
A high-efficiency and long-cycling aqueous indium metal battery enabled by synergistic In3+/K+ interactions†
Songyang
Chang
a,
Wentao
Hou
a,
Amanda
Conde-Delmoral
a,
Irfan
Ullah
a,
Jose Fernando
Florez Gomez
b,
Gerardo
Morell
b and
Xianyong
Wu
 *a
*a
aDepartment of Chemistry, University of Puerto Rico-Rio Piedras Campus, San Juan, PR 00925-2537, USA. E-mail: xianyong.wu@upr.edu
bDepartment of Physics, University of Puerto Rico-Rio Piedras Campus, San Juan, PR 00925-2537, USA
First published on 18th November 2024
Abstract
Aqueous trivalent metal batteries are promising options for energy storage, owing to their ability to transfer three electrons during redox reactions. However, advances in this field have been limited by challenges such as incompatible M3+/M electrode potentials and salt hydrolysis. Herein, we identify the trivalent indium metal as a viable candidate and demonstrate a high-performance indium-Prussian blue hybrid battery using a K+/In3+ mixture electrolyte. Interestingly, there exists a synergistic interaction between K+ and In3+ ions, which enhances the coulombic efficiency and prolongs the cycling life. Specifically, the addition of K+ elevates the In3+/In plating efficiency from 99.3% to 99.6%, due to the decreased electrolyte acidity and enlarged indium particle size. Simultaneously, the presence of In3+ creates an inherently acidic environment (pH ∼3.1), which effectively stabilizes K+ insertion into the Prussian blue framework. Consequently, this hybrid battery delivered a high capacity of 130 mA h g−1, an exceptional rate of 96 A g−1 (∼740 C), and an extraordinary cycling life of 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles. This work offers an innovative approach to develop high-performance hybrid metal batteries.
000 cycles. This work offers an innovative approach to develop high-performance hybrid metal batteries.
 Xianyong Wu | Dr. Xianyong Wu is currently an assistant professor in the Department of Chemistry at the University of Puerto Rico, Rio Piedras. He worked as a postdoc at the University of Washington and Oregon State University from 2016 to 2021, and he obtained his Ph.D. from Wuhan University in 2016. His research focuses on utilizing innovative charge carriers or elements to build next-generation batteries, such as Grotthuss-mechanism-based H+ batteries, rocking-chair NH4+ batteries, reverse dual-ion batteries, transition-metal-sulfur batteries, and post-zinc metal batteries (iron, indium, cadmium, and tin). He has published 60 peer-reviewed papers with citations over 8,500 times. |
Introduction
The development of next-generation battery systems has garnered significant attention, because conventional lithium-ion batteries face challenges in terms of limited resources, high cost, and safety issues.1–3 Recently, aqueous rechargeable batteries have emerged as a competitive contender in the energy storage landscape, due to their intrinsic nonflammability, low cost, and environmental friendliness.4–10 In particular, aqueous multivalent metal batteries represent a mainstream research direction, due to ultrahigh capacities in metal electrodes (450–960 mA h g−1) and the resultant high energy density.11–16 For instance, zinc (Zn) batteries have attracted extensive interest from worldwide researchers, owing to the high capacity (820 mA h g−1), low potential (−0.76 V vs. standard hydrogen electrode, SHE), and low price of the Zn metal.17–19 However, dendrite formation, hydrogen evolution reaction (HER), and electrode passivation significantly retard the development of Zn batteries.20–24 Therefore, there is an emerging trend to develop novel metal batteries to circumvent these challenges.Recently, researchers have started to evaluate other divalent transition metals, such as iron,25–28 nickel,29,30 manganese,31–34 and copper,35–39 as alternative post-zinc battery systems. Nevertheless, much less attention has been paid to trivalent metals (aluminum, gallium, indium, antimony, bismuth), despite their intriguing three-electron transfer reactions with notably high capacities. Among them, aluminum (Al) stands out due to its predominantly high capacity (2980 mA h g−1); however, the very low Al3+/Al potential (−1.66 V vs. SHE) leads to significant water decomposition, posing a considerable challenge.40,41 Other candidates, including gallium (Ga3+), antimony (Sb3+), and bismuth (Bi3+), exhibit higher redox potentials, but they rapidly undergo hydrolysis reactions in aqueous solutions.42–44 Hence, incompatible redox potentials and ion hydrolysis reactions constitute two major challenges for aqueous trivalent metal batteries.
In comparison, indium (In) is a feasible trivalent metal choice, which not only possesses a moderate redox potential (−0.34 V vs. SHE) but also exhibits high solubility in water without hydrolysis.45 Besides, it offers a substantial capacity of ∼700 mA h g−1 by virtue of its 3-electron reaction, approaching that of Zn. Lastly, the In metal demonstrates a spherical, micron-sized, and dendrite-free plating morphology, leading to a high coulombic efficiency of 99.3% in a pristine 1 M InCl3 electrolyte.45 When combined with a low-cost manganese dioxide cathode, the full cell realized stable cycling for 680 cycles. All these results motivate us to further investigate and fine-tune In metal battery performance, particularly regarding the enhancement of In plating efficiency and the development of long-cycling cathode materials.
In this study, we present a high-performance hybrid In metal battery based on the indium-Prussian blue analogue (PBA) reaction chemistry in a hybrid K+/In3+ electrolyte. Importantly, the synergistic effect between In3+ and K+ ions significantly enhances the performance of both the In anode and the PBA cathode. Specifically, the introduction of K+ decreases the acidity of the In3+ electrolyte and promotes the formation of larger In particles, thus raising the In plating efficiency from 99.3% to 99.6%. Meanwhile, the presence of In3+ maintains a moderately acidic environment (pH ∼3.1) and thus stabilizes K+ insertion into the PBA cathode, which increases the capacity retention from 6.45% to 70% after 2000 cycles. Benefitting from this synergistic interaction, the hybrid battery exhibits a superior cycling performance, with 80.7% retention over 10000 cycles and 51.5% over 48000 cycles. Additionally, it supports an ultrahigh rate capability of 96 A g−1 (∼740 C), due to the fast K+ insertion on the cathode and rapid In3+ plating on the anode.
Results and discussion
We aim to fabricate a potassium-based hybrid In metal battery, considering potassium's high abundance, low cost, and high ionic conductivity in water.46–48 We utilized 1.0 m (m: mol kg−1) InCl3 as the baseline electrolyte and studied the effect of KCl concentrations on the battery performance. Given that the maximal solubility of KCl in the hybrid electrolyte is 4.5 m (Fig. S1†), we designed 5 different electrolytes for initial screening: 1 + 0, 1 + 1, 1 + 2, 1 + 3, and 1 + 4.5 m.Fig. 1a shows the impact of KCl additives on the electrolyte properties (pH values and ionic conductivity). As the KCl concentration increases from 0 to 4.5 m, the ionic conductivity markedly rises from 16 mS cm−1 to 149 mS cm−1. It is known that hydrated K+ ions have a small Stokes radius,49–51 leading to high mobility in aqueous electrolytes. Thus, adding K+ can decrease the cell resistance and increase the battery rate performance. Meanwhile, the pH value progressively increases from 2.3 to 3.4 with higher K+ concentrations, suggesting reduced electrolyte acidity. The [H+] concentration decreased by an order of magnitude, because pH is the negative logarithm of proton concentrations (pH = −log[H+]).52 Of note, trivalent cations possess high charge density, resulting in strong coulombic interactions with water molecules.52 This interaction promotes acidic conditions through the chemical equilibrium reaction: [In(H2O)6]3+ (aq) + H2O (l) ⇌ [In(H2O)5OH]2+ (aq) + H3O+ (aq).52 When K+ ions were introduced, the overall In3+ concentration was diluted and decreased, thus lowering the hydronium (H3O+) concentrations and increasing the pH value accordingly.
 | ||
| Fig. 1 Physical and electrochemical characterization of the 1.0 m InCl3 + X m KCl electrolyte (X = 0, 1, 2, 3, and 4.5): (a) the ionic conductivity and pH values; (b) the average plating efficiency; (c) GCD curves of the In||Ti battery in the 1 m InCl3 + 3 m KCl electrolyte; (d) the coulombic efficiency during cycling; (e) the SEM image of the plated In metal in the pristine 1 m InCl3 electrolyte; (f) the SEM image of the plated In metal in the 1 m InCl3 + 3 m KCl electrolyte; (g) the scheme of the electrostatic shielding mechanism. The testing condition is 1 mA cm−2 current for 1 mA h cm−2 capacity. | ||
To evaluate the In plating efficiency, we assembled In||Ti batteries using a chemically stable titanium (Ti) foil as the substrate. Fig. 1b shows the coulombic efficiency with respect to KCl additives. Interestingly, all hybrid electrolytes exhibit higher efficiencies than the pristine InCl3, and the best efficiency was achieved with an intermediate combination of 1 m InCl3 + 3 m KCl (∼99.6%). Fig. 1c displays selected galvanostatic charge/discharge (GCD) curves of this In||Ti battery in the optimal electrolyte, where well-defined plating/stripping curves and minimal polarization (∼2.0 mV) are attainable. The initial efficiency is 94.6%, and it gradually increases to 98.7% in the 10th cycle, 99.5% in the 100th cycle, and reaches 99.7% in the 200th cycle. The cycling performance is presented in Fig. 1d, wherein the battery demonstrates a long calendar life of 1000 hours with an average efficiency of ∼99.6% over 500 cycles. GCD curves and cycling data for In||Ti batteries with other electrolytes are provided in Fig. S2–S5.†
Compared with the pristine 1 m InCl3, the 0.3% efficiency improvement in the 1 + 3 m electrolyte seems to be minor; however, for metal electrodes, the efficiency directly reflects the reaction reversibility and thus plays a critical role in the full cell cycling. From another perspective, the reaction irreversibility decreased from 0.7% to 0.4%, corresponding to 48% improvement. If we assume a full cell with 200 cycles, the capacity retention in the 1 m InCl3 electrolyte will be (0.993)200 = 24.5%. In contrast, the retention will be (0.996)200 = 44.9% in the 1 + 3 m electrolyte, which is 83% higher than the pristine one.
The improved In plating efficiency can be attributed to two primary factors. Firstly, the introduction of K+ ions elevated the pH values of hybrid electrolytes (Fig. 1a), thereby mitigating HER side reactions. Note that a higher pH value means lower [H+] concentrations, leading to a smaller probability of the HER. Secondly, K+ ions influenced the In plating morphology and profoundly increased the metal particle size, which minimized the electrode–electrolyte contact area and benefited the plating efficiency. Fig. 1e, f and Fig. S6† show scanning electron microscopy (SEM) images of the plated In metals. As shown, the In particle size is ∼20 μm in the pristine InCl3, but it increases to 40–60 μm in the 3 m KCl electrolyte (Fig. 1f). Larger particle sizes not only decrease the surface area but also enhance the interconnection between particles, suppressing side reactions and improving plating efficiency.53
Furthermore, we hypothesize that K+ ions may exert an electrostatic shielding effect on In plating (Fig. 1g), a concept originally proposed for the lithium (Li) metal but applicable to other metals.54,55 Considering the significant difference in redox potentials between In3+/In (−0.34 V) and K+/K couples (−2.9 V), K+ ions cannot be reduced in aqueous electrolytes and remain redox-inactive during plating. These positively charged K+ ions will electrostatically absorb on the surface or tips of the growing In particles, which further repel the positive In3+ cations and promote them to plate in adjacent areas, eventually leading to a regulated morphology with larger sizes. Due to these beneficial effects, the In metal can sustain a promising efficiency of 98.7–99.2% at a high plating capacity of 3–10 mA h cm−2 (Fig. S7†).
To fabricate a hybrid battery, it is essential to identify a high-capacity and long-cycling cathode material. Among various materials, Prussian blue analogues (PBAs) represent a promising option,56 due to their easy synthesis, large-open crystal structures, fast ion insertion kinetics, and reversible redox behavior.57,58 In particular, PBA materials are well known for their ionic preference and selectivity in the order of K+ > Na+ > Li+.59 Consequently, K+ insertion features an elevated reaction potential and extended cycling life. Herein, we selected a well-established iron-based PBA material of FeFe(CN)6 as the K-insertion cathode.
This material was prepared by a simple aqueous precipitation method,60–62 and shows a typical dark green color (Fig. S8†). Fig. 2a shows its X-ray diffraction (XRD) pattern, which adopts a face-centered cubic structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) with an index to the standard Fe4[Fe(CN)6]3 compound (JCPDS#01-0239). The diffraction peaks are quite sharp and strong, and there are no extra peaks, indicating high crystalline and material purity. Fig. 2b shows the Fourier-transform infrared (FT-IR) spectrum of this material, wherein two strong peaks at 2172 cm−1 and 2060 cm−1 can be attributed to the Fe3+–CN–Fe3+ and Fe3+–CN–Fe2+ vibrations, respectively.63 The 3635 cm−1 and 1602 cm−1 peaks are attributed to the stretching and bending of O–H bonds that belong to crystal water molecules, respectively.64Fig. 2c displays a SEM image of the PBA material, which exhibits a regular quasi-cube morphology with an average size of ∼500 nm. Energy dispersive spectra (EDS) mapping detects a relatively homogeneous elemental distribution in the sample (Fig. 2d), suggesting high material purity.
m) with an index to the standard Fe4[Fe(CN)6]3 compound (JCPDS#01-0239). The diffraction peaks are quite sharp and strong, and there are no extra peaks, indicating high crystalline and material purity. Fig. 2b shows the Fourier-transform infrared (FT-IR) spectrum of this material, wherein two strong peaks at 2172 cm−1 and 2060 cm−1 can be attributed to the Fe3+–CN–Fe3+ and Fe3+–CN–Fe2+ vibrations, respectively.63 The 3635 cm−1 and 1602 cm−1 peaks are attributed to the stretching and bending of O–H bonds that belong to crystal water molecules, respectively.64Fig. 2c displays a SEM image of the PBA material, which exhibits a regular quasi-cube morphology with an average size of ∼500 nm. Energy dispersive spectra (EDS) mapping detects a relatively homogeneous elemental distribution in the sample (Fig. 2d), suggesting high material purity.
 | ||
| Fig. 2 Physical characterization of the FeFe(CN)6 cathode: (a) the XRD pattern; (b) the FT-IR result; (c) the SEM image; (d) elemental mapping results. | ||
To harness the superior performance of the In anode, we assembled an aqueous hybrid In||FeFe(CN)6 battery. For comparative analysis, we also fabricated a hybrid Zn||FeFe(CN)6 battery. Fig. 3a schematically illustrates the working mechanism. The cathode preferentially works on K+ insertion in the FeFe(CN)6 framework, because PBA shows high ionic selectivity toward bulky monovalent K+ ions. Concurrently, the metal anode undergoes the In3+/In or Zn2+/Zn plating reaction, as K+ ions cannot be reduced in water. Theoretically, the choice of the metal counter electrode should not significantly influence K+ insertion in the cathode, especially considering the large excess of the metal anode (>100 mA cm−2) and the ample electrolyte volume (∼100 μL). Surprisingly, the In||FeFe(CN)6 battery exhibits a superior cycling performance compared to Zn||FeFe(CN)6, suggesting a synergistic interaction between In3+ and K+ ions.
 | ||
| Fig. 3 Electrochemical characterization of hybrid batteries: (a) the working mechanism; (b) GCD curves of the In||FeFe(CN)6 battery at 120 mA g−1; (c) GCD curves of the Zn||FeFe(CN)6 battery at 120 mA g−1; (d) cycling performance comparison at 1.2 A g−1; (e) the SEM image of the cycled electrode in the hybrid In batteries; (f) the SEM image of the cycled electrode in the hybrid Zn batteries; (g and h) EIS comparison of the hybrid battery after cycling. The electrolyte is 1 m InCl3 + 3 m KCl and 1 m ZnCl2 + 3 m KCl for the hybrid In and Zn batteries. | ||
Fig. 3b displays GCD curves of the In||FeFe(CN)6 battery, with two distinct discharge plateaus at ∼1.6 V and 0.85 V, corresponding to the redox reaction of carbon-coordinated and nitrogen-coordinated Fe3+/Fe2+ couples, respectively. The GCD curves are highly consistent and overlap well, and they resemble those in a pure 3 m KCl electrolyte (Fig. S9†), indicating K+ insertion in the cathode structure. The battery delivers a discharge capacity of ∼131 mA h g−1, leading to a specific energy of 141.7 W h kg−1 based on the cathode active mass. In contrast, the Zn-based hybrid battery demonstrates similar GCD curves (Fig. 3c) with two higher discharge plateaus at ∼1.9 and 1.25 V, attributed to the lower Zn2+/Zn redox potential. Despite higher voltages, the Zn||FeFe(CN)6 battery achieves a notable capacity within the first few cycles.
The In||FeFe(CN)6 battery achieves a much better cycling performance than the Zn-based one, highlighting a high compatibility between the In anode and the FeFe(CN)6 cathode. Fig. 3d illustrates the cycling stability of both systems. The In battery capacity fades from 118 mA h g−1 to 95 mA h g−1 after 1000 cycles, corresponding to a capacity retention of 80.5%. After 2000 cycles, the In battery still maintains ∼70% of its original capacity. In stark contrast, the Zn||FeFe(CN)6 battery suffers from drastic capacity fading, with a low capacity retention of ∼15.2% after 1000 cycles and 6.5% after 2000 cycles. Fig. S10† provides the selected GCD curves during cycling for further insights.
To understand the performance disparity, we conducted ex situ SEM and electrochemical impedance spectra (EIS) analyses after 500 cycles at 1.2 A g−1. Fig. 3e shows the SEM image of the FeFe(CN)6 cathode in the In3+/K+ electrolyte, which well retains its original quasi-cube features without noticeable size or morphology changes. Nevertheless, this FeFe(CN)6 cathode experiences an evident morphology change and size decrement in the Zn2+/K+ electrolyte (Fig. 3f), suggesting significant material degradation. EIS studies provide further evidence for their performance differences. As shown in Fig. 3g, the In||FeFe(CN)6 battery exhibits a stable charge-transfer resistance, remaining almost unchanged from ∼0.41 ohms before cycling to ∼0.42 ohms after cycling. This stability indicates a robust insertion process. However, the Zn||FeFe(CN)6 battery shows a substantial increase in charge-transfer resistance, rising from ∼42.0 ohms to ∼72.5 ohms after cycling, reflecting deteriorated electrochemical performance (Fig. 3h).
We reason that the better cycling in the In||FeFe(CN)6 battery may come from two aspects. Firstly, In3+ ions are more densely charged, leading to a more acidic environment than Zn2+ electrolytes. As tested, the pH value for the In3+/K+ and Zn2+/K+ electrolyte is 3.1 and 5.4, respectively. It is well documented that PBA materials favor acidic electrolytes for stable cycling. For instance, Cui et al. reported an outstanding cycling life (40000 cycles) for the KCuFe(CN)6 cathode in an acid-assisted K+ electrolyte (pH∼2.0).65 Secondly, there may exist a crosstalk effect between the cathode and anode. The Zn||FeFe(CN)6 battery is more susceptible to side reactions at the anode. In particular, Zn likely triggers substantial HER side reactions and generates hydroxide (OH−) ions.66 It is plausible that some OH− anions diffuse to the vicinity of the FeFe(CN)6 cathode, contributing to its morphology change and resistance increment. In contrast, the In||FeFe(CN)6 system avoids these issues, as In resists such side reactions and features high efficiency (99.6%), thereby preserving the structural and morphological integrity of the cathode.
The In||FeFe(CN)6 hybrid battery exhibits remarkable ultrahigh-rate capability and exceptional cycling life, distinguishing itself as a superior energy storage solution. As shown in Fig. 4a and Fig. S11,† the discharge capacity is 106, 102, 93, 86, 80, 77, 69, 64 and 56 mA h g−1 at 0.6, 1.2, 3.6, 6, 9.6, 12, 24, 36, and 60 A g−1, respectively. Even at a very high rate of 96 A g−1, the battery still retains a good capacity of 46 mA h g−1, corresponding to a capacity utilization of 43%. Of note, the 96 A g−1 current corresponds to a very high rate of ∼740 C (1 C = 130 mA g−1), meaning that the charge or discharge time is as short as ∼4.9 seconds. Based on these GCD curves, we plotted the Ragone curve to show the energy–power relationship. As shown in Fig. S12,† the specific energy is 115.2 W h kg−1 at 651 W kg−1, which still reaches 33.2 W h kg−1 at a high power of 70348.6 W kg−1.
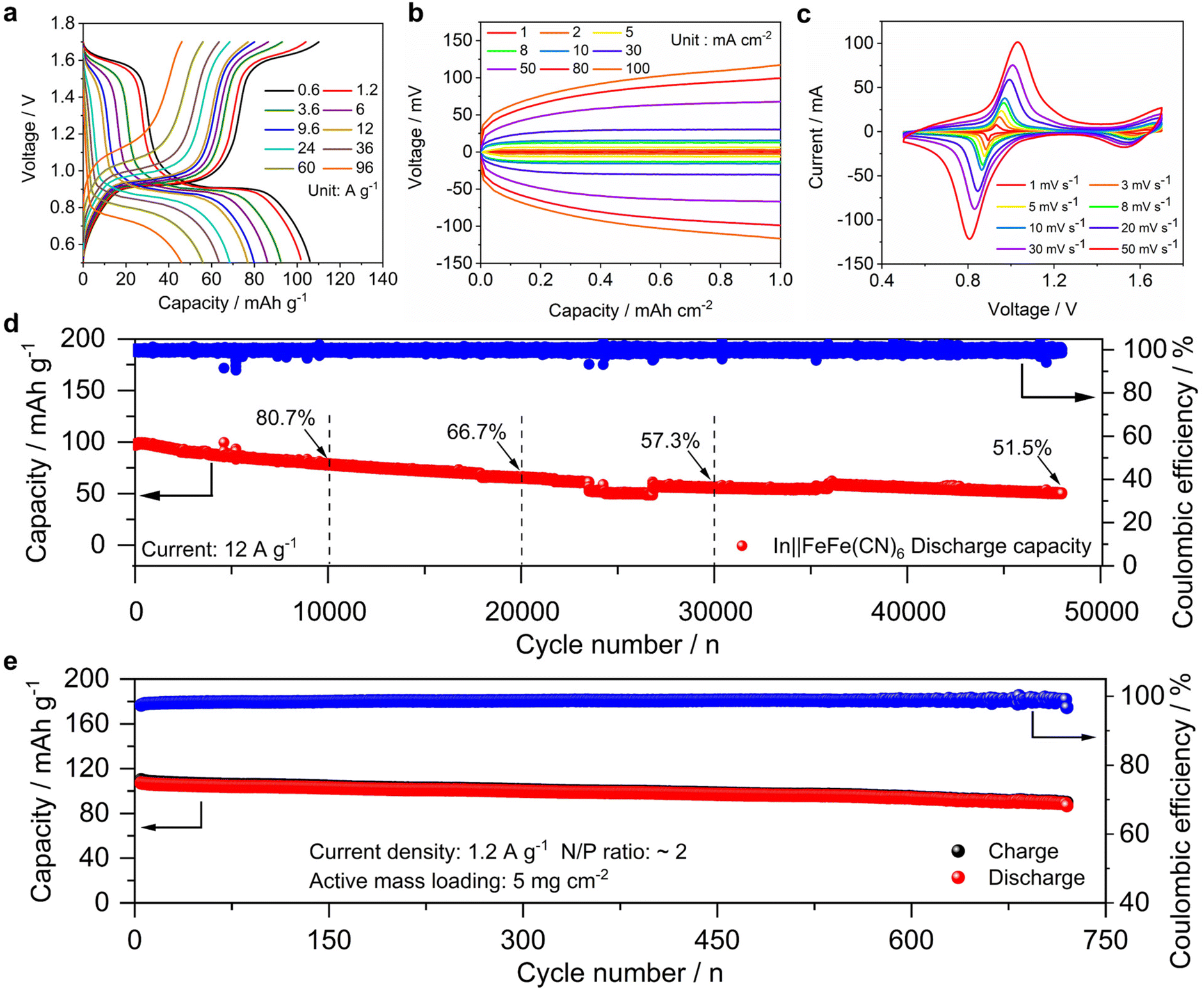 | ||
| Fig. 4 Electrochemical characterization of the In||FeFe(CN)6 hybrid battery: (a) the rate performance; (b) GCD curves of the symmetrical In||In battery at different current densities; (c) CV curves at different scanning rates; (d) the cycling performance at 12 A g−1; (e) the cycling performance of the hybrid full cell at 1.2 A g−1. | ||
Such an excellent rate performance can be attributed to several factors. Firstly, the hybrid electrolyte inherits the high ionic conductivity of K+ ions, which exhibits a high conductivity of 117 mS cm−1 and thus benefits the rate performance. Secondly, the In metal anode demonstrates fast plating/stripping kinetics. In symmetrical In||In batteries (Fig. S13†), the polarization remains remarkably low at approximately 1.5 mV, indicating very fast electrochemical reaction kinetics. Therefore, we tested its rate properties at higher currents. As shown in Fig. 4b and Fig. S14,† the polarization values are 2.5, 6.5, 13, 16, 30, 44, 106 and 130 mV at 2, 5, 8, 10, 30, 50, 80 and 100 mA cm−2, respectively. Thirdly, the PBA cathode features a quasi-capacitive ion insertion process. We recorded cyclic voltammetry (CV) curves of the In||FeFe(CN)6 battery at various scanning rates. At 1 mV s−1, two pairs of oxidation/reduction peaks were observed at 0.93/1.64 and 0.89/1.59 V, corresponding to the redox of carbon-coordinated and nitrogen-coordinated Fe3+/Fe2+ couples. As the scanning rate increases, the cathodic and anodic peak intensities increase accordingly (Fig. 4c). By analyzing the relationship between the logarithm of the peak current [log(i)] and the scanning rate [log(v)], we can derive a slope rate as 0.642 and 0.663 for the cathodic and anodic peaks, respectively (Fig. S15†). These values fall between 0.5 and 1, suggesting a partially capacitive insertion process. Among these factors, the cathode structure should play a major role. In our previous work,60 the FeFe(CN)6 cathode also exhibited an ultrahigh rate capability up to 100 A g−1 in the pristine K2SO4 electrolyte.
Besides the high rate, the hybrid battery delivers an extraordinary cycling life at high current densities. As shown in Fig. 4d, the capacity retention is 80.7%, 66.7%, and 57.3% after 10000, 20000, and 30000 cycles. After 48000 cycles, the capacity retention is 51.6%. Fig. S16† shows selected GCD curves during the long-term cycling. Ex situ SEM characterization reveals the reversible insertion/extraction of K+ ions in the FeFe(CN)6 framework (Fig. S17and S18†), which contributes to the stable cycling life.
To our knowledge, this cycling number has far exceeded the performance of other FeFe(CN)6 based batteries, including Li+, Na+, K+, and NH4+,60,61,64,67–75 as compared in Table S1.† To further push the limit of the cycling life, we assembled a hybrid full cell with a limited negative/positive (N/P) capacity ratio of 2:1. This N/P ratio is higher than those of conventional lithium-ion batteries (1.05–1.1), but it is reasonable in metal based full cells,76,77 because metal electrodes usually exhibit moderate coulombic efficiencies. The cathode exhibits a moderately high active mass loading of ∼5 mg cm−2, leading to an area capacity of ∼0.6 mA h cm−2. The In anode is pre-deposited on the Ti foil with a controlled capacity of 1.2 mA h cm−2. As shown in Fig. 4e and Fig. S19,† this full cell capacity decreases from 107 to 89.5 mA h g−1 after 700 cycles, corresponding to a promising retention rate of 83.6%. The average coulombic efficiency for this hybrid cell is 98.7%. These results clearly indicate the promise of hybrid In metal batteries for energy storage.
Despite these promising cell performances, there are some notable challenges that need to be addressed in future studies. For instance, the K+/In3+ ion concentrations in the hybrid electrolyte will change with the charge/discharge states, especially when the electrode area capacity is high in practical applications. To mitigate this issue, a relatively flooded electrolyte configuration could be used, which can store more ions in the liquid form. In addition, indium is a rare element on Earth, which may restrict its large-scale applications. Therefore, we propose that indium metal batteries might be used in specific cases that prioritize performance over cost, such as miniature applications. For instance, the commercial zinc-silver oxide battery utilizes a rare and expensive silver element, due to the highly stable voltage and high density of silver oxide. It is possible that indium metal batteries could be suitable for wearable devices that require high safety, high performance, and a small battery size (minimal usage of indium).
Conclusions
We developed a high-performance hybrid In metal battery based on an FeFe(CN)6 cathode, an In metal anode, and an In3+/K+ mixture electrolyte. Interestingly, there exists a synergistic effect between In3+ and K+ ions, leading to enhanced In plating efficiency (99.7%) on the anode and stabilized K+ insertion reaction (51.6% over 48000 cycles) on the cathode. Besides, the fast K+ insertion and In3+ plating kinetics concurrently contribute to its ultrahigh rate performance, where 43.4% capacity can be maintained at ∼740 C rate (96 A g−1). This work provides a promising approach to build fast-charging and long-cycling aqueous trivalent metal batteries for energy storage.
Author contributions
All authors have given permission for the final version of this manuscript. Conceptualization: X. Wu; data curation and formal analysis: S. Chang, W. Hou, A. Conde-Delmoral, I. Ullah, and J. Fernando Florez Gomez; visualization: X. Wu; resources: G. Morell and X. Wu; writing–original draft, review, and editing: S. Chang and X. Wu.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Jiayi Xue at Konkuk University for her help with the preparation of Fig. 1g in this paper. The authors acknowledge the financial support from the NSF Center for the Advancement of Wearable Technologies (grant no. 1849243) and NASA MIRO PR-SPRInT (grant no. 80NSSC19M0236).References
- A. Innocenti, D. Bresser, J. Garche and S. Passerini, Nat. Commun., 2024, 15, 4068 CrossRef CAS.
- X. Ji, Energy Environ. Sci., 2019, 12, 3203–3224 RSC.
- Z. Shang, S. Wang, H. Zhang, W. Zhang, S. Lu and K. Lu, Nanoscale, 2022, 14, 14433–14454 RSC.
- J. Shin and J. W. Choi, Adv. Energy Mater., 2020, 10, 2001386 CrossRef CAS.
- L. Li, Q. Zhang, B. He, R. Pan, Z. Wang, M. Chen, Z. Wang, K. Yin, Y. Yao and L. Wei, Adv. Mater., 2022, 34, 2104327 CrossRef CAS.
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2021, 6, 109–123 CrossRef CAS.
- S. Chang, S. Qiu, S. Katiyar, J. F. Florez Gomez, Z. Feng and X. Wu, Batteries, 2023, 9, 349 CrossRef CAS.
- N. Fu, Y.-T. Xu, S. Zhang, Q. Deng, J. Liu, C.-J. Zhou, X.-W. Wu, Y.-G. Guo and X.-X. Zeng, J. Energy Chem., 2022, 67, 563–584 CrossRef CAS.
- X. Zhao, Z. Zhao-Karger, M. Fichtner and X. Shen, Angew. Chem., Int. Ed., 2020, 59, 5902–5949 CrossRef CAS.
- Y. Zhu, R. Zhu, F. Chen, S. Zhang, Y. C. Kuo, P. Guan, M. Li, Y. Liu, Z. Han and T. Wan, Energy Environ. Mater., 2024, 7, e12493 CrossRef CAS.
- S. Qiu, M. Lucero, X. Wu, Q. Wang, M. Wang, Y. Wang, W. S. Samarakoon, M. R. Bolding, Z. Yang and Y. Huang, ACS Mater. Au, 2021, 2, 63–71 CrossRef PubMed.
- L. Ma, Q. Li, Y. Ying, F. Ma, S. Chen, Y. Li, H. Huang and C. Zhi, Adv. Mater., 2021, 33, 2007406 CrossRef CAS.
- H. Jiang, L. Tang, Y. Fu, S. Wang, S. K. Sandstrom, A. M. Scida, G. Li, D. Hoang, J. J. Hong and N.-C. Chiu, Nat. Sustain., 2023, 1–10 Search PubMed.
- S. Chang, L. Lu, I. Ullah, W. Hou, J. F. Florez Gomez, A. Conde-Delmoral, C. M. Font Marin, G. Morell, Z. Chen and X. Wu, Adv. Funct. Mater., 2024, 33, 2407342 CrossRef.
- S. Katiyar, S. Chang, I. Ullah, W. Hou, A. Conde-Delmoral, S. Qiu, G. Morell and X. Wu, Energy Environ. Sci., 2024, 17, 4770–4779 RSC.
- S. Chang, W. Hou, A. Del Valle-Perez, I. Ullah, S. Qiu, J. L. Rodriguez, L. M. Díaz-Vázquez, L. Cunci, G. Morell and X. Wu, Angew. Chem., Int. Ed., 2024 DOI:10.1002/anie.202414346.
- X. Zhao, N. Dong, M. Yan and H. Pan, ACS Appl. Mater. Interfaces, 2023, 15, 4053–4060 CrossRef CAS PubMed.
- F. Wang, E. Hu, W. Sun, T. Gao, X. Ji, X. Fan, F. Han, X.-Q. Yang, K. Xu and C. Wang, Energy Environ. Sci., 2018, 11, 3168–3175 RSC.
- Z. Xing, Y.-P. Deng, S. Sy, G. Tan, A. Li, J. Li, Y. Niu, N. Li, D. Su and J. Lu, Nano Energy, 2019, 65, 104051 CrossRef CAS.
- H. Li, R. Zhao, W. Zhou, L. Wang, W. Li, D. Zhao and D. Chao, JACS Au, 2023, 3, 2107–2116 CrossRef CAS.
- H. Liu, Z. Xin, B. Cao, Z. Xu, B. Xu, Q. Zhu, J. L. Yang, B. Zhang and H. J. Fan, Adv. Funct. Mater., 2024, 34, 2309840 CrossRef CAS.
- J. Hao, L. Yuan, C. Ye, D. Chao, K. Davey, Z. Guo and S. Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 7366–7375 CrossRef CAS PubMed.
- S. Lian, Z. Cai, M. Yan, C. Sun, N. Chai, B. Zhang, K. Yu, M. Xu, J. Zhu and X. Pan, Angew. Chem., Int. Ed., 2024, e202406292 CAS.
- Z. Cai, J. Wang, S. Lian, J. Chen, F. Lang, Z. Li and Q. Li, Adv. Funct. Mater., 2024, 2401367 CrossRef.
- X. Wu, A. Markir, Y. Xu, C. Zhang, D. P. Leonard, W. Shin and X. Ji, Adv. Funct. Mater., 2019, 29, 1900911 CrossRef.
- Z. He, F. Xiong, S. Tan, X. Yao, C. Zhang and Q. An, Mater. Today Adv., 2021, 11, 100156 CrossRef CAS.
- J. Liu, D. Dong, A. L. Caro, N. S. Andreas, Z. Li, Y. Qin, D. Bedrov and T. Gao, ACS Cent. Sci., 2022, 8, 729–740 CrossRef CAS.
- C. Bai, H. Jin, Z. Gong, X. Liu and Z. Yuan, Energy Storage Mater., 2020, 28, 247–254 CrossRef.
- J. Zhao, X. Yang, S. Li, N. Chen, C. Wang, Y. Zeng and F. Du, CCS Chem., 2021, 3, 2498–2508 CrossRef CAS.
- M. Wang, Y. Meng, P. Gao, K. Li, Z. Liu, Z. Zhu, M. Ali, T. Ahmad, N. Chen and Y. Yuan, Adv. Mater., 2023, 35, 2305368 CrossRef CAS PubMed.
- S. Bi, S. Wang, F. Yue, Z. Tie and Z. Niu, Nat. Commun., 2021, 12, 6991 CrossRef CAS.
- Q. Yang, X. Qu, H. Cui, X. He, Y. Shao, Y. Zhang, X. Guo, A. Chen, Z. Chen and R. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202206471 CrossRef CAS.
- S. Bi, Y. Zhang, S. Deng, Z. Tie and Z. Niu, Angew. Chem., Int. Ed., 2022, 61, e202200809 CrossRef CAS.
- M. Wang, Y. Meng, Y. Xu, N. Chen, M. Chuai, Y. Yuan, J. Sun, Z. Liu, X. Zheng and Z. Zhang, Energy Environ. Sci., 2023, 16, 5284–5293 RSC.
- X. Wu, A. Markir, L. Ma, Y. Xu, H. Jiang, D. P. Leonard, W. Shin, T. Wu, J. Lu and X. Ji, Angew. Chem., Int. Ed., 2019, 58, 12640–12645 CrossRef CAS PubMed.
- C. Dai, L. Hu, H. Chen, X. Jin, Y. Han, Y. Wang, X. Li, X. Zhang, L. Song and M. Xu, Nat. Commun., 2022, 13, 1863 CrossRef CAS PubMed.
- J. Zhang, X. Zhang, C. Xu, H. Yan, Y. Liu, J. Xu, H. Yu, L. Zhang and J. Shu, Adv. Energy Mater., 2022, 12, 2103998 CrossRef CAS.
- G. Liang, F. Mo, Q. Yang, Z. Huang, X. Li, D. Wang, Z. Liu, H. Li, Q. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1905873 CrossRef CAS.
- H. Cai, S. Bi, R. Wang, L. Liu and Z. Niu, Angew. Chem., Int. Ed., 2022, 61, e202205472 CrossRef CAS.
- F. Ambroz, T. J. Macdonald and T. Nann, Adv. Energy Mater., 2017, 7, 1602093 CrossRef.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Lin, E. Knipping, W. Peters, J. F. Drillet, S. Passerini and R. Hahn, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed.
- B. W. Zhang, L. Ren, Y. X. Wang, X. Xu, Y. Du and S. X. Dou, Interdiscip. Mater., 2022, 1, 354–372 Search PubMed.
- N. M. Laptash, E. V. Kovaleva, A. A. Mashkovskii, A. Y. Beloliptsev and L. A. Zemnukhova, J. Struct. Chem., 2007, 48, 848–854 CrossRef CAS.
- D. W. Barnum, Inorg. Chem., 1983, 22, 2297–2305 CrossRef CAS.
- S. Chang, J. F. Florez Gomez, S. Katiyar, G. Morell and X. Wu, J. Am. Chem. Soc., 2023, 145, 24746–24754 CAS.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu and H. Li, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- Y. Li, W. Deng, Z. Zhou, C. Li, M. Zhang, X. Yuan, J. Hu, H. Chen and R. Li, J. Mater. Chem. A, 2021, 9, 2822–2829 RSC.
- Y. Xu, T. Ding, D. Sun, X. Ji and X. Zhou, Adv. Funct. Mater., 2023, 33, 2211290 CrossRef CAS.
- X. Zhang, T. Xiong, B. He, S. Feng, X. Wang, L. Wei and L. Mai, Energy Environ. Sci., 2022, 15, 3750–3774 RSC.
- W. Ren, X. Chen and C. Zhao, Adv. Energy Mater., 2018, 8, 1801413 CrossRef.
- D. Su, A. McDonagh, S.-Z. Qiao and G. Wang, Adv. Mater., 2016, 29, 1604007 CrossRef.
- D. C. Harris, Quantitative Chemical Analysis, W. H. Freeman, New York, 8th edn, 2010 Search PubMed.
- J. Zheng, D. C. Bock, T. Tang, Q. Zhao, J. Yin, K. R. Tallman, G. Wheeler, X. Liu, Y. Deng and S. Jin, Nat. Energy, 2021, 6, 398–406 CrossRef CAS.
- J.-G. Zhang, W. Xu, J. Xiao, X. Cao and J. Liu, Chem. Rev., 2020, 120, 13312–13348 CrossRef CAS.
- Y. Liu, Y. Liu, X. Wu and Y.-R. Cho, J. Colloid Interface Sci., 2022, 628, 33–40 CrossRef CAS PubMed.
- J. Peng, W. Zhang, Q. Liu, J. Wang, S. Chou, H. Liu and S. Dou, Adv. Mater., 2022, 34, 2108384 CrossRef CAS PubMed.
- Y. Li, J. Zhao, Q. Hu, T. Hao, H. Cao, X. Huang, Y. Liu, Y. Zhang, D. Lin and Y. Tang, Mater. Today Energy, 2022, 101095 CrossRef CAS.
- H. Yi, R. Qin, S. Ding, Y. Wang, S. Li, Q. Zhao and F. Pan, Adv. Funct. Mater., 2021, 31, 2006970 CrossRef CAS.
- G. Du and H. Pang, Energy Storage Mater., 2021, 36, 387–408 CrossRef.
- X. Wu, Y. Xu, H. Jiang, Z. Wei, J. J. Hong, A. S. Hernandez, F. Du and X. Ji, ACS Appl. Energy Mater., 2018, 1(7), 3077–3083 CrossRef CAS.
- Z. Shadike, D.-R. Shi, M.-H. Cao, S.-F. Yang, J. Chen and Z.-W. Fu, J. Mater. Chem. A, 2017, 5, 6393–6398 RSC.
- A. Zhou, L. Jiang, J. Yue, Y. Tong, Q. Zhang, Z. Lin, B. Liu, C. Wu, L. Suo and Y.-S. Hu, ACS Appl. Mater. Interfaces, 2019, 11, 41356–41362 CrossRef CAS PubMed.
- Z. Ren, D. Hu, X. Zhang, D. Liu and C. Wang, Dalton Trans., 2019, 48, 4058–4066 RSC.
- C. Yang, S. Ding, Y. Zhao, J. Zhou, L. Li and J. Fan, Dalton Trans., 2023, 52, 16984–16992 RSC.
- C. D. Wessells, R. A. Huggins and Y. Cui, Nat. Commun., 2011, 2, 550 CrossRef PubMed.
- X. Liu, Y. Guo, F. Ning, Y. Liu, S. Shi, Q. Li, J. Zhang, S. Lu and J. Yi, Nano-Micro Lett., 2024, 16, 111 CrossRef CAS.
- L. Xiao, H. Lu, Y. Fang, M. L. Sushko, Y. Cao, X. Ai, H. Yang and J. Liu, Adv. Energy Mater., 2018, 8, 1703238 CrossRef.
- C. Yang, Y. Zhao, J. Fan, L. Li, J. Zhou, K. Wang, F. Lu and H. Sun, Mater. Adv., 2024, 5, 5885–5895 RSC.
- S. Li, M. Xia, C. Xiao, X. Zhang, H. Yu, L. Zhang and J. Shu, Dalton Trans., 2021, 50, 6520–6527 RSC.
- X. Wu, M. Shao, C. Wu, J. Qian, Y. Cao, X. Ai and H. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 23706–23712 CrossRef CAS.
- F. Maroni, M. Li, S. Dongmo, C. Gauckler, M. Wohlfahrt-Mehrens, M. Giorgetti and M. Marinaro, ChemElectroChem, 2023, 10, e202201070 CrossRef CAS.
- X. Wu, Y. Luo, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Nano Energy, 2015, 13, 117–123 CrossRef CAS.
- G. Ni, Z. Hao, G. Y. Zou, F. H. Cao, L. Qin and C. G. Zhou, ChemElectroChem, 2022, 9, e202101351 CrossRef CAS.
- G. Ni, M. Sun, Z. Hao, G. Zou, F. Cao, L. Qin, W. Chen and C. Zhou, Mater. Today Energy, 2023, 31, 101204 CrossRef CAS.
- Q. Yang, F. Mo, Z. Liu, L. Ma, X. Li, D. Fang, S. Chen, S. Zhang and C. Zhi, Adv. Mater., 2019, 31, 1901521 CrossRef.
- L. Cao, D. Li, T. Pollard, T. Deng, B. Zhang, C. Yang, L. Chen, J. Vatamanu, E. Hu and M. J. Hourwitz, Nat. Nanotechnol., 2021, 16, 902–910 CrossRef CAS PubMed.
- W. Zhou, D. Zhu, J. He, J. Li, H. Chen, Y. Chen and D. Chao, Energy Environ. Sci., 2020, 13, 4157–4167 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02905d |
| This journal is © The Royal Society of Chemistry 2025 |