
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-(Methyl)phthalimide para functionalized Ni(II)–POCOP pincer complexes. Synthesis, characterization and biological activity†
Andrés Amaya-Flórez a,
Juan S. Serrano-Garcíaa,
Jordi Ruiz-Galindoa,
Antonino Arenaza-Coronaa,
Simón Hernández-Ortegaa,
Adrián L. Orjuelab,
Jorge Alí-Torresb,
Marcos Flores-Alamoc,
Viviana Reyes-Márquezd and
David Morales-Morales*a
a,
Juan S. Serrano-Garcíaa,
Jordi Ruiz-Galindoa,
Antonino Arenaza-Coronaa,
Simón Hernández-Ortegaa,
Adrián L. Orjuelab,
Jorge Alí-Torresb,
Marcos Flores-Alamoc,
Viviana Reyes-Márquezd and
David Morales-Morales*a
aInstituto de Química, Universidad Nacional Autónoma de México, Circuito Exterior, Ciudad de México, CP 04510, Mexico. E-mail: damor@unam.mx; Fax: +52-5556162217; Tel: +52-5556224514
bDepartamento de Química, Universidad Nacional de Colombia -Sede Bogotá, Bogotá DC, Colombia
cFacultad de Química, División de Estudios de Posgrados, Universidad Nacional Autónoma de México, Circuito Exterior, CU, 04510, Mexico
dDepartamento de Ciencias Químico-Biológicas, Universidad de Sonora, Luis Encinas y Rosales s/n, Hermosillo C.P. 83000, Sonora, Mexico
First published on 13th February 2025
Abstract
Pincer compounds have stood out for their great stability, and they have been primarily employed as catalysts. In recent years, they have also been studied to evaluate their biological properties. In this work, three new POCOP–Ni(II) pincer complexes functionalized with a methylphthalimide fragment (1-Ni, 2-Ni, and 3-Ni) were synthesized. These complexes were unequivocally characterized by X-ray diffraction (XRD), and supramolecular studies were conducted using Hirshfeld surfaces. The cytotoxic activity of the complexes was evaluated on six cancer cell lines and one healthy monkey kidney cell line (COS-7). In a preliminary study, 1-Ni was determined to be the most active of the series, with IC50 values ranging from 0.64 μM to 1.38 μM. Additionally, ethidium bromide (EB) displacement studies were conducted, where it was observed that complexes 1-Ni and 3-Ni intercalate into the DNA target. Moreover, molecular docking studies revealed that these complexes intercalate with DNA, as was experimentally corroborated. Furthermore, the POCOP–Ni(II) complexes did not demonstrate antioxidant activity, suggesting that the presence of the phthalimide moiety may block radical inhibition processes.
1. Introduction
Since cisplatin was approved by the FDA as the first metallodrug for cancer treatment, the design of new metal-based compounds has been increasing in recent years.1–4 Among these, organometallic compounds have gained significant attention in the field of medicine, as they have shown promising results for treating various diseases due to their greater stability compared to coordination complexes.5–14Pincer complexes, formed by the union of a tridentate ligand and a metal center, have been attracting attention due to their thermal stability and high catalytic activity. These complexes are characterized by their ability to modulate electronic and steric properties around the metal center.15–18 Due to these qualities, pincer complexes have garnered significant interest in order to explore their biological properties, primarily as antimicrobial19–21 and antitumor agents.22–28 In this context, our research group reported the antibacterial activity of a series of unsymmetrical POCOP–Pd(II) pincer complexes against both Gram-positive and Gram-negative strains, showing activity against the S. aureus strain.29 On the other hand, nickel, being a more abundant metal with isoelectronic properties to platinum (d8), forming square planar geometries, and being present in various active sites of several metalloproteins,30–32 is considered a better option for the development of new metallodrugs against cancer, potentially offering lower toxic effects and greater selectivity towards malignant cells. From this perspective, nickel pincer complexes have demonstrated very promising anticancer properties, where SCS–Ni(II) complexes showed activity against estrogen-sensitive human breast cancer cells (MCF-7) and exhibited a reduction in solid breast cancer tumors (MC4L2) in in vivo studies conducted on female mice.33 Additionally, cytotoxicity studies of POCOP–Ni(II) complexes functionalized with a hydroxyl (–OH) group revealed that these complexes were more active than cisplatin, and through DNA interaction studies, it was suggested that these may interact with DNA via intercalation.34
It is also worth noting that introducing biologically relevant fragments into the skeleton of these pincer complexes could further enhance their biological properties,35 such as the use of pharmacophore fragments like phthalimide. The biological properties of phthalimide are influenced by its hydrophobicity, which facilitates its mobility across biological membranes. It also contains a hydrogen bonding subunit, as well as an electron donor group and an aromatic hydrophobic site, which has allowed it to be used in the development of new drugs, such as immunomodulators (IMIDs) including thalidomide, pomalidomide, and lenalidomide.36
Based on this, we decided to synthesize a series of POCOP–Ni(II) pincer compounds functionalized with a methylphthalimide fragment (1-Ni to 3-Ni) (Fig. 1), which were evaluated using different cancer cell lines, as well as for their antioxidant activity. We also aimed to observe the biological effects that the introduction of a pharmacophore fragment may have, in comparison to the previously reported POCOP–Ni(II) (1–3) precursors functionalized with a hydroxyl fragment.34 Additionally, competitive displacement studies using fluorescence and molecular docking studies were conducted to understand how these compounds interact with the DNA target.
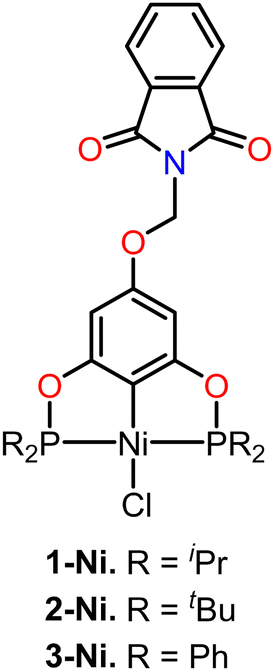 | ||
| Fig. 1 POCOP–Ni(II) pincer complexes functionalized with N-(methyl)phthalimide (1-Ni to 3-Ni). | ||
2. Result and discussion
The synthesis of the para-functionalized pincer compounds (1-Ni and 3-Ni) was carried out in three stages, starting from the POCOP–Ni(II) precursors previously reported by our research group,37 but with slight modifications (Scheme 1). For the preparation of the POCOP-type ligands, phloroglucinol was mixed with 2 eq. of Et3N to deprotonate only two hydroxyl groups, leaving the third one fully protonated. Following this, 2 eq. of the corresponding phosphine chloride were added in toluene at room temperature and stirred for 16 hours. Subsequently, an equimolar amount of anhydrous NiCl2 was added in situ, and the reaction was refluxed for 16 hours to obtain the desired complexes (1 to 3).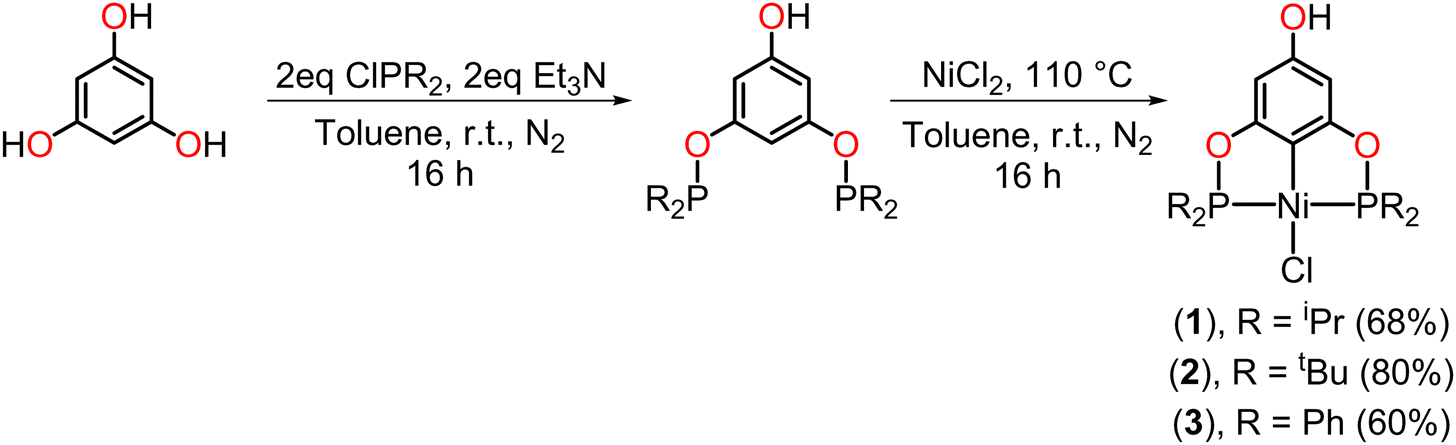 | ||
| Scheme 1 General synthesis of the precursors of POCOP–Ni(II) pincer complexes. | ||
Subsequently, the precursors were dissolved in THF in the presence of NaB(OMe)4 at room temperature and a catalytic amount of 18-crown-6 (18C6) to obtain the corresponding phenolate ion of the POCOP complex. The use of NaB(OMe)4, in addition to providing good yields, was chosen over other bases to avoid the production of water as a by-product and prevent possible hydrolysis reactions. Moreover, this, combined with 18C6, enhances the basicity of the tetramethoxyborate ion for phenolate ion formation, as well as increases its nucleophilicity to facilitate the substitution reaction on N-(chloromethyl)phthalimide, leading to the subsequent formation of NaCl as a by-product. Upon observing a change in coloration in the reaction mixture, N-(chloromethyl)phthalimide was added, and the reaction was allowed to stir at room temperature for 24 h (Scheme 2). The para-functionalized POCOP complexes were isolated as yellow solids with moderate to good yields and were characterized by NMR spectroscopy, FT-IR, mass spectrometry, elemental analysis, and single-crystal X-ray diffraction.
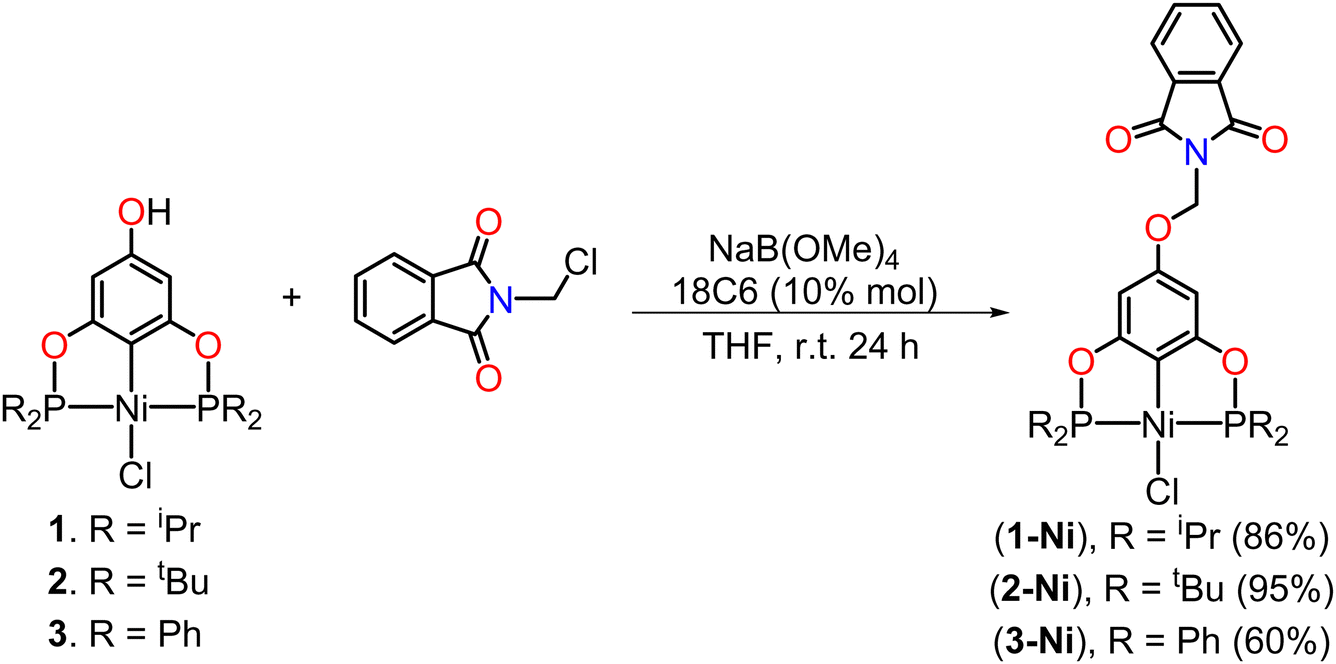 | ||
| Scheme 2 Functionalization reaction of the POCOP–Ni(II) precursors (1–3) with the N-(chloromethyl)phthalimide fragment. | ||
The yields obtained for both the precursors (1–3) and the functionalized POCOP complexes (1-Ni–3-Ni) follow the same trend: tBu > iPr > Ph. This is likely due to electronic effects on the phosphorus atoms, where nucleophilicity is greater with tBu substituents, leading to a more efficient binding to nickel in the case of the precursors (1–3). Similarly, there is an increase in the nucleophilic character of the phenolate ion for the subsequent substitution reaction on the N-(chloromethyl)phthalimide fragment.
The 1H, 13C{1H}, and 31P{1H} NMR spectra of the three complexes exhibited the expected signals for a C2v symmetric POCOP complex. The 1H NMR spectra of these new complexes displayed the characteristic AB pattern for the aromatic fragment of the phthalimide, and a single signal was observed, as expected, for the methylene protons around 5.54–5.58 ppm. In the 13C{1H} spectrum, they exhibited a carbonyl carbon signal at 167.12 ppm for 1-Ni, 169.35 ppm for 2-Ni, and 167.09 ppm for 3-Ni. Additionally, the methylene carbon signal showed a similar chemical shift (∼65 ppm) for all three complexes. All 31P{1H} NMR spectra showed a single signal consistent with a symmetric structure around the phosphorus atoms. Mass spectrometry of the three compounds exhibited peaks corresponding to the molecular ions at 610, 666, and 746 m/z for 1-Ni, 2-Ni, and 3-Ni, respectively.
Finally, the FT-IR spectra of these complexes showed symmetric and asymmetric frequency bands of the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) group between 1722–1730 cm−1.
O) group between 1722–1730 cm−1.
The molecular structures of complexes 1-Ni to 3-Ni were unequivocally determined by X-ray diffraction (Table 1). Single crystals were obtained by slow diffusion of a dichloromethane/methanol mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) at room temperature. 1-Ni and 3-Ni crystallize in a triclinic (P
:3) at room temperature. 1-Ni and 3-Ni crystallize in a triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) system, while 2-Ni in a monoclinic (P21/c) system. Molecular structures were visualized and drawn with OLEX238 v1.5 and they are found in Fig. 2. In all three complexes, the POCOP-type ligands coordinated tridentate to the metal center through two phosphorus donor atoms and the central carbon of the aromatic ring skeleton, with a chloride ligand completing the coordination sphere of the complex adopting a square planar geometry around the nickel atom. Additionally, the functionalized phthalimide fragment is observed in the para position of the aromatic ring. The topology of the ligand leads to the formation of two five-membered metallocycles where the symmetry around the aromatic backbone is evident. Intermolecular distances in the crystal structures of compounds were analyzed with MERCURY39 v2023.3.0. The selected bond distance (Å) and angles (°) for the POCOP–Ni(II) complexes are listed in Table 2.
) system, while 2-Ni in a monoclinic (P21/c) system. Molecular structures were visualized and drawn with OLEX238 v1.5 and they are found in Fig. 2. In all three complexes, the POCOP-type ligands coordinated tridentate to the metal center through two phosphorus donor atoms and the central carbon of the aromatic ring skeleton, with a chloride ligand completing the coordination sphere of the complex adopting a square planar geometry around the nickel atom. Additionally, the functionalized phthalimide fragment is observed in the para position of the aromatic ring. The topology of the ligand leads to the formation of two five-membered metallocycles where the symmetry around the aromatic backbone is evident. Intermolecular distances in the crystal structures of compounds were analyzed with MERCURY39 v2023.3.0. The selected bond distance (Å) and angles (°) for the POCOP–Ni(II) complexes are listed in Table 2.
| 1-Ni | 2-Ni | 3-Ni | |
|---|---|---|---|
| Formula | C27H36ClNNiO5P2 | C31H44ClNNiO5P2 | C39H28ClNNiO5P2 |
| Formula weight | 610.67 | 666.77 | 746.72 |
| Temperature (K) | 120(2) | 120(2) | 298(2) |
| Wavelength | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | Triclinic | Monoclinic | Triclinic |
| Space group | P |
P21/c | P |
| a (Å) | 8.2574(4) | 8.0054(4) | 8.7072(3) |
| b (Å) | 11.5280(8) | 27.8419(12) | 9.4954(4) |
| c (Å) | 15.9268(8) | 14.9413(7) | 22.8387(10) |
| α (°) | 89.739(5) | 90 | 84.987(2) |
| β (°) | 76.201(5) | 103.937(5) | 80.985(2) |
| γ (°) | 76.225(5) | 90 | 67.538(2) |
| V (Å3) | 1427.79(15) | 3232.2(3) | 1722.67(12) |
| Z | 2 | 4 | 2 |
| δcal (g cm−3) | 1.420 | 1.370 | 1.440 |
| μ (mm−1) | 0.922 | 0.821 | 0.780 |
| F(000) | 640 | 1408 | 768 |
| Crystal size (mm3) | 0.550 × 0.350 × 0.290 | 0.450 × 0.290 × 0.250 | 0.369 × 0.231 × 0.155 |
| Theta range (°) | 3.460 to 29.532 | 3.537 to 29.403 | 2.322 to 25.391 |
| Reflaction collected | 21794 |
17128 |
22789 |
| Data/restraints/parameters | 6923/0/342 | 7549/0/382 | 6336/0/442 |
| Goodness-of-fit | 1.083 | 1.054 | 1.088 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0368, wR2 = 0.0792 | R1 = 0.0389, wR2 = 0.0799 | R1 = 0.0459, wR2 = 0.0905 |
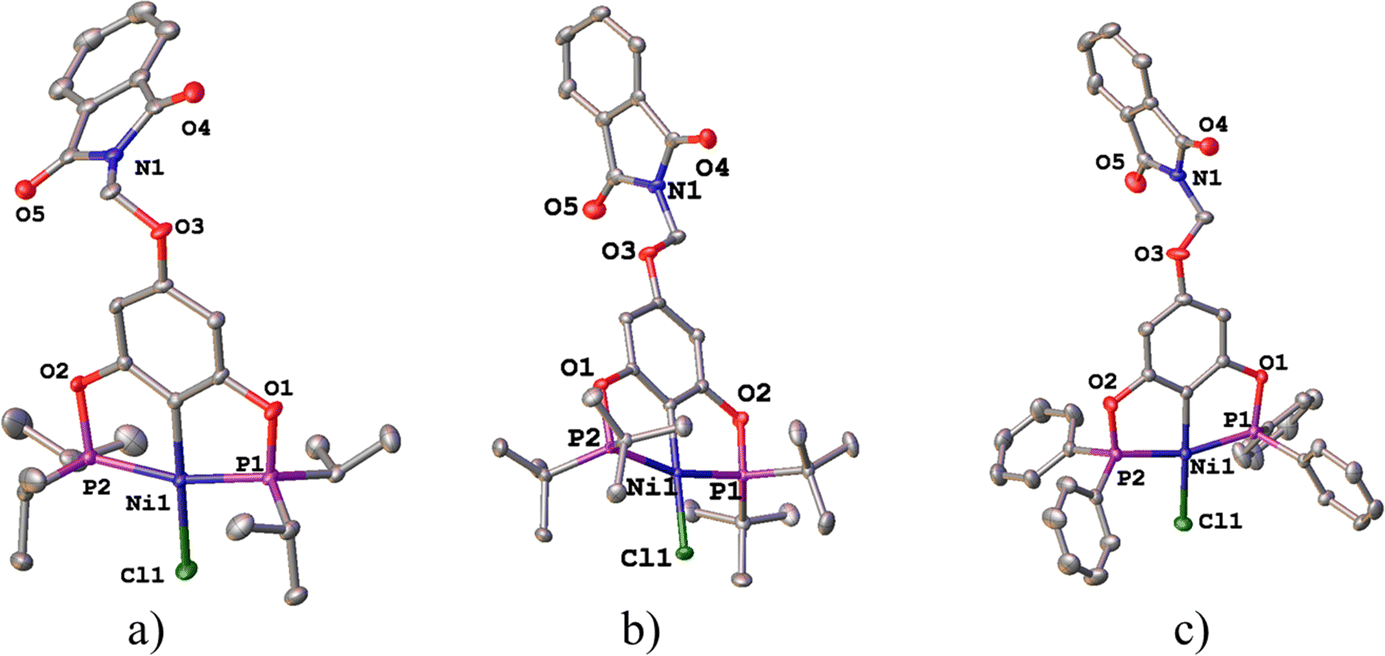 | ||
| Fig. 2 Molecular structures of pincer compound (a) 1-Ni, (b) 2-Ni and (c) 3-Ni. The thermal ellipsoids are represented at 50% of probability and the hydrogen atoms are omitted for clarity. | ||
| Compound | M–C | M–Cl | M–P1 | M–P2 | P1–M–P2 | C–M–Cl |
|---|---|---|---|---|---|---|
| 1-Ni | 1.881(2) | 2.208(6) | 2.161(6) | 2.146(6) | 163.62(2) | 176.66(6) |
| 2-Ni | 1.877(2) | 2.196(8) | 2.185(5) | 2.183(5) | 163.55(3) | 179.19(6) |
| 3-Ni | 1.883(2) | 2.192(7) | 2.159(4) | 2.163(4) | 163.48(3) | 178.16(5) |
The three complexes adopt a molecular geometry close to an ideal square plane, as evidenced by the sum of bond angles around the Ni(II) center: 360.01° (1-Ni), 359.96° (2-Ni), and 359.91° (3-Ni), respectively. The Ni–C bond distances in all complexes are similar of around 1.88 Å. On the other hand, the angles involving P1–Ni–P2 are quite similar in the three complexes, approximately 163.6°. In contrast, the C–Ni–Cl angle varies within the range of 176.66° to 178.16°. These obtained values are consistent with other similar pincer complexes previously reported (Table 2).40,41
3. Supramolecular analysis
Different contacts were found in the pincer derivative compounds to support the supramolecular arrangements via CH⋯π, O⋯H, CH⋯O and CH⋯Cl non-covalent interactions which are summarized in Table 3. Additionally, to simplify these interactions were described as graph set descriptors. The use of graph sets to describe hydrogen-bond patterns in molecular crystals is illustrated in Fig. 3. Molecular arrangement in crystal packing of 1-Ni and 2-Ni are supported by polymeric interactions CH⋯O and CH⋯Cl, respectively (Fig. 3a and b) [graph set descriptor C(12) and C(6)]. A dimeric interaction in complex 3-Ni supported by two reciprocal CH⋯O interactions by the ether bridge additionally the two aromatic phthalimide fragments were π–π stacking for the most stable conformation (Fig. 3c) [graph set descriptor R22(14)]. Despite containing many aromatic fragments in the three complexes, just 3-Ni π–π-staking was found which is further confirmed by Hirshfeld surface analysis (vide infra). All intermolecular parameters of distances and angles were within the expected range reported previously.42–45| Compound | Interaction | Distance (Å) | Distance (Å) | Angle (°) | Symmetry operation |
|---|---|---|---|---|---|
| D–X⋯A | D⋯A | D–X⋯A | |||
| 1-Ni | C22H22⋯O4 | 2.534(2) | 3.456(3) | 153.13(4) | 1 + x, −1 + y,z |
| C8H8B⋯O5 | 2.616(3) | 3.510(2) | 151.76(4) | 1 − x, 1 − y, 1 − z | |
| C24H24C⋯O3 | 2.532(2) | 3.389(3) | 146.12(5) | 2 − x, 1 − y, −z | |
| C25H25⋯O4 | 2.658(2) | 3.424(2) | 133.50(3) | 2 − x, 1 − y, −z | |
| 2-Ni | C22H22B⋯Cl1 | 2.697(3) | 3.677(1) | 179.34(4) | x, −1/2 − y, −1/2 + z |
| C27H27A⋯C13 | 2.816(1) | 3.603(3) | 137.87(5) | −x, 1/2 + y, −1/2 − z | |
| C31H31B⋯C4 | 2.658(2) | 3.562(3) | 153.64(3) | −x, 1/2 + y, −1/2 − z | |
| C31H31B⋯C12 | 2.6868(3) | 3.643(2) | 165.34(5) | −x, 1/2 + y, −1/2 − z | |
| 3-Ni | C6H6⋯O4 | 2.637(1) | 3.559(1) | 171.31(4) | −1 + x, y, z |
| C5H5⋯O3 | 2.628(2) | 3.273(2) | 127.05(5) | 2 − x, −y, 1 − z |
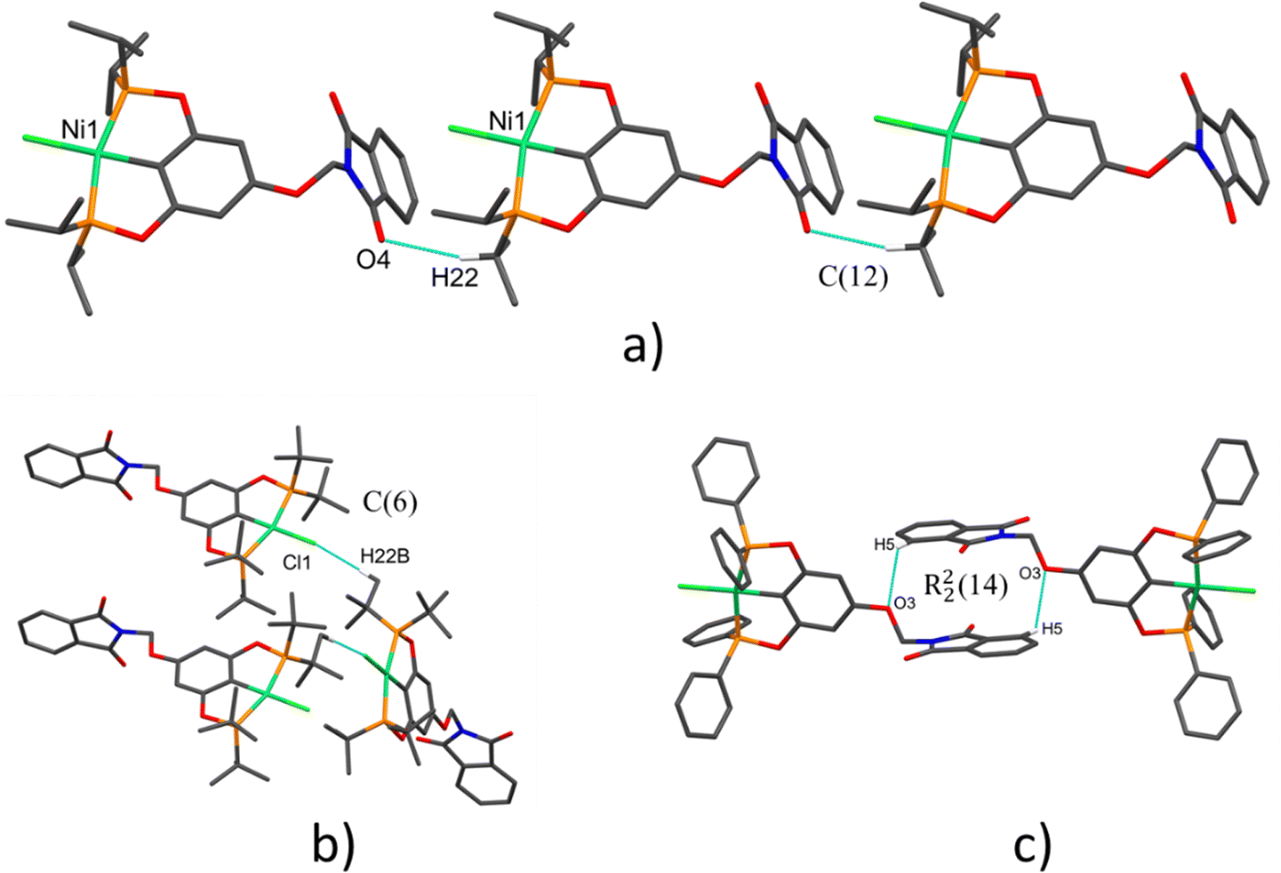 | ||
| Fig. 3 Representatives no-covalent interactions observed in the complexes: (a) graph set descriptor C(12) found in 1-Ni, (b) graph set descriptor C(6) found in 2-Ni and (c) centrosymmetric dimer found in 3-Ni [graph set descriptor R22(14)]. Note: hydrogen atoms not involved in the hydrogen bonds were omitted for clarity. | ||
3.1. Hirshfeld surface
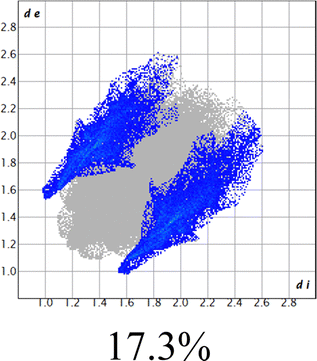
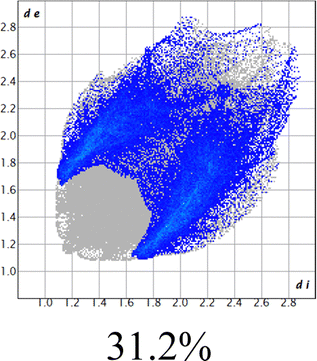
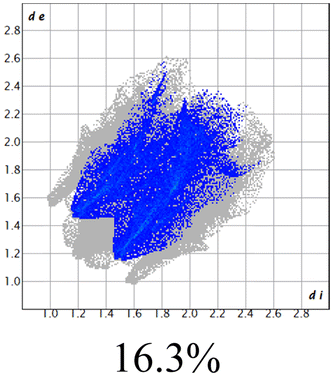
The proposed short interactions in the pincer-type Ni complexes were studied by the Hirshfeld analysis using the Crystal Explorer46 program, it was possible to obtain information about the percentages of non-covalent interactions. Additionally, we determined the two-dimensional fingerprints47 plot derived from the Hirshfeld surface based on our results from the X-ray studies in CIF format. The plots are shown in Fig. 4; the corresponding fingerprints were plotted and are shown in Table 4. The HS over the dnorm function, evident red dots (close contacts) were located over the oxygens of the ether and the carbonyl groups of the phthalimide moiety due to O⋯H/H⋯O contacts (Fig. 4a–c). The shape index shows long red spots revealing mainly CH⋯π contacts (Fig. 4d–f) and red/blue triangles due to π⋯π stacking between phthalimide and phenyl groups (Fig. 5). In their part, fingerprint plots show characteristic appearance due to individual C⋯H/H⋯C, O⋯H/H⋯O, and Cl⋯H/H⋯Cl reciprocal contacts, as two symmetrical wings or tips (see Table 4). However, the majority of contacts were due to H⋯H and C⋯H/H⋯C (over 43% and 17%, respectively) of the total contributing contacts; individual contributions are plotted in Chart 1.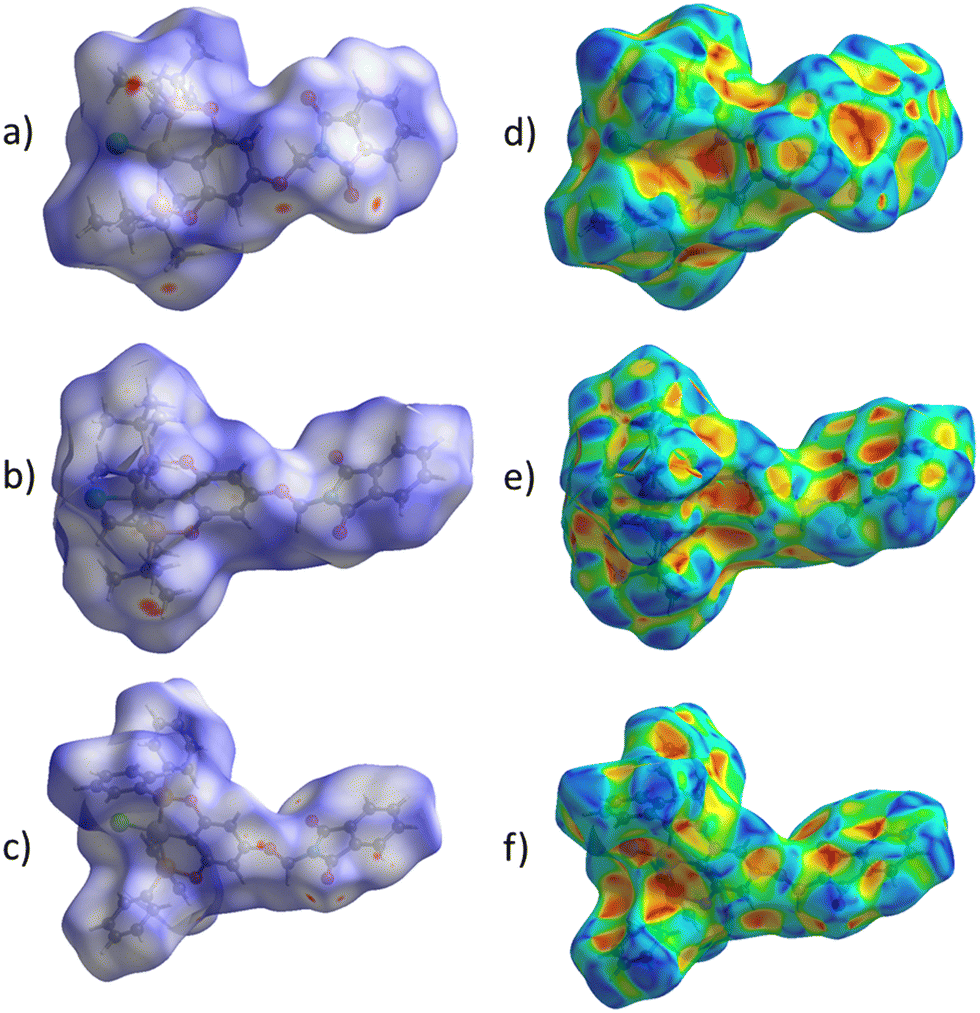 | ||
| Fig. 4 Hirshfeld surface calculated for complexes over the function: dnorm (a) 1-Ni, (b) 2-Ni and (c) 3-Ni; over the shape index (d) 1-Ni, (e) 2-Ni and (f) 3-Ni. | ||
| Contact | 1-Ni | 2-Ni | 3-Ni |
|---|---|---|---|
| All |  |
 |
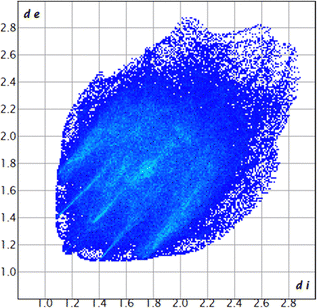 |
| H⋯H | 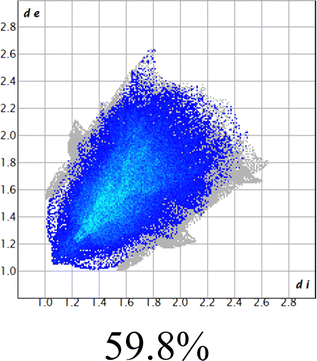 |
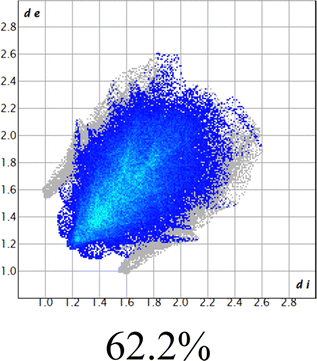 |
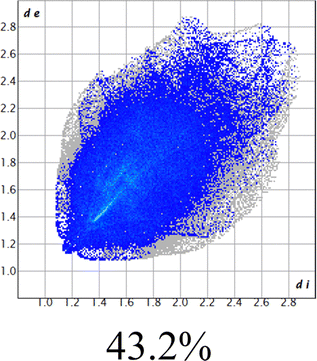 |
| C⋯H/H⋯C |  |
 |
 |
| O⋯H/H⋯O | 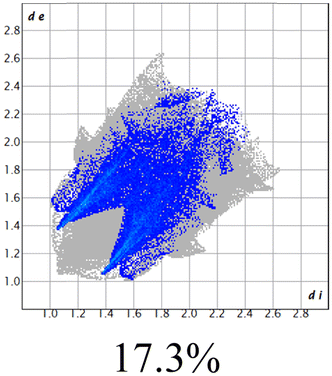 |
 |
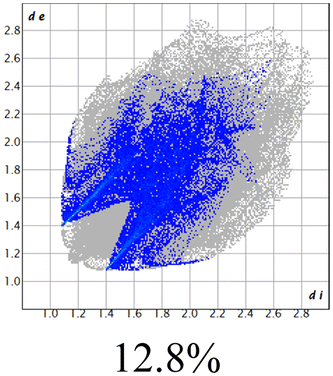 |
| Cl⋯H/H⋯Cl | 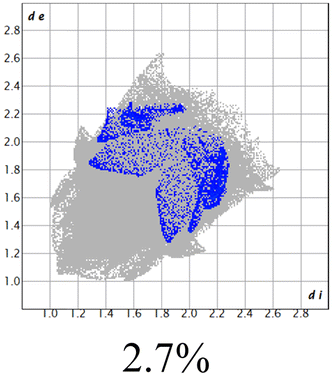 |
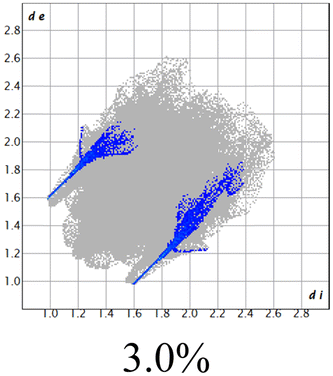 |
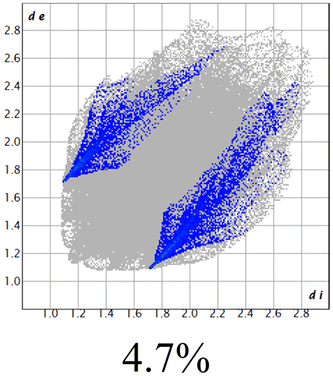 |
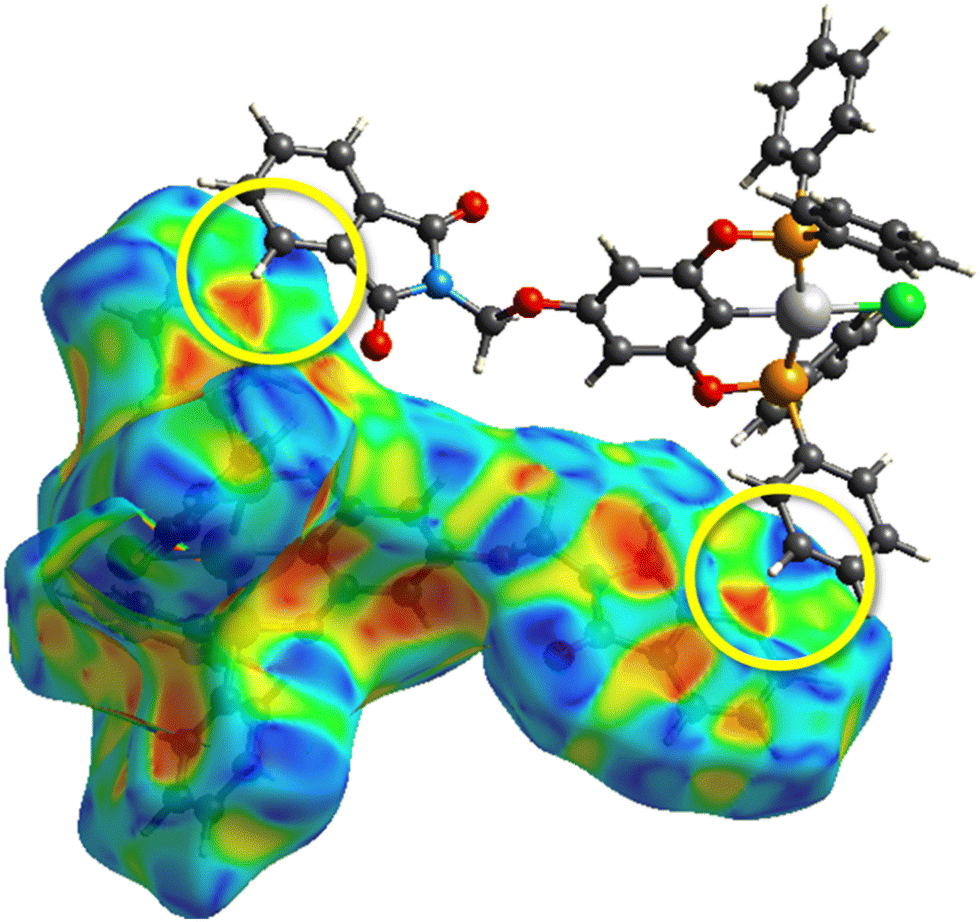 | ||
| Fig. 5 HS over shape index of complex 3-Ni, yellow circle encloses the evident π⋯π stacking. | ||
 | ||
| Chart 1 A plot of percentages of contacts observed in complexes 1-Ni, 2-Ni and 3-Ni. | ||
4. Cytotoxic activity
The three Ni(II) pincer complexes were stable in the presence of air. Additionally, a stability assay was conducted using the 1-Ni complex dissolved in DMSO-d6 and monitored by 1H NMR every 24 hours for three days. It was observed that there were no apparent changes in the signals of the complex during this period (Fig. S10, ESI†). After confirming the stability of the compounds, in vitro cytotoxicity studies were conducted using the sulforhodamine B protocol with a concentration of 10 μM for the complexes, and DMSO was used as the transport vehicle, with an incubation period of 48 hours. Six human cancer cell lines were employed: U251 (human glioblastoma), PC-3 (human prostate adenocarcinoma), K562 (human chronic myelogenous leukaemia), HCT-15 (human colorectal adenocarcinoma), MCF-7 (human breast adenocarcinoma) (these cell lines were donated by the Nacional Cancer Institute, USA), SKLU-1 (human lung adenocarcinoma) (provided by the Cancer Institute of Mexico). Additionally, a healthy monkey kidney cell line (COS7) was included for comparative purpose. Cisplatin was used as a reference standard for evaluation and comparative purposes (Table 5).| Entry | Compound | U251 | K562 | HCT-15 | MCF-7 | SKLU-1 | COS7 |
|---|---|---|---|---|---|---|---|
| 1 | 1-Ni | 100 | 100 | 100 | 100 | 100 | 100 |
| 2 | 2-Ni | 24.5 | 11.1 | — | 43.0 | — | — |
| 3 | 3-Ni | — | — | — | — | — | — |
| 4 | NiCl2 | 9.90 | — | 4.15 | 2.07 | — | — |
| 5 | Cisplatin | 35.7 | 24.4 | 13.3 | 28.8 | 38.9 | 34.7 |
In a previous study conducted by our research group,34 cytotoxicity tests were performed on the POCOP–Ni(II) precursors (1–3). The results showed that complexes 1 and 3 were the most cytotoxic against U251, HCT-15, and MCF-7 cell lines, exhibiting better IC50 values than cisplatin. Based on these results, we decided to incorporate a pharmacophore such as N-(chloromethyl)phthalimide to assess whether selectivity could be improved. In a primary screening, it was observed that the 1-Ni complex was the most cytotoxic in all cell lines, both cancerous and healthy, showing a 100% inhibition rate. This indicates that there was no change in cytotoxic activity regardless of the presence of the methylphthalimide fragment in the pincer complex skeleton. On the other hand, 2-Ni showed lower cytotoxicity, being more selective against MCF-7 (43%) and non-toxic in the healthy cell line. However, this complex exhibited very low solubility in the transport vehicle due to the incorporation of the pharmacophore, making it unsuitable for comparison with precursor 2. Finally, compound 3-Ni did not show cytotoxic activity in any of the cell lines, in contrast to 3, which was active in all cell lines (Table 5).
Based on these primary screening results, we can infer that in the case of 1-Ni, there was no modification in the cytotoxic properties compared to 1, while 2-Ni exhibited a decrease in solubility, affecting the assay results. Compound 3-Ni, however, proved to be the least toxic of all the evaluated compounds, even compared to its counterpart 3, suggesting that the incorporation of such an organic fragment affected the cytotoxic properties. Furthermore, when comparing the cytotoxicities of the complexes to NiCl2, it is evident that there is a synergistic effect between the POCOP ligands and the methylphthalimide on the anticancer properties, enhancing them across the cell lines.
Based on these results, the IC50 assay was conducted with complex 1-Ni, which exhibited higher activity against all cancer cell lines compared to cisplatin but was more toxic than the metallopharmaceutical in the healthy cell line (Table 6). The range of the selectivity index (SI) for the complex is within a range of 0.64 to 0.97, indicating that the pincer complex possesses a high degree of toxicity.
| Compound | U251 | K562 | HCT-15 | MCF-7 | SKLU-1 | COS7 |
|---|---|---|---|---|---|---|
| 1-Ni | 0.74 ± 0.07 | 0.66 ± 0.07 | 1.38 ± 0.01 | 1.0 ± 0.05 | 0.88 ± 0.03 | 0.64 ± 0.03 |
| Cisplatin | 4.7 ± 0.4 | 1.2 ± 0.08 | 17.0 ± 1.6 | 5.5 ± 0.5 | 4.3 ± 0.5 | 7.2 ± 0.6 |
The marked cytotoxic effects observed in these complexes (1-Ni–3-Ni) could be attributed to the substituents present on the phosphorus atom, primarily due to steric factors. A possible explanation for this is that, for the 1-Ni compound, the chloride ion is less hindered from leaving the coordination sphere, thus enabling a coordination site on the Ni(II) center. This allows the center to interact with other biological targets through covalent bonds, thereby increasing its antitumor activity. However, further biochemical studies are required to confirm these hypotheses and to elucidate a potential mode of action for these compounds at the cellular level.
5. Antioxidant activity
Different studies have revealed that the antioxidant activity of certain compounds can help prevent cancerous tumors. It is believed that this disease can be generated by the uncontrolled formation of reactive oxygen species (ROS), which promote the onset of malignant tumor formation. For that reason, the antioxidant activity of the functionalized POCOP–Ni compounds was evaluated. The assays were conducted using the thiobarbituric acid reactive substances (TBARS) protocol, which leads to the formation of ROS from FeSO4 in the presence of lipids obtained from rat brains. The studies were performed using three different concentrations of the evaluated complexes (1, 10, and 100 μM) and using α-tocopherol as a control (Table 7). In a previous study,34 complexes 1–3 were evaluated as potential antioxidant agents, demonstrating very promising activities, especially complex 2, whose IC50 was the highest compared to the other two complexes (1.55 ± 0.08), being a better antioxidant than the organic compound α-tocopherol (6.78 ± 2.16). Here, the electronic properties of the tBu groups have a significant influence on the higher activity, favoring a better charge distribution over the phenolate ion, allowing it to trigger collateral reactions in the formation of free radicals. To compare the antioxidant properties of the precursors (1–3) with the functionalized compounds (1-Ni–3-Ni), the same studies were performed. In a primary screening, it was observed that all three complexes did not exhibit significant antioxidant activity, with complex 1-Ni showing the highest inhibition rate at 100 μM (34.24%), but still lower than that of α-tocopherol at the same concentration (79.09%). Unlike complexes 1–3, the compounds functionalized with the phthalimide fragment did not prove to be good antioxidants. This can be explained by the fact that the formation of the phenolate ion is crucial, as the generation of this species leads to possible radical inhibition by the ROS generated in situ. When phthalimide is attached to the pincer through an ether bond, this bond, being more stable, does not satisfactorily generate the phenolate ion, which implies an inhibition of the antioxidant activity by the POCOP complexes.| Compound | Concentration (μM) | Inhibition (%) |
|---|---|---|
| Homogenized in: PBS; vehicle: DMSO; peroxidation: induced with FeSO4 at 10 μM, 1 h of incubation; EDTA: 2 μM. | ||
| 1-Ni | 1 | −3.40 |
| 10 | 9.92 | |
| 100 | 34.24 | |
| 2-Ni | 1 | 1.85 |
| 10 | 4.39 | |
| 100 | 3.13 | |
| 3-Ni | 1 | 2.02 |
| 10 | −2.12 | |
| 100 | 4.42 | |
| α-Tocopherol | 1 | 21.13 |
| 10 | 59.00 | |
| 100 | 79.09 | |
6. Ethidium bromide (EB) displacement assay
Competitive displacement studies were conducted for complexes 1-Ni and 3-Ni, while complex 2-Ni could not be evaluated due to its low solubility. This assay was used to understand how these compounds can interact with the DNA target. The test involves performing competitive fluorescence titrations with ethidium bromide (EB). EB is a good intercalator with very weak fluorescence, but when intercalated into DNA, it shows an increase in fluorescence. If a compound can intercalate with DNA, it will gradually displace EB from the binding sites, which can be observed as a decrease in the fluorescence intensity of the EB-DNA adduct as the presence of the compound under evaluation increases. As shown in Fig. 6, as the concentration of complexes 1-Ni and 3-Ni increases from 0 to 37 μM (Fig. 6A) and 0 to 10 μM (Fig. 6B), they have the capacity to decrease the fluorescence intensity of the EB-DNA adduct, suggesting that the POCOP–Ni(II) complexes functionalized with the phthalimide fragment intercalate into the DNA double helix, displacing EB from this molecular target.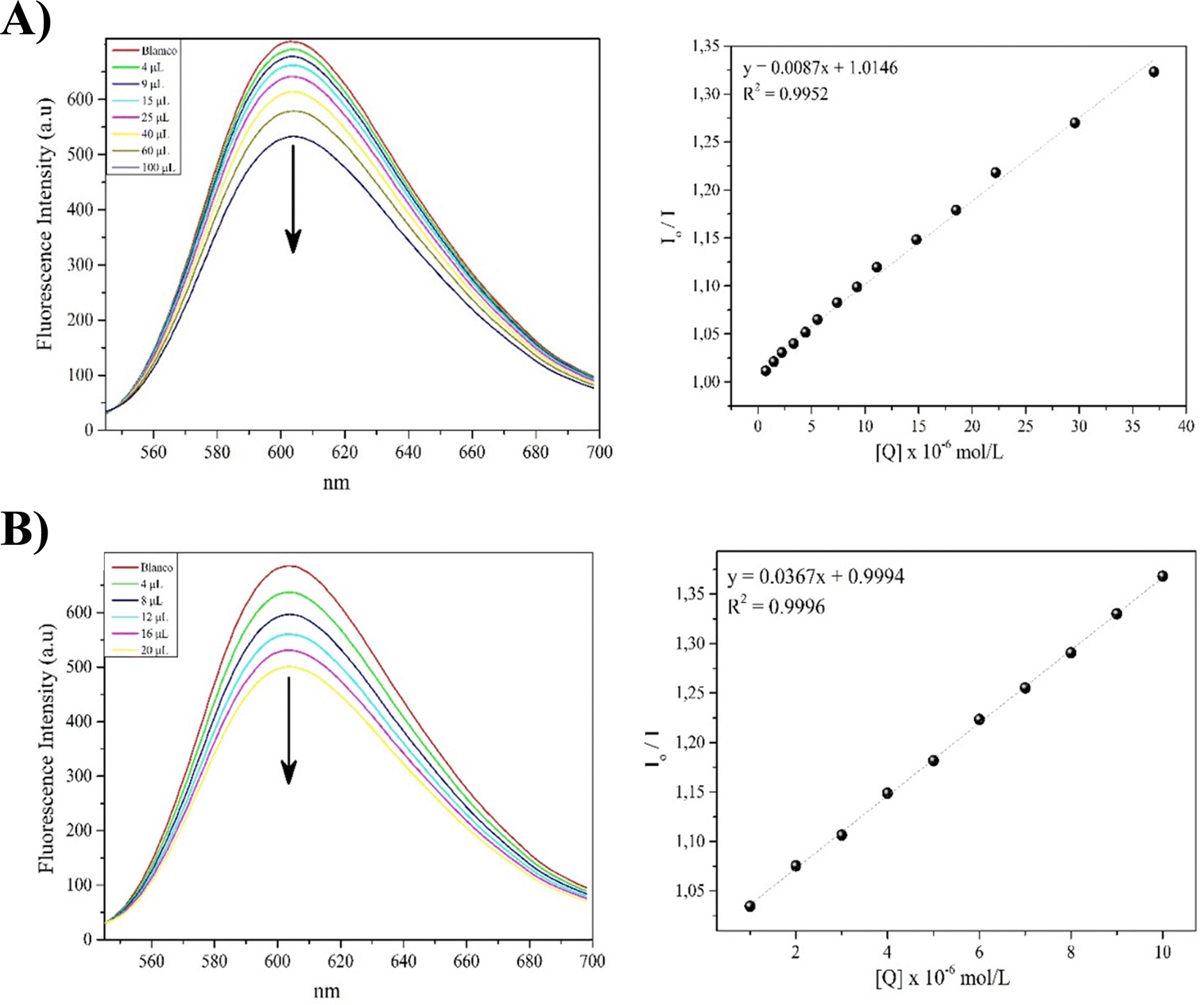 | ||
| Fig. 6 Fluorescence spectrum of EB-DNA in the presence of increasing concentrations of complexes 1-Ni (A) and 3-Ni (B). The arrows indicate changes in emission intensity as a function of the concentration of the complexes. On the right are the Stern–Volmer plots of the fluorescence titration data. | ||
The KSV values for the quenching of the fluorescence intensity of EB bound to DNA for complexes 1-Ni and 3-Ni were determined from the Stern–Volmer plot, which showed good linearity. The quenching constant values for complexes 1-Ni and 3-Ni were found to be 8.7 × 103 and 3.67 × 104 mol L−1, respectively. This suggests that complex 3-Ni intercalates more strongly into DNA than complex 1-Ni, possibly due to the greater number of aromatic rings in complex 3-Ni compared to complex 1-Ni. This could favor a higher number of π–π and σ–π interactions, promoting better intercalation. These suppositions can be clarified through molecular docking studies to strengthen this hypothesis.
7. Computational calculations
To verify the accuracy of the electronic structure method, we made a comparison between the optimized geometry using density functional theory (DFT) and the X-ray crystallography structure. This comparison is crucial as it validates the computational method's reliability in predicting these complexes' structural parameters. As observed in Table 8, the comparison shows no significant differences between the DFT-optimized geometries and the experimentally determined X-ray structures, with low root mean square deviation (RMSD) values consistently. This low RMSD indicates a high level of agreement between the theoretical and experimental geometries, reinforcing the accuracy of the DFT method in modeling these nickel complexes.| Complex | RMSD (Å) | Structure |
|---|---|---|
| 1-Ni | 0.23 |  |
| 2-Ni | 0.31 |  |
| 3-Ni | 0.29 |  |
After the molecular docking simulations, all three complexes present the same conformation in the methylphthalimide group, where hydrogen bonds are found with both carbonyl groups. These intramolecular interactions allow for a better conformation of the methylphthalimide fragment, enabling it to bind with DNA, which could be crucial for more optimal intercalation as well as for the activity of the complexes. As shown in Fig. 7.
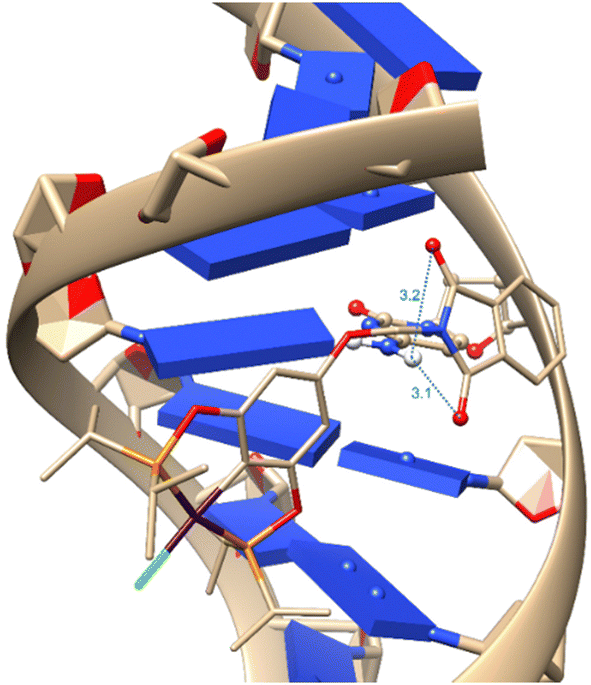 | ||
| Fig. 7 Interaction between DNA and complex 1-Ni, where blue interactions represent hydrogen bonds with distances in Å. | ||
However, the R substituents on the phosphines significantly influence the overall interaction patterns by increasing or decreasing hydrophobic interactions. Specifically, the π–π stacking interactions of the phenyl groups in the 3-Ni complex enhance its stability and binding affinity due to the additional non-covalent interactions (see Table 9). This enhanced interaction indicates the importance of aromatic groups in stabilizing such complexes through π–π stacking, which could be further explored for designing more efficient binding molecules.
| Complex | Binding energy kcal mol−1 | ECR rank | ||
|---|---|---|---|---|
| ATD | Vina | Smina | ||
| 1-Ni | −6.5 | −8.6 | −8.6 | 14.6 |
| 2-Ni | −4.2 | −8.3 | −8.3 | 12.0 |
| 3-Ni | −5.6 | −9.5 | −9.6 | 18.2 |
In contrast, 1-Ni and 2-Ni complexes exhibit repulsion effects. The 2-Ni complex shows notable electrostatic repulsion with DNA. This is attributed to its bulkier nature, which introduces steric hindrance and reduces the effective binding interactions. The electrostatic repulsion observed with the 2-Ni complex, as shown in Fig. 8, highlights the balance required between size and binding efficiency. Too large substituents can disrupt the binding process, leading to lower affinity and potential loss of function.
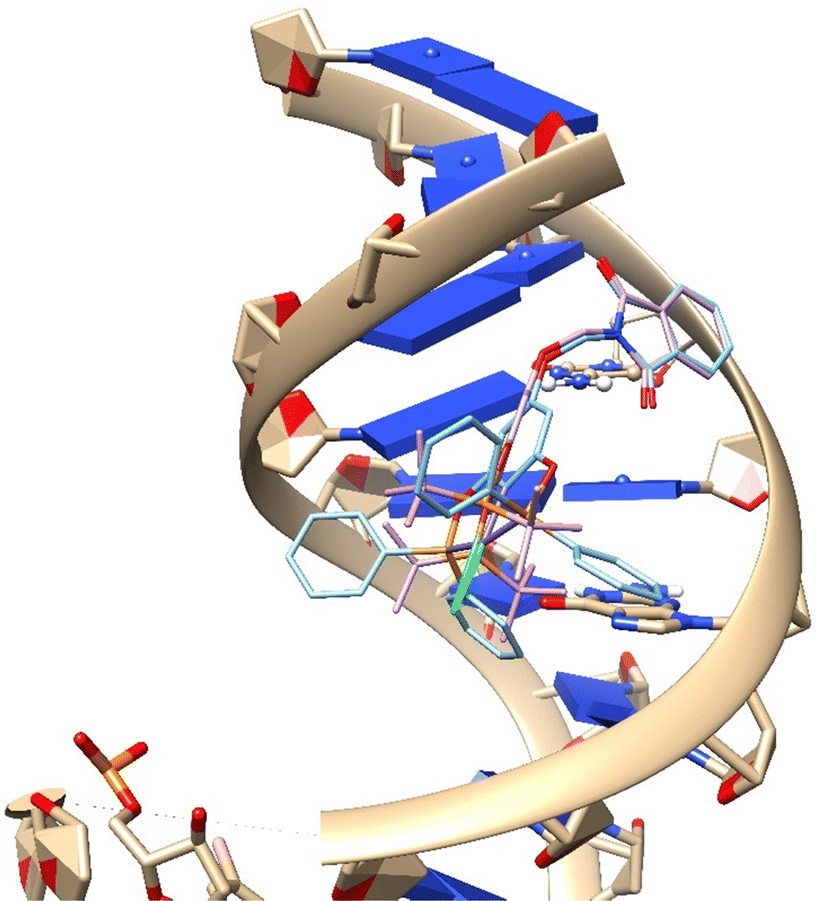 | ||
| Fig. 8 Interaction between DNA and complex 3-Ni (blue), 2-Ni (pink). | ||
Moreover, these findings underscore the role of electrostatic and hydrophobic interactions in the binding process. While the hydrophobic interactions provided by the R substituents can enhance stability and affinity, excessive bulkiness or inappropriate electrostatic environments can lead to repulsion and decreased binding efficiency.
8. Conclusion
Three new functionalized POCOP–Ni(II) pincer complexes with a phthalimide fragment (1-Ni, 2-Ni, and 3-Ni) were synthesized. Single crystals were obtained for three compounds, whose molecular structures were unequivocally elucidated by X-ray diffraction (XRD). The presence of multiple and variable donor and acceptor groups in these compounds allowed for various non-covalent interactions, such as CH⋯π, O⋯H, CH⋯O, CH⋯Cl, and π⋯π, supporting the 1D and 2D polymeric frameworks. Cytotoxicity studies revealed that complex 1-Ni was the most active but also more toxic to the healthy cell line. In competitive displacement assays, complexes 1-Ni and 3-Ni efficiently displaced ethidium bromide (EB), indicating that their planarity and the presence of aromatic groups allowed them to intercalate into the DNA target. Additionally, antioxidant assays revealed that these complexes were not good antioxidants compared to POCOP–Ni(II) complexes functionalized with a hydroxyl group in the para position, suggesting that the phthalimide group blocks processes related to free radical inhibition. All three Ni complexes show consistent conformation with strong DNA binding via the N-(methyl)phthalimide group. Molecular docking simulations indicate that R substituents on phosphines significantly affect binding interactions, with bulkier groups causing electrostatic repulsion. These findings are key for designing effective Ni-based binding agents.9. Experimental section
All reactions were carried out under nitrogen with standard Schlenk techniques unless otherwise stated. All chemical compounds were commercially obtained and used as received without purification. Melting points were recorded on a MELT-TEMP II laboratory devices and are reported without correction. The FT-IR spectra were obtained FT-IR NICOLET IS-50 Thermo Fisher Scientific spectrometer, using attenuated total reflectance (ATR) technique. The 1H, 13C{1H} and 31P{1H} NMR spectra were recorded on a Bruker Advance III 400 MHz spectrometer. Chemical shifts are reported in ppm down field of TMS employing the residual signals in the solvent (CDCl3) as internal standard. Direct analysis real-time (DART+) mass spectra were recorded on a the AccuTOF JMS-T100LC spectrometer. Elemental analyses were performed in a Thermo Scientific Flash 2000 elemental analyzer, using a Mettler Toledo XP6 Automated-S Microbalance and sulfanilamide as standard (Thermo Scientific BN 217826, attained values N = 16.40%, C = 41.91%, H = 4.65%, and S = 18.63%; certified values N = 16.26%, C = 41.81%, H = 4.71%, and S = 18.62%).9.1. General procedure for the synthesis of para-phthalimide functionalized POCOP–Ni(II) pincer complexes
The pincer compounds 1–3 were synthesized following an analogous procedure reported earlier by our research group37 with slight modifications, which include the use of anhydrous NiCl2 (Scheme 1) instead of NiCl2·6H2O. A Schlenk flask was charged with the compounds 1–3 (1 eq.), NaB(OMe)4 (2 eq.) and 18-crown-6 (10% mol, 18-c-6) in dry THF (35 mL) was stirred at room temperature for 2 h under a nitrogen atmosphere. Then, was added N-(chlorometyl)phthalimide (1 eq. ClMePht) to the Schlenk and the reaction mixture was stirred at room temperature for 24 h. After this time, all volatiles were removed under high vacuum. The crude product was purified by chromatographic column using CH2Cl2 as eluent.O), 158.16 (s, 1C, CAr–O), 134.54 (s, 2C, CAr), 131.88 (s, 2C, CAr), 123.90 (s, 2C, CAr), 116.61 (vt, 1C, C–Ni), 93.89 (vt, 2C, JP–C = 6.03 Hz, CHAr), 65.15 (s, 1C, O–C–Pht), 27.79 (s, 4C, –CH(CH3)2), 17.47 (s, 4C, –CH(CH3)2), 16.71 (s, 4C, –CH(CH3)2). 31P{1H} NMR (162 MHz, CDCl3): δ 186.63. MS-DART+, m/z (%): 610 (100) [M+], 574 [M+ − HCl] (65). IR (ATR, υcm−1): 1777 (CO)sym, 1725 (CO)asym. Elem. anal. calc. for C27H36ClNiO5P2 (610.7 g mol−1): C 53.10, H 5.94, N 2.29; found: C 53.32, H 5.93, N 2.31.O), 157.73 (s, 1C, CAr–O), 134.52 (s, 2C, CAr), 131.91 (s, 2C, CAr), 123.89 (s, 2C, CAr), 116.18 (vt, 1C, C–Ni), 93.63 (vt, 2C, JP–C = 6.03 Hz, CHAr), 65.21 (s, 1C, O–C–Pht), 39.24 (vt, 4C, JP–C = 6.03 Hz –C(CH3)3), 27.99 (vt, 12C, JP–C = 3.01 Hz, –C(CH3)3). 31P{1H} NMR (162 MHz, CDCl3): δ 189.21. MS-DART+, m/z (%): 666 (100) [M+]. IR (ATR, υcm−1): 1781 (CO)sym, 1730 (CO)asym. Elem. anal. calc. for C31H44ClNiO5P2 (666.8 g mol−1): C 55.84, H 6.65, N 2.10; found: C 56.15, H 6.62, N 2.07.O), 158.87 (s, 1C, CAr–O), 134.56 (s, 2C, CAr), 132.41 (vt, 4C, JP–C = 24.1 Hz, OP–CAr), 131.92 (s, 2C, CAr), 131.84 (s, 4C, OP–CHAr), 131.80 (vt, 8C, OP–CHAr), 128.77 (vt, 8C, JP–C = 5.02 Hz, OP–CHAr), 123.92 (s, 2C, CAr), 117.72 (vt, 1C, C–Ni), 95.40 (vt, 2C, JP–C = 7.03 Hz, CHAr), 65.26 (s, 1C, O–C–Pht). 31P{1H} NMR (162 MHz, CDCl3): δ 142.31. MS-DART+, m/z (%): 746 (6.92) [M+], 747 (3.81) [M+ + H], 710 (100) [M+ − MeOH]. IR (ATR, υcm−1): 1775 (CO)sym, 1722 (CO)asym. Elem. anal. calc. for C39H28ClNiO5P2 (746.7 g mol−1): C 62.73, H 3.78, N 1.88; found: C 62.07, H 3.79, N 1.88.9.2. Cytotoxic evaluation
The compounds were screened in vitro against human cancer cell lines: HCT-15 (human colorectal adenocarcinoma), MCF-7 (human mammary adenocarcinoma), K562 (human chronic myelogenous leukaemia), U251 (human glioblastoma), PC-3 (human prostatic adenocarcinoma), SKLU-1 (human lung adenocarcinoma), COS-7 (cell line monkey African green kidney) cell lines were supplied by the National Cancer Institute (USA) and were donated by the Cancer Institute of Mexico. The cell lines were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 25 μg mL−1 amphotericin B (Gibco) and 1% non-essential amino acids (Gibco). They were maintained at 37 °C in a humidified atmosphere with 5% CO2.Cytotoxicity after treatment of the tumors cells and normal cells with the test compounds was determined using the protein-binding dye sulforhodamine B (SRB) in a microculture assay to measure cell growth.48 The cultures were exposed for 48 h to the compound at concentrations 25 μM. After the incubation period, cells were fixed to the plastic substratum by addition of 50 μL of cold 50% aqueous trichloroacetic acid. The plates were incubated at 4 °C for 1 h, washed with H2O, and air-dried. The trichloroacetic-acid-fixed cells were stained by the addition of 0.4% SRB. Free SRB solution was the removed by washing with 1% aqueous acetic acid. The plates were then air-dried, and the bound dye was solubilized by the addition of 10 mM unbuffered tris base (100 μL). The plates were placed on and shaken for 10 min, and the absorption was determined at 515 nm using an ELISA plate reader (Bio-Tex Instruments). The inhibitory concentration 50 (IC50) values were calculated on extrapolated fit curves based on doses/response data analysed for each compound through lineal regression analysis.
9.3. Lipid peroxidation inhibition
As an index of lipid peroxidation, TBARS levels were measured using rat brain homogenates according to the method described by Ng et al.,52 with some modifications. Supernatant (375 μL) was added with 50 μL of 20 μM EDTA and 25 μL of each sample concentration solved in DMSO (25 μL of DMSO for control group) and incubated at 37 °C for 30 min. Lipid peroxidation was started adding 50 μL of freshly solution FeSO4 100 μM and incubated at 37 °C for 1 hour. The TBARS content was determined as described by Ohkawa et al.53 with 500 μL of TBA reagent (0.5% 2-thiobarbituric acid in 0.05 N NaOH and 30% trichloroacetic acid, in 1:1 proportion) was added at each tube and cooled on ice for 10 min, centrifugated at 13400 rcf for 5 min and heated at 80 °C in a water bath for 30 min. After cooling at room temperature, the absorbance of 200 μL of supernatant was measured at λ = 540 nm in a Bio-Tek Microplate Reader Synergy HT. Concentration of TBARS was calculated by interpolation in a standard curve of tetra-methoxypropane (TMP) as a precursor of MDA.54 Results were expressed as nmoles of TBARS per mg of protein. The inhibition ratio (IR[%]) was calculated using following formula IR = (C − E) × 100/C, where C is the absorbance of control and E is the absorbance of the test sample. Butylated hydroxytoluene (BHT) and α-tocopherol were used as positive standards.
All data were represented as mean ± standard error (SEM). Data were analyzed by one-way ANOVA followed by Dunnett's test for comparison against control. Values of p ≤ 0.05 (*) and p ≤ 0.01 (**) were considered statistically significant. The inhibitory concentration 50 (IC50), was estimated by means of a linear regression.
9.4. Competitive displacement assay
A 4 mM working solution of salmon sperm DNA (ss-DNA) (SIGMA) was prepared in 5 mM Tris–HCl and 5 mM NaCl buffer at pH 7.4.55 Compounds 1-Ni, 2-Ni and 3-Ni were dissolved in DMSO at concentrations of 10, 6.66 and 3.3 mM respectively. To get insight whether compounds 1-Ni, 2-Ni and 3-Ni may interact with DNA, an ethidium bromide (EB) displacement assay was performed as mentioned in the literature.56 Briefly, a 3 mL buffer containing 5 mM Tris–HCl, 5 mM NaCl buffer at pH 7.4 and 5.0 × 10−5 M EB was mixed in a 1 cm fluorescence cuvette with 2.5 × 10−4 M of ss-DNA. The cuvette was placed in an Agilent Cary Eclipse spectrofluorometer and titrated with different amounts of the stock solution of the compounds 1-Ni, 2-Ni and 3-Ni, after thorough mixing the fluorescence spectra were recorded at 25 °C in the range of 540 and 700 nm (λex = 520 nm).9.5. Data collection and refinement for compound 1-Ni, 2-Ni and 3-Ni
All crystals were grown by slow evaporation of CH2Cl2, then placed on a Bruker Smart Apex II diffractometer with a Mo-target X-Ray source (λ = 0.71073 Å). The detector was placed at 5.0 cm from the crystals and frames were collected with a scan width of 0.5 cm in ω and an exposure time of 10 s per frame. Frames were integrated with the Bruker SAINT software package using a narrow-frame integration algorithm. Non-systematic absences and intensity statistics were used for space group determination of orthorhombic unit cell for 2-Ni. The structures were solved using Patterson methods using the SHELXS-2014/7 program.57 The remaining atoms were located via a few cycles of least squares refinements and difference Fourier maps. Hydrogen atoms were input at calculated positions and allowed to ride on the atoms to which they were attached. Thermal parameters were refined for hydrogen atoms on the phenyl groups using a Ueq = 1.2 Å to precedent atom. The final cycles of refinement were carried out on all non-zero data using SHELXL-2014/7.58 Absorption corrections were applied using the SADABS program.599.6. Computational details
To begin the study of nickel complexes, we utilized reference structures from X-ray crystallography data. The geometry of these complexes was optimized using the B3LYP functional combined with the 6-31+G(d) basis set for all atoms. Following the optimization, frequency calculations were performed to ensure the stability of the optimized geometries. All these calculations were carried out using Gaussian 16 software.60 Additionally, natural bond orbital (NBO) charge calculations were conducted to support the molecular docking studies.61For the molecular docking process, we employed the DNA structure reported in PDB entry 1AIO,62 a well-established reference in various docking studies. The DNA structure was prepared using AutoDock Tools,63 where Gasteiger charges were assigned to the DNA atoms.64 After preparing both ligand and DNA structures, molecular docking was carried out using AutoDock 4, Vina, and Smina software. To achieve a consensus among the different docking results, an exponential consensus ranking (ECR) method was applied.65 Finally, interaction maps were generated using Maestro,66 PyMOL,67 and Chimera software68 to visualize and analyze the docking results.
Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. A.-C. (CVU 824190) express their gratitude to CONAHCyT for the Postdoctoral Fellowships awarded under the “Estancias Posdoctorales por México 2023(1) and 2023(3)” program. J. S. S.-G., A. A.-F. and J. R.-G. are grateful for the Doctoral Fellowships with CVU 997800, 1032866 and 1099989, respectively. D. M. M. acknowledges the financial support for this research provided by PAPIIT-DGAPA-UNAM (PAPIIT IN223323) and CONAHCyT (A1-S-033933). Special thanks are extended to Dr. Patricia Cano-Sánchez for her fundamental contribution to the EB displacement assay, to Antonio Nieto-Camacho and Teresa Ramírez-Apan for their invaluable contributions to the biological assays, and Maria de la Paz Orta for contributions to the elemental analysis assays.References
- B. J. Pages, D. L. Ang, E. P. Wright and J. R. Aldrich-Wright, Dalton Trans., 2015, 44, 3505–3526, 10.1039/C4DT02700K.
- G. Jaouen, A. Vessières and S. Top, Chem. Soc. Rev., 2015, 44, 8802–8817, 10.1039/C5CS00486A.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Shi, F. X. Wang, W. Y. Zhang and Z. Zhang, Chem. Sci., 2020, 11, 12888–12917, 10.1039/D0SC04082G.
- M. Reina, L. F. Hernández-Ayala, M. E. Bravo-Gómez, V. Gómez and L. Ruiz-Azuara, Inorg. Chim. Acta, 2020, 517, 120201, DOI:10.1016/j.ica.2020.120201.
- T. Scattolin, G. Valente, L. Luzietti, M. Piva, N. Demitri, I. Lampronti, R. Gambari and F. Visentin, Appl. Organomet. Chem., 2021, 35, e6438, DOI:10.1002/aoc.6438.
- T. Scattolin, A. Piccin, M. Mauceri, F. Rizzolio, N. Demitri, V. Canzonieri and F. Visentin, Polyhedron, 2021, 207, 115381, DOI:10.1016/j.poly.2021.115381.
- M. Mbaba, T. M. Golding and G. S. Smith, Molecules, 2020, 25, 5276, DOI:10.3390/MOLECULES25225276.
- B. Bertrand, A. De Almeida, E. P. M. Van Der Burgt, M. Picquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 4532–4536, DOI:10.1002/ejic.201402248.
- M. A. Sierra, L. Casarrubios and M. C. de la Torre, Chem. – Eur. J., 2019, 25, 7232–7242, DOI:10.1002/chem.201805985.
- D. L. Ma, D. S. H. Chan and C. H. Leung, Acc. Chem. Res., 2014, 47, 3614–3631, DOI:10.1021/ar500310z.
- T. Völker, F. Dempwolff, P. L. Graumann and E. Meggers, Angew. Chem., Int. Ed., 2014, 53, 10536–10540, DOI:10.1002/anie.201404547.
- Y. C. Ong and G. Gasser, Drug Discovery Today:Technol., 2020, 37, 117–124, DOI:10.1016/j.ddtec.2019.06.001.
- K. Peng, Y. Zheng, W. Xia and Z. W. Mao, Chem. Soc. Rev., 2023, 52, 2790–2832, 10.1039/D2CS00757F.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25, DOI:10.1021/jm100020w.
- J. A. Cruz-Navarro, A. Sánchez-Mora, J. S. Serrano-García, A. Amaya-Flórez, R. Colorado-Peralta, V. Reyes-Márquez and D. Morales-Morales, Catalysts, 2024, 14, 69, DOI:10.3390/catal14010069.
- E. G. Morales-Espinosa, N. Ortiz-Pastrana, V. Gómez-Benítez, R. Reyes-Martínez, H. A. Piñón-Castillo, L. A. Manjarrez-Nevárez, J. M. German-Acacio and D. Morales-Morales, Molbank, 2022, 2022, M1359, DOI:10.3390/M1359.
- C. G. Martínez-De-León, A. Rodríguez-Álvarez, D. Morales-Morales and J. M. Grévy, J. Organomet. Chem., 2024, 1011, 123103, DOI:10.1016/j.jorganchem.2024.123103.
- R. Favela-Mendoza, E. Rufino-Felipe, H. Valdés, R. A. Toscano, S. Hernandez-Ortega and D. Morales-Morales, Inorg. Chim. Acta, 2020, 512, 119920, DOI:10.1016/j.ica.2020.119920.
- S. M. Soliman, S. E. Elsilk and A. El-Faham, Inorg. Chim. Acta, 2020, 510, 119753, DOI:10.1016/j.ica.2020.119753.
- P. Nagarasu, P. Gayathri, S. N. Sri, N. Saisubramanian, P. Dhanaraj, D. Moon, S. P. Anthony and V. Madhu, Inorg. Chem. Commun., 2022, 139, 109316, DOI:10.1016/j.inoche.2022.109316.
- S. M. Soliman, Z. Almarhoon, E. N. Sholkamy and A. El-Faham, J. Mol. Struct., 2019, 1195, 315–322, DOI:10.1016/j.molstruc.2019.05.103.
- S. Wu, Z. Wu, Q. Ge, X. Zheng and Z. Yang, Org. Biomol. Chem., 2021, 19, 5254–5273, 10.1039/D1OB00577D.
- K. Li, T. Zou, Y. Chen, X. Guan and C. M. Che, Chem. – Eur. J., 2015, 21, 7441–7453, DOI:10.1002/chem.201406453.
- T. Chatzisideri, S. Thysiadis, S. Katsamakas, P. Dalezis, I. Sigala, T. Lazarides, E. Nikolakaki, D. Trafalis, O. A. Gederaas, M. Lindgren and V. Sarli, Eur. J. Med. Chem., 2017, 141, 221–231, DOI:10.1016/j.ejmech.2017.09.058.
- B. Bertrand, J. Fernandez-Cestau, J. Angulo, M. M. D. Cominetti, Z. A. E. Waller, M. Searcey, M. A. O’Connell and M. Bochmann, Inorg. Chem., 2017, 56, 5728–5740, DOI:10.1021/acs.inorgchem.7b00339.
- S. G. Churusova, D. V. Aleksanyan, E. Y. Rybalkina, O. Y. Susova, A. S. Peregudov, V. V. Brunova, E. I. Gutsul, Z. S. Klemenkova, Y. V. Nelyubina, V. N. Glushko and V. A. Kozlov, Inorg. Chem., 2021, 60, 9880–9898, DOI:10.1021/acs.inorgchem.1c01138.
- M. Al-Noaimi, F. F. Awwadi, W. H. Talib, S. Atia and H. H. Hammud, J. Mol. Struct., 2019, 1197, 282–291, DOI:10.1016/j.molstruc.2019.07.062.
- M. M. Milutinović, A. Rilak, I. Bratsos, O. Klisurić, M. Vraneš, N. Gligorijević, S. Radulović and Ž. D. Bugarčić, J. Inorg. Biochem., 2017, 169, 1–12, DOI:10.1016/j.jinorgbio.2016.10.001.
- A. Aragón-Muriel, B. A. Aguilar-Castillo, E. Rufino-Felipe, H. Valdés, L. González-Sebastián, R. N. Osorio-Yáñez, Y. Liscano, V. Gómez-Benítez, D. Polo-Cerón and D. Morales-Morales, Polyhedron, 2022, 227, 116115, DOI:10.1016/j.poly.2022.116115.
- J. L. Nevarez, A. Turmo, J. Hu and R. P. Hausinger, ChemCatChem, 2020, 12, 4242–4254, DOI:10.1002/cctc.202000575.
- J. L. Boer, S. B. Mulrooney and R. P. Hausinger, Arch. Biochem. Biophys., 2014, 544, 142–152, DOI:10.1016/j.abb.2013.09.002.
- A. C. Manesis, M. J. O’Connor, C. R. Schneider and H. S. Shafaat, J. Am. Chem. Soc., 2017, 139, 10328–10338, DOI:10.1021/jacs.7b03892.
- M. Hosseini-Kharat, D. Zargarian, A. M. Alizadeh, K. Karami, M. Saeidifar, S. Khalighfard, L. Dubrulle, M. Zakariazadeh, J. P. Cloutier and Z. Sohrabijam, Dalton Trans., 2018, 47, 16944–16957, 10.1039/d3dt90154h.
- A. Amaya-Flórez, J. S. Serrano-García, J. Ruiz-Galindo, A. Arenaza-Corona, J. A. Cruz-Navarro, A. L. Orjuela, J. Alí-Torres, M. Flores-Alamo, P. Cano-Sanchez, V. Reyes-Márquez and D. Morales-Morales, Front. Chem., 2024, 12, 1483999, DOI:10.3389/fchem.2024.1483999.
- A. Sánchez-Mora, E. Briñez, A. Pico, L. González-Sebastián, J. A. Cruz-Navarro, A. Arenaza-Corona, N. Puentes-Díaz, J. Alí-Torres, V. Reyes-Márquez and D. Morales-Morales, Chem. Biodiversity, 2024, 21, e202400995, DOI:10.1002/cbdv.202400995.
- G. F. S. Fernandes, J. R. Lopes, J. L. Dos Santos and C. B. Scarim, Drug Dev. Res., 2023, 84, 1346–1375, DOI:10.1002/ddr.22094.
- M. A. García-Eleno, E. Padilla-Mata, F. Estudiante-Negrete, F. Pichal-Cerda, S. Hernández-Ortega, R. A. Toscano and D. Morales-Morales, New J. Chem., 2015, 39, 3361–3365, 10.1039/C5NJ00052A.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341, DOI:10.1107/S0021889808042726.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235, DOI:10.1107/S1600576719014092.
- A. A. Castillo-García, L. González-Sebastián, L. Lomas-Romero, S. Hernandez-Ortega, R. A. Toscano and D. Morales-Morales, New J. Chem., 2021, 45, 10204–10216, 10.1039/D1NJ01348C.
- B. X. Valderrama-García, E. Rufino-Felipe, H. Valdés, S. Hernandez-Ortega, B. A. Aguilar-Castillo and D. Morales-Morales, Inorg. Chim. Acta, 2020, 502, 119283, DOI:10.1016/j.ica.2019.119283.
- P. K. Thallapally and A. Nangia, CrystEngComm, 2001, 3, 114–119, 10.1039/B102780H.
- L. M. Salonen, M. Ellermann and F. Diederich, Angew. Chem., Int. Ed., 2011, 50, 4808–4842, DOI:10.1002/anie.201007560.
- X. Wang, H. Woo, B. Kiran and L. Wang, Angew. Chem., Int. Ed., 2005, 44, 4968–4972, DOI:10.1002/anie.200501349.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76, DOI:10.1002/1521-3773(20020104)41:1<48::AID-ANIE48>3.0.CO;2-U.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011, DOI:10.1107/S1600576721002910.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392, 10.1039/B203191B.
- V. Vichai and K. Kirtikara, Nat. Protoc., 2006, 1, 1112–1116, DOI:10.1038/nprot.2006.179.
- M. Domínguez, A. Nieto, J. C. Marin, A.-S. Keck, E. Jeffery and C. L. Céspedes, J. Agric. Food Chem., 2005, 53, 5889–5895, DOI:10.1021/jf0504972.
- J. I. Rossato, L. A. Ketzer, F. B. Centurião, S. J. N. Silva, D. S. Lüdtke, G. Zeni, A. L. Braga, M. A. Rubin and J. B. T. Rocha, Neurochem. Res., 2002, 27, 297–303, DOI:10.1023/A:1014907228580.
- O. H. Lowry, N. J. Rosebrough, A. L. Farr and R. J. Randall, J. Biol. Chem., 1951, 193, 265–275 CrossRef CAS PubMed.
- T. B. Ng, F. Liu and Z. T. Wang, Life Sci., 2000, 66, 709–723, DOI:10.1016/S0024-3205(99)00642-6.
- H. Ohkawa, N. Ohishi and K. Yagi, Anal. Biochem., 1979, 95, 351–358, DOI:10.1016/0003-2697(79)90738-3.
- H. Esterbauer and K. H. Cheeseman, Methods Enzymol, Academic Press, 1990, pp. 407–421 DOI:10.1016/0076-6879(90)86134-H.
- G. Backman-Blanco, H. Valdés, M. T. Ramírez-Apan, P. Cano-Sanchez, S. Hernandez-Ortega, A. L. Orjuela, J. Alí-Torres, A. Flores-Gaspar, R. Reyes-Martínez and D. Morales-Morales, J. Inorg. Biochem., 2020, 211, 111206, DOI:10.1016/j.jinorgbio.2020.111206.
- A. Banerjee, J. Singh and D. Dasgupta, J. Fluoresc., 2013, 23, 745–752, DOI:10.1007/s10895-013-1211-0.
- Bruker AXS Inc, 2018.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10, DOI:10.1107/S1600576714022985.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision D.01, 2016.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437, DOI:10.1002/jcc.23266.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652, DOI:10.1038/377649a0.
- G. Morris, R. Huey, W. Lindstrom, M. Sanner, R. Belew, D. Goodsell and A. Olson, J. Comput. Chem., 2009, 30, 2785–2791, DOI:10.1002/jcc.21256.
- J. Gasteiger and M. Marsili, Tetrahedron, 1980, 36, 3219–3288 CrossRef CAS.
- K. Palacio-Rodríguez, I. Lans, C. N. Cavasotto and P. Cossio, Sci. Rep., 2019, 9, 1–14, DOI:10.1038/s41598-019-41594-3.
- Schrödinger Release 2023-2. Maestro. LLC, New York, 2023.
- L. Schrödinger, The PyMOL Molecular Graphics System, Version 1.8, 2015 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612, DOI:10.1002/jcc.20084.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2378802 (1-Ni), 2378803 (2-Ni) and 2378561 (3-Ni). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nj05183a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2025 |