
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced antifungal activity of siRNA-loaded anionic liposomes against the human pathogenic fungus Aspergillus fumigatus†
Yidong
Yu‡
 ab,
Theresa
Vogel‡
c,
Sina
Hirsch
a,
Jürgen
Groll
c,
Krystyna
Albrecht
*c and
Andreas
Beilhack
*a
ab,
Theresa
Vogel‡
c,
Sina
Hirsch
a,
Jürgen
Groll
c,
Krystyna
Albrecht
*c and
Andreas
Beilhack
*a
aDepartment of Internal Medicine II, Center for Experimental Molecular Medicine, Würzburg University Hospital, 97078 Würzburg, Germany. E-mail: beilhack_a@ukw.de
bJSPS International Research Fellow, Endowed Course “Drug Discoveries by Silkworm Models”, Faculty of Pharmaceutical Sciences, Teikyo University, 192-0395 Tokyo, Japan
cDepartment for Functional Materials in Medicine and Dentistry, Institute of Functional Materials and Biofabrication, University of Würzburg, 97070 Würzburg, Germany. E-mail: Krystyna.albrecht@uni-wuerzburg.de
First published on 7th November 2024
Abstract
We developed siRNA-loaded anionic liposomes, co-encapsulating low-dose amphotericin B, to enhance siRNA penetration through the fungal cell wall of Aspergillus fumigatus. Targeting mRNAs of three key genes, these liposomes visibly inhibited fungal growth, demonstrating for the first time the antifungal potential of siRNA against human fungal pathogens.
Introduction
Invasive fungal infections claim over 2.5 million deaths annually.1,2 Current treatment options are limited to three antifungal classes, with no new classes approved in the last two decades.3 Additionally, existing antifungal drugs often cause severe side effects, face increasing drug resistance, and lack effectiveness.4 To enhance the global response to fungal infections and antifungal resistance, the World Health Organization published its first fungal priority pathogens list in 2022 to guide research and policy interventions.5Aspergillus fumigatus, a critical pathogen on this list, causes invasive pulmonary aspergillosis in about 2 million people annually, with mortality rates up to 85%.1 Resistance of A. fumigatus against all clinically available antifungal classes has been reported,6 underscoring the urgent need for novel antifungal solutions.RNA interference (RNAi) is a conserved gene-silencing mechanism in eukaryotes, including fungi.7 Six RNAi-based drugs involving small interfering RNAs (siRNAs) have been approved for genetic diseases,8 and spray-induced gene silencing shows promise against fungal diseases in crops.9 Upon entering eukaryotic cells, an siRNA duplex incorporates into the RNA-induced silencing complex (RISC), a multiprotein component. Within RISC, the siRNA strands separate, and the antisense strand activates the complex, guiding it to bind to complementary target messenger RNA (mRNA) sequences. This leads to mRNA cleavage by the RISC component Argonaute 2 (Ago2), temporarily silencing the target gene's expression.10 The advantages of siRNA-based therapeutics are notable: they have a well-defined mechanism of action and the potential to target virtually all expressed genes at the mRNA level, including those deemed “undruggable” by traditional small molecule drugs at the protein level.11 Furthermore, RNAi might be synergistically combined with conventional antimicrobial drugs to silence genes involved in drug resistance, such as efflux pump genes, thereby enhancing the therapeutic efficacy of antimicrobials, particularly against drug-resistant isolates.12
Few studies have investigated synthetic siRNAs for gene silencing in A. fumigatus. Jöchl et al. found that this fungus could uptake siRNAs from the culture medium, resulting in a 30–60% knockdown of target mRNA levels with no growth inhibition.13 This underscores the need for effective delivery systems to enhance siRNA knockdown efficiency and protect siRNAs from rapid degradation, which is crucial for clinical translation. Previous attempts to encapsulate siRNA into cationic liposomes (LIP) or conjugate siRNA with the cell penetrating peptide PAF26 were ineffective for siRNA delivery into A. fumigatus.14 Consequently, a significant challenge in employing RNAi against human fungal pathogens persists: the complex structure of the fungal cell wall acts as a formidable barrier, preventing siRNAs from effectively entering the cells, thereby limiting their ability to silence fungal gene expression.15 This issue is exacerbated in clinical contexts where fungi form complex 3D structures like nodules, biofilms, and granulomas, complicating the siRNA delivery into fungal cells even further.15
In this study, we explored the potential of anionic LIP for delivering siRNA into A. fumigatus, inspired by the use of anionic LIP in AmBisome® for delivering amphotericin B, a conventional antifungal drug.16
Results and discussion
We formulated anionic LIP with the same components as AmBisome®, including DSPG (1,2-distearoyl-sn-glycero-3-phosphoglycerol), HSPC (hydrogenated soybean phosphatidylcholine), and cholesterol (Chol; Fig. S1, ESI†) in a molar ratio of 0.8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. We synthesized LIP via the “thin film hydration” method,17,18 which involved dissolving lipids in an organic solvent, forming a thin film, rehydrating it to create multilamellar LIP structures, and then converting them to unilamellar structures via ultrasound and extrusion (Fig. S2, ESI†).18,19 Dynamic light scattering analysis showed that five extrusion cycles through a 100 nm polycarbonate membrane produced LIP with a size of 143 ± 2 nm and a Zetasizer count rate within the recommended range.20 Additional extrusion cycles reduced the size (137 ± 3 nm after 11 cycles and 130 ± 2 nm after 21 cycles) without affecting polydispersity or count rate, leading us to select a maximum of 21 cycles for monodisperse LIP (Table S1, ESI†).
:2:1. We synthesized LIP via the “thin film hydration” method,17,18 which involved dissolving lipids in an organic solvent, forming a thin film, rehydrating it to create multilamellar LIP structures, and then converting them to unilamellar structures via ultrasound and extrusion (Fig. S2, ESI†).18,19 Dynamic light scattering analysis showed that five extrusion cycles through a 100 nm polycarbonate membrane produced LIP with a size of 143 ± 2 nm and a Zetasizer count rate within the recommended range.20 Additional extrusion cycles reduced the size (137 ± 3 nm after 11 cycles and 130 ± 2 nm after 21 cycles) without affecting polydispersity or count rate, leading us to select a maximum of 21 cycles for monodisperse LIP (Table S1, ESI†).
To visualize the LIP, we synthesized fluorescently labeled liposomes (RhoB-LIP) by incorporating 2 mol% rhodamine DHPE (rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phospho-ethanolamine, triethylammonium salt; Fig. S1†),21–24 which showed no differences in particle size or zeta-potential compared to unlabeled LIP (Fig. S3, ESI†). We co-incubated RhoB-LIP with A. fumigatus strain A1160p+ (MFIG001)25 in Aspergillus minimal medium (AMM)26 for 16 h at 37 °C. RhoB-LIP accumulated around the hyphal cell wall, but did not enter the fungal cells (Fig. S4, ESI†). This suggested that electrostatic interactions between the positively charged hyphal cell wall13 and the anionic LIP were insufficient for LIP entry. This observation aligns with previous findings where anionic LIP were trapped in the cell wall of another human fungal pathogen, Candida albicans.27 Interestingly, in this previous study, anionic LIP loaded with amphotericin B (AmBisome®) crossed the cell wall and reached the cell membrane due to amphotericin B (AmB)'s high binding affinity for ergosterol, a critical component of the fungal cell membrane. Specifically, while anionic LIP were confined to the junction between the outer and inner cell wall layers, AmBisome® penetrated the inner β-glucan-chitin layer and reached the cell membrane. Anionic LIP loaded with both AmB and gold particles, which do not deform during passage, also successfully reached the cell membrane. In contrast, gold particles alone could not diffuse through the outer and inner cell wall layers, suggesting that AmB-loaded anionic LIP can displace the polysaccharide chains of the cell wall. Brief exposure to AmBisome® increased the cell wall's relative porosity five-fold without significantly affecting fungal cell viability. Notably, in ergosterol-deficient Candida mutants, AmBisome® were trapped at the junction of the outer and inner cell wall layers, similar to anionic LIP, emphasizing the crucial role of the AmB–ergosterol interaction in this process.27 Inspired by these previous findings, we explored incorporating AmB as an adjuvant into anionic LIP to potentially enable siRNA delivery into fungal cells.
To load siRNA into LIP, we incorporated it into the aqueous rehydration solution, as siRNA is hydrophilic. However, the negative charges of siRNA and phospholipids can cause electrostatic repulsion, impeding efficient encapsulation. We used protamine, a protein rich in arginine residues, which confers a high positive charge, to complex with siRNA and neutralize its negative charge.28,29 Protamine sulfate, which is FDA-approved and widely used in medicine, is also a component of neutral protamine Hagedorn (NPH) insulin for long-acting delivery. Its extensive use in diabetic patients has demonstrated minimal toxicity and immunogenicity,30 making it a safe choice for our drug delivery system design. Moreover, protamine sulfate has shown no inhibitory effect on the growth of molds, including Aspergillus.31 Protamine and siRNA were co-incubated at an N/P ratio of 1:1 (N = positively charged amino acids, P = negatively charged phosphate groups on siRNA) before loading into LIP (Fig. S2†). Although free protamine from incomplete incorporation in the siRNA–protamine complex may cause liposome aggregation,32 our experiments with fluorescently labeled non-targeting siRNA (DY547-siRNA; Table S2, ESI†) showed no noticeable changes in LIP characteristics, including size and zeta-potential (Table S3, ESI†). In fact, protamine complexation improved siRNA loading across various batches, compared to those without protamine, resulting in more consistent and higher loading percentages (Table S3†). The concentration of loaded siRNA was quantified using a calibration curve of free DY547-siRNA (Fig. S5, ESI†). Thus, we included protamine as a standard component in the LIP formulation.
Integrating AmB as an adjuvant in siRNA-loaded anionic LIP aimed to keep the AmB content minimal to avoid inhibiting fungal growth. More specifically, binding of AmB with ergosterol in the fungal cell membrane can lead to membrane disruption and ultimately cell death.33,34 While leveraging AmB's ability to enhance siRNA penetration into fungal cells, it is crucial to minimize its potential adverse effects on membrane integrity, particularly to avoid off-target interactions of AmB with cholesterol in mammalian cell membranes.35 We added AmB to the initial organic solution in a molar ratio of 0.8:2:1:0.01 (DSPG:HSPC:Chol:AmB). The concentration of encapsulated AmB was determined using high-performance liquid chromatography (HPLC) according to calibration curves of free AmB (Fig. S6, ESI†). To assess AmB's impact on the interaction between siRNA-loaded LIP and A. fumigatus, we used DY547-siRNA for visualization. After 16 h of co-incubation with A. fumigatus, weak signals were detected on the fungal cell wall with LIP loaded solely with siRNA (LIP × DY547-siRNA). In contrast, siRNA signals were observed inside the fungal hyphae when LIP were co-loaded with AmB and siRNA (LIP × AmB × DY547-siRNA; Fig. S7, ESI†). Importantly, microscopy revealed that hyphal growth remained largely unaffected by AmB (final concentration: 0.43 μg mL−1). These findings indicate that the concentration of AmB in this formulation effectively facilitated siRNA delivery but was likely too low to inhibit fungal growth, explaining the lack of visible antifungal effects. In comparison to previous approaches, cationic multilamellar liposomes used for siRNA encapsulation showed no accumulation of siRNA on the hyphal cell wall of A. fumigatus,14 whereas our anionic LIP formulation (LIP × siRNA) achieved slight accumulation on the hyphal cell wall. Furthermore, using the cationic cell penetrating peptide PAF26 for siRNA conjugation allowed a small amount of PAF26 to penetrate the fungal hyphae, while siRNA was only observed on the fungal cell wall, co-localized with PAF26, indicating that PAF26 cannot translocate siRNA into A. fumigatus.14 In contrast, our LIP × AmB × siRNA formulation successfully delivered siRNA into the fungal hyphae, demonstrating the potential of anionic LIP, coupled with low-concentration AmB as an adjuvant, to facilitate siRNA entry into fungal cells.
Encouraged by these results with non-targeting siRNA, we explored functional siRNAs targeting three transcription factor-encoding genes in A. fumigatus: hapB, hapX, and sreA. The hapB gene encodes a subunit of the CCAAT-binding transcriptional regulatory complex (CBC), regulating over a third of the genome.36 Deletion of hapB results in severe growth reduction37 and attenuated virulence.26 The hapX gene cooperates with the CBC for adaptation to low iron environments, and its deletion also reduces virulence.26,38 While the sreA gene is involved in iron regulation39 and is dispensable for virulence, the double deletion of sreA and hapX is lethal to the fungus.38 Additionally, double deletions of sreA with hapB or hapX are lethal in Aspergillus nidulans, closely related to A. fumigatus.40 These findings led us to target both individual and combined gene silencing for antifungal effects via siRNA treatment.
In our initial screening, individual candidate siRNAs were co-incubated with germinated spores of A. fumigatus for 24 h at 37 °C in AMM, followed by RNA isolation and quantitative real-time PCR (qRT-PCR) assessment of target mRNA levels, using primers listed in Table S4 (ESI†). We selected siRNAs for each target gene (hapB-1, hapX-3, and sreA-1; Table S2†) that achieved 40–50% mRNA knockdown (Fig. S8A, ESI†) but did not visibly inhibit fungal growth. Given the role of these genes in iron regulation, we tested the effect of switching from iron-replete to iron-depleted conditions after 18 h. This led to a transient and ambiguous delay in fungal growth on day 2 for siRNA-treated samples (Fig. S8B†), confirming the siRNAs’ functionality, but without clear growth inhibition.
Next, we co-loaded functional siRNA with AmB into anionic LIP (LIP × AmB × siRNA) and co-incubated siRNA targeting hapB (hapB-1) with germinated spores of A. fumigatus for 18 h, followed by a medium shift to AMM-Fe. On day 2, we observed no visible hyphal growth in samples treated with LIP × AmB × hapB-1, while those treated with scrambled siRNA (LIP × AmB × hapB-S) or LIP × AmB showed comparable growth to PBS-treated controls. By day 3, sporulation (generation of green spores) was evident in controls but not in treated samples, although hyphal growth resumed. By day 4, growth differences disappeared (Fig. 1A). We then tested combinations of individually encapsulated siRNAs (Fig. 1B), finding that dual siRNA treatment extended inhibition to 4 days, and triple siRNA treatment to 5 days. In contrast, scrambled siRNAs or LIP × AmB showed growth similar to controls. These results suggest that targeting hapB, hapX, and sreA simultaneously is more effective than targeting hapB alone or in combination with hapX or sreA.
 | ||
| Fig. 1 Antifungal effect of liposomes co-loaded with amphotericin B and functional siRNA (LIP × AmB × siRNA) on A. fumigatus strain A1160p+. (A) After 6 h pre-incubation of fungal spores in AMM, LIP × AmB × hapB-1 (final concentration: approximately 1.5 nM) was added for an 18 h co-incubation, followed by a medium shift to iron-depleted AMM (AMM-Fe). This delayed hyphal growth until day 2 and sporulation until day 3. Equivalent concentrations of scrambled control (LIP × AmB × hapB-S) and AmB (LIP × AmB) did not inhibit fungal growth. (B) After 6 h pre-incubation of fungal spores in AMM, LIP × AmB × siRNA formulations (final concentration: approximately 1.5 nM each, 4.5 nM in total) were added for an 18 h co-incubation, followed by a shift to AMM-Fe. Targeting hapB, hapX, and sreA simultaneously prolonged the antifungal effect, compared to targeting hapB alone, hapB + hapX, or hapB + sreA. (C) Following the same initial procedure, a second dose of siRNAs (final concentration: approximately 1.5 nM each, 4.5 nM in total) was included upon shifting to AMM-Fe. Two doses of siRNAs targeting hapB, hapX, and sreA showed an antifungal effect for at least 10 days. | ||
Building on these encouraging findings, we tested whether a second dose of siRNAs could prolong antifungal effects. Upon shifting to AMM-Fe, we included a second dose of siRNAs. Two doses of hapB-1 with two scrambled siRNAs extended antifungal effects to 5 days, while two doses of all three functional siRNAs achieved effects lasting at least 10 days (Fig. 1C), underscoring the antifungal efficacy of repeated dosing.
In initial experiments, growth differences were noted between samples and controls even before shifting to AMM-Fe. The medium change, involving a centrifugation step, likely introduced bias: shorter hyphae might be removed from samples, while longer hyphae tend to adhere more strongly to the well bottom in controls. Additionally, the medium change could also account for the temporary variations in growth inhibition observed among triplicates (e.g., Fig. 1C, day 6, in the group treated with LIP × AmB × hapB-1/hapX-S/sreA-S), as it is difficult to completely prevent the removal of short hyphae from each well, leading to some degree of variation among the triplicates. To minimize bias and variation, we conducted subsequent experiments solely in AMM, despite AMM-Fe better mimicking the in vivo iron-restricted conditions encountered by A. fumigatus.41
To better monitor the antifungal effects of siRNAs, we measured the optical density of fungal cultures at 600 nm (OD600).42 Following siRNA treatment, OD600 at 37 °C was monitored for at least 45 h (Fig. 2A). Controls treated with PBS, LIP × AmB, and scrambled siRNAs showed detectable fungal growth at 15 h, reaching saturation (OD600 above 1.0) between 40–45 h. Treatment with hapB-1 and two scrambled siRNAs delayed growth detection to 35 h, while three functional siRNAs extended this to 52 h. Monitoring OD600 was extended to 60 h for these two cases (Fig. 2B): in hapB-1 and scrambled siRNA treatments, OD600 reached 1.0 at 60 h, but stayed below 0.2 with three functional siRNAs, indicating clear fungal growth inhibition even in iron-replete conditions.
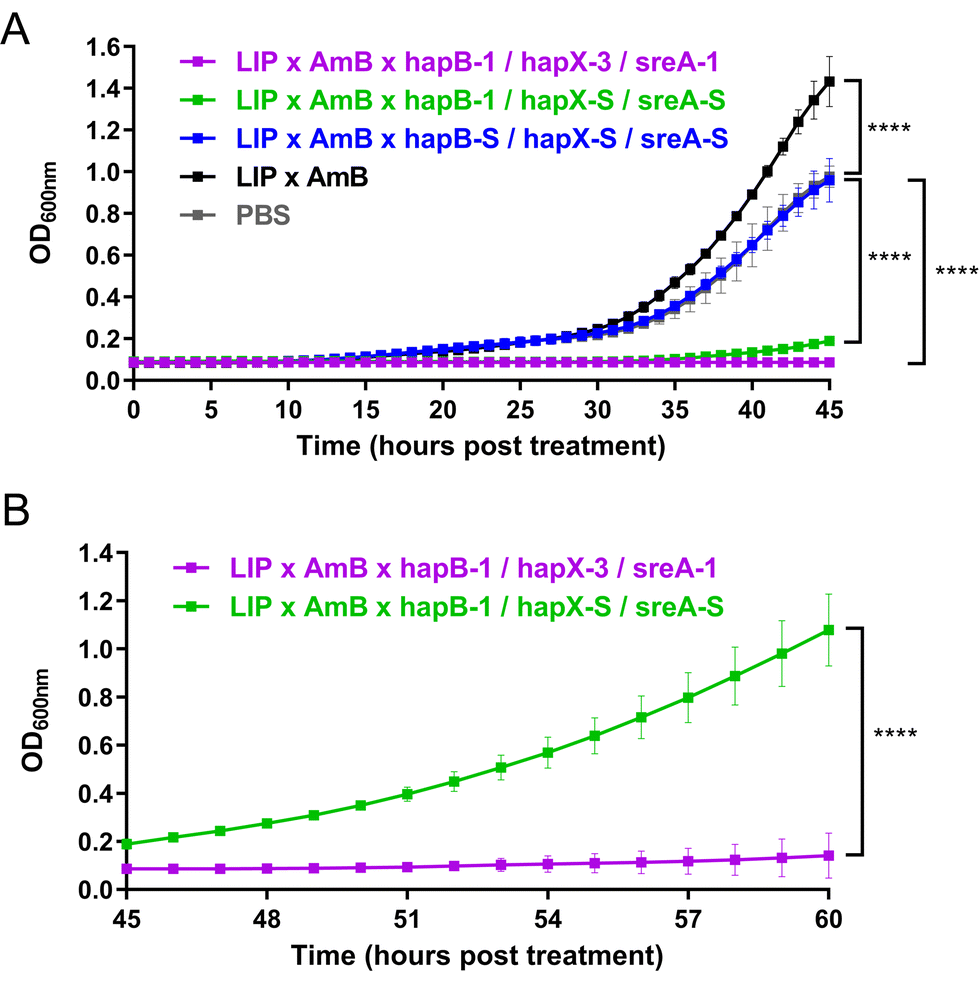 | ||
| Fig. 2 Antifungal effect of LIP × AmB × siRNA under iron-replete conditions. (A) & (B) After 6 h pre-incubation of fungal spores in AMM, LIP × AmB × siRNA formulations (final concentration: approximately 1.5 nM each, 4.5 nM in total) were added. OD600 measurements were taken at 37 °C at 10 min intervals (in triplicates). (B) Due to observed delays in fungal growth, measurements were extended to 60 h to compare the effects of siRNAs targeting hapB, hapX, and sreA simultaneously vs. targeting hapB alone. A two-way ANOVA with multiple comparisons was performed at each time point against the PBS-treated control group in (A) and between the two groups in (B). Significant differences (****: p < 0.0001) are highlighted for the final time points (45 h in A and 60 h in B). No significant difference was found between the PBS group (grey) and the LIP × AmB × hapB-S/hapX-S/sreA-S group (blue) at 45 h (p = 0.67). | ||
In all experiments mentioned above, we had utilized the A. fumigatus strain A1160p+. To ensure broad applicability, we tested the efficacy of LIP × AmB × siRNA on three clinical isolates: Af293, D141, and ATCC46645, compared to A1160p+. These strains are widely used in research and show varied virulence.43 After siRNA treatment, growth inhibition was consistent across all strains on day 2 (Fig. S9, ESI†). By day 3, while growth resumed in A1160p+ and Af293 targeting hapB alone, D141 and ATCC46645 showed minimal growth. Targeting hapB, hapX, and sreA uniformly suppressed hyphal formation across all strains, with the effect lasting until day 4 in D141 and ATCC46645 but resuming in A1160p+ and Af293. Overall, our findings confirmed that the LIP × AmB × functional siRNA formulations are effective across various A. fumigatus strains, albeit with some variation in the duration of RNA knockdown. This variation likely stems from differing growth rates of the tested strains, as evident on day 2 in the PBS-treated controls (Fig. S7†), where green spores appeared in the wells of A1160p+ and Af293, while only mycelial growth occurred in D141 and ATCC46645.
In summary, co-loading siRNA and AmB into anionic LIP effectively delivers siRNA into fungal cells. Minimal AmB facilitates LIP crossing the fungal cell wall, enabling siRNA entry. Targeting hapB, hapX, and sreA inhibited fungal growth across four A. fumigatus strains for 3–4 days under iron-replete conditions, reflecting the temporary nature of RNAi knockdown. This study represents the first in vitro demonstration of siRNA's antifungal potential against A. fumigatus, highlighting RNAi-based gene silencing as a promising new strategy for antifungal therapy. Furthermore, our strategy of targeting multiple regulatory genes simultaneously has proven effective, particularly with combinations that have been shown to be lethal to fungi when deleted concurrently.
Further research should explore additional targets to extend the antifungal effect or achieve a fungicidal outcome through extensive gene expression perturbation. One promising avenue involves targeting the 18 transcription factor-encoding genes that play critical roles in A. fumigatus pathogenicity.44 Our preliminary data suggest that combining multiple target genes could be the most effective strategy to enhance RNAi-based antifungal efficacy. Additionally, due to cytotoxicity concerns with AmBisome® to human cells,45 and the potential off-target effects of siRNAs on human genes, which may not be fully mitigated by targeting fungal-specific genes or avoiding homologous mRNA regions, further optimization of our delivery system is essential. For instance, incorporating a fungal-specific ligand, such as dectin-1 (binds to β-glucans in the fungal cell wall),21 could reduce off-target effects of both AmB and siRNAs on host cells, thereby improving the safety profile of RNAi-based antifungal therapies. Moreover, the need for fungal-specific delivery systems extends beyond RNAi to other RNA knockdown strategies, such as antisense oligonucleotides (ASOs) that induce RNase H-dependent mRNA degradation,46 and CRISPR-mediated interference (CRISPRi) utilizing dCas9 (a nuclease-deactivated form of CRISPR-associated protein 9) to block transcription by binding temporarily to the gene's regulatory region.47 ASO strategies have successfully silenced mRNA in C. albicans; however, they exhibit toxicity to mammalian cells at concentrations exceeding 40 nM,48 indicating unspecific uptake. Similarly, CRISPRi has been successful in gene knockdown in C. albicans, but is predominantly a tool for genetic analysis and requires lithium acetate-based transformation for fungal cell entry.49 Given these challenges, our future research will focus on developing fungal-specific siRNA delivery strategies, which could also improve the clinical applicability of the other RNA-targeting approaches mentioned.
Conclusions
Co-loading siRNA and AmB into anionic LIP effectively delivers siRNA into fungal cells. Targeting key fungal regulatory genes with functional siRNAs achieves a clear growth inhibitory effect, demonstrating the potential of this novel antifungal strategy.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the German Research Foundation (DFG)-TRR 225 “Biofabrication” (project number 326998133, subprojects A06, B07, B08), DFG-GRK/RTG 2157 “3DInfect” (270563345, subproject P01) and DFG-TRR124 “FungiNet” (210879364, subproject A03). Y. Y. received the Postdoctoral Fellowship for Research in Japan from the Japan Society for the Promotion of Science (JSPS) and was supported by funds of the Bavarian State Ministry of Science and the Arts and the University of Würzburg to the Graduate School of Life Sciences (GSLS), University of Würzburg (Germany).References
- D. W. Denning, Lancet Infect. Dis., 2024, 24, 428–438 CrossRef.
- F. Bongomin, S. Gago, R. O. Oladele and D. W. Denning, J. Fungi, 2017, 3, 57 CrossRef.
- M. Hoenigl, R. Sprute, M. Egger, A. Arastehfar, O. A. Cornely, R. Krause, C. Lass-Flörl, J. Prattes, A. Spec, G. R. Thompson, N. Wiederhold and J. D. Jenks, Drugs, 2021, 81, 1703–1729 CrossRef CAS.
- C. M. Hossain, L. K. Ryan, M. Gera, S. Choudhuri, N. Lyle, K. A. Ali and G. Diamond, Encyclopedia, 2022, 2, 1722–1737 CrossRef.
- WHO fungal priority pathogens list to guide research, development and public health action, https://www.who.int/publications/i/item/9789240060241, accessed September 2024.
- Y. Lee, N. Robbins and L. E. Cowen, npj Antimicrob. Resist., 2023, 1, 5 CrossRef PubMed.
- Y. Dang, Q. Yang, Z. Xue and Y. Liu, Eukaryotic Cell, 2011, 10, 1148–1155 CrossRef CAS.
- I. Sehgal, K. Eells and I. Hudson, Pharmacy, 2024, 12, 58 CrossRef PubMed.
- C. W. G. Mann, A. Sawyer, D. M. Gardiner, N. Mitter, B. J. Carroll and A. L. Eamens, Int. J. Mol. Sci., 2023, 24, 12391 CrossRef CAS PubMed.
- H. Dana, G. M. Chalbatani, H. Mahmoodzadeh, R. Karimloo, O. Rezaiean, A. Moradzadeh, N. Mehmandoost, F. Moazzen, A. Mazraeh, V. Marmari, M. Ebrahimi, M. M. Rashno, S. J. Abadi and E. Gharagouzlo, Int. J. Biomed. Sci., 2017, 13, 48–57 CrossRef PubMed.
- R. L. Setten, J. J. Rossi and S. P. Han, Nat. Rev. Drug Discovery, 2019, 18, 421–446 CrossRef CAS PubMed.
- J. A. Edson and Y. J. Kwon, J. Controlled Release, 2014, 189, 150–157 CrossRef CAS PubMed.
- C. Jöchl, E. Loh, A. Ploner, H. Haas and A. Hüttenhofer, RNA Biol., 2009, 6, 179–186 CrossRef PubMed.
- M. van der Torre, PhD thesis, University of Manchester, 2019 Search PubMed.
- A. Bruch, A. A. Kelani and M. G. Blango, Trends Microbiol., 2022, 30, 411–420 CrossRef CAS PubMed.
- N. R. Stone, T. Bicanic, R. Salim and W. Hope, Drugs, 2016, 76, 485–500 CrossRef CAS PubMed.
- M. R. Mozafari, Cell. Mol. Biol. Lett., 2005, 10, 711–719 CAS.
- T. Michel, D. Luft, M. K. Abraham, S. Reinhardt, M. L. Salinas Medina, J. Kurz, M. Schaller, M. Avci-Adali, C. Schlensak, K. Peter, H. P. Wendel, X. Wang and S. Krajewski, Mol. Ther. – Nucleic Acids, 2017, 8, 459–468 CrossRef CAS.
- K. Faber, G. K. Zorzi, N. T. Brazil, M. B. Rott and H. F. Teixeira, Chem. Biol. Drug Des., 2017, 90, 406–416 CrossRef CAS.
- Derived count rate – what is it?, https://www.malvernpanalytical.com/en/learn/knowledge-center/insights/derived-count-rate-what-is-it, accessed September 2024.
- S. Ambati, A. R. Ferarro, S. E. Kang, J. Lin, X. Lin, M. Momany, Z. A. Lewis and R. B. Meagher, mSphere, 2019, 4, e00025-19 CrossRef PubMed.
- S. Ambati, E. C. Ellis, J. Lin, X. Lin, Z. A. Lewis and R. B. Meagher, mSphere, 2019, 4, e00715-19 CrossRef.
- S. J. H. Soenen, D. Vercauteren, K. Braeckmans, W. Noppe, S. De Smedt and M. De Cuyper, ChemBioChem, 2009, 10, 257–267 CrossRef CAS.
- L. Simonsson and F. Höök, Langmuir, 2012, 28, 10528–10533 CrossRef CAS.
- M. G. Fraczek, M. Bromley, A. Buied, C. B. Moore, R. Rajendran, R. Rautemaa, G. Ramage, D. W. Denning and P. Bowyer, J. Antimicrob. Chemother., 2013, 68, 1486–1496 CrossRef CAS.
- Y. Yu, A. K. Wolf, S. Thusek, T. Heinekamp, M. Bromley, S. Krappmann, U. Terpitz, K. Voigt, A. A. Brakhage and A. Beilhack, J. Fungi, 2021, 7, 136 CrossRef CAS PubMed.
- L. Walker, P. Sood, M. D. Lenardon, G. Milne, J. Olson, G. Jensen, J. Wolf, A. Casadevall, J. Adler-Moore and N. A. R. Gow, mBio, 2018, 9, e02383-17 CrossRef.
- W. Alshaer, H. Hillaireau, J. Vergnaud, S. Mura, C. Deloménie, F. Sauvage, S. Ismail and E. Fattal, J. Controlled Release, 2018, 271, 98–106 CrossRef CAS PubMed.
- D. Peer, E. J. Park, Y. Morishita, C. V. Carman and M. Shimaoka, Science, 2008, 319, 627–630 CrossRef CAS.
- F. L. Sorgi, S. Bhattacharya and L. Huang, Gene Ther., 1997, 4, 961–968 CrossRef CAS.
- M. Kamal, T. Motohiro and T. Itakura, Nippon Suisan Gakkaishi, 1986, 52, 1061–1064 CrossRef CAS.
- S. B. Kulkarni, S. R. Dipali and G. V. Betageri, Pharm. Pharmacol. Commun., 1995, 1, 359–362 CAS.
- G. Fujii, J. E. Chang, T. Coley and B. Steere, Biochemistry, 1997, 36, 4959–4968 CrossRef CAS PubMed.
- A. Lewandowska, C. P. Soutar, A. I. Greenwood, E. Nimerovsky, A. M. De Lio, J. T. Holler, G. S. Hisao, A. Khandelwal, J. Zhang, A. M. SantaMaria, C. D. Schwieters, T. V. Pogorelov, M. D. Burke and C. M. Rienstra, Nat. Struct. Mol. Biol., 2021, 28, 972–981 CrossRef CAS.
- A. Noor and C. V. Preuss, Amphotericin B, StatPearls Publishing, 2024, https://www.ncbi.nlm.nih.gov/books/NBK482327/, accessed September 2024 Search PubMed.
- N. van Rhijn, S. Hemmings, I. S. R. Storer, C. Valero, H. B. Shuraym, G. H. Goldman, F. Gsaller, J. Amich and M. J. Bromley, mBio, 2022, 13, e02215-22 CrossRef PubMed.
- F. Gsaller, P. Hortschansky, T. Furukawa, P. D. Carr, B. Rash, J. Capilla, C. Müller, F. Bracher, P. Bowyer, H. Haas, A. A. Brakhage and M. J. Bromley, PLoS Pathog., 2016, 12, e1005775 CrossRef PubMed.
- M. Schrettl, N. Beckmann, J. Varga, T. Heinekamp, I. D. Jacobsen, C. Jöchl, T. A. Moussa, S. Wang, F. Gsaller, M. Blatzer, E. R. Werner, W. C. Nierman, A. A. Brakhage and H. Haas, PLoS Pathog., 2010, 6, e1001124 CrossRef.
- M. Schrettl, H. S. Kim, M. Eisendle, C. Kragl, W. C. Nierman, T. Heinekamp, E. R. Werner, I. Jacobsen, P. Illmer, H. Yi, A. A. Brakhage and H. Haas, Mol. Microbiol., 2008, 70, 27–43 CrossRef CAS.
- P. Hortschansky, M. Eisendle, Q. Al-Abdallah, A. D. Schmidt, S. Bergmann, M. Thön, O. Kniemeyer, B. Abt, B. Seeber, E. R. Werner, M. Kato, A. A. Brakhage and H. Haas, EMBO J., 2007, 26, 3157–3168 CrossRef CAS PubMed.
- T. Ganz, Curr. Opin. Immunol., 2009, 21, 63–67 CrossRef CAS.
- T. Hameed, N. Motsi, E. Bignell and R. J. Tanaka, PLoS Comput. Biol., 2024, 20, e1012105 CrossRef CAS PubMed.
- E. M. Keizer, H. A. B. Wösten and H. de Cock, Front. Microbiol., 2020, 11, 534118 CrossRef.
- N. van Rhijn, PhD thesis, University of Manchester, 2019 Search PubMed.
- C. G. Leon, J. Lee, K. Bartlett, P. Gershkovich, E. K. Wasan, J. Zhao, J. G. Clement and K. M. Wasan, Lipids Health Dis., 2011, 10, 144 CrossRef CAS.
- X. H. Liang, H. Sun, J. G. Nichols and S. T. Crooke, Mol. Ther., 2017, 25, 2075–2092 CrossRef CAS PubMed.
- L. S. Qi, M. H. Larson, L. A. Gilbert, J. A. Doudna, J. S. Weissman, A. P. Arkin and W. A. Lim, Cell, 2013, 152, 1173–1183 CrossRef CAS.
- D. Araújo, D. Mil-Homens, M. E. Rodrigues, M. Henriques, P. T. Jørgensen, J. Wengel and S. Silva, Nanomedicine, 2022, 39, 102469 CrossRef.
- L. Wensing, J. Sharma, D. Uthayakumar, Y. Proteau, A. Chavez and R. S. Shapiro, mSphere, 2019, 4, e00002-19 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental methods and additional figures & tables. See DOI: https://doi.org/10.1039/d4nr03225j |
| ‡ The authors have equally contributed to the manuscript. |
| This journal is © The Royal Society of Chemistry 2025 |