
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular mechanisms behind the anti corona virus activity of small metal oxide nanoparticles†
Björn
Greijer
a,
Alexandra
Nefedova
 b,
Tatiana
Agback
a,
Peter
Agback
a,
Vambola
Kisand
b,
Kai
Rausalu
c,
Alexander
Vanetsev
b,
Gulaim A.
Seisenbaeva
a,
Angela
Ivask
d and
Vadim G.
Kessler
*a
b,
Tatiana
Agback
a,
Peter
Agback
a,
Vambola
Kisand
b,
Kai
Rausalu
c,
Alexander
Vanetsev
b,
Gulaim A.
Seisenbaeva
a,
Angela
Ivask
d and
Vadim G.
Kessler
*a
aDepartment of Molecular Science, BioCenter, Swedish University of Agricultural Sciences, Box 7015, 75007 Uppsala, Sweden. E-mail: vadim.kessler@slu.se
bInstitute of Physics, University of Tartu, Ostwaldi 1, 50411, Tartu, Estonia
cInstitute of Bioengineering, University of Tartu, Nooruse 1, 50411 Tartu, Estonia
dInstitute of Molecular and Cell Biology, University of Tartu, Riia 23, 51010 Tartu, Estonia
First published on 17th January 2025
Abstract
The recent COVID-19 pandemic has set a strong quest for advanced understanding of possible tracks in abating and eliminating viral infections. In the view that several families of “pristine” small oxide nanoparticles (NPs) have demonstrated viricidal activity against SARS-CoV-2, we studied the effect of two NPs, with presumably different reactivity, on two viruses aiming to evaluate two “primary suspect” routes of their antiviral activity, either specific blocking of surface proteins or causing membrane disruption. The chosen NPs were non-photoactive 3.5 nm triethanolamine terminated (surface capped) titania TiO2 NPs (TATT) and ultrasmall (1.1 nm) silicotungstate polyoxometalate (POM) NPs. The former were expected to both, interact with viral surface proteins as well as strongly complex with phosphate groups whereas the latter was not expected to form surface complexes. We demonstrated that expectedly, POM NPs up to 1.25 mM (4.5 mg l−1) had no significant antiviral activity towards neither of the used viruses, an enveloped transmissible gastroenteritis virus (TGEV) belonging to coronaviruses and non-enveloped encelomyocarditis virus (EMCV). At the same time, TATT NPs exhibited statistically significant (p < 0.05) antiviral activity against TGEV starting from 0.125 mM (12 μg ml−1). However, no antiviral activity of TATT against non-enveloped EMCV was detected. The observation that TATT NPs showed activity only against enveloped viruses and at relatively high concentrations suggests that the effect could be related with complexation with phospholipids. Possible chemical mechanism of viral membrane disruption was investigated by a variable temperature NMR study of NP complexation with model organic phosphate molecules, proving TATT to strongly interact with them and POM remain unreacted. Viral membrane disruption by TATT NPs was additionally confirmed by demonstraing RNA leackage from TGEV upon contact with those NPs. Therefore, our study proved a new mechanism of antiviral action of titania NPs in the dark which involved membrane disruption proceeding via direct surface complexation.
Introduction
The recent COVID-19 pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which turned out to be easily mutating and highly contagious, urged the search for both new therapeutic and disinfection tools.1,2 SARS-CoV-2 is an enveloped virus with three transmembrane proteins, which together with membrane form the surface of the viral particle, and a nucleoprotein essential for packing viral RNA.3 Significant focus has been set on potential nanotechnology solutions with stable dispersions of metal or metal oxide nanoparticles (NPs) as anti-SARS-CoV-2 agents.2,4Studies on antiviral NPs have mainly focusing on the following two modes of action: specific blocking of membrane proteins or by destruction of the phospholipid bilayer, commonly via oxidation by NP-generated ROS.1,2 The first of those effects is usually achieved at relatively low, nanomolar to micromolar NP concentrations while the latter takes place usually at the millimolar concentration range.1,2 One of the most studied antiviral NPs is colloidal silver that has been suggested to attack glycoproteins of enveloped viruses such as SARS-CoV-2 and thereby eliminating viral ability to attach to the targeted host cell.5 It has been hypothesized that silver NPs bind to the sulfhydryl groups of the membrane proteins, causing cleavage of the disulfide bonds and disruption of the protein structure. The efficacy of silver NPs in neutralizing viruses is closely tied to their particle size, with NPs ranging from 2 to 15 nm exhibiting the highest affinity for proteins and the most potent virucidal activity.6 It has been proposed that the observed antiviral effect is due to (partial) dissolution of silver NPs and released free Ag+(aq)-ions.7 Similar to silver-based NPs, gold and copper-based NPs express their antiviral activity via membrane protein degradation.8,9
NPs of non-redox active oxides such as silica10 and alumina11 have been reported to have little effect on corona viruses, but were employed as vaccine adjuvants, improving stability of antibodies (and generally) small protein molecules adsorbed on them and stabilizing active component bearing Pickering emulsions. On the contrary, oxide NPs possessing chemical catalytic redox properties, such as iron oxides, were reported to degrade the membranes via lipoperoxidation pathway.12 It is important to note that this kind of behavior is typical for iron oxides containing Fe(II) centers active in Fenton reaction producing peroxide species. The oxides, featuring stabilized Fe(III) cations only turned to possess catalase-like activity, destroying the peroxo-species13 and thus not revealing any cell toxicity or antiviral effects.
Certain NPs utilize photochemically produced oxidative radicals to combat viruses, exemplified by titanium dioxide (TiO2) NPs. When exposed to UV light, holes and free electrons that produce reactive oxygen species (ROS) are generated in TiO2 NPs. These ROS subsequently transform into highly virucidal hydrogen peroxide (H2O2), effectively eradicating viruses.14,15 A separate study of photocatalytically inactive TiO2 NPs (coated by anti-oxidant triethanolamine ligand) showed strong complexation with nsp1 protein of SARS-CoV-2 virus. However, in that work the antiviral activity was not investigated.16
Recently, it has been envisaged that ceria NPs can act as antiviral agent, revealing considerable activity against enveloped viruses, including SARS-CoV-2 and influenza concentrations as low as 20 mg l−1 (30 μM). It was demonstrated that this activity was not caused by dissolution of NPs and release of Ce(III)-cations. The low concentration being enough in this case indicated specific mechanism, most probably destroying the cell membrane proteins in analogy to the effect observed for the precious metals NP.17
The aim of this study was to complement the current knowledge on antiviral efficacy and chemical mode of action of TiO2 NPs, especially in the absence of UV. In the view of earlier obtained evidence of the strong interactions of TiO2 NPs with proteins16 in darkness, we expected an activity of those NPs at nanomolar level. To our surprise, we obtained the antiviral efficacy of TiO2 NPs only at millimolar level. Thus, the hypothesis on the effect of those NPs on membrane proteins was rejected and we further investigated the interactions of TiO2 NPs with phospholipids. The absence of oxidative properties of the TiO2 NPs permitted to suspect direct complexation of NPs with phospholipids – a mode of antiviral action not discussed earlier. To gain an understanding of how such process can occur, we studied complexation of TiO2 NPs with phospholipid-imitating phosphate esters using nuclear magnetic resonance (NMR). As a negative control for TiO2 NPs we analyzed silicotungstate polyoxometalate (POM) NPs that are neither capable of forming complexes with phospholipids, nor possess any redox activity (due to low oxidation potential). In parallel with the potential interaction with phospholipids, antiviral effect of TiO2 and POM NPs was studied using two viruses, an enveloped swine transmissible gastroenteritis virus (TGEV) and a non-enveloped encephalomyocarditis virus (EMCV). Antiviral efficacy was assessed in infectivity assays and with viral RNA release test.
Results and discussion
Antiviral activity of selected NPs
The triethanolamine terminated titania (TATT) nanoparticles used in this study consisted of very uniform spherical TiO2 particles ca. 3.5 nm in diameter, which were stable in aqueous medium. Their solution and solid state characterization details (including TEM, SEM, DLS, UV etc.) are described in literature.16,19 TATT NPs have recently been demonstrated to exert pronounced activity in binding the nsp1 protein of SARS-CoV-2 virus.16 We suspected thus that these NPs might also strongly interact with surface proteins. Within the current work, we were looking both for confirmation of the expected antiviral activity of TATT NPs but also aimed at further elucidating their potential modes of action. Considering the previous literature suggesting that titania may bind strongly to phosphate and phosphonate residues,18 we were expecting to confirm two modes of action of the TATT NPs: (i) a specific protein-blocking mechanism taking place at low concentrations of titania, or (ii) binding of titania NPs to phosphate residues leading to direct membrane disruption (in darkness) occurring at pronouncedly higher concentrations. We assumes that oxidative disruption of phospholipid membrane, which is a well known mechanism for titania NPs under UV light15 was not interfering in our tests as the study was carried out in the dark. Also, the TATT material has been demonstrated not to be UV-activated and photocatalytically active in aqueous medium, likely due to an antioxidant activity of triethanolamine as capping agent.19As negative control not capable to bind strongly to cell membrane another NPs were selected, the silicotungstate polyoxymethalate (POM) ion [SiW12O40]4−(aq).
This species belong to the Keggin family and are, according to formal definition, NPs with actual diameter 1.1 nm. They have been demonstrated to exert pronounced antiviral activity against HIV virus in the acidic form,20 but are essentially inert in pH-neutral media.
POM NPs are weak oxidants and do not form surface complexes with phosphate residues. The solutions of POM were prepared by first complexing it with an amphiphilic peptide, tris-glycine, and then diluted with MES buffer pH = 7.0 generally considered suitable for viral activity tests and free from phosphate capable to compete in complexation with membrane phospholipids.
Antiviral activity of the two selected NPs was tested against two viruses, an enveloped coronavirus TGEV that is surrounded by a phospholipid membrane and three main transmembrane proteins,21,22 and a non-enveloped virus EMCV without phospholipid membrane.22 Before antiviral experiments cytocompatibility of both NPs was tested and no decrease in cell viability of host cells used for viral infection was observed at relevant NP concentrations (Fig. S1†). Infectivity of both viruses was measured after 1 h exposure to both NPs. The results demonstrated that silicotungstate POM is showing no statistically significant antiviral effect at any tested concentration, up to a relatively high concentration of 1.25 mM corresponding to 4.5 mg ml−1, for neither the enveloped nor non-enveloped virus (Fig. 1, statistical analysis in Table S1†). TATT proved itself antiviral against enveloped TGEV virus starting from 0.125 mM corresponding to 12 μg ml−1, but not towards non-enveloped EMCV virus (Fig. 1, statistical analysis in Table S1†), suggesting that this NP is likely affecting phospholipid-containing membranes. As supported by NMR data (see Fig. 3–6 below) indicating interactions between TATT and phospholipid models, we suggest that the mode of action of TATT against the enveloped virus was through interaction with membrane phospholipids.
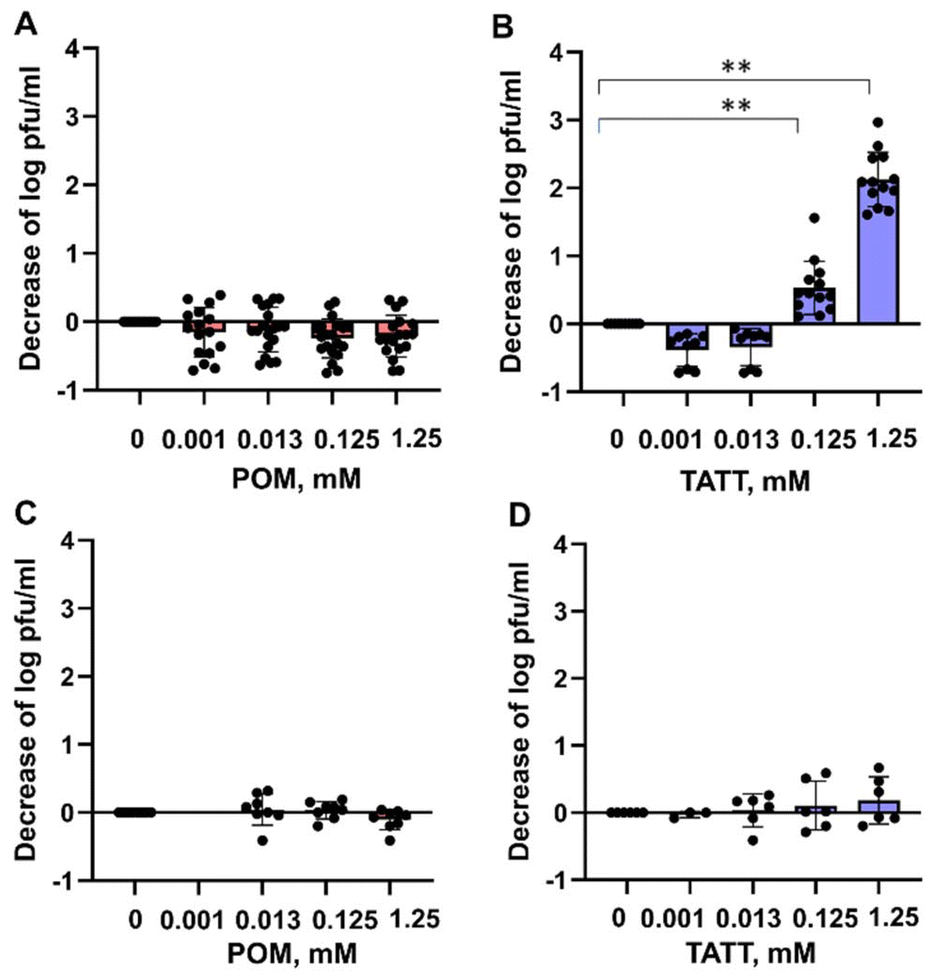 | ||
| Fig. 1 Effect of NPs of silicotungstate POM and TATT on TGEV virus (A and B) and EMCV virus (C and D). The TATT concentration is given with respect to particles, not TiO2 formula unit. Decrease in infectivity (log decrease in plaque forming units (pfu) per mL) in the presence of different concentrations of NPs is shown. Results of all measurements (dots) along with average (column) and standard deviation are displayed. Asterisks mark data points statistically (p < 0.05) different from no exposure conditions according to ANOVA followed by post hoc Tukey's range test. More detailed statistical analysis results with experimental p-values are shown in Table SX.† | ||
When compared with effective concentrations of other types of NPs and antiviral agents, the effective concentration of TATT was among average ones. Known antiviral agents such as Cu ions have been shown to decrease the infectivity of TGEV virus at 0.5 mM,23 CeO2 NPs at 1.5 mM and interestingly, colloidal Ag did not show antiviral effect even at concentrations >1.5 mM.17 However, one must note that the modes of action of all these mentioned compounds may be different and decrease of viral infectivity is an end result of these underlying molecular events.
An additional proof for viral membrane disruption by TATT was gathered by detecting the released RNA of TGEV coronavirus after nanoparticle treatment. As seen from Fig. 2, propidium iodide dye labelled released RNA level was significantly increased in TATT-treated TGEV but not in POM-treated virus indicating that indeed, the viral phospholipid-containing envelope was notably disrupted by the TATT particles. Interestingly, POM treatment decreased the propidium iodide labelled RNA level in viral suspension.
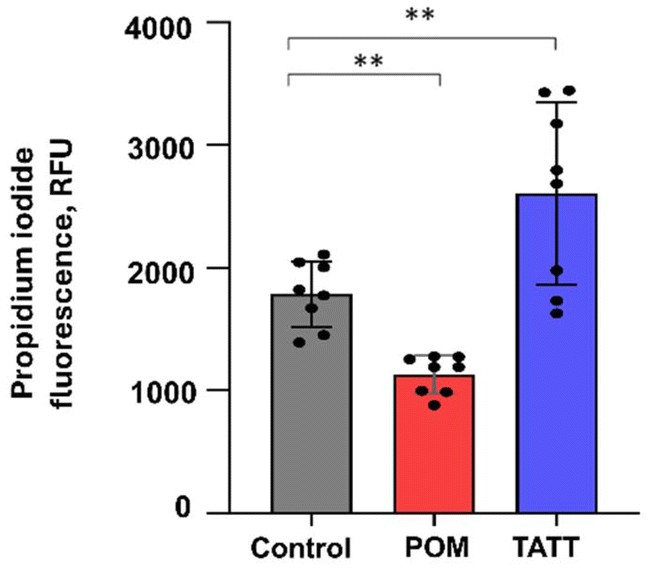 | ||
| Fig. 2 Propidium iodide signal in TGEV virus suspension without any treatment (control), or after 1 h incubation with POM or TATT. Results of all measurements (dots) along with average (column) and standard deviation are shown. Propidium iodide signal was considered as an indicator for released RNA. Asterisks mark statistical (p < 0.05) differences from control in ANOVA statistical analysis. | ||
Binding models for 31P NMR study at different temperature and pH 7.0
Aiming to verify our suggestion that the antiviral effect of TATT NPs against enveloped virus was due to complexation with membrane phospholipids, resulting in potential membrane disruption, we studied the interactions between TATT NPs and phospholipid model compounds. The latter were highly water soluble phosphorylated glucose species, disodium salt of D-glucose-1-phosphate hydrate (glucose-1P; CAS 7558-79-4) and disodium salt of D-glucose-6-phosphate hydrate (glucose-6P; CAS 3671-99-6) (Scheme 1) that are not prone to aggregation/micelle formation. Selecting two slightly different molecules was motivated by the need to rule out any influence of side chain and only be able to observe the complexation of the phosphate group.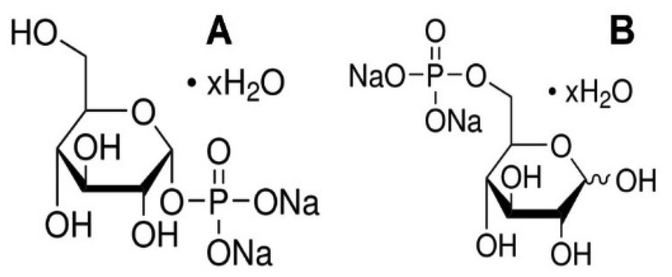 | ||
| Scheme 1 Disodium salt of D-glucose-1-phosphate hydrate (A), disodium salt of D-glucose-6-phosphate hydrate (B). | ||
We have followed the effect of different temperatures on the chemical shift of 31P signal as indication of chemical transformations. The dependence of the 31P chemical shift on the temperature is a complex phenomenon influenced by various factors related to molecular structure, interactions, and environmental conditions. The selected phosphate containing probes allowed us to monitor its interaction with NP in the real temperature-time scale using 1D 31P NMR spectroscopy.
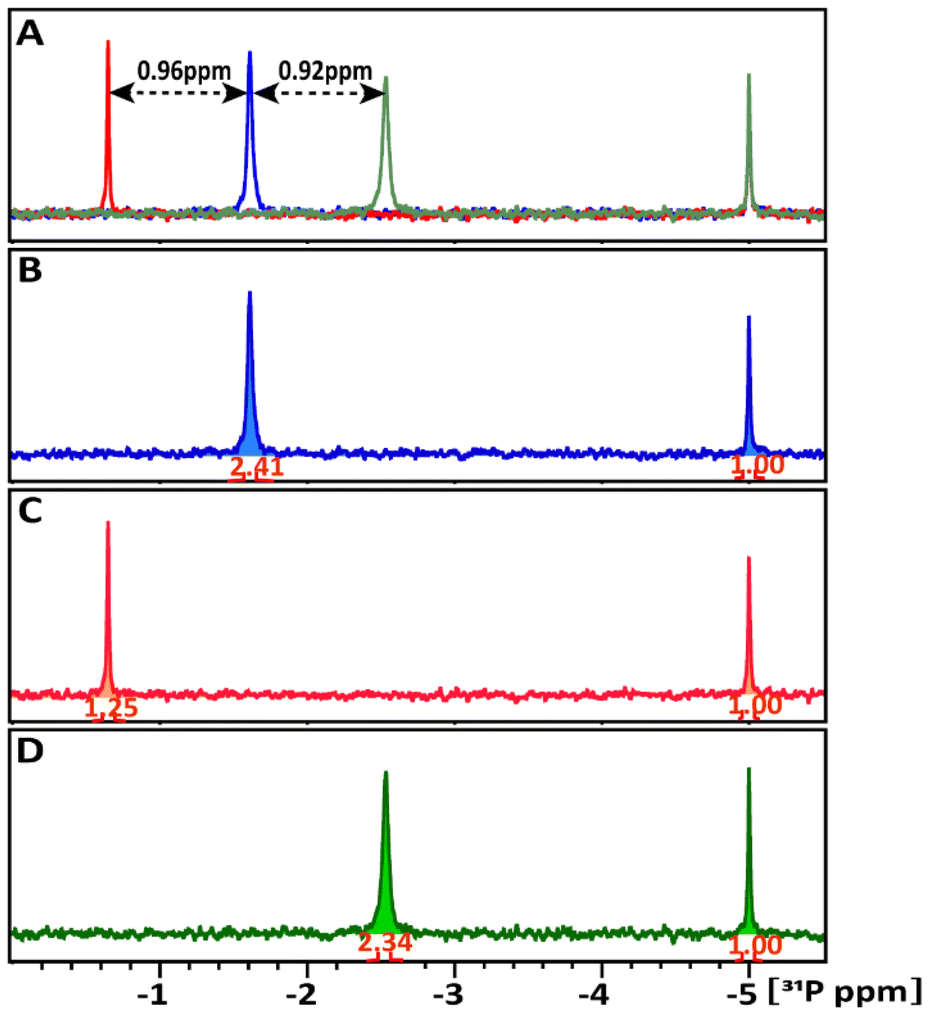 | ||
| Fig. 3 1D 31P NMR spectra of glucose-1P in comparison (A). (B) Glucose-1P only, (C) glucose-1P + TATT NPs, (D) glucose-1P + POM NPs, and (A) the superposition of these spectra. The black arrows indicate the chemical shifts induced by the NPs relative to free glucose-1P. The signal intensities in panels (B) to (D) were normalized relative to the external ERETIC signal with value of 1.00 at −5 ppm. | ||
Though interaction was detected in both cases, the share of NP involved in this interaction is drastically different in the case of TATT and POM. The amount of glucose-1P interacting with NPs at 298 K was estimated by comparing the integrals of the 31P resonance signals of the starting glucose-1P solution and its remaining free signal in the mixture. In Fig. 3B–D, the 31P signal intensities were normalized relative to the external ERETIC signal with value of 1.00 at −5 ppm. In the glucose-1P + TATT mixture, 48% of glucose-1P interacts with NP, whereas in the glucose-1P + POM mixture, only about 3% of it is involved in complexes formation with NP, meaning that the vast majority of POM are not involved in interaction. Presence of a single signal points at quick exchange between bound and free molecules on the NMR scale.
1D 31P spectra at different temperature, ranged between 278–333 K, in D2O buffer solution at pH 7 (A) of the glucose-1P and its mixture with (B) POM and (C) TATT NP's are presented on Fig. 3. Fig. 4A shows that as the temperature increases, the 31P resonance of glucose-1P without NP still shifts upfield with the rate increasing along with increase of the temperature. It is known, that the tumbling time of the molecule depends on the temperature: it decreases with increasing of temperature leading to the narrowing of the 31P resonance line width. However, even at 333 K, the resonance still broadens.
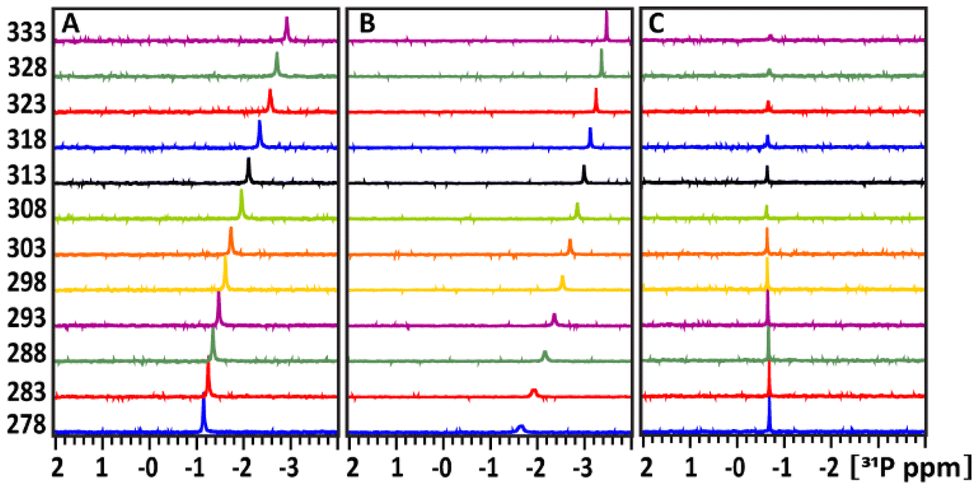 | ||
Fig. 4 Temperature dependence of the 31P chemical shift of the glucose-1P (A), glucose-1P and its mixtures in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with POM NPs (B); and in 1:1 ratio of glucose-1P + TATT NPs (C). The intensities of the signal were normalized to the using an ERETIC signal with value of 1.00 at −5 ppm. :1 ratio with POM NPs (B); and in 1:1 ratio of glucose-1P + TATT NPs (C). The intensities of the signal were normalized to the using an ERETIC signal with value of 1.00 at −5 ppm. | ||
This indicates a conformation change in glucose-1P, likely involving weak internal hydrogen bonding interactions between OH ring groups and 31P nuclei, a temperature-induced anomerization process.24
Adding POM NPs to the glucose-1P solution shows (Fig. 3B) that as the temperature increases, the 31P resonance of glucose-1P also shifts upfield, though with a rate decreasing along with the increase of the temperature, unlike glucose-1P without NP (Fig. 3A). Indeed, already at 278 K, the resonance of the free glucose-1P (97%) is very broad due to a quick exchange between free glucose-1P and a weakly bound complex glucose-1P with NP (3%). The 31P signal belonging to the complex is not observed in the spectrum due to the slow tumbling time caused by the large size of the NP. At 333 K, the increased exchange rate between the bound and unbound states, due to the higher temperature, leads to the narrowing of the 31P resonance. This type of temperature-dependence behaviour of ligand–substance interactions is typical for weakly interacting complexes and is well documented in studies of molecule–NP interactions.25
A completely different temperature-dependent behaviour of the 31P resonance of glucose-1P is observed upon adding TATT, as shown in Fig. 3C. Notably, the exchange between bound and unbound complex is so slow that the 31P resonance which is not involved in complex formation (52%) remains unbroadened even at 278 K. Only above 308 K does the 31P signal start to decrease in intensity, with a slow increase in line broadening and a slight, approximately 0.2 ppm, upfield shift. The transition is not complete even at 333 K. This behaviour is characteristic of strongly bound molecule–NP complexes with a slow exchange rate between the free and bound states.25
:1 intensity ratio, corresponding to two different isomers with alternative orientations of the OH-group at the C(1)-atom. In the glucose-6P and TATT mixture, both 31P signals exhibit downfield shifts, detected at 1.462 and 1.417 ppm (δ31P1 = 1.03 ppm and δ31P2 = 1.11 ppm) respectively, indicating slightly different interactions of the two probes with the NP.
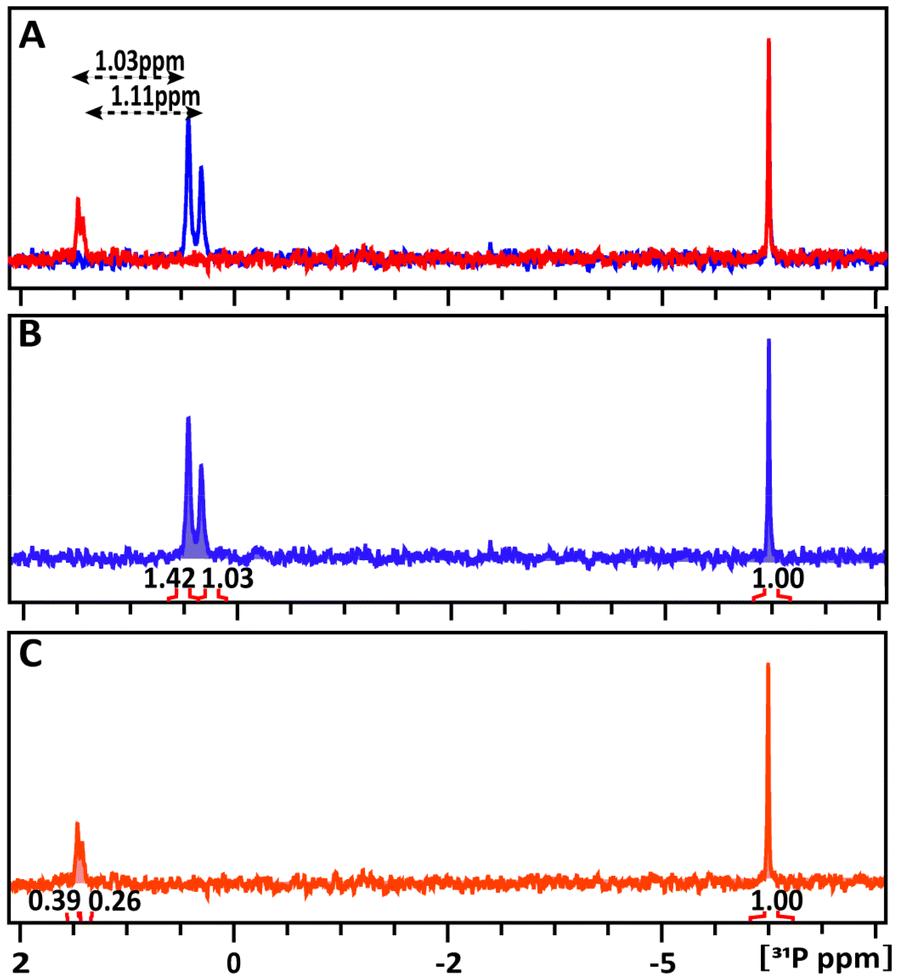 | ||
| Fig. 5 1D 31P NMR spectra of glucose-6P probe with and without NPs in comparison with the superposition of the spectra (A). Free glucose-6P (B), and glucose-6P + TATT NPs (C). The black arrows indicate the chemical shifts induced by the NPs relative to free glucose-6P. The signal intensities in panels (B) and (C) were normalized relative to the external ERETIC signal with value of 1.00 at −5 ppm. | ||
The amount of glucose-6P interacting with TATT at 298 K was estimated by comparing the integrals of the 31P resonance signals of glucose-6P in the solution to the remaining free signals in the mixture. In the glucose-6P and TATT mixture, 73% and 75% of the two different isomers, 31P1 and 31P2, respectively, interact with the NPs, the quick exchange resulting in only one signal for each isomer. This represents approximately twice the loading of glucose-6P in the TATT complex compared to glucose-1P caused possibly by different steric effects in surface complexation of the two molecules.
Fig. 6 presents the 1D 31P spectra at different temperatures, ranging from 278 K to 333 K, in a D2O buffer solution at pH 7 for glucose-6P (A) and its mixture with TATT NPs (B). In Fig. 6A, the 31P resonance of glucose-6P without NPs shifts upfield in a sigmoidal pattern, similar to free glucose-1P (Fig. 3A). This indicates a rearrangement of weak internal hydrogen bonding interactions between the OH ring groups and the 31P nuclei.
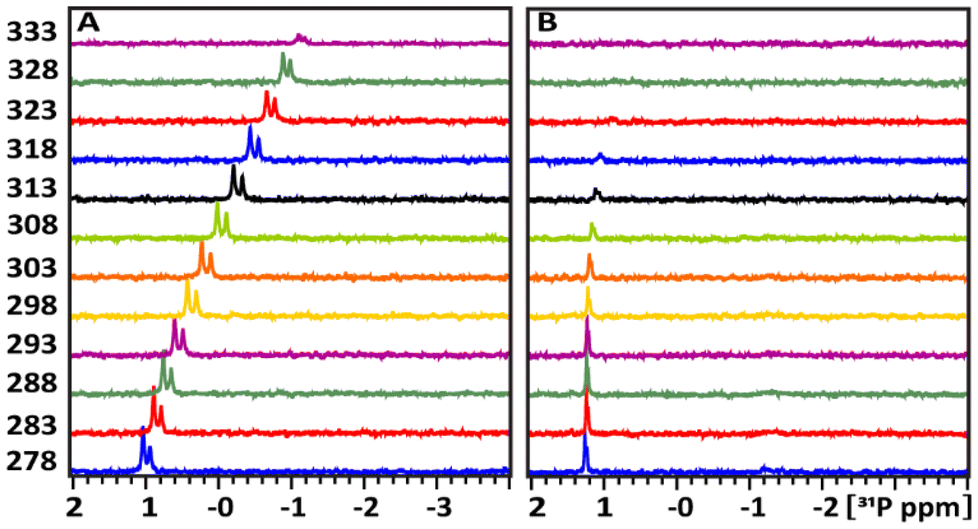 | ||
| Fig. 6 Temperature dependence of the 31P chemical shift in the NMR spectra of the pure glucose-6P (A) and its mixture with TATT (B). The intensities of the signal were normalized to the using an ERETIC signal with value of 1.00 at −5 ppm. | ||
Fig. 6B shows the temperature-dependent behaviour of the 31P resonances of glucose-6P upon adding TATT. The exchange between the bound and unbound complexes is slower compared to glucose-6P without NPs. However, the 31P resonances not involved in complex formation (ca. 26%) begin to show extensive broadening at 303 K, indicating accelerated exchange between the free and bound complexes. Above 303 K, the complex signal is no longer detectable.
In summary, both glucose-1P and glucose-6P show a tendency to form strong complexes with TATT NPs. Notably, the averaging of the 31P chemical shifts between the free and complex forms due to accelerated exchange with increasing temperature occurs at approximately 30 degrees higher for glucose-1P than for glucose-6P. In contrast, the interaction of glucose-1P and glucose-6P with POM NPs is very weak, resulting in low concentration complex formation.
Disappearing of the signal for phosphate ligands turned out in fact to be caused by formation of a gel composed of TATT and adsorbed glucose phosphate.
The gel is rather uniform (featureless and flat) according to AFM data, see Fig. 7, and featureless under SEM, displaying essentially only titania in the EDS analysis as single glucose phosphate molecule was bound to a whole TATT particle containing over 1000 Ti atoms (see Fig. 7).17 Similar process could be the essence of the antiviral action of titania particles observed in this work: with the increase of the temperature TATT particles, including ones bound to the phosphate groups of phospholipids, are forced to aggregate “dragging” the membrane lipids with them in the gel formation.
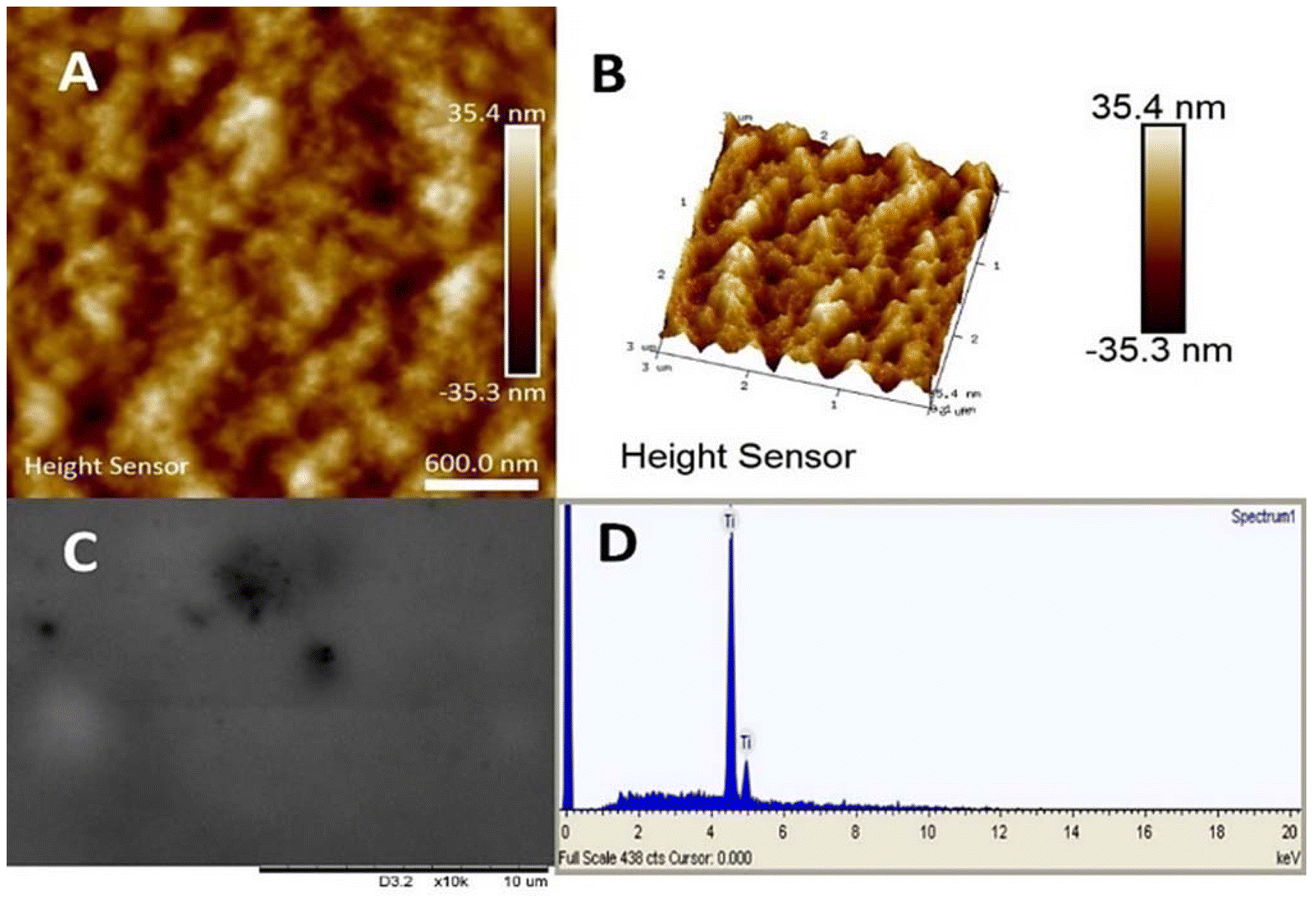 | ||
| Fig. 7 Characterization of the gel formed on interaction of TATT with glucose phosphates. AFM images (A and B), SEM (C) and EDS spectrum (D). | ||
Apparently, the particles are picking up relatively large patches from the membrane, as the coupling of phosphate groups of phospholipids with TATT particles appears to be stronger than interaction between phospholipids in the virus membrane. In other words, one can suggest that the viruses are destroyed via membrane disruption caused by TATT particles aggregation at rather high titania concentrations. This process is visualized in Fig. 8. Direct confirmation for this proposed mechanism was obtained via detection of the RNA leaching from damaged viruses. The temperature when this process is dominating is about the body temperature, starting clearly from 25 °C, which opens a possibility for using nanoparticle dispersions in, for example, personal protection media against corona viruses. Best evidence for the proposed mechanism would be TEM imaging of damaged and non-damaged viruses, but we were unable to achieve high enough concentration necessary for facile identification of the objects to be imaged, to be in the order of 1011 PFU per ml, as has been used in the works of Sadakane et al.26
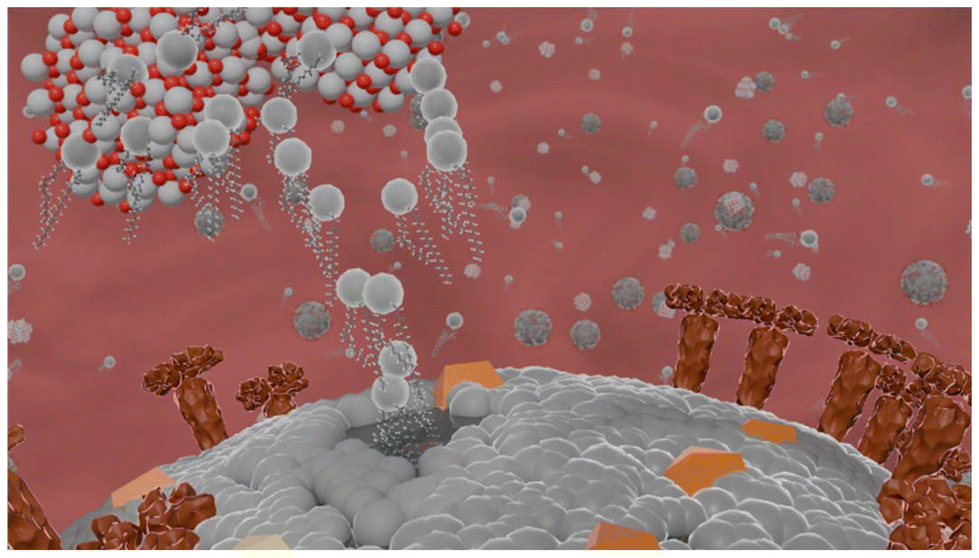 | ||
| Fig. 8 Proposed schematics of the interaction between enveloped virus and TATT TiO2/TATT/TATT titanate nanoparticles (grey color indicates phospholipids, yellow and brown indicate different kinds of membrane proteins). | ||
The particle concentration required for the observation of antiviral effect in our case is just above 1.0 mM value. This correlates well with the proposed non-specific interaction with phospholipids as, according to earlier studies of specific membrane protein interactions with silver NP, the effects were usually observed at 1–10 μM concentration range.27,28
Selective bonding of titania to phospholipids in competition of proteins appears quite logical in the view of mechanisms of chemical bonding to these two classes of compounds. While attachment to proteins occurs usually, in the first hand, on the carboxylate side-groups attached to an α-helix, a relatively weak inner-sphere complexation with two relatively long Ti–O bonds, 2.05–2.09 Å,16,29 the bonding to phosphate or phosphonate group in phospholipids is proceeding at neutral pH via three much stronger and shorter Ti–O bonds, 1.93–1.99 Å (ref. 18) (see Fig. 9).
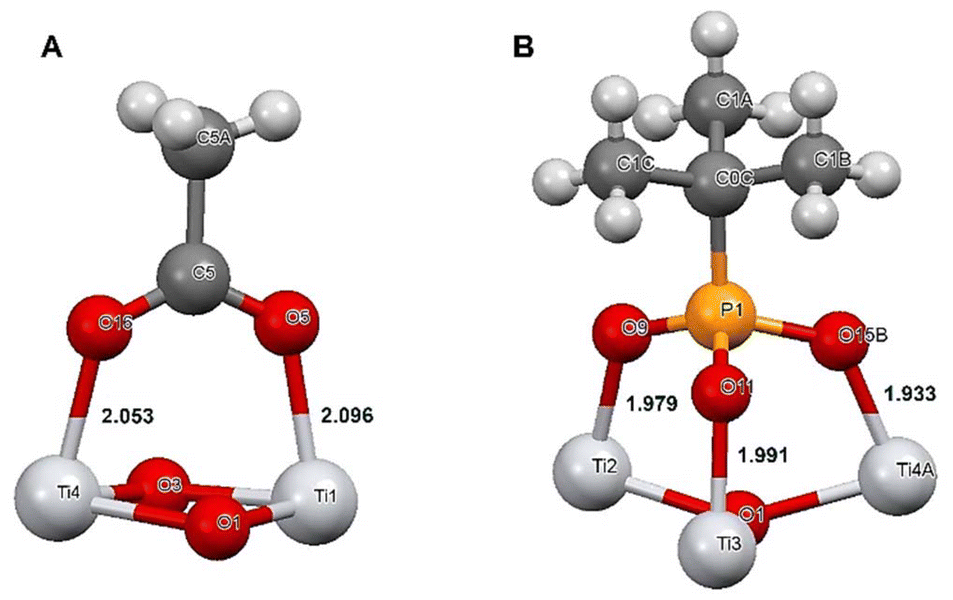
Structure and stability characterization of the reference POM particles
In order to insure that we had the POM as particles (not destroyed through dissociation under pH-neutral conditions) we performed structural characterization of the samples forming in the buffer and of the initial complex used for the experiments. The crystal structures of silicotungstate with triglycine and HEPES buffer molecules (Fig. 10) show extensive H-bonding between ligand species (Fig. S2 and Tables S2, S3†). Triglycine make direct contact through multiple H-bonds with the POM, while HEPES forms only one H-bond, and otherwise displays electrostatic interactions and indirect H-bonding via water molecules. Worth noting are also the approximately 3 Å contacts between the surface oxygen on the silicotungstate in the complex with HEPES. These crystals were formed at neutral pH, while the triglycine complex was produced originally under acidic conditions, but subsequently re-dissolved in a MES buffer with regulated pH. Unlike phosphorous-centered POM analog, the Silicotungstate POM appears to be stable at neutral and slightly basic pH.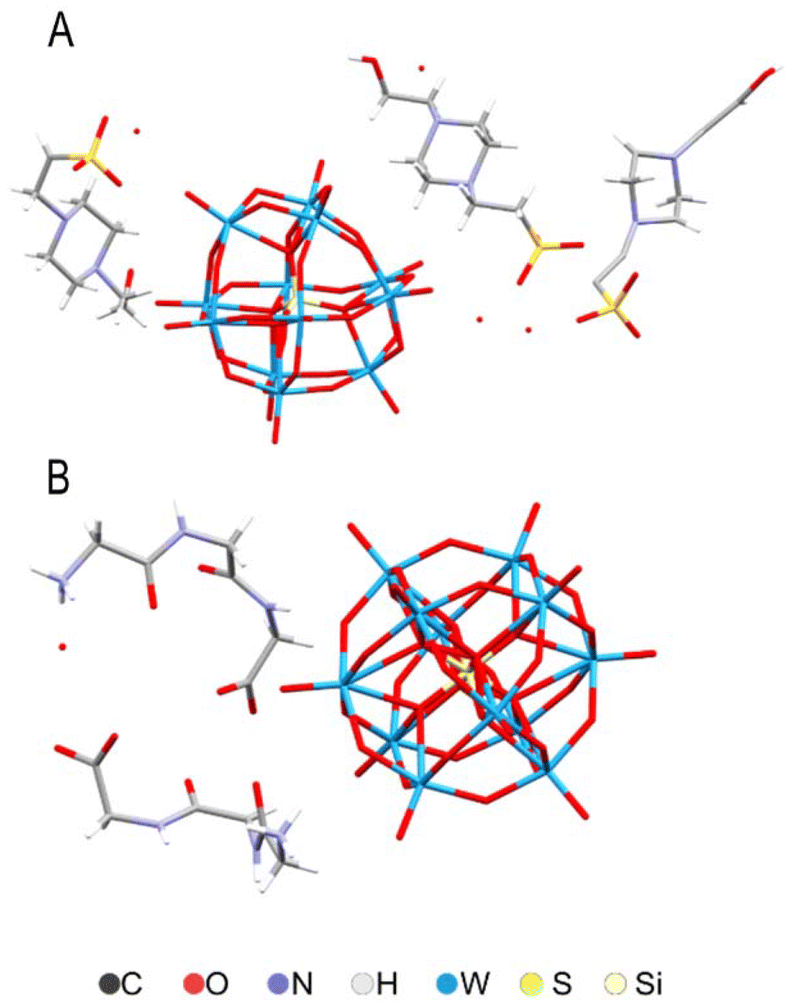 | ||
| Fig. 10 Molecular structures of (HEPES)3(H3O)SiW12O40(H2O) (A) and (HGly3)4SiW12O40(H2O)2 (B) according to X-ray single crystal analysis. | ||
Conclusions
Combination of structural and spectroscopic data along with the antiviral activity studies of TATT titania NPs permit to hypothesize that the mechanism behind antiviral effect of titania in dark conditions, i.e., without UV illumination, is based on the direct membrane disruption via complexation of titania NPs with phospholipids. This further leads to aggregation of titania NPs on phospholipids and to the best of our knowledge, proves a new pathway for metal oxide nanoparticles antiviral action. Our results demonstrating that that TATT titania NPs exhibited antiviral activity towards membrane-containing enveloped viruses and not towards non-enveloped viruses. Allowing to suggest a direct interaction, inner-sphere complex formation, between NPs and the virus membrane, leading to its non-oxidative disruption. The inner-sphere complex formation was proven by studying the interactions of highly water-soluble analogs of phospholipids with titania NPs using NMR spectroscopy. Our observation that interaction of TATT NPs with enveloped viruses resulted in RNA leakage further proved our hypothesis on their direct membrane disruption ability. Importantly, none of the described effects was observed upon exposure to silicotungstate polyoxomethalate (POM) NPs, showing the ability to formation of inner sphere surface complexes to be a crucial factor for this type of mechanism. The obtained results indicate that surfaces coated by titania NPs and potentially by other small oxide NPs with high affinity to phosphate terminated biomolecules (such as iron or aluminium oxides) can be active in destruction of enveloped viruses without the need to be activated by light (UV). Another possible application can be in decontamination of water under emergency conditions by adding TATT and letting the gel formed by NPs and membrane phospholipids to sedimentate.Experimental
All chemicals were purchased from Sigma-Aldrich and used without further purification.Titanium oxide colloids surface stabilized by triethanolamine (TATT) were obtained as reported previously in ref. 15. The sample of TATT TiO2 was produced by modification of titanium ethoxide with dry triethanolamine (mixing 5.0 mL liquid Ti(OEt)4 and 1.5 mL liquid N(C2H4OH)3 until formation of clear yellowish solution) with subsequent dropwise addition on vigorous stirring of a hydrolyzing solution produced by mixing 0.5 M HNO3 (0.5 mL) with EtOH (2.0 mL). This procedure was resulting in NP starting solution with TiO2 concentration 120 mg g−1 as established by TGA. Stabilization of the particles, having uniform size of 3.2 nm according to DLS, occurs in this case, as demonstrated in ref. 15, via protonation of the surface-capping triethanolamine ligand. The size of the particles was also confirmed by high-resolution TEM in the same work. The ligand is partially released on transition into aqueous medium, rendering the resulting particles negatively charged with zeta-potential about −11 mV.
Synthesis of POM model
Samples of POM and POM-peptide complex were produced by dissolution of silicotungstic acid (purchased from Sigma-Aldrich) in either MES or HEPES 0.100 M buffer or crystallizing first the complex (HGly3)4SiW12O40(H2O)2 and dissolving it then in a MES buffer, adjusting pH to 7.0 by addition of 0.10 M NaOH.DLS
Determination of hydrodynamic particle size and surface charge of the particles (zeta-potential) was carried out with Malvern Panalytical Zetasizer Ultra instrument.ESEM-EDS
The measurements were performed with Hitachi TM-1000-μ-DeX instrument, applying the produced coagulated TATT-glucose phosphate gel on a carbon tape applied on a standard aluminium holder.AFM
The measurements were made with Bruker Advance FastScan Bio instrument in a ScanAsyst mode. The sample was prepared by applying the produced coagulated TATT-glucose phosphate gel on a stainless steel holder and drying overnight.X-ray crystallography
Suitable crystals were selected in Nujol oil under microscope and placed on an amorphous cellulose needle onto a goniometer head. The data collection was carried out with Bruker D8 Quest ECO instrument in the 2-theta range 4–58° using Apex-IV program package. Data integration was made in the range 4–50.05° (1.0 Å resolution). Full details of data collection, solution and refinement details are provided in ESI Table 1.†NMR
1D 31P and 1H NMR experiments were acquired on a 600 MHz Bruker Avance III spectrometers equipped with a Smart Probe. Spectra were collected at 278–333 K temperature range with step 5 degree. The equilibration of temperature in sample was 15 min. In 1D 31P the acquisition time, (AQ), of 1.1 s, relaxation delay, (D1) of 1 s, sweep width (SW) 30 ppm and 4096 number scans (NS) with carrier frequency O1 = 0 ppm used. In 1D 1H the AQ = 1.7 s, relaxation delay, D1 = 1 s, SW = 16 ppm, O1 = 4.706 ppm and NS = 32 number scans were used. Spectra were processed by TopSpin4.3.0.All spectra where were acquired in 5 mm NMR tubes (final volume of 0.500 mL) with internal 1H chemical shift standard, 0.1 mM DSS (4,4-dimethyl-4-silapentane-1-sulfonic acid) and 31P chemical shifts were referenced indirectly to the 1H standard using a conversion factor derived from the ratio of NMR frequencies. Additionally, the synthetic ERETIC signal at −5 ppm were implemented as external standard for intensities calibration in every 31P spectrum.
Sample preparations of the phosphorylated glucose with NP's for temperature studies by NMR
1′ and 6′ phosphorylated glucose (glucose-1P and glucose-6P, respectively) were obtained from Sigma Aldrich company and used as received. The following stock solutions were prepared: TATT of 2 mM, POM of 2 mM, glucose 1P and 6P (10 mM) and DSS (25 mM) in D2O. Buffer used in experiments was the composition: 200 mM triethanolamine in D2O pH = 7.0.From the TATT and POM stock solutions, 125 μL was pipetted into separate 1.5 mL Eppendorf vials and freeze dried overnight. To each 250 μL of solution, containing preliminary as 250 μL of buffer and 200 μL of D2O, 2 μL DSS, 50 μL of glucose 1P/or 6P from the stock solution was added. The samples were then put into an ultrasonic bath for 10 min at 25 °C and thereafter transferred into 3 mm NMR tubes. The final ratio of the glucose 1P/or 6P to TATT/POM 1:1 with concentration 1 mM in NMR tube. All experiments were performed at pH 7.
Preparation and characterization of (HEPES)3(H3O)SiW12O40
Crystals were obtained by reverse flow crystallization. 0.1 M HEPES buffer at pH = 6.5 and a solution of 2 g silicotungstic acid and 0.367 g diglycine in 10 mL milliQ water were prepared. 10 mL of the buffer and 2 mL of the SiW/GlyGly solution were added to a fresh, weighed tube. Upon mixing, a milky precipitate formed, and the reaction was allowed to proceed overnight. The precipitate was pelleted by centrifugation and washed with milliQ water, then dried in a 50 °C oven overnight. The tube and pellet were weighed, and the tubes mass subtracted. The mass of the pellet was calculated to 0.2179 g. The theoretical 100% yield was calculated to 0.5039 g, and the yield to 43.24%. A single crystal was selected for structure characterization. Total of 2424 frames were collected. The total exposure time was 13.47 hours. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using a monoclinic unit cell yielded a total of 119465 reflections to a maximum θ angle of 32.49° (0.66 Å resolution), of which 25909 were independent (average redundancy 4.611, completeness = 97.8%, Rint = 9.67%, Rsig = 8.86%) and 17682 (68.25%) were greater than 2σ(F2). The final cell constants of a = 32.191(3) Å, b = 14.8183(12) Å, c = 15.3532(13) Å, β = 91.018(2)°, volume = 7322.6(11) Å3, are based upon the refinement of the XYZ-centroids of 9987 reflections above 20 σ(I) with 4.557° < 2θ < 64.51°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.532. The structure was solved and refined using the Bruker SHELXTL software package, using the space group P21/c, with Z = 4 for the formula unit, C5H9N5O36S3Si3W12. The final anisotropic full-matrix least-squares refinement on F2 with 899 variables converged at R1 = 7.44%, for the observed data and wR2 = 19.97% for all data. The goodness-of-fit was 1.032. The largest peak in the final difference electron density synthesis was 7.463 e− Å−3 and the largest hole was −3.155 e− Å−3 with an RMS deviation of 0.482 e− Å−3. On the basis of the final model, the calculated density was 2.814 g cm−3 and F(000), 5360 e−. For details, please, see Table S2.†
Preparation and characterization of (HGly3)4SiW12O40
Crystals were produced by dissolving 0.0058 g triglycine and 0.028 g silicotungstic acid in 5 mL 0.1 M HCl each. The silicotungstic acid solution was added to the peptide solution, mixed and left to evaporate in a Petri dish. After several days, small colourless plate crystals formed. Quantitative yield. The chemical nature was proved by the single crystal study described below and the phase purity was confirmed by powder X-ray diffraction test. A total of 2424 frames were collected. The total exposure time was 2.02 hours. The frames were integrated with the Bruker SAINT software package using a narrow-frame algorithm. The integration of the data using a triclinic unit cell yielded a total of 16418 reflections to a maximum θ angle of 25.02° (0.84 Å resolution), of which 5757 were independent (average redundancy 2.852, completeness = 96.2%, Rint = 3.77%, Rsig = 4.62%) and 5605 (97.36%) were greater than 2σ(F2). The final cell constants of a = 10.8473(5) Å, b = 12.4152(5) Å, c = 13.1760(6) Å, α = 99.7051(8)°, β = 91.9863(9)°, γ = 103.7460(9)°, volume = 1693.76(13) Å3, are based upon the refinement of the XYZ-centroids of 9993 reflections above 20 σ(I) with 5.181° < 2θ < 69.49°. Data were corrected for absorption effects using the multi-scan method (SADABS). The ratio of minimum to maximum apparent transmission was 0.460. The structure was solved and refined using the Bruker SHELXTL software package, using the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with Z = 1 for the formula unit, C24H44N12O56SiW12. The final anisotropic full-matrix least-squares refinement on F2 with 502 variables converged at R1 = 6.55%, for the observed data and wR2 = 15.70% for all data. The goodness-of-fit was 1.181. The largest peak in the final difference electron density synthesis was 4.514 e− Å−3 and the largest hole was −2.369 e− Å−3 with an RMS deviation of 0.355 e− Å−3. On the basis of the final model, the calculated density was 3.560 g cm−3 and F(000), 1622 e−. For details, please, see Table S3.†
, with Z = 1 for the formula unit, C24H44N12O56SiW12. The final anisotropic full-matrix least-squares refinement on F2 with 502 variables converged at R1 = 6.55%, for the observed data and wR2 = 15.70% for all data. The goodness-of-fit was 1.181. The largest peak in the final difference electron density synthesis was 4.514 e− Å−3 and the largest hole was −2.369 e− Å−3 with an RMS deviation of 0.355 e− Å−3. On the basis of the final model, the calculated density was 3.560 g cm−3 and F(000), 1622 e−. For details, please, see Table S3.†
Antiviral tests
Antiviral assessment was carried out with two viruses, an enveloped porcine transmissible gastroenteritis virus (TGEV)21 (obtained from L. Enjuanes at Department of Molecular and Cell Biology, National Center of Biotechnology, Madrid, Spain) and a non-enveloped encephalomyocarditis virus (EMCV)22 that belongs to family Picornaviridae and is a causative agent for neurological disorders (obtained from A. Kohl MRC-University of Glasgow Centre for Virus Research, Glasgow, Scotland, United Kingdom).TGEV virus was propagated in ST cells (obtained from L. Enjuanes at Department of Molecular and Cell Biology, National Center of Biotechnology, Madrid, Spain) grown using high-glucose DMEM (Corning Inc.) with 5% inactivated fetal bovine serum (FBS, PAN Biotech) and 100 U mL−1/100 μg mL−1 penicillin/streptomycin (Corning). For virus propagation, ST cells were infected with TGEV suspension in virus growth medium, VGM (DMEM supplemented with 0.2% bovine serum albumin (BSA, Sigma) and 100 U mL−1/100 μg mL−1 penicillin/streptomycin). Infected cells were grown for 4 days at 37 °C and 5% CO2, after which cell supernatant was collected, clarified by centrifugation at 3000g for 10 min at +4 °C, filtered through a 0.2 μm filter, aliquoted and stored at −80 °C. For antiviral experiments the viral storage stocks were diluted 20 times with water. The diluted viral suspension was mixed with an equal volume of POM or TATT colloidal solution and left for 1 h at room temperature. As a negative (non-exposed) control, diluted virus stock mixed with sterile water 1:1 v/v was used. Diluted virus exposed to UVC for 10 minutes was used as a positive control and expected to result in 100% viral inactivation. Particle-exposed virus, negative and positive controls were diluted 10 times with VGM and 150 μl of the resulting sample was used to infect fully confluent ST cells grown on 12- or 96-well plates. Infection was carried out at 37 °C, 5% CO2, humidified atmosphere for 1 h with gentle rocking every 15 min. The medium was then removed, and carboxymethylcellulose sodium salt (CMC, ThermoScientific, MW 2500004; DS = 1.2, dissolved in sterile water to 4 mass%):VGM 2:3 v/v mixture was added. Cells were further incubated for 96 h at 37 °C, 5% CO2, CMC/VGM was removed, plates were stained using crystal violet (ThermoFischer, diluted in a mixture of water, ethanol and 37% formaldehyde) and the plaques visually counted. Only wells with at least 3 plaques were considered. Plaque forming units (pfu) per mL for non-exposed viruses, POM and TATT exposed viruses was calculated. Each concentration of NPs was tested at least in two replicates while performing at least three independent experiments.
EMCV virus was propagated in BHK-21 cells grown in GMEM medium (Corning) with 10% heat-inactivated FBS, 2% TPB, 2% Hepes (pH 7.2), 100 U mL−1 penicillin, 100 μg mL−1 streptomycin. For virus propagation, BHK-21 cells were infected with EMCV suspension in VGM (growth medium further supplemented with 0.2% bovine serum albumin (BSA, Sigma)), infected cells were grown for 4 days at 37 °C and 5% CO2 and supernatant was clarified by centrifugation at 3000g for 10 min at +4 °C. Centrifuged extract was then filtered through a 0.2 μm filter, aliquoted and stored at −80 °C. Virus titer was 2.5 × 107 pfu per mL. 10-Fold water-diluted virus suspension was used in antiviral experiments. The diluted virus suspension was then mixed with respective POM and TATT dilutions, or water for negative control and mixtures were incubated at room temperature fir 1 h. For positive control, viral exposure at UVC was used, as described above. TCID50 endpoint dilution method was used to assess the virus titre.30,31 For TCID50 assay virus-containing mixtures were diluted up to 106 times with infection medium (IM); GMEM medium with 0.2% of BSA, 100 U mL−1 penicillin, 100 μg mL−1 streptomycin and 2% Hepes (pH 7.2). 40 μL of the mixtures were then used to infect fully confluent BHK-21 cells in 96-well plates. Each virus dilution was used to infect 7 wells. Non-infected wells with 100 μL IM were used as background control. After 1 h incubation in humidified atmosphere at 37 °C and 5% CO2, the content of all the wells except the background control was aspirated, replaced with 100 μL of IM and cells were further grown for 18–24 h. Then cells were stained with crystal violet as described above. Upon drying, 200 μL ethanol per well was added, the plates were shaken for 10 min and absorbance at 592 nm was measured with Biotek Epok plate reader. Cell viability for each well was calculated as the ratio of absorbance to averaged absorbance of the background control. Wells with cell viability less than 70% of the background were considered as positive, i.e., affected by the virus. The following formula was used to calculate infectious units (I) that were considered equal to pfu:
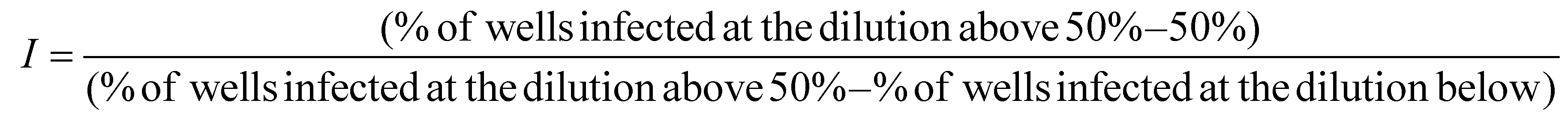
50% tissue culture infectious dose TCID50 (infectious units per mL) was calculated as follows:
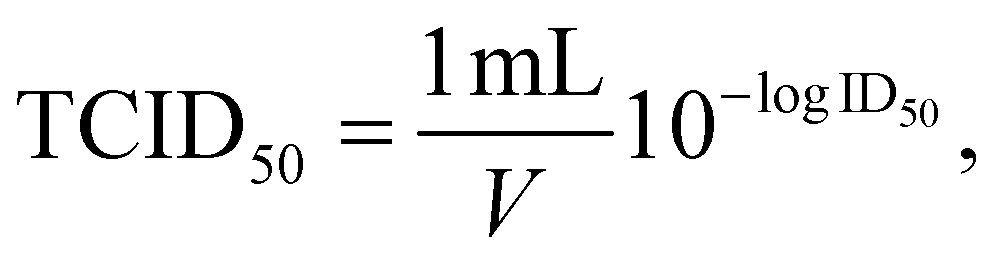
Before antiviral tests, cytotoxicity evaluation of POM and TATT NPs was performed with viral host cell lines at relevant NP concentrations and 1 h contact time using Neutral Red uptake assay.32
To detect the released RNA in treated TGEV samples, TGEV storage stock was diluted 1:10 with sterile water as above, the measured amounts of POM and TATT stock colloids were added to produce the colloidal solutions with the concentration of 1.25 mM. The mixtures were incubated at room temperature for 1 h and then the propidium iodide solution in water (1 μg mL−1) was added. Propidium iodide is a well-known dye binding to nucleic acids, but not penetrating intact membranes. The mixtures were incubated for 20 min after which the fluorescence with the excitation wavelength of 535 nm and the emission wavelength of 617 nm was measured with Tecan Infinite 2000 plate reader. The signal from propidium iodide added to TGEV virus suspension, that was not exposed to nanoparticles, was used as a control.
Statistical analysis
For statistical analysis, log pfu per ml in viral suspensions in non-exposed control and in NP exposures were compared using one-way ANOVA in Origin software (OriginLab). Statistically significant differences (p-values of less than 0.05) were revealed and post hoc Tukey's range test with CLD output was performed.Author contributions
B. G. – synthesis of POM models and their structural characterization, contribution to writing, A. N. – anti-viral effect studies, T. A. – interpretation of NMR data, contribution to discussion and writing, P. A. – acquisition of NMR data, V. K. and K. R. – contribution to interpretation of anti-viral activity data, discussion and writing, A. V. – contributed to conceptualization, discussion and editing, G. A. S. – performed ESEM and AFM studies and contributed to discussion and writing, A. I. – methodology, conceptualization, writing and editing, V. G. K. – methodology, conceptualization, resources, contribution to writing and final editing.Data availability
The results of structure solution and refinement are available under CCDC 2380871 and 2380872 for compounds B and A respectively. All other experimental details are available from the authors on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to express their gratitude to the Swedish Research Council for support to the grant 2022-03971_VR molecular mechanisms in oxide nanoparticle interactions with proteins. This work was partially supported by the Estonian Ministry of Education and Research (TK210) and European Union and Ministry of Education and Research via project TEM-TA55.References
- N. Lina, D. Verma, N. Saini, R. Arbi, M. Munir, M. Jovic and A. Turak, Nano Today, 2021, 40, 101267 CrossRef.
- Y. U. Duan, S. Y. Wang, Q. Z. Zhang, W. W. Gao and L. F. Zhang, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100964 CrossRef CAS.
- B. Boson, V. Legros, B. J. Zhou, E. Siret, C. Mathieu, F. L. Cosset, D. Lavillette and S. Denolly, J. Biol. Chem., 2021, 296, 100111 CrossRef PubMed.
- J. J. Aguilera-Correa, J. Esteban and M. Vallet-Regí, Nanomaterials, 2021, 11, 137 CrossRef PubMed.
- M. Rai, S. D. Deshmukh, A. P. Ingle, I. R. Gupta, M. Galdiero and S. Galdiero, Crit. Rev. Microbiol., 2016, 42, 46–56 CrossRef PubMed.
- K. Siriwardana, A. Wang, M. Gadogbe, W. E. Collier, N. C. Fitzkee and D. Zhang, J. Phys. Chem. C, 2015, 119, 2910–2916 CrossRef PubMed.
- S. Galdiero, A. Falanga, M. Vitiello, M. Cantisani, V. Marra and M. Galdiero, Molecules, 2011, 16, 8894–8918 CrossRef PubMed.
- X. Hang, H. Peng, H. Song, Z. Qi, X. Miao and W. Xu, J. Virol. Methods, 2015, 222, 150–157 CrossRef PubMed.
- A. Tavakoli and M. S. Hashemzadeh, J. Virol. Methods, 2020, 275, 113688 CrossRef PubMed.
- H. Liu, E. Dai, R. Xiao, Z. Zhou, M. Zhang, Z. Bai, Y. Shao, K. Qi, J. Tu, C. Wang and S. Wang, Sens. Actuators, B, 2021, 329, 129196 CrossRef PubMed.
- S. Peng, F. Cao, Y. Xia, X.-D. Gao, L. Dai, J. Yan and G. Ma, Adv. Mater., 2020, 32, 2004210 CrossRef PubMed.
- T. Qin, R. Ma, Y. Yin, X. Miao, S. Chen, K. Fan, J. Xi, Q. Liu, Y. Gu, Y. Yin, J. Hu, X. Liu, D. Peng and L. Gao, Theranostics, 2019, 9, 920–6935 CrossRef PubMed.
- N. G. M. Palmqvist, G. A. Seisenbaeva, P. Svedlindh and V. G. Kessler, Nanoscale Res. Lett., 2017, 12, 631 CrossRef PubMed.
- P. Hajkova, P. Spatenka, J. Horsky, I. Horska and A. Kolouch, Plasma Processes Polym., 2007, 4, S397–S401 Search PubMed.
- S. Akhtar, K. Shahzad, S. Mushtaq, I. Ali, M. H. Rafe and S. M. Fazal-ul-Karim, Mater. Res. Express, 2019, 6, 105409 CrossRef.
- P. Agback, T. Agback, F. Dominguez, E. I. Frolova, G. A. Seisenbaev and V. G. Kessler, Nanoscale Adv., 2022, 4, 1527–1532 RSC.
- A. Nefedova, K. Rausalu, E. Zusinaite, A. Vanetsev, M. Rosenberg, K. Koppel, S. Lilla, M. Visnapuu, K. Smits, T. Tätte and A. Ivask, Sci. Rep., 2022, 12, 18746 CrossRef CAS PubMed.
- F. G. Svensson, G. Daniel, C. W. Tai, G. A. Seisenbaeva and V. G. Kessler, RSC Adv., 2020, 10, 6873–6883 RSC.
- V. G. Kessler, G. A. Seisenbaeva, S. Håkansson and M. Unell, Angew. Chem., Int. Ed., 2008, 47, 8506–8509 CrossRef CAS PubMed.
- C. L. Hill, M. S. Weeks and R. F. Schinazi, J. Med. Chem., 1990, 33, 2767–2772 CrossRef CAS.
- Q. Liu and V. Gerdts, Encyclopedia of Virology, 2021, pp. 850–853 Search PubMed.
- M. Carocci and L. Bakkali-Kassimi, Virulence, 2012, 3, 351–367 CrossRef.
- A. Nefedova, K. Rausalu, E. Zusinaite, V. Kisand, M. Kook, K. Smits, A. Vanetsev and A. Ivask, Heliyon, 2023, 9, e20067 CrossRef CAS PubMed.
- R. S. Balaban and J. A. Ferretti, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 1241–1245 CrossRef CAS.
- Y. R. Perera, R. A. Hill and N. C. Fitzkee, Isr. J. Chem., 2019, 59, 962–979 CrossRef.
- K. Sahiro, Y. Kawato, K. Koike, T. Sano, T. Nakai and M. Sadakane, Sci. Rep., 2012, 12, 7554 CrossRef.
- A. Luceri, R. Francese, D. Lembo, M. Ferraris and C. Balagna, Microorganisms, 2023, 11, 629 CrossRef CAS.
- S. Galdiero, A. Falanga, M. Vitiello, M. Cantisani, V. Marra and M. Galdiero, Molecules, 2011, 16, 8894–8918 CrossRef CAS PubMed.
- G. A. Seisenbaeva, E. Ilina, S. Håkansson and V. G. Kessler, J. Sol–Gel Sci. Technol., 2010, 55, 1–8 CrossRef CAS.
- L. J. Reed and H. Muench, Am. J. Hyg., 1938, 27, 493–497 Search PubMed.
- C. F. Lei, J. Yang, J. Hu and X. L. Sun, Virol. Sin., 2021, 36, 141–144 CrossRef PubMed.
- G. Repetto, A. del Peso and J. L. Zurita, Nat. Protoc., 2008, 3, 1125–1131 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of structure solution and refinement of reference POM compounds. CCDC 2380871 and 2380872. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4nr03730h |
| This journal is © The Royal Society of Chemistry 2025 |