
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlycoconjugate Pd(II) and Cu(II) complexes of fluorinated N,O Schiff base ligands for targeted cancer therapy: synthesis, characterization and in vitro cytotoxic activity evaluation†
María Esther Moreno-Narváez a,
Antonino Arenaza-Coronaa,
Lucero González-Sebastiánb,
Teresa Apan Ramíreza,
Simón Hernández Ortegaa,
J. Antonio Cruz-Navarroa,
Jorge Alí-Torresc,
Adrián L. Orjuelac,
Viviana Reyes-Marquezd,
Leticia Lomas-Romerob and
David Morales-Morales*a
a,
Antonino Arenaza-Coronaa,
Lucero González-Sebastiánb,
Teresa Apan Ramíreza,
Simón Hernández Ortegaa,
J. Antonio Cruz-Navarroa,
Jorge Alí-Torresc,
Adrián L. Orjuelac,
Viviana Reyes-Marquezd,
Leticia Lomas-Romerob and
David Morales-Morales*a
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior s/n, Ciudad Universitaria, C.P. 04510, México, CDMX, Mexico. E-mail: damor@unam.mx; Fax: +52-55-56162217; Tel: +52-55-56224514
bDepartamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Av. San Rafael Atlixco No. 186, Ciudad de México, C.P. 09340, Mexico
cDepartamento de Química, Universidad Nacional de Colombia- Sede Bogotá, Bogotá 111321, Colombia
dDepartamento de Ciencias Químico-Biológicas, Universidad de Sonora, Luis Encinas y Rosales s/n, Hermosillo 83000, Sonora, Mexico
First published on 13th February 2025
Abstract
In the present investigation, we engineered a series of Pd(II) and Cu(II) metal complexes bearing N-aryl fluorinated O-glycoconjugate Schiff base ligands as potential anticancer agents. These novel metal complexes, incorporating CF3 and α-D-glucose groups, were designed, synthesized, and thoroughly characterized using multinuclear NMR, ATR-FTIR spectroscopy, electron paramagnetic resonance (EPR), mass spectrometry, elemental analysis, and single-crystal X-ray diffraction for select compounds. Their effects were subsequently evaluated on six cancer cell lines: human Caucasian prostate adenocarcinoma (PC-3), mammary adenocarcinoma (MCF-7), human colorectal adenocarcinoma (HCT-15), human lung adenocarcinoma (SK-LU-1), myelogenous leukemia (K-562), and human malignant glioblastoma (U-251). Notably, compound Pd(L1G)2 exhibited the highest cytotoxicity against the various cancer cell lines while demonstrating reduced activity against non-cancerous cells. This finding underscores the potential of carbohydrate moieties to enhance selectivity towards malignant cells. Additionally, in silico molecular docking studies were conducted to evaluate interactions between the copper and palladium complexes, revealing binding patterns of the newly synthesized compounds. This analysis indicated that Pd(II) complexes with glycosylated ligands exhibit a stronger DNA binding affinity. Generally, the glycosylated ligands enhance hydrogen bonding interactions, resulting in more stable and effective binding.
1. Introduction
Cancer, a multifactorial disease, ranks as the second leading cause of death globally, according to the World Health Organization.1 The incidence of cancer is expected to increase in the upcoming years, presenting significant challenges. These challenges stem primarily from the limited effectiveness of treatments for specific types of cancer, the emergence of drug resistance, and the discontinuation of treatments due to severe and sometimes irreversible side effects. For example, cisplatin, a well-known metal-based drug, remains a cornerstone in the treatment of various solid tumors; however, its clinical utility is compromised by significant side effects2 and the development of resistance in tumor cells. In response to these issues, the design and synthesis of compounds with enhanced antitumor properties, aimed at improving therapeutic profiles, have become crucial and burgeoning fields of study.In this context, the synthesis of bioactive molecules, such as azomethine or Schiff bases derivatives and their metal complexes, has demonstrated significant cytotoxic activity. These compounds have emerged as alternative anticancer agents that reduce toxicity and enhance efficacy, thus becoming a focal point in the development of new pharmaceuticals. Notably, N,O Schiff bases are among the most attractive ligands for forming thermodynamically stable coordination complexes. This is due to their chelating effect, which allows for binding with a wide variety of metal ions in different oxidation states. The resulting complexes exhibit a broad spectrum of biological activities,3,4 including anticancer, antimicrobial, antioxidant, and anti-inflammatory properties.5 To improve the biological selectivity and reduce the toxicity of these metal complexes, fluorine atoms or fluorinated groups have been strategically incorporated into the ligands, effectively enhancing their biological activity. The introduction of fluorine, the most electronegative atom with the smallest van der Waals radius, imparts unique physicochemical properties to the complexes, such as increased lipophilicity and cellular membrane permeability. These characteristics are critical to modulate the pharmacokinetics and pharmacodynamics of the compounds, often leading to an increase in potency, selectivity, and metabolic stability, thereby enhancing the bioactive profile of the resulting complexes.6–8
Another strategic approach to improving therapeutic properties is the glycosylation of metal complexes.9 Carbohydrates, the most abundant class of biomolecules, are integral to a variety of biological processes including energy provision, cellular signaling, and the formation of complex structures such as glycoproteins and glycolipids. Their role is particularly critical in the metabolic pathways and structural changes associated with cancer cell proliferation and metastasis. The synthesis of carbohydrate-conjugated metal complexes capitalizes on the unique properties of carbohydrates—such as biocompatibility, hydrophilicity, and their ability to interact with specific cellular transporters like glucose transporters (GLUTs). These attributes enhance the solubility, stability, and selectivity of metal-based drugs, positioning them as ideal candidates for the development of targeted therapeutic agents.10,11
In this regard, research into metal-based glycoconjugates involving various metals and ligands has revealed promising diagnostic and therapeutic applications.12
Building on the research outlined above and considering the anticancer properties of copper(II) and palladium(II) complexes with aromatic Schiff base ligands, which interact with DNA base pairs through covalent or non-covalent bonds, here we report the synthesis and characterization of a new series of Pd(II) and Cu(II) complexes. These complexes feature N,O Schiff base ligands that are strategically substituted with carbohydrates and CF3 groups, designed to function as potential anticancer agents. Additionally, we present an in vitro cytotoxicity study conducted on various cell lines.
2. Experimental
2.1. Instrumentation and materials
2,4-Dihydroxybenzaldehyde (98%), 2-(trifluoromethyl)aniline (99%), 3-(trifluoromethyl)aniline (99%), 4-(trifluoromethyl)aniline (99%), copper(II) acetate monohydrate (98%), acetobromo-α-D-glucose (≥95%), potassium carbonate anhydrous (<99) and tetrabutylammonium bromide (≥98.0%) were purchased from Sigma-Aldrich. Solvents were obtained from J.T. Baker and Chemical Co. and used as received without further purification. Palladium(II) acetate (≥99) was obtained from STREM Chemicals, Inc. 1H, 19F{H} and 13C{H} NMR spectra were recorded in DMSO-d6 or CDCl3 on a Bruker 400 MHz Avance III HD or a Bruker Avance 300 MHz. The chemical shift values (δ) are given in parts per million and values for coupling constants (J) in Hz and are referred to the residual peak of the deuterated solvent employed. Electron paramagnetic resonance (EPR) spectra were obtained from acetone solutions at 77 K on a Jeol JES-TE300 spectrometer. Mass spectra were collected by direct analysis in real-time (DART-MS) on a JEOL-JMS-T100LC AccuTOF or a JEOL JMS-700 MStation by fast atom bombardment (FAB) ionization technique in positive mode. ATR-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer in the 4000–500 cm−1 range. Melting points were determined in open capillaries in an electrothermal IA9300 melting point apparatus and are uncorrected. Elemental analyses were performed on a Thermo Scientific™ Flash 2000 using methionine as a verification standard and a Mettler Toledo XP6 microbalance.2.2. Synthetic procedures
All manipulations were carried out under aerobic conditions.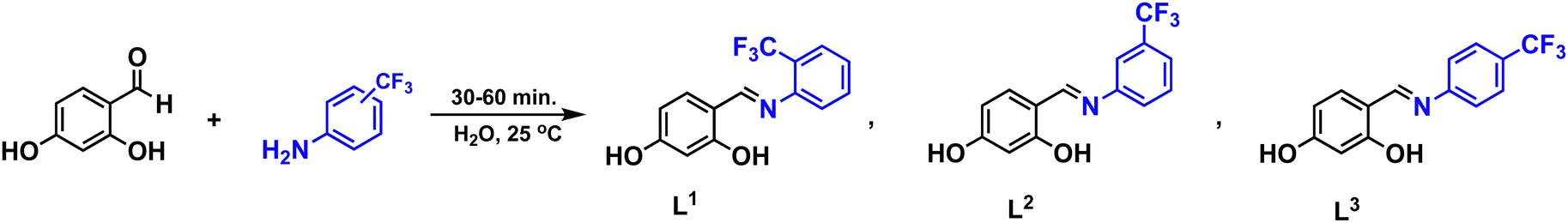 | ||
| Scheme 1 Synthetic route for the ligands L1–L3. | ||
Ligand L1: bright yellow powder. Yield: 95%. Melting point: 189–192 °C. 1H NMR (DMSO-d6, 400 MHz) δ 12.89 (s, 1H), 10.42 (s, 1H), 8.82 (s, 1H), 7.74 (t, J = 8.6 Hz, 1H), 7.56 (d, J = 7.9 Hz, 1H), 7.49 (d, J = 8.5 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 6.44 (dd, J = 8.5, 2.3 Hz, 1H), 6.33 (d, J = 2.2 Hz, 1H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 164.4, 163.3, 162.8, 146.9, 135.1, 134.0, 128.2, 126.32, 126.27, 126.21, 126.16, 126.02, 125.5, 122.8, 122.5, 122.2, 121.9, 120.0, 112.1, 108.2, 102.4. 19F{1H} NMR (DMSO-d6, 376 MHz) δ −59.3. IR (ATR): v/cm−1 = 3256, 3077, 2593, 1620, 1598, 1318, 1186, 1107, 1035, 764. Elemental anal. calcd. (%) for C14H10F3NO2 (MW = 281.207 g mol−1): C, 59.79; H, 3.58; N, 4.98, found C, 59.86; H, 3.24; N, 5.23. DART-MS: m/z 282.04 ([M + H]+ of C14H10F3NO2).
Ligand L2: bright yellow powder. Yield: 98%. Melting point: 165–167 °C. 1H NMR (DMSO-d6, 400 MHz) δ 13.10 (s, 1H), 10.36 (s, 1H), 8.87 (s, 1H), 7.70 (s, 1H), 7.68–7.59 (m, 2H), 7.63–7.55 (m, 1H), 7.47 (d, J = 8.5 Hz, 1H), 6.43 (dd, J = 8.5, 2.3 Hz, 1H), 6.32 (d, J = 2.2 Hz, 1H). 13C{1H} NMR (DMSO-d6, 75 MHz) δ 164.3, 162.9, 149.3, 134.7, 130.5, 130.0, 125.8, 125.4, 122.4, 122.4, 117.7, 117.7, 112.0, 108.1, 102.4. 19F{1H} NMR (DMSO-d6, 282 MHz) δ −61.1. IR (ATR): v/cm−1 = 3064, 298, 2881, 2753, 2624, 1626, 1603, 1582, 1234, 1101, 798, 750, 688, 661. Elemental anal. calcd. (%) for C14H10F3NO2 (MW = 281.07 g mol−1): C, 59.79; H, 3.58; N, 4.98, found C, 59.96; H, 3.61; N, 5.27. DART-MS: m/z 282.08 ([M + H]+ of C14H10F3NO2).
Ligand L3: bright yellow powder. Yield: 98%. Melting point: 143–145 °C. 1H NMR (DMSO-d6, 400 MHz) δ 13.07 (s, 1H), 10.38 (s, 1H), 8.84 (s, 1H), 7.77 (d, J = 8.3 Hz, 2H), 7.50 (dd, J = 15.8, 8.4 Hz, 3H), 7.30 (d, J = 8.2 Hz, 1H), 6.63 (d, J = 8.5 Hz, 1H), 6.43 (dd, J = 8.6, 2.3 Hz, 1H), 6.32 (d, J = 2.3 Hz, 1H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 165.2, 164.5, 163.3, 163.2, 163.1, 152.3, 134.8, 132.8, 126.8, 126.5, 126.3, 126.3, 126.2, 126.2, 122.0, 121.9, 113.0, 108.3, 102.4. 19F{1H} NMR (DMSO-d6, 283 MHz) δ −60.49. IR (ATR): v/cm−1 = 3066, 2871, 2754, 2615, 2510, 2361, 1635, 1601, 1327, 1098, 1067, 829. Elemental anal. calcd. (%) for C14H10F3NO2 (MW = 281.07 g mol−1): C, 59.79; H, 3.58; N, 4.98, found C, 60.24; H, 3.73; N, 5.06. DART-MS: m/z 282.05 ([M + H]+ of C14H10F3NO2).
2.2.2.1. Synthesis of Pd(II) precursors [Pd(L1)2–Pd(L3)2] (1–3). A mixture of palladium(II) acetate (20 mg, 0.089 mmol), the corresponding ligand (50 mg, 0.177 mmol), and 2 mL of THF was subjected to microwave irradiation at 300 W for 40 minutes. The reaction temperature was maintained at 76 °C for the Pd(L1)2 complex and 96 °C for the Pd(L2)2 and Pd(L3)2 complexes. After completion, the products were isolated as yellow powders through recrystallization from a THF/hexane mixture.
Pd(L1)2 (1): yellow powder. Yield: 83%. Decompn. temp.: 189 °C. 1H NMR (DMSO-d6, 400 MHz) δ 9.92 (s, 2H), 7.82–7.67 (m, 6H), 7.57 (t, J = 7.7 Hz, 2H), 7.39 (dd, J = 17.6, 7.8 Hz, 2H), 7.18 (d, J = 8.7 Hz, 2H), 6.00 (dd, J = 8.8, 2.3 Hz, 2H), 5.15 (d, J = 1.9 Hz, 1H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 166.3, 166.3, 164.2, 164.2, 162.3, 162.1, 147.5, 137.0, 136.9, 132.6, 132.5, 128.3, 128.0, 126.5, 126.5, 125.7, 125.7, 125.3, 123.9, 123.7, 123.6, 123.4, 122.6, 113.3, 113.2, 106.5, 106.4, 103.2, 103.2. 19F{1H} NMR (DMSO-d6, 376 MHz) δ −56.57, −56.74. IR (ATR): v/cm−1 = 3296, 1560, 1529, 1315, 1175, 1120, 1057, 1033, 766. Elemental anal. calcd. (%) for C28H18F6N2O4Pd (MW = 666.02 g mol−1): C, 50.43; H, 2.72; N, 4.20, found C, 50.41; H, 3.11; N, 4.18. DART-MS: m/z 666.98 ([M + H]+ of C28H18F6N2O4Pd).
Pd(L2)2 (2): yellow powder. Yield: 93%. Decompn. temp.: 273 °C. 1H NMR (DMSO-d6, 400 MHz) δ 9.94 (s, 2H), 7.89 (s, 2H), 7.75–7.57 (m, 6H), 7.25 (d, J = 8.8 Hz, 2H), 6.06 (dd, J = 8.7, 2.3 Hz, 2H), 5.30 (d, J = 2.2 Hz, 2H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 165.9, 164.3, 162.4, 149.7, 137.1, 129.2, 129.1, 128.9, 128.6, 128.3, 128.2, 125.5, 122.8, 122.5, 122.4, 122.1, 122.0, 120.1, 113.7, 106.7, 103.2. 19F{1H} NMR (DMSO-d6, 376 MHz) δ −60.7. IR (ATR): v/cm−1 = 3105, 1585, 1536, 1325, 1242, 1121, 791, 756, 693, 681. Elemental anal. calcd. (%) for C28H18F6N2O4Pd (MW = 666.02 g mol−1): C, 50.43; H, 2.72; N, 4.20, found C, 50.54; H, 2.91; N, 4.53. DART-MS: m/z 667.02 ([M + H]+ of C28H18F6N2O4Pd).
Pd(L3)2 (3): yellow powder. Yield: 89%. Decompn. temp.: 268 °C. 1H NMR (DMSO-d6, 400 MHz) δ 10.01 (s, 2H), 7.87 (s, 2H), 7.77 (d, J = 8.4 Hz, 4H), 7.52 (d, J = 8.2 Hz, 4H), 7.24 (d, J = 8.7 Hz, 2H), 6.06 (dd, J = 8.7, 2.3 Hz, 2H), 5.30 (d, J = 2.3 Hz, 2H). 13C{1H} NMR (DMSO-d6, 101 MHz) δ 166.0, 164.5, 162.3, 152.7, 137.3, 126.3, 126.1, 125.1, 113.9, 113.1, 106.9, 103.4. 19F{1H} NMR (DMSO-d6, 283 MHz) δ −58.84, −60.33. IR (ATR): v/cm−1 = 3438, 3251, 2617, 1597, 1537, 1320, 1167, 1107, 847. Elemental anal. calcd. (%) for C28H18F6N2O4Pd (MW = 666.02 g mol−1): C, 50.43; H, 2.72; N, 4.20, found C, 50.56; H, 2.99; N, 4.53. DART-MS: m/z 666.98 ([M + H]+ of C28H18F6N2O4Pd).
2.2.2.2. Synthesis of Cu(II) precursors [Cu(L1)2–Cu(L3)2] (4–6). The copper(II) metal complexes were synthesized at room temperature by slowly adding a solution of copper(II) acetate monohydrate (17.74 mg, 0.088 mmol) in methanol to a stirred solution of Schiff base compound (50 mg, 0.177 mmol), with a total volume of 20 mL. As the reaction proceeded, the solution color changed from bright yellow to brown. The reactions were stirred for 1 hour and left to slowly evaporate, yielding products as dark brown crystals.
Cu(L1)2 (4): dark brown crystals. Yield: 80%. Melting point: 244–246 °C. IR (ATR): v/cm−1 = 2962, 2890, 2780, 2712, 2637, 2599, 1608, 1581, 1236, 1184, 1112, 1024, 763. Elemental anal. calcd. (%) for C28H18CuF6N2O4 (MW = 623.05 g mol−1): C, 53.90; H, 2.91; N, 4.49, found C, 53.93; H, 2.81; N, 4.49. DART-MS: m/z 624.03 ([M + H]+ of C28H18CuF6N2O4).
Cu(L2)2 (5): dark brown crystals. Yield: 86%. Melting point: 233–235 °C. IR (ATR): v/cm−1 = 3469, 3040, 2915, 2781, 2661, 1609, 1582, 1548, 1242, 1123, 795, 755, 696, 662. Elemental anal. calcd. (%) for C28H18CuF6N2O4 (MW = 623.05 g mol−1): C, 53.90; H, 2.91; N, 4.49, found C, 53.66; H, 3.27; N, 4.51. DART-MS: m/z 623.99 ([M + H]+ of C28H18CuF6N2O4).
Cu(L3)2 (6): dark brown crystals. Yield %: 88%. Melting point: 180–183 °C. IR (ATR): v/cm−1 = 3055, 1608, 1580, 1537, 1320, 1175, 1109, 1064, 839. Elemental anal. calcd. (%) for C28H18CuF6N2O4 (MW = 623.05 g mol−1): C, 53.90; H, 2.91; N, 4.49, found C, 53.69; H, 3.31; N, 4.63. DART-MS: m/z 624.03 ([M + H]+ of C28H18CuF6N2O4).
2.2.3.1. Synthesis of Pd(II) complexes [Pd(L1G)2–Pd(L3G)2] (7–9). Complexes 7–9 were synthesized using a modified procedure based on the synthesis of acylated phenolic glycosides reported by the Li Y. group.14 The corresponding Pd(Ln)2 precursors (0.05 g, 0.0749 mmol), acetobromo-α-D-glucose (0.0924 g, 0.2247 mmol), and TBAB (0.0120 g, 0.0372 mmol) were dissolved in 1 mL of acetone. Subsequently, 1 mL of an aqueous 10 M K2CO3 solution was added, and the resulting mixture was stirred at 45 °C for varying reaction times. After completion, ethyl acetate was added to the reaction mixture, and the organic phase was washed three times with distilled water, once with brine, dried over anhydrous Na2SO4, filtered, and concentrated. The products were isolated as yellow powders by recrystallization from a CH2Cl2/hexane mixture.
Pd(L1G)2 (7): yellow powder. Yield: 77%. Melting point: 118–121 °C. 1H NMR (300 MHz, CDCl3) δ 7.70 (2, 2H), 7.59 (m, 3H), 7.52 (s, 2H), 7.45 (3, J = 6.5 Hz, 4H), 7.41–7.27 (m, 3H), 7.00 (dd, J = 20.0, 8.8 Hz, 2H), 6.15 (s, 1H), 6.08 (s, 1H), 5.41 (s, 1H), 5.30 (s, 1H), 5.27 (m, 1H), 5.25 (m, 1H), 5.23–5.15 (m, 2H), 4.92 (d, J = 7.9 Hz, 1H), 4.48–4.31 (m, 2H), 4.08 (m, 1H), 3.73 (m, 1H), 3.14 (s, 1H), 2.20–1.98 (m, 24H). 13C{1H} NMR (CDCl3, 101 MHz) δ 170.9, 170.7, 170.5, 170.3, 169.7, 169.5, 169.4, 168.8, 167.2, 167.1, 167.0, 166.9, 162.6, 162.5, 162.0, 156.5, 147.6, 139.4, 136.8, 136.4, 135.8, 132.0, 129.2, 128.4, 127.5, 126.6, 126.5, 126.4, 126.2, 125.4, 117.7, 114.5, 112.3, 110.0, 106.4, 105.2, 104.2, 97.8, 74.3, 72.8, 72.1, 71.0, 68.2, 67.6, 66.5, 61.7, 61.1, 59.0, 29.8, 24.1, 21.4, 20.9, 20.9, 20.8, 20.7, 20.6, 19.8, 13.8. 19F{1H} NMR (CDCl3, 376 MHz) δ −57.76 to −57.82 (m), −57.97 to −58.04 (m). IR (ATR): v/cm−1 = 2963, 2878, 1749, 1601, 1524, 1315, 1217, 1191, 1123, 1056, 1034, 767. Elemental anal. calcd. (%) for C56H54F6N2O22Pd (MW = 1326.21 g mol−1): C, 50.67; H, 4.10; N, 2.11, found C, 50.64; H, 4.10; N, 2.25. FAB-MS: m/z 1327 ([M + H]+ of C56H54F6N2O22Pd).
Pd(L2G)2 (8): yellow powder. Yield: 82%. Melting point: 123–125 °C. 1H NMR (300 MHz, CDCl3) δ 7.77–7.43 (m, 10H), 7.25 (s, 2H), 7.10 (m, 2H), 6.27 (m, 2H), 5.56 (m, 2H), 5.36–5.11 (m, 5H), 4.97 (m, 01H), 4.47–4.30 (m, 2H), 4.03 (d, J = 11.7 Hz, 2H), 3.74 (m, 2H), 3.23 (s, 2H), 2.10–2.01 (m, 24H). 13C{1H} NMR (CDCl3, 75 MHz) δ 170.7, 170.4, 169.5, 169.4, 162.9, 162.3, 149.9, 139.4, 136.6, 136.4, 129.2, 128.2, 122.7, 115.9, 107.4, 105.0, 97.8, 74.3, 72.8, 72.1, 71.1, 68.0, 67.6, 66.5, 61.6, 59.1, 24.2, 20.7, 19.8, 13.8. 19F{1H} NMR (CDCl3, 376 MHz) δ −61.85 to −62.14 (m), −62.22 to −62.32 (m). IR (ATR): v/cm−1 = 2963, 175, 1591, 1526, 1327, 1227, 1123, 799, 700. Elemental anal. calcd. (%) for C56H54F6N2O22Pd (MW = 1326.21 g mol−1): C, 50.67; H, 4.10; N, 2.11, found C, 50.56; H, 4.24; N, 2.53. FAB-MS: m/z 1327 ([M + H]+ of C56H54F6N2O22Pd).
Pd(L3G)2 (9): yellow powder. Yield: 79%. Melting point: 108–110 °C. 1H NMR (300 MHz, CDCl3) δ 7.75–7.64 (m, 6H), 7.60 (s, 1H), 7.55 (s, H), 7.43 (m, 5H), 7.07 (td, J = 9.1, 3.5 Hz, 3H), 6.26 (m, 1H), 6.16 (d, J = 8.6 Hz, 2H), 5.63–5.48 (m, 1H), 5.44 (m, 1H), 5.33–5.12 (m, 4H), 4.97 (dd, J = 7.2, 2.5 Hz, 1H), 4.51–4.18 (m, 4H), 4.03 (m, 1H), 3.71 (m, 1H), 3.25 (m, 1H), 2.16–1.94 (m, 24H). 13C{1H} NMR (CDCl3, 75 MHz) δ 170.3, 169.6, 136.9, 136.4, 125.7, 125.7, 125.5, 107.6, 107.0, 104.3, 97.2, 72.8, 72.1, 71.1, 68.0, 66.5, 61.6, 29.8, 20.8, 20.7. 19F{1H} NMR (CDCl3, 282 MHz) δ −61.65 (d, J = 12.8 Hz), −61.98 (d, J = 11.8 Hz). IR (ATR): v/cm−1 = 2962, 1750, 1597, 1528, 1323, 1223, 1123, 1065, 1040, 848. Elemental anal. calcd. (%) for C56H54F6N2O22Pd (MW = 1326.21 g mol−1): C, 50.67; H, 4.10; N, 2.11, found C, 50.33; H, 4.2; N, 2.58. FAB-MS: m/z 1327 ([M + H]+ of C56H54F6N2O22Pd).
2.2.3.2. Synthesis of Cu(II) complexes [Cu(L1G)2–Cu(L3G)2] (10–12). The Cu(LnG)2 complexes were synthesized using a similar procedure as the Pd(LnG)2 complexes. The corresponding Cu(Ln)2 complex (0.05 g, 0.0801 mmol), acetobromo α-D-glucose (0.0988 g, 0.2403 mmol) and TBAB (0.0120 g, 0.0372 mmol) were dissolved in 1 mL of acetone. Subsequently, 1 mL of an aqueous 10 M K2CO3 solution was added, and the resulting mixture was stirred at 45 °C for 5 h. After completion, 15 mL of ethyl acetate was added to the reaction mixture, and the organic phase was washed three times with distilled water (3 × 5 mL), once with brine (1 × 5 mL), dried over anhydrous Na2SO4, filtered, and concentrated. The products were isolated as light brown powders by recrystallization from a CH2Cl2/hexane mixture.
Cu(L1G)2 (10): light brown powder. Yield: 73%. Melting point: 146–148 °C. IR (ATR): v/cm−1 = 2964, 2878, 1750, 1612, 1598, 1228, 1194, 1120, 1035, 765. Elemental anal. calcd. (%) for C56H54CuF6N2O22 (MW = 1283.24 g mol−1): C, 52.36; H, 4.24; N, 2.18, found C, 52.11; H, 4.32; N, 2.66. FAB-EM(+ev): m/z 1284 ([M+] of C56H54CuF6N2O22).
Cu(L2G)2 (11): light brown powder. Yield: 80%. Melting point: 134–136 °C. IR (ATR): v/cm−1 = 2964, 2879, 1752, 1608, 1588, 1545, 1328, 1227, 1123, 800, 759, 699. Elemental anal. calcd. (%) for C56H54CuF6N2O22 (MW = 1283.24 g mol−1): C, 52.36; H, 4.24; N, 2.18, found C, 51.89; H, 4.69; N, 2.22. FAB-EM(+ev): m/z 1284 ([M+] of C56H54CuF6N2O22).
Cu(L3G)2 (12): light brown powder. Yield: 43%. Melting point: 104–106 °C. IR (ATR): v/cm−1 = 2964, 2878, 1747, 1590, 1538, 1323, 1219, 1178, 1162, 1116, 1065, 1037, 844. Elemental anal. calcd. (%) for C56H54CuF6N2O22 (MW = 1283.24 g mol−1): C, 52.36; H, 4.24; N, 2.18, found C, 51.89; H, 4.52; N, 2.3. FAB-EM(+ev): m/z 1284 ([M+] of C56H54CuF6N2O22).
2.3. Crystal structure analyses
Single crystal X-ray diffraction analyses were performed on a Bruker D8 Venture k-geometry diffractometer 208039-01 with CCD detector and low temperature device with the exception of Cu(L1)2 which was collected on a Bruker APEX-II CCD. The radiation used for ligands was CuKα (λ = 1.54178 Å) and for complexes were MoKa (λ = 0.71073 Å) by performing ω scan frames. Absorption corrections were applied using multi-scan SADABS 2016/215 and refined by full-matrix least-squares treatment against |F|2 in anisotropic approximation with SHELXL-2018/316 in the ShelXle program.17 H-atoms were included in the geometrically calculated positions. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Data were corrected for absorption (semi-empirical from the equivalents method) and polarisation. Molecular structures were visualised and drawn with OLEX2 v1.5.18 Intermolecular distances in the crystal structures of compounds were analysed with MERCURY v2021.3.0.192.4. Cytotoxic activity assays
The in vitro cytotoxic potency of ligands and metal complexes was evaluated by the sulforhodamine B (SRB) assay on 6 cancer cell lines: U-251 (human malignant glioblastoma), PC-3 (human Caucasian prostate adenocarcinoma), K-562 (human myelogenous leukemia), HCT-15 (human colorectal adenocarcinoma), MCF-7 (human mammary adenocarcinoma), and SK-LU-1 (human lung adenocarcinoma). Including a non-cancerous cell line of African green monkey kidney (COS-7) for comparative purposes. Cell lines were purchased from the National Cancer Institute (Bethesda, MD, USA). The cells were seeded into 96-well plates at a density of 5 × 103 cells per well and cultured for 24 h before use. The cells were treated with different concentrations: 25 μM for ligands and Cu(II) complexes and 25, 5 and 1 μM for Pd(II) complexes dissolved in DMSO. After 48 h incubation, the cells were washed with phosphate-buffered saline (PBS) (3×) and treated by the SRB staining assay. The absorbance was detected in a microplate reader at 540 nm and the cell viability was calculated with the formula: survival % (A540nm for the treated cells/A540nm for the control cells), 100%, where the A540nm was the absorbance value. Each assay was repeated three times. The cytotoxicity is presented as inhibition percentage, no IC50 values were determined.2.5. Computational details
To ensure the stability and accuracy of the geometries, we optimized the geometries of the Cu(II) and Pd(II) complexes and compared them with the crystallographic reference structures. For Cu(II), we used the B3LYP functional combined with the 6-31+G(d,p) basis set. For Pd(II), we used the same functional with the LanL2DZ pseudopotentials.20 The geometries were confirmed as minima through frequency calculations. In addition, natural bond orbital (NBO) charge calculations were conducted to provide accurate atomic charges necessary for subsequent molecular docking studies.21 The electronic structure calculations were carried out using the Gaussian 16 suite of programs.22For the molecular docking studies, we used the DNA fragment from the protein data bank (PDB code: 1AIO),23 since it has been proven in previous docking studies.24,25 The DNA structure was prepared using AutoDock Tools,26 where Gasteiger charges were assigned to ensure accurate modeling of electrostatic interactions.27 Both the ligands and DNA were then subjected to molecular docking using AutoDock 4.26
The interactions between the ligands and DNA were visualized and analyzed using Maestro,28 PyMOL,29 and Chimera softwares,30 thereby facilitating a comprehensive interpretation of the docking results and their potential biological implications.
3. Results and discussion
3.1. Synthesis and characterization of ligands L1–L3 and metal precursors N-aryl fluorinated complexes 1–6
The ligands L1–L3 were prepared using an adapted procedure previously reported from commercially available 2,4-dihidroxybenzaldehyde, 2-(trifluoromethyl)aniline (L1), 3-(trifluoromethyl)aniline (L2) and 4-(trifluoromethyl)aniline (L3) in excellent yields (95–98%) outlined in Scheme 1. All ligands were further characterized by 1H, 13C{1H}, 19F{1H} NMR, IR and mass spectrometry analysis and structurally confirmed by comparison of previously reported data (full details are provided in the Experimental section).31With the ligands in hand, the synthesis of the palladium and copper precursor complexes (1–6) was pursued, Scheme 2. The Pd(II) precursor complexes (1–3) were synthesized by reacting ligands L1–L3 with palladium acetate under microwave irradiation, resulting in good yields (83–93%). Meanwhile, the Cu(II) precursor complexes (4–6) were obtained at room temperature, with yields ranging from 80–88%. Pd(II) complexes 1–3 were characterized in solution by NMR, showing a significant downfield shift of the imine protons to around 7.8 ppm compared to the free ligands L1–L3 (≈8.8 ppm). This shift is attributed to coordination with the palladium center, confirming the formation of the Pd(II) complexes. Additionally, the absence of the resonance corresponding to the hydroxyl group proximal to the imine moiety further supports the bidentate coordination of the ligand through the anionic oxygen and nitrogen of the imine group. The presence of the CF3 group was confirmed by a singlet resonance at approximately −60.7 ppm in the 19F{H} NMR spectra of complexes 2 and 3. In contrast, complex 1, which has the CF3 group in the ortho position, exhibited two singlets at 56.6 and 56.7 ppm due to the inequivalence of the CF3 groups.
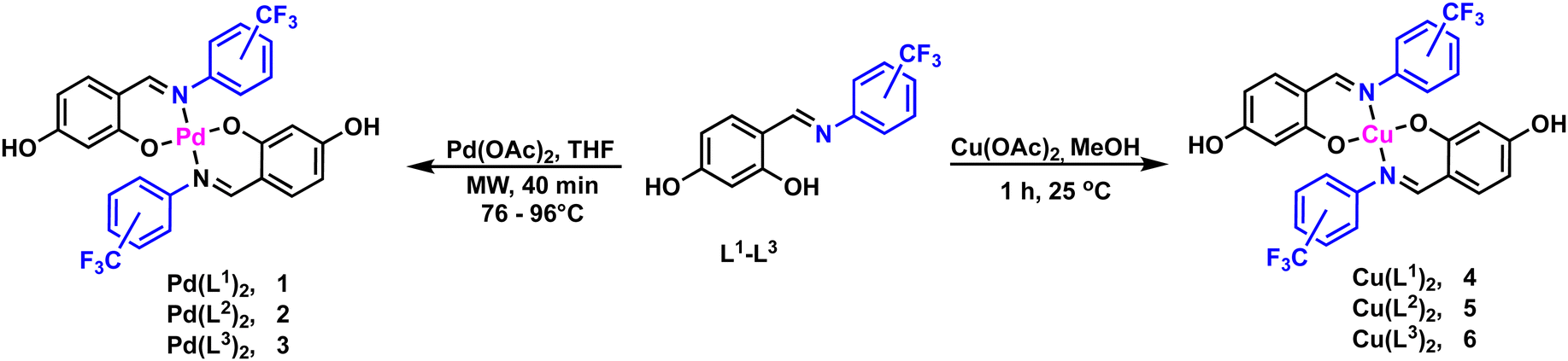 | ||
| Scheme 2 Synthetic route for the metal precursor complexes (1–6). | ||
On the other hand, Cu(II) complexes (3–6) were analyzed by EPR, where visible hyperfine splitting was observed due to the interaction between the unpaired electron of Cu(II) (S = 1/2) and the Cu(II) nucleus spin (I = 3/2). This behavior is commonly observed in Cu(II) complexes with nitrogen-containing ligands, suggesting coordination between the Cu(II) ions and nitrogen atoms. The observed g-value trend (g‖ > g > ge (2.0023)) indicates axial symmetry (gx = gy ≠ gz), suggesting that the unpaired electron resides in the dx2−y2 orbital, characteristic of axially elongated mononuclear copper(II) complexes (Table 1). This anisotropic system implies that the symmetry around the copper centers corresponds to a square planar geometry, consistent with the X-ray diffraction analysis of the Cu(L1)2 and Cu(L2)2 complexes.
| Complexes | g‖ | g⊥ |
|---|---|---|
| Recorded in acetone solutions at 77 K. | ||
| Cu(L1)2 | 2.29 | 2.06 |
| Cu(L2)2 | 2.30 | 2.06 |
| Cu(L3)2 | 2.30 | 2.07 |
| Cu(L1G)2 | 2.29 | 2.05 |
| Cu(L2G)2 | 2.31 | 2.06 |
| Cu(L3G)2 | 2.24 | 2.05 |
In addition, to confirm the coordination of the metal centers to the ligands, FT-IR spectra were recorded. A comparison with the spectra of the free Schiff bases revealed a characteristic absorption band in the 1620–1634 cm−1 region, attributable to the azomethine group (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). Upon complexation, this band shifted to lower frequencies, indicating coordination of the azomethine nitrogen to the palladium (1584–1599 cm−1) and copper (1607–1609 cm−1) centers. This shift is likely due to the donation of electron density from the nitrogen atom to the vacant d-orbitals of the metal center.32 Additionally, the FT-IR spectra of both the ligands and their corresponding complexes displayed characteristic bands of the aromatic rings, including O–H stretching vibrations between 3080–3290 cm−1 and CC stretching bands in the 1500–1600 cm−1 range. The distinctive substitution patterns of the aromatic rings were evident from the out-of-plane bending bands in the fingerprint region and overtones in the 2000–1650 cm−1 region. Moreover, strong bands in the 1350–1050 cm−1 range were assigned to C–F stretching vibrations, with no significant changes observed between the ligands and the metal complexes. The formation of complexes 1–6 was further validated by mass spectrometry. The spectra revealed molecular ion peaks at 666.98 m/z for both Pd(L1)2 and Pd(L3)2, corresponding to the [M+] ion of C28H18F6N2O4Pd, and at 667.02 m/z for Pd(L2)2, representing the [M + 1]+ ion of C28H18F6N2O4Pd. For the Cu(II) complexes, molecular ion peaks were observed at 624.03 m/z, 623.99 m/z, and 624.03 m/z for Cu(L1)2, Cu(L2)2, and Cu(L3)2, respectively, which align with the expected molecular weight of [M+] C28H18CuF6N2O4. Furthermore, the molecular structures of complexes 1–6 are consistent with their elemental analysis data and X-ray diffraction results, corroborating the proposed structures.
N). Upon complexation, this band shifted to lower frequencies, indicating coordination of the azomethine nitrogen to the palladium (1584–1599 cm−1) and copper (1607–1609 cm−1) centers. This shift is likely due to the donation of electron density from the nitrogen atom to the vacant d-orbitals of the metal center.32 Additionally, the FT-IR spectra of both the ligands and their corresponding complexes displayed characteristic bands of the aromatic rings, including O–H stretching vibrations between 3080–3290 cm−1 and CC stretching bands in the 1500–1600 cm−1 range. The distinctive substitution patterns of the aromatic rings were evident from the out-of-plane bending bands in the fingerprint region and overtones in the 2000–1650 cm−1 region. Moreover, strong bands in the 1350–1050 cm−1 range were assigned to C–F stretching vibrations, with no significant changes observed between the ligands and the metal complexes. The formation of complexes 1–6 was further validated by mass spectrometry. The spectra revealed molecular ion peaks at 666.98 m/z for both Pd(L1)2 and Pd(L3)2, corresponding to the [M+] ion of C28H18F6N2O4Pd, and at 667.02 m/z for Pd(L2)2, representing the [M + 1]+ ion of C28H18F6N2O4Pd. For the Cu(II) complexes, molecular ion peaks were observed at 624.03 m/z, 623.99 m/z, and 624.03 m/z for Cu(L1)2, Cu(L2)2, and Cu(L3)2, respectively, which align with the expected molecular weight of [M+] C28H18CuF6N2O4. Furthermore, the molecular structures of complexes 1–6 are consistent with their elemental analysis data and X-ray diffraction results, corroborating the proposed structures.
3.2. Synthesis of Pd(II) and Cu(II) complexes with N-aryl fluorinated O-glycoconjugate Schiff base ligands [M(LnG)2] (7–12)
The synthesis of complexes 7–12 is outlined in Scheme 3. Glycosylation of complexes 1–6 was performed via a 1,2-trans-β-glycosylation of a per-O-acetylated D-glucose donor, leading to the formation of N-aryl fluorinated O-glycoconjugate complexes 7–12 in good yields 73–82%. The identities of these complexes were confirmed through multinuclear NMR (specifically for complexes 7–9), mass spectrometry, infrared spectroscopy (IR), and elemental analysis.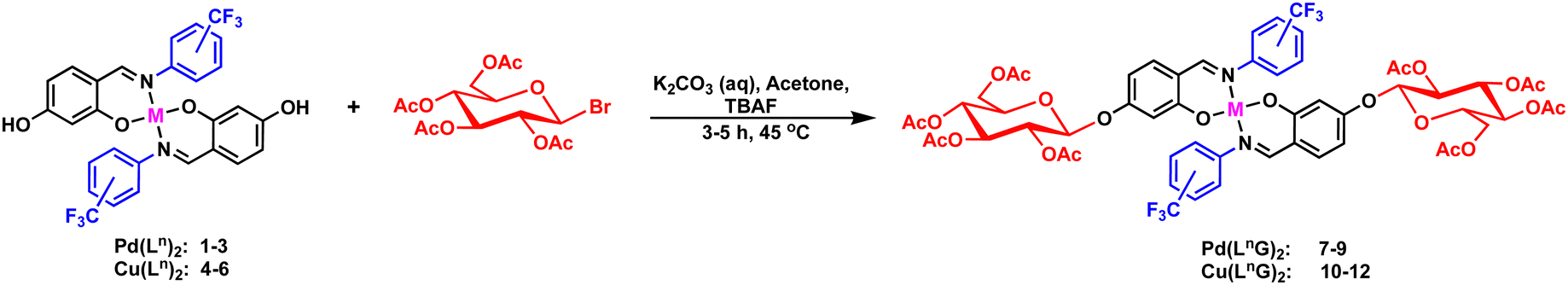 | ||
| Scheme 3 Synthetic route for the metal complexes (7–12). | ||
Comparison of the 1H NMR spectra of palladium complexes 1–3 with their glycosylated analogs revealed a loss of molecular symmetry, broader aromatic proton signals, and new signals corresponding to the O-acetylated D-glucose groups (Fig. 1, see ESI,† Fig. S7 and S8). In addition, the signal for the –OH group was absent in the 1H NMR spectra of complexes 7–9 confirming the deprotonation of this group and the formation of the glycosidic bond. In the 19F{1H} NMR spectra of the glycosylated complexes, two multiplet signals were detected in the range of −57.76 to −62.28 ppm. The emergence of these two signals is likely due to the loss of molecular symmetry, and the observed multiplicity can be attributed to the interaction of fluorine atoms with the surrounding molecular environment, influencing their magnetic equivalence.
 | ||
| Fig. 1 1H NMR spectrum of complex 7 in CDCl3: Pd(L1G)2. | ||
The mass spectra of all palladium complexes Pd(L1G)2, Pd(L2G)2 and Pd(L3G)2 revealed molecular ion peaks at m/z 1327, corresponding to the fragment ion [M + H]+ for C56H54F6N2O22Pd (see Fig. S12 in the ESI†).
Copper complexes 10–12 (Cu(L1)2, Cu(L2)2, and Cu(L3)2) were obtained as light brown powders in moderate to good yields (43–80%). The mass spectra of all the glycosylated copper complexes exhibited a molecular ion peak at 1284 m/z, corresponding to the molecular mass [M]+ (Fig. S15 in the ESI†). Their EPR analysis showed no significant changes in g-values between the hydroxylated complexes (4–6) and their glycoconjugated analogs, suggesting a similar coordination environment (Table 1).
The FTIR spectra of all glycoconjugated compounds displayed the most significant stretching frequencies at 1749–1751 cm−1 (CO), 3080–2300 cm−1 (C–H), and 1500–1600 cm−1 (CC). Additionally, the absence of the –OH vibration band indicate deprotonation of the hydroxylated complexes and the subsequent formation of the glycosidic bond. Selected bond frequencies for the ligands and copper and palladium complexes are shown in Table 2. Furthermore, the molecular structures of complexes 7–12 are consistent with their elemental analysis data corroborating the proposed structures.
| Compounds | v(O–H) | v(CN) |
v(CO) |
|---|---|---|---|
| L1 | 3255 | 1620 | — |
| L2 | 3250 | 1626 | — |
| L3 | 3298 | 1634 | — |
| Pd(L1)2 | 3296 | 1599 | — |
| Pd(L2)2 | 3104 | 1584 | — |
| Pd(L3)2 | 3251 | 1597 | — |
| Pd(L1G)2 | — | 1600 | 1749 |
| Pd(L2G)2 | — | 1591 | 1751 |
| Pd(L3G)2 | — | 1597 | 1750 |
| Cu(L1)2 | 3000 | 1607 | — |
| Cu(L2)2 | 3469 | 1609 | — |
| Cu(L3)2 | 3055 | 1608 | — |
| Cu(L1G)2 | — | 1612 | 1749 |
| Cu(L2G)2 | — | 1607 | 1751 |
| Cu(L3G)2 | — | 1589 | 1746 |
3.3. Single-crystal X-ray diffraction analyses
Suitable crystals of ligands L1 and L2, as well as copper (1–3) and palladium complexes (4–6), were obtained and analyzed by single-crystal X-ray diffraction. The crystallographic data for L1, L2, and the compounds Cu(L1)2, Cu(L2)2, and Cu(L3)2 are presented in the ESI† Table SI-2. Ligand L1 crystallized in the orthorhombic system (space group Pbca), while L2 and Cu(L1)2 were found in monoclinic systems (space groups Pn and P21/c, respectively). In contrast, Cu(L2)2 and Cu(L3)2 crystallized in triclinic systems, both in the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group.
space group.
Ligands L1 and L2 were identified as cis-keto tautomers, due to the influence of the salicylidene moiety, which affects the electronic distribution of the system and drives the equilibrium from the phenol-imine form to the keto-amine form (Fig. 2).33
 | ||
| Fig. 2 Molecular structure of compounds L1 and L2. Ellipsoids are represented at 50% of probability. The intramolecular interactions are shown as dashed red lines. | ||
In the compounds Cu(L1)2 and Cu(L2)2, the deprotonated O-fragment from ligands adopted a trans disposition around of metal center, resulting in a planar square geometry with substituent angles close to 90. In contrast, Cu(L3)2 exhibited a cis arrangement, giving rise to a pseudo-tetrahedral geometry, with τ4 = 0.506 (Fig. 3).34 The selected bond distances and angles are summarized in Table 3.
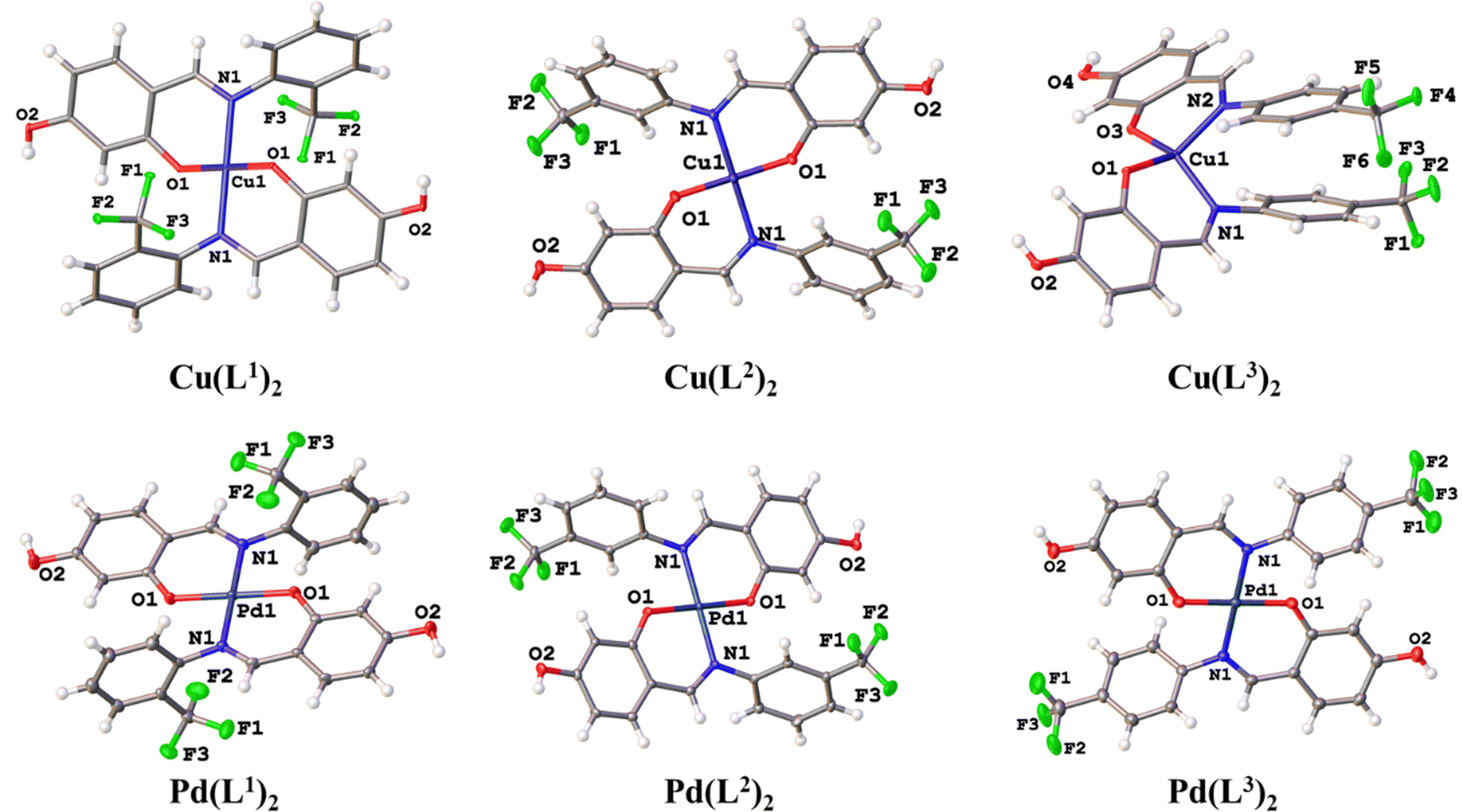 | ||
| Fig. 3 Molecular structure of the compounds Cu(L1)2, Cu(L2)2, Cu(L3)2 Pd(L1)2, Pd(L2)2, and Pd(L3)2. Thermal ellipsoids are shown at 50% of probability. Solvent molecules and disordered parts of Pd(L3)2 were omitted for clarity. | ||
| Angles | Cu(L1)2 | Cu(L2)2 | Cu(L3)2 |
|---|---|---|---|
| O(1)–Cu(1)–O(2) | 180.0 | 180.00(11) | 91.66(6) |
| O(1)–Cu(1)–N(2) | 90.19(5) | 88.51(9) | 94.35(6) |
| N(1)–Cu(1)–O(1) | 89.81(5) | 91.49(9) | 94.35(6) |
| N(1)–Cu(1)–N(2) | 180.0 | 180.00(10) | 100.78(7) |
The crystallographic data for the hydroxylated Pd(II) complexes 4–6 are summarized in the ESI† Table SI-2. All palladium complexes crystallized with two solvent molecules. Pd(L1)2 and Pd(L3)2 crystallized in a monoclinic system (space group P21/c), while Pd(L2)2 crystallized in an orthorhombic system (space group Pbca). In all structures, the palladium center was found to be tetra-coordinated, adopting a planar square geometry. The ligands arranged themselves in the most thermodynamically stable trans position, resulting in half of the molecule being generated by symmetry. This arrangement led to O–Pd–O and N–Pd–N angles of exactly 180°, indicating a planar square geometry, while the bite angles in the O–Pd–N coordination were slightly greater than 90°. Selected bond distances and angles are listed in Table 4. The two rings of the skeleton were not coplanar; they were completely out of phase, with interplanar angles decreasing according to the following trend: 88.62° Pd(L1)2, 66.18° Pd(L2)2, and 48.66° Pd(L3)2. The presence of the CF3 group at the ortho or meta position produced larger angles compared to when the CF3 group is in the para position.35
3.4. In vitro cytotoxic activity
The cytotoxicity of ligands and complexes (1–12) towards PC-3 (human Caucasian prostate adenocarcinoma), MCF-7 (human breast cancer), HCT-15 (human colorectal adenocarcinoma), SK-LU-1 (human lung adenocarcinoma), K-562 (human myelogenous leukemia) and U-251 (human malignant glioblastoma) cancer cell lines was evaluated by sulforhodamine-B (SRB) assay, including a non-cancerous cell line of African green monkey kidney (COS-70), and precursors 2,4-dihydroxybenzaldehyde and acetobromo-α-D-glucose for comparative analysis.Ligands and complexes were initially tested at a concentration of 25 μM (Table 5). The results indicated that the ligands exhibited minimal cytotoxic activity across the different cell lines, comparable to or even lower than that of the precursor 2,4-dihydroxybenzaldehyde. Hydroxylated N-aryl fluorinated Cu(II) complexes also demonstrated modest cytotoxic activity, with the MCF-7 cell line being the most affected, showing inhibition rates between 46% and 55%. Notably, these complexes displayed low toxicity toward the non-cancerous COS-7 cell line. A comparative analysis between hydroxylated Cu(II) complexes and their glycoconjugate counterparts revealed a marked increase in cytotoxic activity for the Cu(L1G)2 complex across various cell lines, particularly K-562, HCT-15, MCF-7, and SK-LU-1. The Cu(L2G)2 and Cu(L3G)2 complexes exhibited variable activity, with enhanced effects on the K-562 and U-251 lines but reduced impact on MCF-7 and SK-LU-1. Importantly, the glycoconjugate complexes were less toxic to the non-cancerous COS-7 cell line compared to their hydroxylated counterparts.
| Compounds (25 μM) | % of inhibition | ||||||
|---|---|---|---|---|---|---|---|
| U251 | PC-3 | K-562 | HCT-15 | MCF-7 | SKLU-1 | COS-7 | |
| Vehicle: DMSO, 25 μM. NC: no cytotoxic. U-251 (human malignant glioblastoma), PC-3 (human Caucasian prostate adenocarcinoma), K-562 (myelogenous leukemia), HCT-15 (human colorectal adenocarcinoma), MCF-7 (human breast cancer), SK-LU-1 (human lung adenocarcinoma), African green monkey kidney (COS-7).a Poor solubility. Data represents the mean of three replica measures. | |||||||
| L1 | NC | NC | 7.2 | 16.0 | 30.6 | 10.3 | 12.5 |
| L2 | 9.1 | NC | 5.8 | 24.6 | 41.3 | 12.6 | 13.0 |
| L3 | 16.8 | 14.0 | 10.7 | 35.4 | NC | NC | 29.0 |
| Pd(L1)2 | 100 | 100 | 95.6 | 100 | 100 | 96.1 | 89.7 |
| Pd(L2)2 | 100 | 79.9 | 75.5 | 83.3 | 97.2 | 94.8 | 92.6 |
| Pd(L3)2a | 92.0 | 91.6 | 100 | 67.7 | 97.0 | 82.2 | 80.5 |
| Pd(L1G)2 | 97.5 | 100 | 95.7 | 100 | 100 | 94.3 | 76.7 |
| Pd(L2G)2 | 98.7 | 95.8 | 92.4 | 100 | 100 | 100 | 94.0 |
| Pd(L3G)2 | 100 | 100 | 95.9 | 100 | 100 | 99.0 | 99.7 |
| Cu(L1)2 | 6.3 | 4.4 | 8.9 | 21.1 | 46.9 | 20.1 | 18.3 |
| Cu(L2)2 | 4.8 | 9.3 | 5.8 | 22.9 | 53.2 | 17.3 | 22.2 |
| Cu(L3)2 | 6.5 | 5.2 | 7.4 | 13.5 | 46.1 | 25.0 | 21.5 |
| Cu(L1G)2 | 33.0 | 34.4 | 47.5 | 58.3 | 62.4 | 44.5 | 15.9 |
| Cu(L2G)2 | 17.4 | 6.2 | 16.7 | 3.5 | 32.5 | 12.7 | 16.0 |
| Cu(L3G)2 | 24.9 | 7.1 | 22.1 | 22.6 | 37.6 | 14.3 | 14.6 |
| 2,4-Dihydroxybenzaldehyde | 19.9 | 5.2 | 5.9 | 46.1 | 60.0 | 10.7 | 28.7 |
| Acetobromo-α-D-glucose | 8.5 | NC | 11.6 | 0.9 | 2.0 | 3.8 | 5.1 |
| Cis-platin | 100 | 100 | 71.6 | 65.4 | 85.5 | 100 | 100 |
Pd(II) complexes activity was also investigated, where both hydroxylated and glycoconjugate variants exhibited high cytotoxic activity across all tested cell lines, including the non-cancerous one (Table 5). Owing to their high activity at 25 μM, we assessed the complexes at 5 μM. However, since the results obtained were similar to those at 25 μM, with all complexes showing high activity over all cell lines, and in order to improve selectivity, we further evaluated the complexes activity at 1 μM (Table 6). At this concentration, only Pd(L1)2 and its glycoconjugate form Pd(L1G)2 maintained substantial activity against the cancer cell lines, particularly for PC-3, K-562, MCF-7, and SK-LU-1 being the latest where both complexes exhibited the highest activity. Notably, both Pd(II) complexes resulted more cytotoxic than cis-platin at the same concentration.
| % of inhibition | |||||||
|---|---|---|---|---|---|---|---|
| Complexes | U251 | PC-3 | K562 | HCT-15 | MCF-7 | SKLU-1 | COS7 |
| Vehicle: DMSO.a 5 μM.b 1 μM. NC: no cytotoxic. U-251 (human malignant glioblastoma), PC-3 (human Caucasian prostate adenocarcinoma), K-562 (myelogenous leukemia), HCT-15 (human colorectal adenocarcinoma), MCF-7 (human breast cancer), SK-LU-1 (human lung adenocarcinoma), African green monkey kidney (COS-7).c Data represents the mean of three replica measures. | |||||||
| Pd(L1)2a | 83.7 | 98.0 | 81.0 | 90.2 | 100 | 100 | 83.4 |
| Pd(L1)2b | 33.7 | 85.5 | 66.8 | 24.0 | 100 | 99.4 | 70.3 |
| Pd(L2)2a | 22.3 | 40.1 | 66.1 | 3.7 | 63.0 | 40.9 | 92.6 |
| Pd(L2)2b | 3.1 | 3.5 | 3.1 | NC | NC | NC | NC |
| Pd(L3)2a | 12.0 | 25.0 | 61.9 | NC | 70.0 | 43.3 | 68.8 |
| Pd(L3)2b | NC | 14 | 20.9 | NC | 0.6 | 2.5 | 8.3 |
| Pd(L1G)2a | 63.7 | 81.2 | 80.0 | 46.6 | 100 | 96.8 | 85.5 |
| Pd(L1G)2b | 14 | 71.4 | 60.0 | NC | 67.3 | 84.2 | 23 |
| Pd(L2G)2a | 50.5 | 71.9 | 85.7 | 19.4 | 100 | 99.1 | 88.9 |
| Pd(L2G)2b | 3.4 | 14.1 | 13.3 | NC | NC | NC | 10 |
| Pd(L3G)2a | 61.9 | 80.0 | 90.58 | 35.0 | 100 | 93.7 | 80 |
| Pd(L3G)2b | NC | 21.1 | 13.2 | NC | 9.1 | 1.6 | NC |
| Cis-platina | 48.7 | 28.7 | 36.6 | 17.8 | 27.0 | 45.0 | 34.3 |
| Cis-platinb | 33.4 | 15.1 | 9.2 | 12.8 | 17.4 | 25.7 | 5.7 |
Interestingly, even Pd(L1)2 complex resulted more active than Pd(L1G)2 complex; the latter displayed significantly reduced cytotoxicity towards the non-cancerous COS-7 cell line (23% vs. 70.3% of Pd(L1)2), a trend similar to the observed in glycoconjugate Cu(II) complexes, where the activity over the healthy cell line (COS-7) was lower than that from hydroxylated Cu(II) complexes. Which demonstrate a substantial selectivity to cancer cells attributable to the presence of carbohydrate moieties which, at the same time, enhanced the biological activity of the glycoconjugate complexes.
Another finding was that the position of the CF3 group in the benzene ring not only influenced the solubility of the complexes but also had a notable impact on their cytotoxic activity. Glycoconjugate complexes with ortho substitution displayed greater activity than those with meta or para positions. However, the mechanisms underlying their selectivity require further investigation.
3.5. Computational results
The DFT-optimized geometries and X-ray structures for the Pd(II) and Cu(II) complexes show little variation (Table 7). Based on these optimized geometries, we proceeded to construct the complexes with α-D-glucose, as these geometries are crucial for conducting a comprehensive molecular docking study.| Pd(II) | Cu(II) | |
|---|---|---|
| (L1)2 | 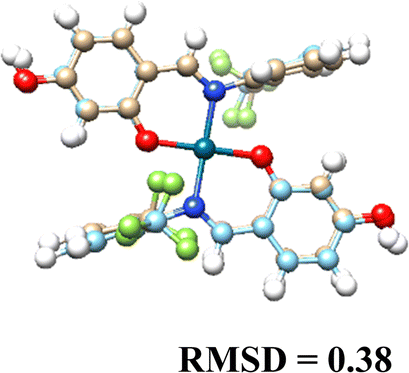 |
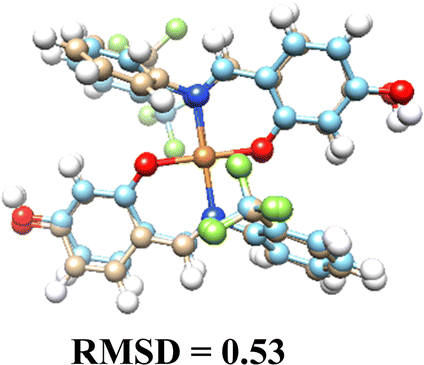 |
| (L2)2 | 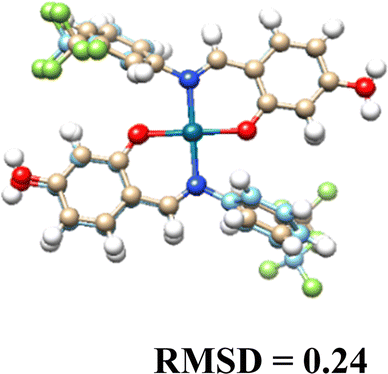 |
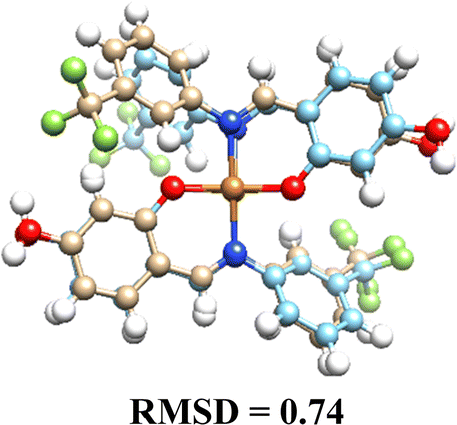 |
| (L3)2 | 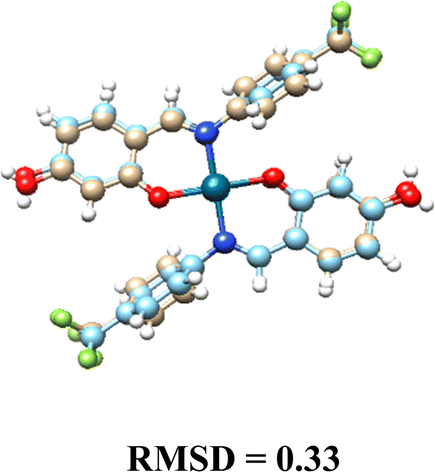 |
 |
The molecular docking results provide insights into the interaction of Cu(II) and Pd(II) complexes with DNA. Pd(II) complexes, especially in their glycosylated forms, exhibited stronger binding affinities, prompting the focus of our analysis on these complexes (Table 8).
| Pd(II) | Cu(II) | |
|---|---|---|
| (L1)2 | −5.34 | −3.42 |
| (L2)2 | −4.73 | −4.63 |
| (L3)2 | −4.23 | −3.75 |
| (L1G)2 | −7.24 | −4.54 |
| (L2G)2 | −6.32 | −3.32 |
| (L3G)2 | −6.21 | −3.48 |
3.6. Binding affinity of Pd(II) complexes
Pd(II) complexes showed significantly higher binding affinities with DNA compared to Cu(II) complexes, particularly when coordinated with glycosylated ligands (L1G)2, (L2G)2, and (L3G)2. These complexes presented affinities of −7.43, −6.32, and −6.21 kcal mol−1, respectively.The inclusion of α-D-glucose in the ligands enhanced binding affinities due to the formation of hydrogen bonds, as illustrated in Fig. 4. The α-D-glucose groups formed stabilizing hydrogen bonds with DNA, while non-glycosylated Pd(II) complexes relied on weaker hydrophobic interactions. These hydrogen bonds are key to the increased stability of the DNA–Pd(II) complex interaction, distinguishing them from the non-glycosylated Pd(II) complexes. In contrast, non-glycosylated Pd(II) complexes primarily relied on hydrophobic interactions.
 | ||
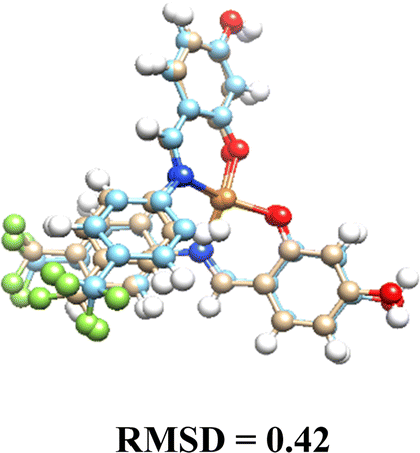
| Fig. 4 Molecular interaction between DNA target and complexes: (A) Pd(L1G)2 (B) Pd(L1)2 – distances shown in angstroms. | ||
The non-glycosylated palladium complexes were found to occupy the same DNA binding site. However, they exhibited a displacement, likely due to repulsion between the fluorine atoms in the ligands and the electronegative groups of the DNA backbone. This fluorine-induced repulsion causes a shift in binding conformation, affecting interaction stability (see Fig. S21, ESI†).
In contrast, the glycosylated Pd(II) complexes displayed no consistent binding mode. This inconsistency is attributed to the bulky α-D-glucose groups and intramolecular fluorine-ligand repulsions. These factors introduce a wider range of binding conformations during molecular docking. The flexibility conferred by the α-D-glucose groups allows Pd(II) complexes to adopt diverse conformations, enhancing interaction with various DNA regions and providing a broader interaction surface. Fig. S22 in the ESI† illustrates the structural versatility of the glycosylated complexes, lacking a fixed binding pattern.
The increased size and complexity of the α-D-glucose ligands suggest that glycosylated Pd(II) complexes may offer enhanced adaptability in interactions with biological targets like DNA, compared to the more rigid binding of non-glycosylated complexes. This flexibility could be crucial for the overall efficacy of Pd(II) complexes in biological systems, where adaptable binding is often key to successful molecular interactions.
3.7. Impact of glycosylation
Glycosylation not only enhanced binding affinity but also introduced hydrogen bonding, resulting in stronger and more stable interactions with DNA. In contrast, the non-glycosylated Pd(II) complexes primarily engaged in hydrophobic interactions, which contributed to binding but were weaker compared to the hydrogen bonds present in the glycosylated complexes.In Cu(II) complexes, glycosylation moderately improved binding affinity, but their interactions with DNA remained less effective compared to Pd(II) complexes. This underscores the inherent advantages of palladium's coordination chemistry for DNA binding.
4. Conclusions
In conclusion, the introduction of carbohydrate fragments into Pd(II) and Cu(II) metal complexes represents a promising strategy for enhancing the selectivity and efficacy of metal-based chemotherapeutics. This study demonstrated that the incorporation of N-aryl fluorinated O-glycoconjugate Schiff base ligands into copper and palladium complexes increases their cytotoxic activity against various cancer cell lines while exhibiting minimal activity against non-cancerous cell lines. Notably, among all tested compounds, the Pd(L1G)2 complex exhibited the highest cytotoxicity, showing reduced activity against non-cancerous cells, underscoring the potential of carbohydrate moieties in improving selectivity toward malignant cells. Molecular docking analysis reveals that Pd(II) complexes, especially those with glycosylated ligands, demonstrate a stronger DNA binding affinity. The glycosylated ligands enhance hydrogen bonding interactions, resulting in more stable and effective binding. While Cu(II) complexes show moderate improvements with glycosylation, their overall binding affinity remains lower compared to Pd(II) complexes.These results highlight the potential of Pd(II) complexes for further investigation, with ligand modifications being crucial for optimizing molecular interactions. The findings underscore the promise of carbohydrate–metal complex conjugates as targeted therapeutic agents, warranting further exploration and development for clinical applications. Future studies should focus on elucidating the precise mechanisms of action to optimize the structural features of these complexes, particularly fluorinated ortho-substituted compounds that demonstrated improved activity over para and meta positions.
Author contributions
Conceptualization, M. E. M.-N. and D. M.-M.; synthesis of ligands and complexes, M. E. M.-N.; cytotoxicity assays M. T. R.-A. and V. R.-M.; X-ray diffraction experiments S. H.-O. and A. A.-C.; investigation M. E. M.-N.; J. A.-T. and A. L. O.; methodology M. E. M.-N.; funding acquisition D. M.-M.; project administration D. M. M.; supervision, D. M.-M.; visualization, M. E. M.-N and D. M.-M.; writing – original draft M. E. M.-N., A. A.-C.; L. G.-S.; L. L.-R.; writing – review & editing, M. E. M.-N., A. A.-C., J. A. C. N. and D. M.-M.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
M. E. M.-N. thanks CONAHCyT for the doctoral scholarship (CVU: 815652). A. A.-C. and J. A. C. is grateful to CONAHCyT for a Postdoctoral Fellowships (estancias posdoctorales por México 2022(1)). We are indebted to María de la Paz Orta (EA), Adriana Romo (ATR-FTIR), Virginia Gómez (EPR), Elizabeth Huerta (NMR), and Francisco Javier Pérez (Mass spectrometry). D. M. M. is grateful for the financial support of this research by DGAPA PAPIIT IN223323.References
- World Health Organization. https://www.who.int/health-topics/cancer#tab=tab_1 (accessed 2024-04-03).
- R. Oun, Y. E. Moussa and N. J. Wheate, The Side Effects of Platinum-Based Chemotherapy Drugs: A Review for Chemists, Dalton Trans., 2018, 47(19), 6645–6653, 10.1039/C8DT00838H.
- C. Boulechfar, H. Ferkous, A. Delimi, A. Djedouani, A. Kahlouche, A. Boublia, A. S. Darwish, T. Lemaoui, R. Verma and Y. Benguerba, Schiff Bases and Their Metal Complexes: A Review on the History, Synthesis, and Applications, Inorg. Chem. Commun., 2023, 150, 110451, DOI:10.1016/j.inoche.2023.110451.
- S. Thakur, A. Jaryal and A. Bhalla, Recent Advances in Biological and Medicinal Profile of Schiff Bases and Their Metal Complexes: An Updated Version (2018–2023), Results Chem., 2024, 7, 101350, DOI:10.1016/j.rechem.2024.101350.
- L. A. Alfonso-Herrera, D. Hernández-Romero, J. A. Cruz-Navarro, Á. Ramos-Ligonio, A. López-Monteon, J. M. Rivera-Villanueva, D. Morales-Morales and R. Colorado-Peralta, Transition Metal Complexes with Tetradentate Schiff Bases (N2O2) Obtained from Salicylaldehyde: A Review of Their Possible Anticancer Properties, Coord. Chem. Rev., 2024, 505, 215698, DOI:10.1016/j.ccr.2024.215698.
- G. Shabir, A. Saeed, W. Zahid, F. Naseer, Z. Riaz, N. Khalil, Muneeba and F. Albericio, Chemistry and Pharmacology of Fluorinated Drugs Approved by the FDA (2016–2022), Pharmaceuticals, 2023, 16(8), 1162, DOI:10.3390/ph16081162.
- E. Henary, S. Casa, T. L. Dost, J. C. Sloop and M. Henary, The Role of Small Molecules Containing Fluorine Atoms in Medicine and Imaging Applications, Pharmaceuticals, 2024, 17(3), 281, DOI:10.3390/ph17030281.
- G. Chandra, D. V. Singh, G. K. Mahato and S. Patel, Fluorine-a Small Magic Bullet Atom in the Drug Development: Perspective to FDA Approved and COVID-19 Recommended Drugs, Chem. Pap., 2023, 77(8), 4085–4106, DOI:10.1007/s11696-023-02804-5.
- S. Yano and Y. Mikata, Recent Progress of Functional Glycoconjugated Metal Complexes, Bull. Chem. Soc. Jpn., 2002, 75(10), 2097–2113, DOI:10.1246/bcsj.75.2097.
- Y. Mikata and S. Yano, Development of Sugar-Based Materials for Biological Devices, Curr. Top. Med. Chem., 2012, 12(3), 145–157, DOI:10.2174/156802612799079017.
- Y. Mikata and M. Gottschaldt, Metal Complexes of Carbohydrate-targeted Ligands in Medicinal Inorganic Chemistry, Ligand Design in Medicinal Inorganic Chemistry, Wiley, 2014, pp. 145–173 DOI:10.1002/9781118697191.ch6.
- G. Bononi, D. Iacopini, G. Cicio, S. Di Pietro, C. Granchi, V. Di Bussolo and F. Minutolo, Glycoconjugated Metal Complexes as Cancer Diagnostic and Therapeutic Agents, ChemMedChem, 2021, 16(1), 30–64, DOI:10.1002/cmdc.202000456.
- V. K. Rao, S. S. Reddy, B. S. Krishna, K. R. M. Naidu, C. N. Raju and S. K. Ghosh, Synthesis of Schiff's Bases in Aqueous Medium: A Green Alternative Approach with Effective Mass Yield and High Reaction Rates, Green Chem. Lett. Rev., 2010, 3(3), 217–223, DOI:10.1080/17518251003716550.
- S. Yan, S. Ren, N. Ding and Y. Li, Concise Total Synthesis of Acylated Phenolic Glycosides Vitexnegheteroin A and Ovatoside D, Carbohydr. Res., 2018, 460, 41–46, DOI:10.1016/j.carres.2018.02.015.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination, J. Appl. Crystallogr., 2015, 48(1), 3–10, DOI:10.1107/S1600576714022985.
- G. M. Sheldrick, Crystal Structure Refinement with SHELXL, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71(1), 3–8, DOI:10.1107/S2053229614024218.
- C. B. Hübschle, G. M. Sheldrick and B. Dittrich, ShelXle: A Qt Graphical User Interface for SHELXL, J. Appl. Crystallogr., 2011, 44(6), 1281–1284, DOI:10.1107/S0021889811043202.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: A Complete Structure Solution, Refinement and Analysis Program, J. Appl. Crystallogr., 2009, 42(2), 339–341, DOI:10.1107/S0021889808042726.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, Mercury 4.0: From Visualization to Analysis, Design and Prediction, J. Appl. Crystallogr., 2020, 53(1), 226–235, DOI:10.1107/S1600576719014092.
- L. E. Roy, P. J. Hay and R. L. Martin, Revised Basis Sets for the LANL Effective Core Potentials, J. Chem. Theory Comput., 2008, 4(7), 1029–1031, DOI:10.1021/ct8000409.
- E. D. Glendening, C. R. Landis and F. Weinhold, NBO 6.0: Natural Bond Orbital Analysis Program, J. Comput. Chem., 2013, 34(16), 1429–1437, DOI:10.1002/jcc.23266.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision D.01, 2016 Search PubMed.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Crystal Structure of Double-Stranded DNA Containing the Major Adduct of the Anticancer Drug Cisplatin, Nature, 1995, 377(6550), 649–652, DOI:10.1038/377649a0.
- G. Backman-Blanco, H. Valdés, M. T. Ramírez-Apan, P. Cano-Sanchez, S. Hernandez-Ortega, A. L. Orjuela, J. Alí-Torres, A. Flores-Gaspar, R. Reyes-Martínez and D. Morales-Morales, Synthesis of Pt(II) Complexes of the Type [Pt(1,10-Phenanthroline)(SArFn)2] (SArFn = SC6H3-3,4-F2; SC6F4-4-H; SC6F5). Preliminary Evaluation of Their in Vitro Anticancer Activity, J. Inorg. Biochem., 2020, 211, 111206, DOI:10.1016/j.jinorgbio.2020.111206.
- D. Canseco-González, I. Rodríguez-Victoria, T. Apam-Ramírez, A. O. Viviano-Posadas, J. S. Serrano-García, A. Arenaza-Corona, A. L. Orjuela, J. Alí-Torres, A. Dorazco-González and D. Morales-Morales, Facile, Single Step Synthesis of a Series of D-Ring Ethisterone Substituted with 1,4-1,2,3-Triazoles. Preliminary Evaluation of Their Cytotoxic Activities, ChemMedChem, 2023, 18, e202200659, DOI:10.1002/cmdc.202200659.
- G. Morris and R. Huey, AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility, J. Comput. Chem., 2009, 30(16), 2785–2791, DOI:10.1002/jcc.21256.AutoDock4.
- J. Gasteiger and M. Marsili, Iterative partial equalization of orbital electronegativity-a rapid access to atomic charges, Tetrahedron, 1980, 36(22), 3219–3288, DOI:10.1016/0040-4020(80)80168-2.
- Schrödinger Release 2023-2. Maestro. LLC: New York, 2023.
- L. Schrödinger, The PyMOL Molecular Graphics System, Version 1.8, 2015 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, UCSF Chimera – A Visualization System for Exploratory Research and Analysis, J. Comput. Chem., 2004, 25(13), 1605–1612, DOI:10.1002/jcc.20084.
- A. Avila-Sorrosa, J. I. Hernández-González, A. Reyes-Arellano, R. A. Toscano, R. Reyes-Martínez, J. Roberto Pioquinto-Mendoza and D. Morales-Morales, Synthesis, Structural Characterization and Biological Activity of Fluorinated Schiff-Bases of the Type [C6H4-1-(OH)-3-(CHNArF)], J. Mol. Struct., 2015, 1085, 249–257, DOI:10.1016/j.molstruc.2014.12.080.
- A. Avila-Sorrosa, A. Y. Bando-Vázquez, V. Alvarez-Alvarez, E. Suarez-Contreras, R. Nieto-Meneses, B. Nogueda-Torres, M. E. Vargas-Díaz, F. Díaz-Cedillo, R. Reyes-Martínez, S. Hernandez-Ortega and D. Morales-Morales, Synthesis, Characterization and Preliminary in Vitro Trypanocidal Activity of N-Arylfluorinated Hydroxylated-Schiff Bases, J. Mol. Struct., 2020, 1218, 128520, DOI:10.1016/j.molstruc.2020.128520.
- S. D. Chatziefthimiou, Y. G. Lazarou, E. Hadjoudis, T. Dziembowska and I. M. Mavridis, Keto Forms of Salicylaldehyde Schiff Bases: Structural and Theoretical Aspects, J. Phys. Chem. B, 2006, 110(47), 23701–23709, DOI:10.1021/jp064110p.
- L. Yang, D. R. Powell and R. P. Houser, Structural variation in copper(I) complexes with pyridylmethylamide ligands: structural analysis with a new four-coordinate geometry index, τ4, Dalton Trans., 2007,(9), 955–964, 10.1039/b617136b.
- L. Á. Turcio-García, H. Valdés, A. Arenaza-Corona, S. Hernández-Ortega and D. Morales-Morales, Electronic Properties and Supramolecular Study of Selenoureas with Fluorinated-NHC Ligands Derived from Imidazo[1,5-a]Pyridines, New J. Chem., 2022, 47(4), 2090–2095, 10.1039/d2nj04699g.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra of: 1H, 19F{1H} and 13C{1H} NMR, ATR-IR, EPR and mass spectra of ligands and metal complexes. CCDC 2404219–2404226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj05181e |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |