Cycloaddition of nitrones with alkenes forms isoxazolidines, which are five-membered heterocycles containing nitrogen and oxygen atoms. This transformation functionalizes alkenes by forming C–C and C–O bonds. The N–O bond in the resultant isoxazolidines is easily cleaved. Additionally, when the cycloaddition is carried out intramolecularly, the regioselectivity of the reaction is influenced by the tether connecting the nitrone and alkene and can differ from the selectivity governed by frontier molecular orbital interaction. These features make the intramolecular cycloaddition of nitrones attractive in the synthesis of complex molecules. In this review, we discuss the intramolecular cycloaddition of nitrones used in the total synthesis of natural products.
Satoshi Yokoshima
Satoshi Yokoshima received his B.S. in 1997, and his PhD in 2002 from The University of Tokyo under the guidance of Professor Tohru Fukuyama. After working for Mitsubishi Pharma Corporation as a medicinal chemist (2002–2004), he joined Professor Fukuyama's group at The University of Tokyo as an assistant professor. He was promoted to lecturer in 2008 and to associate professor in 2011. In 2012 he moved to Nagoya University as an associate professor and was promoted to professor in 2017. His current research interests focus on the synthesis of natural and unnatural molecules with polycyclic systems.
1. Introduction
A nitrone is a 1,3-dipole consisting of carbon, nitrogen, and oxygen atoms arranged in that sequence.1 It is usually depicted as a structure in which carbon and nitrogen atoms form a double bond, with the oxygen atom negatively charged and the nitrogen atom positively charged (Scheme 1). A nitrone undergoes cycloaddition with an alkene to form an isoxazolidine.2 In this transformation, the C–C double bond is functionalized to form C–O and C–C bonds. The N–O bond in the isoxazolidine is readily cleaved. These observations show that the cycloaddition of a nitrone is a useful method to introduce oxygen and nitrogen functionalities with concomitant formation of a C–C bond.
Scheme 1 Cycloaddition of nitrone.
In the cycloaddition step, there is an issue of regioselectivity, which can be understood by frontier molecular orbital (FMO) interaction.1a,h,3 Typical examples of intermolecular cycloaddition of a nitrone with various alkenes are summarized in Table 1. Reactions of nitrone 1 with electron rich alkenes produce 5-substituted isoxazolidines A (entries 1–3).4 These can be understood by the matching of the largest coefficients both in the LUMOnitrone–HOMOalkene and HOMOnitrone–LUMOalkene interactions (Fig. 1a). In the reaction of 1 with nitroethylene (entry 5),5 the HOMOnitrone–LUMOalkene interaction is dominant, selectively producing 4-substituted isoxazolidine B (Fig. 1b). In the case of ethyl acrylate (entry 4),6 which is a less electron-deficient alkene than nitroethylene, the less energetically preferred LUMOnitrone–HOMOalkene also contributes to the regioselectivity in addition to the HOMOnitrone–LUMOalkene interaction, leading to the formation of a mixture of 5- and 4-substituted isoxazolidines.
Table 1Regioselectivity in cycloaddition of nitrones
Fig. 1 HOMO–LUMO interaction of cycloaddition of nitrone. (a) Cycloaddition with an electron-rich alkene. (b) Cycloaddition with an electron-deficient alkene.
These regioselectivities are often changed in the case of intramolecular cycloaddition where the nitrone and alkene are connected by a tether. This tether regulates the approach between the nitrone and the alkene, determining the selectivity.7
In the case of intramolecular cycloaddition, the stereoselectivity, including facial and endo/exo-selectivities, tends to also be controlled by the tether. This feature makes the intramolecular cycloaddition of nitrones attractive for the synthesis of complex molecules, including natural products.
This review summarizes intramolecular cycloaddition reactions of nitrones in natural product syntheses reported between 2015 and 2024.8–11 The preparation of the nitrones and the transformations after the cycloadditions are also described.
2. Construction of isoxazolidine rings in natural products
Although some natural products contain isoxazolidine rings (Fig. 2), such natural products are rare.1e The biosynthetic pathway might involve the generation of a nitrone and its cycloaddition.12 In this section, three syntheses of natural products containing an isoxazolidine ring are demonstrated.
Fig. 2 Selected Natural products containing an isoxazolidine ring.
2.1. Synthesis of alsmaphorazine B
Alsmaphorazine B (15) is a hexacyclic indole alkaloid containing an isoxazolidine ring (Scheme 2). Although a biosynthetic pathway of alsmaphorazine B from an akuammicine-type molecule via multiple stepwise oxidation and rearrangements had been proposed in a paper reporting isolation of alsmaphorazine B,13 Vanderwal and coworkers independently devised an alternative biosynthetic pathway, along which they achieved the synthesis of alsmaphorazine B (racemic, 15 steps).14
Scheme 2 Synthesis of alsmaphorazine B.
They first prepared akuammicine (7) from tryptamine in nine steps. One of the key reactions is base-mediated cycloaddition of Zincke aldehyde 4 to afford tetracyclic compound 5. After the conversion that included introducing an iodoalkene unit onto the nitrogen atom, the resultant unsaturated ester 6 was subjected to an intramolecular Heck reaction to give akuammicine (7).
The next task was to convert the ethylidene into a ketone moiety. After extensive investigations, they established a three-step sequence involving osmium-mediated dihydroxylation, oxidation with Dess–Martin periodinane (DMP) in the presence of tert-butanol, and reduction of α-ketol 8 with samarium(II) iodide. The products—a 1.5:1 mixture of alstolucines B (9) and F (10)—were oxidized with dimethyldioxirane (DMDO) to afford N-oxide 11, which was heated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) under an oxygen atmosphere. Under these conditions, base-mediated E1cb elimination or Cope elimination occurred to liberate an enone and a hydroxylamine.15 Oxidation of the hydroxylamine with gaseous oxygen furnished nitrone 13, which underwent intramolecular cycloaddition with the enone moiety to give cycloadduct 14 in 49% yield. The structure of 14 was confirmed by X-ray crystallographic analysis. In this transformation, a nitrone isomer was also isolated in 29% yield. Further oxidation of the vinylogous carbamate moiety by treatment with lithium hexamethyldisilazide (LHMDS) and Davis oxaziridine afforded alsmaphorazine B (15).
2.2. Synthesis of virosaine A
Virosaine A (18) is a pentacyclic natural product containing an isoxazolidine ring (Scheme 3). In the proposed biosynthetic pathway, the isoxazolidine ring is constructed at the end of the biosynthesis via intramolecular cycloaddition of a five-membered cyclic nitrone 16.16 Hughes and Gleason reported a synthesis of virosaine A (enantioselective, 15 steps),17,18 in which the formation of the isoxazolidine ring was conducted in an early stage of the synthesis via cycloaddition of a five-membered cyclic nitrone.
Scheme 3 A proposed biosynthetic pathway of virosaine A via intramolecular cycloaddition of a five-membered cyclic nitrone.
An asymmetric Diels–Alder reaction of 2-bromoacrolein (19) with furan (20) catalyzed by oxazaborolidinone 21 yielded bicyclic aldehyde 22,19 which was reacted with organolithium reagent 23 to furnish a 2.7:1 diastereomeric mixture of alcohols 24 (Scheme 4). The diastereomeric ratio and the enantiomeric excess could be improved by recrystallization. Acidic hydrolysis of the acetal moiety, followed by a reaction of the resultant cyclic acetal with O-TBS hydroxylamine (TBS = tert-butyldimethylsilyl), afforded O-silyl oxime 25. Treatment of 25 with sodium hydride induced epoxide formation to give 26. When 26 was heated in acetic acid at 120 °C under microwave irradiation, the nitrogen atom intramolecularly attacked the epoxide to form nitrone 27, which underwent cycloaddition to give pentacyclic compound 28 in 92% yield.
Scheme 4 Synthesis of virosaine A.
The remaining task for the synthesis of virosaine A was functionalization of C14 for the construction of the butenolide moiety. The first attempts involved the intramolecular C–H insertion of carbene or nitrene at C14, but the desired products were not obtained. The insertion reactions of carbene or nitrene occurred at undesired positions. Finally, the authors found a site-directed deprotonation was effective for the transformation. Thus, reaction of the hydroxy group in 28 with 1,1′-carbonyldiimidazole (CDI) and then with n-butylamine afforded carbamate 29. Selective lithiation at C14 by treatment with sec-butyllithium, followed by addition of bromine, yielded bromide 30. Radical allylation under Keck's conditions introduced an allyl group at the bridgehead position.20 Reductive cleavage of the carbamate moiety with lithium aluminum hydride liberated alcohol 32. Ozonolysis of the terminal alkene followed by oxidation of the resultant hemiacetal with DMP furnished lactone 33, which was exposed to activated neutral Al2O3 to afford virosaine A (18).
2.3. Synthesis of lycojaponicumin A
Lycojaponicumin A (35) is an alkaloid containing a bicyclo[2.2.1] system within which an isoxazolidine ring is embedded as a 1-aza-7-oxabicyclo[2.2.1]heptane (Scheme 5).21 A biosynthetic pathway of lycojaponicumin A has been proposed, in which the bicyclo[2.2.1] system is formed via intramolecular cycloaddition of nine-membered cyclic nitrone 34. Zhang, Tu and coworkers reported a synthesis of lycojaponicumin A (racemic, 23 steps).22 They constructed the 1-aza-7-oxabicyclo[2.2.1] system via cycloaddition of an NH nitrone and subsequent intramolecular alkylation.
Scheme 5 A proposed biosynthetic pathway of lycojaponicumin A via intramolecular cycloaddition of a nine-membered cyclic nitrone.
A reaction of amide 36 with triflic anhydride formed a keteniminium salt, which underwent intramolecular [2 + 2] cycloaddition with the terminal alkene moiety to give cyclobutanone 37 (Scheme 6). Introduction of a dihydrofuran unit afforded tertiary alcohol 38. Upon treatment of 38 with BF3·OEt2, a ring expansion reaction occurred via selective migration of the quaternary carbon to furnish cyclopentanone 39 with a good diastereoselectivity (11:1 dr at C4). After a five-step conversion, the tetrahydrofuran ring in 40 was reductively cleaved by treatment with samarium(II) iodide. The resulting enolate was transformed into silyl enol ether 41via addition of TBS triflate and subsequent thermal isomerization. Phenylselenylation of the silyl enol ether 41, followed by one-pot oxidative elimination afforded Z-enone 42, which was subjected to photoinduced isomerization with an LED lamp (395 nm), giving, after cleavage of the TBS group with TBAF, E-enone 43. A three-step conversion afforded aldehyde 44, which was reacted with hydroxylamine hydrochloride and sodium bicarbonate in toluene at 100 °C. Under these conditions, NH nitrone 45 was formed and underwent intramolecular cycloaddition with the enone moiety.23 Subsequent intramolecular SN2 reaction between the nitrogen atom in the resulting isoxazolidine ring and the mesylate moiety furnished pentacyclic compound 47. Sequential oxidation of the cyclohexene ring in four steps produced lycojaponicumin A (35).
Scheme 6 Synthesis of lycojaponicumin A.
3. Synthesis via intramolecular cycloaddition of acyclic nitrones
Acyclic nitrones for the intramolecular cycloaddition can be classified into two groups depending on whether the unit containing the alkene moiety is attached to the carbon atom or the nitrogen atom of the nitrone. The former (C-linked unsaturated nitrone, 48) can be basically prepared from aldehydes bearing an alkene moiety by condensation with a hydroxylamine, and its cycloaddition produces bridged and/or fused isoxazolidines (49A and/or 49B, Scheme 7). The latter (N-linked unsaturated nitrone, 50) can be derived from hydroxylamines bearing an alkene moiety, leading to bridged isoxazolidines (51A and/or 51B). Isoxazolidines 49A and 51A are 5-substituted isoxazolidines similar to isoxazolidine A described in Table 1, whereas isoxazolidines 49B and 51B are 4-substituted isoxazolidines resembling isoxazolidine B. This section presents syntheses via intramolecular cycloaddition of acyclic nitrones using C-linked unsaturated nitrones followed by N-linked unsaturated nitrones.
Scheme 7 C-linked and N-linked nitrones.
3.1. Syntheses via cycloaddition of C-linked unsaturated nitrones
3.1.1. Synthesis of himalensine A. Gao and coworkers reported a synthesis of himalensine A (racemic, 29 steps; Scheme 8).24 Heating aldehyde 52 with hydroxylamine 53 yielded a nitrone, which underwent intramolecular cycloaddition via a transition state 54 to give cis-fused isoxazolidine 55. Reductive cleavage of the N–O bond with zinc in acetic acid, followed by basic treatment, produced lactam 56. A two-step conversion of compound 56 including protection of the secondary alcohol with a TBS group and removal of the 1,3-dithiane group afforded ketone 57, which underwent Pd-mediated intramolecular vinylation to produce tricyclic compound 58. Ring-closing metathesis constructed a seven-membered ring, and Nazarov cyclization of 59 led to a cyclopentenone ring. The selective reduction of the lactam in 60 with Vaska's catalyst gave himalensine A (61).25
Scheme 8 Synthesis of himalensine A.
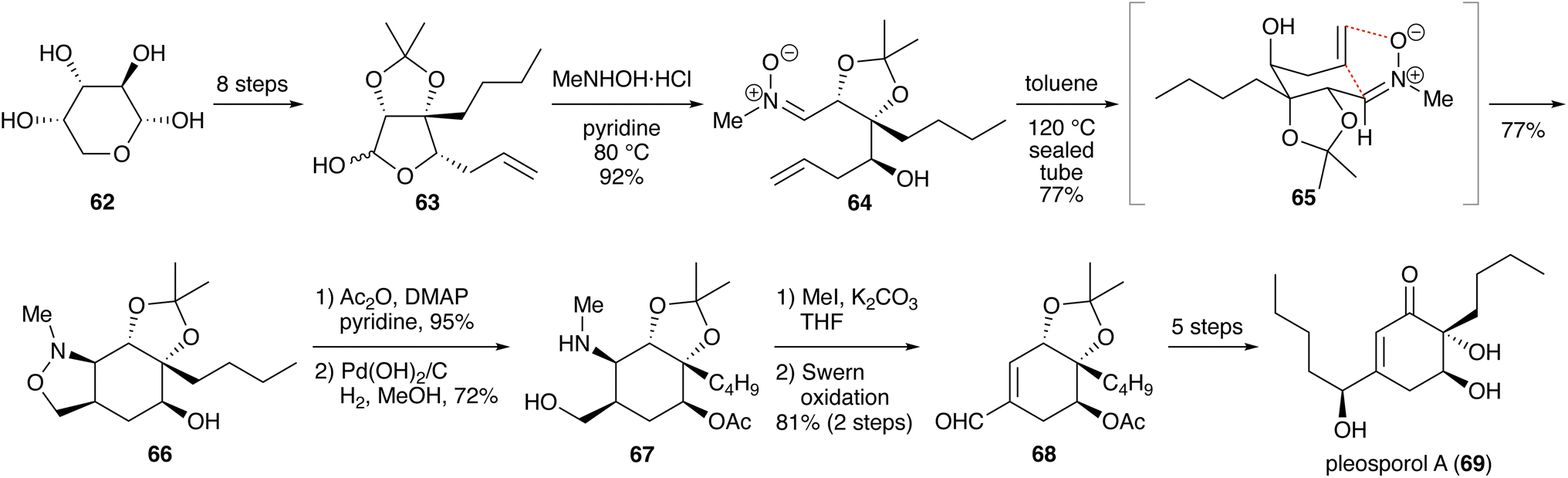
3.1.2. Synthesis of pleosporol A. Xu and coworkers reported a synthesis of pleosporol A (19 steps; Scheme 9).26 Starting from L-arabinose (62), compound 63 was prepared. Treatment with N-methylhydroxylamine hydrochloride in pyridine at 80 °C opened the lactol moiety to furnish nitrone 64 in 92% yield. Cycloaddition of the nitrone was induced by heating in toluene at 120 °C in a sealed tube to give cis-fused isoxazolidine 66 in 77% yield. The transition state 65 explains the stereoselectivity of the cycloaddition. After acetylation of the hydroxy group, the N–O bond was cleaved by hydrogenolysis in the presence of Pearlman's catalyst. Formation of the corresponding quaternary ammonium salt by treatment with iodomethane, followed by Swern oxidation, gave α,β-unsaturated aldehyde 68, which was converted into pleosporol A (69) in five steps.
Scheme 9 Synthesis of pleosporol A.
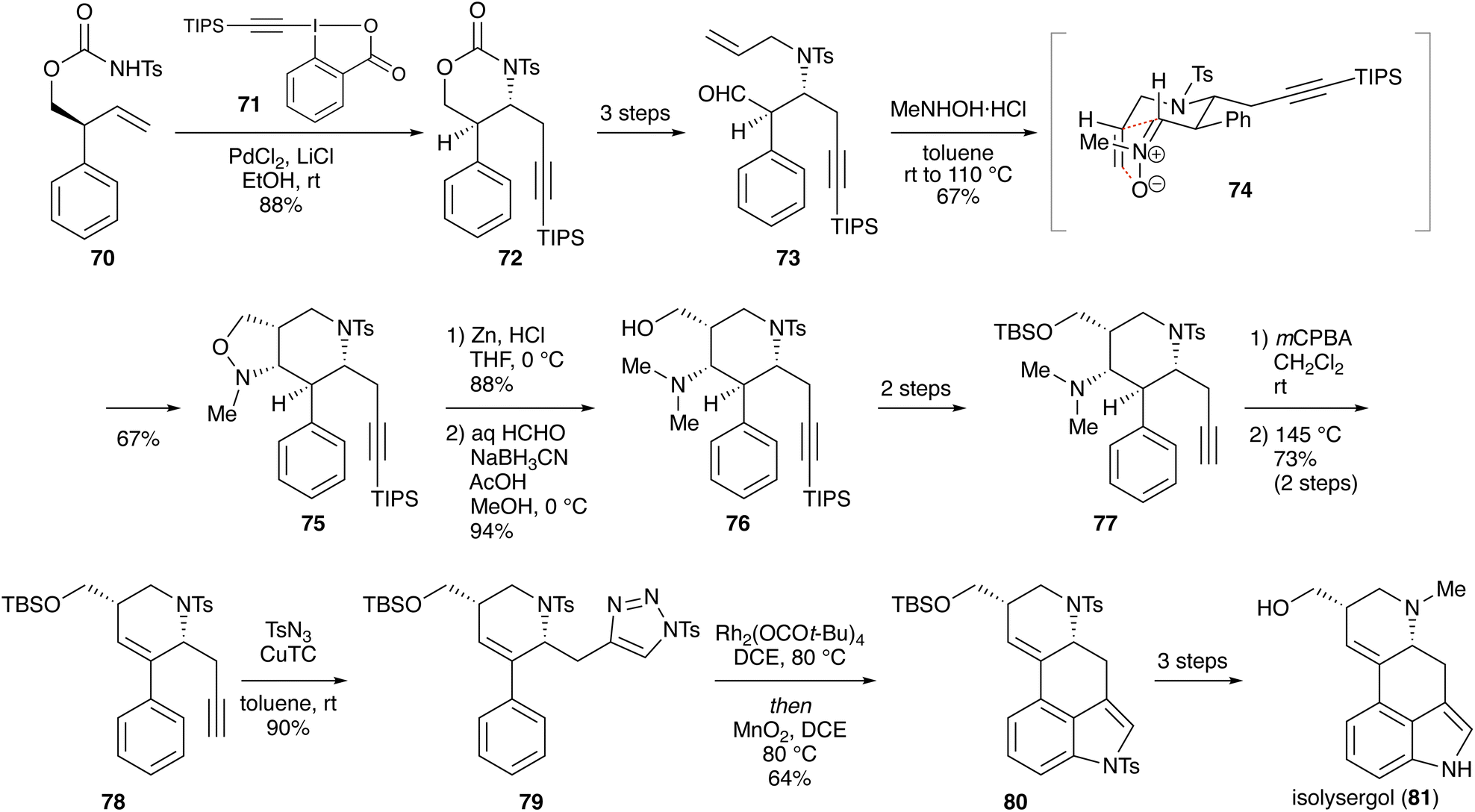
3.1.3. Synthesis of isolysergol. Wang and coworkers reported a synthesis of an ergot alkaloid, isolysergol (18 steps from (2R)-phenyloxirane; Scheme 10).27 Palladium-catalyzed aminoalkynylation of carbamate 70 with 1-[(triisopropylsilyl)ethynyl]-1,2-benziodoxol-3(1H)-one (TIPS-EBX, 71) proceeded stereoselectively to afford 4,5-trans-substituted product 72,28 which was converted into aldehyde 73via three steps including N-allylation. Treatment of 73 with N-methylhydroxylamine led to nitrone formation, and subsequent cycloaddition via transition state 74 gave cis-fused isoxazolidine 75 in 67% yield. Reductive cleavage of the N–O bond with zinc, followed by reductive amination with formalin, furnished compound 76 with a dimethylamino group. After a two-step conversion including removal of the TIPS group and protection of the primary alcohol with a TBS group, the dimethylamino group was oxidized with m-chloroperbenzoic acid (mCPBA) to produce the corresponding N-oxide, which was heated at 145 °C in neat conditions to induce syn-elimination (Cope elimination), giving 78 in 73% yield over two steps. Copper-catalyzed Huisgen cycloaddition with p-toluenesulfonyl azide (TsN3) afforded N-tosyl-1,2,3-triazole 79, which was treated with a rhodium catalyst in 1,2-dichloroethane at 80 °C to give, after oxidation with manganese(IV) oxide, 3,4-fused indole 80.29 Deprotection and N-methylation afforded isolysergol (81).
Scheme 10 Synthesis of isolysergol.
3.1.4. Synthesis of thebainone A. Metz and coworkers reported a synthesis of thebainone A (racemic, 22 steps; Scheme 11).30 An intramolecular Heck reaction of 82 afforded spiro lactone 83. After cleavage of the acetal moieties by treatment with a mild Lewis acid, Pd(MeCN)2Cl2, the resultant keto-aldehyde 84 was treated with N-methylhydroxylamine without purification. The formation of a nitrone and subsequent cycloaddition proceeded at low temperature with high diastereoselectivity via a transition sate 85. Reduction of the ketone and the lactone moieties in 86 with lithium aluminum hydride, followed by reductive cleavage of the N–O bond by treatment with RANEY® nickel under a hydrogen atmosphere, furnished aminotetraol 88, which could be converted into compound 89via an intramolecular Mitsunobu reaction between the amino group and the primary alcohol moiety. Elaboration of the oxygen functionalities on the cyclohexane ring in eight steps produced thebainone A (90).
Scheme 11 Synthesis of thebainone A.
3.2. Syntheses via cycloaddition of N-linked unsaturated nitrones
3.2.1. Synthesis of lasubine II. Sato, Chida and coworkers reported a synthesis of lasubine II (racemic, 7 steps; Scheme 12).31 3,4-Dimethoxybenzaldehyde (91) was converted into N-methoxylamine 92via the formation of an oxime ether and its reduction. Three-component coupling of 92 with aldehyde 93 and allyltributylstannane in the presence of Gd(OTf)3 produced the desired product 94 in 93% yield. Oxidation with mCPBA selectively yielded nitrone 95 without forming the other isomer. When nitrone 95 in toluene was heated at 130 °C in a sealed tube, intramolecular cycloaddition occurred to form isoxazolidine 97 in 74% yield, along with two other diastereomers in 10% yield. Reduction with zinc in hydrochloric acid cleaved the N–O bond and the acetal, and cyclization via reductive amination also occurred to furnish quinolizidine 98. Inversion of the secondary alcohol moiety via a Mitsunobu reaction, followed by methanolysis, produced lasubine II (99).
Scheme 12 Synthesis of lasubine II.
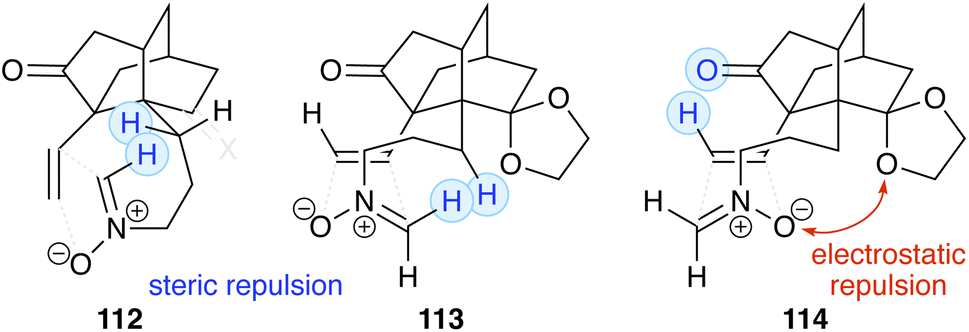
3.2.2. Synthesis of palhinines A and D. Palhinine A (111, Scheme 13) is an alkaloid characterized by a unique skeleton that includes a nitrogen-containing nine-membered ring. Fan and coworkers successfully constructed the nine-membered ring via nitrone cycloaddition, thereby completing the synthesis of palhinine A (racemic, 23 steps).32 Starting from readily available enone 100, compound 101 was prepared through 6 steps including Hosomi–Sakurai allylation and Nozaki–Hiyama–Kishi coupling.33 Treatment of 101 with trimethylsilyl triflate and triethylamine produced siloxy-diene 102, which was heated in p-xylene at 180 °C to induce an intramolecular Diels–Alder reaction that gave tricyclic compound 103 in 65% yield. After conversion in six steps including a Wittig olefination, the resultant aldehyde 104 was transformed into hydroxylamine 105 through the formation and reduction of an oxime. Condensation of the hydroxylamine with aqueous formaldehyde in a biphasic system afforded nitrone 106, which was subjected to the subsequent step without further purification because of its instability. Thus, nitrone 106 was heated in o-dichlorobenzene at 150 °C under microwave irradiation. The cycloaddition of nitrone occurred intramolecularly, and isoxazolidine 108 was obtained in 52% yield as a sole isomer. Fan and coworkers attributed the high regio- and stereoselectivity to the minimal steric interaction and the favorable dipole–dipole attraction in the transition state (Fig. 3). N-Methylation, followed by reductive cleavage of the N–O bond with zinc, yielded aminoalcohol 109. Inversion of the secondary alcohol in 109 was induced by an oxidation/reduction sequence. Acidic hydrolysis of the cyclic acetal afforded palhinine A (111). When N-allylation of 108 was carried out instead of N-methylation, palhinine D (117) was also synthesized according to the same sequence (Scheme 14). The allyl group was removed by treatment with ruthenium(III) chloride in a mixture of acetonitrile and water.
Scheme 13 Synthesis of palhinine A.
Fig. 3 Discussion on regio- and stereoselectivity.
Scheme 14 Synthesis of palhinine D.
3.2.3. Synthesis of (19Z)-taberpsychine, isodihydrokoumine, and isodihydrokoumine N4-oxide. Kerkovius and Kerr reported a synthesis of (19Z)-taberpsychine (racemic, 11 steps; Scheme 15).34 Diol 118 was prepared through a sequence including hydrostannylation of an alkyne, Stille coupling, and copper-catalyzed conjugate addition. A Mitsunobu reaction of diol 118 with N,O-bis(Boc)-hydroxylamine (Boc = tert-butoxycarbonyl) occurred selectively at the allylic position to furnish protected hydroxylamine 119 in 83% yield.35 Cleavage of the Boc groups by treatment with trifluoroacetic acid (TFA), followed by condensation with aldehyde 120 in the presence of triethylamine, afforded nitrone 121, which was heated in refluxing benzene to give isoxazolidine 123 (2:1 ratio about the 2-hydroxyethyl chain, 1:1 ratio about the indoline). To understand the reactivity of the hydroxylamine, the reader should note that when the hydroxylamine salt was neutralized with triethylamine in the absence of aldehyde 120, N-hydroxypyrrolidine 129 was produced via a Cope-type hydroamination (Scheme 16).36
Scheme 15 Synthesis of (19Z)-taberpsychine, isodihydrokoumine, and isodihydrokoumine N4-oxide.
Scheme 16 Cope-type hydroamination of hydroxylamine.
After cleavage of the Ts group, the resultant compound was subjected to Swern oxidation. Under these conditions, simultaneous oxidation of the primary alcohol and the indoline occurred; the aldehyde was then protected as its dimethyl acetal. At this stage, the diastereomers could be separated. The N–O bond was cleaved by a reaction with samarium(II) iodide. Upon treatment with BF3·OEt2 in acetonitrile, the dimethyl acetal moiety in 124 was activated and then intramolecularly attacked by the primary alcohol. Subsequent Friedel–Crafts reaction with the indole nucleus gave 125. Reductive methylation of the secondary amine afforded (19Z)-taberpsychine (126). Treatment of (19Z)-taberpsychine (126) with trimethylsilyl iodide led to a Conia-ene-type reaction between the indole and the ethylidene moiety to furnish isodihydrokoumine (127). Oxidation with mCPBA produced isodihydrokoumine N4-oxide (128).
4. Synthesis via intramolecular cycloaddition of cyclic nitrones
Intramolecular cycloaddition of mono-cyclic nitrones produces tricyclic isoxazolidines, beneficial for the synthesis of complex molecules (Scheme 17). Although preparation of cyclic nitrones has some difficulties, such as introducing multiple functional groups in a molecule, various methods to generate cyclic nitrones have been explored. This section presents syntheses via intramolecular cycloaddition of cyclic nitrones.
Scheme 17 Intramolecular cycloaddition of cyclic nitrones.
4.1. Synthesis of 19-hydroxyibogamine
Alkayar and Coldham reported a formal synthesis of 19-hydroxyibogamine (racemic, 16 steps; Scheme 18).37 Aldehyde 132 was prepared via malonic ester synthesis and alkylation of a 1,3-dithiane. Oxime formation, followed by intramolecular alkylation, formed six-membered cyclic nitrone 134, which underwent intramolecular cycloaddition to give isoxazolidine 135. Hydrolysis of the 1,3-dithiane moiety under oxidative conditions afforded ketone 136. Reductive cleavage of the N–O bond with zinc and acetic acid afforded aminoalcohol 137, which was condensed with indoleacetic acid to give 138. Conversion of the ketone moiety into an acetal, followed by acetylation of the hydroxy group, gave the reported intermediate 139, which could be converted into 19-hydroxyibogamine (140) in three steps.
Scheme 18 Synthesis of 19-hydroxyibogamine.
4.2. Synthesis of N-methyl-euphococcine
Kürti and coworkers developed a method for C-allylation of unprotected oximes and applied the method to the synthesis of N-methyl-euphococcine (racemic, 6 steps; Scheme 19).38 Oxime 141 was treated with diisopropyl allylboronate in the presence of 3,5-difluorophenylboronic acid as a catalyst to give hydroxylamine 142. After acidic hydrolysis of the acetal moiety and subsequent basic workup, the resultant six-membered cyclic nitrone was heated in toluene at 110 °C to produce isoxazolidine 144 in 57% yield in two steps. N-methylation, followed by reduction with zinc, cleaved the N–O bond, and oxidation of the resultant secondary alcohol 146 with DMP afforded N-methyl-euphococcine (147).
Scheme 19 Synthesis of N-methyl-euphococcine.
4.3. Synthesis of cylindricine C
Sato, Chida and coworkers developed a method to synthesize cyclic nitrones from N-hydroxylactams, and applied it to the synthesis of cylindricine C (asymmetric, 12 steps; Scheme 20).39O-Methylation of known lactam 148 with Meerwein's reagent yielded imidate 149, which was converted into N-hydroxylactam 150via oxidation with mCPBA, acidic hydrolysis of the resultant oxaziridine, and lactam formation under basic conditions. After the hydroxy group was protected with a 2-(trimethylsilyl)ethoxymethyl (SEM) group, the addition of hexenyllithium and subsequent acidic treatment afforded five-membered cyclic nitrone 152. When heated in tert-butylbenzene, 152 underwent intramolecular cycloaddition to give a 1.3:1 mixture of regioisomers 155 and 156. After separation of the isomers, the N–O bond in 156 was reductively cleaved with zinc in aqueous acetic acid. The resultant alcohol was converted into aldehyde 158 in the presence of the unprotected secondary amine under the conditions developed by Iwabuchi and coworkers.40 Addition of an acetylide to aldehyde 158 produced 159, which was oxidized again under Iwabuchi's conditions, where intramolecular Michael addition also proceeded to furnish dihydro-4-pyridone 160. Removal of the tert-butyldiphenylsilyl (TBDPS) group, followed by stereoselective reduction of the enamine moiety directed by the hydroxy group, afforded cylindricine C (161).
Scheme 20 Synthesis of cylindricine C.
4.4. Synthesis of sarain A
Yokoshima, Fukuyama and coworkers reported a formal synthesis of sarain A (enantioselective; Scheme 21).41 Claisen–Ireland rearrangement of 162 selectively constructed the contiguous stereogenic centers to produce 163. After conversion of 163 into alcohol 164, a hydroxylamine unit was introduced via a Mitsunobu reaction. The p-methoxyphenyl (PMP)-protected alcohol was transformed into α,β-unsaturated aldehyde 167via steps including oxidative cleavage of the PMP group, Swern oxidation, a Mannich reaction, and a methylation-E1cb elimination process. Treatment of 167 with TFA cleaved the two Boc groups to liberate the hydroxylamine moiety. Upon neutralization with pyridine, condensation of the hydroxylamine with the aldehyde moiety proceeded intramolecularly to form 8-membered cyclic nitrone 168, which underwent intramolecular cycloaddition with the alkene moiety, giving tricyclic compound 169. This intermediate could be converted into an advanced intermediate (compound 170) in Overman's synthesis of sarain A.42
Scheme 21 Synthesis of sarain A.
4.5. Synthesis of kopsone
Yokoshima and coworkers reported a synthesis of kopsone (enantioselective, 19 steps; Scheme 22).43 Claisen–Ireland rearrangement and subsequent methylation produced compound 173. Reduction of the ester moiety in 173, followed by a Mitsunobu reaction with N,O-bis(Boc)-hydroxylamine, furnished 174. The TBS-protected alcohol was converted into α,β-unsaturated aldehyde 176 in three steps. Upon treatment of 176 with TFA, the two Boc groups in 176 were cleaved. Neutralization with triethylamine induced the formation of eight-membered cyclic nitrone 177, which underwent intramolecular cycloaddition to give tricyclic isoxazolidine 178 in 76% yield. The diastereomeric compound 176′, which was prepared from Z-isomer of 172 by an analogous sequence, could also be converted into tricyclic isoxazolidine 178′ in 57% yield. After reductive cleavage of the N–O bond in 178 with zinc and acetic acid, the resultant secondary amine was protected with a Boc group. Hydrogenation of the C–C double bond and hydrogenolysis of the benzyl ether were carried out under a hydrogen atmosphere in the presence of palladium(II) hydroxide. The resultant 1,2-diol was oxidatively cleaved to yield aldehyde 180, which was converted into ketone 181 by treatment with sodium hydride and oxygen.44 Cleavage of the Boc group and reductive methylation afforded kopsone (182).
Scheme 22 Synthesis of kopsone.
4.6. Synthesis of stephadiamine A
Stephadiamine (196) is a densely-functionalized pentacyclic natural product (Scheme 23). Zhu and coworkers used nitrone cycloaddition to efficiently construct the skeleton and introduce functional groups.45 Asymmetric Michael addition of phenol 183 to nitroethylene mediated by Takemoto catalyst 184 produced ketone 185 with a quaternary stereocenter.46 Oxidative cleavage of the terminal alkene formed an aldehyde, which was reductively condensed with Meldrum's acid in the presence of Hantzsch ester and L-proline. After 1,4-reduction of the enone moiety in 186 with L-selectride, a reaction with Eschenmoser's salt in methanol afforded α,β-unsaturated ester 188. Upon treatment of 188 with an excess amount of sodium dithionite in a mixed solvent system of methanol and water at 80 °C, a domino sequence involving reduction of the nitro group into a hydroxylamine, nitrone formation, and intramolecular cycloaddition with the unsaturated ester moiety gave isoxazolidine 190 in 63% yield.
Scheme 23 Synthesis of stephadiamine.
Cleavage of the N–O bond with zinc in acetic acid liberated aminoalcohol 191, which was converted into acyl azide in three steps. When heated in allyl alcohol, 192 underwent Curtius rearrangement to produce compound 193. Benzylic oxidation was carried out using a photocatalytic method reported by Yoon and coworkers,47 giving ketone 194, which was stereoselectively reduced under Luche's conditions to afford an alcohol. The formyl group was also reduced with sodium borohydride in THF in the presence of cerium(III) chloride and TFA to yield 195. Lactone formation under basic conditions, followed by palladium-catalyzed cleavage of the allyloxycarbonyl (Alloc) group, produced stephadiamine (196). The synthesis was completed in 18 steps.
4.7. Synthesis of putative melognine
Irie and Yokoshima reported a synthesis of putative melognine (racemic, 29 steps; Scheme 24).48 Compound 197 was prepared using malonic ester synthesis and Sonogashira coupling. Reduction of the ester moieties with diisobutylaluminum hydride (DIBAL), followed by selective protection of the 1,3-diol as its benzylidene acetal, furnished 198. After mesylation of the primary alcohol moiety in 198, the resultant mesylate was heated with potassium carbonate in N,N-dimethylformamide (DMF) at 100 °C to induce an intramolecular SN2 reaction, giving ten-membered cyclic alkyne 199. Enyne metathesis with ethylene in the presence of Hoveyda–Grubbs second-generation catalyst (HG II) yielded 1,3-diene 200.49 Cleavage of the Ns group liberated the aniline. In this step, the diastereomers derived from the benzylidene acetal could be separated. Treatment of 201 with hydrogen peroxide in the presence of sodium tungstate formed ten-membered cyclic nitrone 202,50 which underwent intramolecular cycloaddition to give isoxazolidine 203 as a sole isomer. Considering the stereochemistry of the product (compound 203), the nitrone formed in situ must be the Z-isomer (Scheme 25). The diastereomeric product (compound 210), which is derived from the E-isomer, was not detected. The regioisomeric product (compound 211) was also not obtained. Interestingly, in the absence of the constraint imposed by the ten-membered ring, the opposite regioselectivity has been observed.51
Scheme 24 Synthesis of putative melognine.
Scheme 25 Stereo- and regioselectivity in the cycloaddition.
When the oxidation of 201 was carried out in methanol,52 isoxazolidine 203 and N-oxide 213 were obtained in 23% and 39% yields, respectively (Scheme 26). The latter is speculated to have formed via a Cope-type hydroamination of hydroxylamine 212.36
Scheme 26 Cope-type hydroamination of hydroxylamine.
Cleavage of the N–O bond in 203 and protection of the resultant hydroxy group with an acetyl group furnished compound 204, which was heated in the presence of Shvo's catalyst to induce isomerization at the α-position of the nitrogen atom.53 A seven-step transformation of compound 205 gave aldehyde 206, which was reacted with N-allylglycine in DMF at 130 °C. Under these conditions, the formation of an azomethine ylide and its cycloaddition proceeded to yield pentacyclic compound 207. Ring-closing metathesis with HG II constructed a piperidine ring, and further transformation yielded putative melognine (209).
5. Discussion on regioselectivity
5.1. Intramolecular cycloaddition of acyclic nitrones
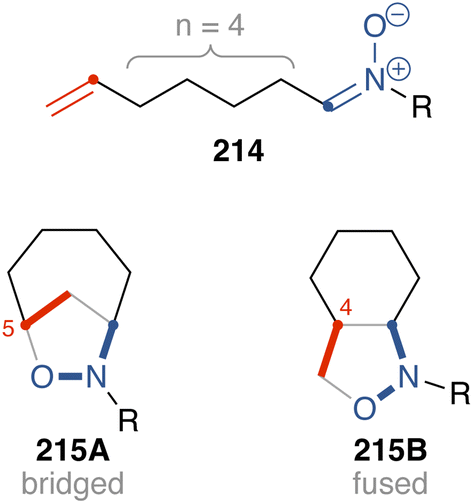
In this review, acyclic nitrones for intramolecular cycloaddition are classified into two groups: C-linked unsaturated nitrones and N-linked unsaturated nitrones. The C-linked unsaturated nitrones discussed in Section 3 (nitrones 54, 65, 74, and 85) are classified as having a four-carbon tether, with a structure like 214 (Fig. 4), and their cycloaddition produced fused isoxazolidines, represented by 215B (4-substituted isoxazolidine). There are many examples of producing fused isoxazolidines as shown in this review. However, formation of bridged isoxazolidine 215A (5-substituted isoxazolidine) has also been reported.54 The regioselectivity is not primarily determined by the tether length. Instead, it depends on the substituents on the nitrone, alkene, and tether.
Fig. 4 C-linked unsaturated nitrone.
Cycloaddition of N-linked unsaturated nitrones can produce two types of regio isomers (51Avs.51B, Scheme 7). For a two-carbon tether (216, n = 2; Fig. 5), 217A (5-substituted isoxazolidine) is typically the major or sole product,55 consistent with the results observed in the synthesis of lasubine II (Scheme 12). For a three-carbon tether (218, n = 3), the regioselectivity varies depending on the substituents.51,55a,56 In the synthesis of isodihydrokoumine (Scheme 15), a 4-substituted isoxazolidine, corresponding to 219B (4-substituted isoxazolidine) was obtained. The nitrone used in the synthesis of palhinines (Scheme 13) is formally designated as 220 (n = 5), and the product of the cycloaddition was a 5-substituted isoxazolidine, corresponding to 221A. Fan and coworkers explain the regioselectivity using steric and electrostatic repulsion (Fig. 3).
Fig. 5 N-linked unsaturated nitrone.
5.2. Intramolecular cycloaddition of cyclic nitrones
In contrast to acyclic nitrones, cyclic nitrones have an additional linkage that imposes constraints on the interaction between the nitrone and the alkene during intramolecular cycloaddition (Fig. 6), which can affect regioselectivity.
Fig. 6 Cyclic nitrones.
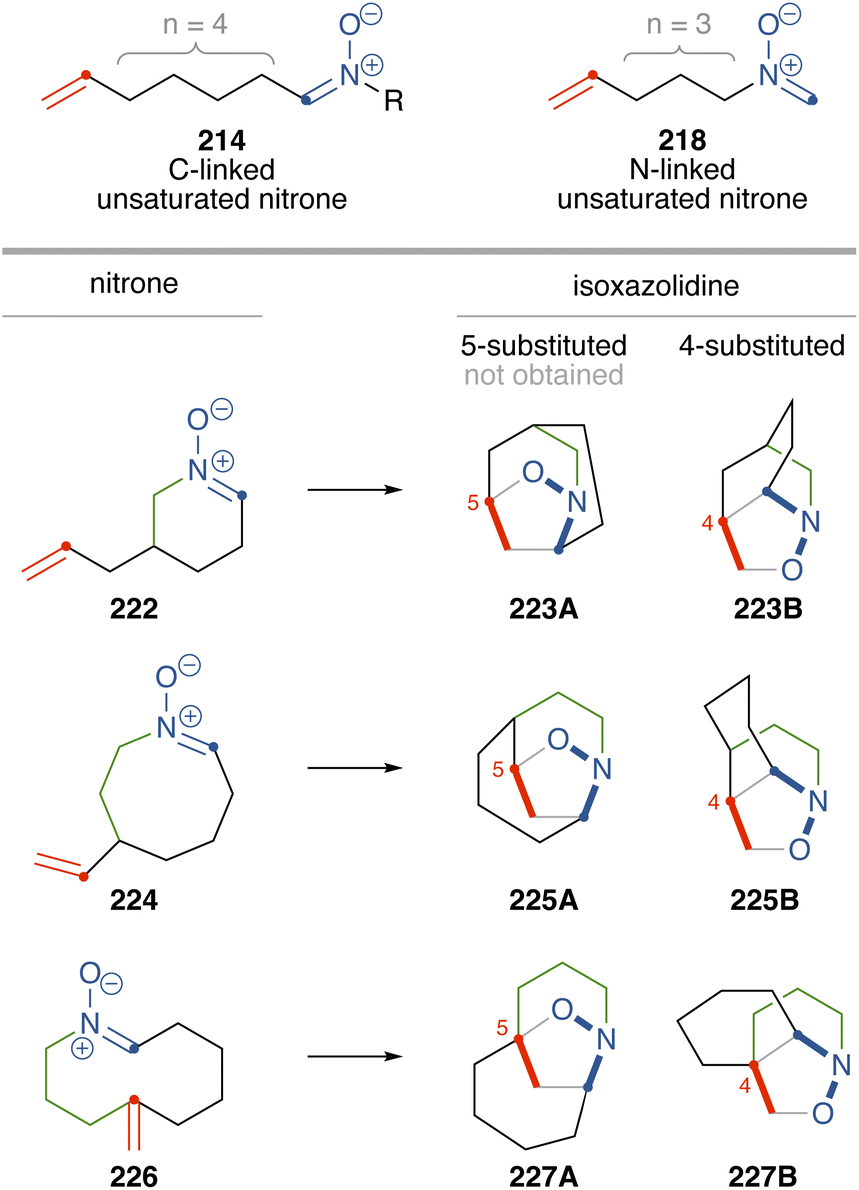
Nitrones 134, 168, 177, and 202 are considered as both C-linked unsaturated nitrones with a four-carbon tether (214) and N-linked unsaturated nitrones with a three-carbon tether (218). Although the cycloaddition of acyclic unsaturated nitrones 214 or 218 can yield both regioisomers (215A/B or 219A/B) depending on substituents, cycloaddition of cyclic nitrones 134, 168, 177, and 202 produced sole regioisomers, corresponding to 223B, 225B, and 227B (4-substituted isoxazolidines, Scheme 27).
Scheme 27 Cycloaddition of cyclic nitrones.
Fig. 7 illustrates the transition states involved in the cycloaddition of acyclic C-linked unsaturated nitrone 214. Nitrone 214 has two alkyl chains attached to the carbon and nitrogen atoms of the nitrone. When the two alkyl chains are positioned on opposite sides, the configuration is referred to as “trans”, while when they are positioned on the same side, it is referred to as “cis”. In the transition state 228 from trans-nitrone, the nitrone and alkene moieties can smoothly interact to produce bridged isoxazolidine 215A, whereas in the cis-nitrone, interaction between the nitrone and alkene moieties such as to form bridged isoxazolidine 215A is geometrically restricted. This makes transition state 231 unfavorable. On the other hand, cis-nitrone undergoes intramolecular cycloaddition to form fused isoxazoline 215B through the transition state 232 or 233.57 Since cyclic nitrones 222 and 224 are cis-nitrones, cycloaddition through a transition state such as 231 to form 223A or 225A is unfavorable. However, cycloaddition through a transition state such as 232 is expected to proceed. This discussion explains the selective formation of 223B or 225B in the cycloaddition of 222 or 224. The transition states of these cycloaddition (222TS and 224TS) can be depicted by adding a linkage to 232 (Fig. 8). The transition state 222TS adopts a boat-shaped six-membered conformation.
Fig. 7 Transition states in cycloaddition of C-linked unsaturated nitrone 214.
Fig. 8 Transition states in cycloaddition of cyclic nitrones.
Cyclic nitrone 226 is a trans-nitrone, and therefore the above argument cannot be adopted to explain the selective formation of 227B. In a transition state forming 227B (226TS, Fig. 8), newly formed rings other than the isoxazolidines are six-membered rings, while in the transition state to yield the regioisomers (227A), newly formed rings become seven-membered rings, causing strains in the transition state of the cycloaddition.
Nitrone 189 is considered as both C-linked unsaturated nitrones with a three-carbon tether (234) and N-linked unsaturated nitrones with a five-carbon tether (220), represented by 235 (Scheme 28). In the cycloaddition of acyclic nitrone 234, the formation of a bridged isoxazolidine from a cis-nitrone is unfavorable, as is the transition state 231, while in the transition state to form a fused isoxazolidine, the nitrone and alkene moieties can sufficiently interact. Since cyclic nitrone 235 is a cis-nitrone, this explains why the formation of bridged (5-substituted) isoxazolidine 236A is unfavorable, leading to the selective formation of 236B in the cycloaddition of 235.58
Scheme 28 Cycloaddition of cyclic nitrones.
Nitrone 152 is a 5-membered cyclic nitrone, and can be considered as a C-linked unsaturated nitrone with a four-carbon tether (214, Fig. 4 and Scheme 27). Since the alkenyl chain is attached to the carbon atom of the nitrone, the geometry of the nitrone is necessarily trans, allowing for the formation of bridged and fused isoxazolidines (Fig. 7). Indeed, cycloaddition of nitrone 152 produced a mixture of bridged and fused isoxazolidines (155 and 156 in Scheme 20).59
Nitrone 143 is regarded as a N-linked unsaturated nitrone with a two-carbon tether (216, Fig. 5). The cycloaddition of 143 regioselectively produces isoxazolidine 144, a 5-substituted isoxazolidine. This is consistent with the regioselectivity in cycloaddition of 216 to yield 217A (5-substituted isoxazolidine, Fig. 5) as the major or sole product.60
6. Conclusions
Various ring systems of natural products have been constructed via the intramolecular cycloaddition of nitrones. The cycloaddition of nitrones can be carried out under simple conditions, typically heating in a solvent. This reaction enables the functionalization of alkenes through the formation of both C–O and C–C bonds, making it possible to construct even quaternary carbon centers. As discussed in Section 5, regioselectivity of intramolecular cycloaddition is often different from that of intermolecular cycloaddition of nitrones. In particular, intramolecular cycloaddition of cyclic nitrones tends to proceed in a regioselective manner as the additional bond forming the cyclic nitrone may limit the access between the nitrone and the alkene.
The preparation of nitrones is also an important issue in this review. Condensation of hydroxylamines with aldehydes or ketones is a simple method for the preparation of nitrones. In addition, the oxidation of amines (Schemes 2, 12 and 24), the N-alkylation of oximes (Schemes 4 and 18), and the transformation of an N-hydroxylactam (Scheme 20) are included in this review.61 For the preparation of hydroxylamines, various methods have been demonstrated, including reduction or C-allylation of an oxime (Schemes 12, 13 and 19), introduction of a hydroxylamine unit via a Mitsunobu reaction (Schemes 15, 21 and 22), and reduction of a nitro group (Scheme 23). After the cycloaddition, reduction with zinc, samarium(II) iodide, or RANEY® nickel is widely used to cleave the N–O bond in the isoxazolidine, yielding versatile 1,3-amino alcohols. Oxidative cleavage of the N–O bond has also been reported but is not included in this review.62 Further accumulation of results related to the cycloaddition of nitrones could lead to the synthesis of various molecules with complex structures.
7. Conflicts of interest
There are no conflicts to declare.
8. Acknowledgements
This work was supported by JSPS KAKENHI (Grant Numbers JP23H02602 and JP24H01767) and by the Research Support Project for Life Science and Drug Discovery [Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)] from the Japan Agency for Medical Research and Development (AMED) under Grant Number JP24ama121044.
9. Notes and references
For selected review on nitrones, see:
(a) P. N. Confalone and E. M. Huie, in Organic Reactions, 2004, p. 1 Search PubMed;
(b) V. Nair and T. D. Suja, Tetrahedron, 2007, 63, 12247 CrossRefCAS;
(c) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, Chem.–Eur. J., 2009, 15, 7808 CrossRefCASPubMed;
(d) J. M. B. Adam and C. Iain, Curr. Org. Synth., 2010, 7, 312 CrossRef;
(e) M. Berthet, T. Cheviet, G. Dujardin, I. Parrot and J. Martinez, Chem. Rev., 2016, 116, 15235 CrossRefCASPubMed;
(f) A. Padwa and S. Bur, in Advances in Heterocyclic Chemistry, ed. E. F. V. Scriven and C. A. Ramsden, Academic Press, 2016, vol. 119, p. 241 Search PubMed;
(g) L. L. Anderson, Asian J. Org. Chem., 2016, 5, 9 CrossRefCAS;
(h) A. Brandi, F. Cardona, S. Cicchi, F. M. Cordero and A. Goti, in Organic Reactions, 2017, p. 1 Search PubMed;
(i) S.-I. Murahashi and Y. Imada, Chem. Rev., 2019, 119, 4684 CrossRefCASPubMed;
(j) C. Kouklovsky, Vietnam J. Chem., 2020, 58, 20 CrossRefCAS;
(k) S. Thakur, A. Das and T. Das, New J. Chem., 2021, 45, 11420 RSC;
(l) O. Tamura, Chem. Pharm. Bull., 2024, 72, 731 CrossRefCASPubMed;
(m) K. Tanaka, Eur. J. Org Chem., 2024, e202400202 CrossRef.
(a) N. A. LeBel, M. E. Post and J. J. Whang, J. Am. Chem. Soc., 1964, 86, 3759 CrossRefCAS;
(b) W. Oppolzer and K. Keller, Tetrahedron Lett., 1970, 11, 1117 CrossRef.
(a) K. V. Gothelf and K. A. Jørgensen, Chem. Commun., 2000, 1449 RSC;
(b) T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366 CrossRefCASPubMed.
(a) F. Casuscelli, U. Chiacchio, A. Rescifina, R. Romeo, G. Romeo, S. Tommasini and N. Uccella, Tetrahedron, 1995, 51, 2979 CrossRefCAS;
(b) G. Floresta, C. Talotta, C. Gaeta, M. De Rosa, U. Chiacchio, P. Neri and A. Rescifina, J. Org. Chem., 2017, 82, 4631 CrossRefCASPubMed;
(c) P. DeShong, C. M. Dicken, R. R. Staib, A. J. Freyer and S. M. Weinreb, J. Org. Chem., 1982, 47, 4397 CrossRefCAS.
(a) J. Sims and K. N. Houk, J. Am. Chem. Soc., 1973, 95, 5798 Search PubMed;
(b) A. Padwa, L. Fisera, K. F. Koehler, A. Rodriguez and G. S. K. Wong, J. Org. Chem., 1984, 49, 276 CrossRefCAS.
T. Shimizu, M. Ishizaki and N. Nitada, Chem. Pharm. Bull., 2002, 50, 908 Search PubMed.
(a) L. R. Domingo, M. Ríos-Gutiérrez, A. I. Adjieufack, I. M. Ndassa, C. N. Nouhou and J. K. Mbadcam, ChemistrySelect, 2018, 3, 5412 CrossRefCAS;
(b) M. Bakthadoss and M. Mushaf, Org. Biomol. Chem., 2020, 18, 9653 RSC;
(c) A. Mondal, L. R. Domingo and N. Acharjee, J. Phys. Org. Chem., 2024, 37, e4574 CrossRefCAS.
For natural product syntheses using intermolecular cycloaddition of nitrone, see:
(a) T. Ueda, M. Inada, N. Morita and O. Tamura, Heterocycles, 2015, 90, 1179 CrossRefCASPubMed;
(b) T. Hirai, K. Shibata, Y. Niwano, M. Shiozaki, Y. Hashimoto, N. Morita, S. Ban and O. Tamura, Org. Lett., 2017, 19, 6320 CrossRefCASPubMed;
(c) M. Pieczykolan, B. Furman and M. Chmielewski, J. Antibiot., 2017, 70, 781 CrossRefCASPubMed;
(d) C. Wang, Z. Wang, X. Xie, X. Yao, G. Li and L. Zu, Org. Lett., 2017, 19, 1828 CrossRefCASPubMed;
(e) T. Pecchioli, F. Cardona, H.-U. Reissig, R. Zimmer and A. Goti, J. Org. Chem., 2017, 82, 5835 CrossRefCASPubMed;
(f) F. Xue, H. Lu, L. He, W. Li, D. Zhang, X.-Y. Liu and Y. Qin, J. Org. Chem., 2018, 83, 754 CrossRefCASPubMed;
(g) T. Malatinský, B. Otočková, L. Dikošová and R. Fischer, ChemistrySelect, 2019, 4, 4233 CrossRef;
(h) B. D. A. Shennan, P. W. Smith, Y. Ogura and D. J. Dixon, Chem. Sci., 2020, 11, 10354 RSC;
(i) L. Dikošová, B. Otočková, T. Malatinský, J. Doháňošová, M. Kopáčová, A. Ďurinová, L. Smutná, F. Trejtnar and R. Fischer, RSC Adv., 2021, 11, 31621 RSC;
(j) X. Tian, T. Xuan, J. Gao, X. Zhang, T. Liu, F. Luo, R. Pang, P. Shao, Y.-F. Yang and Y. Wang, Nat. Commun., 2024, 15, 6429 CrossRefCASPubMed.
For selected synthetic studies of natural product using intramolecular cycloaddition of nitrone, see:
(a) S. R. Paladugu and R. E. Looper, Tetrahedron Lett., 2015, 56, 6332 CrossRefCASPubMed;
(b) E. Ideue, J. Shimokawa and T. Fukuyama, Org. Lett., 2015, 17, 4964 CrossRefCASPubMed;
(c) J. Rautschek and P. Metz, Heterocycles, 2017, 95, 1106 Search PubMed;
(d) J. S. Cannon, Org. Lett., 2018, 20, 3883 Search PubMed;
(e) R.-J. Zhou, G.-Y. Dai, X.-H. Zhou, M.-J. Zhang, P.-Z. Wu, D. Zhang, H. Song, X.-Y. Liu and Y. Qin, Org. Chem. Front., 2019, 6, 377 Search PubMed;
(f) J. Zhong, H. He and S. Gao, Org. Chem. Front., 2019, 6, 3781 RSC.
For a synthesis of natural product analogs using intramolecular cycloaddition of nitrone, see: M. Yamaguchi, A. Matsuda and S. Ichikawa, Org. Biomol. Chem., 2015, 13, 1187 Search PubMed.
For scalable syntheses using intramolecular cycloaddition of nitrone, see:
(a) S. P. Kolis, M. M. Hansen, E. Arslantas, L. Brändli, J. Buser, A. C. DeBaillie, A. L. Frederick, D. W. Hoard, A. Hollister, D. Huber, T. Kull, R. J. Linder, T. J. Martin, R. N. Richey, A. Stutz, M. Waibel, J. A. Ward and A. Zamfir, Org. Process Res. Dev., 2015, 19, 1203 Search PubMed;
(b) P. Garcia-Losada, A. C. DeBaillie, J. Eugenio de Diego, S. J. Green, M. M. Hansen, C. Jaramillo, M. Johnson, T. Kaoudi, J. Li, P. J. Lindsay-Scott, C. Mateos, D. J. Mergott, J. A. Rincon, R. R. Rothhaar, K. D. Seibert, B. M. Watson, L. L. Winneroski, S. Gangula, D. Jing, H. Sun, L. Zhang and M. O. Frederick, Org. Process Res. Dev., 2020, 24, 306 Search PubMed.
P. P. Painter, R. P. Pemberton, B. M. Wong, K. C. Ho and D. J. Tantillo, J. Org. Chem., 2014, 79, 432 CrossRefCASPubMed.
K. Koyama, Y. Hirasawa, A. E. Nugroho, T. Hosoya, T. C. Hoe, K.-L. Chan and H. Morita, Org. Lett., 2010, 12, 4188 CrossRefCASPubMed.
A. Y. Hong and C. D. Vanderwal, J. Am. Chem. Soc., 2015, 137, 7306 CrossRefCASPubMed.
For the Cope elimination to occur, the oxygen atom of the N-oxide and the α-proton of the ketone must be in a cis-relationship.
B.-X. Zhao, Y. Wang, D.-M. Zhang, X.-J. Huang, L.-L. Bai, Y. Yan, J.-M. Chen, T.-B. Lu, Y.-T. Wang, Q.-W. Zhang and W.-C. Ye, Org. Lett., 2012, 14, 3096 CrossRefCASPubMed.
(a) J. M. E. Hughes and J. L. Gleason, Angew. Chem., Int. Ed., 2017, 56, 10830 CrossRefCASPubMed;
(b) J. M. E. Hughes and J. L. Gleason, Tetrahedron, 2018, 74, 759 CrossRefCAS.
For the syntheses of virosaines A and B reported before 2015, see:
(a) H. Wei, C. Qiao, G. Liu, Z. Yang and C.-c. Li, Angew. Chem., Int. Ed., 2013, 52, 620 CrossRefCASPubMed;
(b) H. Miyatake-Ondozabal, L. M. Bannwart and K. Gademann, Chem. Commun., 2013, 49, 1921 RSC.
(a) E. J. Corey and T.-P. Loh, Tetrahedron Lett., 1993, 34, 3979 CrossRefCAS;
(b) C. S. Schindler, C. R. J. Stephenson and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 8852 Search PubMed;
(c) H. F. Zipfel and E. M. Carreira, Org. Lett., 2014, 16, 2854 CrossRefCASPubMed.
G. E. Keck and J. B. Yates, J. Am. Chem. Soc., 1982, 104, 5829 Search PubMed.
X.-J. Wang, G.-J. Zhang, P.-Y. Zhuang, Y. Zhang, S.-S. Yu, X.-Q. Bao, D. Zhang, Y.-H. Yuan, N.-H. Chen, S.-g. Ma, J. Qu and Y. Li, Org. Lett., 2012, 14, 2614 CrossRefCASPubMed.
H. Shao, K. Fang, Y.-P. Wang, X.-M. Zhang, T.-M. Ding, S.-Y. Zhang, Z.-M. Chen and Y.-Q. Tu, Org. Lett., 2020, 22, 3775 Search PubMed.
(a) A. Hassner, R. Maurya, A. Padwa and W. H. Bullock, J. Org. Chem., 1991, 56, 2775 Search PubMed;
(b) A. Hassner, R. Maurya, O. Friedman, H. E. Gottlieb, A. Padwa and D. Austin, J. Org. Chem., 1993, 58, 4539 Search PubMed.
J. Zhong, K. Chen, Y. Qiu, H. He and S. Gao, Org. Lett., 2019, 21, 3741 CrossRefCASPubMed.
H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, J. Am. Chem. Soc., 2017, 139, 17755 Search PubMed.
H. Mao, P.-M. Wang and J. Xu, Tetrahedron, 2021, 81, 131913 CrossRefCAS.
J.-T. Lu, Y. Zong, X. Yue and J. Wang, J. Org. Chem., 2023, 88, 8761 Search PubMed.
S. Nicolai, C. Piemontesi and J. Waser, Angew. Chem., Int. Ed., 2011, 50, 4680 CrossRefCASPubMed.
T. Miura, Y. Funakoshi and M. Murakami, J. Am. Chem. Soc., 2014, 136, 2272 CrossRefCASPubMed.
(a) Y. Wang, A. Hennig, T. Küttler, C. Hahn, A. Jäger and P. Metz, Org. Lett., 2020, 22, 3145 CrossRefCASPubMed;
(b) T. Erhard, G. Ehrlich and P. Metz, Angew. Chem., Int. Ed., 2011, 50, 3892 CrossRefCASPubMed;
(c) J. Rautschek, A. Jäger and P. Metz, Org. Lett., 2018, 20, 832 CrossRefCASPubMed.
T. Yokoyama, Y. Fukami, T. Sato and N. Chida, Chem.–Asian J., 2016, 11, 470 CrossRefCASPubMed.
F.-X. Wang, J.-Y. Du, H.-B. Wang, P.-L. Zhang, G.-B. Zhang, K.-Y. Yu, X.-Z. Zhang, X.-T. An, Y.-X. Cao and C.-A. Fan, J. Am. Chem. Soc., 2017, 139, 4282 CrossRefCASPubMed.
G.-B. Zhang, F.-X. Wang, J.-Y. Du, H. Qu, X.-Y. Ma, M.-X. Wei, C.-T. Wang, Q. Li and C.-A. Fan, Org. Lett., 2012, 14, 3696 CrossRefCASPubMed.
J. K. Kerkovius and M. A. Kerr, J. Am. Chem. Soc., 2018, 140, 8415 CrossRefCASPubMed.
M. A. Staszak and C. W. Doecke, Tetrahedron Lett., 1993, 34, 7043 CrossRefCAS.
A. M. Beauchemin, Org. Biomol. Chem., 2013, 11, 7039 RSC.
Z. T. I. Alkayar and I. Coldham, Org. Biomol. Chem., 2019, 17, 66 RSC.
J. H. Siitonen, P. V. Kattamuri, M. Yousufuddin and L. Kürti, Org. Lett., 2020, 22, 2486 CrossRefCASPubMed.
S. Hiraoka, T. Matsumoto, K. Matsuzaka, T. Sato and N. Chida, Angew. Chem., Int. Ed., 2019, 58, 4381 Search PubMed.
Y. Sasano, S. Nagasawa, M. Yamazaki, M. Shibuya, J. Park and Y. Iwabuchi, Angew. Chem., Int. Ed., 2014, 53, 3236 Search PubMed.
T. Higo, T. Ukegawa, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2015, 54, 7367 CrossRefCASPubMed.
M. H. Becker, P. Chua, R. Downham, C. J. Douglas, N. K. Garg, S. Hiebert, S. Jaroch, R. T. Matsuoka, J. A. Middleton, F. W. Ng and L. E. Overman, J. Am. Chem. Soc., 2007, 129, 11987 CrossRefCASPubMed.
K. Tomoya, K. Komiya, D. Nakajima, N. Umekubo and S. Yokoshima, Org. Lett., 2023, 25, 2718 Search PubMed.
S. A. Shipilovskikh, A. E. Rubtsov and A. V. Malkov, Org. Lett., 2017, 19, 6760 CrossRefCASPubMed.
B. Yang, G. Li, Q. Wang and J. Zhu, J. Am. Chem. Soc., 2023, 145, 5001 Search PubMed.
(a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672 CrossRefCASPubMed;
(b) S.-G. Wang, X.-J. Liu, Q.-C. Zhao, C. Zheng, S.-B. Wang and S.-L. You, Angew. Chem., Int. Ed., 2015, 54, 14929 Search PubMed.
B. J. Lee, K. S. DeGlopper and T. P. Yoon, Angew. Chem., Int. Ed., 2020, 59, 197 Search PubMed.
Y. Irie and S. Yokoshima, J. Am. Chem. Soc., 2024, 146, 9526 Search PubMed.
(a) S. T. Diver and A. J. Giessert, Chem. Rev., 2004, 104, 1317 CrossRefCASPubMed;
(b) S. Kotha, M. Meshram and A. Tiwari, Chem. Soc. Rev., 2009, 38, 2065 RSC.
S. Murahashi, H. Mitsui, T. Shiota, T. Tsuda and S. Watanabe, J. Org. Chem., 1990, 55, 1736 CrossRefCAS.
(a) S. L. Gómez Ayala, E. Stashenko, A. Palma, A. Bahsas and J. M. Amaro-Luis, Synlett, 2006, 2006, 2275 CrossRef;
(b) S. Gómez-Ayala, J. A. Castrillón, A. Palma, S. M. Leal, P. Escobar and A. Bahsas, Bioorg. Med. Chem., 2010, 18, 4721 Search PubMed;
(c) L. M. Acosta, A. Palma and A. Bahsas, Tetrahedron, 2010, 66, 8392 CrossRefCAS;
(d) L. M. Acosta Quintero, A. Palma, M. Nogueras and J. Cobo, Synthesis, 2012, 44, 3765 CrossRef;
(e) S. A. Guerrero, J. E. Ramírez, C. M. Sanabria, L. M. Acosta, J. Cobo, M. Nogueras and A. Palma, Synthesis, 2020, 53, 1315 Search PubMed;
(f) Y. Li, J. S. Ng, B. Wang and S. Chiba, Org. Lett., 2021, 23, 5060 Search PubMed.
H. Mitsui, S.-i. Zenki, T. Shiota and S.-I. Murahashi, J. Chem. Soc., Chem. Commun., 1984, 874 RSC.
(a) Y. Shvo, D. Czarkie, Y. Rahamim and D. F. Chodosh, J. Am. Chem. Soc., 1986, 108, 7400 CrossRefCAS;
(b) B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294 CrossRefCASPubMed.
For selected examples of C-linked unsaturated nitrones, see:
(a) J. R. Hwu and J. A. Robl, J. Chem. Soc., Chem. Commun., 1986, 704 RSC;
(b) K. Vanhessche, C. G. Bello and M. Vandewalle, Synlett, 1991, 1991, 921 Search PubMed;
(c) S. Saito, T. Ishikawa and T. Moriwake, J. Org. Chem., 1994, 59, 4375 Search PubMed;
(d) N. A. Lebel and N. Balasubramanian, Tetrahedron Lett., 1985, 26, 4331 Search PubMed;
(e) A. Arnone, G. Broggini, D. Passarella, A. Terraneo and G. Zecchi, J. Org. Chem., 1998, 63, 9279 CrossRefCAS;
(f) E. M. Beccalli, G. Broggini, C. La Rosa, D. Passarella, T. Pilati, A. Terraneo and G. Zecchi, J. Org. Chem., 2000, 65, 8924 Search PubMed;
(g) Q. Cheng, W. Zhang, Y. Tagami and T. Oritani, J. Chem. Soc., Perkin Trans. 1, 2001, 452 RSC;
(h) D. Basso, G. Broggini, D. Passarella, T. Pilati, A. Terraneo and G. Zecchi, Tetrahedron, 2002, 58, 4445 Search PubMed;
(i) J. Marcus, J. Brussee and A. van der Gen, Eur. J. Org Chem., 1998, 1998, 2513 Search PubMed;
(j) Y. Liu, A. Maden and W.
V. Murray, Tetrahedron, 2002, 58, 3159 CrossRefCAS;
(k) T. K. M. Shing, W. F. Wong, T. Ikeno and T. Yamada, Org. Lett., 2007, 9, 207 CrossRefCASPubMed;
(l) M. Würdemann and J. Christoffers, Synlett, 2011, 2841 Search PubMed;
(m) F. Rabasa-Alcañiz, A. Asensio, M. Sánchez-Roselló, M. Escolano, C. del Pozo and S. Fustero, J. Org. Chem., 2017, 82, 2505 Search PubMed;
(n) T. Tanino, M. Yamaguchi, A. Matsuda and S. Ichikawa, Eur. J. Org Chem., 2014, 2014, 1836 CrossRefCAS.
(a) W. Oppolzer, S. Siles, R. L. Snowden, B. H. Bakker and M. Petrzilka, Tetrahedron, 1985, 41, 3497 CrossRefCAS;
(b) A. Budzińska and W. Sas, Tetrahedron, 2001, 57, 2021 CrossRef;
(c) N. Saha and S. K. Chattopadhyay, J. Org. Chem., 2012, 77, 11056 CrossRefCASPubMed.
(a) H. A. Dondas, M. Frederickson, R. Grigg, J. Markandu and M. Thornton-Pett, Tetrahedron, 1997, 53, 14339 Search PubMed;
(b) Z. Xiang, Comput. Theor. Chem., 2012, 992, 128 CrossRefCAS.
The transition state 230, where a trans-fused isoxazolidine is produced from a trans-nitrone, is also not preferred.
A. J. M. Burrell, I. Coldham and N. Oram, Org. Lett., 2009, 11, 1515 Search PubMed.
R. Saruengkhanphasit, D. Collier and I. Coldham, J. Org. Chem., 2017, 82, 6489 Search PubMed.
(a) R. E. Looper and R. M. Williams, Tetrahedron Lett., 2001, 42, 769 Search PubMed;
(b) R. E. Looper, M. T. C. Runnegar and R. M. Williams, Tetrahedron, 2006, 62, 4549 Search PubMed.
Reduction of N-hydroxyamide and transformation of nitro compounds have also been reported for the synthesis of nitrones
(a) S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, Bull. Chem. Soc. Jpn., 2017, 90, 893 Search PubMed;
(b) S. Katahara, S. Kobayashi, K. Fujita, T. Matsumoto, T. Sato and N. Chida, J. Am. Chem. Soc., 2016, 138, 5246 CrossRefCASPubMed;
(c) G. Bartoli, E. Marcantoni, M. Petrini and R. Dalpozzo, J. Org. Chem., 1990, 55, 4456 Search PubMed;
(d) H. Shimizu, K. Yoshinaga and S. Yokoshima, Org. Lett., 2021, 23, 2704 CrossRefCASPubMed.
For selected examples on oxidative cleavage of isoxazolidines, see:
(a) J. J. Tufariello, G. B. Mullen, J. J. Tegeler, E. J. Trybulski, S. C. Wong and S. A. Ali, J. Am. Chem. Soc., 1979, 101, 2435 CrossRefCAS;
(b) J. J. Tufariello and J. M. Puglis, Tetrahedron Lett., 1986, 27, 1489 CrossRefCAS;
(c) T. Ishiwata, T. Hino, H. Koshino, Y. Hashimoto, T. Nakata and K. Nagasawa, Org. Lett., 2002, 4, 2921 Search PubMed;
(d) K. Nagasawa, T. Ishiwata, Y. Hashimoto and T. Nakata, Tetrahedron Lett., 2002, 43, 6383 CrossRefCAS;
(e) S.-H. Yang and V. Caprio, Synlett, 2007, 1219 CAS;
(f) S.-H. Yang, G. R. Clark and V. Caprio, Org. Biomol. Chem., 2009, 7, 2981 RSC;
(g) Y. Fukahori, Y. Takayama, T. Imaoka, O. Iwamoto and K. Nagasawa, Chem.–Asian J., 2013, 8, 244 Search PubMed.

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*






![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: