OP), 165.1 (s, OC–O), 151.8 (s, C–O), 135.9 (s, Cipso), 132.6 (s, CHAr), 132.0 (s, CHAr), 129.6 (s, CHAr), 128.7 (s, CHAr), 128.5 (s, CHAr), 127.9 (s, CHAr), 126.9 (s, CHAr), 126.8 (s, CHAr), 125.4 (s, CHAr), 122.0 (t, 1JC–P = 21 Hz, CNi), 99.6 (t, 3JC–P = 6 Hz), 27.9 (t, 1JC–P = 11 Hz, –CH–), 17.6 (s, CH3), 16.8 (s, CH3). 31P{1H} NMR (202 MHz, CDCl3): δ 187.20. MS (DART +): 605 m/z [M]+. IR (ATR, cm−1): 2960 (m), 2870 (w), 1722 (s), 1267 (m), 479 (m). Elem. anal. calcd for C29H37ClNiO4P2: C, 57.81; H, 6.16. Found: C, 57.83; H, 6.15.
Synthesis of 8
Yield: 108 mg (81%). M.p. 250–251 °C. 1H NMR (500 MHz, CDCl3) δ 8.73 (s, 1H, CHAr), 8.15 (dd, 3JH–H = 8.6 Hz, 4JH–H = 1.6 Hz, 1H, CHAr), 7.98 (d, 3JH–H = 8.1 Hz, 1H, CHAr), 7.94 (d, 3JH–H = 8.9 Hz, 1H, CHAr), 7.91 (d, 3JH–H = 8.1 Hz, 1H, CHAr), 7.65–7.61 (m, 1H, CHAr), 7.60–7.56 (m, 1H, CHAr), 6.41 (s, 2H, CHAr), 1.51 (m, 36H, –CH3). 13C{1H} NMR (126 MHz, CDCl3) δ 169.1 (t, 2JC–P = 9 Hz, C–OP), 165.2 (s, OC–O), 151.5 (s, C–O), 135.9 (s, Cipso), 132.6 (s, CHAr), 132.0 (s, CHAr), 129.6 (s, CHAr), 128.7 (s, CHAr), 128.5 (s, CHAr), 127.9 (s, CHAr), 126.9 (s, CHAr), 125.5 (s, CHAr), 121.5 (t, 1JC–P = 21 Hz, CNi), 99.2 (t, 3JC–P = 6 Hz), 35.5 (t, 1JC–P = 7 Hz, –C(CH3)3), 28.13 (s, CH3). 31P{1H} NMR (202 MHz, CDCl3): δ 189.7. MS (DART+): 661 m/z [M]+. IR (ATR, cm−1): 2961 (m), 2866 (w), 1735 (s), 1259 (m), 475 (m). Elem. aanal. calcd for C33H45ClNiO4P2: C, 59.89; H, 6.85. Found: C, 59.95; H, 6.81.
Synthesis of 9
Yield: 110 mg (85%). M.p. 276–277 °C. 1H NMR (500 MHz, CDCl3) δ 8.76 (s, 1H, CHAr), 8.16 (dd, 3JH–H = 8.6 Hz, 4JH–H = 1.7 Hz, 1H, CHAr), 8.05–7.98 (m, 9H, CHAr), 7.94 (d, 3JH–H = 8.7 Hz, 1H, CHAr), 7.92 (d, 3JH–H = 8.2 Hz, 1H, CHAr), 7.66–7.61 (m, 1H, CHAr), 7.60–7.56 (m, 1H, CHAr), 7.56–7.46 (m, 12H, CHAr), 6.41 (s, 2H, CHAr). 13C{1H} NMR (126 MHz, CDCl3) δ 166.9 (t, 2JC–P = 12 Hz, C–OP), 165.2 (s, OC–O), 152.6 (s, C–O), 135.9 (s, Cipso), 132.6 (s, CHAr), 132.5 (s, CHAr), 132.3 (s, CHAr), 132.1 (s, CHAr), 132.0 (s, CHAr), 131.9 (s, CHAr), 129.6 (s, CHAr), 129.0 (s, CHAr), 128.9 (s, CHAr), 128.8 (s, CHAr), 128.5 (s, CHAr), 127.9 (s, CHAr), 127.0 (s, CHAr), 126.6 (s, CHAr), 125.5 (s, CHAr), 123.2 (t, 1JC–P = 24 Hz, CNi), 101.2 (t, 3JC–P = 7 Hz), 31P{1H} NMR (202 MHz, CDCl3): δ 142.7. MS (DART +): 741 m/z [M]+. IR (ATR, cm−1): 2960 (m), 2855 (w), 1734 (s), 1260 (m), 474 (m). Elem. anal. calcd for C41H29ClNiO4P2: C, 66.39; H, 3.94. Found: C, 66.37; H, 3.90.
Catalytic experiments
Under nitrogen atmosphere, a solution of KOtBu (0.25 mmol) and the corresponding nickel catalyst (1 mol%) in DMF (5 mL) was stirred at ambient temperature for 10 min. Then, iodobenzene (0.25 mmol) and the respective thiol (0.25 mmol) were added to the solution. The reaction was heated at 110 °C for the desired time. The reaction mixture was cooled to room temperature and the organic phase analyzed by gas chromatography (GC-MS) (quantitative analyses were performed on an Agilent 6890 N GC with a 30.0 m DB-1MS capillary column coupled to an Agilent 5973 Inert Mass Selective detector).
Data collection and refinement for 4, 8 and 9
Crystals of 4, 8 and 9 were mounted on glass fibers, then placed on a Bruker Smart Apex II diffractometer with a Mo-target X-ray source (λ = 0.71073 Å). The detector was placed at a distance of 5.0 cm from the crystals and frames were collected with a scan width of 0.5 in ω and an exposure time of 10 s frame−1. Frames were integrated with the Bruker SAINT software package using a narrow-frame integration algorithm.57 Non-systematic absences and intensity statistics were used in a monoclinic system and P21/n space groups, in all cases. The structures were solved using Patterson methods using the SHELXS-2014/7 program.58 The remaining atoms were located via a few cycles of least squares refinements and difference Fourier maps. Hydrogen atoms were inputted at calculated positions and allowed to ride on the atoms to which they are attached. Thermal parameters were refined for hydrogen atoms on the phenyl groups using a Ueq = 1.2 Å to the precedent atom. The final cycles of refinement were carried out on all non-zero data using SHELXL-2014/7. Absorption corrections were applied using a SADABS program.59 In compound 8, the fragment tBu2P and one molecule of toluene are disordered and were modelled and refined anisotropically in two positions using a variable site occupational factor (SOF), the ratio of SOF was 0.5/0.5.
Data availability
The data supporting this article has been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We would like to thank Chem. Eng. Luis Velasco Ibarra, Dr Francisco Javier Pérez Flores, Q. Eréndira García Ríos, M.Sc. Lucia de Carmen Márquez Alonso, M.Sc. Lucero Ríos Ruiz, M.Sc. Alejandra Núñez Pineda (CCIQS), Q. María de la Paz Orta Pérez, Q. Rocío Patiño-Maya and PhD Nuria Esturau Escofet for technical assistance. E. R. F. would like to thank Programa de Becas Posdoctorales-DGAPA-UNAM for postdoctoral scholarships (Oficio: CJIC/CTIC/4851/2021). The financial support of this research by PAPIIT-DGAPA-UNAM (PAPIIT IN223323) and CONACYT A1-S-033933 is gratefully acknowledged.
References
- D. Morales-Morales, Rev. Soc. Quím. Mex., 2004, 48, 338–346 CAS.
-
D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, 2007 Search PubMed.
- D. Morales-Morales, Mini-Rev. Org. Chem., 2008, 5, 141–152 CrossRef CAS.
- J. Serrano-Becerra and D. Morales-Morales, Curr. Org. Synth., 2009, 6, 169–192 CrossRef CAS.
- D. F. Brayton, P. R. Beaumont, E. Y. Fukushima, H. T. Sartain, D. Morales-Morales and C. M. Jensen, Organometallics, 2014, 33, 5198–5202 CrossRef CAS.
- M. Asay and D. Morales-Morales, Dalton Trans., 2015, 44, 17432–17447 RSC.
-
D. Morales-Morales, Pincer Compounds Chemistry and Applications, Elsevier, 2018 Search PubMed.
- H. Valdés, M. A. García-Eleno, D. Canseco-Gonzalez and D. Morales-Morales, ChemCatChem, 2018, 10, 3136–3172 CrossRef.
- L. González-Sebastián and D. Morales-Morales, J. Organomet. Chem., 2019, 893, 39–51 CrossRef.
- H. Valdés, E. Rufino-Felipe and D. Morales-Morales, J. Organomet. Chem., 2019, 898, 120864 CrossRef.
- A. Mukherjee and D. Milstein, ACS Catal., 2018, 8, 11435–11469 CrossRef CAS.
- V. Arora, H. Narjinari, P. G. Nandi and A. Kumar, Dalton Trans., 2021, 50, 3394–3428 RSC.
- Z. Wang and N. Liu, Eur. J. Inorg. Chem., 2012, 901–911 CrossRef CAS.
- N. Selander and K. J. Szabó, Chem. Rev., 2011, 111, 2048–2076 CrossRef CAS PubMed.
- A. Kasera, J. P. Biswas, A. Ali Alshehri, S. Ahmed Al-Thabaiti, M. Mokhtar and D. Maiti, Coord. Chem. Rev., 2023, 475, 214915 CrossRef CAS.
- G. van Koten, J. Organomet. Chem., 2013, 730, 156–164 CrossRef CAS.
- H. Valdés, L. González-Sebastián and D. Morales-Morales, J. Organomet. Chem., 2017, 845, 229–257 CrossRef.
- M. A. W. Lawrence, K.-A. Green, P. N. Nelson and S. C. Lorraine, Polyhedron, 2018, 143, 11–27 CrossRef CAS.
- J. W. J. Knapen, A. W. V. D. Made, J. C. D. Wilde, P. W. N. M. V. Leeuwen, P. Wijkens, D. M. Grove and G. V. Koten, Nature, 1994, 372, 659–663 CrossRef CAS.
- G. D. Batema, M. Lutz, A. L. Spek, C. A. V. Walree, C. D. M. Donegá, A. Meijerink, R. W. A. Havenith, J. Pérez-Moreno, K. Clays, M. Büchel, A. V. Dijken, D. L. Bryce, G. P. M. V. Klink and G. V. Koten, Organometallics, 2008, 27, 1690–1701 CrossRef CAS.
- W. J. Sommer, K. Yu, J. S. Sears, Y. Ji, X. Zheng, R. J. Davis, C. D. Sherrill, C. W. Jones and M. Weck, Organometallics, 2005, 24, 4351–4361 CrossRef CAS.
- H. P. Dijkstra, M. D. Meijer, J. Patel, R. Kreiter, G. P. M. van Klink, M. Lutz, A. L. Spek, A. J. Canty and G. V. Koten, Organometallics, 2001, 20, 3159–3168 CrossRef CAS.
- M. Albrech and G. V. Koten, Adv. Mater., 1999, 11, 171–174 CrossRef.
- J. W. J. Knapen, A.
W. van der Made, J. C. de Wilde, P. W. N. M. van Leeuwen, P. Wijkens, D. M. Grove and G. V. Koten, Nature, 1994, 372, 659–663 CrossRef CAS.
- W. T. S. Huck, F. C. J. M. V. Veggel, S. S. Sheiko, M. Möller and D. N. Reinhoudt, J. Phys. Org. Chem., 1998, 11, 540–545 CrossRef CAS.
- M. Albrecht and G. v Koten, Adv. Mater., 1999, 11, 171–174 CrossRef CAS.
- M. Albrecht, R. A. Gossage, M. Lutz, A. L. Spek and G. V. Koten, Chem. – Eur. J., 2000, 6, 1431–1445 CrossRef CAS PubMed.
- H.-J. V. Manen, R. H. Fokkens, N. M. M. Nibbering, F. C. J. M. V. Veggel and D. N. Reinhoudt, J. Org. Chem., 2001, 66, 4643–4650 CrossRef.
- G. Rodríguez, M. Albrecht, J. Schoenmaker, A. Ford, M. Lutz, A. L. Spek and G. V. Koten, J. Am. Chem. Soc., 2002, 124, 5127–5138 CrossRef.
- K. Yu, W. Sommer, M. Weck and C. Jones, J. Catal., 2004, 226, 101–110 CrossRef CAS.
- H. P. Dijkstra, M. D. Meijer, J. Patel, R. Kreiter, G. P. M. V. Klink, M. Lutz, A. L. Spek, A. J. Canty and G. V. Koten, Organometallics, 2001, 20, 3159–3168 CrossRef CAS.
- D. E. Bergbreiter, Philip L. Osburn and Y.-S. Liu, J. Am. Chem. Soc., 1999, 121, 9531–9538 CrossRef CAS.
- M. Q. Slagt, D. A. P. v Zwieten, A. J. C. M. Moerkerk, R. J. M. K. Gebbink and G. V. Koten, Coord. Chem. Rev., 2004, 248, 2275–2282 CrossRef CAS.
- M. A. García-Eleno, E. Padilla-Mata, F. Estudiante-Negrete, F. Pichal-Cerda, S. Hernández-Ortega, R. A. Toscano and D. Morales-Morales, New J. Chem., 2015, 39, 3361–3365 RSC.
- M. A. Garcia-Eleno, M. Quezada-Miriel, R. Reyes-Martinez, S. Hernandez-Ortega and D. Morales-Morales, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2016, 72, 393–397 CrossRef CAS.
- A. A. Castillo-García, L. González-Sebastián, L. Lomas-Romero, S. Hernandez-Ortega, R. A. Toscano and D. Morales-Morales, New J. Chem., 2021, 45, 10204–10216 RSC.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Cryst., 2021, 54, 1006–1011 CrossRef CAS.
- A. Parkin, G. Barr, W. Dong, C. J. Gilmore, D. Jayatilaka, J. J. McKinnon, M. A. Spackman and C. C. Wilson, CrystEngComm, 2007, 9, 648–652 RSC.
- K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 5 CrossRef PubMed.
- G. D. Martino, M. C. Edler, G. L. Regina, A. Coluccia, M. C. Barbera, D. Barrow, R. I. Nicholson, G. Chiosis, A. Brancale, E. Hamel, M. Artico and R. Silvestri, J. Med. Chem., 2006, 49, 947–954 CrossRef.
- G. Evano, C. Theunissen and A. Pradal, Nat. Prod. Rep., 2013, 30, 1467–1489 RSC.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS.
- C. F. Lee, Y. C. Liu and S. S. Badsara, Chem. – Asian J., 2014, 9, 706–722 CrossRef CAS PubMed.
- D. A. Boyd, Angew. Chem., Int. Ed., 2016, 55, 15486–15502 CrossRef CAS PubMed.
- Y.-C. Wong, T. T. Jayanth and C.-H. Cheng, Org. Lett., 2006, 8, 5613–5616 CrossRef CAS PubMed.
- M.-T. Lan, W.-Y. Wu, S.-H. Huang, K.-L. Luo and F.-Y. Tsai, RSC Adv., 2011, 1, 1751–1755 RSC.
- D. Andrada, S. Soria-Castro, D. Caminos, J. Argüello and A. Peñéñory, Catalysts, 2017, 7, 388 CrossRef.
- W.-K. Huang, W.-T. Chen, I. J. Hsu, C.-C. Han and S.-G. Shyu, RSC Adv., 2017, 7, 4912–4920 RSC.
- T. Itoh and T. Mase, Org. Lett., 2004, 6, 4587–4590 CrossRef CAS PubMed.
- M. A. Fernández-Rodríguez, Q. Shen and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 2180–2181 CrossRef.
- Y. Zhang, K. C. Ngeow and J. Y. Ying, Org. Lett., 2007, 9, 3495–3498 CrossRef CAS.
- X.-B. Xu, J. Liu, J.-J. Zhang, Y.-W. Wang and Y. Peng, Org. Lett., 2013, 15, 550–553 CrossRef CAS.
- V. Gómez-Benítez, O. Baldovino-Pantaleón, C. Herrera-Álvarez, R. A. Toscano and D. Morales-Morales, Tetrahedron Lett., 2006, 47, 5059–5062 CrossRef.
- J. Zhang, C. M. Medley, J. A. Krause and H. Guan, Organometallics, 2010, 29, 6393–6401 CrossRef CAS.
- G. T. Venkanna, H. D. Arman and Z. J. Tonzetich, ACS Catal., 2014, 4, 2941–2950 CrossRef CAS.
- J. M. Serrano-Becerra, H. Valdés, D. Canseco-González, V. Gómez-Benítez, S. Hernández-Ortega and D. Morales-Morales, Tetrahedron Lett., 2018, 59, 3377–3380 CrossRef CAS.
-
Bruker Programs: APEX3, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2018 Search PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, SADABS 2016/2, J. Appl. Cryst., 2015, 48, 3–10 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2062939–2062941. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj03776f |
| ‡ Current address: Facultad de Farmacia, Universidad Autónoma del Estado de Morelos, Av. Universidad 1001, Chamilpa, Cuernavaca, Morelos, CP 62209, Mexico. |
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Juan S.
Serrano-García
a,
Juan S.
Serrano-García