
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpacer engineering in nanoparticle–peptide conjugates boosts targeting specificity for tumor-associated antigens†
María de los Angeles
Ramírez
a,
Jolie
Bou-Gharios
 bc,
Barbara
Freis
a,
Julien
Draussin
bfg,
Clémence
Cheignon
d,
Loic J.
Charbonnière
d,
Sophie
Laurent
e,
Thomas
Gevart
e,
Adeline
Gasser
bfg,
Sebastian
Jung
bfgh,
Fabien
Rossetti
i,
Olivier
Tillement
i,
Georges
Noel
bc,
Xavier
Pivot
bfg,
Alexandre
Detappe
*bfg,
Sylvie
Bégin-Colin
*a and
Sébastien
Harlepp
*bfg
bc,
Barbara
Freis
a,
Julien
Draussin
bfg,
Clémence
Cheignon
d,
Loic J.
Charbonnière
d,
Sophie
Laurent
e,
Thomas
Gevart
e,
Adeline
Gasser
bfg,
Sebastian
Jung
bfgh,
Fabien
Rossetti
i,
Olivier
Tillement
i,
Georges
Noel
bc,
Xavier
Pivot
bfg,
Alexandre
Detappe
*bfg,
Sylvie
Bégin-Colin
*a and
Sébastien
Harlepp
*bfg
aUniversité de Strasbourg, CNRS, Institut de Physique et de Chimie des Matériaux de Strasbourg (IPCMS) UMR 7504, 67087 Strasbourg, Cedex 2, France. E-mail: sylvie.begin@unistra.fr
bInstitut de Cancérologie Strasbourg Europe, 67000 Strasbourg, France. E-mail: a.detappe@icans.eu; s.harlepp@icans.eu
cLaboratory of Engineering, Informatics and Imaging (ICube), Integrative multimodal imaging in healthcare (IMIS), UMR 7357, University of Strasbourg, 67000 Strasbourg, France
dEquipe de Synthèse Pour l'Analyse, Institut Pluridisciplinaire Hubert Curien (IPHC), UMR 7178 CNRS/Université de Strasbourg, Strasbourg, Cedex 2 67087, France
eService de Chimie Générale, Organique et Biomédicale, Laboratoire de NMR et d'imagerie moléculaire, Université de Mons, Avenue Maistriau 19, 7000 Mons, Belgium
fEquipe Labellisée Ligue Contre le Cancer, France
gStrasbourg Drug Discovery and Development Institute (IMS), 67000 Strasbourg, France
hUniversity of Strasbourg, CNRS, ISIS & icFRC, 8 allée Gaspard Monge, Strasbourg 67000, France
iInstitut Lumière-Matière, UMR 5306, Université Lyon1-CNRS, Université de Lyon, Villeurbanne Cedex 69100, France
First published on 4th February 2025
Abstract
Developing and synthesizing nano-objects capable of enabling early targeted diagnosis and ensuring effective tumor treatment represents a significant challenge in the theranostic field. Among various nanoparticles (NPs), iron oxide nanoparticles (IONPs) have made significant contributions to advancing this field. However, a key challenge lies in achieving selective recognition of specific cell types. In oncology, the primary goal is to develop innovative strategies to enhance NP uptake by tumors, primarily through active targeting. This involves adding targeting ligands (TL) to the NP surface to facilitate tumor accumulation and increase retention within the tumor microenvironment. Despite biofunctionalization strategies, overall tumor uptake remains modest at only 5–7% of the injected dose per gram. In this work, we demonstrate the effect of spacing between the NPs and the TL to improve their availability and thus the tumor uptake of the complex. This proof-of-concept study targets the epidermal growth factor receptor (EGFR) using a peptide as a targeting ligand. Specifically, we characterized the PEG-peptide coupled to dendronized IONPs, including the density of grafted TL. These nano-objects underwent in vitro evaluation to assess their ability to specifically target and be internalized by tumor cells. Therapeutically, compared to non-functionalized NPs, the presence of the TL with a PEG linker enhanced targeting efficacy and increased internalization, leading to improved photothermal efficacy.
Introduction
Precision therapy relies on the intricacies of delivering therapeutics to the tumor microenvironment (TME), where nanoparticles (NPs) play a pivotal role as transport vectors. Upon their administration into the bloodstream, these NPs dynamically acquire a distinctive biological identity by interacting with various organs, which underscores the complexity of their journey towards the targeted site. The success of a tumor targeting strategy requires these NPs to adeptly navigate through tumors while concurrently minimizing off-target distribution in healthy organs. As a result, their transport dynamics are influenced by their physicochemical properties, size, shape, and surface chemistry.1,2 The optimization of tumor uptake involves strategic functionalization,3–5 wherein monoclonal antibodies (mAbs) and peptides have emerged as key players. Surprisingly, despite the implementation of biofunctionalization strategies, the pharmacokinetic (PK) parameters of these NPs exhibit minimal changes when compared to their unmodified counterparts.6,7 This suggests that the biofunctionalization process maintains the NPs’ inherent pharmacological properties. While mAb biofunctionalization strategies exhibit promising preclinical efficacy, challenges persist in their application such as issues with heterogeneous functionalization, batch-to-batch variability, and complexities in maintaining a consistent orientation.4 In response to these challenges, the use of fragments of mAbs and peptides offers a more accessible route for functionalization, alleviating concerns related to replication and antigen orientation.8 However, the intricate interplay between NPs and the biological milieu post-functionalization raises questions about the formation of a protein corona, potentially impacting the binding efficiency of these tailored carriers.9,10The targeting strategy is often linked to the tumor type. The epithelial growth factor receptor (EGFR) is an illustrative example and targeting this receptor holds significant promise due to its status as a transmembrane protein that is frequently overexpressed or mutated in a variety of cancers.11–15 Several studies have established the involvement of EGFR in diverse phenotypic cascades, including cell proliferation, differentiation, invasion, angiogenesis, and metastasis, among others.16 This has led to the development and approval of mAbs and tyrosine kinase inhibitors (TKIs) for clinical use, such as cetuximab (Erbitux), erlotinib (Tarceva), gefitinib (Iressa), osimertinib (Tagrisso), dacomitinib (Vizimpro), lapatinib (Tykerb), mobocertinib (Exkivity), vandetanib (Caprelsa), panitumumab (Vectibix), and necitumumab (Portrazza). However, the development of resistance to such treatments requires the implementation of novel innovative strategies for improved delivery to cancer tissue and a more effective drug targeting approach. Of interest, it is well established that nanomedicine has demonstrated a preclinical capability to overcome multidrug resistance (MDR) mechanisms due to its distinct internalization process, differing from that of small molecules.17,18
Here, we aimed to use a biofunctionalized dendronized iron oxide NPs (DNPs) with EGFR-targeted agents presenting a compelling alternative to potentially address the challenge of drug delivery while mitigating MDR mechanisms, offering an effective solution in cancer therapeutics. The use of these DNPs additionally provides theranostic properties due to the intrinsic superparamagnetic properties of iron oxide.19–22 Indeed, spherical DNPs with a mean size of 18 nm (saturation magnetization (Ms) of 89 emu g−1) were shown to display SAR values of 147 W gFe−1 under laser irradiation for photothermal therapy (808 nm, 0.3 W cm−2) and around 100 W gFe−1 under magnetic hyperthermia (100 kHz, 32 kA m−1).23 In another study, dendronized 22 nm nanospheres and 19 nm nanocubes presented promising theranostic properties (respectively, r2 = 416 s−1 mM−1, SARMH = 580 W g−1, SARPTT = 800 W g−1; and r2 = 407 s−1 mM−1, SARMH = 899 W g−1, SARPTT = 300 W g−1).1 DNPs with a mean size of 12 nm and a Ms value of 56 emu g−1 did not heat at 300 G and 395 kHz (requiring a higher frequency of 536 kHz, which is outside the clinical limit, to generate heat) but displayed high heating power under photothermal conditions.1 Recently, we demonstrated that DNPs with few defects and a mean size of 26 nm, along with a saturation magnetization of 88 emu g−1, were shown to efficiently combine both magnetic hyperthermia and photothermia.24 Thus, DNPs which are well-known contrast agents for MRI regardless of their size and shape1,25–28 were shown to be good photothermal agents as well. Higher sizes were optimal to combine both magnetic hyperthermia and photothermia properties. To avoid the issue of full mAb functionalization,29 we focused our work on small peptide biofunctionalization strategies, allowing for improved orientation and functionalization control during the synthesis steps. The P22 peptide was chosen for the biofunctionalization strategy due to its specific advantages. While nanoparticle biofunctionalization with full monoclonal antibodies or fragments—regardless of the synthesis method—has been reported to improve tumor uptake and retention,6,7 large-scale synthesis and batch-to-batch consistency remain challenging with these methods. The use of a peptide targeting a tumor-associated antigen, such as EGFR, offers several benefits: peptides are smaller, more cost-effective, highly specific, more stable, and have reduced immunogenicity due to the absence of Fc-mediated effects. Additionally, in this specific study, the P22 peptide targets a different region of EGFR than cetuximab, potentially allowing for synergistic applications or serving as a follow-up therapy after cetuximab treatment.
The dodecapeptide GE11 (Tyr-His-Trp-Tyr-Gly-Tyr-Thr-Pro-Gln-Asn-Val-Ile) has demonstrated selectivity and specificity towards the EGFR allosteric ligand (kd = 22 nM).30 Previously reported studies have demonstrated the binding avidity of this GE11 peptide post-functionalization to various molecules including polymers, micelles, liposomes, and drugs such as doxorubicin, paclitaxel, or gemcitabine through the N- or C-terminus of the peptide, suggesting that terminal modifications did not alter the binding capacity to the receptor.31,32 When coupled to NPs, the resulting GE11-DNPs complex behaves similarly to previously reported active-targeting NPs with improved tumor retention and a similar PK profile to the naked NPs.6 Interestingly, through the introduction of the QE9 point mutation in the dodecapeptide GE11 sequence, the derived peptide, referred to as P22”, demonstrated superior specificity in comparison to GE11, with a marked affinity for targeting EGFR receptors.33 Our prior research demonstrated that DNPs coupled to P22 effectively targeted EGFR on head and neck cancer cell lines, but the NPs internalization levels remained quite low (≈10 and 1.6 pg of iron per HPV-FaDu cell with DNPs bearing P22 and without P22, respectively), while the the HPV+ 93-VU cell line exhibited minimal internalization.34 Indeed, these experiments have strongly suggested that the conformation of the peptide at the surface of DNPs was not optimal, with P22 tending to fold inward and become buried in dendron PEG chains. Therefore, a challenge remained in optimizing the orientation and accessibility of the peptide to the EGFR antigen.
This study aims to address this critical challenge by improving the availability of P22 and avoiding its folding into PEG chains. By adjusting the distance between NPs and peptides, we hypothesize that this approach can enhance tumor associated antigens (TAA) binding efficiency. The outcomes of this research hold the potential to advance the field of targeted nanomedicine, offering nuanced insights that could significantly refine the design and performance of nanoscale drug delivery systems. Therefore, in this research, we aimed to assess the influence of a spacer on the surface of the DNP, designed to enhance the presentation and conformation of the P22 peptide and improve its binding efficacy.
Results and discussion
Synthesis of the dendron-coated iron oxide nanoparticles (DNPs)
After their synthesis by the thermal decomposition method using a well-established protocol,35–37 the iron oxide NPs (IONPs) are covered with hydrophobic oleic acid molecules and then further coated with the dendron molecule through a reproducible ligand exchange process in THF, as described previously.38,39 The dendron is a bifunctional branched molecule, which bears at its focal point two phosphonic acid tweezers to ensure strong binding to the IONPs surface, while its periphery is designed to allow further conjugations to targeting ligands or fluorescent dyes.26,40,41 Specifically, the dendron exhibits three polyethylene glycol (PEG) chains, with a longer PEG6 chain in the middle compared to the two shorter side chains (PEG4) (Fig. 1 and ESI Fig. 1A–C†). The middle PEG chain bears a carboxylate end group, which serves as a coupling site for molecules such as fluorophores, chelating agents, or targeting ligands via a carbodiimidation reaction.34,42–44 Classical characterization of the IONPs through transmission electron microscopy (TEM) revealed a spherical morphology with an average diameter of 13.1 ± 1.3 nm (ESI Fig. 1D and E†). The magnetization curve at 300 K is characteristic of IONPS displaying superparamagnetic behavior, with a saturation magnetization of 53 emu g−1, consistent with already reported values for similar DNPs (ESI Fig. 1F†). The structural characterization by X-Ray diffraction confirmed the formation of a spinel iron oxide phase with a lattice parameter of 8.38 Å, intermediate between those of magnetite and maghemite phases (8.395 Å and 8.347 Å, respectively). The IR spectroscopy showed Fe–O bands in the range of 800–400 cm−1, at a position intermediate between those characteristic of maghemite (638 cm−1) and magnetite (571 cm−1), confirming the formation of IONPs with a composition consisting of an oxidized magnetite phase (ESI Fig. 1G and H†).35 The comparison of IR spectra between the dendron, oleic acid-coated IONPs, and dendronized IONPs revealed that the IR bands of oleic acid disappeared in the DNPs spectrum, while those corresponding to the dendron are clearly identified.20,25,26,34,45 The dynamic light scattering measurement confirmed a monomodal size distribution in water, with a mean hydrodynamic size of 17.9 ± 5.1 nm and a polydispersity index (PDI) of 0.19 (ESI Fig. 1I†). This increase in size aligns with the addition of dendron molecules, whose length is about 1.5–2 nm. This result additionally confirms the excellent colloidal stability of the dendron-coated IONPs (DNPs). Moreover, the dendron quantity per IONP was determined via inductively coupled plasma mass spectrometry (ICP-MS), indicating an approximate density of 1.4 ± 0.2 dendron molecules per nm2, consistent with both previously reported findings34 and theoretical calculations (the surface area of the dendron being approximately 72 Å2), corresponding to approximately 700 dendron molecules per DNP.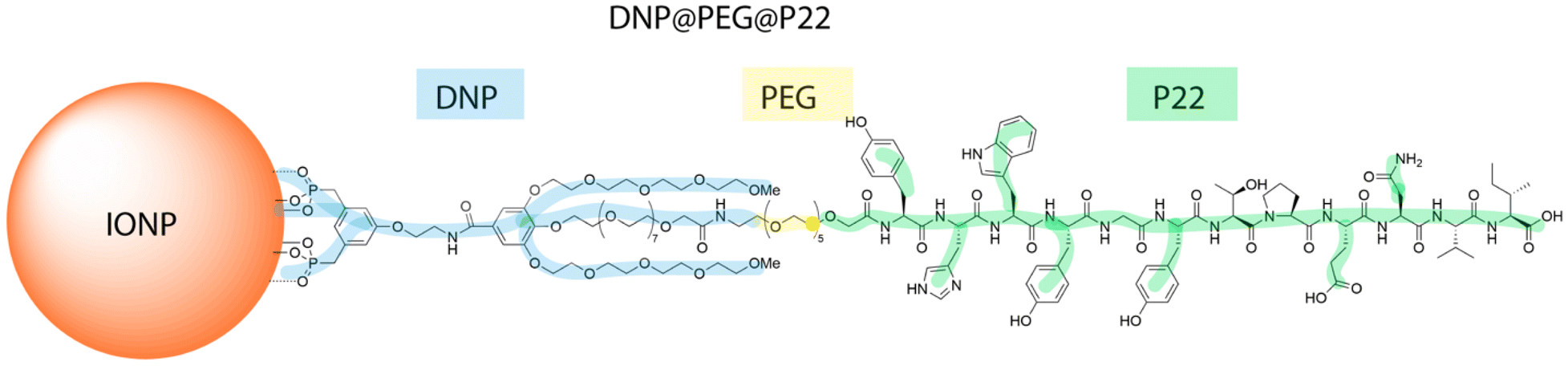 | ||
| Fig. 1 Structural characterizations of the DNP@PEG@P22: schematic and chemical representation of the dendronized iron oxide nanoparticle with the different layers of chemicals surrounding the iron oxide core. First, the dendron in blue, followed by the PEG linker in yellow and concluded by the peptide P22 in green. | ||
Functionalization strategy of the DNPs with PEG-P22 peptide for enhanced EGFR targeting
In addition to the labeling of DNPs with P22 and to improving the effectiveness and accessibility of P22-DNPs at the surface of DNPs, we sought to add a spacer - a PEG6 chain - to limit the folding of P22 within dendron PEG chains, as suggested in our previous study.34 However, such a spacer should also provide sufficient distance to avoid interference from the protein corona, reduce steric hindrance, and provide sufficient flexibility for P22 to bind the TAA. In this context, the PEG6 chain was added to the N-terminus of P22, resulting in PEG@P22 (NH2-PEG6-YHWYGYTPENVI), which was further coupled to the surface of the DNPs by a carbodiimidation reaction. The amount of coupled PEG@P22 was difficult to determine through IR spectroscopy, as the strong overlap of P22 bands with those of the dendron made it impossible to unambiguously identify the P22 IR bands.34 To quantify the amount of P22 and PEG@P22 grafted onto the surface of DNPs, we performed indirect quantification by high performance liquid chromatography coupled with UV spectroscopy (HPLC-UV). After synthesis, the supernatant was weighed to ascertain the combined mass of unbound P22 and PEG@P22 using the HPLC standard curve, specifically by evaluating the area under the peaks at 280 nm (ESI Fig. 1J†). The binding efficiency was subsequently determined by subtracting the predetermined quantity of unbound PEG@P22 or P22 from the total intial amount. The results revealed 33% and 37.5% of PEG@P22 or P22, respectively, were covalently linked to the dendrons. Thus, on average, each DNP is coated with a first layer of approx. 700 carboxylate groups on the dendrons and a second layer consisting of 230 PEG@P22 or 260 P22 molecules attached to the dendrons. This results in an increase in the mean hydrodynamic size up to 25.8 ± 8.3 and 17.9 ± 5.1 nm after the coupling of PEG@P22 and P22 respectively (ESI Fig. 1I†). The larger increase in the mean hydrodynamic diameter with DNPs@PEG@P22 compared with DNPs and DNPs@P22 strongly suggests that P22 is positioned outside the DNPs rather than being buried withinthe dendron PEG chains.The addition of a PEG spacer increased the specificity of EGFR targeting in vitro
After successfully functionalizing DNPs with P22, both with and without a PEG spacer in between, we assessed whether the addition of the PEG chain on the dendrons of the DNPs affected the specificity of P22 in binding to EGFR. Previously conducted viability assays with DNP@P22 demonstrated no cytotoxicity up to 100 μg Fe per mL in head and neck cancer cell lines.34 Similarly, we did not observe any signs of cytotoxicity at this concentration in the breast cancer cell lines used in this study. Subsequently, we first sought to validate that the addition of P22 led to improved uptake in EGFR+ cells (MDA-MB-231) (ESI Fig. 2A and B†). These cells were incubated at 37 °C overnight with either DNPs, DNP@P22, or DNP@PEG@P22. Following incubation, the cells underwent three successive washes with PBS, and we quantified their uptake of iron using the Prussian blue staining.34 As expected, minimal internalization was observed with EGFR-MCF-7 cells, but statistically significant differences were observed between the different NPs (Fig. 2A) with MDA-MB-231 cells. As expected, the enhancement in internalization between DNPs and DNP@P22 was quite low (p-value = 0.17, one-way ANOVA test), whereas a 10-fold increase in uptake was observed were DNPs were coupled with PEG@P22 (p-value = 0.003, one way ANOVA test). This increased uptake resulted in higher cytotoxicity at high concentrations in these EGFR+ cells compared to both DNPs@P22 and DNPs alone. Importantly, no differences were observed between these constructs in EGFR− cells (MCF7). This cell internalization study strongly suggests that the PEG spacer facilitates a suitable conformation of P22 at the surface of DNPs, thereby promoting its interactions with EGFR+ MDA-MB-231 cells (Fig. 2B).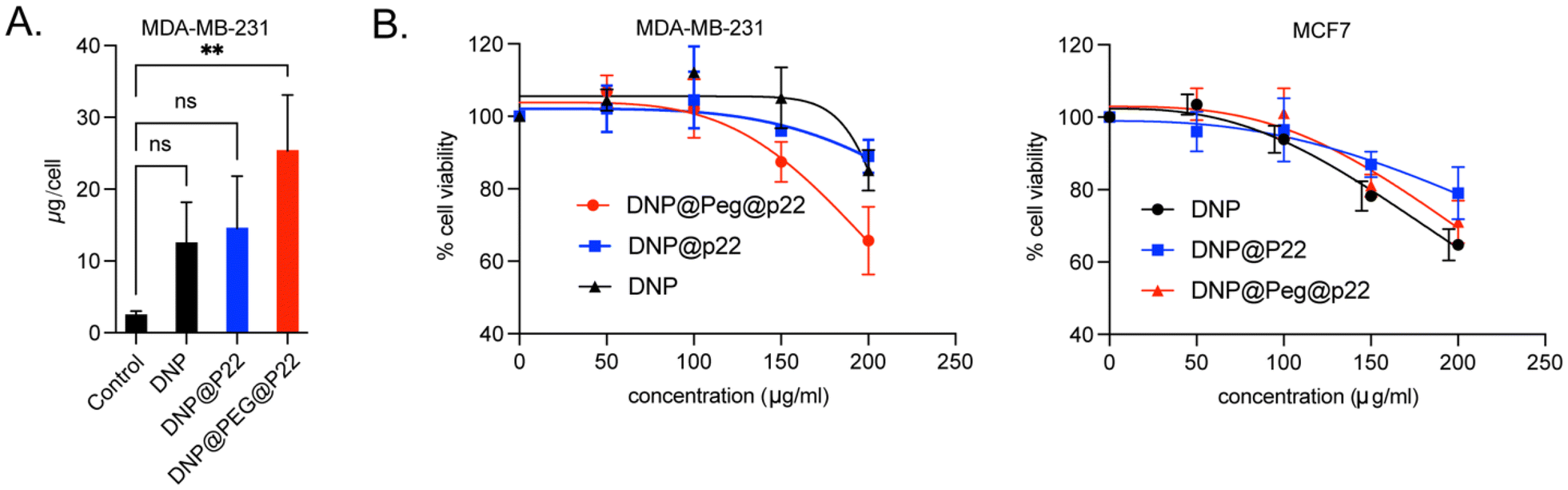 | ||
| Fig. 2 A. Fe uptake (pg per cell) in MDA-MB-231 for the different types of nanoparticles. B. Alamar blue cell viability assay for MDA-MB-231 and MCF-7 cell lines using different DNPs. Fe concentrations varied from 0 to 200 μg mL−1. | ||
With the observed enhancement in NP uptake prompted by the presence of the PEG linker surrounding P22, our research shifted focused to elucidating the cellular effects of these DNP@PEG@P22s. Considering the low cell internalization of DNPs@P22 and our objective to specifically study the interaction of P22 with cells while minimizing artefacts from any potentially exposed P22 at the surface of DNPs@P22, we narrowed this study to DNPs and DNPs@PEG@P22. We first confirmed the internalization of DNP@PEG@P22 by confocal microscopy. Specifically, our analysis focused on visualizing the adherence patterns of untargeted DNPs and targeted DNP@PEG@P22 on both MDA-MB-231 cells and MCF-7 cells (Fig. 3A and B). As anticipated, untargeted DNPs did not bind to MCF-7 cells, even when coupled with the PEG@P22. However, an increase in binding was observed with MDA-MB-231 cells when PEG@P22 was added (Fig. 3A). A competition binding study was then conducted on MDA-MB-231 cells using fluorescently labeled cetuximab, a clinically approved monoclonal antibody targeting EGFR. Interestingly, we observed that DNP@PEG@P22 demonstrated comparable binding activity to cetuximab on MDA-MB-231 cells, confirming that both cetuximab and P22 exhibited non-competitive interaction. This suggests that the two agents target distinct domains of the EGFR, allowing the possibility to use the former as either a companion biomarker for the latter or in a therapeutic combination.
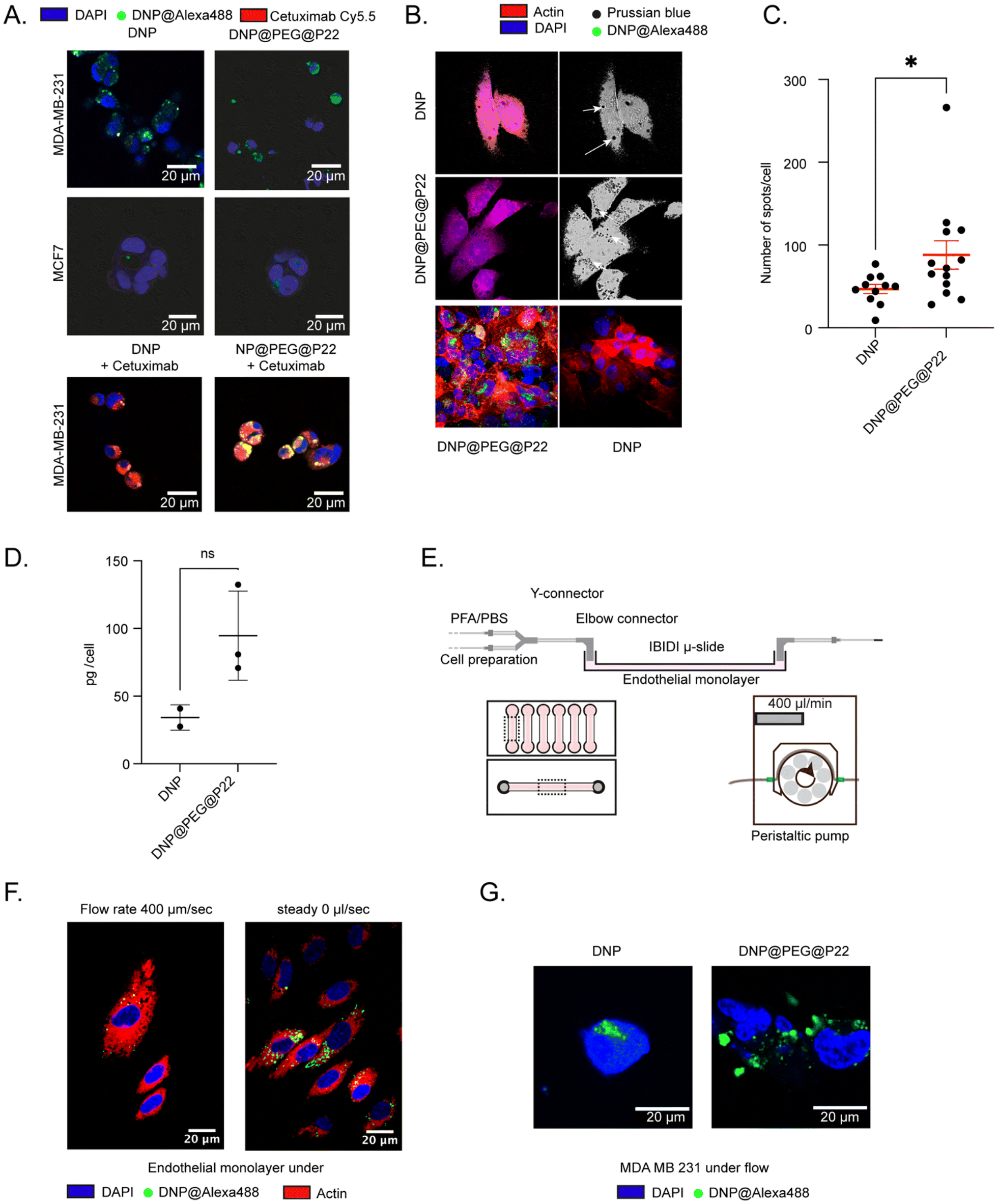 | ||
| Fig. 3 DNPS@PEG@P22 affinity to different cell lines with or without hydrodynamic flow. A. Fluorescence confocal images of MDA-MB-231 and MCF7 cells. Nuclei are labelled with DAPI, NPs labelled with Alexa-488 and Cy5.5-cetuximab labels EGFR proteins. B. Confocal images of MDA-MB-231 cells treated with DNP or DNP@PEG@P22. Nuclei are labeled with DAPI, actin is labeled with Alexa Fluor™ 532 Phalloidin, and the NPs are labeled with Prussian blue. Arrows indicate the DNPs stained with Prussian blue C. Graphical representation of the number of dots counted in different cells. D. ICP-MS quantification on MDA-MB-231 cells for the different synthesized NPs (pg per cell). E. Schematic representation of the microfluidic system developed to perform experiments under flow conditions. F. Confocal images obtained on endothelial cells incubated with Alexa488-DNP@PEG@P22 under flow conditions or in steady conditions. G. Confocal images obtained on MDA-MB-231 cells incubated with Alexa488-DNP or Alexa488-DNP@PEG@P22 under flow conditions. | ||
To further characterize the cellular uptake of DNPs@PEG@P22, we initially employed a classical 2D culture system. MDA-MB-231 cells were incubated with DNPs and DNPs@PEG@P22 formulations at a concentration of 100 μg mL−1 for 24 hours. Following incubation and washing steps, Prussian blue staining was performed and subsequently imaged and quantified to determine the number of DNPs per cell, taking advantage of the specificity of this staining method toward the iron core of the DNPs.1,46,47 Subsequently, imaging was conducted on fluorescently labeled DNPs to discern differences in internalization between the DNPs and DNP@PEG@P22 formulations (Fig. 3B). While some non-specific internalization of passively targeted DNPs could not be entirely avoided, the specific interaction with the MDA-MB-231 cells significantly increased in the presence of PEG@P22. Notably, the inclusion of the PEG chain between the DNPs and the P22 resulted in a 2-fold increase in uptake compared to passive targeting (p-value = 0.02, Mann–Whitney test) (Fig. 3C). This enhanced uptake was further confirmed by ICP-MS measurements, where the use of the PEG chain increased the amount of intracellular iron content from 34 pg to 95 pg per cell (Fig. 3D).
Encouraged by these results, we next sought to assess the binding affinity of DNP@PEG@P22 under a 2D microfluidic system to replicate, as closely as possible, the in vivo bloodstream environment preceding tumor internalization (i.e., EGFR+ cells accessible only upon migration through the endothelial monolayer understeady flow rate condition) (Fig. 3E). As expected, the presence of the flow decreased the total amount of DNP@PEG@P22 internalized by the endothelial cells, thereby reducing off-target binding (Fig. 3F). Consecutively, the targeting approach significantly increased uptake in the MDA-MB-231 cells compared to non-targeted DNPs, confirming that the presence of a spacer between the NPs and the peptide creates a proper environment for effective TAA binding efficacy and enhances uptake efficiency (Fig. 3G).
Enhanced in vitro photothermal therapy efficacy was observed when incorporating a spacer between DNPs and the targeting peptide
The main advantage of using DNPs is their ability to release heat after absorbing infrared irradiation.48–51 This phenomenon, called photothermal therapy (PTT), is well-known as an emerging cancer therapy option.52–55 To confirm this effect, we used MDA-MB-231 cells treated with 100 μg mL−1 of DNPs and DNP@PEG@P22, which were then irradiated at 1064 nm at 2 W cm−2 for 15 minutes, one hour after DNPs incubation (ESI Fig. S3†). This protocol is known to be non-toxic to cells in the absence of NPs.56,57 Under these conditions, the IC50 values decreased from 40 μg ml−1 when non-irradiated to 16 μg ml−1 and 6 μg ml−1 for DNPs and DNPs@PEG@P22 when irradiated, respectively. This finding demonstrates that the targeted approach enhances the efficacy of PTT. To thoroughly evaluate the biological impact of these DNP@PEG@P22 formulations upon PTT, we quantified the DNA double-strand breaks (DSBs) and DNA DSBs repair via 53BP1 foci, analyzing both induced and residual DSBs at two timepoints - 30 min and 24 h, respectively (Fig. 4A and B). As expected, the number of foci remained stable in MCF7 across all timepoints. In contrast, a significant increase was observed in MDA-MB-231 cells when the DNPs were associated with PTT (p-values = 0.04 for γH2AX and 0.0003 for 53BP1 with Kruskal–Wallis tests). This increase became noticeable after 30 minutes and was significantly more pronounced 24 h post-therapy. The targeted binding of DNPs to tumor cells, coupled with PTT, triggers DNA damage through two main mechanisms: local heating due to the interaction between the DNPs and the infrared light, and an amplified release of reactive oxygen species (ROS) in the vicinity of the DNPs, both of which contributed to increased cell death.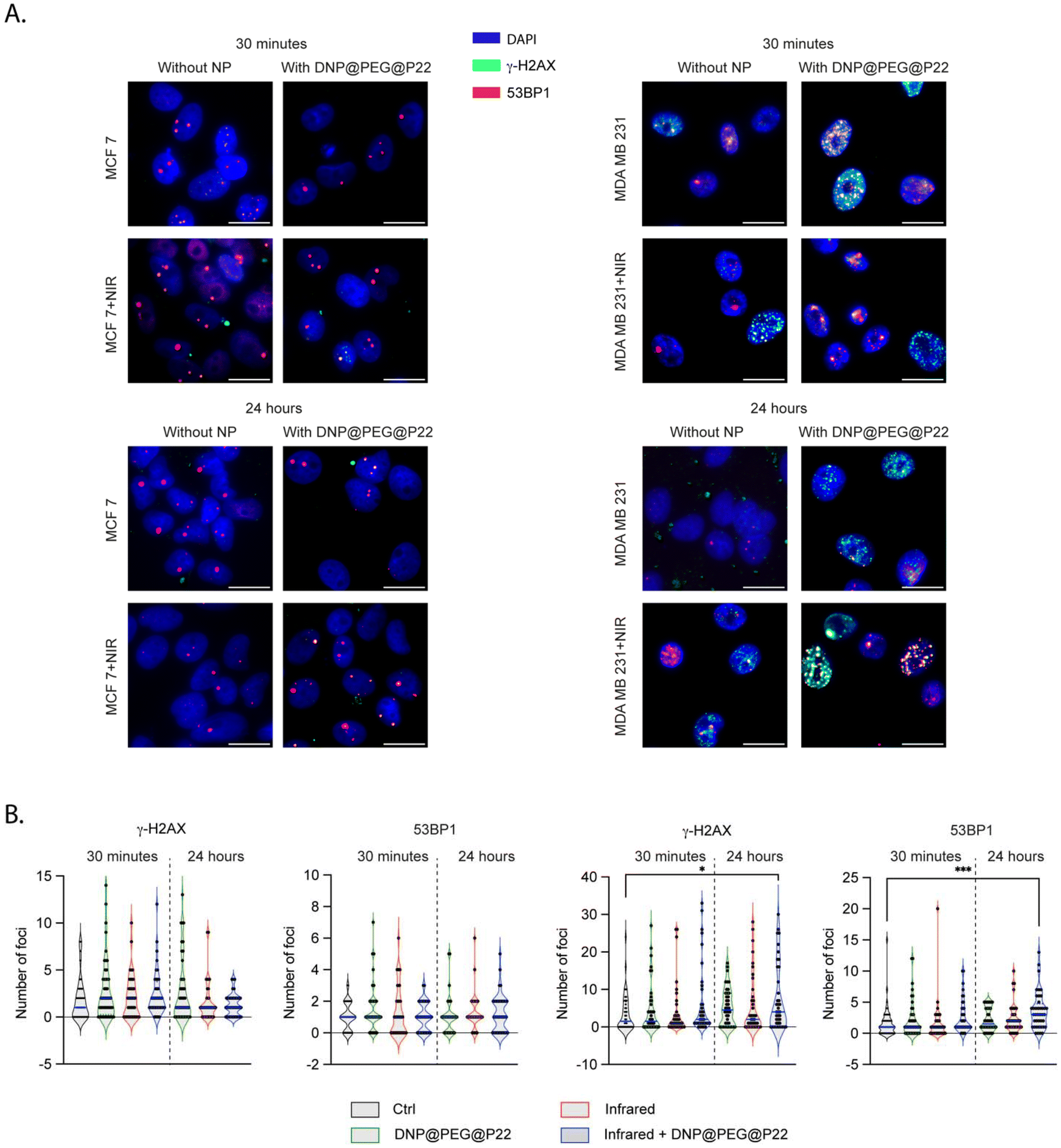 | ||
| Fig. 4 Cell viability and affinity for the different treatments with and without DNP@PEG@P22. A. Immunofluorescence images of γH2AX (green) and 53BP1 (red) foci in MDA-MB-231 and MCF7 cells at 30 minutes and 24 hours post-treatments. B. Foci quantification under the different treatments. | ||
Conclusion
In summary, this research underscores the significant challenge of efficiently targeting tumor cells with theragnostic nanoparticles for effective cancer treatment. The findings indicate that the mere addition of targeting ligands to nanoparticle surfaces is insufficient to ensure therapeutic efficacy. Recent insights advocate for the incorporation of a spacer between the targeting ligand and the nanoparticle surface to provide a more suitable conformation of the targeting ligand. This spacer is crucial, as it prevents ligands from being buried in molecular coatings, enhances flexibility for receptor interaction, and mitigates the impact of protein corona formation.The binding efficiencies observed with the P22 peptide were consistent with those previously reported in vitro for other cell lines, indicating reliable target engagement across different biological contexts.31–34 However, the introduction of the linker significantly improved these binding results, highlighting the critical role of spatial optimization in nanoparticle design. The increased efficiency confirms that the presence of the peptide alone at the nanoparticle surface can induce steric hindrance, limiting its accessibility to the target. The enhanced performance with the linker underscores the necessity of such structural modifications to mitigate steric effects and optimize biofunctionalization for therapeutic applications.
Functionalizing DNPs with the EGFR-targeting peptide P22, along with a PEG spacer, yielded promising results. The presence of the peptide enhanced cytotoxicity, allowing for lower doses without compromising efficacy. Initial experiments with EGFR+ breast cancer cells demonstrated increased internalization of DNPs bearing a targeting ligand with a PEG spacer, confirming its role in improving the accessibility of the binding agent to the TAA. Further characterization, including confocal imaging and Prussian blue staining, substantiated these findings. Importantly, DNPs with PEG@P22 exhibited enhanced efficacy in photothermal treatment upon cellular internalization. These results highlight the importance of targeting ligand conformation and accessibility in improving the therapeutic performance of targeted nanoparticles in cancer treatment.
The PEG6 linker was selected as the optimal linker length due to its ability to balance several critical factors in nanoparticle biofunctionalization. The PEG6 chain provides sufficient spatial separation between the nanoparticle core and the P22 peptide, minimizing steric hindrance and allowing for effective EGFR binding without interference from the nanoparticle surface. This spacing ensures that the peptide can access the target antigen while maintaining a compact overall nanoparticle size, which is crucial for favorable pharmacokinetics and biodistribution. We speculated that a shorter PEG chain might not provide adequate separation to overcome steric hindrance, potentially compromising the binding efficiency of the peptide. Conversely, longer PEG chains could further reduce steric effects but would likely increase nanoparticle size, potentially impairing tumor penetration and clearance profiles. Furthermore, the flexibility of PEG6 enables the peptide to adopt the most favorable orientation for binding, thereby improving specificity and affinity. PEG chains are also well-known for their ability to confer stealth properties by reducing immune recognition and protein adsorption. PEG6 provides a balance between providing sufficient immune evasion while preventing overly prolonged circulation, which could lead to off-target accumulation and systemic toxicity.
While other spacer molecules, such as alkyl chains or dendrimers, could theoretically be used, PEG offers superior solubility, biocompatibility, and well-established performance in nanoparticle systems. Non-PEG-based alternatives might present challenges in achieving the same level of hydrophilicity and flexibility. Thus, PEG6 represents an ideal choice for this biofunctionalization strategy, offering a combination of steric shielding, flexibility, and biocompatibility, which are difficult to achieve with other linkers or alternative chain lengths.
This study, which demonstrates the effect of a spacer on NPs cell internalization underscores the importance of the availability of targeting ligands at the NP surfaces to enhance specific internalization marking a critical advancement in nanoparticle-based cancer therapies.
Materials and methods
Synthesis of 10 nm iron oxide nanoparticles
Iron stearate (FeSt3) with a minimum purity of 60% stearic acid, 5.8–7% iron, and a maximum of 10% free acid was procured from TCI. Acetone (99.8% purity), chloroform (99% purity), and THF (99.5% purity) were obtained from Carlo Erba. Dioctyl ether (OE) with a purity of 99%, HEPES buffer (99.5% purity), N-hydroxy sulfosuccinimide (sulfo-NHS, 98% purity), sodium carbonate (Na2CO3, 99.5% purity), thiazolyl blue tetrazolium bromide (MTT, 98% purity), and 1-ethyl-3-(3′-dimethylaminopropyl)carbodiimide hydrochloride (EDC-HCl, 99% purity) were purchased from Sigma Aldrich. Sodium hydrogen carbonate (NaHCO3, 99.5% purity) was obtained as a Bioreagent. Oleic acid (OA) with a purity of 99% was sourced from Alfa Aesar. Dimethyl sulfoxide (DMSO, 99% purity) and Alexa Fluor™ 488 Cadaverine were acquired from Fisher Scientific. The dendron D1-2P was supplied by Superbranche SAS.IONPs were synthesized via thermal decomposition of an iron stearate precursor, following a reported procedure.35 In summary, 2.2 mmol of FeSt3 and 4.4 mmol of OA were mixed with 20 mL of dioctyl ether in a 100 mL two-neck round-bottom flask. The mixture was heated to 120 °C for 60 min until complete reagent dissolution. Subsequently, a condenser was attached, and the mixture was heated to 290 °C at a rate of 5 °C min−1. Refluxing at 290 °C occurred over 120 min, resulting in the formation of black IONPs suspended in OA (IONPs@OA). After cooling to 100 °C, washing steps were initiated by adding 10 mL of chloroform to the IONPs@OA, followed by placement in a flask containing 400 mL of acetone. This mixture was heated at 60 °C for 1 h under mechanical stirring in a thermal bath. The IONPs@OA were collected with a magnet, and the process was repeated. The final IONPs@OA were collected, and after discarding the supernatant, they were resuspended in 40 mL of THF for storage until further use.
Dendronization of IONPs
The dendron D1-2P was provided by Superbranche company. The ligand exchange between OA and the dendron (D1-2P) was conducted in THF.38 In the initial step, IONPs were combined with the dendron at a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7 [Fe:dendron] overnight at room temperature. The resulting suspension was purified through ultrafiltration, and a second ligand exchange was performed (at a ratio of 5:5) to optimize dendronization. Subsequently, the suspension of dendronized nanoparticles (DNPs) was mixed with hexane (volume ratio 1/3:2/3) to induce DNP precipitation. To further enhance the precipitation of DNPs, the suspension was centrifuged at 8000 rpm for 5 minutes, and the supernatant was discarded. Finally, the DNPs were collected, dispersed in deionized water, and stored in water at 4 °C.
:7 [Fe:dendron] overnight at room temperature. The resulting suspension was purified through ultrafiltration, and a second ligand exchange was performed (at a ratio of 5:5) to optimize dendronization. Subsequently, the suspension of dendronized nanoparticles (DNPs) was mixed with hexane (volume ratio 1/3:2/3) to induce DNP precipitation. To further enhance the precipitation of DNPs, the suspension was centrifuged at 8000 rpm for 5 minutes, and the supernatant was discarded. Finally, the DNPs were collected, dispersed in deionized water, and stored in water at 4 °C.
Coupling reaction
Peptide 22 (P22) and NH2-PEG(6)-YHWYGYTPENVI (PEG-P22) with a purity of 98.7% were procured from polypeptide SAS, and the coupling process followed the protocol developed by Freis et al.58 The reaction involved two steps. In the first step, DNPs were suspended in HEPES 0.1 M, pH 6.5, at a concentration of 5 mg Fe per mL. EDC was then added at a 20-fold molar excess compared to the carboxylate groups of the dendron. The reaction was stirred for 10 minutes, followed by the addition of sulfo-NHS (also at a 20-fold molar excess compared to carboxylate groups on dendron molecules) for an additional 20 minutes. The activated DNPs are washed with 0.1 M carbonate buffer, pH 9.2, using a centrifugal filter unit (10 minutes at 8000 rpm) to eliminate excess EDC, sulfo-NHS, and other possible reaction intermediates. The filtrate was promptly recovered and mixed with 0.9 molar equivalents of PEG-P22 (compared to dendron molecules) in carbonate buffer. The reaction proceeded overnight at a final iron concentration of 0.5 mg mL−1 before purification with centrifugal filter units (Amicon®, MW cutoff: 30 kDa in cycles of 10 minutes at 8000 rpm). Multiple washes were performed, and each supernatant was retained for indirect quantification (of unbound PEG-P22) by HPLC. For the coupling with Alexa, the same protocol was used.Characterization of the nanoparticles
Transmission electron microscopy (TEM) was employed to characterize the sizes and structures of the IONPs. The analysis was conducted using a JEOL 2100 microscope operating at 200 kV with a point resolution of 0.18 nm. Size distribution estimation was performed using ImageJ software.X-ray diffraction (XRD) patterns were collected at room temperature using a Bruker D8 Discover diffractometer in Bragg Brentano geometry. The system was equipped with a monochromatic copper radiation source (Kα1 = 0.154056 nm) and an energy-resolved Lynx-Eye XE-T detector in the 25–65° (2θ) range with a scan step of 0.03°. High purity silicon powder (a = 0.543082 nm) served as an internal standard. LeBail's method59 was applied for further refinement of diffraction patterns using Fullprof software.60 The background, modelled as a linear function based on 20 experimental points, was refined, along with the zero shift. Peaks were modelled using the modified Thompson–Cox–Hasting (TCH) pseudo-Voigt profile function.
Dynamic light scattering (DLS) measurements and zeta potential assessments were performed using a MALVERN (nano ZetaSizer) instrument to evaluate the colloidal stability and mean hydrodynamic diameter of the IONPs suspension in water after ligand exchange.
High-performance liquid chromatography-UV spectroscopy (HPLC-UV) analysis was carried out on an Agilent 1100 system equipped with a diode-array detector (DAD). For PEG-P22 quantification, 10 μL of samples (supernatant and washes obtained after the coupling reaction) were injected into the column (Zorbax Extend-C18, 150 × 4.6 mm, 3.5 μm, Agilent) at room temperature. Ultrapure water, HPLC-grade solvents, and additives were used for the analysis. The elution gradient was performed with trifluoroacetic acid (TFA) 0.1% (mobile phase A) and acetonitrile acidified with TFA 0.1% (mobile phase B) at a flow rate of 1 mL min−1. The DAD simultaneously detected at 220 ± 5 nm and 280 ± 5 nm. Integration of the peak area on the chromatogram, corresponding to the analyte of interest, was compared to calibration areas obtained at different concentrations of PEG@P22.
Cell lines
MDA-MB-231 and MCF-7 cell lines were purchased from ATCC and cultured according to the manufacturer's instructions. Prior to usage, mycoplasma tests confirmed the absence of contamination in both cell lines. Specifically, the MDA-MB-231 cells were cultivated in DMEM medium supplemented with 10% fetal bovine serum FBS, 1% penicillin–streptomycin, and MCF7 in EMEM medium supplemented with 10% FBS, 0.01 mg mL−1 human recombinant insulin and 4 μg mL−1 filtered blasticidin. Incubation took place at 37 °C with 5% CO2 under fully humidified conditions.Cell viability assay
Cell viability assays were conducted using the Alamar blue test. MDA-MB-231 or MCF7 cells were seeded in their specific medium 96-well opaque plates at 15000 cells per well and incubated for 24 hours. DNPs, diluted in fresh medium, were added at various concentrations to achieve a final concentration ranging from 0 μg mL−1 to 200 μg mL−1 Fe. The mixture of DNPs and cells was incubated for 24 hours before being washed 3 times with PBS and addition of 100 μL of culture medium supplemented with 10% of Alamar blue. After 4 more hours of incubation, the fluorescence was collected from each well (excitation 545 nm, emission 600 nm).
In ESI Fig. 3† the cell viability was performed in a 96 well plate where 5000 cells were seeded in medium complemented only with 1% FBS to avoid any cell growth for 24 hours. Then the nanoparticles were added at 100 mg ml−1 for 15 minutes before insolation for 15 minutes at 2 W cm−2. The cells were additionally incubated for 24 hours before CTG analysis.
Iron uptake quantification
The internalization of DNPs and DNPs@PEG-P22 into cells was quantified using the Prussian blue reaction developed by Boutry et al.42 For these assays, a concentration of 100 μg mL−1 Fe was employed based on viability tests. MDA-MB-231 or MCF7 cells were seeded in a 6-well plate and incubated for 24 h at 37 °C and 5% CO2 to allow for attachment (∼80% confluence reached). The following day, the medium was replaced with fresh medium containing 100 μg mL−1 of DNPs or DNPs@PEG-P22. The incubation time with the NPs was 24 h. After this period, the cells were trypsinized, gently recovered from the wells in a 1:5 volume ratio of trypsin to cell medium and counted in a KOVA chamber. The cells were then centrifuged and washed three times with PBS 1× before being digested with 5 N HCl for 24 h at 37 °C. HCl also digested the cell-internalized DNPs, leading to the formation of Fe3+. The addition of potassium ferrocyanide trihydrate, a salt that reacts with Fe3+, forms a colored complex called Prussian blue. In parallel, 100 μL of DNPs suspensions at different concentrations were exposed to the same conditions as previously described to obtain the calibration curve. All suspensions were shaken on an orbital stirrer for 15 min, and the absorbance was read on a microplate reader (Xenius XC spectrophotometer, SAFAS) at 630 nm (Fig. 2B). On Fig. 3C, ICP-MS experiments were performed to quantify the mass of iron up taken by the cells.
Immunofluorescence imaging
Cells were cultured on 4-well Lab-Tek chambers (Nunc, Thermo Fisher) at a density of 25 × 103 cells per well. They were then treated with 12.5 mg mL−1 IONPs for 24 hours and subsequently subjected to 15 minutes of 2 W cm−2 infrared (IR) irradiation or left untreated. After IR irradiation, cells were returned to the incubator and fixed either 30 minutes or 24 hours post-laser with 4% paraformaldehyde for 10 minutes at room temperature. Subsequently, cells were permeabilized using 0.5% Triton-X-100 for 5 minutes and blocked with 1% BSA in 0.1% Triton-X-100 and PBS solution for 20 minutes at room temperature. Next, cells were incubated with primary antibodies against γH2AX (ser139) (Merck Millipore, 05-636-I) and 53BP1 (Novus, NB100-304) in the blocking solution overnight at 4 °C. Subsequently, secondary antibodies, goat anti-mouse IgG (Alexa Fluor 488, Invitrogen A-11029), and goat anti-rabbit IgG (Alexa Fluor 568, Invitrogen A-11036), were applied for 1 h at room temperature. Slides were counterstained using FluoromountG with DAPI (Invitrogen 00-4959-52), and γH2AX and 53BP1 foci were counted and analyzed using a Nikon Eclipse 80-i fluorescence microscope equipped with a digital camera.For the competition study between cetuximab and the P22 peptide, the cetuximab was labeled using reaction between amine groups of the mAb and NHS ester group. The cetuximab was purchased at the ICANS Pharmacy and labeled with Cy5.5 NHS ester (#27020 Lumiprobe). The reagents were incubated in a 1:5 molar ratio of cetuximab and dye overnight at 4 °C. Purification was performed using dye removal columns (#22858 ThermoFisher). The labeled mAbs were stored at 4 °C after purification.
Statistical analysis
Data are represented as mean ± standard deviation. All results were analyzed using GraphPad Prism 9.5.1. Statistical analysis was performed using unpaired non-parametric t-test for multiple groups comparison at a specific time point (Mann–Whitney test) or a one-way ANOVA for group comparison. Bonferroni post hoc test was performed to compare the different conditions. Significance was determined at the following cutoff points: *P < 0.05, **P < 0.01, and ***p < 0.001.Author contributions
M. A. R. and B. F. designed and characterized the nanoparticles, performed the cellular uptake experiments with Prussian blue. J. D. labeled cetuximab with the Cyanine 5.5 and imaged the cells. T. G. performed the Alamar blue experiments F. R. and O. T. performed and analyzed the ICP-MS experiments. C. C., M. A. R. and B. F. performed the HPLC-UV analysis. J. B.-G. performed the immunofluorescence experiments and their analysis. A. G. extracted the proteins and quantified by western blot the amount of EGFR proteins. S. H. performed the imaging experiments. S. J. designed the molecular constructs. X. P., G. N., A. D., S. H., S. L. and S. B. designed the study and analyzed the results. M. A. R., X. P., S. H., A. D., and S. B. wrote the manuscript.Data availability
The main data supporting the results in this study are available within the paper and the ESI.† The raw data are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
A. D. acknowledges funding by, in part, the European Research Council (ERC) Starting Grant TheranoImmuno (grant agreement #95010), ITMO cancer of Aviesan within the framework of the 2021–2030 Cancer Control Strategy, on funds administered by Inserm, and the labelisation by the Ligue Contre le Cancer. S. H. acknowledges the Strasbourg Drug Discovery and Development Institute (IMS) for funding this study, as part of the Interdisciplinary Thematic Institute (ITI) 2021–2028 program of the University of Strasbourg, CNRS and Inserm, IdEx Unistra (ANR-10-IDEX-0002), and by the SFRI-STRAT'US project (ANR-20-SFRI-0012) under the framework of the French Investments for the Future Program. A. D. and S. H. also acknowledge support from the Institut of Cancerologie de Strasbourg Europe. B. F. acknowledges support from The Region Alsace, France, and the University of Mons for the doctoral fellowship. S. B. acknowledges funding from ANR (EURONANOMED2020-121 – THERAGET) under the umbrella of the ERA-NET EuroNanoMed (grant agreement #723770) of the EU Horizon 2020 and from Alsace Contre le Cancer. Additionally, the authors acknowledge support from Pascal Kessler at the PIC-STRA imaging platform from the University of Strasbourg. SuperBranche is thanked for providing dendron molecules.References
- B. Freis, M. D. L. A. Ramirez, C. Kiefer, S. Harlepp, C. Iacovita, C. Henoumont, C. Affolter-Zbaraszczuk, F. Meyer, D. Mertz, A. Boos, M. Tasso, S. Furgiuele, F. Journe, S. Saussez, S. Bégin-Colin and S. Laurent, Pharmaceutics, 2023, 15, 1104 CrossRef CAS PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139–2147 CrossRef CAS PubMed.
- D. C. Julien, S. Behnke, G. Wang, G. K. Murdoch and R. A. Hill, mAbs, 2011, 3, 467–478 CrossRef PubMed.
- M. Brückner, J. Simon, K. Landfester and V. Mailänder, Nanoscale, 2021, 13, 9816–9824 RSC.
- H. S. Choi, W. Liu, F. Liu, K. Nasr, P. Misra, M. G. Bawendi and J. V. Frangioni, Nat. Nanotechnol., 2010, 5, 42–47 CrossRef CAS PubMed.
- V. Mittelheisser, P. Coliat, E. Moeglin, L. Goepp, J. G. Goetz, L. J. Charbonnière, X. Pivot and A. Detappe, Adv. Mater., 2022, 34, 2110305 CrossRef CAS PubMed.
- L. Carmès, M. Banerjee, P. Coliat, S. Harlepp, X. Pivot, O. Tillement, F. Lux and A. Detappe, Adv. Ther., 2023, 6, 2300019 CrossRef.
- A. Accardo, D. Tesauro and G. Morelli, Polym. J., 2013, 45, 481–493 CrossRef CAS.
- T. Kopac, Int. J. Biol. Macromol., 2021, 169, 290–301 CrossRef CAS PubMed.
- M. Mahmoudi, M. P. Landry, A. Moore and R. Coreas, Nat. Rev. Mater., 2023, 8, 422–438 CrossRef PubMed.
- T. Troiani, E. Martinelli, A. Capasso, F. Morgillo, M. Orditura, F. De Vita and F. Ciardiello, Curr. Drug Targets, 2012, 13, 802–810 CrossRef CAS PubMed.
- R. Roskoski, Pharmacol. Res., 2014, 79, 34–74 CrossRef CAS PubMed.
- M.-H. Chien, Y.-C. Yang, K.-H. Ho, Y.-F. Ding, L.-H. Chen, W.-K. Chiu, J.-Q. Chen, M.-C. Tung, M. Hsiao and W.-J. Lee, Cell Death Dis., 2024, 15, 82 CrossRef CAS PubMed.
- Z. Abrehdari-Tafreshi, E. Arefian, N. Rakhshani and S. M. A. Najafi, Biochem. Genet., 2024, 62(6), 4929–4951 CrossRef CAS PubMed.
- H.-P. Lee, C.-J. Li and C.-C. Lee, Head Neck, 2024, 46(6), 1294–1303 CrossRef PubMed.
- M. L. Uribe, I. Marrocco and Y. Yarden, Cancers, 2021, 13, 2748 CrossRef CAS PubMed.
- S. Kunjachan, A. Błauż, D. Möckel, B. Theek, F. Kiessling, T. Etrych, K. Ulbrich, L. van Bloois, G. Storm, G. Bartosz, B. Rychlik and T. Lammers, Eur. J. Pharm. Sci., 2012, 45, 421–428 CrossRef CAS PubMed.
- S. Kunjachan, B. Rychlik, G. Storm, F. Kiessling and T. Lammers, Adv. Drug Delivery Rev., 2013, 65, 1852–1865 CrossRef CAS PubMed.
- C. Blanco-Andujar, A. Walter, G. Cotin, C. Bordeianu, D. Mertz, D. Felder-Flesch and S. Begin-Colin, Nanomedicine, 2016, 11, 1889–1910 CrossRef CAS PubMed.
- G. Cotin, C. Blanco-Andujar, F. Perton, L. Asín, J. M. de la Fuente, W. Reichardt, D. Schaffner, D.-V. Ngyen, D. Mertz, C. Kiefer, F. Meyer, S. Spassov, O. Ersen, M. Chatzidakis, G. A. Botton, C. Hénoumont, S. Laurent, J.-M. Greneche, F. J. Teran, D. Ortega, D. Felder-Flesch and S. Begin-Colin, Nanoscale, 2021, 13, 14552–14571 RSC.
- B. Freis, G. Cotin, F. Perton, D. Mertz, S. Boutry, S. Laurent and S. Begin-Colin, in Magnetic Nanoparticles in Human Health and Medicine, ed. C. Caizer and M. Rai, Wiley, 1st edn, 2021, pp. 380–429 Search PubMed.
- G. Cotin, F. Perton, C. Blanco-Andujar, B. Pichon, D. Mertz and S. Bégin-Colin, in Nanomaterials for Magnetic and Optical Hyperthermia Applications, Elsevier, 2019, pp. 41–60 Search PubMed.
- C. Lozano-Pedraza, E. Plaza-Mayoral, A. Espinosa, B. Sot, A. Serrano, G. Salas, C. Blanco-Andujar, G. Cotin, D. Felder-Flesch, S. Begin-Colin and F. J. Teran, Nanoscale Adv., 2021, 3, 6490–6502 RSC.
- B. Freis, C. Kiefer, M. D. L. A. Ramirez, S. Harlepp, D. Mertz, B. Pichon, C. Iacovita, S. Laurent and S. Begin, Nanoscale, 2024, 16, 20542–20555 RSC.
- G. Cotin, C. Blanco-Andujar, D.-V. Nguyen, C. Affolter, S. Boutry, A. Boos, P. Ronot, B. Uring-Lambert, P. Choquet, P. E. Zorn, D. Mertz, S. Laurent, R. N. Muller, F. Meyer, D. Felder-Flesch and S. Begin-Colin, Nanotechnology, 2019, 30, 374002 CrossRef CAS PubMed.
- A. Walter, A. Garofalo, A. Parat, J. Jouhannaud, G. Pourroy, E. Voirin, S. Laurent, P. Bonazza, J. Taleb, C. Billotey, L. Vander Elst, R. N. Muller, S. Begin-Colin and D. Felder-Flesch, J. Mater. Chem. B, 2015, 3, 1484–1494 RSC.
- B. Basly, D. Felder-Flesch, P. Perriat, G. Pourroy and S. Bégin-Colin, Contrast Media Mol. Imaging, 2011, 6(3), 132–138 CrossRef CAS PubMed.
- B. Basly, G. Popa, S. Fleutot, B. P. Pichon, A. Garofalo, C. Ghobril, C. Billotey, A. Berniard, P. Bonazza, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2013, 42, 2146–2157 RSC.
- S. Richard, M. Boucher, A. Šarić, A. Herbet, Y. Lalatonne, P. Petit, S. Meriaux, D. Boquet and L. Motte, J. Mater. Chem. B, 2017, 5, 2896–2907 RSC.
- Z. Li, R. Zhao, X. Wu, Y. Sun, M. Yao, J. Li, Y. Xu and J. Gu, FASEB J., 2005, 19, 1978–1985 CrossRef CAS PubMed.
- D. Li, S. Ma, D. Xu, X. Meng, N. Lei, C. Liu, Y. Zhao, Y. Qi, Z. Cheng and F. Wang, Nanomedicine, 2023, 50, 102669 CrossRef CAS PubMed.
- I. Paiva, S. Mattingly, M. Wuest, S. Leier, M. R. Vakili, M. Weinfeld, A. Lavasanifar and F. Wuest, Mol. Pharmaceutics, 2020, 17, 1470–1481 CrossRef CAS PubMed.
- H. Hossein-Nejad-Ariani, E. Althagafi and K. Kaur, Sci. Rep., 2019, 9, 2723 CrossRef PubMed.
- B. Freis, M. D. L. Á. Ramírez, S. Furgiuele, F. Journe, C. Cheignon, L. J. Charbonnière, C. Henoumont, C. Kiefer, D. Mertz, C. Affolter-Zbaraszczuk, F. Meyer, S. Saussez, S. Laurent, M. Tasso and S. Bégin-Colin, Int. J. Pharm., 2023, 635, 122654 CrossRef CAS PubMed.
- W. Baaziz, B. P. Pichon, S. Fleutot, Y. Liu, C. Lefevre, J.-M. Greneche, M. Toumi, T. Mhiri and S. Begin-Colin, J. Phys. Chem. C, 2014, 118, 3795–3810 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, M. Boero, B. Özdamar, A. Bouzid, G. Ori, C. Massobrio, D. Begin, B. Pichon, D. Mertz and S. Begin-Colin, ACS Appl. Nano Mater., 2018, 1, 4306–4316 CrossRef CAS.
- G. Cotin, C. Kiefer, F. Perton, D. Ihiawakrim, C. Blanco-Andujar, S. Moldovan, C. Lefevre, O. Ersen, B. Pichon, D. Mertz and S. Bégin-Colin, Nanomaterials, 2018, 8, 881 CrossRef PubMed.
- A. Walter, A. Garofalo, P. Bonazza, F. Meyer, H. Martinez, S. Fleutot, C. Billotey, J. Taleb, D. Felder-Flesch and S. Begin-Colin, ChemPlusChem, 2017, 82, 647–659 CrossRef CAS PubMed.
- C. Bordeianu, A. Parat, C. Affolter-Zbaraszczuk, R. N. Muller, S. Boutry, S. Begin-Colin, F. Meyer, S. Laurent and D. Felder-Flesch, J. Mater. Chem. B, 2017, 5, 5152–5164 RSC.
- A. Parat, C. Bordeianu, H. Dib, A. Garofalo, A. Walter, S. Bégin-Colin and D. Felder-Flesch, Nanomedicine, 2015, 10, 977–992 CrossRef CAS PubMed.
- A. Garofalo, A. Parat, C. Bordeianu, C. Ghobril, M. Kueny-Stotz, A. Walter, J. Jouhannaud, S. Begin-Colin and D. Felder-Flesch, New J. Chem., 2014, 38, 5226–5239 RSC.
- S. Boutry, D. Forge, C. Burtea, I. Mahieu, O. Murariu, S. Laurent, L. Vander Elst and R. N. Muller, Contrast Media & Molecular Imaging, 2009, 4, 299–304 Search PubMed.
- B. Basly, G. Popa, S. Fleutot, B. P. Pichon, A. Garofalo, C. Ghobril, C. Billotey, A. Berniard, P. Bonazza, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2013, 42, 2146–2157 RSC.
- C. Bordeianu, A. Parat, S. Piant, A. Walter, C. Zbaraszczuk-Affolter, F. Meyer, S. Begin-Colin, S. Boutry, R. N. Muller, E. Jouberton, J.-M. Chezal, B. Labeille, E. Cinotti, J.-L. Perrot, E. Miot-Noirault, S. Laurent and D. Felder-Flesch, Mol. Pharmaceutics, 2018, 15, 536–547 CrossRef CAS PubMed.
- A. Walter, A. Garofalo, A. Parat, H. Martinez, D. Felder-Flesch and S. Begin-Colin, Nanotechnol. Rev., 2015, 4(6), 581–593 CAS.
- W. Sun, S. Mignani, M. Shen and X. Shi, Drug Discovery Today, 2016, 21, 1873–1885 CrossRef CAS PubMed.
- M. Filippi, D.-V. Nguyen, F. Garello, F. Perton, S. Bégin-Colin, D. Felder-Flesch, L. Power and A. Scherberich, Nanoscale, 2019, 11, 22559–22574 RSC.
- X. Ma, H. Tao, K. Yang, L. Feng, L. Cheng, X. Shi, Y. Li, L. Guo and Z. Liu, Nano Res., 2012, 5, 199–212 CrossRef CAS.
- A. Espinosa, R. Di Corato, J. Kolosnjaj-Tabi, P. Flaud, T. Pellegrino and C. Wilhelm, ACS Nano, 2016, 10, 2436–2446 CrossRef CAS PubMed.
- Z. Zhou, Y. Sun, J. Shen, J. Wei, C. Yu, B. Kong, W. Liu, H. Yang, S. Yang and W. Wang, Biomaterials, 2014, 35, 7470–7478 CrossRef CAS PubMed.
- X. Song, H. Gong, S. Yin, L. Cheng, C. Wang, Z. Li, Y. Li, X. Wang, G. Liu and Z. Liu, Adv. Funct. Mater., 2014, 24, 1194–1201 CrossRef CAS.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17, 657–674 CrossRef PubMed.
- M. Rahman, O. Afzal, S. N. M. N. Ullah, M. Y. Alshahrani, A. G. Alkhathami, A. S. A. Altamimi, S. S. Almujri, W. H. Almalki, E. M. Shorog, M. A. Alossaimi, A. K. Mandal, A. Abdulrahman and A. Sahoo, ACS Omega, 2023, 8(51), 48625–48649 CrossRef CAS PubMed.
- Y. Liu, B. M. Crawford and T. Vo-Dinh, Immunotherapy, 2018, 10, 1175–1188 CrossRef CAS PubMed.
- E. Carrasco, B. del Rosal, F. Sanz-Rodríguez, Á. J. de la Fuente, P. H. Gonzalez, U. Rocha, K. U. Kumar, C. Jacinto, J. G. Solé and D. Jaque, Adv. Funct. Mater., 2015, 25, 615–626 CrossRef CAS.
- P. Peidaee, N. Almansour, R. Shukla and E. Pirogova, Comput. Struct. Biotechnol. J., 2013, 6, e201303015 CrossRef CAS PubMed.
- B. K. Najafabad, N. Attaran, M. Mahmoudi and A. Sazgarnia, Photodiagn. Photodyn. Ther., 2023, 43, 103648 CrossRef CAS PubMed.
- B. Freis, M. D. L. Á. Ramírez, S. Furgiuele, F. Journe, C. Cheignon, L. J. Charbonnière, C. Henoumont, C. Kiefer, D. Mertz, C. Affolter-Zbaraszczuk, F. Meyer, S. Saussez, S. Laurent, M. Tasso and S. Bégin-Colin, Int. J. Pharm., 2023, 635, 122654 CrossRef CAS PubMed.
- A. Le Bail, H. Duroy and J. L. Fourquet, Mater. Res. Bull., 1988, 23, 447–452 CrossRef CAS.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55–69 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr02931c |
| This journal is © The Royal Society of Chemistry 2025 |