
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent insights into the biosynthesis and biological activities of the peptide-derived redox cofactor mycofactocin†
Mark Ellerhorst
 a,
Vadim Nikitushkin
a,
Walid K. Al-Jammal
b,
Lucas Gregor
b,
Ivan Vilotijević
b and
Gerald Lackner
*a
a,
Vadim Nikitushkin
a,
Walid K. Al-Jammal
b,
Lucas Gregor
b,
Ivan Vilotijević
b and
Gerald Lackner
*a
aChair of Biochemistry of Microorganisms, University of Bayreuth, Germany. E-mail: gerald.lackner@uni-bayreuth.de
bInstitute for Organic Chemistry and Macromolecular Chemistry, Friedrich Schiller University Jena, Germany
First published on 16th May 2025
Abstract
Covering: 2011 to 2025
The importance of redox cofactors like nicotinamide adenine dinucleotide or flavin adenine dinucleotide as cofactors for enzymatic reactions in living organisms is widely known. However, many microbial species also employ unusual redox cofactors such as the coenzyme F420 or the peptide-derived pyrroloquinoline quinone (PQQ). In this review, we introduce the reader to the recently discovered bacterial redox cofactor mycofactocin (MFT), a valine-tyrosine-derived small molecule of the class of ribosomally synthesized and post-translationally modified peptides (RiPPs) with remarkable biosynthetic and functional similarities to PQQ. The cofactor plays an important role in the reoxidation of non-exchangeable nicotinamide redox cofactors of specialized oxidoreductases in mycobacteria and related actinobacteria. We highlight the bioinformatic discovery of the mycofactocin gene cluster and its auxiliary genes, present strategies for the chemical synthesis of the cofactor, and take a detailed look at the biosynthesis of the glycosylated molecule. Subsequently, the diverse mycofactocin-inducing conditions and associated oxidoreductase families are reviewed, and a potential electron transfer route from high-energy alcohols via mycofactocin to oxygen as a final electron acceptor is presented. The review concludes with a comparison of the physiological roles of PQQ and MFT, and an outlook for future research questions and potential biotechnological applications of mycofactocin.
 Mark Ellerhorst | Mark Ellerhorst obtained his BSc and MSc degrees from the Friedrich-Schiller University of Jena in 2018 and 2020, respectively, and worked during his master's thesis on the role of mycofactocin in the antibiotic-producing bacterium Saccharopolyspora erythraea in the group of Gerald Lackner at the Leibniz Institute for Natural Product Research and Infection Biology. Subsequently, he began his PhD work in the same group, focusing on the elucidation of mycofactocin methylation and its physiological role in Mycolicibacterium smegmatis and other Actinobacteria. In 2024 he moved to the University of Bayreuth, Campus Kulmbach, where he continues his PhD project. |
 Vadim Nikitushkin | Vadim Nikitushkin obtained his PhD in the Russian Academy of Sciences (A. N. Bach Institute of Biochemisty). During doctoral studies he investigated molecular mechanisms of reactivation of dormant mycobacteria under the influence of mycobacterial peptidoglycan cleavage products (muropeptides). Applying bioanalytical methods, he revealed the structures of porphyrin molecules accumulated by dormant forms of mycobacteria. In 2022, he joined Prof. Dr Gerald Lackner's group at the Leibniz Institute for Natural Product Research and Infection Biology in Jena, where he worked on mycobacteria under different physiological conditions and stress factors. Current interests comprise application of omics-based approaches for the investigation of cofactor metabolism. |
 Walid K. Al-Jammal | Walid Al-Jammal earned his BSc and MSc in Chemistry from Jordan University of Science and Technology, Jordan where he focused on the regioselective synthesis of iodinated aromatics via Suzuki–Miyaura cross-coupling. In 2019, he began his PhD with Prof. Dr Ivan Vilotijević at Friedrich Schiller University Jena, working on the total synthesis of mycofactocins and paleofuran A, metal-carbenoid reactions for the synthesis of C–P, C–B, and C–Si compounds, and the development of asymmetric phosphine catalysis. As a postdoctoral researcher in the group of Prof. Dr Ivan Vilotijević, he is currently developing modified lipopeptide mediators to study polymicrobial interaction. |
 Lucas Gregor | Lucas Gregor earned his BSc and MSc degrees in chemistry from the Friedrich Schiller University Jena in 2021 and 2023, respectively. He completed his bachelor's thesis in the group of Prof. Dr Arndt and his master's thesis on the total synthesis of mycofactocins in the group of Prof. Dr Vilotijević. Currently pursuing his PhD in the same group, he continues working on the synthesis of mycofactocins and explores novel strategies for regioselective radical functionalizations through hydrogen atom transfer (HAT). |
 Ivan Vilotijević | Ivan Vilotijević studied chemistry at University of Belgrade, Ohio State University and University of Illinois Urbana-Champaign. He earned a PhD in organic chemistry from Massachusetts Institute of Technology working on epoxide-opening cascade reactions aimed at synthesis of ladder polyether natural products. After a postdoctoral appointment at Max Planck Institute of Colloids and Interfaces working on synthesis of glycosylphosphatidylinositols and GPI-anchored proteins, he joined Friedrich Schiller University Jena where he currently serves as a professor for organic synthesis methods. His team develops novel catalytic methods for synthesis of complex organic molecules and pursues target-oriented synthesis of bioactive molecules and biological probes. |
 Gerald Lackner | Gerald Lackner studied biochemistry at Friedrich Schiller University in Jena, Germany. He earned his PhD in 2011 at the Leibniz Institute for Natural Product Research and Infection Biology (Leibniz-HKI). As a Feodor Lynen fellow at ETH Zurich, his research centered on unculturable bacteria producing bioactive compounds. In 2016, he became junior group leader at the Leibniz-HKI focusing on the biosynthesis of the cofactor mycofactocin and its biological activities. In 2023, he was appointed as a full professor for Biochemistry of Microorganisms at the University of Bayreuth based at the Campus Kulmbach, where he continues his work on bioactive natural products. |
1. Peptide-derived redox cofactors
Life, as we know it, is impossible without enzymes that catalyse specific metabolic reactions. An important class of enzymes are oxidoreductases which facilitate a broad range of redox reactions both in dissimilatory as well as in assimilatory metabolic pathways. The limited chemical variety of amino acids in oxidoreductases necessitates the employment of redox-active cofactors to perform redox catalysis and electron transport. Typical redox cofactors are metals (especially iron, manganese, nickel and copper),1 iron-sulphur clusters,2 or small organic molecules like flavins or quinones.3,4 Organic cofactors exhibit a remarkable structural diversity,4 and the metabolic pathways leading to their formation are equally varied. Yet, a notable subset of cofactors shares a common biosynthetic origin, as they are derived from peptides. The most studied example of a peptide-derived redox cofactor is pyrroloquinoline quinone (PQQ), which was initially associated with the activity of glucose dehydrogenases5 and methanol dehydrogenases.6–8 PQQ is a quinone cofactor that non-covalently binds to the active site of a corresponding dehydrogenase. These enzymes, usually referred to as quinoproteins, are present in the periplasmic space of predominantly Gram-negative bacteria and perform oxidations of an alcohol substrate to its corresponding carbonyl product with the concomitant formation of the reduced PQQH2 cofactor (Fig. 1).10–14 | ||
| Fig. 1 Schematic visualisation of PQQ-dependent alcohol oxidation. As an example, a dehydrogenase complex from Gluconobacter oxydans is shown (PDB: 8GY2), consisting of three subunits: a small cofactorless subunit (green), participating in membrane binding, a large subunit (yellow), containing the PQQ cofactor (blue) and a heme c (red) binding site, where alcohol oxidation occurs. The third subunit (red) is the cytochrome c subunit, binding three heme c molecules, participating in electron transfer to the ubiquinone (Q/QH2) pool (purple). The reduced quinone pool is regenerated by the ubiquinol oxidase9 (AlphaFold 2 structure of UniProt: Q5FSK4). Source of “membrane-2d-bluelight” graphic: Servier (https://smart.servier.com/) licensed under CC-BY 3.0 Unported (https://creativecommons.org/licenses/by/3.0/). | ||
1.1 The biosynthesis of PQQ
The elucidation of the biosynthesis of PQQ demonstrated that it follows the logic of ribosomally synthesized and post-translationally modified peptides (RiPPs, Fig. 2).15 The biosynthesis of a RiPP starts with the translation of a short precursor peptide (Fig. 2), typically consisting of a leader and a core region and, in some cases, a follower peptide located at the C-terminus. Maturation enzymes install post-translational modifications at the core region. Afterwards, a dedicated peptidase typically excises the modified core peptide from the precursor peptide backbone, resulting in the liberation of the modified core, i.e. the final RiPP natural product, leaving the leader and follower peptides as byproducts. In case of PQQ, the biosynthesis is accomplished by the gene products of the PQQ biosynthesis genes pqqA-F/G, which can be encoded as a biosynthetic gene cluster (BGC) (Fig. 3A).11,16 PqqA is the precursor peptide, which harbours a core region located between the leader and a short follower peptide, framed by Glu and Tyr residues (Fig. 3B). A crucial step in the PQQ biosynthesis is the crosslinking of the C9–C9a atoms of the Glu and Tyr residues within the PqqA core region (Fig. 3C).11,16 This challenging reaction is performed by the radical S-adenosyl-L-methionine (rSAM) enzyme PqqE.16,18 These enzymes coordinate [4Fe–4S]/[2Fe–2S] clusters and are capable of cleaving SAM to methionine and a 5′-deoxyadenosine radical.18,19 However, for the successful cross-linking of the Glu and Tyr residues another protein is required, namely PqqD – a chaperone or RiPP recognition element (RRE), which was shown to form a trimeric complex with PqqA and PqqE (Fig. 3C).16,20–23 To generate a free PQQ precursor molecule, the cross-linked Tyr–Glu core needs to be excised from the modified PqqA precursor peptide. This excision reaction is performed by the PqqF/G heterodimer if it is encoded in a particular genome.24 Alternatively, the excision is achieved by non-specific cell-associated proteases.24 The liberated diamino acid precursor is further oxidized by a specific iron-dependent hydroxylase PqqB25 and spontaneously undergoes sequential condensation to the bicyclic intermediate AHQQ (3a-(2-amino-2-carboxyethyl)-4,5-dioxo-4,5,6,7,8,9-hexahydroquinoline-7,9-dicarboxylic acid).16,25 The final oxidative steps are catalysed by PqqC – a cofactorless oxidase that performs an oxidation of AHQQ to PQQ, consuming three oxygen equivalents.25,26 The resulting PQQ molecule (Fig. 3C) serves as a redox cofactor in PQQ-dependent dehydrogenase reactions as shown in Fig. 1.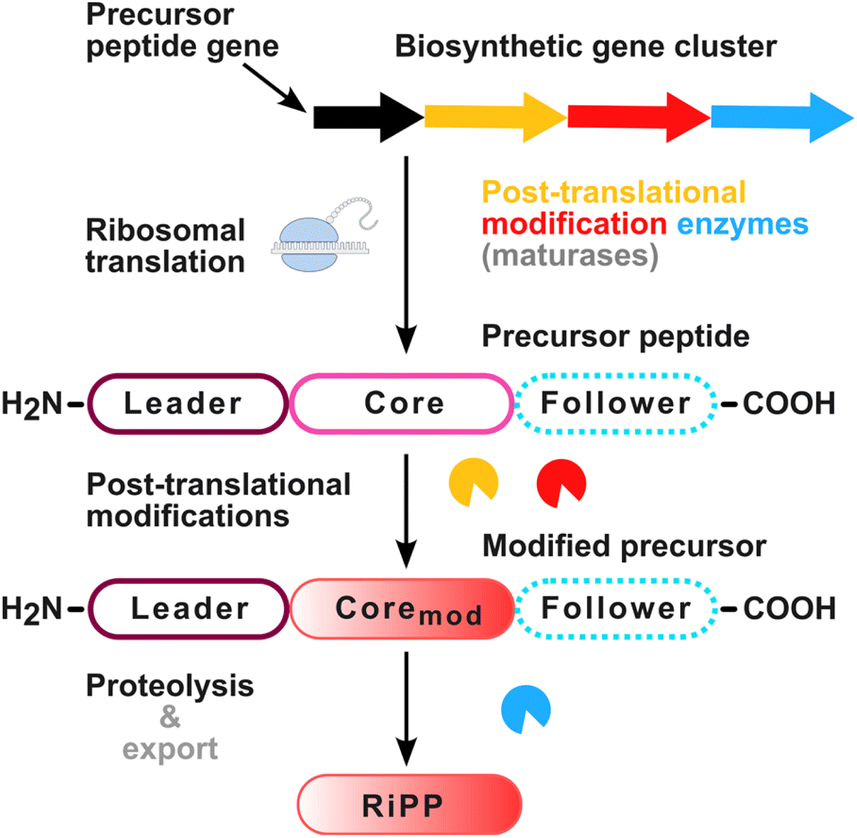 | ||
| Fig. 2 General biosynthetic scheme of ribosomally synthesized and post-translationally modified peptides (RiPPs). The precursor peptide gene, e.g. pqqA, is transcribed and ribosomally translated. It usually contains an N-terminal leader sequence, a core region, and in some cases a follower peptide at the C-terminus of the core region. Maturases, e.g. PqqB-G, often encoded clustered with the precursor gene in a biosynthetic gene cluster, post-translationally modify the precursor peptide in its core region. Proteases then release the modified core from the peptide, which forms the final RiPP. Source of ribosome graphic: NIAID Visual & Medical Arts. (07/10/2024, https://bioart.niaid.nih.gov/bioart/448). | ||
 | ||
| Fig. 3 The PQQ biosynthetic gene cluster and PQQ biosynthesis. (A) Biosynthetic gene clusters of PQQ in different organisms. (B) Sequence alignment of the PqqA peptide shows conserved glutamate (E) and tyrosine (Y) residues essential for PQQ core formation. Alignment generated in Geneious Prime 2024.0.7 (https://www.geneious.com). (C) PQQ biosynthesis. PqqA, the PQQ precursor peptide, is bound by the PqqDE complex consisting of the chaperone PqqD and the radical SAM enzyme PqqE. The PqqF/G heterodimer promotes liberation of the core peptide but seems to be non-essential for the PQQ biosynthesis in some organisms. The metallolactamase PqqB and the oxidase PqqC perform sequential oxidation and cyclisation.16 Visualisation of the PqqADE complex was achieved with AlphaFold 2. The model with the highest parameter values for pLDDT, pTM and iPTM was selected.17 Wavy lines indicate peptide bond connections to the PqqA backbone peptide. SAM – S-adenosyl-L-methionine, 5′-dAdo – 5′-deoxyadenosine, Met – methionine. | ||
1.2 Enzyme backbone-derived quinone cofactors
Besides PQQ, a couple of further peptide-derived quinone cofactors have been discovered, namely trihydroxyphenylalanine quinone (TPQ),27 tryptophan tryptophylquinone (TTQ),28 lysyl tyrosine quinone (LTQ),29 and cysteine tryptophyl quinone (CTQ).30 A commonality among this family of quinone cofactors is the extensive modification of tryptophan and tyrosine aromatic rings of the protein backbone resulting in corresponding quinones, followed by the formation of new C–N, C–S or C–C bonds (in case of TTQ, LTQ and CTQ).11 It should be noted that these cofactors in contrast to PQQ are formed in situ on the polypeptide chain of the apoenzyme, and therefore can be attributed to the group of protein-derived cofactors, whereas PQQ originates from its own precursor peptide, not from its cognate dehydrogenase.A more recently discovered peptide-derived redox cofactor, mycofactocin (MFT), is the focus of this review. Mycofactocin can be regarded as an analog to PQQ, sharing a striking number of similarities. Although their biosynthetic gene clusters are not directly related, both PQQ and mycofactocin are products of RiPP pathways. Their biosyntheses involve rSAM-catalysed cyclization reactions as a first step and ultimately produce redox-active cofactors. In the following sections, we will recapitulate the discovery of the cofactor and explore recent advances concerning the structure elucidation, biosynthesis, and physiological roles of mycofactocin. We will finish with an outlook into the possible medical and biotechnological applications of the mycofactocin system.
2. Discovery of mycofactocin
The mycofactocin biosynthesis genes came into focus as a result of a comparative genomics study, aimed at the identification of potential novel cofactors.31 To this end, an approach developed during a previous genomic profiling study that linked coenzyme F420 biosynthesis genes to genes encoding F420-dependent enzymes was exploited as a strategy to discover unknown cofactors.32 In particular, it was speculated that cofactor biosynthesis might involve radical S-adenosyl methionine (rSAM) domain-containing enzymes as known from PqqE (PQQ biosynthesis) or FbiC (coenzyme F420 biosynthesis). Furthermore, it was assumed that the presence of this rSAM gene should be tightly linked, i.e. co-occur genome-wide, with further biosynthetic genes and that the occurrence of these biosynthesis genes should correlate with gene families encoding putative cofactor-dependent enzymes. Using this Partial Phylogenetic Profiling strategy, the study revealed a gene encoding an rSAM enzyme belonging to the IPR023913 family (MftC, Table 1).31 The gene strictly clustered locally with several other putative biosynthetic genes, one of which appeared to be a short open reading frame encoding for a putative precursor peptide with a conserved Val–Tyr carboxy terminus (MftA, IPR023988). As expected for a redox cofactor, its BGC was found to have a significant tendency to co-occur with oxidoreductases, in this case with members of three oxidoreductase families (IPR023985, IPR023921, IPR001670; Table 1). The finding that the presence of these oxidoreductases strictly depends on the presence of biosynthetic genes strongly supported the idea that they might require the product of the putative BGC as a cofactor. The identified BGC mainly occurred in mycobacteria, including the causative agent of tuberculosis, Mycobacterium tuberculosis.31 The predicted product of the BGC was named mycofactocin, a portmanteau of mycobacteria, PQQ cofactor and bacteriocins, with which it presumably shared a common function and biosynthetic logic, respectively.31| Description | In vitro studied examples | TIGRFAM/PFAM/NCBIFam | InterPro database entry |
|---|---|---|---|
| a Full protein sequence not available. The list of in vitro studied examples contains only proteins studied in isolated form and is non-exhaustive due to lack of sequence data for some studied proteins. | |||
| MftA – mycofactocin precursor peptide | P0DUE9 (Mycobacterium ulcerans Agy99)36 | TIGR03969 | IPR023988 |
| MftB – mycofactocin chaperone | A0PM48 (M. ulcerans Agy99)36 | TIGR03967 | IPR023850 |
| MftC – mycofactocin rSAM maturase | A0PM49 (M. ulcerans Agy99)36 | TIGR03962 | IPR023913 |
| MftD – pre-mycofactocin synthase | A0PM50 (M. ulcerans Agy99)37 | TIGR03966 | IPR023989 |
| MftE – mycofactocin precursor peptide peptidase | A0PM51 (M. ulcerans Agy99)38 | TIGR03964 | IPR023871 |
| MftF – mycofactocin glycosyltransferase | — | TIGR03965 | IPR023981 |
| MftM – mycofactocin oligosaccharide methyltransferase | — | NF041255 | — |
| MftR – mycofactocin transcriptional regulator | A0QSB5 (M. smegmatis mc2 155)39 | TIGR03968 | IPR023851 |
| P9WMB7 (M. tuberculosis H37Rv)40 | |||
| MftG – glucose-methanol-choline oxidoreductase, bacteria | A0QSC2 (M. smegmatis mc2 155)41 | TIGR03970 | IPR023978 |
| NF038210 (restricted to genus Dietzia) | |||
| TIGR04542 (restricted to genus Gordonia) | |||
| Mycofactocin-dependent oxidoreductase/SDR_subfam_1 | A0QSA5 (M. smegmatis mc2 155)37 | TIGR03971 | IPR023985 |
| B1MLR7 (PDB: 3S55, Mycobacteroides abcessus ATCC 19977)42 | |||
| A0A0H2ZWY3 (PDB: 3TC7, Mycobacterium avium 104)42 | |||
| A0A0H2ZTN5 (PDB: 3UVE, M. avium 104)42 | |||
| A0A0H2ZU39 (PDB: 4RGB, M. avium 104)42 | |||
| A0A0M3KKT7 (PDB: 3PXX, M. avium 104)42 | |||
| A0A0H2ZYS9 (PDB: 5EJ2, M. avium 104)42 | |||
| Q73SC8 (PDB: 3PGX, Mycolicibacterium paratuberculosis K-10)42 | |||
| Q73W00 (PDB: 3SX2, M. paratuberculosis K-10)42 | |||
| Q73X99 (PDB: 3TSC, M. paratuberculosis K-10)42 | |||
| Q9RA05 (Rhodococcus erythropolis DCL14)43 | |||
| Q53062 (R. erythropolis NI86/21)44 | |||
| Alcohol dehydrogenase, zinc-type, ADH_Zn_actinomycetes | P80175 (Amycolatopsis methanolica 239)45,46 | TIGR03989 | IPR023921 |
| P81747a (Rhodococcus opacus DSM 1069)47 | |||
| NDMA-dependent methanol dehydrogenase, NDMA_methanol_DH | Q9RCG0 (A. methanolica 239)48,49 | TIGR04266 | IPR026338 |
| C5MRT8 (Mycobacterium sp. JC1)50 | |||
| A0R5M3 (M. smegmatis mc2 155)51 | |||
| Q53062 (R. erythropolis NI86/21)44 | |||
| A5LHA1 (R. erythropolis N9T-4)52 | |||
| Alcohol dehydrogenase, iron-type, ADH_Fe | Q9RCG0 (A. methanolica 239)48,49 | PF00465 | IPR001670 |
| C5MRT8 (Mycobacterium sp. JC1)50 | |||
| A0R5M3 (M. smegmatis mc2 155)51 | |||
| Q53062 (R. erythropolis NI86/21)44 | |||
| A5LHA1 (R. erythropolis N9T-4)52 | |||
| CHP03996, oxidoreductase/OYE_1 | — | TIGR03996 | IPR023967 |
| CHP03977, oxidoreductase/OYE_2 | — | TIGR03977 | IPR023987 |
| Mycofactocin-dependent oxidoreductase/SDR_subfam_2 | — | TIGR04504 | IPR030981 |
| Mycofactocin-dependent oxidoreductase/SDR_subfam_3 | — | NF040490 | — |
| Mycofactocin-dependent oxidoreductase/SDR_subfam_4 | — | NF040491 | — |
3. Structure and chemical synthesis of mycofactocin
The term mycofactocin designates a group of closely related compounds consisting of a peptide-derived aglycone, i.e. a redox-active core, and an oligosaccharide. The structure of mycofactocin mMFT-8, shown in Fig. 4A, is illustrative of the general structure of mycofactocins where the redox core structure is non-variable while the attached oligosaccharide varies in length and modifications with numerous different glycoforms shown to exist in vivo.53 A table of known and hypothetical mycofactocin structures including their SMILES, sum formulas, and theoretical monoisotopic mass is provided in the ESI (ESI, mycofactocin_SMILES.xlsx).† | ||
| Fig. 4 Structural diversity and nomenclature of mycofactocins. (A) Structure of mycofactocin mMFT-8 in its oxidized form depicting the redox active core structure and the variable oligosaccharide. (B) Mycofactocin-related aglycones. (C) Structures of glycosylated mycofactocins from M. smegmatis extracts: (m)MFT-n(H2): “m” indicates the presence of methyl ether on the second glucose unit; “n” indicates the number of glucose residues in β-1,4-glucan oligosaccharide with n = 0 to 9; “H2” indicates the reduced form of the α-keto-γ-butyrolactam redox core. Stereochemical assignment for aglycones and hypothesized biosynthetic precursors is unknown, but theoretical stereogenic centres have been marked with asterisks for clarity. | ||
The redox-active aglycone core structure, MFT-0, is a highly substituted α-keto-γ-butyrolactam carrying two geminal methyl substituents at the C4 position and a 4-hydroxybenzyl substituent at C5 (Fig. 4B). The reduced aglycone, MFT-0H2, formerly also named premycofactocinol PMFTH2, where H2 indicates the reduced form, features an alcohol instead of the ketone in C3 position of the γ-butyrolactam. Structurally, these aglycones appear to be derivatives of a Tyr–Val dipeptide that has undergone decarboxylation and cyclization by bond formation between the C3 of valine and C2 of tyrosine which creates the C4–C5 bond of the resulting γ-butyrolactam core structure. The α-amino function apparently is further converted either by substitution to form the alcohol in MFT-0H2 or by oxidation to form the ketone in MFT-0. This hypothesis is further corroborated by identification of the related aglycons AHDP-0 and GAHDP-0 that feature an α-amino or α-amido instead of α-keto or α-hydroxy substituent in the γ-butyrolactam core (Fig. 4B). Absolute stereochemical assignment of natural MFT-0 as well as the relative stereochemistry of MFT-0H2, AHDP and GAHDP remain unknown.
The phenol oxygen in both MFT-0 and MFT-0H2 can carry a β-1,4-glucan consisting of up to nine glycosyl residues resulting in mycofactocins MFT-n and MFT-nH2 respectively, where “n” indicates the number of glucose residues in the oligosaccharide in a range from 1 to 9. Both the oxidized (MFT-n) and reduced (MFT-nH2) glycosylated forms (Fig. 4C) have been identified in natural extracts.53,54 Further derivatives that have been isolated and characterized carry an O-methyl ether in C2 position of the second glucose unit, counting from the core structure. The methylation results in mMFT-n and mMFT-nH2, where “m” indicates the presence of this methyl ether (Fig. 4C).53 The general term used for oxidized mycofactocins is mycofactocinones and that for reduced mycofactocins is mycofactocinols, following the nomenclature of quinones and quinols as used, for example, for PQQ.
Structural assignments of both aglycones and different glycoforms has been established based on extensive tandem MS and NMR studies of material isolated from Mycolicibacterium smegmatis mc2 155. Confirmation of the identities of MFT-0 and MFT-0H2 was provided by chemical synthesis by the teams of Latham and Michel,55 and of Vilotijević and Lackner.54 The reported retrosynthetic approach to MFT-0 and MFT-0H2 (Scheme 1A) is based on late stage α-oxidation of the γ-butyrolactam 1 which was envisioned from the nitro ester 2 via reduction of the nitro group and cyclization via amid formation. The required precursor 2 could be prepared via Michael addition of the anion derived from the nitroalkane 4 to the acrylate 3.
 | ||
| Scheme 1 A) Retrosynthetic analysis of mycofactocins MFT-0 and MFT-0H2 (protecting groups are omitted). (B) Synthesis of mycofactocins MFT-0H2 and MFT-0. (a) Methyl-3,3-dimethyl acrylate 3, TBAF, THF, 40 °C, 80 h, 47%; (b) H2, Raney-Ni, MeOH, 60 °C, 12 h, 7a: 51%, 7b: 34%; (c) H2, Pd/C, MeOH, rt, 12 h, 96%; (d) Boc2O, Et3N, DMAP, CH2Cl2, rt, 12 h, 73%; (e) Davis reagent, LDA, THF, −78 °C to rt, 4 h, 38%; (f) BBr3, CH2Cl2, 0 °C, 2 h, 30%; (g) (COCl)2, DMSO, Et3N, CH2Cl2, −78 °C, 2 h, 72%; (h) BBr3, CH2Cl2, 0 °C, 2 h, 21%. | ||
The synthesis of MFT-0 and MFT-0H2 is described in Scheme 1B.54 The suitable nitroalkane 5 is available in two steps via Henry reaction of the corresponding benzaldehyde and nitromethane followed by hydrogenation of the alkene. Nitroalkane 5 undergoes Michael addition to methyl 3,3-dimethylacrylate 3 promoted by tetrabutylammonium fluoride. Subsequent reduction of the nitro group in 6 to amine triggers a cyclization resulting in the γ-butyrolactam 7a and the corresponding enamide 7b which is easily reduced to 7a by hydrogenation in the presence of palladium on carbon. Boc protection followed by Davis' oxidation completes the α-hydroxy-γ-butyrolactam core of the protected MFT-0H2 8. MFT-0H2 was obtained through removal of Boc and methyl protecting groups by treatment with boron tribromide. Oxidation of alcohol 8 to the corresponding ketone followed by deprotection yields MFT-0. Comparison of the synthetic material to material isolated from natural sources confirmed the identities of MFT-0H2 and MFT-0. It should be mentioned, however, that the core molecules MFT-0 and MFT-0H2 as well as ADHP-0 have not yet been observed in bacterial extracts, but only appear after cellulase treatment of glycosylated mycofactocins, thereby liberating the core structures (Fig. 4B). Previous evidence for the existence of the core aglycone structures in bacterial extracts appears to be a mass spectrometry in-source fragment artifact of glycosylated mycofactocin.53,54 Future research will have to focus on the investigation of the relative and absolute configuration of mycofactocin and their potential effects on biologically-relevant catalytic activities.
4. Biosynthesis of mycofactocin
4.1 The biosynthetic pathway of mycofactocin
Experimental and bioinformatic efforts revealed that the mycofactocin BGC comprises the genes mftABCDEF and in approximately thirty percent of MFT BGC-encoding bacteria also the gene mftM (Fig. 5A).54 The MFT BGC can be found primarily in mycobacteria but also other actinobacteria and even archaea. For a detailed description of the phylogenetic distribution of this gene cluster, we refer the reader to the first review published on mycofactocin by Ayikpoe et al.57 Fig. 7 depicts the current hypothesis of mycofactocin biosynthesis catalysed by the gene products of the MFT BGC mftABCDEF(M). The order of the biosynthetic steps, however, is ambiguous with currently available data. Thus, multiple scenarios will be discussed in this chapter along with the individual functions of the MFT BGC genes, starting with the hypothetical pathway that would result in the redox active core structure MFT-0.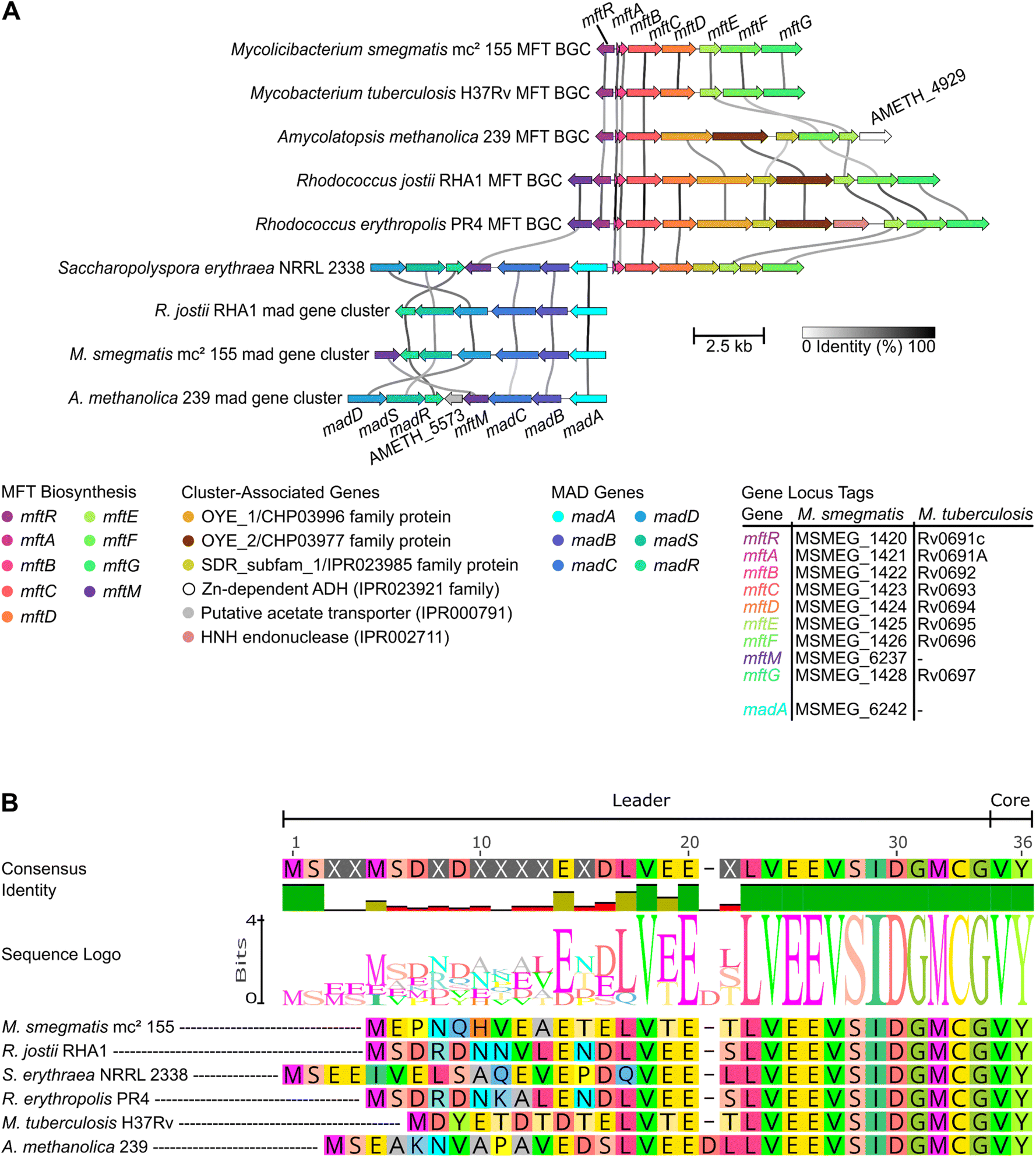 | ||
| Fig. 5 The genetic landscape of mycofactocin and the conservation of MftA. (A) The mycofactocin-dependent alcohol degradation (mad) gene cluster can be but is not necessarily encoded adjacent to the mycofactocin BGC. Species encoding a gene cluster for methylated mycofactocin, i.e. encoding an mftM homolog aside the regular mftABCDEF gene cluster, also encode madA.54 Cluster-associated genes are not present in every MFT or MAD locus. Line darkness indicates gene nucleic acid sequence similarity. Arrow length represents gene length. Colours highlight homologous genes. Plot created with clinker.56 (B) Shown is a multiple sequence alignment of different MftA highlighting the strong conservation of the C-terminal VY-core, the precursor structure of mycofactocin. Alignment generated in Geneious Prime 2024.0.7 (https://www.geneious.com). | ||
In vitro studies showed that MftA is first bound by MftB, its chaperone or RRE. Afterwards, it is modified by the [4Fe–4S]-binding enzyme MftC, the rSAM enzyme that led to the discovery of the mycofactocin BGC. MftC catalyses a two-step oxidative decarboxylation and cyclization of the highly conserved C-terminal VY core peptide of MftA (Fig. 5B) which results in the formation of MftA*, containing a valine–tyrosine crosslink (Fig. 6).36 The modified core is cleaved off of MftA* by the mycofactocin peptidase MftE between the C-terminal glycyl and (modified) valyl residues,58 forming AHDP, i.e. AHDP-0 since no glucose moieties are attached (Fig. 7).38 AHDP, which carries a free amine originating from valine, can subsequently undergo oxidative deamination by the flavin mononucleotide (FMN)-dependent deaminase MftD to produce the redox active MFT core structure with a redox potential (midpoint potential) of −255 mV vs. a standard hydrogen electrode (SHE) determined for MFT-0 via cyclic voltammetry.37 This AHDP-0-derived redox pair is called MFT-0 (oxidized form)/MFT-0H2 (reduced form, Fig. 4C).
 | ||
| Fig. 6 The two-step MftC-mediated decarboxylation and cyclization of MftA. The mycofactocin precursor peptide MftA is bound by its chaperone (RRE) MftB and modified in a radical S-adenosyl methionine-dependent two-step reaction by MftC to the cyclized mycofactocin precursor MftA*. During the reaction, the apparently unstable intermediate MftA** is formed.36 SAM – S-adenosyl-L-methionine, 5′-dAdo – 5′-deoxyadenosine, Met – methionine. | ||
 | ||
| Fig. 7 Mycofactocin biosynthesis. Possible biosynthetic routes to mycofactocin (MFT-n) and methylmycofactocin (mMFT-n) are shown (final products in black boxes). Green arrows mark likely in vivo routes. The GAHDP pathway (light grey) represents a shunt pathway whose product carries an additional glycine as a result from an unspecific cleavage of the cyclized precursor MftA*. Molecules observed in natural extracts are underlined. MftA – mycofactocin precursor peptide, MftA* – MftA modified by MftC, MftA′ – MftA* N-terminus after MftE cleavage, MftA′′ – MftA* N-terminus after unspecific cleavage resulting in the liberation of GAHDP-0/GAHDP-n, MftB – mycofactocin chaperone, MftC – mycofactocin radical SAM maturase, MftD – premycofactocin synthase, MftE – mycofactocin peptidase, MftF – mycofactocin glycosyltransferase, MftM – mycofactocin SAM-dependent methyltransferase, NDP-Glc – nucleotide diphosphate glucose, SAM – S-adenosyl methionine, SAH – S-adenosyl homocysteine, 5′-dAdo – 5′-deoxyadenosine, Met – methionine. Number of glucose moieties attached to the core through β-1,4-glycosydic bonds denoted by “n” with 1 ≤ n ≤ 9. A complete list of hypothetical and observed mycofactocin congeners and precursors including their corresponding SMILES is provided in the ESI† “mycofactocin_SMILES.xlsx”. | ||
Investigation of bacterial mycofactocin additionally revealed that the core structure was glycosylated with up to nine glucose moieties with β-1,4-linkage attached to the hydroxyl group of the former tyrosyl residue forming MFT-n (Fig. 4C and 7).53 This glycosylation was shown to be mediated by MftF, a member of the glycosyl transferase family IPR023981, whose gene is an integral part of the mycofactocin gene locus.31 Enzymatic redox studies with natural mycofactocin from Mycolicibacterium smegmatis mc2 155, the model organism for mycofactocin research, provided support for the existence of redox active MFT-n with a two-electron hydride transfer occurring on the core structure as shown by in vitro studies described above (Fig. 4C and 8).53
 | ||
| Fig. 8 Mycofactocin-mediated NAD(P) cofactor recycling. An example of the hypothetical mycofactocin-mediated internal NAD(P)+ cofactor recycling is shown. A substrate, here L-carveol, is oxidized by an NAD+-dependent dehydrogenase possessing an internal non-exchangeable NAD+ as electron acceptor, e.g. LimC from R. erythropolis DCL14 (see Section 6.1). During the dehydrogenation reaction, two reducing equivalents are transferred onto NAD+ leading to a reduction of the internal cofactor. To maintain enzyme functionality, the internal cofactor needs to be reoxidized from NADH to NAD+. According to the mycofactocin hypothesis, this re-oxidation is achieved through a two-reducing-equivalents-transfer onto (methyl)mycofactocinone (m)MFT-n resulting in NAD(P)+ and (methyl)mycofactocinol (m)MFT-nH2. Reducing equivalents are highlighted in green. Both NAD+ and NAD(P)+ have been found as non-exchangeable cofactors of mycofactocin-dependent enzymes. ADP – adenosine diphosphate. | ||
As described in chapter 3, the oligosaccharide can further be singly methylated on the second glucose unit resulting in methylmycofactocinone-n (mMFT-n) and methylmycofactocinol-n (mMFT-nH2). The evidence for methylated mycofactocin derivatives implied the existence of a methyltransferase capable of catalysing this very specific 2-O-methylation reaction. In the mycofactocin BGC of M. smegmatis, however, no such putative methyltransferase was found (Fig. 5A), and neither was one predicted to be part of the MFT BGC by Daniel H. Haft's seminal bioinformatics study.31 In 2022, the groups of Lackner and Vilotijević identified the gene responsible for this methylation in M. smegmatis, mftM, belonging to the novel mycofactocin-specific protein family NF041255.54 The gene is not located in the vicinity of the mycofactocin BGC in the genome of this strain. However, it was found to be clustered with the putative mycofactocin-dependent alcohol dehydrogenases MSMEG_6239 (madD) and MSMEG_6242 (madA). In several Rhodococcus strains, the mftM gene was subsequently found to be encoded directly upstream of the mycofactocin BGC in reverse orientation (Fig. 5A). In species lacking an MSMEG_6242 homolog, e.g. M. tuberculosis, the mftM gene was missing, too. In accordance with this finding, no methylated mycofactocin species were found in these madA- and mftM-negative organisms.54
4.2 The order of the mycofactocin biosynthetic steps
While all genes of the mycofactocin BGC, mftABCDEF(M), were functionally described, the order of the biosynthetic steps in vivo is not yet fully elucidated. It is reasonable to assume that the MftE peptidase reaction should occur before the MftD reaction that converts the amino group of the modified valine of AHDP-n, which is only accessible after MftE-catalysed release of the modified RiPP core from MftA*-n (Fig. 7). However, it is unclear which mycofactocin congener or precursor is glycosylated, how the glucan chain length is regulated, and at which point the methylation takes place. It is possible that already the unmodified MftA precursor peptide is glycosylated since the hydroxyl group of the C-terminal tyrosyl residue does not appear to be directly involved in core maturation.36,37 The glycosylation might also happen on the cyclized MftA* structure after the MftC reaction took place. However, neither of the two hypothesized peptides, i.e. MftA-n nor MftA*-n, were observed in vivo so far, independent of their glycosylation state. Metabolome data corroborated the hypothesis that the MftF reaction occurs before the MftD deamination reaction since glycosylated AHDP was found in vivo and a deletion of mftF resulted in complete absence of MFT-0(H2),53 i.e. upon loss of the MftF reaction, all potential downstream products of MftD were absent. The fact that intracellular AHDP-0 was strongly reduced as well might imply that MftF acts on a mycofactocin peptide precursor, before the MftE cleavage reaction took place. Possibly, the MftF protein might be structurally essential for the function of MftD and MftE in vivo, e.g. as an integral part of an enzyme complex. Alternatively, glycosylation could be essential for the stabilization or correct trafficking of biosynthetic intermediates in vivo. Recent work on the mycofactocin system of Rhodococcus jostii RHA1 supports the central role of MftF, since a ΔmftF mutant of this strain exhibited the same growth defect as a ΔmftAB strain of the same organism on ethylene glycol as the sole carbon source while both strains showed no growth defect on glucose,59 i.e. mycofactocin-mediated utilization of ethylene glycol as a carbon and energy source is lost in both cases which allows for the hypothesis that functional mycofactocin is not available in these strains. Finally, the methylation reaction mediated by MftM appears to occur before or independent of the MftD reaction, since methylated and glycosylated AHDP, i.e. mAHDP-n, was found in vivo.53Thus, a likely biosynthetic route starts with the ribosomal translation of the MftA precursor, followed by cyclization by MftC, glycosylation by MftF and optional methylation by MftM. This modified glycosylated peptide is then cleaved between the glycyl and modified valyl-tyrosyl residues of MtfA*-n and ends with the installation of the redox functionality by MftD by oxidative deamination of AHDP-n resulting in (m)MFT-n (Fig. 7).
4.3 The potential role of the side-chain modifications
A second open question concerns the functions of the glycosylation and methylation modifications. While it was shown that the methylation of the sugar chain confers resistance against cellulases in vitro,54 it remains unclear which physiological roles these two unusual modifications play and how the glycosylation is regulated in vivo. The protective function might be directed towards host-derived cellulases, such as MSMEG_6752, a cellulase shown to degrade cellulose to cellobiose in vitro,60 which might be co-expressed with the mycofactocin BGC. The role of the glucose methylation thus would be a protection against cofactor degradation by the host. This system, however, would only be functional in mftM-positive bacteria while the situation in bacteria that do not methylate mycofactocins needs to be further clarified. Further functions of mycofactocin methylation could be the modulation of cofactor solubility or the modulation of cofactor–enzyme interactions.The glycosylation itself might aid in protein–cofactor interaction through hydrogen bonds between the glucose-derived hydroxyl groups and hydrophilic residues of mycofactocin-dependent enzymes. Similar functions are described for many cofactors' side chains, such as the nucleotide moiety of FAD.61 Further examples are folates and coenzyme F420, which carry an oligoglutamate side chain that strengthens their binding to enzymes.62–64 Another function of the oligoglycosylation could be to prevent the leakage of the cofactor from the cell. The small and relatively apolar cyclic core molecules of many cofactors, such as the riboflavin core of FAD or the deazariboflavin core of coenzyme F420, are often found to cross the cytoplasmic membrane.65,66 This hypothesis is supported by the fact that mycofactocin-related aglycons, especially AHDP-0, can be detected in the supernatant of M. smegmatis.54 A possible function of the glycosyl chain as a solubility-modulating part of mycofactocin awaits experimental investigation. Previous reports showed that increasing chain lengths of β-1,4-linked glucans are negatively correlated with water solubility, already for short-chain β-1,4-linked glucose oligosaccharides as present in mycofactocins.67
5. Physiological functions and regulation of mycofactocin
While the structure and biosynthesis of mycofactocin are fascinating from a chemical point of view, its physiological implications are equally intriguing. As outlined in the previous chapter, the mycofactocin system was found in part via co-occurrence of the BGC with oxidoreductases. Strikingly, one of the oxidoreductase families, encoding for short-chain dehydrogenases (SDR, SDR_subfam_1 IPR023985), includes a carveol dehydrogenase from Rhodococcus erythropolis DCL14, which had been experimentally characterized long before mycofactocin was postulated (Table 1). It was shown to bind one non-exchangeable nicotinamide adenine dinucleotide (NAD) molecule per subunit in a tight non-covalent mode.43 This usage of NAD as a tightly bound prosthetic group, which is reduced during oxidation of carveol to carvone, necessitates an external (diffusible) electron acceptor to reoxidize the internal nicotinamide cofactor. For in vitro experiments, an artificial electron acceptor, 2,6-dichlorophenol indophenol (DCPIP), was used to substitute the natural electron acceptor, which was unknown at the time of the study.43 In the context of more recently published experiments discussed in chapter 6.1, it is highly likely that the cryptic electron acceptor is mycofactocin. A hypothetical schematic representation of this mycofactocin-mediated NAD cofactor recycling reaction is presented in Fig. 8. This early finding already suggested a role of the cofactor in alcohol dehydrogenation reactions.5.1 The role of mycofactocin in ethanol metabolism
Inspired by a previous study,68 Krishnamoorthy et al.69 attempted to study a potential role of mycofactocin in the utilization of cholesterol as a carbon source by different mycobacterial strains, such as M. smegmatis or M. tuberculosis, the causative agent of tuberculosis. Their investigations showed that it was not cholesterol that was used in a mycofactocin-dependent manner but it was instead the ethanol that was used to prepare cholesterol stock solutions, shifting the focus of mycofactocin-related metabolic processes towards ethanol utilization. Transcriptome profiling of M. smegmatis on medium containing ethanol versus glucose as the sole carbon source revealed upregulation of a gene encoding for the putative alcohol dehydrogenase MSMEG_6242 (madA, previously called mdo or mno). A knock-out mutant of this gene exhibited the same phenotype on ethanol as the mycofactocin knock-out mutants of M. smegmatis. However, it was able to grow on acetate as a sole carbon source, a likely product of ethanol oxidation (Fig. 9A).69 We could further show that not only madA but also the mycofactocin biosynthesis is upregulated in M. smegmatis on ethanol versus glucose as the sole carbon source53 and also that acetate accumulates in liquid medium when grown on ethanol.41 These results led to the proposed mycofactocin-dependent ethanol utilization pathway depicted in Fig. 9A. Accordingly, ethanol is oxidized by madA to acetaldehyde in an MFT-dependent dehydrogenation reaction, followed by a second oxidation step from acetaldehyde to acetate by an as-yet unidentified acetaldehyde dehydrogenase. One candidate for the elusive acetaldehyde dehydrogenase could be the zinc-dependent aldehyde dehydrogenase MscR (MSMEG_4340). It was shown to be able to oxidize formaldehyde, acetaldehyde, and propionaldehyde in vitro and is upregulated under aldehyde stress.70,73 However, MscR is merely one candidate of more than 50 putative aldehyde dehydrogenases encoded in the genome of M. smegmatis mc2 155. Whether the acetaldehyde oxidation is mycofactocin-dependent remains to be elucidated. The following acetate utilization, however, is not strictly mycofactocin-dependent as demonstrated by mycofactocin biosynthesis gene knock-outs able to grow on acetate as the sole carbon source.69 | ||
| Fig. 9 Hypothesized mycofactocin-dependent mad-mediated alcohol utilization pathways. (A) Proposed ethanol assimilation pathway in M. smegmatis. Ethanol is oxidized to acetaldehyde in an MFT-dependent reaction by MadA (A0R5M3, MSMEG_6242) and further oxidized to acetate by an unknown acetaldehyde dehydrogenase, potentially MscR (A0R0C7, MSMEG_4340).69,70 The acetate might either be excreted as an overflow metabolite71 or further metabolized via acetyl-CoA. (B) Proposed and simplified ethylene glycol assimilation pathway for R. jostii RHA1. Ethylene glycol is oxidized in a mycofactocin-dependent reaction by MadA (Q0S3Q2, RHA1_ro06057) followed by oxidation of the resulting glycolaldehyde to glycolate by AldA1 (Q0S3M8, RHA1_ro06081) and/or AldA2 (Q0RXM5, RHA1_ro08917) and further to glyoxylate by GlcD (Q0SBQ6, RHA1_ro03227). Glyoxylate might be further metabolized via the glyoxylate shunt.59,72 mMFT-n – methylmycofactocinone-n, mMFT-nH2 – methylmycofactocinol-n, MadA – mycofactocin-dependent alcohol dehydrogenase, MscR – S-nitrosomycothiol reductase MscR, AldA1 – aldehyde dehydrogenase A1, AldA2 – aldehyde dehydrogenase A2. | ||
5.2 The mycofactocin-dependent alcohol degradation gene cluster
MadA from M. smegmatis mc2 155 (MSMEG_6242, Table 2) was not only indirectly shown to be involved in the utilization of short-chain primary alcohols. It was also directly demonstrated in vitro to possess primary alcohol (C1–C4) dehydrogenase as well as formaldehyde dismutase activities in the presence of the artificial electron acceptor N,N-dimethyl-p-nitrosoaniline (NDMA).51 In M. smegmatis, madA is encoded in a putative gene cluster comprising the genes MSMEG_6236-6242. Interestingly, this gene cluster is located directly upstream of mftM, to which, as outlined in chapter 4.2, madA is tightly linked via strict co-occurrence in a genome-wide context. It should be noted that these two genes are not always encoded in the same locus, albeit encoded in the same genome (Fig. 5A). The genes included in the MSMEG_6236-6242 cluster, despite MSMEG_6242, are MSMEG_6239 (madD) – a putative IPR001670-family 1,3-propandiol dehydrogenase, MSMEG_6240 – a putative von Willebrand factor type A (VWA) domain (PF05762)-containing protein of unknown function, MSMEG_6241 – a putative MoxR-type AAA+ ATPase domain (PTHR42759)-containing protein of unknown function, and MSMEG_6236 (madR)/MSMEG_6238 (madS) – a two-component system regulating the expression of MSMEG_6239 and MSMEG_6242 in the presence of methanol, 1,3-propandiol, and under starvation conditions, i.e. absence of a carbon source.39,40 MSMEG_6240 and MSMEG_6241 might be involved in maturation of MSMEG_6239 and MSMEG_6242, which is most likely dependent on cations as described in chapter 6.3. Notably, MoxR-type ATPase domain proteins are known to co-occur and co-locate with VWA-domain proteins74 and can act as chaperones aiding in the maturation of metal-dependent proteins, e.g. during the insertion of non-heme iron.75 Two independent studies recently discovered a gene cluster homologous to the M. smegmatis MSMEG_6236-6242 cluster in R. jostii RHA1,59,72 where it is involved in ethylene glycol catabolism. Roccor et al. demonstrated that an R. jostii RHA1 ΔmadA strain failed to utilize ethylene glycol as the sole carbon source and the wildtype was shown to convert ethylene glycol to glycolaldehyde (Fig. 9B).72 These results suggest that the MSMEG_6242 cluster and homologs thereof confer mycofactocin-dependent alcohol utilization capabilities which therefore were named mycofactocin-dependent alcohol degradation (mad) pathway. Thus, the MSMEG_6236-6242 cluster and homologous gene clusters are further addressed in the naming convention proposed by Roccor et al. as the madABCDSR cluster.72 Table 2 provides an overview of the M. smegmatis mc2 155 and R. jostii RHA1 mad gene clusters with gene names and linked loci as depicted in Fig. 5A. In the system of R. jostii, two aldehyde dehydrogenases were proposed to convert glycolaldehyde to glycolate, AldA1 (RHA1_ro06081/RHA1_RS29725) and AldA2 (RHA1_ro08917/RHA1_RS39755).72 Both do not share sequence homology with MSMEG_4240 (MscR).| R. jostii RHA1 gene name (locus) | M. smegmatis mc2 155 gene name (locus) |
|---|---|
| madA (RHA1_ro06057) | mno/mdo (MSMEG_6242) |
| madB (RHA1_ro06058) | MSMEG_6241 |
| madC (RHA1_ro06059) | MSMEG_6240 |
| madD (RHA1_ro06060) | MSMEG_6239 |
| madS (RHA1_ro06061) | mnoS (MSMEG_6238) |
| madR (RHA1_ro06062) | mnoR (MSMEG_6236) |
In summary, the evidence suggests that one of the physiological roles of mycofactocin in mycofactocin methylating strains, encoding mftM in combination with the mad gene cluster, is the oxidation of short-chain alcohols, such as ethanol. Mycofactocin supposedly acts as an external electron acceptor for the non-exchangeable nicotinamide adenine dinucleotide (phosphate) of madA (and potentially MadD) as outlined in Fig. 8.
5.3 The physiology of mftM-negative mycofactocin producers
In contrast to the mycofactocin-dependent ethanol utilization in mycofactocin methylating strains, little is known about the physiological role of mycofactocin in non-methylating strains, such as M. tuberculosis H37Rv. Mycofactocin methylating strains maintain the ability to grow on ethanol as the sole carbon source when mftM is knocked out, albeit with a longer lag phase, as demonstrated for M. smegmatis.54Notably, non-methylating strains systematically lack the madA gene and therefore also the alcohol utilization pathways proposed in Fig. 9. This suggests the existence of an alternative, yet unknown, ethanol assimilation pathway in these strains. M. marinum, a species of the non-methylating group, was shown to grow on ethanol as the sole carbon source.69 Growth was inhibited when mftD was deleted, indicating that the ethanol utilization in M. marinum is MFT-dependent. A second line of evidence for an alternative madA-independent ethanol degradation pathway involves M. tuberculosis H37Rv. Here, the maximum cell density in vitro was halved when grown on ethanol and cholesterol as carbon sources upon deletion of mftD.69 Notably, Daniel Haft identified three and nine potential mycofactocin-dependent oxidoreductases in M. tuberculosis H37Rv and M. marinum strain M, respectively.31 These data sets indicate the presence of a madA-independent but mycofactocin-dependent ethanol utilization pathway in this group of mycofactocin producing organisms. Besides alcohol utilization, mycofactocin might also play a role in the biosynthesis of the cell wall of these strains as we will describe in chapter 6.2.
5.4 Regulation of mycofactocin biosynthesis
In the previous chapters, we described how mycofactocin is involved in the short chain alcohol metabolism of bacteria and how mycofactocin biosynthesis is elevated upon presence of different alcohols such as ethanol or ethylene glycol. However, not only alcohols but also oxygen appears to play a role in the regulation of mycofactocin biosynthesis. It was shown that the expression levels of the mycofactocin biosynthesis genes mftC and mftD are highly increased in low oxygen conditions in M. tuberculosis.76 This trend is supported by findings for M. smegmatis where elevation in mycofactocin biosynthesis correlated with decreasing oxygen transfer rates in liquid culture.71 Proteome data for M. tuberculosis during in vitro dormancy, a state of survival induced by low oxygen availability, showed increased MftD levels.77 The increased expression of mftD under low oxygen conditions might be a compensation mechanism for its oxygen-dependency, as the MftD reaction consumes one oxygen molecule (Fig. 7). Another reason for the increased mycofactocin biosynthesis during low oxygen conditions might be an increased reliance of the bacteria on energy gained through mycofactocin-dependent substrate oxidation pathways. For example, ethanol oxidation via acetaldehyde to acetate as proposed in Fig. 9A could be such an energy source. This hypothesis is supported by data for M. smegmatis where increasing mycofactocin concentrations correlated with increasing acetate concentrations in ethanol-containing liquid medium upon lowering oxygen availability.71 Thus, the energy and carbon metabolism under low O2 conditions appear to be fuelled mostly by the first steps of ethanol oxidation instead of downstream acetate utilization, potentially necessitated by a reduced proton gradient-driven ATP generation as an effect of the lower oxygen availability.Mycofactocin biosynthesis is induced by short-chain primary alcohols and low oxygen concentration. However, the molecular regulatory network behind this induction is unknown. The mycofactocin biosynthetic gene cluster sensu latu not only includes the genes mftABCDEF(M) but also mftR, encoding the transcriptional regulator MftR. As depicted in Fig. 5A, the regulator is usually encoded upstream of mftA in reverse orientation and has been shown to recognize a palindromic sequence upstream of the mftA gene (5′-T-N2-GGCA-N5-TGCC-N2-A-3′) acting as a transcriptional repressor for mftA in dimeric form.78 Despite all metabolic evidence presented so far pointing towards a function of mycofactocin in alcohol utilization, it was shown that MftR binds long-chain acyl coenzyme A esters (acyl-CoA), particularly oleoyl-CoA. Binding of ligand induces a confirmational change in the MftR dimer leading to dissociation of the repressor from the bound DNA, resulting in derepression of the mycofactocin BGC.78,79 A direct link between the induction of mycofactocin biosynthesis by ethanol or low oxygen conditions, and derepression of mycofactocin biosynthesis by binding long-chain acyl-CoA to MftR remains to be established. In chapter 6 we will examine how not only the MFT biosynthesis but also the expression of potentially mycofactocin-dependent enzymes is induced upon presence of ethanol and a variety of other molecules (overview presented in Fig. 10).
 | ||
| Fig. 10 Mycofactocin-system inducing agents. The listed molecules and conditions were shown to induce either mycofactocin biosynthesis directly or to induce the expression of putatively mycofactocin-dependent enzymes. The list is not exhaustive but comprises major classes of reported inducing agents, such as linear primary alcohols, secondary alcohols, aldehydes, thiocarbamates and acyl-CoA esters. CoA – coenzyme A. | ||
6. Mycofactocin-dependent dehydrogenases
Chapter 2 introduced the mycofactocin BGC to be co-occurring with originally three families of putatively mycofactocin-dependent oxidoreductases postulated to use the redox cofactor to reoxidize their internal non-exchangeable NAD(P)H (Fig. 8 and Table 1). These were the short-chain dehydrogenase family SDR_subfam_1 IPR023985, the putatively zinc-dependent alcohol dehydrogenase family IPR023921 (ADH_Zn_actinomycetes), and lastly the divalent cation-dependent alcohol dehydrogenase family IPR001670, whose members were not exclusively associated with the mycofactocin BGC as it represents a superfamily with members from the bacterial and fungal kingdom.31 Since this first study in 2011, additional putatively mycofactocin-dependent protein families were defined via hidden Markov models (HMMs) including the 4-nitroso-N,N-dimethylaniline (NDMA)-dependent methanol dehydrogenase family IPR026338 (NDMA_methanol_DH), the two conserved hypothetical protein (CHP) families CHP03977 and CHP03996, and the short-chain dehydrogenase families SDR_subfam_2, SDR_subfam_3, and SDR_subfam_4 (InterPro version 104.0).35,80 Currently, no experimental data is available for any member of these newly defined families, except for NDMA_methanol_DH. However, several members of the originally proposed families and of IPR026338 NDMA_methanol_DH were experimentally investigated, albeit almost exclusively with artificial electron acceptors.6.1 The short-chain dehydrogenase family IPR023985
One family of oxidoreductases that was shown to genomically co-occur with the mycofactocin cluster is the SDR_subfam_1 IPR023985. The first studied member of this family, and to date the only biochemically characterised enzyme of this group, is the carveol dehydrogenase LimC from R. erythropolis DCL14 (UniProt: Q9RA05). It was shown to bind one NAD(H) molecule per subunit of its homotetramer, but does not bind metal ions (Fig. 11A).43 The enzyme catalyses oxidation of L-carveol to L-carvone, the first step of a proposed limonene degradation pathway of R. erythropolis DCL14, hence LimC.82 However, this reaction was conducted with an artificial external electron acceptor, 2,6-dichlorophenol indophenol (DCPIP), in vitro, with the natural redox partner remaining unknown at the time. Already in this study by van der Werf et al. from 1999 it was proposed that the internal NAD(H) is hindered from diffusing out of the protein by a ten amino acid long insertion loop located on primary sequence level towards the N-terminus and adjacent to the NAD(H) cofactor insertion site.43 These results were supported by a crystallography study conducted on other mycobacterial members of the IPR023985 SDR_subfam_1 family which showed non-exchangeable binding of NAD(H) coordinated by the insertion loop as proposed for LimC (Fig. 11B) but also a second disordered loop located towards the C-terminus of the proteins, which together almost entirely bury the cofactor inside each subunit of the homotetramer.42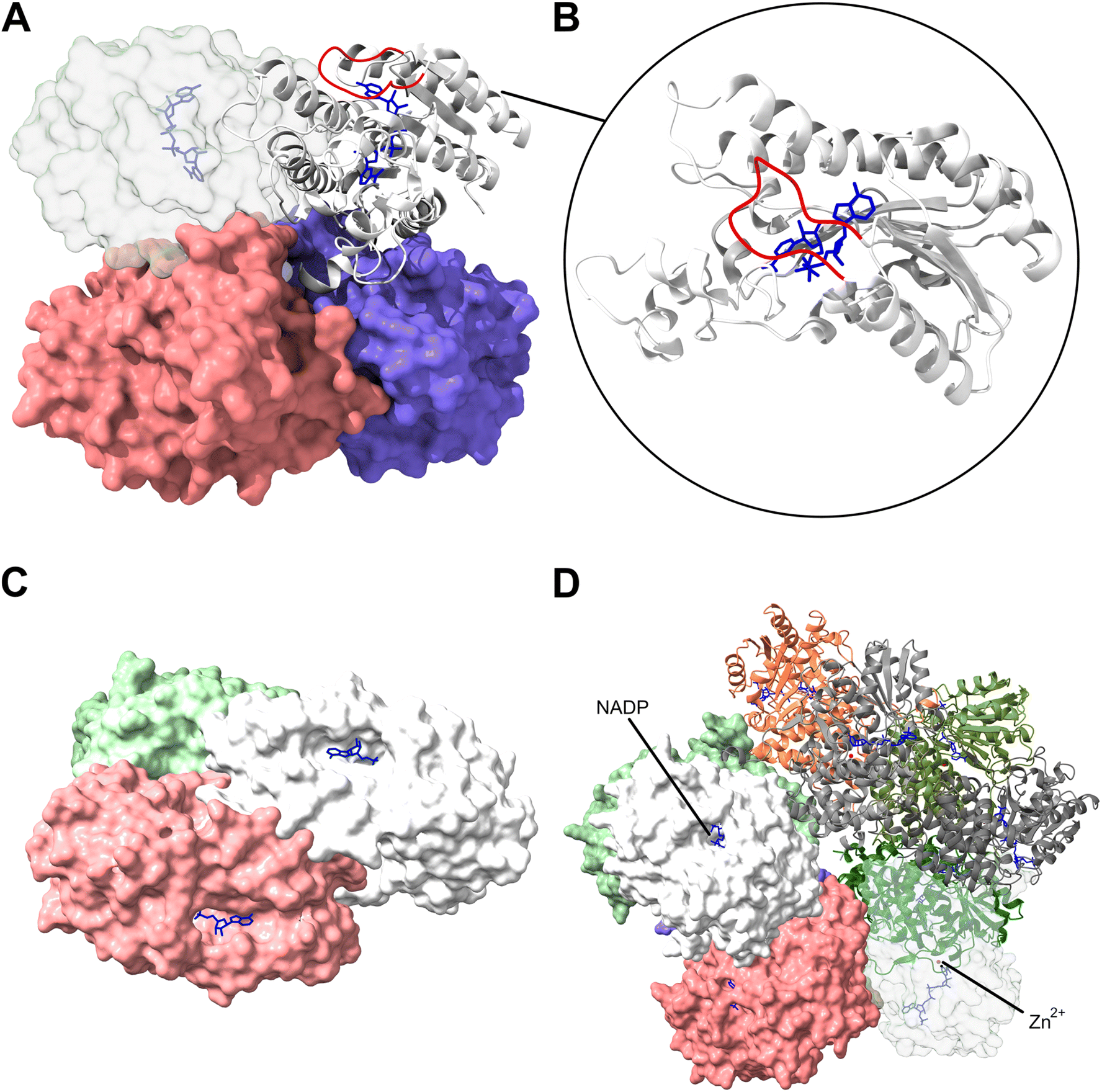 | ||
| Fig. 11 Protein structures of studied mycofactocin-dependent dehydrogenases. (A) Shows the crystal structure of the homotetrameric structure of the putative carveol dehydrogenase B1MLR7 (PDB: 3S55) from Mycobacteriodes abscessus belonging to the SDR_subfam_1 family with one subunit depicted in detail in (B).42 The insertion loop hindering the internal NAD(H) from diffusing, here ENSDVVGYPLA (amino acids 41–51), is highlighted in red for this subunit. (C) Structural model of the homotrimeric NDMA-dependent alcohol dehydrogenase P80175 from Amycolatopsis methanolica 239 belonging to the IPR023921 ADH_Zn_actinomycetes family. (D) Structural model of the decameric NADP(H)-binding NDMA-dependent alcohol dehydrogenase Q9RCG0 from A. methanolica 239 closely resembling the reported electron micrographs of this five-fold symmetric decameric enzyme as shown by Bystrykh et al. which belongs to the NDMA_methanol_DH family IPR026338 with Zn2+ ions highlighted in red.49 Typically, 1–2 zinc and magnesium ions and one NADP(H) are bound per subunit. All cofactors are buried deeply within the structure. The NAD(P) cofactors are highlighted in blue for all structures. Structure models C and D were constructed using AlphaFold 3.81 | ||
First evidence for an interaction of mycofactocin with a putatively mycofactocin-dependent dehydrogenase of SDR_subfam_1 was presented for the LimC homolog MSMEG_1410 (UniProt: A0QSA5) from M. smegmatis mc2 155 which was shown to reduce the mycofactocin aglycon MFT-0 during carveol oxidation to MFT-0H2 in vitro.37 Similarly, heterologously produced LimC from R. erythropolis DCL14 reduced all observable mycofactocin species, including glycosylated congeners, present in metabolome extracts of M. smegmatis when carveol was offered as a substrate.53 However, kinetic studies with pure cofactors, particularly mycofactocins with defined glycosyl side chains, are missing to date, as well as a crystal structure showing the interaction of SDR_subfam_1 enzymes with the mycofactocin cofactor. This is true for all putatively mycofactocin-dependent enzymes described in this chapter.
6.2 The putatively zinc-dependent dehydrogenase family IPR023921
The second originally proposed protein family associated with the mycofactocin BGC is the family of putatively zinc-dependent alcohol dehydrogenases IPR023921 ADH_Zn_actinomycetes.31 Within this family, one enzyme from Amycolatopsis methanolica 239 (UniProt: P80175) and one from Rhodococcus opacus DSM 1069 (UniProt: P81747) have been studied in vitro. The former was shown to be a homotrimeric, the latter to be a homotetrameric alcohol dehydrogenase with one NAD(H) bound per subunit (Fig. 11C). In vitro, the enzymes exhibited NDMA-dependent dehydrogenase activities towards a wide range of alcohols, such as ethanol, propan-1-ol, and butan-1-ol, up to octan-1-ol, but also isobutanol, cyclohexanol, benzyl alcohol, vanillyl alcohol, or 3-phenylpropan-1-ol,47 which also induced expression of their genes.46 These enzymes, unlike those of the NDMA_methanol_DH family, did not display methanol dehydrogenase, nor aldehyde dismutase or aldehyde reductase activities. Despite different primary structures as well as higher-order structures, this family shares key similarities with enzymes from the SDR_subfam_1 family in that it contains one non-exchangeable NAD(H) per subunit and oxidizes alcohol groups in dehydrogenation reactions dependent on an external redox molecule.Another putative dehydrogenase of this family, Rv3086 (UniProt: P9WQB9), encoded in the genome of M. tuberculosis H37Rv, appears to play a role in the biosynthesis of the mycobacterial cell wall, specifically in the biosynthesis of mycolic acids as part of the mymA operon (Rv3083–Rv3089). This operon is induced under acidic conditions and during infection of macrophages. Disruption of the mymA operon and its regulation resulted in a reduced bacterial infection load in guinea pig spleens as well as a higher susceptibility to antimycobacterial drugs. However, no function could be attributed to this gene so far.83 All three studied proteins of this family were not investigated regarding their metal content and dependencies. Thus, it remains to be elucidated whether this family is zinc dependent as inferred from sequence homology.46
6.3 NDMA-dependent methanol dehydrogenase family IPR026338
The last family of experimentally investigated enzymes of the potentially mycofactocin-dependent alcohol dehydrogenase families is the NDMA_methanol_DH family IPR026338, with one prominent example being the methanol:NDMA oxidoreductase Q9RCG0 (UniProt) from A. methanolica 239 (previously known as Mno), which is one of the defining members of this enzyme family while it also appears to be part of the divalent cation-dependent IPR001670 superfamily. This enzyme, along with an apparent homolog from Mycobacterium gastri MB19, was shown to be a homodecamer, binding non-exchangeable NADP(H) and containing Mg2+ and Zn2+ ions (Fig. 11D).49 Both enzymes were further shown to possess dehydrogenase capabilities for a wide range of alcohols, such as methanol, ethanol, or butan-1-ol, but also ethylene glycol or isobutanol. Interestingly they also possess formaldehyde dismutase and NADH-dependent primary aldehyde (C1–C4) reductase activities.48,84 In the methylotrophic Mycobacterium sp. JC1, a similar protein, C5MRT8 (UniProt), was identified, which exhibited NDMA-dependent dehydrogenase activities for methanol, ethanol, and formaldehyde but not for propan-1-ol or butan-1-ol. The protein was further shown to be essential for growth on methanol as the sole carbon source in this strain.50 Another member of this family, ThcE (UniProt: Q53062), was found in R. erythropolis NI86/21, which showed an NDMA-dependent methanol dehydrogenase activity and was expressed upon presence of herbicides such as atrazine or the thiocarbamates S-ethyl dipropylcarbamothioate (EPTC) and vernolate but also ethanolamine or ethanol as carbon sources (Fig. 10).44,85 In this strain, the enzyme appears to be part of a cytochrome P450 system involved in the degradation of these herbicides.86 During the herbicide degradation process, aldehydes like propionaldehyde in case of EPTC are formed,83 which hypothetically could be metabolized by ThcE in aldehyde dismutase or reductase reactions as shown for its homologs from A. methanolica and M. gastri described above. Furthermore, a member of this family from R. erythropolis N9T-4 (UniProt: A5LHA1) was shown to be highly induced under oligotrophic conditions, surprisingly together with an NAD-dependent formaldehyde dehydrogenase (UniProt: A5LHA2). The latter is a homolog of the proposed aldehyde dehydrogenases AldA1 (Q0S3M8) and AldA2 (Q0RXM5) of R. jostii RHA1, potentially involved in glycolaldehyde oxidation (Fig. 9B), exhibiting 95% primary sequence identity to both proteins. This NDMA_methanol_DH family member of R. erythropolis N9T-4 (A5LHA1) exhibited formaldehyde dismutase activity as shown for its homologs described above.52 Intriguingly, also madA from R. jostii RHA1 (UniProt: Q0S3Q2) and M. smegmatis mc2 155 (UniProt: A0R5M3) belong to the NDMA_methanol_DH family, which, as outlined above, are involved in the oxidation of ethylene glycol and ethanol, respectively.59,69,72 madA from M. smegmatis mc2 155, besides its ethanol dehydrogenase activity, was also shown to possess dehydrogenase activities for methanol, propan-1-ol, butan-1-ol, and NADH-dependent reductase activity towards their corresponding aldehydes in vitro and was shown to be required for growth of the strain on methanol as the sole carbon source.51 The enzyme was further demonstrated to possess NADH- but not NADPH-dependent aldehyde reductase activity as known for the homologs from M. gastri and A. methanolica.87 Overall, it appears that MadA and its homologs of the NDMA_methanol_DH family are dependent on divalent cations, use NADP(H) as an internal non-exchangeable cofactor and accept a wide range of rather short-chained alcohols in dehydrogenation reactions but might also contribute to the detoxification of aldehydes via aldehyde dismutase and reductase activities. However, a direct interaction of these enzymes with mycofactocin remains to be investigated as all studies on these enzymes used the artificial electron acceptors NDMA or DCPIP instead of their elusive bona fide electron acceptor, which likely is mycofactocin.In summary, all mycofactocin-dependent alcohol dehydrogenase families studied so far share certain characteristics such as the dependence on an external redox cofactor, most likely mycofactocin, as well as the use of a non-exchangeable nicotinamide during the dehydrogenation reaction. However, the three families also exhibit clear differences, which can be summarized as follows: (1) SDR_subfam_1 – metal-independent homotetrameric NAD(H)-binding short chain alcohol dehydrogenases described by IPR023985, (2) ADH_Zn_actinomycetes – NAD(H)-employing dehydrogenases with a broad alcohol substrate spectrum described by IPR023921, and (3) NDMA_methanol_DH – divalent cation-dependent homodecameric and NADP(H)-utilizing alcohol dehydrogenases, with a potential secondary function as aldehyde dismutase and reductase, described by IPR026338. Enzymes of these three families appear to be involved in a wide range of pathways for alcohol utilization but also seem to play a role during cell wall biosynthesis and might even be indirectly involved in carbon fixation as proposed by Ohhata et al. for R. erythropolis N9T-4.52 Proteins of this group of potentially mycofactocin-dependent enzyme families thereby mediate a high metabolic flexibility and stress resistance enabling their cognate hosts to thrive in versatile environments. Future studies will have to investigate a direct interaction between the enzymes and their presumable redox cofactor mycofactocin as well as experimentally characterize members of the recently defined putative mycofactocin-dependent enzyme families described in the introduction of chapter 6.
7. The role of mycofactocin in respiration
With the substrate oxidation side of the mycofactocin energy conservation landscape covered, we need to take a closer look at how reoxidation of mycofactocinols, i.e. (m)MFT-nH2 to mycofactocinones, i.e. (m)MFT-n, is achieved. First, we will revisit the electron flow in PQQ systems, to which the mycofactocin system appears to be functionally related. Subsequently, we will propose a hypothetical scheme of electron flow from ethanol via mycofactocin to a terminal electron acceptor and discuss, how the reoxidation of accumulated mycofactocinols could be coupled with energy conservation in the electron transport chain (ETC).7.1 The electron flow in PQQ systems
Acetic acid bacteria utilizing PQQ-dependent alcohol dehydrogenases demonstrate additional entry points for electrons into the ETC. Besides the classical bacterial ETC, comprising an NADH-dehydrogenase, a quinone pool, a terminal oxidase and an ATP synthase, located in the cell membrane, acetic acid bacteria possess a so-called oxidative fermentation pathway. In this pathway, PQQ-dependent dehydrogenases, located in the periplasmic space and the cell membrane, obtain electrons from reduced substrates such as ethanol or glucose. During the reaction, the PQQ cofactor, a prosthetic group of the same enzyme, is reduced to PQQH2. The cofactor is subsequently reoxidized to PQQ by cytochrome c subunits that facilitate electron transfer to the ubiquinone pool.9 The reduced ubiquinols are reoxidized by a terminal oxidase – ubiquinol oxidase – thereby contributing to the proton motive force, which is harnessed by ATP synthase to generate ATP (Fig. 1).9 Thus, the PQQ system allows for an additional electron entry point besides the classical NADH-dehydrogenase for electron shuttling from alcohols directly to the quinone pool.7.2 MftG is involved in the electron transfer from mycofactocinol to the electron transport chain
In the mycofactocin system, MFT is supposed to serve as a freely diffusible electron acceptor of primary dehydrogenases. However, the cofactor reoxidation step would require an additional enzyme or subunit that would reoxidize mycofactocin, potentially transferring the electrons to the ETC. This enzyme remained unknown until we recently investigated mftG, a previously uncharacterized gene encoded in the majority (ca. 75%) of mycofactocin BGCs (Fig. 5A).41 Its gene product is a flavoenzyme belonging to the superfamily of the glucose–methanol–choline (GMC) oxidoreductases.88 Our recent observations demonstrated that deletion of the mftG gene (MSMEG_1428) in M. smegmatis resulted in a growth defect on ethanol as a sole carbon source, comparable to the phenotype of a mutant deficient in mycofactocin biosynthesis on ethanol. Moreover, ΔmftG cells demonstrated a starvation phenotype inferred from an ADP/ATP ratio evaluation, and a differential transcriptomic analysis of M. smegmatis cells lacking the mftG gene revealed a dramatic reorganization of the ETC.41,89 Strikingly, metabolome analyses revealed that ΔmftG mutants accumulated reduced mycofactocin species, pointing towards impaired cofactor reoxidation as a cause of the growth deficit. Indeed, experiments using isolated mycobacterial membranes from M. smegmatis wildtype, incubated with either MFT-0H2 or mMFT-2H2, demonstrated stimulation of respiration. Membranes isolated from the corresponding ΔmftG strain did not show a comparable respiration rate. These findings were corroborated by LC-MS analyses of the incubated membranes exhibiting indeed an oxidation of mycofactocinols by wildtype membranes. This suggested that MftG is involved in the electron transfer from mycofactocinols to the ETC of mycobacteria as depicted schematically in Fig. 12. The direct interaction of MftG with mycofactocin was further shown by cell-free enzymatic assays with semi-purified MftG that promoted conversion of MFT-0H2 and mMFT-2H2 into their respective oxidized forms. These experiments showed the ability of MftG to interact with and reoxidize reduced mycofactocins. However, the particular electron acceptor of MftG and the entry point of the electrons into the respiratory chain, i.e. the quinone pool or cytochromes, remain unclear. In conclusion, a hypothetical mycofactocin-dependent energy conservation pathway would follow the scheme outlined in Fig. 12. | ||
| Fig. 12 Hypothetical mycofactocin-mediated electron shuttling pathway. Alcohol oxidation is performed by mycofactocin-dependent alcohol dehydrogenase (MadA, MSMEG_6242). This protein represents a decameric complex – AlphaFold 3 prediction of the structure is shown. Autodock Vina90 was used for visualisation of NADPH binding to the internal pocket of one subunit (coloured in yellow). Similarly, docking of mMFT-8H2 was performed (sky blue); the binding mode of cofactors is an illustration only and requires experimental confirmation. Reoxidation of the reduced MFT-molecules is performed by MftG (MSMEG_1428). Docking of FAD (green) and mMFT-8H2 (dark blue) to the receptor is shown (AlphaFold 2 structural prediction). The binding of a heme c (red) can be similarly expected. A further interaction partner of MftG remains unknown, but a flow of electrons to the mycobacterial ETC through the menaquinone pool (MQ/MQH2) may be hypothesised. | ||
7.3 A comparison of the electron flow in PQQ and mycofactocin systems
As described above, mycofactocin serves as the redox cofactor transferring electrons from alcohols to the mycobacterial ETC in a similar fashion as PQQ serves as a redox cofactor in alcohol oxidation pathways of acetic acid bacteria. The reduced PQQH2 is oxidized by the cytochrome c subunits of PQQ-dependent dehydrogenases using ubiquinone as the electron acceptor, thus functioning as an entry point into the ETC. Mycofactocin, however, appears to require the activity of MftG for its reoxidation, a flavoprotein that delivers electrons to an unknown acceptor, most likely menaquinones or cytochromes. One important difference between the two cofactors is that PQQ is tightly bound to its enzymes, thus acting as a prosthetic group, while mycofactocin is a diffusible molecule, acting as a co-substrate. The redox cofactor thus represents an integrating redox pool for reducing equivalents originating from a variety of MFT-dependent dehydrogenation reactions, as demonstrated e.g. by the oxidation of ethanol and carveol. This system, as opposed to the prosthetic PQQ system, might circumvent the necessity for MFT-dependent dehydrogenases to carry an additional dedicated subunit for coenzyme reoxidation. Instead, the mobile coenzyme may diffuse freely towards MftG, which thus bundles electrons from several MFT-dependent primary oxidases.8. Open questions and future directions
This review provides a comprehensive overview of mycofactocin research, tracing its progression from initial bioinformatic predictions to its discovery in vivo. The biosynthetic pathways and chemical synthesis strategies for mycofactocin were discussed in detail and recent biological advances, including the elucidation of an MftG-mediated electron transfer pathway linking mycofactocin to the mycobacterial respiratory chain, were highlighted. Over the past 14 years, experimental efforts have established mycofactocin as a redox cofactor with intriguing parallels to PQQ. These peculiar similarities comprise a biosynthesis via RiPP pathways involving rSAM-mediated cyclization reactions as well as an involvement in electron transfer during aerobic alcohol metabolism. In the absence of (detectable) shared ancestry of most enzymes involved in each biosynthetic system, a common ancestry of the PQQ and mycofactocin systems, which are predominantly found in Gram-negative and Gram-positive bacteria, respectively, is unlikely. Therefore, the two cofactors might represent an extraordinary example of parallel evolution. Besides the obvious selective pressure to evolve alternative hydride carriers, the fundamental question remains: what biological advantage is conferred by their production? Besides an optimized redox potential, alternative cofactors might offer more flexibility, in particular independence of the NAD(H) pool as well as alternative regulation and localization. It also remains open whether further examples of as-of-yet uncharacterized peptide-derived cofactors exist.Looking at the mycofactocin system in particular, several critical questions remain unanswered, presenting excellent opportunities for future research. In recent years, mycofactocin-dependent alcohol oxidation reactions were investigated both in vivo and in vitro. However, the direct mode of interaction between the protein and the mycofactocin molecules remains to be investigated. Future studies will have to focus on in-depth biochemical investigation using purified enzymes and cofactors as well as crystallographic studies of mycofactocin-dependent dehydrogenases in complex with their cofactor. Attention should be given to the difference between methylated and non-methylated mycofactocins and structural studies will need to define the cofactor's absolute stereochemistry. These experiments should be accompanied by synthetic approaches to the different stereoisomers, followed by thorough biochemical characterization and comparison.
Another important task will be to find the exact electron transfer pathway from mycofactocinol to a terminal electron acceptor during energy conservation. Moreover, future investigation is necessary on how a lack of MftG is compensated in organisms not encoding a member of this protein family. It was speculated that enzymes of related families could be able to act in replacement of MftG but no functional homolog has been found so far. Another prospect is the experimental search and investigation of predicted and potentially undiscovered mycofactocin-dependent enzymes through approaches such as chemical proteomics or classical activity-guided protein purification. Such dehydrogenases might be useful for specific alcohol oxidation reactions, as demonstrated for the stereoselective conversion of L-carveol to L-carvone by LimC, and might turn out valuable for synthetic biology approaches to increase the substrate spectrum of naturally mycofactocin-negative organisms. It could be envisioned, for example, to degrade environmentally critical plastic waste like polyethylene terephthalate (PET) via PET hydrolases to ethylene glycol as one main product91 and subsequently use ethylene glycol dehydrogenases as proposed by Roccor et al. to fuel the bacterial carbon and energy metabolism to biologically produce compounds of interest in bacterial hosts.72
Not only biotechnological use cases are possible. As seen in Chapter 6.2, mycofactocin may also play a critical role in the mycobacterial cell wall biosynthesis through involvement in the cell wall biosynthesis-related mymA operon in M. tuberculosis H37Rv, making it a potential target for infection and antibiotic research to find new ways to fight infections caused by this pathogenic species. A related example for an antimycobacterial agent linked to cofactor metabolism is the prodrug pretomanid, which is activated by a coenzyme-F420-dependent oxidoreductase. The activation reaction transforms the compound into a molecule that subsequently impairs the mycobacterial cell wall biosynthesis thereby reducing its fitness under infection conditions.92
In summary, mycofactocin not only provides an interesting landscape for academic research in the fields of microbiology and biochemistry but also has the potential to serve in biotechnological approaches and potentially in the development of novel antimycobacterial drugs.
9. Data availability
No primary research results, software or code have been included and no new data were generated as part of this review.10. Conflicts of interest
There are no conflicts to declare.11. Acknowledgments
W. K. A-J. thanks the German Academic Exchange Service (DAAD) for a graduate fellowship. L. G. thanks the State of Thuringia for a graduate fellowship (Landesgraduiertenstipendium). Funded by the Open Access Publishing Fund of the University of Bayreuth.12. References
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, J. Biol. Inorg. Chem., 2008, 13, 1205–1218 CrossRef CAS PubMed.
- D. C. Johnson, D. R. Dean, A. D. Smith and M. K. Johnson, Annu. Rev. Biochem., 2005, 74, 247–281 CrossRef CAS PubMed.
- M. Richter, Nat. Prod. Rep., 2013, 30, 1324–1345 RSC.
- J. D. Fischer, G. L. Holliday, S. A. Rahman and J. M. Thornton, J. Mol. Biol., 2010, 403, 803–824 CrossRef CAS PubMed.
- J. G. Hauge, J. Biol. Chem., 1964, 239, 3630–3639 CrossRef CAS PubMed.
- J. A. Duine, J. Biosci. Bioeng., 1999, 88, 231–236 CrossRef CAS PubMed.
- S. A. Salisbury, H. S. Forrest, W. B. Cruse and O. Kennard, Nature, 1979, 280, 843–844 CrossRef CAS PubMed.
- C. Anthony and L. J. Zatman, Biochem. J., 1967, 104, 960–969 CrossRef CAS PubMed.
- Y. He, Z. Xie, H. Zhang, W. Liebl, H. Toyama and F. Chen, Front. Microbiol., 2022, 13, 879246 CrossRef PubMed.
- M. Matsutani and T. Yakushi, Appl. Microbiol. Biotechnol., 2018, 102, 9531–9540 CrossRef CAS PubMed.
- J. P. Klinman and F. Bonnot, Chem. Rev., 2014, 114, 4343–4365 CrossRef CAS PubMed.
- Y. Q. Shen, F. Bonnot, E. M. Imsand, J. M. RoseFigura, K. Sjolander and J. P. Klinman, Biochemistry, 2012, 51, 2265–2275 CrossRef CAS PubMed.
- C. Anthony, Antioxid. Redox Signaling, 2001, 3, 757–774 CrossRef CAS PubMed.
- P. M. Goodwin and C. Anthony, Adv. Microb. Physiol., 1998, 40, 1–80 CrossRef CAS PubMed.
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Sussmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Nat. Prod. Rep., 2013, 30, 108–160 RSC.
- W. Zhu and J. P. Klinman, Curr. Opin. Chem. Biol., 2020, 59, 93–103 CrossRef CAS PubMed.
- M. Mirdita, K. Schutze, Y. Moriwaki, L. Heo, S. Ovchinnikov and M. Steinegger, Nat. Methods, 2022, 19, 679–682 CrossRef CAS PubMed.
- W. Zhu, L. M. Walker, L. Tao, A. T. Iavarone, X. Wei, R. D. Britt, S. J. Elliott and J. P. Klinman, J. Am. Chem. Soc., 2020, 142, 12620–12634 CrossRef CAS PubMed.
- S. R. Wecksler, S. Stoll, H. Tran, O. T. Magnusson, S. P. Wu, D. King, R. D. Britt and J. P. Klinman, Biochemistry, 2009, 48, 10151–10161 CrossRef CAS PubMed.
- W. Zhu, A. T. Iavarone and J. P. Klinman, ACS Cent. Sci., 2024, 10, 251–263 CrossRef CAS PubMed.
- I. Barr, J. A. Latham, A. T. Iavarone, T. Chantarojsiri, J. D. Hwang and J. P. Klinman, J. Biol. Chem., 2016, 291, 8877–8884 CrossRef CAS PubMed.
- J. A. Latham, A. T. Iavarone, I. Barr, P. V. Juthani and J. P. Klinman, J. Biol. Chem., 2015, 290, 12908–12918 CrossRef CAS PubMed.
- R. L. Evans III, J. A. Latham, Y. Xia, J. P. Klinman and C. M. Wilmot, Biochemistry, 2017, 56, 2735–2746 CrossRef PubMed.
- A. M. Martins, J. A. Latham, P. J. Martel, I. Barr, A. T. Iavarone and J. P. Klinman, J. Biol. Chem., 2019, 294, 15025–15036 CrossRef CAS PubMed.
- E. M. Koehn, J. A. Latham, T. Armand, R. L. Evans III, X. Tu, C. M. Wilmot, A. T. Iavarone and J. P. Klinman, J. Am. Chem. Soc., 2019, 141, 4398–4405 CrossRef CAS PubMed.
- F. Bonnot, A. T. Iavarone and J. P. Klinman, Biochemistry, 2013, 52, 4667–4675 CrossRef CAS PubMed.
- S. M. Janes, D. Mu, D. Wemmer, A. J. Smith, S. Kaur, D. Maltby, A. L. Burlingame and J. P. Klinman, Science, 1990, 248, 981–987 CrossRef CAS PubMed.
- W. S. McIntire, D. E. Wemmer, A. Chistoserdov and M. E. Lidstrom, Science, 1991, 252, 817–824 CrossRef CAS PubMed.
- S. X. Wang, M. Mure, K. F. Medzihradszky, A. L. Burlingame, D. E. Brown, D. M. Dooley, A. J. Smith, H. M. Kagan and J. P. Klinman, Science, 1996, 273, 1078–1084 CrossRef CAS PubMed.
- S. Datta, Y. Mori, K. Takagi, K. Kawaguchi, Z. W. Chen, T. Okajima, S. Kuroda, T. Ikeda, K. Kano, K. Tanizawa and F. S. Mathews, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14268–14273 CrossRef CAS PubMed.
- D. H. Haft, BMC Genomics, 2011, 12, 21 CrossRef CAS PubMed.
- J. D. Selengut and D. H. Haft, J. Bacteriol., 2010, 192, 5788–5798 CrossRef CAS PubMed.
- J. D. Selengut, D. H. Haft, T. Davidsen, A. Ganapathy, M. Gwinn-Giglio, W. C. Nelson, A. R. Richter and O. White, Nucleic Acids Res., 2007, 35, D260–D264 CrossRef CAS PubMed.
- W. Li, K. R. O'Neill, D. H. Haft, M. DiCuccio, V. Chetvernin, A. Badretdin, G. Coulouris, F. Chitsaz, M. K. Derbyshire, A. S. Durkin, N. R. Gonzales, M. Gwadz, C. J. Lanczycki, J. S. Song, N. Thanki, J. Wang, R. A. Yamashita, M. Yang, C. Zheng, A. Marchler-Bauer and F. Thibaud-Nissen, Nucleic Acids Res., 2021, 49, D1020–D1028 CrossRef CAS PubMed.
- T. Paysan-Lafosse, M. Blum, S. Chuguransky, T. Grego, B. L. Pinto, G. A. Salazar, M. L. Bileschi, P. Bork, A. Bridge, L. Colwell, J. Gough, D. H. Haft, I. Letunic, A. Marchler-Bauer, H. Mi, D. A. Natale, C. A. Orengo, A. P. Pandurangan, C. Rivoire, C. J. A. Sigrist, I. Sillitoe, N. Thanki, P. D. Thomas, S. C. E. Tosatto, C. H. Wu and A. Bateman, Nucleic Acids Res., 2023, 51, D418–D427 CrossRef CAS PubMed.
- B. Khaliullin, R. Ayikpoe, M. Tuttle and J. A. Latham, J. Biol. Chem., 2017, 292, 13022–13033 CrossRef CAS PubMed.
- R. S. Ayikpoe and J. A. Latham, J. Am. Chem. Soc., 2019, 141, 13582–13591 CrossRef CAS PubMed.
- R. Ayikpoe, J. Salazar, B. Majestic and J. A. Latham, Biochemistry, 2018, 57, 5379–5383 CrossRef CAS PubMed.
- A. A. Dubey and V. Jain, Appl. Environ. Microbiol., 2019, 85, e00535 CrossRef CAS PubMed.
- R. Plocinska, K. Strus, M. Korycka-Machala, P. Plocinski, M. Kuziola, A. Zaczek, M. Slomka and J. Dziadek, Front. Cell. Infect. Microbiol., 2024, 14, 1427829 CrossRef CAS PubMed.
- A. P. Graça, V. Nikitushkin, M. Ellerhorst, C. Vilhena, T. E. Klassert, A. Starick, M. Siemers, W. K. Al-Jammal, I. Vilotijevic, H. Slevogt, K. Papenfort and G. Lackner, eLife, 2025, 13, RP97559 CrossRef.
- D. H. Haft, P. G. Pierce, S. J. Mayclin, A. Sullivan, A. S. Gardberg, J. Abendroth, D. W. Begley, I. Q. Phan, B. L. Staker, P. J. Myler, V. M. Marathias, D. D. Lorimer and T. E. Edwards, Sci. Rep., 2017, 7, 41074 CrossRef CAS PubMed.
- M. J. van der Werf, C. van der Ven, F. Barbirato, M. H. Eppink, J. A. de Bont and W. J. van Berkel, J. Biol. Chem., 1999, 274, 26296–26304 CrossRef CAS PubMed.
- I. Nagy, S. Verheijen, A. De Schrijver, J. Van Damme, P. Proost, G. Schoofs, J. Vanderleyden and R. De Mot, Arch. Microbiol., 1995, 163, 439–446 CrossRef CAS PubMed.
- A. Norin, S. R. Piersma, J. A. Duine and H. Jornvall, Cell. Mol. Life Sci., 2003, 60, 999–1006 CrossRef CAS PubMed.
- P. W. Van Ophem, J. Van Beeumen and J. A. Duine, Eur. J. Biochem., 1993, 212, 819–826 CrossRef CAS PubMed.
- P. Schenkels and J. A. Duine, Microbiology, 2000, 146(Pt 4), 775–785 CrossRef CAS PubMed.
- L. V. Bystrykh, N. I. Govorukhina, P. W. Van Ophem, H. J. Hektor, L. Dijkhuizen and J. A. Duine, J. Gen. Microbiol., 1993, 139, 1979–1985 CrossRef CAS.
- L. V. Bystrykh, J. Vonck, E. F. J. Van Bruggen, J. Van Beeumen, B. Samyn, N. I. Govorukhina, N. Arfman, J. A. Duine and L. Dijkhuizen, J. Bacteriol., 1993, 175, 1814–1822 CrossRef CAS PubMed.
- H. Park, H. Lee, Y. T. Ro and Y. M. Kim, Microbiology, 2010, 156, 463–471 CrossRef CAS PubMed.
- A. A. Dubey, S. R. Wani and V. Jain, J. Bacteriol., 2018, 200 DOI:10.1128/jb.00288-18.
- N. Ohhata, N. Yoshida, H. Egami, T. Katsuragi, Y. Tani and H. Takagi, J. Bacteriol., 2007, 189, 6824–6831 CrossRef CAS PubMed.
- L. Peña-Ortiz, A. P. Graça, H. J. Guo, D. Braga, T. G. Köllner, L. Regestein, C. Beemelmanns and G. Lackner, Chem. Sci., 2020, 11, 5182–5190 RSC.
- M. Ellerhorst, S. A. Barth, A. P. Graça, W. K. Al-Jammal, L. Peña-Ortiz, I. Vilotijevic and G. Lackner, ACS Chem. Biol., 2022, 17, 3207–3217 CrossRef CAS PubMed.
- M. L. Ellinwood, Master’s thesis, University of Denver, 2020.
- C. L. M. Gilchrist and Y. H. Chooi, Bioinformatics, 2021, 37, 2473–2475 CrossRef CAS PubMed.
- R. Ayikpoe, V. Govindarajan and J. A. Latham, Appl. Microbiol. Biotechnol., 2019, 103, 2903–2912 CrossRef CAS PubMed.
- N. A. Bruender and V. Bandarian, J. Biol. Chem., 2017, 292, 4371–4381 CrossRef CAS PubMed.
- T. Shimizu, K. Suzuki and M. Inui, Appl. Microbiol. Biotechnol., 2024, 108, 58 CrossRef CAS PubMed.
- N. Van Wyk, D. Navarro, M. Blaise, J. G. Berrin, B. Henrissat, M. Drancourt and L. Kremer, Glycobiology, 2017, 27, 392–399 CrossRef CAS PubMed.
- O. Dym and D. Eisenberg, Protein Sci., 2001, 10, 1712–1728 CrossRef CAS PubMed.
- H. Qi, I. Atkinson, S. Xiao, Y. J. Choi, T. Tobimatsu and B. Shane, Adv. Enzyme Regul., 1999, 39, 263–273 CrossRef CAS PubMed.
- B. Ney, C. R. Carere, R. Sparling, T. Jirapanjawat, M. B. Stott, C. J. Jackson, J. G. Oakeshott, A. C. Warden and C. Greening, Front. Microbiol., 2017, 8, 1902 CrossRef PubMed.
- C. Greening, F. H. Ahmed, A. E. Mohamed, B. M. Lee, G. Pandey, A. C. Warden, C. Scott, J. G. Oakeshott, M. C. Taylor and C. J. Jackson, Microbiol. Mol. Biol. Rev., 2016, 80, 451–493 CrossRef CAS PubMed.
- C. A. Abbas and A. A. Sibirny, Microbiol. Mol. Biol. Rev., 2011, 75, 321–360 CrossRef CAS PubMed.
- D. Last, M. Hasan, L. Rothenburger, D. Braga and G. Lackner, Metab. Eng., 2022, 73, 158–167 CrossRef CAS PubMed.
- J. B. Taylor, Trans. Faraday Soc., 1957, 53, 1198–1203 RSC.
- J. E. Griffin, J. D. Gawronski, M. A. Dejesus, T. R. Ioerger, B. J. Akerley and C. M. Sassetti, PLoS Pathog., 2011, 7, e1002251 CrossRef CAS PubMed.
- G. Krishnamoorthy, P. Kaiser, L. Lozza, K. Hahnke, H. J. Mollenkopf and S. H. E. Kaufmann, mBio, 2019, 10, e00190–e00119 CrossRef CAS PubMed.
- S. R. Wani and V. Jain, Protein Sci., 2022, 31, 628–638 CrossRef CAS PubMed.
- L. Peña-Ortiz, I. Schlembach, G. Lackner and L. Regestein, Front. Bioeng. Biotechnol., 2020, 8, 593781 CrossRef PubMed.
- R. Roccor, M. E. Wolf, J. Liu and L. D. Eltis, Appl. Environ. Microbiol., 2024, 90, e0041624 CrossRef PubMed.
- D. Vargas, S. Hageman, M. Gulati, C. J. Nobile and M. Rawat, IUBMB Life, 2016, 68, 621–628 CrossRef CAS PubMed.
- K. S. Wong and W. A. Houry, J. Struct. Biol., 2012, 179, 211–221 CrossRef CAS PubMed.
- M. Kahle, S. Appelgren, A. Elofsson, M. Carroni and P. Ädelroth, BMC Biol., 2023, 21, 47 CrossRef CAS PubMed.
- G. Krishnamoorthy, P. Kaiser, P. Constant, U. Abu Abed, M. Schmid, C. K. Frese, V. Brinkmann, M. Daffe and S. H. E. Kaufmann, mBio, 2021, 12, e0166521 CrossRef PubMed.
- V. Nikitushkin, M. Shleeva, D. Loginov, F. F. Dycka, J. Sterba and A. Kaprelyants, PLoS One, 2022, 17, e0269847 CrossRef CAS PubMed.
- A. Mendauletova and J. A. Latham, J. Biol. Chem., 2022, 298, 101474 CrossRef CAS PubMed.
- F. Peng, Z. H. Ke, H. R. Jin, W. Wang, H. R. Zhang and Y. Li, FASEB J., 2024, 38, e23724 CrossRef CAS PubMed.
- M. Blum, A. Andreeva, L. C. Florentino, S. R. Chuguransky, T. Grego, E. Hobbs, B. L. Pinto, A. Orr, T. Paysan-Lafosse, I. Ponamareva, G. A. Salazar, N. Bordin, P. Bork, A. Bridge, L. Colwell, J. Gough, D. H. Haft, I. Letunic, F. Llinares-Lopez, A. Marchler-Bauer, L. Meng-Papaxanthos, H. Mi, D. A. Natale, C. A. Orengo, A. P. Pandurangan, D. Piovesan, C. Rivoire, C. J. A. Sigrist, N. Thanki, F. Thibaud-Nissen, P. D. Thomas, S. C. E. Tosatto, C. H. Wu and A. Bateman, Nucleic Acids Res., 2025, 53, D444–D456 CrossRef PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C. C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Zemgulyte, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Zidek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- M. J. van der Werf and A. M. Boot, Microbiology, 2000, 146(Pt 5), 1129–1141 CrossRef CAS PubMed.
- A. Singh, S. Jain, S. Gupta, T. Das and A. K. Tyagi, FEMS Microbiol. Lett., 2003, 227, 53–63 CrossRef CAS PubMed.
- H. J. Hektor and L. Dijkhuizen, FEMS Microbiol. Lett., 1996, 144, 73–79 CrossRef CAS.
- I. Nagy, G. Schoofs, F. Compernolle, P. Proost, J. Vanderleyden and R. De Mot, J. Bacteriol., 1995, 177, 676–687 CrossRef CAS PubMed.
- I. Nagy, F. Compernolle, K. Ghys, J. Vanderleyden and R. De Mot, Appl. Environ. Microbiol., 1995, 61, 2056–2060 CrossRef CAS PubMed.
- A. A. Dubey and V. Jain, Biochem. Biophys. Res. Commun., 2019, 516, 1073–1077 CrossRef CAS PubMed.
- D. R. Cavener, J. Mol. Biol., 1992, 223, 811–814 CrossRef CAS PubMed.
- M. Berney and G. M. Cook, PLoS One, 2010, 5, e8614 CrossRef PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed.
- N. F. S. K. Anuar, F. Huyop, G. Ur-Rehman, F. Abdullah, Y. M. Normi, M. K. Sabullah and R. A. Wahab, Int. J. Mol. Sci., 2022, 23, 12644 CrossRef PubMed.
- K. A. Abrahams, S. M. Batt, S. S. Gurcha, N. Veerapen, G. Bashiri and G. S. Besra, Nat. Commun., 2023, 14, 3828 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5np00012b |
| This journal is © The Royal Society of Chemistry 2025 |