DOI:
10.1039/D4NJ04035J
(Paper)
New J. Chem., 2025,
49, 1145-1158
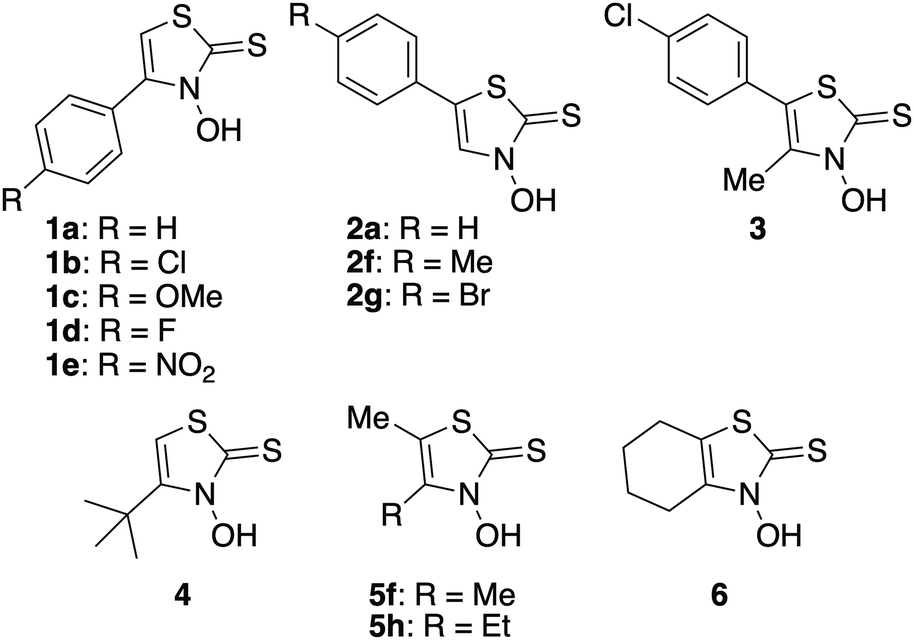
Synthesis, crystal structure, and insulin-mimetic activity of zinc(II) complexes with 4-alkyl- and 4,5-dialkyl-3-hydroxythiazole-2(3H)-thiones as a new class of hypoglycemic agent candidates†
Received
14th September 2024
, Accepted 6th November 2024
First published on 12th November 2024
Abstract
The development of potent hypoglycemic zinc complexes is important for treating diabetes mellitus. In this study, 3-hydroxythiazole-2(3H)-thiones (3,2-HTTs) with alkyl groups at the 4- and 4,5-positions were synthesized, and the insulin-mimetic activities of their zinc(II) complexes were evaluated by determining the 50% inhibitory concentration (IC50) of free fatty acids released from isolated rat adipocytes treated with epinephrine. The structures of the complexes were unambiguously determined by means of X-ray crystallographic analyses. The IC50 values of the newly synthesized complexes with 4,5-dialkyl-3,2-HTTs ranged from 16 to 24 μM, indicating higher inhibitory activities than those of previously reported 4-aryl-3,2-HTT-zinc(II) complexes (30–44 μM). The correlation between some properties of the complexes showed that complexes with smaller sizes tended to have higher insulin-mimetic activity. Besides, for all complexes, the stability constant were estimated to be less than 10.5, confirming the usefulness of the stability constant as a parameter for identifying zinc(II) complexes with high insulin-mimetic activity.
Introduction
Diabetes mellitus (DM) is a metabolic disease characterized by chronic hyperglycemia, which can result in various complications, such as nerve damage, retinopathy, kidney damage, atherosclerosis, and, in the worst case, death. The prevalence of diabetes is increasing annually, with projections indicating that by 2024, more than 12% of the global population aged 20–79 will be affected.1 DM is generally classified into types 1 and 2.2 In type 1 DM, there is an absolute shortage of insulin secretion owing to the destruction of insulin-secreting beta cells in the pancreas. Therefore, type 1 diabetics inevitably rely on insulin injections for their treatment. However, these injections impose significant financial and physical burdens. Type 2 DM, which accounts for >90% of diabetic patients,3 results from a deficiency in insulin action due to genetic predisposition and environmental and lifestyle factors, such as aging and obesity. The treatment of type 2 diabetes involves the administration of synthetic drugs, along with exercise and diet therapy. Currently, the most commonly used hypoglycemic agents include alpha-glycosidase inhibitors, which suppress the postprandial increase in blood glucose levels by degrading carbohydrates to delay their absorption; sulfonylureas, which stimulate insulin secretion; and thiazolidinediones, which improve insulin resistance by acting on insulin receptors.4 However, even with these drugs, blood glucose control is difficult and requires treatment with burdensome insulin injections.5 Therefore, it is imperative to develop a new drug that works via a different mechanism from conventional drugs and can be administered orally without burdensome insulin injections. Zinc ions have thus recently attracted considerable attention as novel drug candidates for DM. Zinc is an essential trace metal and is the second most abundant metal in the human body after iron.6 The optimal concentration range of zinc ions in the human body is wide and the risk of overdose poisoning is low.7–9 This makes the development of zinc ion-based drugs a highly attractive area of research. In 1980, Coulston and Dandona reported that zinc(II) ions stimulated lipogenesis in rat adipocytes in a manner similar to the action of insulin.10 Subsequently, it was found that oral administration of inorganic zinc salts, such as ZnCl2 and ZnSO4, could regulate glucose levels in blood plasma. However, the low bioavailability of inorganic zinc compounds, such as ZnCl2 and ZnSO4, necessitates higher doses, which can have adverse effects.11 In response to this, based on the idea that an organic zinc(II) complex may increase the absorption rate of zinc ions, organic zinc(II) complexes were orally administered to rats and found to increase their bioavailability compared with inorganic ZnCl2.11,12 This discovery led to the synthesis of several organozinc complexes to investigate their hypoglycemic effects and mechanisms of action. Sakurai et al. (2004) reported that a zinc(II) complex with picolinic acids as ligands exhibits insulin-mimetic activity by activating the insulin receptor and phosphatidylinositol 3-kinase. They also reported that zinc(II) complexes with maltol ligands exhibits insulin-mimetic activity by activating the glucose transporter GLUT4, which mediates insulin-stimulated glucose transport in adipocytes.13 In 2011, Yasui et al. demonstrated that a zinc(II) complex with 1-hydroxy-2(1H)-pyridinethione exhibited high insulin-mimetic activity because of the phosphorylation activation of Akt/PKB.14 According to Yoshikawa et al., a zinc(II) complex with hinokitiol ligands promoted Akt/PKB phosphorylation, exhibiting high insulin-mimetic activity.15 These ligands were not designed specifically for zinc(II) complexes but are mainly known to complex with other metal ions, such as iron complexes. Hence, they are either commercially available, naturally occurring, or synthesized using established methods. To clarify the mechanism of action and develop more potent compounds, more detailed studies on the relationship between the structure of the ligands and the insulin-mimetic activity of their zinc(II) complexes are required. For this purpose, it is necessary to expand the compound library; therefore, the development of new synthetic ligands is imperative. Against this background, our research group focused on developing novel organic bidentate ligands for zinc(II) complexes. Recently, we developed a new class of zinc(II) complexes with insulin-mimetic activity by demonstrating the insulin-mimetic activity of several zinc(II) complexes with 4-(p-substituted)phenyl-3-hydroxythiazole-2(3H)-thiones (1, Fig. 1) as ligands for the first time.16 Then, in the present study, which is part of our ongoing project involving the development of chemotherapeutics based on zinc(II) complexes of 3-hydroxythiazole-2(3H)-thione (3,2-HTT) derivatives, we synthesized several new zinc(II) complexes using 5-(p-substituted)phenyl-3,2-HTTs (2 and 3) and 4,5-dialkyl-3,2-HTTs (4–6) as ligands (Fig. 1). We also evaluated the in vitro insulin-mimetic activities of the new zinc(II) complexes and found that the new complexes with 4,5-dialkyl-3,2-HTTs exhibited increased insulin-mimetic activity.
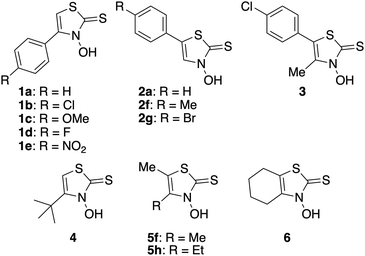 |
| | Fig. 1 Structurs of 3-hydroxythiazole-2(3H)-thione (3,2-HTT) derivatives. | |
Results and discussion
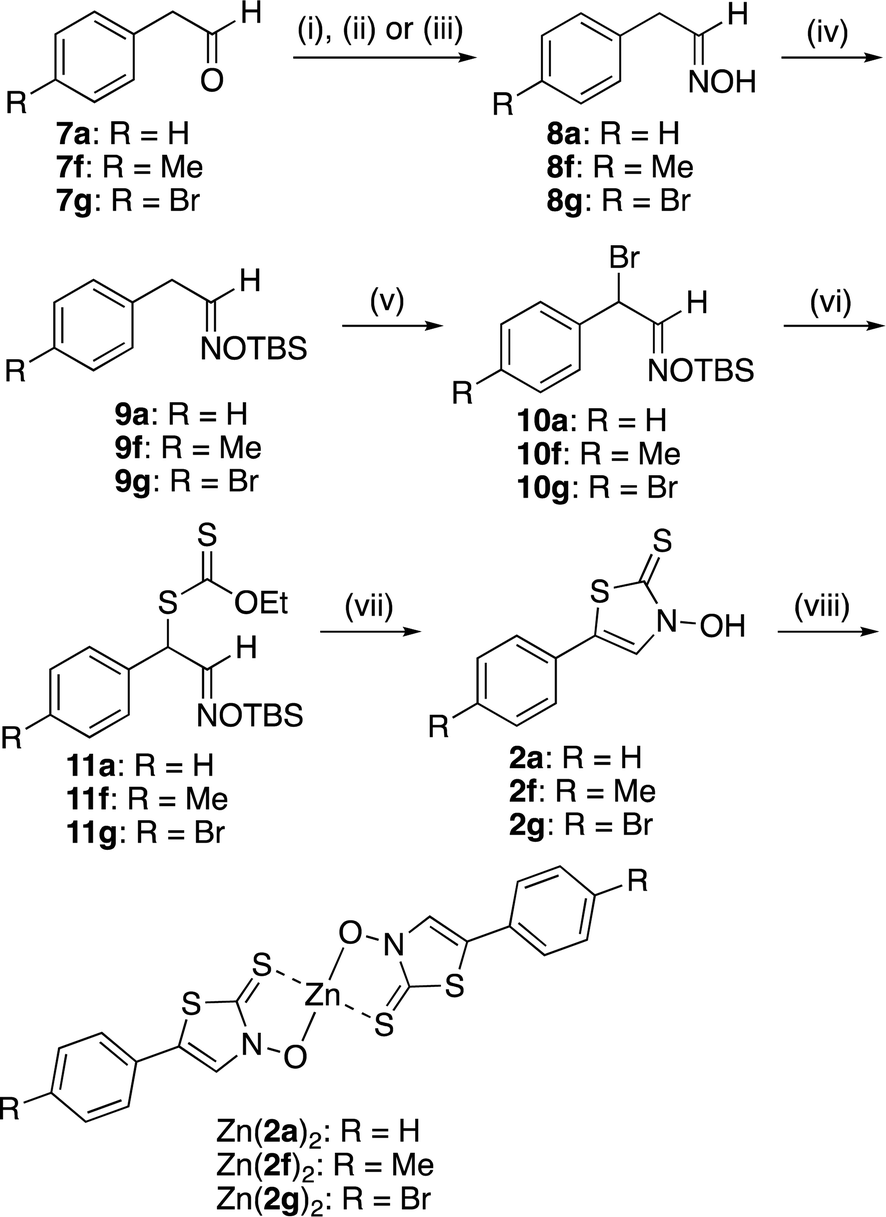
Zinc(II) complexes with the new ligand 2, which are phenyl regioisomers of Zn(1)2 complexes with high insulin-mimetic activity, were prepared according to Scheme 1. First, aldehydes 7a, 7f, and 7g were reacted with hydroxylamine hydrochloride to obtain the corresponding oximes 8a, 8f, and 8g, respectively. The oxime hydroxyl group was protected by TBSCl. AIBN-initiated radical bromination reactions of TBS-protected oximes 9a, 9f, and 9g yielded the corresponding bromides 10a, 10f, and 10g, respectively. The bromides were then reacted with potassium O-ethyl dithiocarbonate to obtain the corresponding xanthates (11a, 11f, and 11g). Intramolecular cyclocondensation of 11a, 11f, and 11g was achieved using potassium hydroxide to form new ligands 2a, 2f, and 2g, respectively. The zinc(II) complexes 2a, 2f, and 2g were furnished by reacting 11a, 11f, and 11g, respectively, with zinc acetate in the presence of lithium hydroxide.
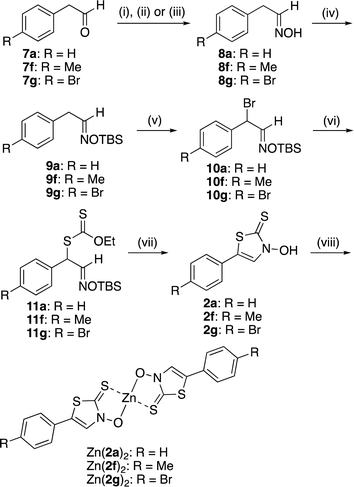 |
| | Scheme 1 Reagents and conditions: (i) NH2OH·HCl, Na2CO3, 50% methanol aq., room temperature (r.t.), 2.5 h (for 7a); (ii) NH2OH·HCl, pyridine, ethanol, 85 °C, 4 h (for 7f); (iii) NH2OH·HCl, pyridine, methanol, r.t., 4 h (for 7g); (iv) TBSCl, imidazole, CH2Cl2, 0 °C to r.t., 3–4 h; (v) NBS, AIBN, CCl4, reflux, 3 h; (vi) potassium O-ethyl dithiocarbonate, acetone, r.t., 16–26 h; (vii) ZnCl2, Et2O, 0 °C to r.t., 6–8 d, then HCl aq.; (viii) Zn(OAc)2, LiOH, H2O/EtOH, r.t., 1–3 h. | |
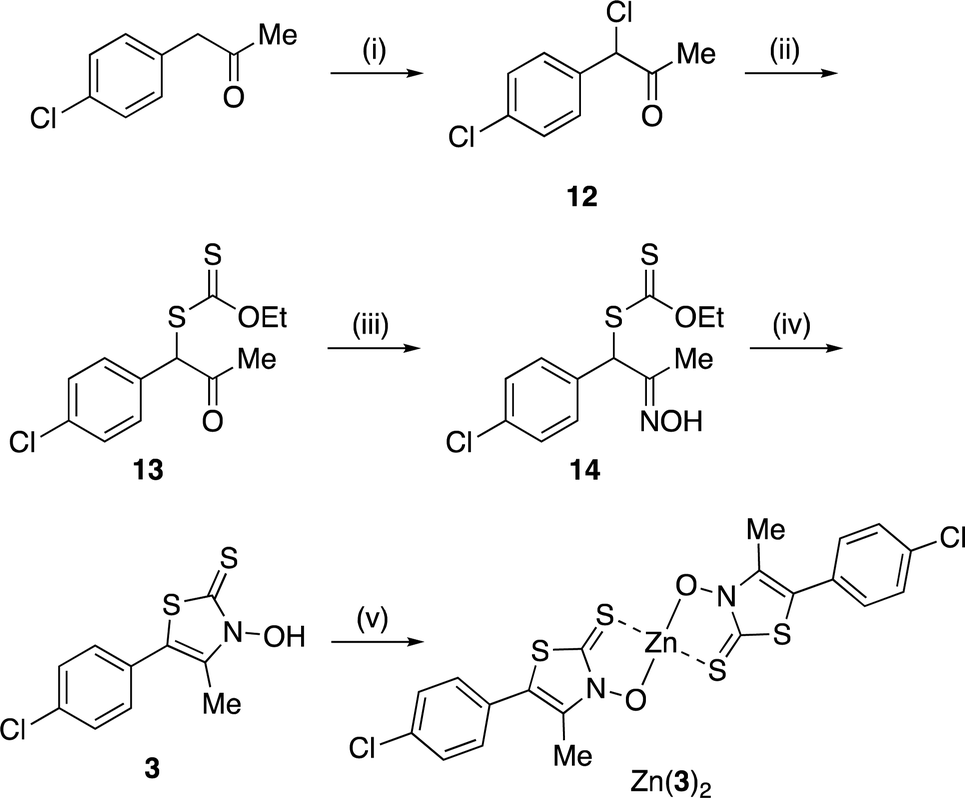
A zinc(II) complex of 3 (Zn(3)2) was obtained, as shown in Scheme 2. α-Chlorination of (p-chlorophenyl)acetone with sulfuryl chloride,17 followed by nucleophilic substitution with potassium O-ethyl dithiocarbonate, yielded xanthate 13, which was converted into the corresponding oxime 14 with hydroxylamine hydrochloride.17 The same cyclocondensation reaction used in the synthesis of 2 yielded the targeted 3,2-HTT 3.17 Subsequently, 3 was reacted with zinc perchlorate, yielding Zn(3)2. Notably, Zn(3)2 was obtained in high yield, even in the absence of lithium hydroxide.
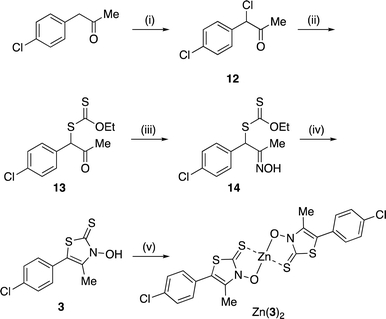 |
| | Scheme 2 Synthetic route to zinc(II) Zn(3)2. Reagents and conditions: (i) SO2Cl2, CH2Cl2, 0 °C to r.t., 22 h, quant.; (ii) potassium O-ethyl dithiocarbonate, acetone, r.t., 4 h, quant.; (iii) NH2OH·HCl, aqueous ethanol, r.t., 21 h, 91%; (iv) KOH, aqueous CH2Cl2, r.t., 48 h, 29%; (v) Zn(ClO4)2·6H2O, aqueous ethanol, r.t., 1 h, 75%. | |
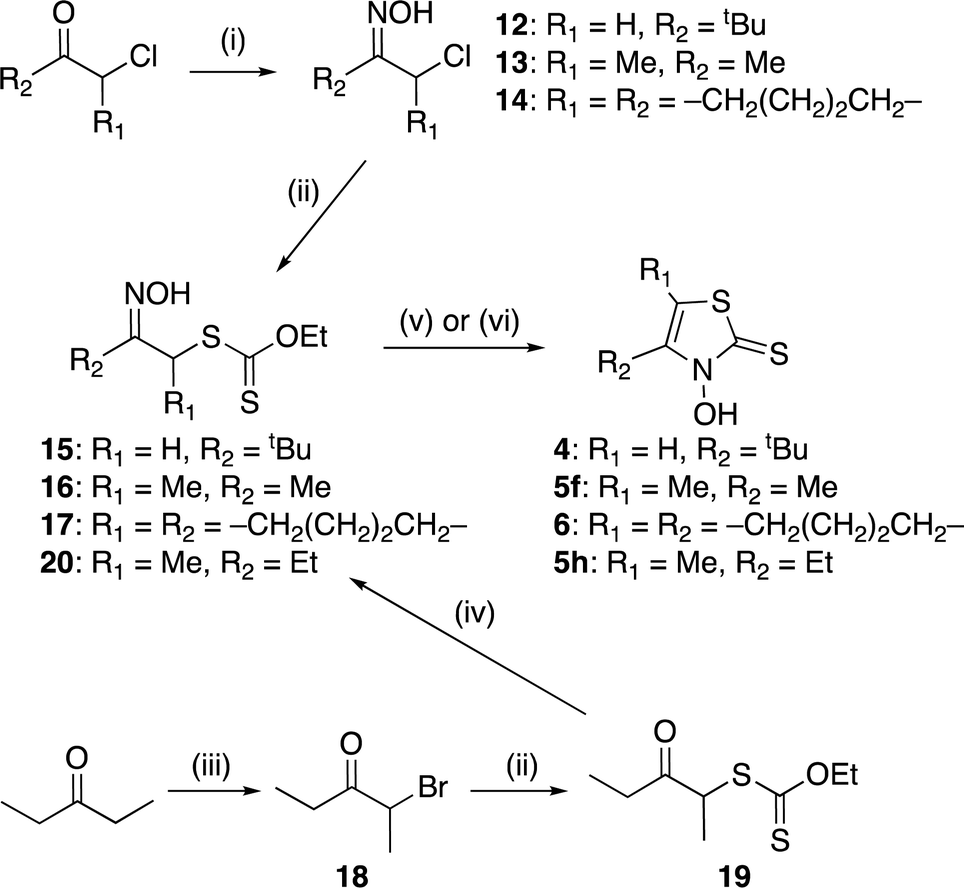
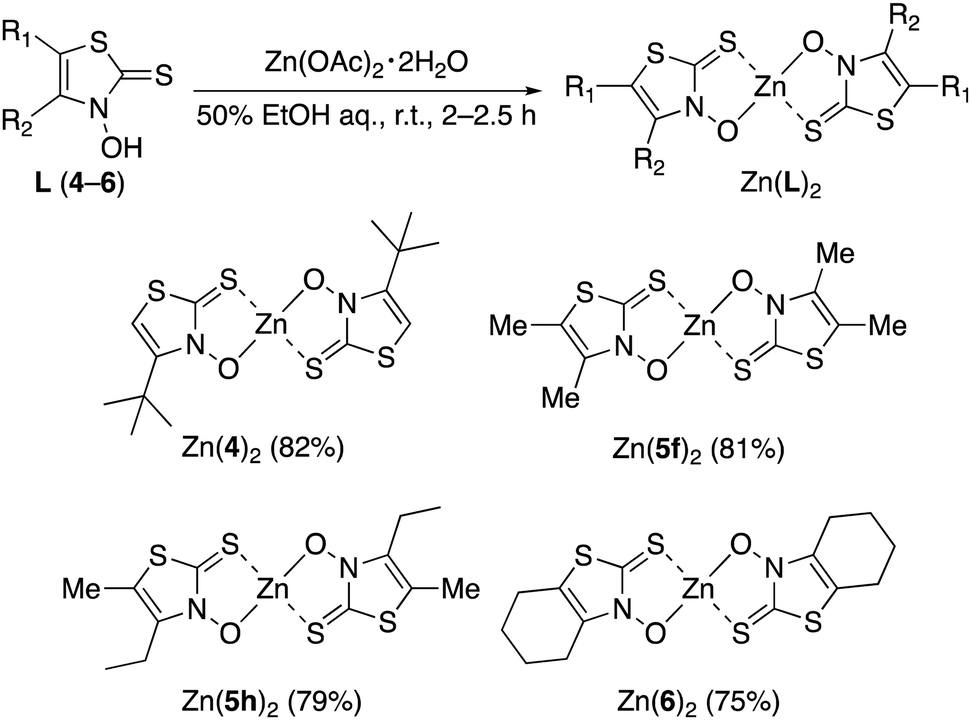
Ligands 4, 5f, 5h, and 6 were synthesized from the appropriate α-chloroketones, which were converted into the corresponding oximes 12–14 using hydroxylamine hydrochloride (Scheme 3). In contrast to the reactions in Schemes 1 and 2, these reactions require low-temperature conditions.18 When these oxidation reactions were performed under the same conditions as those in Schemes 1 and 2, multiple unisolable products were formed, and the desired chloride was not detected. Oximes 12–14 were then reacted with O-ethyl dithiocarbonate to obtain xanthates 15–17. Meanwhile, 2-bromo-3-pentanone (18), which was prepared from 3-pentanone and bromine,19,20 was treated with O-ethyl dithiocarbonate to obtain xanthate 19.17,21 Subsequently, 19 was converted into the corresponding oxime 20. In this case, the reaction conditions used for the preparation of 8g were employed. The obtained xanthates (15–17 and 20) were subjected to cyclocondensation reactions, yielding the targeted 3,2-HTTs 4–6 (Scheme 3).
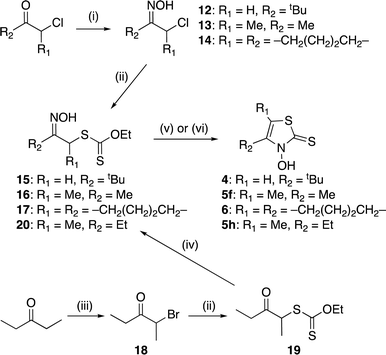 |
| | Scheme 3 Synthetic route to zinc(II) Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2. Reagents and conditions: (i) NH2OH·HCl, CH3COONa, water, −20 °C, 19–20 h; (ii) potassium O-ethyl dithiocarbonate, acetone, r.t., 2.5–19.5 h; (iii) Br2, Et2O, r.t., 45 min; (iv) NH2OH·HCl, pyridine, methanol, r.t., 11 h; (v) ZnCl2, Et2O, 0 °C to r.t., 43–66 h, then HCl aq. (for 15–17); (vi) KOH, CH2Cl2, 0 °C to r.t., 2.5 h, then HCl aq. (for 20); (vii) Zn(OAc)2·2H2O, H2O/EtOH, r.t., 2–2.5 h. | |
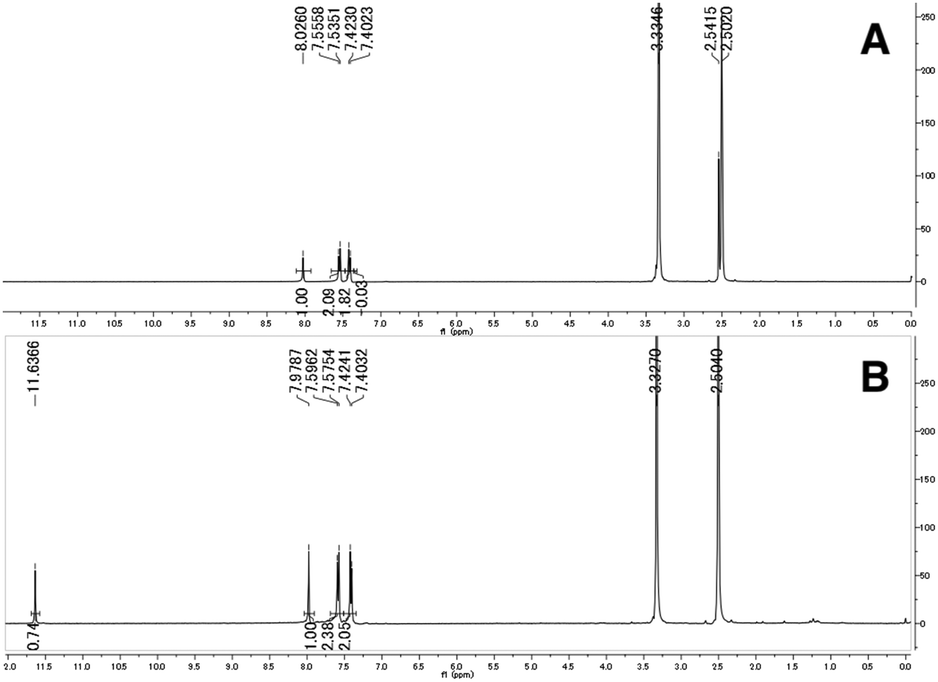
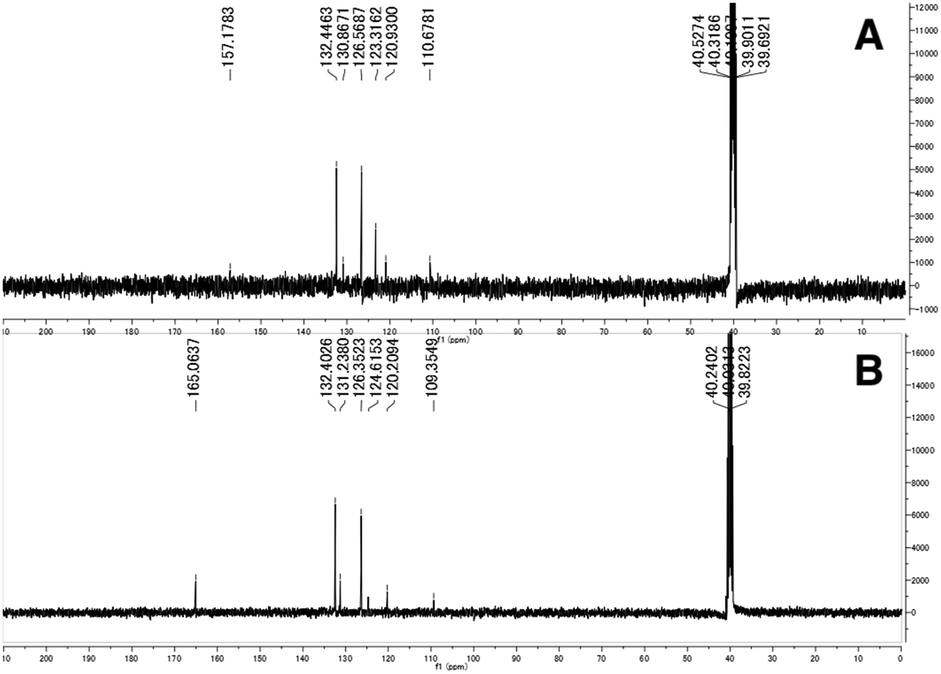
The synthesized 3,2-HTTs (4, 5f, 5h, and 6) were reacted with half the equimolar amount of zinc acetate dihydrate to form the desired zinc(II) complexes in high yield (Fig. 2). Structural determination of these complexes was performed via1H- and 13C-NMR, infrared (IR) spectroscopy, and combustion analysis. Upon complexation with zinc(II) ions, the OH signals of the ligands in 1H-NMR disappeared (Fig. 3), and all the carbon signals of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S groups shifted upfield (Fig. 4). In the IR spectra, for all the ligands, the peaks corresponding to the CS stretching vibrations, which are generally observed at approximately 1600 cm−1, of the thiocarbonyl groups were shifted to lower wavenumbers upon complexation (see Experimental section). Similar spectral changes were observed upon complexation of 1a–e with zinc(II) ions.16
S groups shifted upfield (Fig. 4). In the IR spectra, for all the ligands, the peaks corresponding to the CS stretching vibrations, which are generally observed at approximately 1600 cm−1, of the thiocarbonyl groups were shifted to lower wavenumbers upon complexation (see Experimental section). Similar spectral changes were observed upon complexation of 1a–e with zinc(II) ions.16
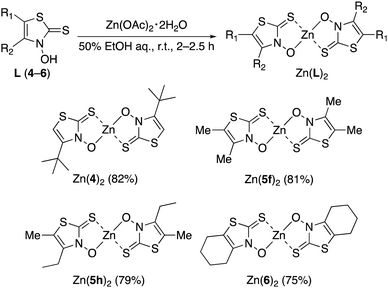 |
| | Fig. 2 Complexation reactions for the synthesis of zinc(II) complexes having 3,2-HTT ligands. | |
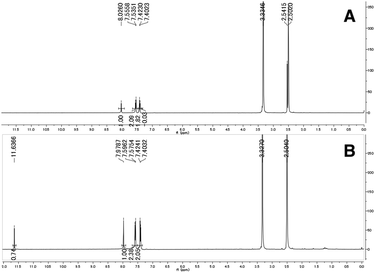 |
| | Fig. 3 Selected 1H-NMR spectra of 2f (A) and Zn(2f)2 (B) in dimethyl sulfoxide-d6. | |
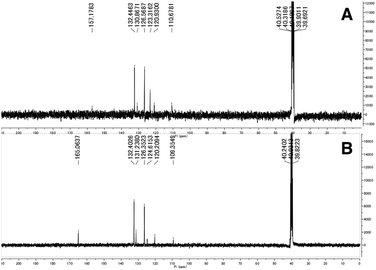 |
| | Fig. 4 Selected 13C-NMR spectra of 2f (A) and Zn(2f)2 (B) in dimethyl sulfoxide-d6. | |
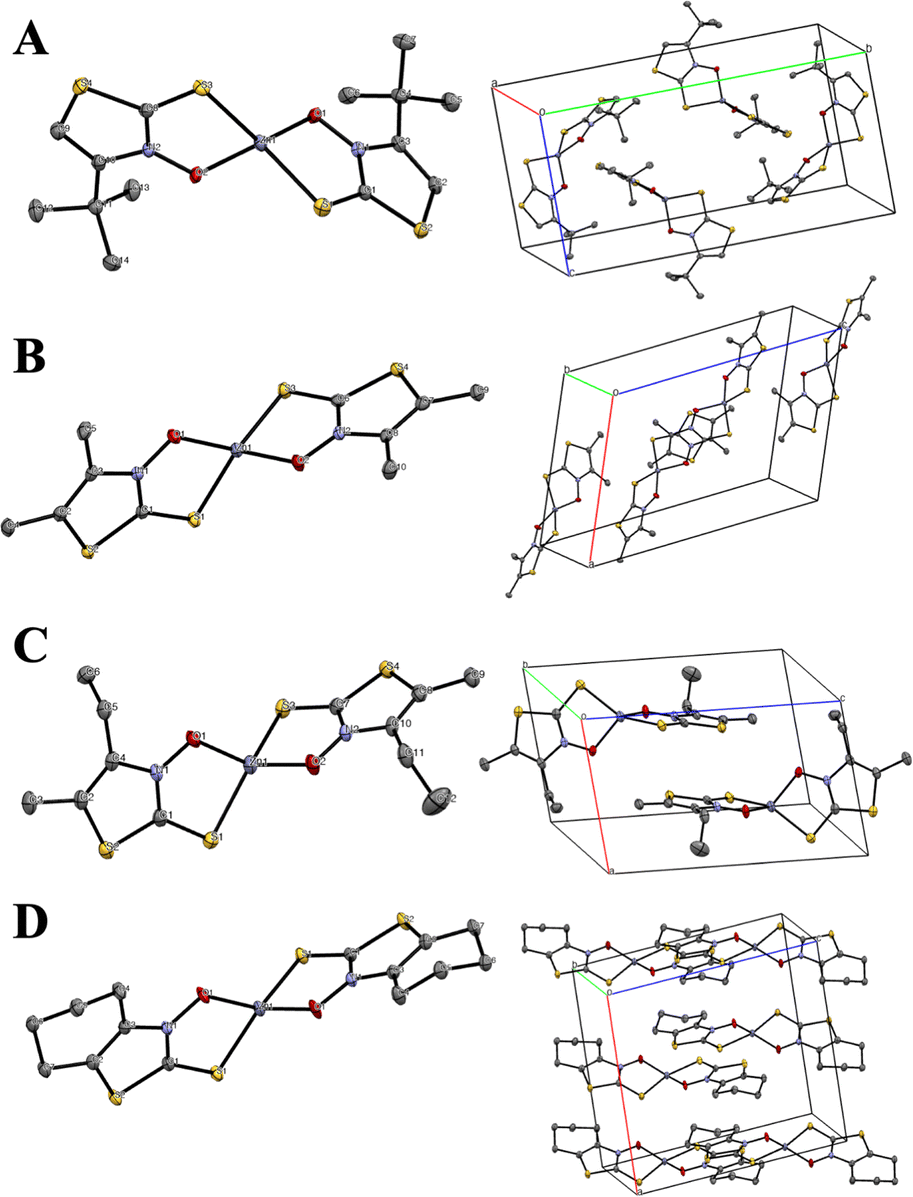
The zinc-ligand stoichiometry and binding modes of all the complexes were unambiguously determined via X-ray crystallographic analysis. The zinc(II) complexes Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2 were soluble in organic solvents, and single crystals suitable for X-ray diffraction analysis were obtained from a dichloromethane/n-hexane binary solvent system.22 The crystallographic data are summarized in Table 1. The crystals of Zn(4)2, Zn(5f)2, and Zn(6)2 were monoclinic, with space groups P21/n, P21/c, and C2/c, respectively. Zn(5h)2 crystallized in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (Table 1). Their solid-state structures are shown in Fig. 5, proving that the 4,5-dialkyl-3,2-HTT ligands coordinate to zinc(II) ions with a 1
(Table 1). Their solid-state structures are shown in Fig. 5, proving that the 4,5-dialkyl-3,2-HTT ligands coordinate to zinc(II) ions with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal-to-ligand ratio and in the trans S2O2 mode. In all cases, the coordination geometries around the central zinc(II) ions were tetrahedral. Selected bond distances and angles are presented in Table 2. The bond angles reveal that one of the two siemens–Zn–O angles is nearly 90° for all the complexes, whereas the other S–Zn–O, S–Zn–S, and O–Zn–O angles exhibit considerable variation (109.87° to 129.29°). The central angle of a tetrahedron is approximately 109.5°. Therefore, the tetrahedral coordination of these zinc(II) complexes was highly distorted. In contrast, Zn(2a)2, Zn(2f)2, Zn(2g)2, and Zn(3)2, which are composed of ligands containing phenyl rings, are poorly soluble in conventional organic solvents, and attempts to obtain single crystals for X-ray crystallographic analysis were unsuccessful in all but one case. The only exception was Zn(3)2, for which single crystals were obtained from dimethyl formamide with pyridine as an auxiliary solvent. The solved crystal data details for Zn(3)2 are compiled in Table 1, and the molecular structure of Zn(3)2 is depicted in Fig. 6(A).
:2 metal-to-ligand ratio and in the trans S2O2 mode. In all cases, the coordination geometries around the central zinc(II) ions were tetrahedral. Selected bond distances and angles are presented in Table 2. The bond angles reveal that one of the two siemens–Zn–O angles is nearly 90° for all the complexes, whereas the other S–Zn–O, S–Zn–S, and O–Zn–O angles exhibit considerable variation (109.87° to 129.29°). The central angle of a tetrahedron is approximately 109.5°. Therefore, the tetrahedral coordination of these zinc(II) complexes was highly distorted. In contrast, Zn(2a)2, Zn(2f)2, Zn(2g)2, and Zn(3)2, which are composed of ligands containing phenyl rings, are poorly soluble in conventional organic solvents, and attempts to obtain single crystals for X-ray crystallographic analysis were unsuccessful in all but one case. The only exception was Zn(3)2, for which single crystals were obtained from dimethyl formamide with pyridine as an auxiliary solvent. The solved crystal data details for Zn(3)2 are compiled in Table 1, and the molecular structure of Zn(3)2 is depicted in Fig. 6(A).
Table 1 Selected crystallographic data for Zn(3)2, Zn(4)2, Zn(5f)2, Zn(5h)2 and Zn(6)2
| Complex |
Zn(4)2 |
Zn(5f)2 |
Zn(5h)2 |
Zn(6)2 |
Zn(3)2 (with pyridine) |
Zn(5f)2 (with pyridine) |
| Formula |
C10H12N2O2S6Zn |
C10H12N2O2S4Zn |
C12H16N2O2S4Zn |
C14H16N2O2S4Zn |
C25H19Cl2N3O2S4Zn |
C15H17N3O2S4Zn |
|
F
w
|
441.93 |
385.83 |
413.88 |
437.90 |
657.97 |
464.93 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/c |
P |
C2/c |
P21/c |
P21 |
| Temperature(K) |
120 |
120 |
120 |
120 |
93 |
120 |
|
a/Å |
8.6523(3) |
11.6997(5) |
7.5733(16) |
11.9955(7) |
13.8856(3) |
8.3806(3) |
|
b/Å |
21.5296(8) |
7.5824(3) |
8.7066(19) |
10.9525(7) |
15.5863(4) |
13.3652(4) |
|
c/Å |
10.1015(4) |
17.5946(7) |
13.442(3) |
12.7788(8) |
12.3909(3) |
9.0229(3) |
|
α/deg |
90 |
90 |
78.741(2) |
90 |
90 |
90 |
|
β/deg |
103.2029(6) |
108.7288(6) |
74.963(2) |
94.2060(10) |
92.948(2) |
103.7934(4) |
|
γ/deg |
90 |
90 |
88.673(3) |
90 |
90 |
90 |
|
Z
|
4 |
4 |
2 |
4 |
4 |
2 |
|
μ(MoKα)/mm−1 |
1.805 |
2.293 |
1.964 |
1.974 |
1.459 |
1.69 |
| Max. and min. Trans. |
0.746 and 0.678 |
0.746 and 0.656 |
0.746 and 0.638 |
0.746 and 0.643 |
0.926 and 0.655 |
0.752 and 0.551 |
| 2θmax/deg. |
51.996 |
51.998 |
52.000 |
51.988 |
54.998 |
55.800 |
|
R
1 [I > 2σ(I)] on F |
0.0223 |
0.0299 |
0.0376 |
0.0232 |
0.0486 |
0.0168 |
| wR2 (all data) on F2 |
0.0570 |
0.0865 |
0.0963 |
0.0600 |
0.0696 |
0.0445 |
| Goodness-of-fit on F2 |
1.030 |
1.045 |
1.006 |
1.033 |
1.027 |
1.064 |
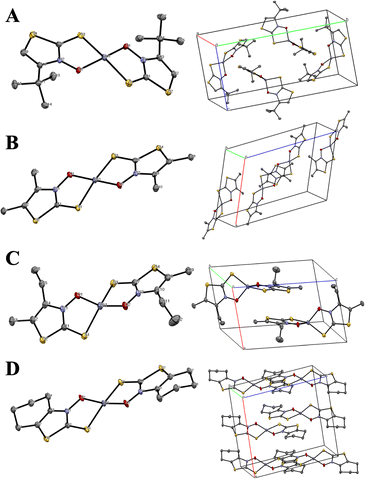 |
| | Fig. 5 Molecular structures with atomic numbering scheme (right side) and packing diagrams (left side) for (A) Zn(4)2, (B) Zn(5f)2, (C) Zn(5h)2, and (D) Zn(6)2. The thermal ellipsoids of non-H atoms are depicted at the 50% probability level. Hydrogen atoms are omitted for clarity. | |
Table 2 Selected bond length (d) and bond angles (ω) for Zn(3)2, Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2
| Bond |
d [Å] |
| Zn(4)2 |
Zn(5f)2 |
Zn(5h)2 |
Zn(6)2 |
Zn(3)2 (with pyridine) |
Zn(5f)2 (with pyridine) |
| Zn1–S1 |
2.2979 (5) |
2.3258 (7) |
2.3138(11) |
2.3163 (5) |
2.4121(6) |
2.3747(4) |
| Zn1–S3 |
2.3104 (5) |
2.3057 (7) |
2.3191(11) |
2.3163 (5) |
2.3961(6) |
2.3933(4) |
| Zn1–O1 |
1.9772 (12) |
1.974 (2) |
1.993(3) |
1.9790 (14) |
2.0758(13) |
2.0857(11) |
| Zn1–O2 |
1.9984 (12) |
1.9907 (19) |
1.967(3) |
1.9790 (14) |
2.0492(14) |
2.0892(11) |
| Bond angle |
ω [°] |
| O1–Zn1–O2 |
109.87 (5) |
110.03 (8) |
113.37(11) |
102.62 (9) |
165.18(6) |
175.50(5) |
| O1–Zn1–S3 |
124.56 (4) |
127.49 (6) |
114.08(8) |
129.51 (4) |
91.42(4) |
85.80(4) |
| O2–Zn1–S3 |
89.34 (3) |
90.21 (6) |
90.57(8) |
89.92 (4) |
86.28(4) |
93.14(3) |
| S1–Zn1–O1 |
90.43 (3) |
89.89 (6) |
89.75(8) |
89.92 (4) |
84.98(4) |
92.68(3) |
| S1–Zn1–O2 |
114.09 (4) |
120.25 (6) |
116.50(8) |
129.51 (4) |
89.48(4) |
84.78(3) |
| S1–Zn1–S3 |
129.181 (17) |
121.50 (3) |
133.87(4) |
118.96 (3) |
149.12(2) |
132.211(15) |
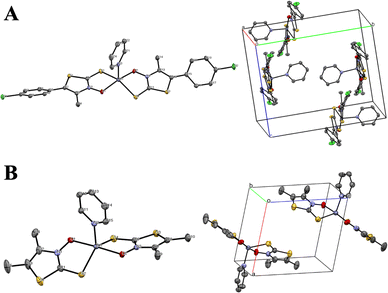 |
| | Fig. 6 Molecular structures with the atomic numbering scheme (right side) and packing diagrams (left side) for (A) Zn(3)2 and (B) Zn(5f)2 crystallized from pyridine-containing solvents. The thermal ellipsoids of non-H atoms are depicted at the 50% probability level. | |
Zn(3)2 crystallizes in the monoclinic space group P21/c. As shown in Fig. 6(A), Zn(3)2 has a square pyramidal structure with a 1:2 metal-to-ligand ratio and a trans S2O2 mode. In this case, a pyridine molecule is bound to the central zinc(II) ion at the apical position. Similar examples of solvent molecules coordinating to zinc(II) ions from apical positions have been observed in Zn(1b)2 and Zn(1d)2.16 These results suggest that the 3,2-HTTs/Zn complexes have one coordinating solvent molecule apically when a coordinating solvent is present in the medium. To confirm this, we recrystallized Zn(5f)2 from a pyridine/n-hexane binary solvent system and subjected the resulting single crystals to X-ray crystallographic analysis. As a result, apical coordination of pyridine to the central zinc atom was observed (Fig. 6(B)). From these results, it can be concluded that water molecules coordinate apically in vivo and assume a square pyramidal state, as observed in a zinc(II) complex with 4-methyl-3,2-HTT ligands reported by Bond et al.23
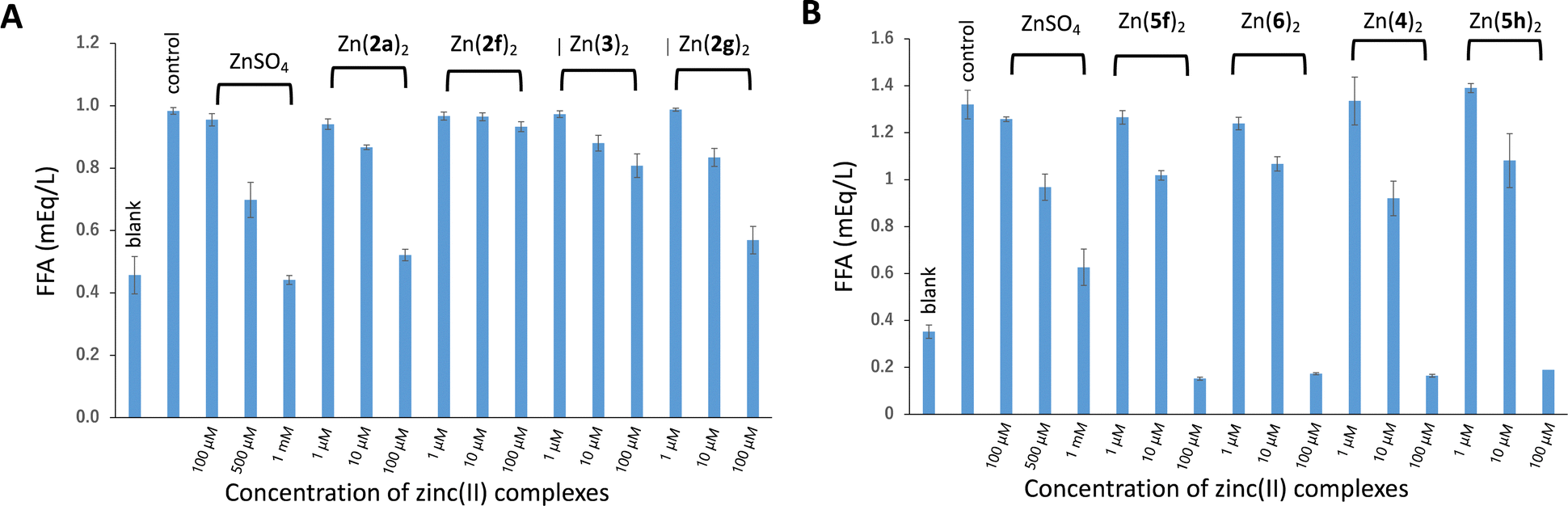
The insulin-mimetic activities of the synthesized zinc(II) complexes Zn(2)2–Zn(6)2in vitro were evaluated by measuring the effects of the complexes on free fatty acid (FFA) release from rat adipose cells stimulated with epinephrine. This is a simple and reliable method for assessing the insulin-mimetic activity of materials.24–27 The results are shown in Fig. 7. Of the nine complexes tested, six exhibited inhibitory activity toward the FFA release in a dose-dependent manner: Zn(2a)2, Zn(2g)2, Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2. This indicated that these complexes had insulin-mimetic activity. No significant inhibitory effects were observed for Zn(2f)2 or Zn(3)2. According to these results, the inhibitory activities of the six complexes were quantified by calculating the IC50 value, which represents the concentration of a complex at which FFA release from isolated rat adipocytes was inhibited by 50%. The results are summarized in Table 3. The insulin-mimetic activities (i.e., IC50 values) of the complexes having mono- and di-alkyl-3,2-HTT ligands (4–6) ranged from 16–24 μM, all of which were higher than those of Zn(2a)2 and Zn(2f)2, which have phenyl groups at the C5 position of the 3,2-HTT skeleton. These results indicate that the introduction of a more hydrophobic phenyl group reduced the insulin-mimetic activity of zinc(II) complexes with 3,2-HTT ligands. However, it has been reported that zinc complexes with 1a–e, which have phenyl groups, exhibit high insulin-mimetic activity. As these experiments were not conducted simultaneously, a direct comparison of the IC50 values was not feasible. Thus, to compare these values more accurately, the relative IC50 values were calculated using the IC50 value of zinc sulfate—the reference material—for both the present and previous experiments. The calculated relative IC50 values are presented in Table 3 for analysis and comparison. Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2, i.e., the zinc(II) complexes having the alkyl-substituted 3,2-HTT ligands, were found to have more than 4-fold higher inhibitory activity than the previously reported Zn(1a)2–Zn(1e)2 and 10-fold higher inhibitory activity than Zn(2a)2 and Zn(2f)2. Zn(5f)2 with a 3,2-HTT ligand with two methyl groups at the 4- and 5-positions was identified as the most potent among the 3,2-HTT-based zinc(II) complexes investigated. Thus, a complex with smaller and less hydrophobic functional groups on the 3,2-HTT skeleton exhibited the highest inhibitory activity, suggesting that the hydrophobicity and molecular size of the complexes affect the insulin-mimetic activity. Therefore, an attempt was made to quantify the partition coefficient (logP value), which is a lipophilic parameter of a compound and one of the most important measures in evaluating the pharmacokinetics of a drug, using reversed-phase high-performance liquid chromatography (HPLC). Among the 11 complexes exhibiting insulin-mimetic activity, Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2 were soluble in conventional organic solvents, and their logP values were successfully determined; however, the logP values of other complexes with phenyl groups could not be determined, because of their low solubility in the HPLC mobile phase. The obtained logP values are presented in Table 3, all of which are <5 and satisfy Lipinski's Rule of Five, i.e., logP < 5. However, these data are insufficient for meaningful comparisons. The calculated logP values (ClogP) for the 11 complexes are also presented in Table 3. These results indicated that Zn(4)2, Zn(5f)2, Zn(5h)2 and Zn(6)2, which were estimated to be less hydrophobic than the complexes with phenyl groups, tended to exhibit higher insulin-mimetic activity.
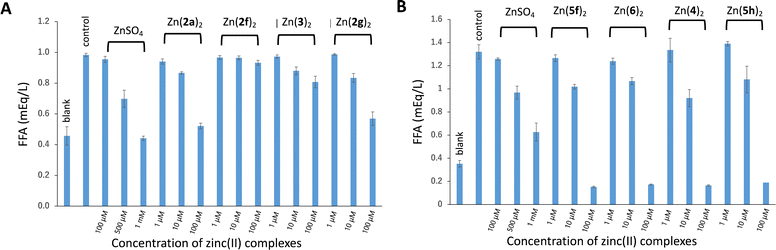 |
| | Fig. 7 Inhibitory effects of zinc(II) complexes on stimulated FFA release from rat adipocytes treated with epinephrine: (A) for Zn(2a)2, Zn(2f)2, Zn(2g)2, and Zn(3)2; (B) for Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2. Each column represents the mean ± standard deviation for three experiments. “Blank” refers to normal adipocyte, and “control” refers to epinephrine-stimulated rat adipocyte. | |
Table 3
In vitro insulin-mimetic activities, partition coefficients (logP), and overall stability constants (logβ) for Zn(2a)2, Zn(2f)2, Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2
| Compound |
IC50/μM |
Ratioa |
pIC50b |
pIC50,relc |
LogP |
C![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) LogP LogP |
Logβ |
|
Relative insulin-mimetic activity index based on the IC50 value of ZnSO4 as a standard.
The ordinary logarithmic representation of IC50 is defined as follows: pIC50 = −log (IC50).
Relative pIC50 values of zinc(II) complexes against zinc sulfate.
Values obtained in the experiment shown in Fig. 7(A).
Value obtained in the experiment shown in Fig. 7(B).
Data were obtained from ref. 11.
These values were calculated using the IC50 value of ZnSO4 (0.47 mM) measured in the same experiment.16
Because of the polarized electronic structure of the nitro groups, the calculated values were implausibly hydrophilic and were eliminated.
|
| Zn(2a)2 |
228 |
4.29 |
3.64 |
1.21 |
|
4.41 |
|
| Zn(2f)2 |
289 |
3.38 |
3.54 |
1.18 |
|
5.29 |
|
| Zn(4)2 |
19 |
60.9 |
4.73 |
1.61 |
4.34 |
3.37 |
14.40 |
| Zn(5f)2 |
16 |
70.7 |
4.79 |
1.63 |
3.27 |
1.56 |
12.26 |
| Zn(5h)2 |
21 |
55.5 |
4.69 |
1.59 |
2.21 |
2.08 |
12.76 |
| Zn(6)2 |
24 |
47.6 |
4.62 |
1.60 |
2.57 |
1.85 |
12.10 |
| ZnSO4 |
977d |
1 |
3.01d |
1 |
|
|
|
| 1,138e |
2.94e |
| Zn(1a)2 |
37f |
12.7g |
4.43 |
1.33 |
|
5.37 |
|
| Zn(1b)2 |
30f |
15.7g |
4.52 |
1.36 |
|
6.59 |
|
| Zn(1c)2 |
37f |
12.7g |
4.43 |
1.33 |
|
5.15 |
|
| Zn(1d)2 |
44f |
10.7g |
4.36 |
1.31 |
|
5.50 |
|
| Zn(1e)2 |
36f |
13.1g |
4.44 |
1.34 |
|
—h |
|
Next, we focused on the size of the complexes, as the results in Table 3 indicated that smaller molecules had higher insulin-like activity. To examine the effect of the molecular size on the insulin-mimetic activity, Verloop's sterimol parameters (L, B1, and B5) of the ligand moiety of the complex were estimated. These parameters are frequently used in quantitative structure–activity relationship studies of bioactive materials to quantify and compare the steric bulkiness of different substituents attached to a molecule. The descriptors L, B1, and B5 represent the length, minimum width, and maximum width of the substituents, respectively, and the estimated values are presented in Table 4. To examine the correlation between these parameters and insulin-like activity, we employed the pIC50 value—a commonly used index of inhibitory activity in structure–activity relationship studies of drug candidates—which is defined as follows:
Table 4 Physicochemical parameters of the 3-hydroxythiazole-2(3H)-thiones (3,2-HTTs) studied
| Compound |
L
|
B
1
|
B
5
|
pKa |
|
Total length of a ligand in bond direction.
Shortest distance from L axis when a ligand is projected on a plane perpendicular to L.
Longest distance from L axis when a ligand is projected on a plane perpendicular to L.
Each value was obtained by extrapolating a plot of the pKa values measured in 40.0%, 50.0%, and 66.7% aqueous THF.
Data were obtained from ref. 11.
|
|
4
|
5.5303 |
2.1439 |
5.7094 |
5.64d |
|
5f
|
6.5525 |
1.8709 |
4.3106 |
4.78d |
|
5h
|
6.3156 |
1.8543 |
5.0319 |
4.91d |
|
6
|
6.3006 |
1.9235 |
5.7004 |
4.68d |
|
1a
|
5.1222 |
2.4933 |
7.1821 |
4.01e |
|
1b
|
5.1150 |
2.4699 |
8.2762 |
4.91e |
|
1c
|
5.1173 |
2.4726 |
9.0764 |
4.63e |
|
1d
|
5.1145 |
2.4677 |
7.6386 |
4.80e |
|
1e
|
5.1132 |
2.4733 |
8.5457 |
3.92 |
The relative pIC50 values of the zinc(II) complexes with 3,2-HTT ligands against the pIC50 of zinc sulfate were plotted against the above descriptors, as shown in Fig S1 (ESI†). A negative correlation was observed between the relative pIC50 values and B5, and a weak negative correlation was observed with L. These results suggest that the molecular size affects the insulin-like activity, with smaller complexes tending to yield better results.
In addition to the lipophilicity and the molecular size, the stability constant (logβ) of the complexes is an important factor affecting insulin-mimetic activity, as it has been reported that high insulin-mimetic activity is observed in zinc(II) complexes with logβ values not exceeding 10.5, while the activity tends to be lower for complexes with logβ exceeding 10.5.28 Prior to studying the correlation between the stability constant of the complex and the insulin-mimetic activity, the acid dissociation constants (pKa values), which are directly related to the stability of the complexes, of 4, 5f, 5h, and 6 were quantified. Because the ligands are insoluble in water, the pKa values were measured in a tetrahydrofuran (THF)–water binary solvent with various THF ratios, and the obtained values were plotted against the THF content (volume%) and extrapolated to estimate the pKa value in 100% water.16 Other ligands could not be measured, because they were insoluble, even in this solvent. The pKa values of 4, 5f, 5h, and 6 were estimated to be within the range of 4.6–5.6, indicating that 3,2-HTTs are acidic and comparable to acetic acid (Table 4). The disparity in the pKa values of the ligands is attributed to the electron-donation strength of the alkyl groups. The stability constants (logβ values) of Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2 were determined using the Bjerrum method.29–32 All measurements were conducted in 50% THF, and the pKa values of the corresponding ligands in 50% THF were used to calculate the logβ values. The results are summarized in Table 3. The logβ values of the four zinc complexes were in the range of 12–14. As reported by Irving and Rossotti,33 the stability constant of metal complexes is approximately proportional to the pKa value of the ligand measured under the same conditions. In addition, data reported by Mui et al. indicated a proportional relationship between the stability constants of metal complexes in organic solvents with varying water contents and the pKa values of the ligands in the corresponding organic solvents with the same water content.34 Considering these findings, the stability constants of the complexes Zn(4)2, Zn(5f)2, Zn(5h)2, and Zn(6)2 in water in this study were estimated to be approximately 8.7, 8.8, 10.4, and 9.0, respectively, satisfying the condition of <10.5.
Conclusions
We demonstrated that newly synthesized zinc(II) complexes with 4-alkylated and 4,5-dialkylated 3,2-HTT ligands exhibit higher insulin-like activity than those with 3,2-HTT ligands with phenyl substituents. Zn(5f)2, which has two methyl groups at positions 4 and 5, is the most potent among the zinc(II) complexes with 3,2-HTT ligands synthesized to date. The lipophilicity and molecular size of the complexes were identified as factors exerting a degree of influence on the observed increase in activity. The experimental logP values of the complexes with alkylated ligands were <5. A comparison of the experimental logP and ClogP values of the complexes with alkylated 3,2-HTT ligands suggested that the logP values for complexes with phenyl groups were >5. Thus, higher activity was observed for complexes with logP values of <5 owing to the smaller alkyl groups. Regarding the effect of the stability constant of the complex on the insulin-mimetic activity, the logβ was maintained within a range not exceeding 10.5, strengthening the versatility of the guideline of the stability constants required for complexes that exhibit high insulin-mimetic activity. The relationship between the hydrophobicity and molecular size of the complexes and their insulin-mimetic activity has not been sufficiently clarified. Thus, the findings of this study provide valuable guidance for the development of more potent hypoglycemic zinc complexes.
Experimental
General
1H-NMR spectra were recorded on an ECP-400 spectrometer (JEOL Ltd, Japan). Chemical shifts (δ) are reported in ppm using tetramethylsilane or an undeuterated solvent as internal standards in the deuterated solvent used. Coupling constants (J) are given in Hz. 13C-NMR spectra were recorded on an ECP-400 spectrometer (JEOL Ltd, Japan). Chemical shift multiplicities are reported as s = singlet, d = doublet, t = triplet, q = quintet, m = multiplet. Infrared (IR) spectra were obtained using FT/IR-4100, FT/IR-660plus, or FT/IR-460plus spectrophotometers (JASCO, Ltd, Japan). Fast-atom-bombardment (FAB) mass spectra were taken on a JMS-600-H mass spectrometer (JEOL Ltd, Japan). Xenon was used as a bombardment gas, and all analyses were carried out in positive mode with the ionization energy and the accelerating voltage set at 70 eV and 3 kV, respectively. A mixture of dithiothreitol (DTT) and α-thioglycerol (TG) (1:1) was used as a liquid matrix. All conventional chemicals used in the present study are commercially available and were used as received. Column chromatography was carried out on silica gel (particle size; 46–50 μM; Fuji Silysia Chemical Ltd, Japan). Compounds 3, 12–14, 18, and 19 were synthesized by the method reported in the literature cited in the main text.
Synthesis
2-Phenylacetaldehyde oxime (8a).
To a suspension of 2-phenylacetaldehyde (7a, 1.99 g, 16.6 mmol) and hydroxylamine hydrochloride (1.16 g, 16.6 mmol) in 50% aqueous methanol (12 mL), an aqueous solution of sodium carbonate (881 mg, 8.31 mmol) was slowly added. The resulting mixture was stirred at room temperature for 2.5 h, and then methanol was evaporated. H2O (20 mL) was added to the reaction mixture followed by extraction with Et2O (3 × 50 mL). The organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting pale-yellow residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (4/1, v/v) as eluents to give 8a as a white solid (1.23 g, 55%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 3.54 (d, 2H, J = 6.1 Hz, Z-isomer), 3.75(d, 2H, J = 5.3 Hz, E-isomer), 6.90 (t, 1H, J = 5.3 Hz, E-isomer), 7.21–7.35 (m, 10H, E-, and Z-isomer), 7.54 (t, 1H, J = 6.1 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 31.8, 36.0, 126.8, 127.0, 128.9, 128.9, 128.9, 136.2, 136.7, 150.8, 151.0. The spectroscopic data for this compound are consistent with data available in the literature.35
2-(p-Tolyl)acetaldehyde oxime (8f).
Hydroxylamine hydrochloride (317 mg, 4.57 mmol) was added to a solution of 2-(p-tolyl)acetaldehyde (7f, 670 mg, 3.76 mmol) in methanol (15 mL) followed by pyridine (0.30 mL, 3.79 mmol). The mixture was refluxed for 2 h. After evaporation of the solvent, H2O (20 mL) was added to the reaction mixture followed by extraction with dichloromethane (2 × 20 mL). The organic phase was washed with 1.0 M HCl (13 mL), and then dried over sodium sulfate. After evaporation of the solvent, the resulting pale yellow residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (4/1, v/v) as eluents to give 8f as a white solid (407 mg, 56%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 2.33 (s, 6H, E- and Z-isomer), 3.49 (d, 2H, J = 6.2 Hz, Z-isomer), 3.70, (d, 2H, J = 5.2 Hz, E-isomer), 6.89 (t, 1H, J = 5.2 Hz, E-isomer), 7.09–7.14 (m, 8H, E- and Z-isomer), 7.52 (t, 1H, J = 6.2 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 21.10, 31.22, 128.7, 128.8, 129.5, 133.6, 136.3, 151.5. The spectroscopic data for this compound are consistent with data available in the literature.36
2-(4-Bromophenyl)acetaldehyde oxime (8g).
This material was prepared from 7g (871 mg, 4.37 mmol) with hydroxylamine hydrochloride (370 mg, 5.33 mmol) in a procedure analogous to that for obtaining 8f. The product was obtained as a white solid (468 mg, 41%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 3.48 (d, 2H, J = 6.1 Hz, Z-isomer), 3.68 (d, 2H, J = 5.4 Hz, E-isomer), 6.85 (t, 1H, J = 5.4 Hz, E-isomer), 6.94 (s, 1H, OH), 7.07(s, 1H, OH), 7.10(d, 4H, J = 7.9 Hz, E- and Z-isomer), 7.44 (d, 4H, J = 7.9 Hz, E- and Z-isomer), 7.49 (t, 1H, J = 6.1 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 31.01, 35.33, 130.6, 130.6, 131.9, 131.9, 150.3. The spectroscopic data for this compound are consistent with data available in the literature.37
2-Phenylacetaldehyde O-(tert-butyldimethylsilyl)oxime (9a).
To a solution of 8a (1.23 g, 9.14 mmol) and tert-butylchlorodimethylsilane (2.09 g, 13.9 mmol) in dichloromethane (20 mL), imidazole (1.25 g, 18.4 mmol) was slowly added at 0 °C. The resulting mixture was stirred at room temperature for 4 h. H2O (100 mL) was added to the reaction mixture, and the organic materials were extracted with chloroform (2 × 100 mL). The organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting pale yellow residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (9/1, v/v) as eluents to give 9a as a pale yellow liquid (2.16 g, 94%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.18 (s, 6H, Z-isomer), 0.21 (s, 6H, E-isomer), 0.9 (s, 9H, Z-isomer), 0.96 (s, 9H, E-isomer), 3.53 (d, 2H, J = 6.4 Hz, Z-isomer), 3.74 (d, 2H, J = 5.2 Hz, E-isomer), 7.02 (t, 1H, J = 5.2 Hz, E-isomer), 7.20–7.33 (m, 10H, E- and Z-isomer), 7.58 (t, 1H, J = 6.4 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.3, 25.8, 26.2, 126.6, 128.8, 128.9, 137.2, 154.2. This material was used directly in the next step without further purification.
3-hydroxy-5-phenylthiazole-2(3H)-thione (2a) (via10a and 11a).
To a slurry of 9a (2.16 g, 8.67 mmol) and N-bromosuccinimide (1.55 g, 8.71 mmol) in tetrachloromethane (40 mL), 2,2′-azodiisobutyronitrile (83.7 mg, 0.588 mmol) was slowly added. The mixture was then refluxed for 3 h. After cooling to room temperature, the reaction mixture was passed through a filter and the filtrate was washed successively with a 10% sodium thiosulfate solution (50 mL) and water (2 × 50 mL), and the organic phase was dried over sodium sulfate. After evaporation of the solvent, 10a was obtained as an orange oily material (3.10 g). This material was directly used in the next step without further purification. 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.15 (s, 6H, E-isomer), 0.17 (s, 6H, Z-isomer), 0.92 (s, 9H, E-isomer), 0.92 (s, 9H, Z-isomer), 5.70 (d, 1H, J = 8.9 Hz, E-isomer), 6.29 (d, 1H, J = 7.7 Hz, Z-isomer), 7.30–7.47 (m, 10H, E- and Z-isomer), 7.42 (d, 1H, J = 7.7 Hz, Z-isomer) 7.92 (d, 1H, J = 8.9 Hz, E-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.2, 18.3, 26.0, 26.1, 40.3, 48.6, 127.9, 128.1, 128.8, 128.9, 129.0, 138.0, 138.1, 152.7, 153.4. To a slurry of potassium O-ethyldithiocarbonate (1.47 g, 9.18 mmol) in acetone (45 mL) was added a solution of 10a (3.00 g, 9.16 mmol) over a period of 15 min at room temperature. The reaction mixture was stirred for 26 h at room temperature. After evaporation of the solvent, H2O (50 mL) was added to the residue, and organic materials were extracted with ethyl acetate (3 × 50 mL). The combined organic phase was dried over sodium sulfate. After evaporation of the solvent, 11a was obtained as an orange liquid (4.77 g). This material was directly used in the next step without further purification. 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.16 (s, 6H, Z-isomer), 0.16 (s, 6H, E-isomer), 0.89 (s, 9H, Z-isomer), 0.91 (s, 9H, E-isomer), 1.33–1.44 (m, 6H, E- and Z-isomer), 4.56–4.68 (m, 4H, E- and Z-isomer), 5.58 (d, 1H, J = 6.5 Hz, E-isomer), 6.15 (d, 1H, J = 6.7 Hz, Z-isomer), 7.21 (d, 1H, J = 6.7 Hz, Z-isomer), 7.29–7.35 (m, 10H, E- and Z-isomer), 7.71 (d, 1H, J = 6.5 Hz, E-isomer).13C NMR (chloroform-d, 100 MHz) δ/ppm 13.7, 13.8, 18.1, 18.3, 25.9, 26.1, 52.7, 53.9, 128.0, 128.2, 128.3, 128.5, 128.9,136.2, 136.4, 152.2, 152.7, 211.2, 211.5. Zinc chloride (6.04 g, 47.1 mmol) was added to a solution of 11a (4.77 g, 12.9 mmol) in diethyl ether (30 mL, anhydrous) at such a rate that the solvent did not boil constantly, and then the mixture was stirred for 8 days at room temperature. 1.0 M HCl (21 mL) was added dropwise to the mixture, and then the reaction mixture was stirred for 30 min at 0 °C to room temperature. The solution was filtered and the filtrate was extracted with chloroform (3 × 60 mL). The organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting brown residue was purified by column chromatography on silica gel with chloroform/ethyl acetate (1/1, v/v) as eluents to give 2a as a brown solid (282 mg, 29%). 1H NMR (chloroform-d, 400 MHz) δ/ppm 7.18 (s, 1H), 7.30–7.40 (m, 5H). 13C NMR (chloroform-d, 100 MHz) δ/ppm 115.6, 119.5, 124.9, 125.2, 128.4, 129.2, 130.6. IR (KBr): 3126 (νC–H), 1647 (νCS), 1509 (νN–O), 1348 (νN–O), and 756 cm−1 (δC–H). Anal. calcd for C9H7NOS2·0.2H2O: C, 50.77; H, 3.50; N, 6.58%. Found: C, 50.81; H, 3.28; N, 6.54%.
2-(p-Tolyl)acetaldehyde O-(tert-butyldimethylsilyl) oxime (9f).
This material was prepared from 8f (643 mg, 4.31 mmol) with tert-butylchlorodimethylsilane (979 mg, 6.49 mmol) in a procedure analogous to that for obtaining 9a. The product (9f) was obtained as pale-yellow oily material (908 mg, 80%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.18 (s, 6H, Z-isomer), 0.20 (s, 6H, E-isomer), 0.94 (s, 9H, Z-isomer), 0.96 (s, 9H, E-isomer), 2.32 (s, 6H, E- and Z-isomer), 3.48 (d, 2H, J = 6.4 Hz, Z-isomer), 3.69 (d, 2H, J = 5.1 Hz, E-isomer), 6.99 (t, 1H, J = 5.1 Hz, E-isomer), 7.08–7.13 (m, 8H, E- and Z-isomer), 7.55 (t, 1H, J = 6.4 Hz, Z-isomer)). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.24, 21.10, 26.18, 31.79, 35.64, 128.7, 129.4, 134.1, 136.1, 154.2, 154.6. This material was used directly in the next step without further purification.
3-Hydroxy-5-(p-tolyl)thiazole-2(3H)-thione (2f) (via10f and 11f).
Bromide 10f (2.28 g, orange oily material) was prepared from 9f (665 mg, 2.02 mmol) with N-bromosuccinimide (388 mg, 2.18 mmol) and 2,2′-azodiisobutyronitrile (20.9 mg, 0.127 mmol) in a procedure analogous to that for obtaining 10a, and was directly used in the next step without further purification. 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.15 (s, 6H, E-isomer), 0.17 (s, 6H, Z-isomer), 0.92 (s, 9H, E-isomer), 0.93 (s, 9H, Z-isomer), 2.34 (s, 6H, E- and Z-isomer), 5.69 (d, 1H, J = 9.0 Hz, E-isomer), 6.28 (d, 1H, J = 7.7 Hz, Z-isomer), 7.17 (d, 2H, J = 7.7 Hz, E-isomer), 7.34 (d, 2H, J = 7.7 Hz, E-isomer), 7.38 (d, 1H, J = 7.7 Hz, Z-isomer), 7.22 (d, 2H, J = 7.6 Hz, Z-isomer), 7.42 (d, 2H, J = 7.6 Hz, Z-isomer), 7.93 (d, 1H, J = 9.0 Hz, E-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.21, 21.28, 26.05, 40.29, 48.69, 127.7, 127.9, 129.7, 135.2, 139.0, 152.7, 153.4. The obtained crude 10f (2.28 g, 6.66 mmol (crude), orange oily material) was converted into 11f (brown liquid, 2.55 g) by reacting with O-ethyldithiocarbonate (1.11 g, 6.94 mmol) in acetone (48 mL) using the same procedure that for obtaining 11a. The obtained 11f was directly used in the next step without further purification; 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.16 (s, 6H, E-isomer), 0.17 (s, 6H, Z-isomer), 0.90 (s, 9H, Z-isomer), 0.92 (s, 9H, E-isomer), 1.36–1.44 (m, 6H, E- and Z-isomer), 2.33 (s, 6H, E- and Z-isomer), 4.57–4.66 (m, 4H, E- and Z-isomer), 5.54 (d, 1H, J = 6.6 Hz, E-isomer), 6.13 (d, 1H, J = 6.9 Hz, Z-isomer), 7.19 (d, 1H, J = 6.9 Hz, Z-isomer), 7.11–7.31 (m, 8H, E- and Z-isomer), 7.71 (d, 1H, J = 6.6 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.96, 23.28, 23.45, 26.40, 31.15, 31.32, 52.98, 57.59, 75.31, 133.3, 133.6, 133.9, 134.8, 138.2, 138.5, 143.0, 143.3, 157.5, 158.0. Ligand 2f was then prepared by reacting the crude 11f (1.14 g, 2.96 mmol) with zinc chloride (4.10 g, 2.99 mmol) in diethyl ether (5 mL, anhydrous) in a procedure analogous to that for obtaining 2a, and obtained as a gray solid (285 mg, 19%, 3 steps). 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm 2.30 (s, 3H), 7.20 (d, 2H, J = 8.0 Hz), 7.34 (d, 2H, J = 8.0 Hz) 11.6 (s, 1H). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 21.28, 112.1, 121.8, 124.6, 128.7, 130.1, 137.6, 165.1. IR (KBr): 3159 (νC–H), 1663 (νCS), 1508 (νN–O), 1414 (νN–O), and 780 cm−1 (δC–H). HR-ESIMS calcd for C10H10NOS2+ [M + H]+ 224.0198. Found 224.0227.
Synthesis of 2-(4-bromophenyl)acetaldehyde O-(tert-butyldimethylsilyl) oxime (9g)
This material was prepared from 8g (468 mg, 2.19 mmol) with tert-butylchlorodimethylsilane (687 mg, 4.56 mmol) in a procedure analogous to that for obtaining 9a. The product (9g) was obtained as a pale-yellow oily material (665 mg, 93%). 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.17 (s, 6H, Z-isomer), 0.20 (s, 6H, E-isomer), 0.93 (s, 9H, Z-isomer), 0.95 (s, 9H, E-isomer), 3.47 (d, 2H, J = 6.4 Hz, Z-isomer), 3.68 (d, 2H, J = 5.2 Hz, E-isomer), 6.97 (t, 1H, J = 5.2 Hz, E-isomer), 7.07 (d, 2H, J = 8.0 Hz, Z-isomer), 7.08 (d, 2H, J = 8.0 Hz, E-isomer), 7.43 (d, 4H, J = 8.0 Hz, E- and Z-isomer), 7.56 (t, 1H, J = 6.4 Hz, Z-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 18.23, 26.08, 31.60, 65.55, 120.0, 130.8, 131.7, 136.6, 153.5. This material was used directly in the next step without further purification.
5-(4-Bromophenyl)-3-hydroxythiazole-2(3H)-thione (2g) (via10g and 11g).
Bromide 10g was prepared from 8g (665 mg, 2.02 mmol) with N-bromosuccinimide (388 mg, 2.18 mmol) and 2,2′-azodiisobutyronitrile (20.9 mg, 0.127 mmol) in a procedure analogous to that for obtaining 8f. The product (9g) was obtained as an orange oily material (1.09 g). This crude material was directly used in the next step without further purification; 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.17 (s, 6H, E-isomer), 0.19 (s, 6H, Z-isomer), 0.91 (s, 9H, E-isomer), 0.92 (s, 9H, Z-isomer), 2.17 (s, 6H, E and Z-isomer), 5.64 (d, 1H, J = 8.5 Hz, E-isomer), 6.23 (d, 1H, J = 7.5 Hz, Z-isomer), 7.32 (d, 2H, J = 8.2 Hz, E-isomer), 7.33 (d, 2H, J = 8.2 Hz, Z-isomer), 7.36 (d, 1H, J = 7.5 Hz, Z-isomer), 7.50 (d, 4H, J = 8.2 Hz, E- and Z-isomer) 7.86(d, 1H, J = 8.5 Hz, E-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 23.45, 16.52, 31.28, 45.42, 53.92, 133.0, 134.9, 140.4, 144.3, 157.9, 158.6. From the obtained crude 10g (1.09 g, 2.67 mmol), 11g was prepared using potassium O-ethyldithiocarbonate (478 mg, 2.98 mmol) in a procedure analogous to that for obtaining 10a, and was obtained as a yellow oil (908 mg). This material was directly used in the next step without further purification; 1H NMR (chloroform-d, 400 MHz; a mixture of E- and Z-isomer) δ/ppm 0.15 (s, 6H, E-isomer), 0.16 (s, 6H, Z-isomer), 0.91 (s, 9H, Z-isomer), 0.92 (s, 9H, E-isomer), 1.38 (t, 6H, J = 7.0 Hz, E- and Z-isomer), 4.59–4.65 (m, 4H, E- and Z-isomer), 5.53 (d, 1H, J = 6.2 Hz, E-isomer), 6.09 (d, 1H, J = 6.6 Hz, Z-isomer), 7.17 (d, 1H, J = 6.6 Hz, Z-isomer), 7.22–7.24 (m, 4H, E- and Z-isomer), 7.45–7.50 (m, 4H, E- and Z-isomer), 7.68 (d, 1H, J = 6.2 Hz, E-isomer). 13C NMR (chloroform-d, 100 MHz) δ/ppm 13.77, 14.28, 18.25, 21.13, 25.92, 26.09, 47.65, 52.25, 60.47, 70.36, 122.2, 122.9, 130.3, 131.9, 132.0, 135.7, 151.6, 151.9. Ligand 2g was then prepared by reacting the crude 11g (343 mg, 0.764 mmol) with zinc chloride (519 mg, 3.81 mmol) in diethyl ether (10 mL, anhydrous) in a procedure analogous to that for obtaining 2a, and obtained as a gray solid (124 mg, 45%, 3 steps). 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm. 7.41 (d, 2H, J = 8.3 Hz), 7.58 (d, 2H, J = 8.3 Hz), 7.98 (s, 1H), 11.64 (s, 1H). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 110.7, 120.9, 123.3, 126.6, 130.9, 132.4, 165.1. IR (KBr): 3087 (νC–H), 1653 (νCS), 1474 (νN–O), 1401 (νN–O), and 768 cm−1 (δC–H). ESIMS calcd for C9H579BrNS2 [M − OH]−: 269.9052 and C9H581BrNS2 [M − OH]−: 271.9031. Found: 269.9615 and 271.9545, respectively. Anal. calcd for C9H6BrNOS2·0.25EtOAc: C, 38.72; H, 2.60; N, 4.52%. Found: C, 38.88; H, 2.32; N, 4.98%.
Bis[3-hydroxy-5-phenylthiazole-2(3H)-thionolato]zinc(II): Zn(2a)2.
To a solution of 2a (23.7 mg,134 mmol) in 50% aqueous ethanol (2 mL) was successively added LiOH·2H2O (5.4 mg, 129 mmol) and a solution of Zn(OAc)2·2H2O (14.7 mg, 67.0 mmol) in H2O (1.0 mL). The mixture was then stirred at room temperature for 1 hour. The resulting precipitate was collected by filtration and washed with H2O and THF to give Zn(2a)2 as a white solid (16.6 mg, 34.6 μmol, 52%). 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm 7.24 (t, 1H, J = 7.3 Hz), 7.36 (dd, 2H, J = 7.3, 7.6 Hz), 7.45 (d, 2H, J = 7.6 Hz), 7.89 (s, 1H). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 110.9, 123.9, 124.6, 127.6, 129,5, 132.0, 165.2. IR (KBr): 3076 (νC–H), 1579 (νCS), 1491 (νN–O), 1363 (νN–O), and 750 cm−1 (δC–H). Anal. calcd for C18H12N2O2S4Zn·1.6H2O: C, 42.32; H, 3.00; N, 5.49% found: C, 42.14; H, 2.80; N, 5.35%.
Bis[3-hydroxy-5-(p-tolyl)thiazole-2(3H)-thionolato]zinc(II): Zn(2f)2.
The complex Zn(2f)2 was prepared from 11f (48.0 mg, 0.215 mmol) with Zn(OAc)2·2H2O (18.2 mg, 82.9 μmol) in a procedure analogous to that used for the preparation of Zn(2a)2, with the exception that the reaction time was 3 hours. Zn(2f)2 was obtained as white solid (28.1 mg, 55.4 μmol, 67%). 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm 2.29 (s, 3H), 7.18 (d, 2H, J = 7.9 Hz), 7.35 (d, 2H, J = 7.9 Hz), 7.88 (s, 1H). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 21.26, 110.9, 124.1, 123.3, 124.5, 129.1, 130.1, 137.0, 164.7. IR (KBr): 3087 (νC–H), 1575 (νCS), 1508 (νN–O), 1410 (νN–O), and 783 cm−1 (δC–H). Anal. calcd for C20H16N2O2S4Zn·0.3H2O: C, 46.60; H, 3.25; N, 5.44%. Found: C, 46.68; H, 3.49; N, 5.14%.
Bis[5-(4-bromophenyl)-3-hydroxythiazole-2(3H)-thionolato]zinc(II): Zn(2g)2.
The complex Zn(2g)2 was prepared by reacting 11g (123.8 mg, 0.430 mmol) with Zn(OAc)2·2H2O (47.5 mg, 0.216 mmol) in 75% aqueous ethanol (4.0 mL) at room temperature for 3 h using the same procedure as that used for the preparation of Zn(2a)2. Zn(2g)2 was obtained as a white solid. 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm 7.42 (d, 2H, J = 8.4 Hz), 7.55 (d, 2H, J = 8.4 Hz), 8.04 (s, 1H). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 109.3, 120.2, 124.7, 126.4, 131.2, 132.4, 165.1. IR (KBr): 3089 (νC–H), 1586 (νCS), 1486 (νN–O), 1408 (νN–O), and 797 cm−1 (δC–H). Anal. calcd for C18H10Br2N2O2S4Zn·0.7H2O: C, 33.14; H, 1.76; N, 4.30%. Found: C, 33.00; H, 2.05; N, 4.13%.
Bis[5-(4-chlorophenyl)-3-hydroxy-4-methylthiazole-2(3H)-thionolato]zinc(II): Zn(3)2.
The complex Zn(3)2 was prepared from 3 (51.1 mg, 198 mmol) with Zn(ClO4)2·6H2O (35.9 mg, 97.0 mmol) in a procedure analogous to that used for the preparation of Zn(2a)2, with the exception that lithium hydroxide was absent. Zn(3)2 was obtained as a white solid (41.9 mg, 72.4 μmol, 75%). 1H NMR (dimethyl sulfoxide-d6, 400 MHz) δ/ppm 2.42 (s, 3H, CH3), 7.54 (d 2H, J = 8.4 Hz), 7.58 (d, 2H, J = 8.4 Hz). 13C NMR (dimethyl sulfoxide-d6, 100 MHz) δ/ppm 13.66, 118.3, 129.8, 130.0, 130.1, 133.9, 146.4, 164.4. Anal. calcd for C18H12N2O2S4Zn: C, 41.50; H, 2.44; N, 4.84%. Found: C, 41.46; H, 2.63; N, 4.80%.
1-Chloro-3,3-dimethylbutan-2-one oxime (12).
Hydroxylamine hydrochloride (2.21 g, 31.8 mmol) and sodium acetate (2.41 g, 29.4 mmol) were dissolved in water (9 mL), and 1-chloro-3,3-dimethylbutan-2-one (3.00 g, 22.3 mmol) was added to the aqueous solution with stirring. The resulting mixture was stirred vigorously for 20 minutes at room temperature, and then stirring was continued for 20 hours at room temperature. Ethyl acetate was added to the reaction mixture and organic materials were separated. The organic phase was dried over sodium sulfate. After evaporation of the solvent, 12 was obtained as a white solid (2.95 g, crude, 89%). This material was directly used in the next step without further purification. 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.21 (9H, s), 4.15 (2H, s), 8.24 (1H, br, OH). The spectral data were in good accordance with those reported previously.38
3-Chlorobutan-2-one oxime (13).
The oxime 13 was prepared from 3-chloro-2-butanone (5.00 g, 46.9 mmol) with hydroxylamine hydrochloride (3.56 g, 51.2 mmol) and sodium acetate (4.10 g, 50.0 mmol in a procedure analogous to that for obtaining 12, with the exception that the reaction mixture was allowed to stand at −20 °C for 19 hours after the initial vigorous stirring at room temperature for 20 min, and then again stirred vigorously for 20 minutes at room temperature. The product (13) was obtained as a colorless oil (5.45 g, 96%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.65 (3H, d, J = 6.8 Hz), 1.98 (3H, s), 4.62 (1H, q, J = 6.8 Hz), 8.02 (1H, br, OH). The spectral data were in good accordance with those reported previously.17
O-Ethyl S-(2-(hydroxyimino)-3,3-dimethylbutyl) carbonodithioate (15).
To a suspension of potassium O-ethyldithiocarbonate (432 mg, 2.70 mmol) in acetone (6 mL) was added a solution of 1-chloro-3,3-dimethylbutan-2-one oxime (356 mg, 2.38 mmol) and acetone (6 mL) over a period of 30 min at room temperature. The reaction mixture was stirred for 3 hours at room temperature. The resulting solution was filtered through a Celite bed, and the filtrate was concentrated under reduced pressure. Water (15 mL) was added to the resulting residue, and organic materials were extracted with chloroform (3 × 10 mL). The organic phase was dried over sodium sulfate After evaporation of the solvent, the resulting yellow residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (3/1, v/v) as eluents to give 15 as a white solid (492 mg, 88%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.18 (9H, s), 1.43 (3H, t, J = 7.2 Hz) 4.10 (2H, s), 4.68 (2H, q, J = 7.2 Hz). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 13.9, 27.6, 29.1, 37.5, 70.4, 162.0, 214.7. HR-ESIMS calcd for C9H17NNaO2S2+ [M + Na]+ 258.0593. Found 258.0564.
O-Ethyl S-(3-(hydroxyimino)butan-2-yl) carbonodithioate (16).
The xanthate 16 was prepared from 13 (135 mg, 1.11 mmol) with potassium O-ethyldithiocarbonate (180 mg, 1.12 mmol) in a procedure analogous to that for obtaining 15, with the exception that the reaction mixture was 19.5 hours. The product (16) was obtained as a yellow oil (153 mg, 66%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.42 (3H, t, J = 7.1 Hz), 1.51 (3H, d, J = 7.2 Hz), 1.94 (3H, s), 4.50 (1H, q, J = 7.2 Hz), 4.65 (2H, d, J = 7.1 Hz). The spectral data were in good accordance with those reported previously.17
Synthesis of O-ethyl S-(3-(hydroxyimino)pentan-2-yl) carbonodithioate (20).
Hydroxylamine hydrochloride (1.10 g, 15.8 mmol) was added to a solution of O-ethyl S-(3-oxopentan-2-yl)carbonodithioate (2.70 g, 13.1 mmol) in methanol (7 mL) followed by addition of pyridine (1.3 mL, 16.3 mmol). The mixture was stirred for 11 hours at room temperature. After evaporation of the solvent, water (20 mL) and 4 M HCl aq. (20 mL) were added to the reaction mixture, and organic materials were extracted with ether (50 mL). The organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting pale-yellow residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (5/1, v/v) as eluents to give 20 as a white solid (1.12 g, 39%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.16 (3H, t, J = 7.6 Hz), 1.43 (3H, t, J = 7.1 Hz), 1.54 (3H, d, J = 7.1 Hz), 2.43 (2H, dq, J = 13.4, 7.6 Hz), 4.54 (1H, q, J = 7.1 Hz), 4.66 (2H, q, J = 7.1 Hz), 8.18 (1H, br s). The spectral data were in good accordance with those reported previously.39
Synthesis of 4-(tert-butyl)-3-hydroxythiazole-2(3H)-thione (4).
Zinc chloride (5.2 g, 38.2 mmol) was added to a solution of 15 (3.00 g, 12.7 mmol) in absolute ether (7.5 mL) at such a rate that the solvent did not boil constantly, and then the mixture was stirred at room temperature for 43 hours. Then, 4.0 M HCl (12 mL) was added dropwise to the mixture at 0 °C, and the mixture was stirred for 30 min at room temperature. The organic materials were extracted with chloroform (3 × 20 mL), and the organic phase was dried over sodium sulfate. After evaporation of the solvent, 0.5 M NaOH (18 mL) was added, and the aqueous solution was washed with chloroform, and then acidified with 4 M HCl (4 mL) to pH = 2. Organic materials were then extracted with chloroform The resulting organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting crude product was purified by recrystallization using the dichloromethane/n-hexane binary solvent system to afford 4 as beige plates (1.05 g, 72%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.42 (9H, s), 6.27 (1H, s), 10.1 (1H, br s). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 27.8, 34.4, 100.6, 147.4, 172.0. IR (KBr): ν/cm−1 3142, 2971, 2938, 2871, 1561, 1434, 1342, 1241, 1096, 994, 760. Anal calcd for C7H11NOS2: C, 44.42; H, 5.86; N, 7.40%. Found: C, 44.28; H, 5.62; N, 7.37%.
3-Hydroxy-4,5-dimethylthiazole-2(3H)-thione (5f).
Zinc chloride (10.3 g, 75.6 mmol) was added to a solution of 16 (5.07 g, 24.5 mmol) in absolute ether (15.5 mL) at such a rate that the solvent did not boil constantly, and then the mixture was stirred for 49 hours at room temperature. 4.0 M HCl aq. (25 mL) was added dropwise to the mixture at 0 °C, and then the reaction mixture was stirred for 30 min at room temperature. The solution was extracted with CHCl3 (3 × 20 mL). The organic phase was dried over Na2SO4. After evaporation of the solvent, the resulting green residue was purified by column chromatography on silica gel with n-hexane/ethyl acetate (2/3, v/v) as eluents to give 5f as blueish-green solid (2.85 g, 72%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 2.23 (3H, s), 2.29 (3H, s), 9.78 (1H, br s). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 11.1, 11.8, 114.9, 130.9, 168.5. IR (KBr): ν/cm−1 3189, 2881, 2787, 1628, 1539, 1444, 1436, 1371, 1223, 1011. Anal. calcd for C5H7NOS2Zn 0.2H2O: C, 36.43; H, 4.52; N, 8.50%. Found: C, 36.11; H, 4.46; N, 8.39%.
4-Ethyl-3-hydroxy-5-methylthiazole-2(3H)-thione (5h).
A solution of 2039 (1.12 g, 5.06 mmol) in dichloromethane (18 mL) was added to an ice-cooled solution of potassium hydroxide (1.13 g, 20.1 mmol) in water (20 mL), and the mixture was stirred for 2.5 hours at room temperature. After completion of the reaction, water (5 mL) was added to the reaction mixture, and the organic phase was separated. The aqueous phase was washed with n-hexane (2 × 10 mL), and acidified with conc. HCl until pH = 2. Then, the organic materials were extracted with dichloromethane (3 × 10 mL), and the combined organic phase was dried over sodium sulfate. After evaporation of the solvent, the resulting crude product was purified by recrystallization from dichloromethane/n-hexane binary solvent system to afford 5h as blueish-green plates (695 mg, 78%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.22 (3H, t, J = 7.5 Hz), 2.23 (3H, s), 2.69 (2H, q, J = 7.5 Hz), 9.81 (1H, br s). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 11.6, 12.5, 19.2, 114.5, 136.0, 168.6. IR (KBr): ν/cm−1 2973, 2824, 2739, 2656, 1612, 1458, 1371, 1108, 1019, 985, 960, 799. Anal calcd for C5H9NOS2: C, 41.12; H, 5.18; N, 7.99%. Found: C, 41.15; H, 5.03; N, 8.02%.
3-Hydroxy-4,5,6,7-tetrahydrobenzo[d]thiazole-2(3H)-thione (6) (via14 and 17).
Oxime 14 was prepared from 2-chlorocyclohexanone (2.80 g, 21.1 mmol) with hydroxylamine hydrochloride (1.80 g, 25.9 mmol) and sodium acetate (1.92 g, 23.4 mmol) in a procedure analogous to that for obtaining 12. The product (14) was obtained as a pale-yellow oil (2.83 g) and was directly used in the next step without further purification. Then, 14 (2.83 g, 19.2 mmol) was reacted with potassium O-ethyldithiocarbonate (3.31 g, 22.7 mmol) in a procedure analogous to that used for the preparation of 16, with the exception that the reaction time was 2.5 hours, to afford 17 as a yellow oil (4.14 g), which was directly used in the next step without further purification. The obtained 17 was then converted into 6 with zinc chloride (8.01 g, 58.8 mmol) in a procedure analogous to that for obtaining 5f, with the exception that the reaction time was 66 hours and n-hexane/chloroform/ethyl acetate ternary solvent system (2/2/1, v/v/v) was used as eluents for the column chromatography. The product (6) was obtained as a blueish-green solid (1.76 g, 45% in 3 steps). 1H-NMR (chloroform-d, 400 MHz): δ/ppm,1.88 (4H, quint, J = 2.8 Hz), 2.53–2.56 (2H, m), 9.73 (1H, br s). 13C-NMR (chloroform-d, 100 MHz): δ/ppm, 21.1, 22.4, 22.8, 23.3, 100.0, 117.8, 133.4. IR (KBr): ν/cm−1 2941, 2663, 1621, 1438, 1373, 1357, 1206, 977. Anal. calcd for C7H9NOS2: C, 44.89; H, 4.84; N, 7.48%. Found: C, 44.74; H, 4.78; N, 7.47%.
Synthesis of bis[4-(tert-butyl)-3-hydroxythiazole-2(3H)-thionolato]zinc(II): Zn(4)2
To a solution of 4 (950 mg, 5.02 mmol) in 50% aqueous ethanol (40 mL) was added Zn(OAc)2·2H2O (687 mg, 3.13 mmol) in water (12 mL), and the mixture was stirred for 2 hours at room temperature. The resulting precipitate was collected by filtration and successively washed with water and THF to give white solids, which were purified by recrystallization from dichloromethane/n-hexane to give Zn(5f)2 as colorless crystals (908 mg, 82%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.45 (9H, s), 6.51 (1H, s). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 27.6, 34.9, 103.6, 152.1, 160.5. IR (KBr): ν/cm−1 3135, 3081, 2957, 1558, 1359, 1245, 1032, 685. Anal calcd for C14H20N2O2S4Zn: C, 38.05; H, 4.56; N, 6.34%. Found: C, 38.01; H, 4.35; N, 6.36%.
Bis[3-hydroxy-4,5-dimethyl-2-thioxo-3-thiazololato]zinc(II): Zn(5f)2.
The complex Zn(5f)2 was prepared from 5f (1.00 g, 6.20 mmol) with Zn(OAc)2·2H2O (775 mg, 3.53 mmol) in a procedure analogous to that used for the preparation of Zn(4)2, and was obtained as colorless crystals. (965 mg, 81%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 2.28 (3H, s), 2.31 (3H, s). 13C-NMR (chloroform-d, 100 MHz): δ/ppm, 12.0, 12.4, 117.4, 136.5, 155.6. IR (KBr): ν/cm−1 3445, 2955, 2918, 1616, 1441, 1396, 1320, 1228, 1093, 1035. Anal. calcd for C10H12N2O2S4Zn: C, 31.13; H, 3.13; N, 7.26%. Found: C, 31.02; H, 3.20; N, 7.14%.
Bis[4-ethyl-3-hydroxy-5-methylthiazole-2(3H)-thionolato]zinc(II): Zn(5h)2.
The complex Zn(5h)2 was prepared from 5h (400 mg, 2.28 mmol) with Zn(OAc)2·2H2O (277 mg, 1.26 mmol) in a procedure analogous to that used for the preparation of Zn(4)2 and was obtained as colorless needles (373 mg, 79%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.21 (3H, t, J = 7.5 Hz), 2.29 (3H, s), 2.76 (2H, q, J = 7.5 Hz). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 12.2, 12.5, 19.9, 117.3, 141.6, 155.7. IR (KBr): ν/cm−1 2969, 2929, 2870, 1610, 1452, 1391, 1312, 1223, 1065, 1038, 1001, 804. Anal calcd for C14H16N2O2S4Zn·0.15 H2O: C, 34.59; H, 3.94; N, 6.73%. Found: C, 34.51; H, 3.91; N, 6.73%.
Bis[3-hydroxy-4,5,6,7-tetrahydrobenzo[d]thiazole-2(3H)-thionolato]zinc(II): Zn(6)2.
The complex Zn(6)2 was prepared from 6 (380 mg, 2.03 mmol) with Zn(OAc)2·2H2O (256 mg, 1.17 mmol) in a procedure analogous to that used for the preparation of Zn(5f)2, and was obtained as a pale green solid (332 mg, 75%). 1H-NMR (chloroform-d, 400 MHz): δ/ppm, 1.85 (4H, quint, J = 2.7 Hz), 2.57–2.61 (4H, m), 2.71–2.74 (4H, m). 13C NMR (chloroform-d, 100 MHz): δ/ppm, 21.3, 23.0, 23.4, 23.8, 120.8, 138.5, 156.4. IR (KBr): ν/cm−1 2946, 1614, 1439, 1387, 1300, 1206, 1033, 1011. Anal calcd for C14H16N2O2S4Zn 0.15 H2O: C, 38.16; H, 3.73; N, 6.36%. Found: C, 38.07; H, 3.62; N, 6.25%.
X-ray crystallographic analysis
X-ray data for Zn(3)2 with pyridine were collected on a Rigaku XtaLAB P200 diffractometer with multi-layer mirror monochromated MoKa (λ = 0.71073 Å) and a hybrid photon counting detector (PILATUS 200K). The crystal structure was solved by direct methods (SHELXT Version 2014/5)40 and refined by full-matrix least-squares SHELXL-2014/7.41 All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located from difference electron density maps.
X-ray data for Zn(4)2, Zn(5f)2, Zn(5h)2, Zn(6)2, and Zn(5h)2 with pyridine were collected on a Bruker SMART APEX II ULTRA diffractometer equipped with graphite monochromated MoKα radiation (λ = 0.71073 Å) generated by a rotating anode. The cell parameters for the compounds were obtained from a least-squares refinement of the spot. Data collection, data reduction and semi-empirical absorption correction were carried out using the software package of APEX2.42 All of the calculations for the structure determination were carried out using the SHELXTL package.41 In all cases, nonhydrogen atoms were refined anisotropically and hydrogen atoms were placed in idealized positions and refined isotropically in a riding manner along with their respective parent atoms.
In vitro insulin-mimetic activity
Male Wistar rats were sacrificed under anesthesia with ether. The adipose tissues were removed, chopped with scissors and digested with collagenase for 60 min at 37 °C in Krebs Ringer bicarbonate buffer (120 mM NaCl, 1.27 mM CaCl2, 1.2 mM MgSO4, 4.75 mM KCl, 1.2 mM KH2PO4, 24 mM NaHCO3: pH 7.4) containing 2% bovine serum albumin. The adipocytes were then separated from undigested tissues and washed with the buffer three times. The isolated adipocyte solutions were preincubated at 37 °C for 30 min with various concentrations of the zinc(II) complexes or ZnSO4 in saline containing 5 mM glucose. Then, an epinephrine solution was added to the reaction mixtures (final concentration; 10 mM), and the resulting solutions were incubated at 37 °C for 3 h. The reactions were stopped by soaking in ice water, and the mixtures were centrifuged at 3000 rpm at 4 °C for 10 min. FFA levels in the outer solution of the cells were determined with an FFA kit (NEFA C-test Wako, Wako Pure Chemicals, Osaka, Japan). All animal experiments in the present study were approved by the Experimental Animal Research of Kyoto Pharmaceutical University (KPU) and were performed according to the Guideline for Animal Experimentation of KPU.
Determination of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P values by reversed-phase HPLC analysis
P values by reversed-phase HPLC analysis
An HPLC analysis was performed on a Mightysil RP-18 GP II column (particle size: 5 μm, 4.6 mm i.d. × 250 mm, KANTO CHEMICAL Co., Inc., Japan) with an SSC-3461 pump (SENSHU SCIENTIFIC Co., Ltd, Japan), SSC-2300 column oven (SENSHU SCIENTIFIC Co., Ltd, Japan), and 875-UV Intelligent UV/VIS detector (JASCO Co., Ltd, Japan). Eluents: 80%(v/v) methanol in water, Flow rate: 0.7 mL/min, Detection: 254 nm, and Column oven temperature: 35 °C. Tartrazine was used as an unretained compound to determine the dead time (t0). A series of standard compounds including phenol (logP = 1.46), benzene (logP = 2.13), bromobenzene (logP = 2.99), naphthalene (logP = 3.37), biphenyl (logP = 3.98), phenanthrene (logP = 4.57) were analyzed to generate a calibration curve (Fig. S2, ESI†).43
Determination of acid dissociation constants (pKa) of the ligands
The pH values of aqueous solutions containing the ligands were measured using a composite electrode GST-5841C (DKK-TOA Corp., Japan) and a multi-water quality meter MM-60R (DKK-TOA Corp., Japan). Compounds were dissolved in a THF–water mixture in 20 ml (Adjusted to be approximately 0.004 M) and neutralization titrated with 0.01 M sodium hydroxide solution (f = 1.001, KANTO CHEMICAL CO., INC.) prepared in any THF concentration. The pKa was determined from the half-equivalence point. Titrations were performed with different THF–water ratios (THF: 40.0, 50.0, 66.7%), and the pKa in water was determined by three-point extrapolation.
Determination of the stability constants (logK) of the zinc(II) complexes
The pH was measured using a composite electrode GST-5841C (DKK-TOA Corp., Japan) and a multi-water quality meter MM-60R (DKK-TOA Corp., Japan). A THF solution of the ligand is mixed with an aqueous solution of zinc ions and the pH is measured. The overall stability constant was determined by calculation from the hydrogen ion concentration. All THF concentrations were 50%. The acid dissociation constant “Ka” at that concentration was determined experimentally.
Author contributions
R. Saito: writing – original draft, conceptualization, data curation, funding acquisition, investigation, methodology, project administration, resources, supervision, visualization. Y. Naito: data curation, formal analysis, investigation, resources, validation, visualization, writing – review and editing. Y. Yoshikawa: data curation, formal analysis, investigation, resources, validation, visualization, writing – review and editing. H. Yasui: methodology, project administration, resources, supervision, validation, writing – review and editing. S. Kikkawa: data curation, formal analysis, investigation, resources, validation, visualization, writing – review and editing. R. Ohba: data curation, formal analysis, investigation, visualization. T. Miyamoto: data curation, formal analysis, investigation, visualization. S. Sano: data curation, formal analysis, investigation, visualization. K. Maeda: data curation, formal analysis, investigation, visualization. M. Tamura: data curation, formal analysis, investigation, visualization. I. Azumaya: investigation, resources, validation, writing – review and editing.
Data availability
Relevant crystal data collection and refinement data for the crystal structures are summarized in Table S1 (ESI†). CCDC 2379130 (for Zn(3)2), 2383333 (for Zn(4)2), 2383335 (for Zn(5f)2), 2383331 (for Zn(5f)2 + pyridine), 2383324 (for Zn(5h)2), and 2383338 (for Zn(6)2) contain the supplementary crystallographic data for this paper.
Conflicts of interest
The authors declare that they have no competing interests that could have appeared to influence this study.
Acknowledgements
This research was supported by JSPS KAKENHI Grant Number JP25410179. The authors would like to thank Editage (https://www.editage.jp) for English language editing.
References
- H. Sun, P. Saeedi, S. Karuranga, M. Pinkepank, K. Ogurtsova, B. B. Duncan, C. Stein, A. Basit, J. C. N. Chan, J. C. Mbanya, M. E. Pavkov, A. Ramachandaran, S. H. Wild, S. James, W. H. H. Herman, P. Zhang, C. Bommer, S. H. Kuo, E. J. J. Boyko and D. J. Magliano, Diabetes Res. Clin. Pract., 2022, 183, 109119 CrossRef PubMed.
- World Health Organ. Tech. Rep. Ser., 1985, vol. 727, pp. 1–113 Search PubMed.
- A. F. Amos, D. J. McCarty and P. Zimmet, Diabet. Med., 1997, 14, S7–S85 CrossRef.
- B. Lorenzati, C. Zucco, S. Miglietta, F. Lamberti and G. Bruno, Pharmaceuticals, 2010, 3, 3005–3020 CrossRef CAS.
- E. Standl and M. Füchtenbusch, Diabetologia, 2003, 46, M30–M36 CrossRef CAS.
-
W. Kaim and B. Schwederski, Bioinorganic Chemistry: Inorganic Elements in the Chemistry of Life: An Introduction and Guide, John Wiley & Sons Ltd, Chichester, UK, 1994 Search PubMed.
- G. J. Fosmire, Am. J. Clin. Nutr., 1990, 51, 225–227 CrossRef CAS.
-
J. Wolf, H. H. Sandstead and L. Rink, in Handbook on the Toxicology of Metals, ed. G. F. Nordberg and M. Costa, Academic Press, 5th edn, 2022, ch. 38, pp. 963–984, DOI:10.1016/B978-0-12-822946-0.00034-9.
- L. I. Stiles, K. Ferrao and K. J. Mehta, Clin. Exp. Med., 2024, 24, 38–56 CrossRef CAS.
- L. Coulston and P. Dandona, Diabetes, 1980, 29, 665–667 CrossRef CAS.
- M. Nishida, H. Sakurai, J. Kawada, M. Koyama and J. Takada, Naturwissenschaften, 1989, 76, 220–222 CrossRef CAS.
- J. Fugono, H. Yasui and H. Sakurai, J. Pharm. Pharmacol., 2002, 54, 611–615 CrossRef CAS.
- Y. Yoshikawa, E. Ueda, Y. Kojima and H. Sakurai, Life Sci., 2004, 75, 741–751 CrossRef CAS.
- Y. Yoshikawa, A. Murayama, Y. Adachi, H. Sakurai and H. Yasui, Metallomics, 2011, 3, 686–692 CrossRef CAS PubMed.
- Y. Naito, Y. Yoshikawa and H. Yasui, Bull. Chem. Soc. Jpn., 2011, 84, 298–305 CrossRef CAS.
- M. Yamaguchi, R. Saito, Y. Adachi, Y. Yoshikawa, H. Sakurai and A. Katoh, Heterocycles, 2007, 73, 603–615 CrossRef CAS.
- A. Grogss, N. Schneiders, K. Daniel, T. Gottwald and J. Hartung, Tetrahedron, 2008, 64, 10882–10889 CrossRef.
- J. M. Hatcher, M. C. Kohler and D. M. Coltart, Org. Lett., 2011, 13, 3810–3813 CrossRef CAS PubMed.
- D. H. R. Barton, D. Crich and G. Kretzschmar, J. Chem. Soc., Perkin Trans. 1, 1986, 39–53, 10.1039/p19860000039.
- S. Lou and G. C. Fu, J. Am. Chem. Soc., 2010, 132, 1264–1266 CrossRef CAS PubMed.
- K. K. K. Goh, S. Kim and S. Z. Zard, Org. Lett., 2013, 15, 4818–4821 CrossRef CAS PubMed.
- Z. R. He, Z. Y. Zhang, K. Asare-Yeboah and S. Bi, Mater. Adv., 2023, 4, 769–786 RSC.
- A. D. Bond, F. Benevelli and W. Jones, J. Mater. Chem., 2002, 12, 324–332 RSC.
- M. Nakai, H. Watanabe, C. Fujiwara, H. Kakegawa, T. Satoh, J. Takada, R. Matsushita and H. Sakurai, Biol. Pharm. Bull., 1995, 18, 719–725 CrossRef CAS.
- M. Yamaguchi, K. Wakasugi, R. Saito, Y. Adachi, Y. Yoshikawa, H. Sakurai and A. Katoh, J. Inorg. Biochem., 2006, 100, 260–269 CrossRef CAS.
- H. Kawarada, Y. Yoshikawa, H. Yasui, S. Kuwahara, Y. Habata and R. Saito, Metallomics, 2011, 3, 675–679 CrossRef CAS PubMed.
- R. Saito, M. Tamura, S. Kawano, Y. Yoshikawa, A. Kato, K. Sasaki and H. Yasui, New J. Chem., 2017, 41, 5572–5581 RSC.
- Y. Yoshikawa, E. Ueda, Y. Suzuki, N. Yanagihara, H. Sakura and Y. Kojima, Chem. Pharm. Bull., 2001, 49, 652–654 CrossRef CAS PubMed.
- M. Calvin and K. W. Wilson, J. Am. Chem. Soc., 1945, 67, 2003–2007 CrossRef CAS.
- H. Freiser, R. G. Charles and W. D. Johnston, J. Am. Chem. Soc., 1952, 74, 1383–1385 CrossRef CAS.
-
J. Bjerrum, Metal Ammine Formation in Aqueous Solution: Theory of the Reversible Step Reactions, ed. P. Haase, 1957 Search PubMed.
- K. Suzuki, M. Yasuda and K. Yamasaki, Bull. Chem. Soc. Jpn., 1957, 75, 229–231 Search PubMed.
- H. M. Irving and H. S. Rossotti, J. Chem. Soc., 1941, 2904–2910 Search PubMed.
- K. K. Mui, W. A. E. Mcbryde and E. Nieboer, Can. J. Chem., 1974, 52, 1821–1833 CrossRef CAS.
- S. Castellano, D. Kuck, M. Viviano, J. Yoo, F. López-Vallejo, P. Conti, L. Tamborini, A. Pinto, J. L. Medina-Franco and G. Sbardella, J. Med. Chem., 2011, 54, 7663–7677 CrossRef CAS PubMed.
- I. T. Papadas, S. Fountoulaki, I. N. Lykakis and G. S. Armatas, Chem. – Eur. J., 2016, 22, 4600–4607 CrossRef CAS.
- T. Yasukawa, K. Sakamoto, Y. Yamashita and S. Kobayashi, ACS Catal., 2022, 12, 5887–5893 CrossRef CAS.
- R. Buchman, R. A. Komoroski, K. M. Kauppila, J. J. Mannion and L. Gehrlein, J. Agric. Food Chem., 1985, 33, 896–907 CrossRef CAS.
- EP249328, 1987.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
APEX2 Version 2009.1-0 Data collection and Processing Software, Bruker AXS Inc., Madison, WI, 2008 Search PubMed.
- R. Saito, A. Ohno and E. Ito, Tetrahedron, 2010, 66, 583–590 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Yuki
Naito
*ab,
Yuki
Naito