
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent highlights of the total synthesis of cyclic peptide natural products
Takayuki Doi *a,
Masaya Kumashiroa and
Kosuke Ohsawaab
*a,
Masaya Kumashiroa and
Kosuke Ohsawaab
aGraduate School of Pharmaceutical Sciences, Tohoku University, 6-3 Aza-aoba, Aramaki, Aoba-ku, Sendai 980-8578, Japan. E-mail: doi_taka@mail.pharm.tohoku.ac.jp
bFaculty of Pharmacy, Juntendo University, 6-8-1 Hinode, Urayasu, 279-0013, Japan
First published on 14th May 2025
Abstract
Covering: 2020 to 2022
This review described the total synthesis of naturally occurring cyclic peptides with unique structures covering 2020 to 2022, i.e., darobactin A, pyritide A2, decatransin, mannopeptimycin β, α- and β-amanitins, orfamide A, and MA026, paying particular attention to the construction of their unique structures via macrocyclization.
1 Introduction
Cyclic peptides, whose molecular weights lie between those of small molecules and proteins, are garnering considerable attention in drug discovery, and they are expected to have potent activity with high target specificity.1 Peptides are easily hydrolyzed by proteases and are not membrane-permeable, which hinders their application as oral drugs. However, recent advancements in lipophilic cyclic peptides containing unnatural amino acids with side-chain modification, N-alkylamino acids, D-amino acids, lipophilic hydroxycarboxylic acids, and other bridging structures, have significantly improved their pharmacokinetics, such as stability and membrane permeability, characteristics essential for oral drugs.2–5This review highlights recent advancements in the synthetic methodologies for interesting cyclic peptide natural products reported in 2020–2022 and focuses on strategies for building their complex structures. Key developments include the formation of novel structural motifs bridged by side chains within cyclic peptides, such as darobactin A and α- and β-amanitins. The unique construction of a pyridine ring within the cyclic peptide framework, as achieved in the synthesis of pyritide A2, is also discussed. For decatransin, the formation of a depsipeptide containing contiguous N-alkylamino acids and subsequent macrolactone cyclization via the Mitsunobu reaction are discussed. Furthermore, for the synthesis of glycopeptide mannopeptimycin β, the difficult macrocyclization at hydroxyenduracididine was achieved by the Ser/Thr ligation, a method originally developed for protein synthesis. This review also focuses on the structure determination of decatransin through a total synthesis and the structural revisions of orfamide A and MA026 based on synthetic evidence. In addition, highlighting methodologies for optimizing amidation reactions important for peptide bond formation and macrocyclization, emphasizes the importance of careful selections of conditions for these reactions, and achieving the total synthesis through a final deprotection step provides guidance on the use of the protecting groups. By offering insights into both the design and execution of advanced synthetic strategies, this review aims to inform and inspire further progress in the synthesis of cyclic peptides and their analogues.
2 Total synthesis of cyclic peptide natural products
2.1 Darobactin A
Darobactin A (1), a bicyclic peptide natural product isolated from Photorhabdus microbiome, shows little activity against Gram-positive bacteria but potent antibacterial activity against various Gram-negative bacteria.6 Darobactin A is a novel antibiotic that functions via an unprecedented mechanism of action, which involves mimicking the β-sheet structure and binding to the bacterial insertase complex BamA to inhibit its activity.7,8 Unprecedent structure of 1 is characterized by the two rings linked by the indole moieties of two tryptophans with C–O and C–C bonds, respectively. The challenging total synthesis of 1 was reported simultaneously by two groups using intramolecular palladium-catalyzed Larock indole synthesis as key reactions.The Patel, Petrone, and Sarlah group initially attempted the double palladium-catalyzed intramolecular Larock indole synthesis of linear peptide 2. However, although this unique double macrocyclization proceeded, the undesired product 3 was obtained in 18% yield, in which the eastern macrocycle was the atropisomer. This is due to the fact that the construction of the western macrocycle occurred first. Thus, they decided to start their synthetic route by preparing the eastern macrocycle. The more reactive phenyl iodide was then introduced into precursor 4, which underwent selective formation of the eastern macrocycle by the Larock indole synthesis, leading to the desired product 5 and its atropisomer in 39% and 13% yields, respectively. Subsequent conversion of 5 to compound 6, followed by a second Larock indole synthesis, provided the desired product 7 in 51% yield. The nine protecting groups in 7 were removed via treatment with TMSBr/TFA/PhSMe, and subsequent addition of ethylenediamine in one pot furnished 1, completing the total synthesis of darobactin A (1) (Scheme 1).9
 | ||
| Scheme 1 The total synthesis of darobactin A (1) by the Patel, Petrone, and Sarlah group.9 | ||
The Baran group also investigated the construction of the eastern macrocycle via Larock indole synthesis.10 As shown in Scheme 2, the reaction of triethylsilylated alkyne 8b required 3.5 equivalents of Pd(Pt-Bu3)2. Although cyclization yielded 9b in 41% yield, there was a problem of reproducibility. In contrast, the cyclization of terminal alkyne 8a proceeded in the presence of 0.3 equivalents of Pd(Pt-Bu3)2 catalyst, furnishing product 9a as a single atropisomer in an improved yield of 61% on a gram scale. Product 9a was then converted to cyclization precursor 10 with a western macrocycle, from which the desired 11 was obtained in 67% yield via reduction of the nitro group followed by a second Larock indole synthesis. Various condensation agents were investigated for the coupling of carboxylic acid 12 with the dipeptide H-Ser(Bn)-Phe-Ot-Bu. Notably, only EDCI/HOAt allowed the reaction to proceed, and treatment with NH3/MeOH and deprotection with a TMSOTf/TFA/p-cresol/1,2-ethanedithiol cocktail furnished darobactin A (1).
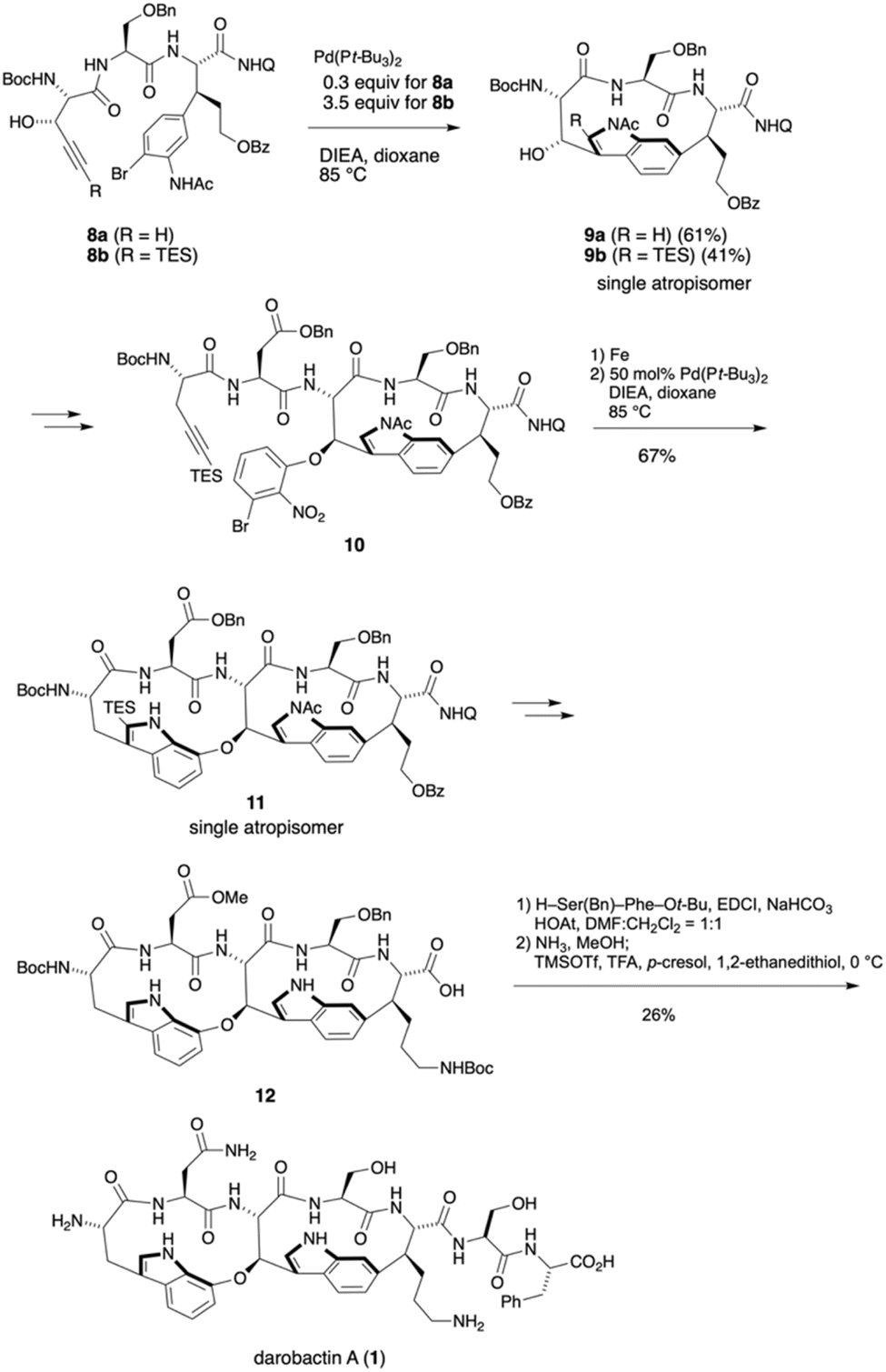 | ||
| Scheme 2 The total synthesis of darobactin A (1) by the Baran group.10 | ||
It should be noted that the intramolecular Larock indole synthesis is worthwhile to construct an indole ring within a macrocyclic peptide. Particularly, in the synthesis of darobactin A using this method, the eastern macrocycle needs to be constructed prior to the western one to control stereoselective formation of the desired atropisomer.
2.2 Pyritide A2
Pyritides A1 and A2 (13) were discovered by the Mitchell group through chemoenzymatic synthesis using putative synthetic intermediates based on genome mining.11 The pyritide possesses a unique structure that includes 6-(1-amino-4-guanidinobutyl)pyridine-2,5-dicarboxylic acid as a nonproteinogenic amino acid component. One of its carboxyl groups along with its amino group participates in the formation of a macrocyclic peptide, while the other carboxyl group is attached to a tripeptide side chain. Notably, it is intriguing how two amide bonds, one of which forms the macrocyclic peptide, are generated from the pyridine-2,5-dicarboxylic acid, which is presumed to have low reactivity. Their total synthesis was reported by the Sarlah group, as shown in Scheme 3.12 They found that using freshly prepared DMDO was crucial to afford unstable tricarbonyl compound 15 via oxidative cleavage of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P bond of diacylylide 14. Next, the reaction of 15 and the amidrazone prepared from hydrazine and ethyl thioamido oxalate was performed to provide triazine 16, which was directly subjected to an aza-Diels–Alder reaction with 2,5-norboradiene to construct a pyridine ring, yielding 17 in 86% yield. Subsequently, a tetrapeptide was condensed to the amino terminus of 17 using EDCI/HOAt to afford 18. Although acid deprotection resulted in degradation, TASF was used to convert the TMSE ester to a carboxylic acid, which was then treated with TFA to remove the Boc group, leading to a cyclization precursor. Macrolactamization of the resulting pyridine carboxylic acid with the N-terminal glycine residue was challenging. Conventional reagents such as HATU, PyBOP, BOP-Cl, and PyAOP led to the formation of oligomers, failing to afford the desired cyclic peptide 19. In contrast, T3P proved effective for macrolactamization, providing 19 in 48% overall yield over three steps. Attempts to hydrolyze the pyridine carboxylic acid ethyl ester using NaOH, LiOH, Ba(OH)2, and TMSOK resulted in the removal of the Cbz group of the guanidyl group in all cases. In contrast, using Me3SnOH, only the ethyl ester was successfully converted to carboxylic acid 20, which was probably facilitated by the nitrogen atom of the pyridine ring. Finally, coupling of 20 with tripeptide 21 followed by hydrogenolysis completed the total synthesis of pyritide A2 (13).
P bond of diacylylide 14. Next, the reaction of 15 and the amidrazone prepared from hydrazine and ethyl thioamido oxalate was performed to provide triazine 16, which was directly subjected to an aza-Diels–Alder reaction with 2,5-norboradiene to construct a pyridine ring, yielding 17 in 86% yield. Subsequently, a tetrapeptide was condensed to the amino terminus of 17 using EDCI/HOAt to afford 18. Although acid deprotection resulted in degradation, TASF was used to convert the TMSE ester to a carboxylic acid, which was then treated with TFA to remove the Boc group, leading to a cyclization precursor. Macrolactamization of the resulting pyridine carboxylic acid with the N-terminal glycine residue was challenging. Conventional reagents such as HATU, PyBOP, BOP-Cl, and PyAOP led to the formation of oligomers, failing to afford the desired cyclic peptide 19. In contrast, T3P proved effective for macrolactamization, providing 19 in 48% overall yield over three steps. Attempts to hydrolyze the pyridine carboxylic acid ethyl ester using NaOH, LiOH, Ba(OH)2, and TMSOK resulted in the removal of the Cbz group of the guanidyl group in all cases. In contrast, using Me3SnOH, only the ethyl ester was successfully converted to carboxylic acid 20, which was probably facilitated by the nitrogen atom of the pyridine ring. Finally, coupling of 20 with tripeptide 21 followed by hydrogenolysis completed the total synthesis of pyritide A2 (13).
 | ||
| Scheme 3 Total synthesis of pyritide A2 (13). | ||
2.3 Decatransin
Decatransin (22), a 30-membered cyclodecadepsipeptide that was isolated from saprophyte fungus Chaetosphaeria tulasneorum in 2015.13 It exhibits potent growth inhibition against HCT116 cells with an IC50 value of 140 nM through inhibition of the translocon channel Sec61. Since other Sec61 inhibitors have been studied as anticancer, immunosuppressive, and antiviral agents,14 22 is expected to serve as a potential lead for drug discovery. In 2023, cryo-EM analysis elucidated the binding structures of the Sec61 complex with various inhibitors, including decatransin.15 Once efficient synthetic methods are established, structure-based drug design will enable further structural modification to optimize its biological activity. Decatransin consists of 2-hydroxy-5-methylhexanoic acid and nine amino acids including nonproteinogenic amino acids, such as three pipecolic acids, homoleucine, two methylhomoleucines, methylthreonine, alanine, and methylisoleucine. Accordingly the backbone amides are highly N-alkylated, a key feature contributing to cell permeability of cyclic peptides. Genome sequencing analysis revealed that 22 is biosynthesized by a non-ribosomal peptide synthetase (NRPS). The Doi group narrowed down the possible structures of the natural product to two stereoisomers based on the presumed biosynthetic pathway, completed the total synthesis, and determined the stereochemical configurations of decatransin as 22 by comparing the spectral and biological data of the synthetic product with those of the isolated natural product.16As shown in Scheme 4, the Cbz group of dipeptide 23 containing a TMSE ester was removed via hydrogenolysis in the presence of HCl, and the resulting hydrochloride salts were treated with 24 and PyBroP/CH2Cl2 with slow addition of DIEA. Using this method, tripeptide 25 was obtained without forming a diketopiperazine. Although a t-butyl ester is usually used to avoid the formation of diketopiperazines, it could not be used in this case because the N-methylamide bond was cleaved under the acidic conditions used for the hydrolysis of the t-butyl ester corresponding to pentapeptide 26. The pentapeptide TMSE ester 26 was prepared via subsequent elongation to the N-methylamino terminus with MeHle and Hle using PyBroP/DIEA/CH2Cl2. Thus, removal of the TMSE ester in pentapeptide 26 with TBAF/THF proceeded without cleavage of the N-methylamide bonds, and the resulting carboxylic acid was coupled with tripeptide 27 using EDCI/HOAt/DIEA to provide octapeptide 28 in 92% yield. After removal of the Cbz group, coupling the resulting amine with (1αR)-29a and (1αS)-29b was performed using HATU/DIEA/CH2Cl2, respectively. (1αR)-30a was obtained in 59% yield with 5% epimerization, whereas (1αS)-30b was provided in 70% yield with 22% epimerization, which was reduced to 10% when COMU/DIEA/CH2Cl2 was used. After the removal of the two silyl groups with TBAF, attempts to achieve macrolactonization using either MNBA or TCBC were unsuccessful. Notably, however, the Mitsunobu reaction of the seco acid derived from (1αR)-30a with DIAD/PPh3/THF proceeded with inversion of configuration, leading to the cyclized product (1αS)-31 in 37% yield. Similarly, the reaction of the seco acid from (1αS)-30b furnished (1αR)-22 in 44% yield. The yield was further improved to 65% when P(4-F-C6H4)3 was used instead of PPh3. The spectral data and cytotoxicity against HCT-116 cells of (1αR)-22 were in good agreement with those of isolated decatransin, confirming that the total synthesis of decatransin was accomplished and its structure is (1αR)-22.
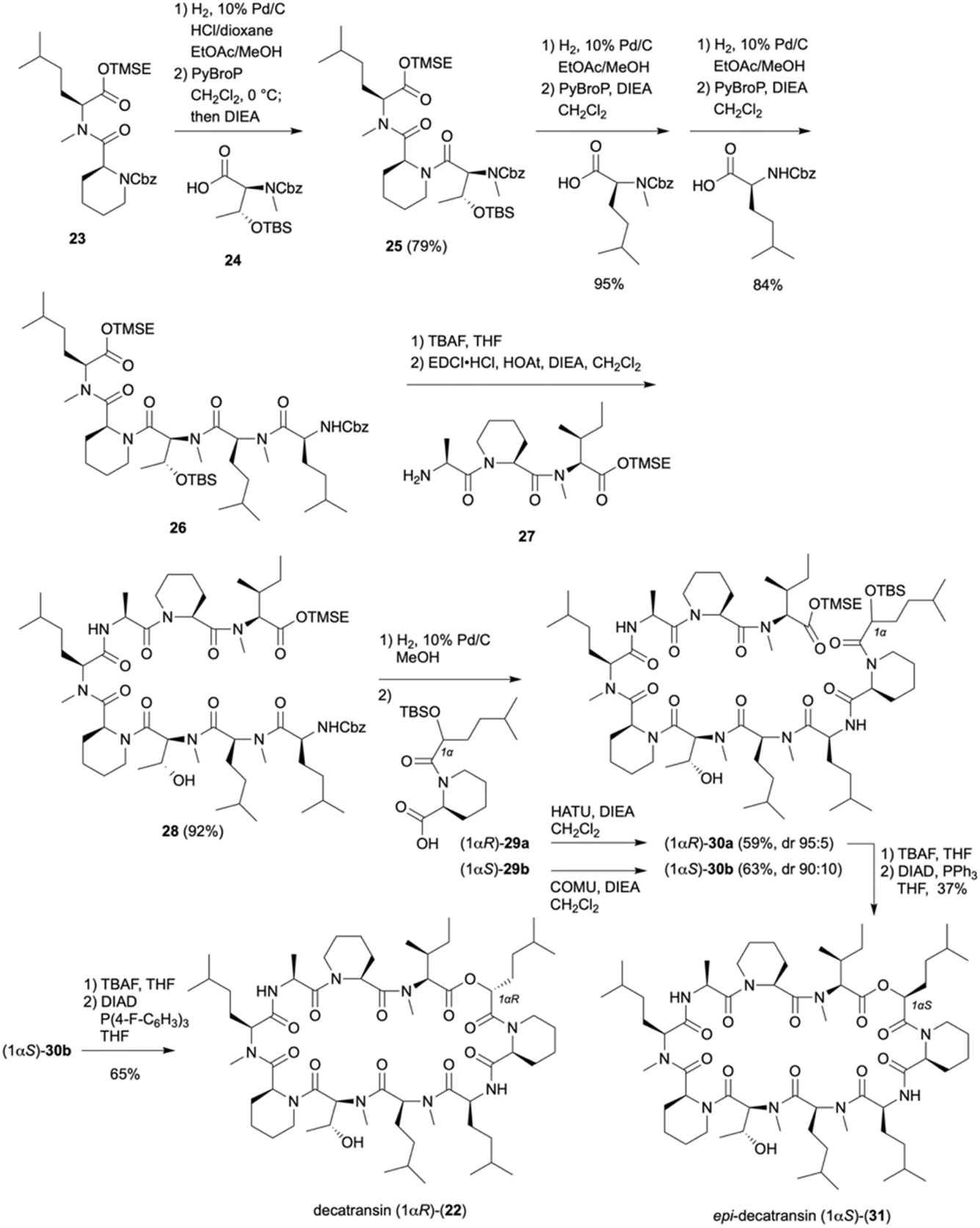 | ||
| Scheme 4 Total synthesis of decatransin (22). | ||
This total synthesis is considered to have paved the way for the further study on the structure–activity relationships (SAR) of decatransin and its potential in drug development.
2.4 Mannopeptimycin β
Mannopeptimycins are cyclic peptide natural products that were isolated from Streptomyces hygroscopicus LL-AC98 and contain Tyr, Gly, Ser, and nonproteinogenic amino acids such as L- and D-β-hydroxyenduracididines (βhEnd) and β-methylphenylalanine. The guanidyl group of D-βhEnd is N-mannosylated in mannopeptimycins α and β, and Tyr is O-dimannosylated in mannopeptimycin α.17,18 The high antimicrobial activity of mannopeptimycins against Gram-positive bacteria was initially demonstrated, and it was later found that they also possess antimicrobial activity against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE). The total synthesis of their aglycone reported by the Fuse and Doi group corrected the stereochemical configuration of the β-position of β-methylphenylananine,19 and the Chen group developed the total synthesis of mannopeptimycins α and β.20 The Li group succeeded in the total synthesis of mannopeptimycin β (32) using an originally developed Ser/Thr ligation method for the challenging formation of the macrocycle.21In a prior solution-phase synthesis, serious epimerization at the α-position was observed upon condensation of the protected N-α-Man-D-βhEnd-OH at the N-terminus of the tetrapeptide. Moreover, the purification of the desired compound from the mixture of diastereomers was difficult. Conversely, condensation with protected N-α-Man-D-βhEnd-OH and dipeptide H-Ser(Bn)-Gly-OAll proceeded without any problem. Thus, the solid-phase synthesis using the obtained tripeptide 36 was investigated.
As shown in Scheme 5, Boc-D-Tyr(Bn)-OH (34) was condensed with hydroxycinnamic acid-immobilized AM resin 33 using PyBOP/DIEA/DMF. After the removal of the Boc group, condensation with Boc-β-MePhe-OH (35) was performed. Similarly, mannosylated tripeptide 36 was condensed to provide polymer-supported pentapeptide 37. In these couplings, the excess amount of the activated form of a carboxylic acid was hydrolyzed to recover the acid. After the removal of the acetonide and the Boc group in 37, condensation of the resulting free amino alcohol with L-βhEnd 38 was performed, but the corresponding product was obtained in low yield. When a large excess of 38 was used, dimerization of the activated ester of 38 afforded a cyclic diester. This problem was solved by performing the inversed addition of 38 in a portion-wise manner, which afforded a sufficient conversion (70%) when three equivalents of 38 were used. The removal of the three Boc groups in the resulting 39 followed by ozonolysis provided the linear precursor salicylaldehyde ester at the C-terminus. Macrocyclization using an originally developed Ser/Thr ligation was performed in pyridine/AcOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) under high dilution conditions of 2 mM to provide the desired product 40. The benzyl group was removed via hydrogenolysis in the presence of Pd(OAc)2 instead of the conventionally used Pd/C to avoid absorption of the product on activated charcoal. Finally, the N,O-acetal was hydrolyzed to furnish mannopeptimycin β (32) in 11% overall yield from the polymer-supported starting material. The Ser/Thr ligation method using the salicylaldehyde ester was also applicable to β-hydroxyenduracididine and enabled the otherwise difficult to conduct cyclization reaction.
:1) under high dilution conditions of 2 mM to provide the desired product 40. The benzyl group was removed via hydrogenolysis in the presence of Pd(OAc)2 instead of the conventionally used Pd/C to avoid absorption of the product on activated charcoal. Finally, the N,O-acetal was hydrolyzed to furnish mannopeptimycin β (32) in 11% overall yield from the polymer-supported starting material. The Ser/Thr ligation method using the salicylaldehyde ester was also applicable to β-hydroxyenduracididine and enabled the otherwise difficult to conduct cyclization reaction.
 | ||
| Scheme 5 The total synthesis of mannopeptimycin β (32). | ||
2.5 Amanitin
α-Amanitin (41) and β-amanitin (42) produced by the death cap mushroom (Amanita phalloides) are toxins that inhibit RNA polymerase II. They possess a cyclooctapeptide structure in which the sulfur atom in cysteine is linked at the 2′ position of 6′-hydroxytryptophan, forming tryptathionine, and is oxidized to (R)-sulfoxide. They also contain other nonproteinogenic amino acids such as hydroxyproline and 4,5-dihydroxyisoleucine.22 Due to its potent toxicity, amanitin has been studied as a payload of an antibody–drug conjugate (ADC). The total synthesis of 41 was first accomplished by the Perrin group in 2018 (ref. 23) and later by the Süssmuth24,25 and Müller26 groups. The latter is presented in this section.As reported by the Perrin group, hydroxyproline was loaded onto the solid phase via a THP linker. Starting from 43, the palladium-catalyzed conversion of the allyl ester to a carboxylic acid, followed by condensation at the C-terminus with (3R,4R)-4,5-diacetoxyisoleucine t-butyl ester 44 (PyBOP/HOBt/DIEA) under microwave irradiation [35 W, 50 °C, 10 min]. Then, the N-terminus was elongated by repeated removal of the Fmoc group (20% piperidine) and condensation with amino acids using PyBOP under microwave irradiation as shown in Method A (Scheme 6). Thus, Asp(OAll), Cys(Trt), Gly, Ile, Gly, and 45 were elongated to provide linear octapeptide 46. Treatment of 46 with TFA led to cleavage from the solid phase and removal of the t-butyl ester, Boc, and trityl groups, and a first cyclization via the Savige–Fontana reaction afforded the desired tryptathionine 47 in 47% yield.27 In a second cyclization, macrolactamization of 47 (DPPA/DIEA/2-propanol/6 mM) was conducted followed by ammonia treatment to remove the acetyl groups and conversion of the allyl ester to a primary amide. Oxidation with mCPBA yielded a 2:1 mixture of diastereomeric sulfoxides. The desired α-amanitin (41) was isolated by preparative reversed-phase HPLC in 54% yield. After the second cyclization, the allyl ester was converted to a carboxylic acid using Pd(PPh3)4/HCO2H/TEA, and treatment with ammonia followed by oxidation under the conditions used for the synthesis of 41 furnished β-amanitin (42) (Scheme 6). Notably, these synthetic methods led to the development of anti-BCMA ADC DHP-101, which is undergoing clinical studies.28
 | ||
| Scheme 6 Total synthesis of α-amanitin (41) and β-amanitin (42). | ||
In addition, the Perrin group investigated the reaction conditions for the (R)-selective sulfoxidation and achieved the stereoselective synthesis of (R)-5′-OH-6′-deoxyamanitin (71%, dr 19:1), which was found to be as potent as 41. Application of this method using UHP/Ti(Oi-Pr)4/L-DET (1.2:1:8) and CH2Cl2/MeOH (3:1) to the synthesis of 41 gave a stereoselectivity of >50:1.29 Therefore, highly efficient and stereoselective synthetic methods for amanitins have been established.
2.6 Orfamide A
Orfamide A (48) is a cyclic lipodepsipeptide isolated from Pseudomonas protegens Pf-5 showing an antifungal activity.30 It is a cyclodepsipeptide bearing an ester linkage formed between the hydroxyl group of Thr and the carboxylic acid of Val. The amino group of the Thr is further acylated with a fatty acid-linked dipeptide. In this context, installation of the ester bond after N-acylation is expected to be challenging due to significant steric hindrance. Conversely, the pre-prepared ester bond may lead to undesired O→N acyl migration during the subsequent N-acylation step. These synthetic challenges must be carefully considered in the design of its synthesis. Arndt group achieved the total synthesis of 48 and corrected the stereochemical configuration. Initially, compounds with L-Leu1, L-Leu5, and 3′S- or 3′R-configuration were synthesized. However, the retention times observed in a LC/MS analysis and the 1H NMR spectra did not match those of the natural product. Analysis of the NRPS biosynthesis genes of orfamide A suggested that the condensation–epimerization domains in modules 1 and 5 may undergo epimerization of Leu1 and/or Leu5. Thus, all possible stereoisomers were synthesized. Initial attempts on on-resin macrolactonization resulted in epimerization. Therefore, solid-phase peptide synthesis was conducted using didepsipeptide 50 with a pre-prepared ester linkage. The side-chain hydroxyl group of Ser was loaded on trityl resin and sequentially condensed with amino acids at the N-terminus to form hexapeptide 49 using HBTU/HOBt/DIEA/DMF/NMP. After the removal of the Fmoc group, condensation with didepsipeptide 50 using HATU/HOAt/2,4,6-collidine/DMF afforded octadepsipeptide 51 along with 3% of a dehydrobutyrine residue. Then, the allyl and Alloc groups were removed by treatment with Pd(PPh3)4/PhSiH3/CH2Cl2. On-resin macrolactamization was performed using the optimized conditions, i.e., HATU/HOAt/2,4,6-collidine, to provide 52. The Fmoc group in 52 was carefully removed by two 30 s treatments with 2% DBU/2% piperidine/DMF to reduce N,O-acyl migration to 4%. In contrast, 54% N,O-acyl migration was observed when 20% piperidine/DMF was used. Notably, 86% N,O-acyl migration occurred in linear peptide 51, suggesting that fixing the conformation by constructing the ring structure would suppress the N,O-acyl migration. Then, the dipeptide side chain was introduced using HBTU/HOBt/DIEA/DMF. Subsequent condensation with TBS-protected carboxylic acid under similar conditions furnished protected 48, from which orfamide A (48) was obtained in 35% overall yield after removal of the TBS and t-Bu groups (1% TIS/0.1 M HCl/HFIP). Similarly, seven of its diastereomers were also synthesized. The reversed-phase HPLC analysis, 1H NMR spectra, and biological response of the synthesized 48 were in good agreement with those of the natural product. Therefore, the correct structure of orfamide A was established (Scheme 7).31 | ||
| Scheme 7 Total synthesis of the revised structure of orfamide A (48). | ||
2.7 MA026
MA026 (53), a 25-membered cyclic depsipeptide isolated from Pseudomonas sp. RtIB026, possesses anti-infectious hematopoietic necrosis virus activity.32 In 53, a Ser in place of the Thr found in orfamide A (48), forms the macrolactone linkage via its hydroxyl group. The amino group of the Ser is further acylated with a peptide side chain. This structural arrangement raises synthetic challenges similar to those encountered in the synthesis of 48. Therefore, the bond-forming steps should be carefully planned in the synthesis of 53. The Hayashi group performed a total synthesis of a reported structure that includes an L-Leu10–D-Gln11 moiety. However, the HPLC retention times of the synthesized product and MA026 did not match, and the former did not show tight junction-opening activity unlike MA026. Then, the linear peptide hydrolyzed at the ester bond of MA026 and the corresponding linear peptide with L-Leu10–D-Gln11 prepared separately were compared via MS/MS analysis, finding that the true structure contains L-Leu and D-Gln in the reversed order. In fact, synthesized 53 with D-Gln10–L-Leu11 was identical to MA026. The total synthesis of the revised structure of MA026 (53) was performed as depicted in Scheme 8. | ||
| Scheme 8 Total synthesis of the revised structure of MA026 (53). | ||
The side-chain carboxylic acid of Fmoc-D-Glu-OAll was loaded on SAL resin. From 54, SPPS was performed to afford hexapeptide 55. The didepsipeptide 56 with a pre-prepared ester linkage was condensed using DIC/HOBt/CH2Cl2 to provide octadepsipeptide. After removing the Alloc and allyl groups using Pd(PPh3)4/PhSiH3, on-resin macrolactamization was performed using DIC/HOBt/DMF to afford 57 without formation of its dimer. During the removal of the Fmoc group, N,O-acyl migration could be reduced to 5.2% by shortening the treatment time with 20% piperidine to 5 min. After sequential condensation of amino acids, coupling with (R)-3-TBSoxydecanoic acid yielded protected 53. Removal of the protecting groups with 95% TFA/H2O afforded MA026 (53) in 11% overall yield.33 Furthermore, single-crystal X-ray crystallography and circular dichroism analyses revealed that 53 has a left-handed α-helical structure, and a SAR study was also performed.34
3 Summary
This review describes the total syntheses of cyclic peptide natural products, including darobactin A, pyritide A2, decatransin, mannopeptimycin β, α- and β-amanitins, orfamide A, and MA026. The focus is on constructing their unique structures, providing detailed solutions and optimized reaction conditions. While solid-phase peptide synthesis is a versatile and powerful method, the incorporation of an unnatural amino acid, a hydroxycarboxylic acid, or a cyclic structure requires careful optimization of the coupling reaction conditions to ensure high reactivity without epimerization. The various reaction conditions discussed in this review offer potential solutions to these challenges. The achievement of total synthesis not only enables detailed SAR studies of analogues and ensures a sufficient supply for drug discovery research, but also paves the way for further applications such as ADCs. In pursuit of these studies, unambiguous structural elucidation through total synthesis plays a pivotal role.4 Abbreviations
| ADC | Antibody–drug conjugate |
| Boc | Tert-butyloxycarbonyl |
| BOP-Cl | Bis(2-oxo-3-oxazolidinyl)phosphorodiamidic chloride |
| Cbz | Benzyloxycarbonyl |
| COMU | (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholinocarbenium hexafluorophosphate |
| DBU | 1,8-Diazabicyclo[5.4.0]-7-undecene |
| DET | Diethyl tartrate |
| DIAD | Diisopropyl azodicarboxylate |
| DIC | N,N′-Diisopropylcarbodiimide |
| DIEA | N,N-Diisopropylethylamine |
| DMBA | 1,3-Dimethylbarbituric acid |
| DMDO | Dimethyldioxirane |
| DMF | N,N-Dimethylformamide |
| DMP | Dimethylphenyl |
| DPPA | Diphenylphosphoryl azide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| Fmoc | 9-Fluorenylmethoxycarbonyl |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| HBTU | 1-[Bis(dimethylamino)methylene]-1H-benzotriazolium 3-oxide hexafluorophosphate |
| HFIP | 1,1,1,3,3,3-Hexafluoropropan-2-ol |
| HOAt | 1-Hydroxy-7-azabenzotriazole |
| HOBt | 1-Hydroxybenzotriazole |
| IBCF | Isobutyl chloroformate |
| mCPBA | Meta-chloroperbenzoic acid |
| MNBA | 2-Methyl-6-nitrobenzoic anhydride |
| NMM | N-Methylmorpholine |
| NMP | N-Methyl-2-pyrrolidone |
| NRPS | Nonribosomal peptide synthetase |
| PyAOP | (7-Azabenzotriazol-1-yloxy)trispyrrolidinophosphonium hexafluorophosphate |
| PyBOP | Benzotriazol-1-yloxy-tris-pyrrolidino-phosphonium hexafluorophosphate |
| PyBroP | Bromo-tris-pyrrolidino-phosphonium hexafluorophosphate |
| SAR | Structure–activity relationships |
| T3P | Propanephosphonic acid anhydride |
| TASF | Tris(dimethylamino)sulfur trimethylsilyl difluoride |
| TBAF | Tetrabutylammonium fluoride |
| TBS | t-Butyldimethylsilyl |
| TCBC | 2,4,6-Trichlorobenzoyl chloride |
| TEA | Triethylamine |
| TFA | Trifluoroacetic acid |
| TIS | Triisopropylsilane |
| TMP | 2,2,6,6-Tetramethylpiperidine |
| TMS | Trimethylsilyl |
| TMSE | 2-(Trimethylsilyl)ethyl |
| UHP | Urea hydrogen peroxide |
5 Conflicts of interest
There are no conflicts to declare.6 Notes and references
- S. You, G. McIntyre and T. Passioura, Expet Opin. Drug Discov., 2024, 19, 961–973 CrossRef CAS PubMed.
- A. Ohta, M. Tanada, S. Shinohara, Y. Morita, K. Nakano, Y. Yamagishi, R. Takano, S. Kariyuki, T. Iida, A. Matsuo, K. Ozeki, T. Emura, Y. Sakurai, K. Takano, A. Higashida, M. Kojima, T. Muraoka, R. Takeyama, T. Kato, K. Kimura, K. Ogawa, K. Ohara, S. Tanaka, Y. Kikuchi, N. Hisada, R. Hayashi, Y. Nishimura, K. Nomura, T. Tachibana, M. Irie, H. Kawada, T. Torizawa, N. Murao, T. Kotake, M. Tanaka, S. Ishikawa, T. Miyake, M. Tamiya, M. Arai, A. Chiyoda, S. Akai, H. Sase, S. Kuramoto, T. Ito, T. Shiraishi, T. Kojima and H. Iikura, J. Am. Chem. Soc., 2023, 145, 24035–24051 CrossRef CAS PubMed.
- T. J. Tucker, M. W. Embrey, C. Alleyne, R. P. Amin, A. Bass, B. Bhatt, E. Bianchi, D. Branca, T. Bueters, N. Buist, S. N. Ha, M. Hafey, H. He, J. Higgins, D. G. Johns, A. D. Kerekes, K. A. Koeplinger, J. T. Kuethe, N. Li, B. Murphy, P. Orth, S. Salowe, A. Shahripour, R. Tracy, W. Wang, C. Wu, Y. Xiong, H. J. Zokian, H. B. Wood and A. Walji, J. Med. Chem., 2021, 64, 16770–16800 CrossRef CAS PubMed.
- R. W. Busby, M. M. Kessler, W. P. Bartolini, A. P. Bryant, G. Hannig, C. S. Higgins, R. M. Solinga, J. V. Tobin, J. D. Wakefield, C. B. Kurtz and M. G. Currie, J. Pharmacol. Exp. Ther., 2013, 344, 106–206 CrossRef PubMed.
- R. S. Cvetković and G. L. Plosker, Drugs, 2005, 65, 99–107 CrossRef PubMed.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O'Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D'Onofrio and K. Lewis, Nature, 2019, 576(7787), 459–464 CrossRef CAS PubMed.
- H. Kaur, R. P. Jakob, J. K. Marzinek, R. Green, Y. Imai, J. R. Bolla, E. Agustoni, C. V. Robinson, P. J. Bond, K. Lewis, T. Maier and S. Hiller, Nature, 2021, 593(7857), 125–129 CrossRef CAS PubMed.
- X. Li, S. Ma and Q. Zhang, Tetrahedron, 2023, 116, 154337 CrossRef CAS.
- M. Nesic, D. B. Ryffel, J. Maturan, M. Shevlin, S. R. Pollack, D. R. Gauthier Jr, P. Trigo-Mouriño, L.-K. Zhang, D. M. Schultz, J. M. M. Dunn, L.-C. Campeau, N. R. Patel, D. A. Petrone and D. Sarlah, J. Am. Chem. Soc., 2022, 144, 14026–14030 CrossRef CAS PubMed.
- Y.-C. Lin, F. Schneider, K. J. Eberle, D. Chiodi, H. Nakamura, S. H. Reisberg, J. Chen, M. Saito and P. S. Baran, J. Am. Chem. Soc., 2022, 144, 14458–14462 CrossRef CAS PubMed.
- G. A. Hudson, A. R. Hooper, A. J. DiCaprio, D. Sarlah and D. A. Mitchell, Org. Lett., 2021, 23, 253–256 CrossRef CAS PubMed.
- A. R. Hooper, A. Oštrek, A. Milian-Lopez and D. Sarlah, Angew. Chem., Int. Ed., 2021, 61, e202212299 CrossRef PubMed.
- T. Junne, J. Wong, C. Studer, T. Aust, B. W. Bauer, M. Beibel, B. Bhullar, R. Bruccoleri, J. Eichenberger, D. Estoppey, N. Hartmann, B. Knapp, P. Krastel, N. Melin, E. J. Oakeley, L. Oberer, R. Riedl, G. Roma, S. Schuierer, F. Petersen, J. A. Tallarico, T. A. Rapoport, M. Spiess and D. Hoepfner, J. Cell Sci., 2015, 128, 1217–1229 CAS.
- E. Pauwels, R. Schülein and K. Vermeire, Int. J. Mol. Sci., 2021, 22, 12007 CrossRef CAS PubMed.
- S. Itskanov, L. Wang, T. Junne, R. Sherriff, L. Xiao, N. Blanchard, W. Q. Shi, C. Forsyth, D. Hoepfner, M. Spiess and E. Park, Nat. Chem. Biol., 2023, 19, 1063–1071 CrossRef CAS PubMed.
- K. Ohsawa, S. Fukaya and T. Doi, Org. Lett., 2022, 24, 5552–5556 CrossRef CAS PubMed.
- S. E. De Voe and M. P. Kunstmann, Antibiotic AC98 and production, US Pat., 3495004, 1970 Search PubMed.
- H. He, R. T. Williamson, B. Shen, E. I. Graziani, H. Y. Yang, S. M. Sakya, P. J. Petersen and G. T. Carter, J. Am. Chem. Soc., 2002, 124, 9729–9736 CrossRef CAS PubMed.
- S. Fuse, H. Koinuma, A. Kimbara, M. Izumikawa, Y. Mifune, H. He, K. Shin-ya, T. Takahashi and T. Doi, J. Am. Chem. Soc., 2014, 136, 12011–12017 CrossRef CAS PubMed.
- B. Wang, Y. Liu, R. Jiao, Y. Feng, Q. Li, C. Chen, L. Liu, G. He and G. Chen, J. Am. Chem. Soc., 2016, 138, 3926–3932 CrossRef CAS PubMed.
- J. Wang, D. Lin, M. Liu, H. Liu, P. Blasco, Z. Sun, Y. C. Cheung, S. Chen and X. Li, J. Am. Chem. Soc., 2021, 143, 12784–12790 CrossRef CAS PubMed.
- T. Wieland and U. Gebert, Justus Liebigs Ann. Chem., 1967, 700, 157–173 CrossRef PubMed.
- K. Matinkhoo, A. Pryyma, M. Todorovic, B. O. Patrick and D. M. Perrin, J. Am. Chem. Soc., 2018, 140, 6513–6517 CrossRef CAS PubMed.
- M.-A. J. Siegert, C. H. Knittel and R. D. Süssmuth, Angew. Chem., Int. Ed., 2020, 59, 5500–5504 CrossRef CAS PubMed.
- G. Yao, C. H. Knittel, S. Kosol, M. T. Wenz, B. G. Keller, H. Gruß, A. C. Braun, C. Lutz, T. Hechler, A. Pahl and R. D. Süssmuth, J. Am. Chem. Soc., 2021, 143, 14322–14331 CrossRef CAS PubMed.
- C. Lutz, W. Simon, S. Werner-Simon, A. Pahl and C. Müller, Angew. Chem., Int. Ed., 2020, 59, 11390–11393 CrossRef CAS PubMed.
- J. P. May and D. M. Perrin, Chem.–Eur. J., 2008, 14, 3404–3409 CrossRef CAS PubMed.
- V. Figueroa-Vazquez, J. Ko, C. Breunig, A. Baumann, N. Giesen, A. Pálfi, C. Müller, C. Lutz, T. Hechler, M. Kulke, C. Müller-Tidow, A. Krämer, H. Goldschmidt, A. Pahl and M. S. Raab, Mol. Cancer Ther., 2021, 20, 367–378 CrossRef CAS PubMed.
- A. Pryyma, K. Matinkhoo, A. A. W. L. Wong and D. M. Perrin, Chem. Sci., 2020, 11, 11927–11935 RSC.
- H. Gross, V. O. Stockwell, M. D. Henkels, B. Nowak-Thompson, J. E. Loper and W. H. Gerwick, Chem. Biol., 2007, 14, 53–63 CrossRef CAS PubMed.
- Y. Bando, Y. Hou, L. Seyfarth, J. Probst, S. Götze, M. Bogacz, U. A. Hellmich, P. Stallforth, M. Mittag and H.-D. Arndt, Chem.–Eur. J., 2022, 28, e202104417 CrossRef CAS PubMed.
- M. Ishima, T. Yoshida, T. Yamazaki, F. Sugawara, K. Hatta, M. Shimojoe and K. Masaki, WO02062831, 2002.
- C. Uchiyama, A. Fukuda, M. Mukaiyama, Y. Nakazawa, Y. Kuramochi, K. Muguruma, M. Arimoto, A. Ninomiya, K. Kako, Y. Katsuyama, S. Konno, A. Taguchi, K. Takayama, A. Taniguchi, Y. Nagumo, T. Usui and Y. Hayashi, Angew. Chem., Int. Ed., 2021, 60, 8792–8797 CrossRef CAS PubMed.
- M. Mukaiyama, C. Uchiyama, A. Fukuda, Y. Nakazawa, Y. Kuramochi, Y. Shibata, S. Konno, K. Muguruma, N. Matsugaki, T. Asari, K. Ogawa, A. Taguchi, K. Takayama, A. Taniguchi, Y. Nagumo, T. Senda, Y. Hayashi and T. Usui, J. Med. Chem., 2023, 66, 8717–8724 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |