
Organic semiconductor bulk heterojunctions for solar-to-chemical conversion: recent advances and challenges
Yuri
Kim†
 a,
Hoon
Kim†
b,
Hyeongyu
Lee
c,
Tack Ho
Lee
*a and
Han-Hee
Cho
*bd
a,
Hoon
Kim†
b,
Hyeongyu
Lee
c,
Tack Ho
Lee
*a and
Han-Hee
Cho
*bd
aDepartment of Chemistry Education, Graduate Department of Chemical Materials, Institute for Plastic Information and Energy Materials, Sustainable Utilization of Photovoltaic Energy Research Center, Pusan National University, Busan 46241, Republic of Korea. E-mail: tackho@pusan.ac.kr
bDepartment of Materials Science and Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea. E-mail: hhcho@unist.ac.kr
cSchool of Energy and Chemical Engineering, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
dGraduate School of Carbon Neutrality, Ulsan National Institute of Science and Technology (UNIST), Ulsan 44919, Republic of Korea
First published on 17th December 2024
Abstract
Solar fuel production involving the conversion of solar energy directly into chemical fuels such as hydrogen and valuable chemicals using photoelectrochemical (PEC) cells and photocatalysts (PCs) offers a promising avenue for sustainable energy while reducing carbon emissions. However, existing PEC cells and PCs fall short of economic viability due to their low solar-to-chemical (STC) conversion efficiency associated with the employed semiconductors, highlighting the clear need for identifying ideal semiconductor materials. Organic semiconductors (OSs), π-conjugated carbon-based materials, have emerged as promising candidates for enhancing STC conversion efficiency due to their remarkable optoelectrical properties, which can be readily adjustable through molecular engineering. In particular, the use of OS bulk heterojunctions (BHJs) consisting of intermixed electron-donating and electron-accepting OSs facilitates efficient charge generation under illumination, thereby contributing to enhanced STC conversion efficiency. This review explores the recent advancements in the rational design of OS materials and approaches aimed at enhancing the performance of BHJ-based PEC cells and PCs for solar-driven production of hydrogen and valuable chemicals. The discussion also introduces new perspectives to address the remaining challenges in this field.
 Tack Ho Lee | Tack Ho Lee is Assistant Professor in the Department of Chemical Materials, Pusan National University. His research has addressed organic and hybrid semiconductors for solar energy conversion since the start of his higher education career at Ulsan National Institute of Science and Technology. He has fostered an interest in organic semiconductors from photovoltaics to solar-driven fuel synthesis, and finally in time-resolved spectroscopy after moving to the Durrant group at Imperial College London. His group now develops photoelectrodes and photocatalysts for green hydrogen synthesis from carbon-based materials. He has published 37 research papers, which have been cited over 1700 times. |
 Han-Hee Cho | Han-Hee Cho studied at Korea Advanced Institute of Science and Technology (KAIST) where he completed a PhD in 2017 under the supervision of Prof. Bumjoon J. Kim. He then joined the Laboratory for Molecular Engineering of Optoelectronic Nanomaterials (LIMNO, led by Prof. Kevin Sivula) at École Polytechnique Fédérale de Lausanne (EPFL) as a postdoctoral researcher. He has cultivated his research career in the field of solar energy conversion using organic semiconductors and currently he is an assistant professor of Materials Science and Engineering at Ulsan National Institute of Science and Technology (UNIST). His research interests include organic semiconductor-based photoelectrochemical cells and photocatalysts for solar fuel production. |
1. Introduction
Solar fuel production via artificial photosynthesis where solar energy is directly converted into the energy of chemical fuels has been recognized as a promising alternative to achieve a circular energy economy while reducing carbon dioxide (CO2) emissions.1 Specifically, solar-driven water splitting and CO2 reduction to produce green hydrogen (H2) and value-added chemicals (e.g., methanol or ethanol), respectively, using photoelectrochemical (PEC) cells or heterogeneous photocatalysts (PCs), are the leading technologies to obtain eco-friendly solar fuels in a scalable and economically feasible manner.2 Indeed, the recent demonstration of large-scale photocatalytic water splitting for green H2 evolution, utilizing a 100 m2 array of panel reactors based on inorganic semiconductors, highlights significant progress.3 However, despite this advancement, state-of-the-art PEC cells and PCs have not yet met the requirements for economic viability due to their poor solar-to-chemical (STC) conversion efficiency and high manufacturing costs and instability originating from the nature of the semiconductors employed. Thus, the identification of robust and effective light-harvesting semiconductors remains a challenge to address the given limitations in PEC cells and PCs.4–6Organic semiconductors (OSs) which contain π-conjugated carbon bonds are distinct candidates for applications in solar energy conversion compared with their inorganic counterparts, primarily due to their exceptional absorption ability which extends up to near infrared and because their energy levels can be easily tuned by molecular engineering. OSs have found extensive application in solar-driven reactions, including solar water splitting, CO2 photoreduction, and solar-driven alternative oxidation with the aim of the production of value-added chemicals.7–13 Among various types of OSs, conjugated polymers or small molecules have emerged as superior semiconductor materials for PEC cells and PCs due to their high absorption coefficients and solution-processability.14–19 This capability allows the economical fabrication of high-performance large-scale PEC cells or scalable particulates at low temperatures.
After Yanagida et al. reported the initial attempts to use a conjugated polymer, poly(p-phenylene), as a PC for solar H2 production, there have been substantial advancements in developing OS-based PEC cells and PCs for diverse solar fuel production.11,20 Indeed, some recent reports in which a single-component OS is solely employed as a photoactive material have showed noticeable performance in solar fuel production through molecular engineering to enhance wettability and optoelectrical properties.7,11,14,18,21–30 However, further improvements in performance are hindered by a strong propensity for exciton recombination within single OSs. Photogenerated hole–electron pairs, which are termed excitons, in OSs should be effectively separated into free charge carriers to maximize the number of charges participating in the desired reactions before recombination occurs. In single-component PEC cells and PCs, excitons are strongly bound due to low dielectric constants of OSs, resulting in recombination rather than effective separation into free charge carriers.19 Thus, a bulk heterojunction (BHJ), where an electron-donating OS is well-intermixed with an electron-accepting one on a nanometer scale, has been utilized in OS-based optoelectronic devices to address unfavorable exciton recombination.31–35 This BHJ structure, formed via solution processing, facilitates the generation of free charge carriers at the donor/acceptor interface under illumination by physically separating photogenerated electrons in the acceptor from the holes in the donor, thereby ensuring effective light-harvesting capability. Indeed, BHJ blends have been extensively utilized in the field of organic photovoltaics (OPVs) where solar energy is converted into electrical energy to leverage these advantages.36,37 Similarly, the STC conversion efficiency of OS-based PEC cells (both photocathodes and photoanodes) and PCs has been further enhanced by leveraging this BHJ concept, leading to new benchmark performances due to efficient free charge carrier generation.38,39
In PEC cells using BHJ blends, photogenerated holes in the highest unoccupied molecular orbital (HOMO) of OS donors within a photoanode and electrons in the lowest unoccupied molecular orbital (LUMO) of OS acceptors within a photocathode can drive oxidation and reduction reactions, respectively, when they have sufficient driving forces for these reactions (Fig. 1a). Building on this working principle, employing two different BHJ structures with complementary absorption spectra in photocathodes and photoanodes provides a promising strategy to simultaneously enhance the performance of the solar-driven hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) in PEC tandem devices. Meanwhile, counter charges in both the photocathode and photoanode are balanced through the external circuit. Similarly, in BHJ-based PCs, the solar-driven HER takes place by utilizing photogenerated electrons in the LUMO of OS acceptors, whereas photogenerated holes are consumed by sacrificial agents or alternative oxidation reactions such as methanol oxidation (Fig. 1b). However, BHJ-based PCs whose energy levels straddle both HER and OER potentials simultaneously have been seldom reported, as low bandgap semiconducting polymer donor and non-fullerene acceptors are generally adopted to maximize light harvesting ability. Nevertheless, since numerous OSs and their heterojunction combination can be readily prepared by molecular engineering, OS-based BHJ PEC cells and PCs can potentially advance solar fuel production while meeting the requirements for industrial viability.39,40
 | ||
| Fig. 1 Illustration of working mechanisms of (a) BHJ-based PEC cells and (b) BHJ-based PCs. PEC cells consist of a BHJ photocathode for reduction reactions and a BHJ photoanode for oxidation reactions. BHJ PCs typically comprise a single BHJ blend to drive both reduction and oxidation reactions simultaneously. Ox: oxidized species, Red: reduced species, SO: sacrificial oxidation, AO: alternative oxidation. | ||
In this review, we present an overview of recent progress in STC conversion using OS BHJs and discuss strategies to address the challenges remaining in OS-based PEC cells and PCs for efficient and durable production of solar fuels. In particular, this minireview highlights the advancements in BHJ-based PEC cells and PCs over the past five years, a period marked by intensive exploration of the BHJ concept. The review categorizes and discusses various STC energy conversion systems across five subsections: (i) BHJ-based photocathodes for solar-driven H2 production, (ii) BHJ-based photoanodes for solar-driven O2 production and alternative oxidation reactions, (iii) BHJ-based unassisted PEC cells for overall reactions, (iv) BHJ-based PCs for solar-driven H2 production, and (v) BHJ-based PCs for alternative oxidation (Fig. 1). This approach provides a comprehensive overview of recent progress in the field of OS-based PEC cells and PCs.
2. BHJ-based PEC cells
2.1. BHJ photocathodes
Electron-donating OSs can be easily blended with electron-accepting OSs to form BHJ structures. This BHJ structure offers a significant advantage over single-component systems by facilitating charge dissociation at the donor/acceptor interface, thereby efficiently producing photogenerated electrons for solar-driven HER (Fig. 1). Recent advances in non-fullerene acceptors for BHJ structures have greatly enhanced photogenerated charge production upon illumination, which boosted the performance of BHJ-based photocathodes.11,41 In addition to OS material development, various approaches for further improving the operational stability of BHJ-based photocathodes have also been established. The following subsections review recent strategies designed to improve the performance and operational stability of OS BHJ-based photocathodes. The molecular structures and energy levels of the BHJ materials used in photocathodes are shown, respectively, in Fig. 2 and 3. | ||
| Fig. 2 Molecular structures of conjugated polymers and small molecules for BHJ photocathodes. | ||
 | ||
| Fig. 3 Energy level alignment of conjugated polymers and small molecules for BHJ photocathodes with redox potential of H+/H2 at pH 0 and Eu3+/Eu2+ (sacrificial agent). | ||
Shi et al. reported photocathodes based on a BHJ composed of a low band gap donor, PTB7, and PCBM to enhance light-harvesting capabilities (Fig. 2 and 3).46 A hole-transporting layer, CuOx, was utilized to reduce charge recombination by efficiently extracting photogenerated holes in the PTB7-based BHJ, and an electron-transporting layer, TiOx, was employed to improve the injection of photogenerated electrons from the BHJ to a Pt co-catalyst while blocking the back-migration of holes (Fig. 4a). The linear sweep voltammetry (LSV) curve of the final FTO/CuOx/PTB7:PCBM/TiOx/Pt photocathode showed a notable photocurrent density (Jph) of −7.27 mA cm−2 at 0 VRHE in an electrolyte buffered at pH 2 for solar-driven HER with an onset potential of 0.63 VRHE, demonstrating that the use of other OS BHJs beyond the P3HT:PC61BM BHJ is essential for substantially enhancing the performance of OS-based photocathodes (Fig. 4b and Table 1). However, the chronoamperometry (CA) curve of the final PTB7-based photocathode displayed a 75% decay in PEC activity after 1 h of operation even in the presence of a protection layer of TiOx grown by atomic layer deposition (ALD) as shown in Fig. 4c. This may be attributed to the instability of PCBM that undergoes photodegradation through a dimerization process.47
 | ||
| Fig. 4 (a) Energy level diagram of the FTO/CuOx/PTB7:PCBM/TiOx/Pt photocathode. (b) Optimal LSV curve of the PTB7:PCBM photocathode and (c) stability test for the PTB7-based BHJ photocathodes with an applied potential of 0 VRHE in 0.1 M H2SO4 buffer with 0.1 M Na2SO4 (pH 2) under 1 sun illumination. Adapted with permission.46 Copyright 2019, American Chemical Society. (d) Energy level alignments of PTB7-Th, PDI-V, and PCBM. (e) LSV curves and (f) CA behavior at 0 VRHE of the PDI-V-based photocathode and PCBM-based photocathode in 1.2 M Eu3+ based aqueous electrolyte (pH 4.2) under 1 sun illumination. Adapted with permission.35 Copyright 2020, American Chemical Society. | ||
| Device architecturea | Co-catalyst | J ph/faradaic efficiency (operating time)b | Stabilityb/ηSTC (half-cell) | Max. EQE | Reaction medium, illumination conditionc | Ref. |
|---|---|---|---|---|---|---|
| a Device structures with interlayers and/or protection layers. b The longest time over which continuous solar fuel production was reported, not the maximum stability of each photoelectrode. c Photocurrent under simulated 1 sun illumination (100 mW cm−2 of irradiance under AM 1.5 G condition) unless otherwise specified. d A faradaic efficiency was calculated using gas chromatograph (GC) data with calibration conducted by a Pt electrode. e STC efficiency of tandem cells are recorded in Table 3. | ||||||
| CuOx/PTB7:PCBM/TiOx | Pt | −7.27 mA cm−2 @ 0 VRHE/— | 0% (∼2 min)/— | — | 0.1 M H2SO4 with 0.1 M Na2SO4 (pH 2) | 46 |
| CuOx/PTB7:PCBM/ALD-TiOx | Pt | −3.50 mA cm−2 @ 0 VRHE FE = ∼100% (1 h) | 25% (1 h)/1.5% @ 0.3 VRHE | |||
| PEDOT:PSS/P3HT:PCBM | — | −21 μA cm−2 @ 0 VRHE/— | 50.5% (10 h)/0.03% @ 0 VRHE | — | 0.1 M H2SO4 (pH 1) | 51 |
| NiOx/PBDB-T:ITIC/TiOx | Pt | −8.90 mA cm−2 @ 0 VRHE/— | — | 52.2% at 620 nm @ 0 VRHE | 0.1 M H2SO4 (pH 1) | 50 |
| MoO3/PBDB-T:ITIC/TiOx | Pt | −9.10 mA cm−2 @ 0 VRHE/— | — | 54.8% at 620 nm @ 0 VRHE | ||
| CuOx/PBDB-T:ITIC/TiOx | Pt | −11.7 mA cm−2 @ 0 VRHE/— | ∼10% (30 min)/4.1% @ 0.68 VRHE![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e e |
63.5% at 620 nm @0 VRHE | ||
| CuOx/PBDB-T:ITIC-NiDDC/TiOx/ALD-TiOx |
Pt | −9.9 mA cm−2 @ 0 VRHE/FE = ∼100% (30 min) | ∼10% (75 min)/— | — | ||
| Cu film/Cu NSA/CuI/P3HT:PCBM | — | −1.0 mA cm−2 @ 0 VRHE FE = 97.5% (4 h) | 4% (300 s)/—e | 5.7% at 520 nm @ 0 VRHE | 0.5 M Na2SO4 (pH 6.8) | 54 |
| PM6:Y6/TiOx | Pt | −12 mA cm−2 @ 0 VRHE/— | ∼85% (5 min)/2.1% @ 0.31 VRHE | 69% at 620 nm @ 0 VRHE | 0.06 ppm H2PtCl6 with 0.5 M Na2SO4 (pH 1) | 49 |
| PM6:Y6 | RuO2 | −15 mA cm−2 @ 0 VRHE FE = ∼100% (∼1 h) | ∼50% (0.5 h)/3.7% @ 0.4 VRHE | 76% at 620 nm @ 0 VRHE | ||
| J71:N2200(Pt) | RuO2 | −9.0 mA cm−2 @ 0 VRHE/— | 71% (1 h)/2.52% @ 0.5 VRHE | 66% at 520 nm @ 0 VRHE | 0.1 M H2SO4 with 0.5 M Na2SO4 (pH 1) | 56 |
| MoO3/PTB7-Th:PDI-V | MoS3 | −5.3 mA cm−2 @ 0 VRHE/— | ∼100% (1 h)/— | — | 0.3 M H2SO4 with 0.5 M Na2SO4 (pH 1) | 35 |
| MoP | −1.1 mA cm−2 @ 0 VRHE/— | — | ||||
| MoS3:MoP (80:20) |
−8.7 mA cm−2 @ 0 VRHE/— | 91% (1 h), & <10% (8 h)/— | ||||
| RuO2 | −8.2 mA cm−2 @ 0 VRHE FE = 101 ± 2% (3 h)d | 85% (8 h), & 67% (20 h)/2.17% @ 0.26 VRHE | 49% at 610 nm @ 0 VRHE | |||
| CuOx/PBDB-T:ITIC/TiO2 | Pt | −7.50 mA cm−2 @ 0 VRHE/— | 16% (30 min)/— | 46% at 640 nm @ 0 VRHE | 0.1 M H2SO4 (pH 1) | 48 |
| PEDOT:PSS/PBDB-T:ITIC/PDINO/Ag/C | Pt/C | −12.35 mA cm−2 @ 0 VRHE/— | 84% (10 h)/2.02% @ 0.27 VRHE | |||
| CuOx/PBDB-T:ITIC:PCBM/TiO2 | Pt | −8.07 mA cm−2 @ 0 VRHE/— | 88% (30 min)/— | 48% at 645 nm @ 0 VRHE | ||
| PEDOT:PSS/PBDB-T:ITIC:PC71BM/PDINO/Ag/C | Pt/C | −13.84 mA cm−2 @ 0 VRHE/FE = ∼100% (15 h) | 97% (15 h)/2.84% @ 0.31 VRHE | |||
| 2PACz/PBDTTTPD:PNDIHDT | RuO2 | −4.6 mA cm−2 @ 0 VRHE FE = 102 ± 5% (1 h)d | ∼90% (6 h)/—e | — | 0.1 M NaBi buffer (pH 9) | 32 |
To mitigate the instability of PCBM under continuous illumination, extensive research has been conducted on BHJ blends incorporating non-fullerene acceptors for their application in photocathodes. Yao et al. corroborated that replacing the photodegradable fullerene-based electron acceptors with a perylene diimide-based electron-accepting OS, PDI-V (molecular structures are shown in Fig. 2), is imperative for establishing the operational stability of organic photocathodes (Fig. 4d–f).35 They compared BHJ photocathodes composed of a polymer donor, PTB7-Th, and either a fullerene-based acceptor, phenyl-C71-butyric acid (PC71BM), or a non-fullerene acceptor, PDI-V. For the sacrificial photoreduction of Eu3+, both BHJs exhibited similar initial Jph values around −4 mA cm−2 at 0 VRHE (Fig. 4e). However, the PC71BM-based photocathode showed a significant decrease in the Jph within the first 10 min of continuous operation, leading to complete device failure. In contrast, the PDI-V-based photocathode displayed no observable degradation for at least 20 min under continuous operation (Fig. 4f). Although the electron-accepting OSs in their study, PDI-V and PC71BM, have similar energy level alignments, the performance and operational stability of the PDI-V-based photocathode were notably enhanced, likely due to improved generation of free charge carriers and better photostability of PDI-V compared with PC71BM.
Interestingly, robust BHJ-based photocathodes with remarkably high Jph have been demonstrated very recently even with the use of PC71BM. In this recent study, PC71BM was introduced as the second acceptor material into the PBDB-T:ITIC BHJ (binary BHJ) blend to construct a ternary BHJ.48 The energy levels of three components, PBDB-T, ITIC, and PC71BM (see Fig. 2 for their chemical structures), were beneficial for establishing a cascade energy level that meets the thermodynamic requirements for solar-driven HER in an electrolyte buffered at pH 1 (Fig. 5a). As PC71BM possesses higher electron mobility due its isotropic charge transport behavior compared with ITIC, the presence of PC71BM in the ternary BHJ enhanced the overall electron mobility (μe) of the BHJ layers, as determined by space-charge-limited current (SCLC) measurements. This led to a decrease in the ratio of hole mobility (μh) to μe from 21.12 to 2.45 for the binary BHJ and ternary BHJ blends, respectively (Fig. 5b). Moreover, intensity-modulated photocurrent spectroscopy (IMPS) measurements clearly showed that the charge transfer efficiency (ηtrans) was dramatically increased by ∼60% and the charge recombination rate constant (krec) was sharply decreased by ∼45% due to the presence of PC71BM. Consequently, the optimized ternary BHJ-based photocathode (FTO/CuOx/BHJ/TiOx/Pt) exhibited an initial Jph of −8.2 mA cm−2 at 0 VRHE and stable operation for 30 min without any noticeable degradation in the pH 1 electrolyte, whereas the binary BHJ-based photocathode produced an initial Jph of −7.7 mA cm−2 at 0 VRHE and retained only ∼50% of the initial Jph after 30 min of operation (Fig. 5c, d and Table 1). This improvement was attributed to the suppression of charge accumulation within the BHJ layers.
 | ||
| Fig. 5 (a) Energy level diagram of PBDB-T, ITIC, and PC71BM in ternary photocathode. (b) μh and μe of the binary BHJ without PC71BM and ternary BHJ with PC71BM films. (c) J–V curves of the binary and ternary BHJ-based photocathode under 1 sun illumination in 0.1 M H2SO4 (pH = 1.0). (d) CA tests at 0 VRHE for the binary and ternary BHJ-based photocathode. Adapted with permission.48 Copyright 2022, Wiley-VCH GmbH. | ||
Therefore, recent scientific findings regarding BHJ engineering conclude that the use of photostable acceptors with suitable energy levels for the solar-driven HER and the alleviation of charge accumulation within BHJ layers are crucial prerequisites for attaining high performance and operational stability in BHJ photocathodes.
It has been experimentally demonstrated that suppressing charge accumulation is a significant challenge for achieving operational stability in BHJ PEC cells. Yao et al. examined the sacrificial photoreduction of Eu3+ by photocathodes based on a BHJ of PTB7-Th and PDI-V (denoted as an all-polymer BHJ) instead of the HER.35 Their in-depth study uncovered that photogenerated electron accumulation at the organic/water interface at levels around 1 μC cm−2 gives rise to the rapid and irreversible degradation of the all-polymer BHJ whereas the alleviation of this accumulation to the order of 1 nC cm−2 ensures the stable operation of BHJ photocathodes. A similar observation was also made through electrochemical impedance spectroscopy (EIS) analysis of P3HT:PCBM-based photocathodes.51 Consequently, it is noted that efficient charge-transporting layers to minimize charge accumulation are highly desired.
Poly(3,4-ethylenedioxythiophene)-poly(styrenesulfonate), commonly referred to as PEDOT:PSS, is one of the most widely used HTLs in organic electronics. However, its application in OS PEC cells is limited owing to its hydrophilic and hygroscopic properties which lead to dissolution when in direct contact with an electrolyte. To replace the vulnerable PEDOT:PSS layer in BHJ photocathodes, metal oxide HTLs are straightforward alternatives. Indeed, Shi et al. introduced various well-known metal oxide layers, CuOx, NiOx, and MoO3, which have similar work functions, as HTLs in BHJ photocathodes (Fig. 6a).50 In their study, a BHJ composed of the electron donor PBDB-T and the non-fullerene acceptor ITIC was used to overcome the aforementioned limitations associated with fullerene-based BHJs (see Fig. 2 for their chemical structures). Hole mobilities measured using the SCLC method for the CuOx-, NiOx-, and MoO3-based devices (HTLs/BHJ/MoO3/Au) were determined to be 1.5 × 10−4, 1.0 × 10−6, and 9.0 × 10−5 cm2 V−1 s−1, showing better hole-transporting capability in the CuOx-based photocathode. Stored charges estimated via chopped light chrono-amperometry measurements were found to be 20.44, 0.67, and 11.85 μC cm−2 for the CuOx-, NiOx-, and MoO3-based photocathodes, respectively, further representing the enhanced hole extraction of the CuOx-based BHJ photocathode. Moreover, a TiOx layer was employed as an electron-transporting layer (ETL) to mitigate charge recombination by reducing the back migration of photogenerated holes within the BHJ photocathodes. The fully assembled FTO/CuOx/BHJ/TiOx/Pt afforded a remarkable Jph of −11.7 mA cm−2 at 0 VRHE for the HER in a pH 1 electrolyte with an onset potential as positive as 0.8 VRHE while the NiOx- and MoO3-based photocathodes exhibited initial Jph values of −8.9 and −9.1 mA cm−2 at 0 VRHE, respectively (Fig. 6b and Table 1). This improvement was ascribed to the improved hole extraction and reduced charge recombination resulting from the incorporation of the CuOx and TiOx layers. However, photooxidation of OS BHJ photocathodes was observed due to degradation via free radical reactions, which has also been noted in related organic electronics.52,53 To alleviate the photooxidation of OS-based photocathodes, a common antioxidant, nickel(II) dibutyl dithiocarbamate (NiDDC), was incorporated into the PBDB-T:ITIC BHJ. Consequently, the FTO/CuOx/BHJ/TiOx/Pt photocathode modified with the incorporation of NiDDC presented an extended performance decay time, reaching up to 75 min due to the suppressed photooxidation of BHJ (Fig. 6c).
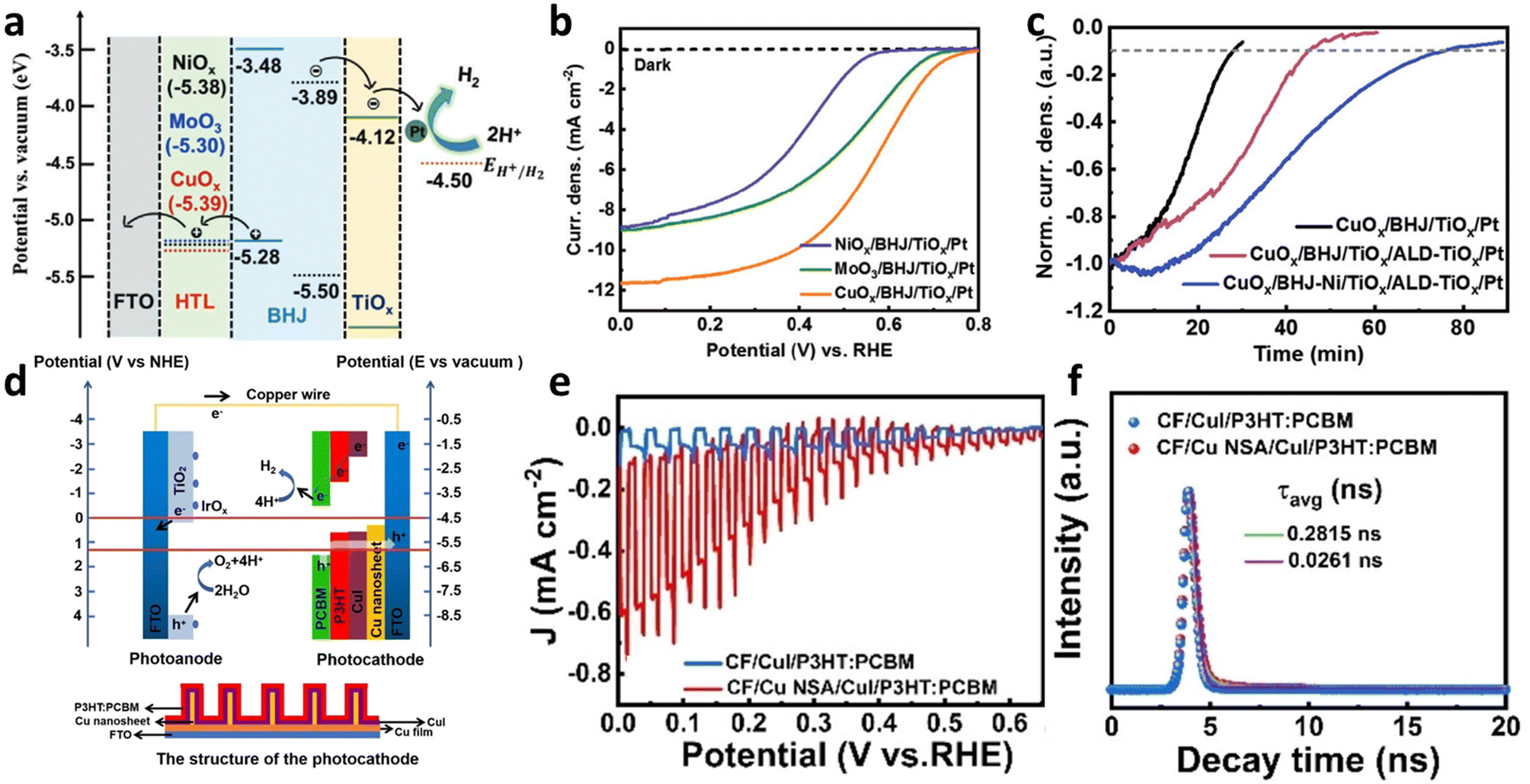 | ||
| Fig. 6 (a) Energy level diagram of the photocathode based on the non-fullerene BHJ (PBDB-T:ITIC). (b) LSV curves of the photocathodes with the various HTLs and (c) stability test of the different photocathode at 0 VRHE in 0.1 M H2SO4 buffer adjusted to pH 1. Adapted with permission. Adapted with permission.50 Copyright 2020, Wiley-VCH GmbH. (d) Energy level alignment of Cu nanosheet (Cu NSA)-fabricated photocathode and photoanode (above) with the structure of the photocathode (below). (e) Chopped J–V curves and (f) time-resolved transient PL spectra of the photocathodes with and without Cu NSA. Adapted with permission.54 Copyright 2021, Wiley-VCH GmbH. | ||
In a parallel approach, Zhang et al. incorporated a nano-structured HTL to facilitate hole transport while preventing back electron transfer.54 A copper iodide (CuI) layer was chosen as the HTL due to its suitable energy levels for selectively transporting holes from P3HT:PCBM BHJ to the substrate. In addition, a copper nanosheet array (Cu NSA) was introduced as an underlying substrate of the CuI layer to further enhance performance by creating a cascade energy level and enlarging the interfacial area between the P3HT:PCBM BHJ and the CuI layer (Fig. 6d).54 Whereas a photocathode without both the CuI and Cu NSA layers exhibited a negligible Jph of −61 μA cm−2 at 0 VRHE and an onset potential of 0.263 VRHE, the incorporation of the CuI layer improved the Jph and anodically shifted the onset potential (−92 μA cm−2 at 0 VRHE and 0.497 VRHE, respectively) (Fig. 6e). Furthermore, the optimized BHJ photocathode based on the Cu NSA layer, which was vertically formed by an electrodeposition method onto a Cu film, displayed a significantly enhanced Jph of −1 mA cm−2 at 0 VRHE with a higher onset potential of 0.650 VRHE (Table 1). The transport resistance of the photocathode (Rbulk) estimated by EIS was decreased from 117.1 to 61.7 Ω by the insertion of the Cu NSA and CuI layers. Moreover, the photoluminescence (PL) spectrum of the P3HT:PCBM BHJ blend was more quenched, and an average charge carrier lifetime (τavg), estimated by transient PL, was remarkably elongated from 0.0261 to 0.2815 ns after introducing the Cu NSA layer (Fig. 6f). These observations revealed that the recombination of photogenerated charge carriers was effectively suppressed by the incorporation of the nanostructured CuI HTL.
The impact of efficient charge extraction on the performance and operational stability of BHJ photocathodes is clearly demonstrated in these significant achievements. However, most studies are constrained to the use of metal oxide-based charge transport layers. These layers often require thermal annealing at high temperatures, which can potentially damage BHJs during the device fabrication. They are also stable within a limited window of applied bias in the given electrolyte, as determined by their Pourbaix diagrams, unless additional protection layers are applied. Thus, the development of novel HTLs and ETLs for BHJ photocathodes remains a key challenge for further boosting performance and operational stability by reducing unfavorable charge recombination and accumulation.
Among the possible co-catalysts, platinum (Pt) is frequently adopted in BHJ PEC cells due to its excellent catalytic activity and ease of preparation. Pt nanoparticles can be deposited onto BHJ layers through photoassisted electrodeposition where BHJ-based devices are immersed in an electrolyte containing H2PtCl6, and electrodeposition is conducted at a constant potential under illumination.46,50,57,58 However, the Pt nanoparticle layer often provides poor coverage on BHJ devices, leading to inefficient extraction of photogenerated charges for the HER. This is evidenced by a decrease in HER performance in BHJ-based devices even with a higher loading of Pt nanoparticles. Moreover, the detachment of Pt nanoparticles caused by the formation of H2 bubbles further contributes to the gradual degradation of BHJ photocathodes.
Ruthenium(IV) oxide (RuO2) nanoparticles have been widely utilized as co-catalysts in BHJ PEC cells by virtue of their outstanding catalytic activity for the HER in acidic media and solution processibility. Wu et al. introduced BHJ photocathodes consisting of PM6:Y6 BHJ coupled with RuO2 layers for the HER in a pH 1 electrolyte.49 The RuO2 layer employed in their work was found to be a porous layer of nanoparticle aggregates, leading to an increased surface area and reduced charge diffusion length for the HER. Consequently, the RuO2 layer served as an interfacial layer as well as a co-catalyst layer, yielding an impressive Jph of −15 mA cm−2 at 0 VRHE with an onset potential of 0.8 VRHE for the optimized FTO/PM6:Y6/RuO2 photocathodes (Fig. 7a and Table 1). This performance was superior to that of FTO/PM6:Y6/Pt and FTO/PM6:Y6/TiO2/Pt photocathodes, highlighting the RuO2 layer as a promising alternative co-catalyst to Pt. Furthermore, the RuO2-based BHJ photocathodes displayed slightly improved operational stability, maintaining approximately 50% of the initial photocurrent over 30 min while the FTO/PM6:Y6/TiO2/Pt photocathode experienced a 90% loss within 5 min (Fig. 7b). Although delamination occurred in all BHJ photocathodes regardless of the overlayer type, the exposed RuO2 after exfoliation continued to facilitate the HER, whereas the detachment of the TiO2 layer in the FTO/PM6:Y6/TiO2/Pt photocathode resulted in an irreversible loss of the co-catalyst layer. However, similar to the deposition of Pt nanoparticles, achieving homogeneous coverage of hydrophilic RuO2 layers on hydrophobic BHJ layers remains a critical obstacle to further enhancing both performance and operational stability. Wu et al. developed a Pt nanoparticle-assisted RuO2 layer deposition method in their subsequent study.56 To mitigate the incompatibility between hydrophobic BHJ layers and hydrophilic RuO2 co-catalyst layers, Pt nanoparticles were deposited directly onto BHJ layers by a photoassisted electrodeposition technique prior to the deposition of the RuO2 co-catalyst layer (Fig. 7c). In their in-depth study using dynamic cycling contact angle (DCCA) measurements, they observed a significant increase in water contact angle (WCA) hysteresis (Δθ) from 16° for the unmodified BHJ surface to 47° for the Pt-decorated BHJ surface, implying that the Pt nanoparticle decoration rendered the BHJ surface more hydrophilic (Fig. 7d). Consequently, the Pt-decorated BHJ film (denoted as BHJ(Pt)) prevented shrinkage of RuO2 solution droplets during spin-coating, resulting in a uniform RuO2 thin film with complete surface coverage. The optimized FTO/J71:N2200(Pt)/RuO2 photocathodes afforded a notably improved Jph of −9.0 mA cm−2 at 0 VRHE compared with −2.8 mA cm−2 for the FTO/J71:N2200/Pt photocathode (Fig. 7e and Table 1). This improvement was attributed to not only the improved coverage of RuO2 layers but also to promoted charge transfer as evidenced by the remarkable reductions in charge-carrier transfer resistance (Rct) and surface charge-chemical conversion resistance (Rsc) determined through EIS measurements (Fig. 7f). Additionally, the Pt nanoparticle-assisted RuO2 deposition was also successfully applied to two different BHJ pairs, PM6:BTP-BO-4Cl and J71:ITIC (see Fig. 2 for their chemical structures), demonstrating the versatility of this approach.
 | ||
| Fig. 7 (a) The J–V curves of FTO/BHJ (PM6:Y6), FTO/BHJ/Pt, FTO/BHJ/TiO2/Pt and FTO/BHJ/RuO2 photocathodes and (b) stability test of FTO/BHJ/TiO2/Pt and FTO/BHJ/RuO2 photocathodes at 0.1 VRHE under AM 1.5 G illumination with chopped light in 0.5 M Na2SO4 solution (pH 1). Adapted with permission.49 Copyright 2022, Elsevier B.V. (c) A scheme of photocathode fabrication procedure by Pt nanoparticles-assisted homogeneous surface engineering method. (d) The dynamic WCA of FTO/BHJ (J71:N2200) before (left) and after (right) Pt deposition. (e) The chopped J–V curves and (f) Nyquist plots of different photocathodes of FTO/BHJ, FTO/BHJ/Pt, and FTO/BHJ(Pt)/RuO2 photocathodes 1 sun illumination in 0.5 M Na2SO4 with 0.1 M H2SO4. Adapted with permission.56 Copyright 2023, Wiley-VCH GmbH. | ||
To replace co-catalysts based on rare-earth elements, various co-catalyst layers composed of Earth-abundant materials have been investigated. Yao et al. investigated photocathodes based on a BHJ of PTB7-Th and PDI-V (denoted as an all-polymer BHJ) with the incorporation of molybdenum (Mo)-based co-catalysts.35 They found that the performance of all-polymer BHJ photocathodes is strongly affected by the uniformity of the interface between the all-polymer BHJ and co-catalyst nanoparticles. Amorphous MoS3 nanoparticles were deposited uniformly onto the all-polymer BHJ layer, while micron-sized particles of crystalline MoP gave a poor interface, resulting in the improved performance for the BHJ photocathodes based on the MoS3 co-catalysts. Indeed, the optimized photocathodes with the device architecture of FTO/MoO3/PTB7-Th:PDI-V/MoS3 produced an Jph of −5.3 mA cm−2 at 0 VRHE whereas the FTO/MoO3/PTB7-Th:PDI-V/MoP photocathodes exhibited a limited Jph of −1.1 mA cm−2 at 0 VRHE (Fig. 8a and Table 1). Moreover, as shown in Fig. 8b, the BHJ/MoO3 photocathodes demonstrated stable operation for 1 h while the BHJ/MoP photocathodes showed a severe decrease in the Jph, which was attributed to charge accumulation caused by poor coverage of the MoP layers. This suggests again that the uniform coverage of co-catalyst layers on BHJ films plays an important role in determining performance and operational stability. Interestingly, their further attempt to use a blend of the MoS3 and MoP co-catalysts resulted in a considerably increased Jph of −8.7 mA cm−2 at 0 VRHE, slightly exceeding a Jph of −8.2 mA cm−2 at 0 VRHE for the BHJ photocathodes based on the well-known RuO2 co-catalyst (Fig. 8c and Table 1). However, this optimized BHJ photocathode showed slow degradation due to the gradual catalyst detachment under the operational conditions (Fig. 8d).
 | ||
| Fig. 8 (a) LSV scans and (b) CA curves of the FTO/MoO3/PTB7-Th:PDI-V/MoO3 and FTO/MoO3/PTB7-Th:PDI-V/MoP photocathodes. (c) LSV scans of the BHJ photocathodes based on the MoS3:MoP co-catalyst blends. (d) CA curve of the BHJ photocathodes with the MoS3:MoP (20:80 wt%) co-catalyst layer. LSV scans and CA curves were recorded in pH 1 aqueous electrolyte under 1 sun illumination. Adapted with permission.35 Copyright 2020, American Chemical Society. | ||
Consequently, it can be concluded that establishing robust and homogeneous coverage of Earth-abundant co-catalyst layers on BHJ layers is fundamental to enhancing both the performance and stability of BHJ photocathodes for the HER under operational conditions.
In their follow-up study, a carbon plate which has high conductivity and a rough surface was utilized to construct robust BHJ photocathodes.48 A carbon plate coupled with Pt/C co-catalysts was directly delivered on BHJ photocathodes based on a ternary blend of OSs (PBDB-T, ITIC, and PC71BM), resulting in encapsulated photocathodes (denoted as t-BHJ-C–Pt/C) (Fig. 9a). The optimized t-BHJ-C–Pt/C photocathodes generated an initial Jph of −13.8 mA cm−2 at 0 VRHE and achieved stable operation for a long duration of 15 h (Fig. 9b, c and Table 1). Furthermore, the faradaic efficiency for the continuous operation of t-BHJ-C–Pt/C photocathodes was close to 100%, demonstrating the stable production of solar-driven H2 (Fig. 9c). This represents a noticeable achievement, yet further improving the operation stability of OS BHJ photocathodes remains a challenge.
 | ||
| Fig. 9 (a) Schematic diagram of the carbon-encapsulated ternary BHJ (PBDB-T:ITIC:PC71BM) photocathode with PEDOT:PSS and PDNIO as HTL and ETL, respectively. (b) The J–V curves and (c) CA curves of the b-BHJ (PBDB-T:ITIC)-C–Pt/C and t-BHJ-C–Pt/C photocathodes in 0.1 M H2SO4 solution (pH 1.0) at 0 VRHE under AM 1.5 G illumination. Adapted with permission.48 Copyright 2022, Wiley-VCH GmbH. | ||
2.2. BHJ photoanodes
The overall solar water splitting in PEC cells is completed when photogenerated holes and electrons in the photoanode and photocathode drive the O2 and H2 evolution reactions, respectively, while the remaining counter charges are recombined efficiently (Fig. 1a). This implies that developing high-performance and robust photoanodes for solar-driven OER is as crucial as developing efficient photocathodes to enhance the overall performance of solar water splitting. Given the high anodic potential (1.23 VRHE) required for the solar-driven O2 production and the complexity of the four-hole involved water oxidation mechanisms, the identification of suitable OS BHJs and device engineering strategies to address challenges posed by this sluggish water oxidation reaction is imperative for demonstrating high-performance and robust photoanodes.60–62 The following subsections summarize and discuss recent approaches aimed at enhancing the performance and operational stability of OS BHJ-based photoanodes. The molecular structures and energy levels of the BHJ materials incorporated into BHJ photoanodes are shown in Fig. 10 and 11. | ||
| Fig. 10 Molecular structures of conjugated polymers and small molecules for photoanode. | ||
 | ||
| Fig. 11 Energy level alignment of conjugated polymers and small molecules for photoanode with redox potential of dehydroascorbic acid (DHA)/ascorbic acid (AA), SO32−/SO3˙− (sacrificial agents) and H2O/O2. | ||
 | ||
| Fig. 12 (a) Schematic energy band diagram of OS-based photoanodes (ΔEi is the energy offset between the HOMO of acceptor and donor. ΔEii is the energy offset between the HOMO of donor and the oxidation potential). (b) Optimal LSV curves of the BHJ photoanodes in 1 M phosphate buffer and 1.5 M Na2SO3 sacrificial reductant (pH 6.7) under 1 sun illumination. (c) Incident and absorbed photon-to-current efficiency spectra at 1.23 VRHE of the P1:A1 (left) and the P2:A2 (right) BHJ photoanodes. Adapted with permission.63 Copyright 2022, American Chemical Society. | ||
Sekar et al. reported a simple yet effective method to achieve BHJ photoanodes operating over a broad pH range by adjusting the energy levels of the donor and acceptor materials. The optimal energy level alignment and operating conditions were determined using thiophenedicarboximide (TPD)-benzodithiophene (BDT)-based donor polymers (P1 and P2) and rylene diimide-based acceptor homologues (A1 and A2) (Fig. 10). Notably, P2 possessed a deeper HOMO level than P1 due to the incorporation of electronegative chlorine atoms, while A2 displayed a deeper LUMO level compared to A1 owing to the extended structure through the linkage at the bay positions.64–67 Given the energy levels of the OSs, P2 was capable of driving water oxidation over a wide pH range from 0 to 14 and exhibited a sufficient LUMO level offset when blended with A2, leading to efficient exciton separation in the P2:A2 BHJ photoanodes.34,68,69 To observe the effect of energy level modulation in the direct exposure of OS-based photoanodes to the aqueous electrolyte, the photocurrent was measured under 1 sun illumination in the presence of 1.5 M Na2SO3 (sacrificial agent) without co-catalysts. The use of sulfite as a sacrificial agent has been suggested as an alternative to water oxidation, with a thermodynamic redox potential of 0.73 VRHE, providing a greater driving force than water oxidation (1.23 VRHE). A maximum photocurrent of 4.1 mA cm−2 was observed from the P2:A2 photoanode for the sacrificial oxidation at 1.23 VRHE (Fig. 12b and Table 2). In addition, the P2:A2 BHJ photoanode exhibited improved incident photon-to-current efficiency (IPCE), indicative of better exciton separation and/or charge collection (Fig. 12c).
| Device architecture | Co-catalyst | J ph/faradaic efficiency (operating time)a | Stabilitya/ηSTC (half-cell) | Max. EQE | Reaction medium, illumination conditionb | Ref. |
|---|---|---|---|---|---|---|
| a The longest time over which continuous STC conversion was reported, not the maximum stability of each photoelectrode. b Photocurrent under simulated 1 sun illumination (100 mW cm−2 of irradiance under AM 1.5 G condition) unless otherwise specified. c A faradaic efficiency was calculated using GC data with calibration conducted by a Pt electrode. | ||||||
| pSnO2/mSnO2/P2:A2 |
— | 4.1 mA cm−2 @ 1.23 VRHE | 92.7% (3 h)/— | ∼25% at 530 nm @ 1.23 VRHE | 1 M phosphate buffer with 1.5 M Na2SO3 (pH 6.7) | 63 |
| ZnO/PM6:Y6/Au | NiFeOOH | ∼1 mA cm−2 @ 1.23 VRHE/— | 10% (500 s) | — | 0.1 M KOH (pH 13) | 33 |
| ZnO/PM6:Y6/PM6/Au | 4 mA cm−2 @ 1.23 VRHE/FE = 103 ± 10.7% (1400 s) | 50% (1 h)/1.87% @ 1.08 VRHE | 25% at 770 nm @ 1.23 VRHE | |||
| ZnO/PM6:Y6/PM6 | — | 5.0 mA cm−2 @ 1.23 VRHE/— | 18.2% (1000 s)/— | — | 1 M borate buffer with 2 M ascorbic acid (pH 5.5) | 76 |
| ZnO/PM6:Y6 | 6.6 mA cm−2 @ 1.23 VRHE FE = 76.1 ± 4.4% (1280 s)c | 60% (2500 s)/— | 17.8% at 850 nm @ 1.23 VRHE | |||
| pZnO/PBDTTTPD:PNDITCVT | — | 3.5 mA cm−2 @ 1.23 VRHE/— | 32% (1.5 h)/— | — | 1.0 M KPi buffer with 1.5 M Na2SO3 (pH 9) | 34 |
| mZnO/PBDTTTPD:PNDITCVT | 3.3 mA cm−2 @ 1.23 VRHE/— | ∼98%% (6 h)/— | — | 0.5 M KPi buffer with 1.5 M Na2SO3 (pH 9) | ||
| mZnO/PBDTTTPD:PNDITCVT | Li-IrO2 | 1.9 mA cm−2 @ 1.23 VRHE/— | 66% (10 min)/— | — | 0.1 M NaBi buffer (pH 9) | |
| mZnO/PBDTTTPD:PNDITCVT/PTAA | 2.3 mA cm−2 @ 1.23 VRHE/FE = 100 ± 7% (30 min)c | 32.6% (1 h)/1.23% @ 0.28 VRHE | 27% at 610 nm @ 1.23 VRHE | |||
Facilitating water oxidation on a photoanode requires not only designing OSs with appropriate energy levels for the OER, but also configuring interlayers that enables effective extraction of photogenerated charges. Cho et al. reported a polymer BHJ photoanode with carefully selected OSs and interlayers to achieve high performance during solar water oxidation (Fig. 13a).34 Interestingly, their in-depth study which exploited Na2SO3 as a sacrificial agent for the oxidation reaction identified key factors significantly affecting the performance and operational stability of OS-based photoanodes: (i) the rational selection of OS-based BHJs to efficiently generate free charges and (ii) the mitigation of charge accumulation at the interfaces between the OS-based BHJ layer and the neighboring layers. Specifically, the benzodithiophene-based polymer, referred to as PBDTTTPD, and the naphthalenediimide-based polymer, coded as PNDITCVT, were utilized as a donor and an acceptor to create BHJ and generate free charge carriers efficiently (molecular structures are shown in Fig. 10). Furthermore, to reduce the accumulation of photogenerated charges and facilitate charge extraction, a layer of poly(triarylamine) (PTAA) was added as an HTL. Then, an OER catalyst, iridium oxide modified by lithium hydroxide nanoparticles (LIO), was spray-coated onto the PTAA layer for solar water oxidation (Fig. 13b). Finally, the optimized FTO/mZnO/BHJ/PTAA/LIO photoanode achieved an impressive Jph of up to 2.3 mA cm−2 at 1.23 VRHE under 1 sun illumination (Fig. 13c and Table 2), with the faradaic efficiency of 100% for solar-driven oxygen production (Fig. 13d). As a result, the energy level alignment from the BHJ to the electrolyte (considering pH) is a key factor to improve the performance of BHJ photoanodes by the fine modifications of molecular structures, interlayers, and electrolytes.
 | ||
| Fig. 13 (a) Schematic of the FTO/mZnO/BHJ (PBDTTTPD:PNDITCVT)/PTAA/LIO photoanode. (b) Energy level diagram of the polymer donor (PD, PBDTTTPD), polymer acceptor (PA, PNDITCVT), PTAA, and water oxidation potential at pH 9. (c) LSV scans of the FTO/mZnO/BHJ/PTAA/LIO and FTO/mZnO/BHJ/LIO photoanodes for solar water oxidation in 0.1 M NaBi electrolyte buffer (pH 9) under 1 sun illumination. (d) Comparison of the measured amount and the estimated amount of evolved O2 by GC detector response. Adapted with permission.34 Copyright 2021, Springer Nature. | ||
Generation of long-lived charges in the OS-based photoanodes is a key to overcoming the kinetic challenges for chemical reactions (including four-hole transfer reactions for water oxidation).72–75 Spatial separation of electrons and holes has been demonstrated by device engineering such as introduction of polymer overlayers and unipolar charge pathway to prevent charge recombination. While carrier lifetime is important to drive PEC reactions, only a few publications have discussed the carrier lifetime at the OS/electrolyte interface in the photoanode.
Lee et al. demonstrated that the incorporation of PM6 interlayers in BHJ photoanodes suppressed the recombination of the accumulated holes by blocking electron back transfer, thereby enabling efficient solar water oxidation (Fig. 14a).33 Consequently, a remarkable Jph of 4 mA cm−2 at 1.23 VRHE (0.1 M KOH, pH 13) was obtained for the ITO/ZnO/Y6:PM6/PM6/Au/NiFeOOH (denoted as Y6:PM6/PM6 photoanode), while the ITO/ZnO/Y6:PM6/Au/NiFeOOH (denoted as Y6:PM6 photoanode) recorded unstable PEC activities with a Jph of ∼1 mA cm−2 at 1.23 VRHE (Fig. 14b and Table 2). The optimized Y6:PM6/PM6 photoanode exhibited an IPCE of 25% at 770 nm illumination and a wide range of photoresponses up to 950 nm (Fig. 14c). Furthermore, the introduction of the PM6 interlayer reduced the onset potential of the photoanode from 0.8 to 0.4 VRHE by suppressing interfacial recombination losses. Whereas the number of recombining charges increased for the Y6:PM6 photoanode at high potentials close to 1.23 VRHE, it was considerably suppressed for the Y6:PM6/PM6 photoanode (Fig. 14d). As a result, the dual functional PM6 interlayer allowed the first demonstration of near-infrared-absorbing photoanodes for solar water oxidation.
 | ||
| Fig. 14 (a) Energy levels of the Y6:PM6/PM6 photoanode, describing the PM6 layer blocking the electron back transfer from the Y6:PM6 BHJ. (b) LSV scans of the Y6:PM6/PM6 and the Y6:PM6 photoanodes in 0.1 M KOH solution (pH 13) under 1 sun illumination. (c) IPCE spectrum of the Y6:PM6/PM6 photoanode in 0.1 M KOH solution (pH 13) at 1.23 VRHE. (d) The amount of recombining charge as a function of applied bias, obtained by integration of the negative transient current. Adapted with permission.33 Copyright 2022, Wiley-VCH GmbH. | ||
More recently, Lee et al. eliminated metal and co-catalyst layers from the BHJ photoanode architecture by employing a sacrificial agent, which allowed them to directly investigate the role played by a PM6 polymer overlayer at the OS/electrolyte interface (Fig. 15a)76 Under operational conditions, the introduction of the PM6 overlayer resulted in improved accumulation of photogenerated charges. Furthermore, the lifetime of hole polarons was significantly prolonged, lasting several seconds, as evidenced by the comparison of photoinduced absorption kinetics between the Y6:PM6/PM6 and the Y6:PM6 photoanodes (Fig. 15b). These long-lived polarons have a significant impact on the performance of the photoanode and the coupled H2 evolution at the counter electrode, as supported by the performance measurement in the presence of sacrificial agent, ascorbic acid (AA). A Jph of 6.6 mA cm−2 was achieved under conditions promoting long-lived charge generation with the PM6 overlayer, compared with a Jph of 5.0 mA cm−2 without the PM6 overlayer (Fig. 15c and Table 2). Moreover, the Y6:PM6/PM6 photoanode showed a remarkable external quantum efficiency (EQE) of 17.8% measured under 850 nm illumination (Fig. 15d), indicating the importance of material selection in creating efficient BHJs. Although the PM6 overlayer extended the maximum absorption range to 600–650 nm, the EQE in this range remained relatively lower than that of Y6 (700–900 nm), highlighting the major role played by the PM6 overlayer in extending charge lifetime rather than enhancing light absorption. Thus far, the carrier lifetime in BHJ photoanodes has been elongated through the introduction of functional interlayers, suggesting that newly designed OSs could further generate long-lived carriers in polar environments, such as those found in aqueous electrolytes.77–80
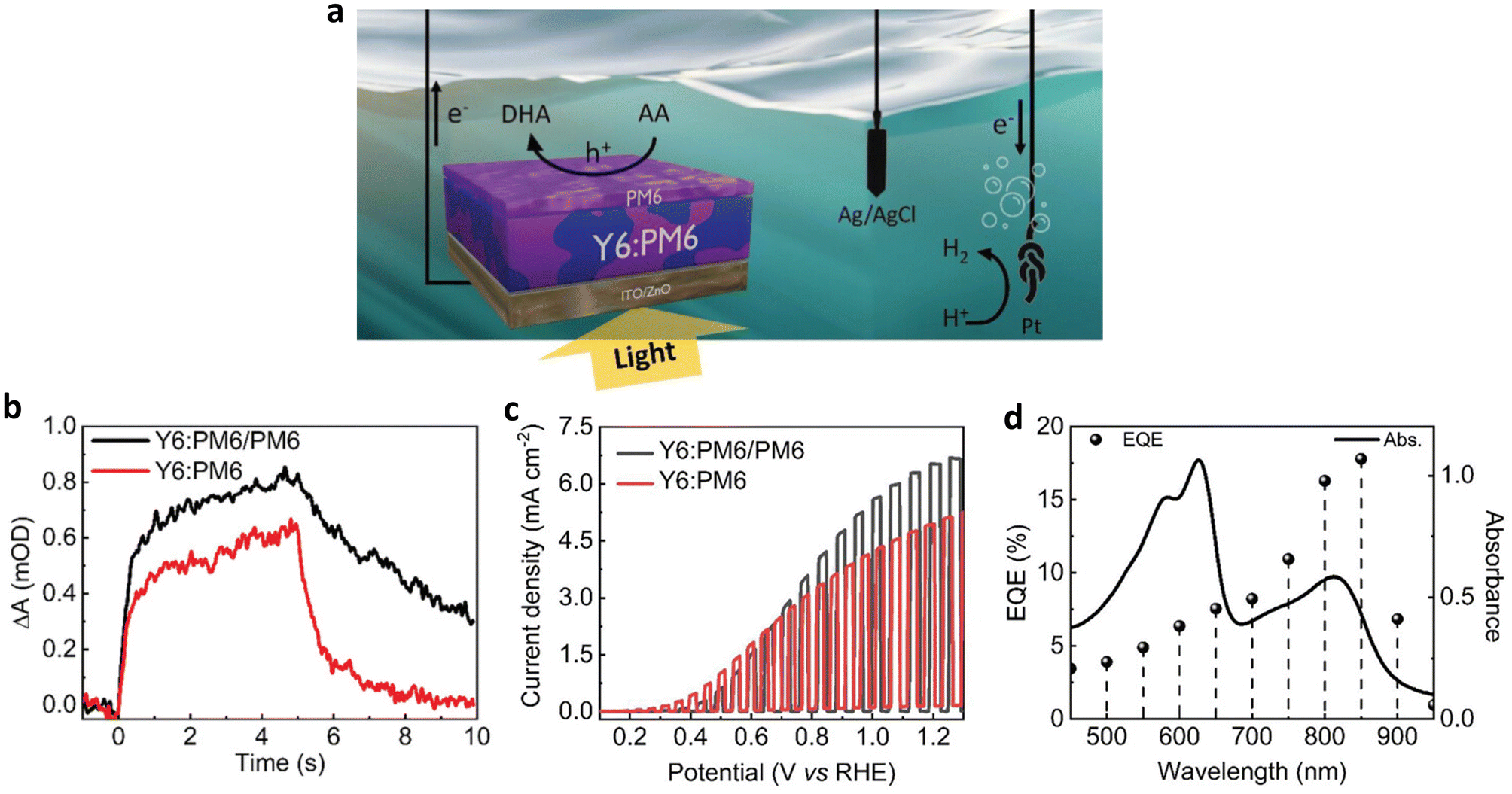 | ||
| Fig. 15 (a) Schematic image of a photoelectrochemical cell with a three-electrode configuration: the Y6:PM6/PM6 photoanode as the working electrode (AA oxidation to DHA), a Pt mesh counter electrode (coupled hydrogen evolution), and an Ag/AgCl reference electrode. (b) Photoinduced absorption kinetics of the Y6:PM6/PM6 and the Y6:PM6 photoanodes probing at 950 nm in 1 M borate buffer (pH 8.1) without AA. A 365 nm LED (40 mW cm−2) illuminates the sample for 5 s before being switched off. (c) LSV scans of the Y6:PM6/PM6 and the Y6:PM6 photoanodes in 0.2 M AA in 1 m borate buffer (pH 5.5) under chopped 1 sun illumination. (d) External quantum efficiency of the Y6:PM6/PM6 photoanode and absorption spectrum of the Y6:PM6/PM6 film. Adapted with permission.76 Copyright 2023, Wiley-VCH GmbH. | ||
Cho et al. improved adhesion by applying mesoporous ZnO (mZnO, solution-processed by nanoparticle dispersion) as an ETL to increase the interfacial area between the BHJ layer and the substrate. This modification resulted in better adhesion of the BHJ layer to the substrate (Fig. 16a).34 The photoanode with planar ZnO (pZnO, processed by sol–gel method) as the ETL exhibits weaker stability as evidenced by rapid photocurrent decay under operational conditions as shown in Fig. 16b. In addition, the original performance of the pZnO device was not recovered even after rinsing the surface of the electrode and refreshing the electrolyte (asterisks in the middle of the CA measurements in Fig. 16b). After long-term operation, the cross-section of the photoanodes was investigated by scanning electron microscopy (SEM) measurements (Fig. 16c). While the BHJ film on top of pZnO was delaminated from the substrate, the film on top of mZnO retained its structure. Interestingly, Lee et al. reported similar results, demonstrating improved operational stability through the use of ZnO nanoparticles (Fig. 16d).33 Mesoporous tin oxide (mSnO2) was also employed by Sekar et al. to ensure good contact between the BHJ layers and the ETL, leading to enhanced stability of BHJ photoanodes.63 To further improve the adhesion between the inorganic ETL and the organic BHJ, benzoic acid was used to form a self-assembled monolayer (SAM) on top of the mSnO2. This approach improved the compatibility of the organic BHJ on the inorganic ETL, thereby enhancing the photocurrent density (Fig. 16e).
 | ||
| Fig. 16 (a) Schematic of OS-based BHJ photoanode architecture and the role played by interlayers for operational stability. (b) CA curves (under 1 sun illumination) at 1.23 VRHE of the FTO/pZnO/BHJ (PBDTTTPD:PNDITCVT) (in 1 M KPi buffer adjusted to pH 9) and FTO/mZnO/BHJ photoanodes (in 0.5 M KPi buffer adjusted to pH 9) in the presence of 1.5 M Na2SO3 as sacrificial agent. At the indicated times (*), the photoanodes were rinsed with deionized water and the electrolyte was refreshed. (c) Cross-sectional SEM images of the FTO/pZnO/BHJ and FTO/mZnO/BHJ photoanodes before and after the CA measurements (scale bars, 200 nm). Adapted with permission.34 Copyright 2021, Springer Nature. (d) Normalized CA curves of ITO/ZnO/Y6:PM6/PM6/Au/NiFeOOH photoanodes with sol–gel ZnO and ZnO nanoparticles under 1 sun illumination in 1 M KOH solution (pH 14) at 1.23 VRHE. Adapted with permission.33 Copyright 2022, Wiley-VCH GmbH. (e) LSV curves of the P2:A2 (1:1) BHJ photoanode with different ETL architectures are shown at pH 6.7 with 1 M phosphate buffer and 1.5 M Na2SO3 sacrificial agent. Adapted with permission.63 Copyright 2022, American Chemical Society. | ||
In general, photoanodes for solar water oxidation operate in alkaline solution. However, the well-known HTLs, such as MoO3 and NiOx, are not stable under high pH conditions.34,81,82 Cho et al. improved the stability of BHJ photoanodes by introducing an organic HTL, PTAA, between the BHJ and the co-catalysts as an alternative to metal–oxide HTLs.34 After the PTAA-deposited BHJ photoanodes were immersed in a pH 9 electrolyte for 4 h, no significant changes in the surface properties or the performance were observed, as confirmed by WCA (Fig. 17a) and LSV measurements (Fig. 17b). In the CA measurement, the photocurrent decayed at a slower rate when the PTAA layer was introduced (Fig. 17c). This improvement was attributed to the facilitated hole transport, which prevented the unfavorable oxidation of the BHJ layers by suppressing hole accumulation.
 | ||
| Fig. 17 (a) Contact angle of a water drop on the FTO/mZnO/BHJ (PBDTTTPD:PNDITCVT)/PTAA photoanode before and after the immersion in 1 M KPi buffer electrolyte (pH 9) for 4 h. (b) LSV scans (in the presence of 1.5 M Na2SO3 as sacrificial agent) of the FTO/mZnO/BHJ/PTAA photoanode before and after the immersion in 1 M KPi buffer solution (pH 9). (c) CA curves (under 1 sun illumination) of the FTO/mZnO/BHJ/LIO and FTO/mZnO/BHJ/PTAA/LIO photoanodes for solar water oxidation in 0.1 M NaBi electrolyte buffered at pH 9. Adapted with permission.34 Copyright 2021, Springer Nature. (d) Schematic device architecture of the ITO/ZnO/Y6:PM6/PM6/Au/NiFeOOH photoanode. (e) Normalized CA curves of the ITO/ZnO/Y6:PM6/PM6/Au/NiFeOOH (denoted as Y6:PM6/PM6) and ITO/ZnO/Y6:PM6/PM6/Au/NiFeOOH (denoted as Y6:PM6) photoanodes under 1 sun illumination in 0.1 M KOH solution (pH 13) at 1.23 VRHE. Adapted with permission.33 Copyright 2022, Wiley-VCH GmbH. | ||
Hydrophobic polymer semiconductors can be used as waterproof layers to enhance the underwater stability of OS-based photoanodes. Lee et al. introduced an additional hydrophobic polymer layer, PM6, on top of a BHJ layer consisting of PM6:Y6 (molecular structures are shown in Fig. 10).33 To couple the OS-based hydrophobic photoactive layer with an OER catalyst, NiFeOOH, a thin Au layer was thermally evaporated (Fig. 17d). The introduction of the PM6 interlayer improved the underwater operational stability of the BHJ photoanodes due to its hydrophobic nature as shown in Fig. 17e.
Consequently, the operational stability of BHJ photoanodes can be further enhanced with physically fixed layers to prevent unnecessary water ingress, as well as chemically stabilized layers to withstand harsh conditions for long-term operation.
2.3. BHJ-based unassisted PEC cells
Despite recent advances in OS-based PEC cells for the half reactions, the development of tandem systems for overall reactions remains a challenge as all components in the tandem systems must be carefully chosen to attain high-performance and stable operation in the same electrolyte.7,83 There is a strong demand for OS-based unassisted PEC tandem cells that operate without sacrificial agents to enable large-scale solar fuel production in an economically feasible manner.2,84–86 Recent research has predominantly focused on hybrid tandem systems, where BHJ-based PEC cells are utilized for photoreduction reactions, while their metal oxide-based counterparts facilitate photocatalytic oxidation reactions.46,50,54 Indeed, BHJ photocathodes based on PBDB-T:ITIC and P3HT:PCBM blends combined with BiVO4- and TiO2-based photoanodes, respectively, yielded Jph values of 1.00 and 0.11 mA cm−2 for overall solar water splitting without the need for external bias and sacrificial agents (Table 3).50,54| Device configuration | Photoactive layer | Co-catalyst | J ph | Faradaic efficiency (operating time)a | Stabilitya/ηSTC | Reaction medium, illumination conditionb | Ref. |
|---|---|---|---|---|---|---|---|
| a The longest time over which continuous STC conversion was reported, not the maximum stability of each photoelectrode. b Photocurrent under simulated 1 sun illumination (100 mW cm−2 of irradiance under AM 1.5 G condition) unless otherwise specified. c A faradaic efficiency was calculated using GC data with calibration conducted by a Pt electrode. d The performance of photocathode and photoanode was measured under illumination intensity of 65 mW cm−2 and 75 mW cm−2, respectively. | |||||||
| Photocathode | PBDB-T:ITIC | Pt | 1.00 mA cm−2 @ 0.68 VRHE | — | ∼42% (2 h)/1.1% @ 0.68 VRHE | 0.2 M KPi (pH 7) | 50 |
| Photoanode | BiVO4 | NiFeOx | |||||
| Photocathode | P3HT:PCBM | — | 0.11 mA cm−2 @ 0.42 VRHE | 97.5% (4 h) | ∼100% (300 s)/0.05% @ 0.42 VRHE | 0.5 M Na2SO4 (pH 6.8) | 54 |
| Photoanode | TiO2 | IrOx | 92.0% (4 h) | ||||
| Photocathode | PBDB-T:ITIC:PCBM | Pt | 3.5 mA cm−2 @ 0.6 VRHE | ∼100% (2500 s) | ∼100% (200 min)/4.3% @ 0.6 VRHE | 0.2 M KPi (pH 7.0) | 87 |
| Photoanode | BiVO4 | Co4O4 cubane | ∼100% (2500 s) | ||||
| Photocathode | PBDTTTPD:PNDIHDT | RuO2 | 0.64 mA cm−2 @ 0.64 VRHE | 102 ± 5% (1 h)c | 39% (2 h)/0.3% @ 0.64 VRHE | 0.1 M NaBi (pH 9) | 32 |
| Photoanode | PBDTTTPD:PNDITCVT | Li-IrO2 | 91 ± 9% (1 h)c | ||||
| Photocathode | PTB7-Th:PDV-V | RuO2 | 23.8 μA cm−2 @ unbiased | — | — | 1.0 M HI (pH 0), under low light-intensityd | 88 |
| Photoanode | CPN:SnO2 | — | |||||
Inspired by natural photosynthesis, Ye et al. assembled PEC tandem cells comprising an inorganic BiVO4-based photoanode and a BHJ-based photocathode (PBDB-T:ITIC:PC71BM denoted as PIP) with multi-mediators (Fig. 18a–c).87 While inefficient charge transfer between a photocathode and a photoanode in unassisted PEC tandem cells has restricted the performance for solar water splitting, the multi-mediators in their study constructed a cascade electrochemical potential alignment which facilitated the orderly transfer of photogenerated charges and suppressed charge recombination. As a result, the optimized hybrid PEC tandem cell (the Co4O4/pGO/BiVO4/SnOx-Pt/TiOx/PIP/CuOx system) delivered a solar-to-hydrogen (STH) conversion efficiency as high as 4.3% through the multi-meditator modulation and the complementary absorption of BiVO4 and PIP (Fig. 18b).
 | ||
| Fig. 18 (a) Schematic of the unassisted PEC tandem cell with the Co4O4/pGO/BiVO4/SnOx photoanode (front) connected to the Pt/TiOx/PIP (PBDB-T:ITIC:PC71BM)/CuOx photocathode (behind) for overall solar water splitting. (b) UV–vis spectra of BiVO4 and PIP showing complementary light absorption. (c) Schematic illustration displaying the device structures of BiVO4-based photoanode and PIP-based photocathode. Adapted with permission.87 Copyright 2021, American Chemical Society. | ||
More recently, Zhang et al. reported the pioneering demonstration of bias-free OS BHJ-based PEC tandem cells, wherein both photoanode and photocathode employ OS BHJs as light-harvesting layers.32 To develop a photocathode that can be coupled with the previously established BHJ-based photoanode by the same group, an SAM of the organic small molecule, 2PACz (Fig. 19a), was introduced as a stable hole extraction layer in pH 9 electrolyte. Furthermore, a polymer BHJ blend of PBDTTTPD:PNDIHDT (denoted as BHJ-1 in Fig. 19a) was employed as the light-harvesting layer to offer sufficient driving force for solar water reduction. The resulting photocathode with the structure of FTO/2PACz(SAM)/BHJ-1/RuO2 exhibited a photocurrent density of 4.6 mA cm−2 at 0 VRHE and maintained a stable photocurrent density with only a 10% decrease over 6 hours of operation (Fig. 19b). Finally, an OS-based PEC tandem cell where this OS BHJ-based photocathode was coupled with the previously established OS BHJ-based photoanode (FTO/mZnO/BHJ-3/PTAA/Li-IrO2) delivered an operational photocurrent density of 0.2–0.3 mA cm−2 without an applied potential for overall solar water splitting, as confirmed by gas chromatography at ∼100% faradaic efficiency (Fig. 19c, d and Table 3). Although an STH was estimated to be 0.3% after the stabilization period, this achievement represents a remarkable benchmark in that it is the first successful demonstration of bias-free organic PEC tandem cells.
 | ||
| Fig. 19 (a) Molecular structures of 2PACz, PBDTTTPD, and PNDIHDT. (b) CA curves at 0 VRHE of the FTO/2PACz(SAM)/BHJ-1 (PBDTTTPD:PNDIHDT)/RuO2 photocathode in 0.1 M NaBi buffer adjusted to pH 9. (c) Schematic illustration of the OS-based PEC tandem cell with the FTO/SAM/BHJ-1/RuO2 photocathode and the FTO/mZnO/BHJ-3 (PBDTTTPD:PNDITCVT)/PTAA/Li-IrO2 photoanode for overall solar water splitting. (d) Photocurrent without an applied potential and STH efficiency of the OS-based PEC tandem cell in 0.1 M NaBi buffer adjusted to pH 9 under 1 sun illumination. Adapted with permission.32 Copyright 2022, Wiley-VCH GmbH. | ||
Beyond overall solar water splitting, OS-based PEC tandem cells for solar H2 production coupled with alternative oxidation reactions have been recently demonstrated. Yao et al. presented an organic PEC tandem cell that combined an OS BHJ-based photocathode with a hybrid BHJ photoanode composed of a covalent polymer network and metal oxide to drive an alternative overall reaction, splitting HI into H2 and I3−.88 The precursor materials, azide-containing perylene diimide (PDI-DA) and terminal alkyne-functionalized triazine (Triazine-TA), were initially infiltrated into SnO2 nanoparticles (optimized thickness of 1.77 μm), subsequently followed by a thermal azide–alkyne cycloaddition (TAAC) networking reaction to form hybrid covalent polymer network (CPN):SnO2 BHJs (Fig. 20a). These photoanodes operated directly in an aqueous electrolyte (1 M HI, pH 0) without any other co-catalysts or protective layers. The hybrid CPN:SnO2 photoanodes effectively promoted iodide oxidation, achieving a Jph of 3.3 mA cm−2 at the iodide oxidation potential (+0.54 V vs. NHE) as shown in Fig. 20b and Table 3, and demonstrated operational stability with only a 12% decrease in Jph over 27 h of continuous operation. By coupling this photoanode with a PTB7-Th:PDI-V/RuO2 photocathode, the first all-organic semiconductor-based PEC cell for unassisted solar-to-chemical conversion was realized (Fig. 20c). At a photocurrent onset potential of +0.43 V vs. NHE, the side-by-side photocathode/photoanode configuration yielded a stable photocurrent of 10 μA, which was maintained for over 10 min in 1 M HI under simulated solar illumination (Fig. 20d).
 | ||
| Fig. 20 (a) Molecular structures of PDI-DA and Triazine-TA and a schematic reaction for generating the CPN. (b) LSV scans of CPN:SnO2 photoanodes in 1 M HI (pH 0) under chopped 1 sun illumination. (c) Schematic of the side-by-side configuration of bias-free solar HI splitting with all OS-based PEC cell. (d) CA measurement obtained in 1 M HI electrolyte (pH 0) from unassisted tandem organic PEC cell, under the illumination intensity of 65 mW cm−2 for the photocathode and 75 mW cm−2 for the photoanode, respectively. Adapted with permission.88 Copyright 2021, The Royal Society of Chemistry. | ||
Overall, a few research groups have recently begun to demonstrate various types of BHJ-based tandem cells, achieving noteworthy benchmarks. However, improvements in the performance of BHJ-based PEC tandem cells remain modest, and further progress primarily requires the establishment of their operational stability.
3. BHJ-based photocatalysts
3.1. Solar-driven H2 production by BHJ photocatalysts
Recent studies employing single OSs in PCs have achieved remarkable performance in solar-driven water reduction through molecular engineering approaches such as the use of bulky and twisted monomers to create microporous structures or the incorporation of hydrophilic side chains to enhance wettability.24–26,28,89–94 These single-component systems are effective at elucidating the complex interactions between OSs and electrolytes, thereby providing scientific insights into the molecular engineering of OSs.7,22–30,95 However, since excitons generated upon illumination are strongly bound due to the intrinsically low dielectric constants of OSs, charge separation in single-component PCs for the HER is finite. Thus, employing the BHJ concept has been recognized as a major research strategy to promote exciton dissociation (Fig. 1). Indeed, Kosco et al. demonstrated that OS PCs based on donor/acceptor BHJs can effectively dissociate excitons into long-lived charge carriers, even in the absence of electron/hole scavengers or platinum co-catalysts.61 Nonetheless, despite the application of the BHJ concept, several critical factors must be considered to achieve high-performance and robust OS-based PCs. The following subsections summarize and discuss recent approaches aimed at improving the performance and operational stability of OS BHJ-based PCs. | ||
| Fig. 21 Molecular structures of conjugated polymers and small molecules for photocatalysts. | ||
 | ||
| Fig. 22 Energy level alignment of conjugated polymers and small molecules for photocatalysts with redox potential of H+/H2, DHA/AA, and O2/H2O. | ||
 | ||
| Fig. 23 (a) Cryo-TEM image of the PTB7-Th:EH-IDTBR nanoparticle after the Pt deposition. The upper and lower insets are magnified images of the areas in the right and left rectangles, respectively. The spacings along the diagonal lines in the upper and lower insets correspond to the lamellar stacking distances of PTB7-Th (2.1 nm) and EH-IDTBR (1.6 nm), respectively. (b) External quantum efficiency at different wavelengths (red dot) and absorption spectrum of the PTB7-Th:EH-IDTBR nanoparticle PCs. (c) Average H2 evolution rates of the PTB7-Th:EH-IDTBR, TiO2, CNxHy, and F8BT nanoparticle catalysts measured over 16 h. Adapted with permission.31 Copyright 2020, Springer Nature. (d) Nano-precipitation process used to prepare the DANHs and scheme representing high-throughput screening process for photocatalytic activity. (e) H2 production rated for A1/D1 NADHs with various metal precursors and (f) photocatalytic hydrogen production rate for A1 (PCBM)/D1 (PCDTBT) NADHs as a function of A1 fraction for 2 h. Adapted with permission.96 Copyright 2020, Wiley-VCH GmbH. | ||
Another pioneering study by Yang et al. established key principles in the design of BHJ PCs by constructing a library of 237 different BHJ PCs. In this work, conjugated polymer donors were intermixed with either fullerene or non-fullerene molecular acceptors, allowing for a systematic exploration of the factors influencing the performance of BHJ PCs (Fig. 23d).96 First, BHJ PCs outperformed those based on pure donor particles, pure acceptor particles, and physical mixtures of donor and acceptor particles, demonstrating efficient charge generation enabled by the BHJ concept. Second, consistent with findings in PEC cells, BHJ PCs incorporating non-fullerene acceptors surpassed those using fullerene derivatives. Notably, Pt emerged as the most efficient co-catalyst among the 11 candidates tested (Fig. 23e). Interestingly, the majority of BHJ PCs achieved their maximum hydrogen production rates at high acceptor ratios, typically between 70–80% (acceptor/donor, by weight) (Fig. 23f). Among the PC combinations screened using a high-throughput approach, the polymer donor, PCDTBT (D1), and non-fullerene acceptor, ITIC-2F (A4), pair exhibited the highest HER rate of 166.8 ± 7.9 mmol h−1 g−1 (Table 4). Their morphological study using SEM characterizations, UV-vis spectroscopy, and 1H NMR spectroscopy suggested that the operational stability of BHJ PCs may be more closely linked to colloidal stability rather than chemical degradation of the BHJ blends. Additionally, a subset of ternary BHJ blend systems demonstrated enhanced performance relative to their binary counterparts, potentially leading to a multicomponent optimization challenge. Similar observations were found in a ternary blend system comprising two conjugated polymer donors (PFBT (D1) and PFODTBT (D2)) and one small molecule acceptor (ITIC) to construct panchromatic BHJ PCs. Liu et al. investigated energy and charge transfer dynamics within this ternary BHJ blend.97 Indeed, energy transfer from excited PFBT (D1) to PFODTBT (D2) and ITIC arose within 600 fs, and charge transfer between PFODTBT (D2) and ITIC occurred within 200 fs upon illumination (Fig. 24a). Due to the favorable morphology and efficient energy and charge transfer, the optimized PCs based on the ternary BHJ blends exhibited a HER rate of 60.8 ± 6.7 mmol h−1 g−1 for the first 24 h with an EQE up to 7% at 600 nm (Fig. 24b and Table 4). Thus, PCs based on ternary BHJ blends hold significant promise for improving photocatalytic performance by leveraging energy and charge transfer among OS materials.
 | ||
| Fig. 24 (a) Scheme of photophysical pathways of D1 (PFBT)D2 (PFODTBT)/ITIC ternary BHJ PCs. (b) Recycling experiment of hydrogen evolution. A 35 mg amount of ascorbic acid was added after cycle 1 and cycle 3, and the final pH was adjusted to pH 4 with 2 M KOH solution. Adapted with permission.97 Copyright 2021, American Chemical Society. (c) Size distribution of BHJ (PBDT-T:ITIC) nanoparticles with varied SDBS concentration. (d) PL spectra excited at 520 nm of PBDB-T, ITIC, and BHJ nanoparticles fabricated using single solvent (CF) and binary solvent mixture (CF/CB). (e) The stability test of the BHJ nanoparticles during a long-term photocatalytic hydrogen production. Adapted with permission.98 Copyright 2022, Elsevier. Ltd. | ||
| OS BHJ | Co-catalyst/surfactant | Solar fuel production rate (mmol h−1 g−1) (with product) | Stabilitya (retained production rate (%)) | EQE (%) (λ (nm)) | Reaction medium/illumination conditionb | Ref. |
|---|---|---|---|---|---|---|
| a The longest time over which continuous solar fuel production was reported, not the maximum stability of each photocatalyst. b Photocurrent under simulated 1 sun illumination (100 mW cm−2 of irradiance under AM 1.5 G condition) unless otherwise specified. c mmolH2 h−1 gH2ase−1. d SDS = sodium dodecyl sulfate, TEBS = 2-(3-thienyl)ethyloxybutylsulfonate sodium salt, AA = ascorbic acid, MeOH = methanol, SDBS = sodium dodecyl benzene sulfonate, TEOA = triethanolamine, MV = methyl viologen, PS-PEG-COOH = polystyrene grafted with ethylene oxide and carboxyl groups. | ||||||
| PM6:Y6 | Pt/TEBSd | 43.6 (H2) | 72 h (92) | 4.3 (400) | 0.2 M AAd (pH 2.4)/AM 1.5 G solar simulator (1 sun) | 61 |
| 4.5 (620) | ||||||
| 5.0 (800) | ||||||
| PM6:PCBM | 73.7 (H2) | 72 h (71) | 8.7 (400) | |||
| 8.2 (470) | ||||||
| 7.7 (560) | ||||||
| 6.6 (620) | ||||||
| PTB7-Th:ET-IDTBR | 5 wt% Pt /SDSd | 3.044 ± 0.322 (H2) | — | — | 0.2 M AAd (pH 2.0)/300 W Xe lamp (350 < λ < 800 nm) | 31 |
| 5 wt% Pt/TEBSd | 28.133 ± 3.067 (H2) | 4.3 (620) | ||||
| 10 wt% Pt/TEBSd | 64.426 ± 7.022 (H2) | 5.6 (660) | ||||
| 6.2 (700) | ||||||
| PCDTBT:PCBM | 9 wt% Pt/— | 171.4 (H2) | 18 h (35.9) | 3.72 (420) | 0.2 M AAd (pH 2.0)/300 W Xe lamp (λ > 420 nm) | 96 |
| 3.43 (490) | ||||||
| 3.16 (515) | ||||||
| PCDTBT:ITIC-2F | 166.8 ± 7.9 (H2) | 10 h (∼0) | — | |||
| PFBT:PFODTBT:ITIC | 6 wt% Pt/PS-PEG-COOHd | 60.8 ± 6.7 (H2) | 120 h (50) | 7.1 (600) | 0.5 M AAd (pH 4 adjust by 2 M KOH)/50 mW cm−2 LED light source (420 < λ < 750 nm) | 97 |
| PBDT-T:ITIC | Pt/SDBSd | 257 (H2) | 48 h (13) | 7.9 (400) | 0.2 M AAd (pH 3)/300 W Xe lamp (1.5 sun)(320 < λ < 780 nm) | 98 |
| 8.6 (450) | ||||||
| 9.9 (650) | ||||||
| 9.0 (700) | ||||||
| PFODTBT:ITIC (28:82) |
6 wt% Pt/PS-PEG-COOHd | ∼40.8 (H2) | 24 h (72.8) | 2.1 (650) | 0.2 M AAd (pH 4)/300 W Xe lamp (λ > 420 nm) | 99 |
| PIDT-T8BT:Y6 (1:6.7) |
2 wt% Pt/TEBSd | 6.0 ± 0.2 (H2) | — | 0.10 | 0.2 M AAd/300 W Xe lamp (λ > 420 nm) | 100 |
| glDTBT:olDTBR | 10 wt% Pt/SDSd | 18.5 (H2) | 72 h (81) | 5.3 (400) | 0.2 M AAd (pH 2.0)/AM 1.5 G solar simulator (1 sun) | 102 |
| 2.9 (620) | ||||||
| 2.8 (660) | ||||||
| PM6:Y6 | 15 wt% Pt/TEBSd | 64.3 (H2) | — | 3.7 (600) | 0.2 M AAd/300 W Xe lamp (330 < λ < 1100 nm) | 104 |
| 6.7 (700) | ||||||
| 7.1 (800) | ||||||
| PM6:BTP-FOH | 78.4 (H2) | 68 h (∼73) | 6.6 (600) | |||
| 8.0 (700) | ||||||
| 8.4 (800) | ||||||
| PM6:BTP-2OH | 102.1 (H2) | 17 h (∼37) | 8.6 (600) | |||
| 9.0 (700) | ||||||
| 9.2 (800) | ||||||
| P1 (F8T2):P2 | 3LX4 ([FeFe]-H2ase)/PS-PEG-COOHd | 88.46c (H2) | 150 h (17) | 1.1 (405) | 20% TEOAd with 5 mM MV2+d and 35 pmol H2ase (pH 7)/50 mW cm−2 LED lamp (λ > 420 nm) |
111 |
| PFBT:PCBM | PS-PEG-COOH | 188 (H2O2, CHOO−) | 75 min (47) | 30 (450, 5 min) 14 (450, 75 min) | 1 M KOH, 5 MeOHd (pH 14)/50 mW cm−2 LED lamp (420 < λ < 750 nm) | 115 |
| PFBT-T:PCBM | PS-PEG-COOH | 0.865 (H2O2, CHOO−) | — | 0.75 (450) | Water/MeOH (4/1, v/v) (pH 7.4)/50 mW cm−2 LED lamp (420 < λ < 750 nm) | 117 |
Yang et al. provided valuable insights into the influence of particle size, the D/A ratio, and solvent choice on the performance of BHJ PCs in their follow-up study.98 Dynamic light scattering (DLS) analysis revealed that the primary factor controlling average particle size was the concentration of surfactant. Specifically, increasing the concentration of sodium dodecyl benzene sulfonate (SDBS) resulted in a reduction in average particle size, which had a more significant effect than the D/A ratio or binary solvent fraction (Fig. 24c). While an optimal concentration of surfactant produced smaller BHJ particles, which is beneficial for shortening the charge diffusion distance from the bulk to the surface, excessive surfactant concentrations inhibited the HER of BHJ PCs owing to the insulating nature of surfactants. In addition, the use of a binary solvent system led to pronounced PL quenching, indicative of a reduced decay lifetime and more efficient extraction of photogenerated electrons by Pt (Fig. 24d). The optimized PCs fabricated with PBDT-T:ITIC (40:60 w/w), CF:CB (60:40 v/v), and SDBS (1.0 mg ml−1) exhibited a HER rate of 257 mmol h−1 g−1 and retained their photocatalytic activity over a 48 h period (Fig. 24e and Table 4). Although the D/A ratio and solvent volume fraction had no impact on nanoparticle size control, they influenced exciton dissociation and charge carrier separation efficiency. Increasing the mass ratio of the acceptor facilitated more efficient charge transfer at the BHJ interface between the donor and acceptor, thereby extending the exciton lifetime through spatial separation. This finding aligns with recent studies that systematically examined the photocatalytic and photovoltaic performance of BHJ blends by varying the donor-to-acceptor mass ratio. For instance, Liu et al. proposed that enhancing the crystallinity of the BHJ composed of PFODTBT (donor polymer) and ITIC (small molecule acceptor) as well as achieving optimal phase segregation between the donor and acceptor, by increasing the ITIC mass ratio, can prolong the charge carrier lifetime and mitigate charge recombination (Fig. 25a).99 This conclusion was firmly supported by their comprehensive steady-state absorption and time-resolved photoluminescence spectroscopy (TRPL) measurements (Fig. 25b and c). Similarly, Dolan et al. observed that BHJ PCs composed of the amorphous polymer donor PIDT-T8BT and the non-fullerene acceptor Y6 exhibited an optimal HER rate of 6.0 ± 0.2 mmol h−1 g−1 at a donor-to-acceptor ratio of 1:6.7 (Fig. 25d and Table 4).100 This improvement was attributed to reduced charge recombination losses at higher acceptor mass ratios. More importantly, their in-depth study suggested that the long-range ambipolar charge transport properties of Y6 contributed to the increased charge-carrier lifetime in the BHJ PCs, thereby enhancing overall HER performance. These pioneering studies indicate that the use of non-fullerene acceptors is critical for improving the HER performance of BHJ PCs and unconventional donor-to-acceptor mass ratios should be explored to minimize charge recombination while extending carrier lifetimes.
 | ||
| Fig. 25 (a) Amount of H2 evolved for the binary BHJ (PFODTBT:ITIC) PCs over 24 h as a function of the mass ratio of A. Reaction conditions: 60 μg with 6 wt% of Pt in 2 mL of a 0.2 M ascorbic acid solution, pH 4, in a 9 mL air-tight vial. (b) Normalized UV-vis absorption spectra of binary BHJ PCs with various amounts of A. (c) TRPL spectra under excitation at 550 nm of D/A binary with A 51 wt%. Adapted with permission.99 Copyright 2022, The Royal Society of Chemistry. (d) Hydrogen evolution rate relative to the total photocatalyst mass for all PIDT-T8BT:Y6 and neat Y6 nanoparticles under identical experimental conditions.100 Copyright 2024, Wiley-VCH GmbH. | ||
 | ||
| Fig. 26 (a) Water contact angle measurements performed on thin films of IDTBT (above) and gIDTBT (below). (b) H2 evolution versus time of nanoparticles containing a D/A heterojunction formed of a 1:1 blend by mass of a donor polymer matched with either oIDTBR or PC71BM electron acceptors. (c) Stability and recyclability test of gIDTBT:oIDTBR nanoparticles. Adapted with permission.102 Copyright 2021, Wiley-VCH GmbH. (d) TRPL results of PM6:BTP-4F, PM6:BTP-FOH, and PM6:BTP-2OH nanoparticles with Pt loading. The three samples exhibit step-by-step reduced PL lifetimes with values of 0.67, 0.45, and 0.36 ns, respectively, revealing the more effective electron transfer to Pt after introducing more hydroxyl groups. (e) Average HER rates at the beginning 10 h of the PM6:acceptor nanoparticles under the 4.52 cm2 illumination area. Adapted with permission.104 Copyright 2023, The Royal Society of Chemistry. | ||
The photovoltage generated by BHJ blends is generally proportional to the energy level difference between the HOMO levels of OS donors and the LUMO levels of OS acceptors. In line with findings in PEC cells, a higher LUMO level of acceptor materials within BHJ PCs is beneficial for enhancing the driving force for the HER. Liu et al. recently proposed a molecular design strategy that incorporates hydroxyl groups into OS acceptors in BHJ PCs.104 By functionalizing OS acceptors with hydroxyl groups, the LUMO levels were gradually elevated from −4.10 eV for the control acceptor (BTP-4F), to −4.03 eV for the acceptor with one hydroxyl group (BTP-FOH), and to −3.98 eV for the acceptor with two hydroxyl groups (BTP-2OH) (Fig. 21 and 22). TRPL measurements for PM6:BTP-4F, PM6:BTP-FOH, and PM6:BTP-2OH PCs decorated with Pt co-catalysts showed a progressive reduction in PL lifetimes with values of 0.67, 0.45, and 0.36 ns, respectively (Fig. 26d). This implies that the elevated LUMO levels resulting from the incorporation of hydroxyl groups facilitated stronger driving forces for photogenerated electron transfer from the OS acceptors to the Pt co-catalysts. Furthermore, the hydrophilic property of the hydroxyl group led to a high affinity with the Pt co-catalysts and the sacrificial hole scavenger (AA). Accordingly, the PM6:BTP-2OH and PM6:BTP-FOH PCs displayed HER rates of 102.1 and 78.4 mmol h−1 g−1, respectively, which represented a noticeable enhancement compared with the HER rate of 64.3 mmol h−1 g−1 for the PM6:BTP-4F PCs (Fig. 26e and Table 4).
 | ||
| Fig. 27 (a) A model structure of HydA1 [FeFe]-hydrogenase generated from its crystal structure (PDB ID:3LX4). (b) Energy diagram summarizing the photophysical processes involved with biohybrid assembly during photocatalysis. The cyan highlighted portion represents processes of photogeneration and charge separation within BHJ PCs (ET – electron transfer, HT – hole transfer). (c) Photocatalytic data for P1:P2 BHJ PCs (16 μg mL−1), MV2+ (5 mM), and hydrogenase (158 pmol) in the presence of various amounts of TEOA (10, 19, 30% vol, pH 7 adjusted with HCl). Adapted with permission.111 Copyright 2022, Wiley-VCH GmbH. | ||
Overall, the utilization of BHJs between electron donors and acceptors has greatly advanced the performance of PCs for solar-driven HER by effectively promoting the generation of free charge carriers that participate in catalytic reactions. However, in the case of PCs, it is noted that most BHJs in PCs are adapted from the OPV community, and the catalyst selection is still limited to Pd or Pt. Therefore, the identification of ideal BHJs for PCs and the demonstration of organic PCs coupled with a wide range of co-catalysts remain challenges to enhancing the performance of OS-based PCs and ensuring long-term operation stability.
3.2. Solar-driven alternative oxidation by BHJ photocatalysts
Instead of generating oxygen through solar-driven water oxidation, alternative oxidation reactions can yield value-added products with favourable kinetics.112–114 Wang et al. reported on BHJ PCs capable of producing H2O2, which can be used as a clean energy carrier, via photooxidation.115 In this study, BHJ PCs were prepared using a heterojunction formed by PFBT (F8BT) as the electron donor and PCBM as the electron acceptor. Polystyrene grafted with carboxyl group-functionalised ethylene oxide (PS-PEG-COOH) was used as a surfactant, making PCs hydrophilic. The PFBT:PCBM PCs performed the oxidation of MeOH and the reduction of oxygen to form formate (CHOO−) and H2O2 through a series of mechanisms in alkaline conditions (Fig. 28a). In the suggested mechanisms, MeOH efficiently takes the photogenerated holes and reacts with superoxide (O2˙−) (the photoreduced form of the oxygen (O2)) generating unstable intermediates that rearrange to produce deprotonated hydrogen peroxide (H2O−) and formaldehyde (CH2O). Thus, these MeOH oxidation and oxygen reduction reactions in alkaline conditions are essential for the photocatalytic production of H2O2. The oxidation of MeOH was carried out by PFBT+˙, which was previously generated by charge transfer between PFBT and PCBM. In addition, the reduction of oxygen was via PCBM˙−, which was generated by accepting electrons from PFBT (the efficient PFBT emission quenching by PCBM is shown in Fig. 28b). Interestingly, the concentration decay of 9,10-anthracenediyl-bis(methylene)dimalonic acid (ABDA) by the generation of singlet oxygen (1O2) in various pH values (Fig. 28c) illustrated that an increased generation rate of 1O2 at a pH below 7 inhibits the production of H2O2 during photocatalysis.116 Therefore, alkaline conditions (pH 14) were required for the efficient generation of H2O2. Finally, the PFBT:PCBM PCs (20 μg mL−1) at pH 14 showed a high H2O2 generation rate of up to 188 mmol h−1 g−1 (Fig. 28d and Table 4) along with an EQE of 30% at 5 min and 14% at 75 min under 450 nm illumination (Fig. 28e), outperforming other organic PCs. More recently, in their follow-up study, Wang et al. reported BHJ PCs capable of performing alternative oxidations under neutral conditions (pH ∼ 7). The BHJ PCs were produced by introducing 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO) as an organic molecular catalyst to the polymer PFBT backbone (PFBT-T) and forming a BHJ with PCBM.117 The MeOH oxidation reaction was accelerated even under neutral conditions, achieving a formaldehyde generation rate of 865 μmol h−1 gPCs−1 at pH 7.4, with an EQE of 0.75% at 450 nm. Furthermore, PFBT-T:PCBM BHJ PCs were able to demonstrate the oxidation of other alcohols at neutral pH, such as the conversion of ethanol to H2O2/acetaldehyde and benzyl alcohol to H2O2/benzaldehyde. The introduction of TEMPO allowed effective charge extraction within the BHJ PC system, which suppressed the formation of singlet oxygen and improved operational stability. This illustrates that incorporating molecular catalysts into the backbone of OSs is an effective strategy to enable photocatalytic alcohol oxidation in neutral conditions. | ||
| Fig. 28 (a) The proposed photocatalytic H2O2 and formate production with PFBT-PCBM BHJ PCs. (b) Absorbance and PL spectra of PFBT-PCBM binary BHJ PCs, PFBT PCs, and PCBM PCs. (c) ABDA degradation with PFBT-PCBM binary BHJ PCs of 20 μg mL−1 in 5 M MeOH under various pH values. (d) Photocatalytic H2O2 generation experiment with 3 mL of 20 μg mL−1 PCs under LED (420–750 nm, 50 mW cm−2) illumination. (e) EQE measurement with 3 mL 100 μg mL−1 PFBT-PCBM BHJ PCs in 1 M KOH and 5 M MeOH, excited at 450 nm (2.9 mW cm−2). Adapted with permission.115 Copyright 2022, Wiley-VCH GmbH. | ||
4. Summary and outlook
This report summarizes the recent advancements in PEC cells and PCs based on OS BHJs for STC conversion. As OSs are emerging materials in STC conversion systems, identifying ideal OSs under operational electrolyte conditions and elucidating the working mechanisms for various STC reactions have become the centre of research interest. Suitable HOMO and LUMO energy levels of BHJ blends for HER, OER, and alternative oxidation reactions have been primarily investigated to provide photogenerated charges with sufficient electrochemical driving force in both BHJ-based PEC cells and PCs. Considering the significantly different timescales from charge generation to electrochemical reactions occurring at the surface of PEC cells and PCs, diverse strategies have been developed to extend charge lifetimes, suppress charge accumulation, and facilitate charge transfer from OSs to co-catalysts. In BHJ PEC cells, engineering co-catalysts and interlayers such as charge transporting layers is essential for mitigating unwanted charge accumulation and boosting charge transfer. Furthermore, the incorporation of protective layers was studied to further improve operational stability. In BHJ PCs, research approaches such as the use of various surfactants have been explored to produce uniform particulates. The ratio of OS donors to acceptors and BHJ morphology were further optimized to enhance the STC conversion efficiency by attaining efficient charge generation. Alongside the identification of appropriate BHJs, a range of co-catalysts have been introduced into both BHJ-based PEC cells and PCs to replace co-catalysts based on rare-earth elements and improve charge transfer at the OS/co-catalyst interface. However, despite significant research and development efforts, there remains considerable potential for further advancing current BHJ-based PEC cells and PCs. The following outlines specific needs in material and device engineering:(1) Development of novel organic semiconductors for PEC cells and PCs: recent studies have demonstrated that BHJs beyond the commonly used P3HT:PCBM combination can noticeably improve the performance and operational stability of BHJ-based PEC cells and PCs since the PCBM-based BHJ shows serious photoinduced degradation.31,35,47 Furthermore, a series of in-depth studies revealed that suppressing charge accumulation is critical for establishing the operational stability of OS BHJ-based PEC cells.34,35 However, the OSs currently employed have primarily been those already demonstrated to perform well in OPVs. While this has contributed to achieving high STC conversion efficiencies in PEC cells and PCs from the outset, it has also limited further advancements due to the different working mechanisms and device architectures involved.31,33–35,46,50,76 In OPVs, OS selection typically considers the energy levels of widely used charge transporting layers and electrodes relative to the vacuum level. In PEC cells and PCs, however, OSs need to be judiciously selected by considering their energy level alignment with the Nernstian behavior of HER and OER potentials. Therefore, to enhance the performance of PEC cells and PCs, it is essential to develop novel OSs capable of efficiently producing photogenerated charges with sufficient electrochemical potential to drive specific oxidation or reduction reactions.
(2) Development of efficient charge-transporting layers for BHJ PEC cells: the incorporation of efficient charge-transporting layers in BHJ PEC cells is required to optimize their performance for solar-driven HER and OER. These layers enable the complete extraction of photogenerated charges with reduced recombination, while improving chemical stability by suppressing side reactions caused by charge accumulation.33,34 However, as mentioned earlier, most research has been restricted to metal oxide-based charge-transport layers that often require high-temperature thermal annealing and operate within a limited range of applied bias in specific electrolytes, which may damage the BHJ layers when these transport layers are applied as overlayers during device fabrication.32,43,46,49 Therefore, there is a clear need for novel HTLs and ETLs for BHJ PEC cells that can suppress charge recombination and accumulation while ensuring compatibility with organic BHJ layers. Emerging candidates for addressing these challenges include low-temperature-processable charge-transporting polymers and self-assembled monolayers, and could significantly enhance both the efficiency and stability of BHJ PEC cells.118,119
(3) Advancement of BHJ PEC tandem cells: the development of tandem systems is greatly needed for achieving overall reactions without requiring an external bias potential.120 Although several BHJ-based PEC tandem cells have been introduced, the STC conversion efficiency and stability of BHJ PEC tandem cells have remained particularly poor.32,50,54,87 In particular, state-of-the-art all-BHJ PEC tandem cells for overall solar water splitting, where both photocathodes and photoanodes are based on BHJ blends, lag significantly behind their inorganic counterparts. This shortfall is largely attributed to the tandem cell's limited light-harvesting capability, resulting from the use of the same electron-donating polymer in both electrodes.32 A straightforward solution to this challenge is the development of wide-bandgap OSs with suitable energy levels for reduction or oxidation reactions, while providing sufficient thermodynamic driving force. Such materials would enable complementary absorption when paired with low-bandgap OS materials in the counter photoelectrode of a tandem configuration, thereby enhancing overall light harvesting.120,121
Moreover, most studies have demonstrated BHJ PEC tandem cells using a specific device architecture, where the photocathode and photoanode are arranged in parallel and connected by wiring, which increases manufacturing costs and hinders the fabrication of large-area devices. To address these challenges and enhance economic viability, the integration of photocathodes and photoanodes into wireless monolithic BHJ PEC tandem cells should be pursued.122 In addition to simplifying the design, monolithic devices offer the advantage of separate production of H2 and O2 on either side, thereby ensuring stable and controlled generation of both gases.123 This separation not only enhances overall efficiency but also contributes to safe and consistent operation of the system during solar water splitting.
(4) Demonstration of sacrificial agent-free BHJ PCs for overall reactions: while redox mediators are typically used to couple two different PCs in Z-scheme tandem systems, their use should be avoided due to several limitations. First, it is difficult to balance both half-reactions when charger transfer between the mediator and the PCs is inefficient. Second, mass transfer may be limited by the slow diffusion kinetics of redox mediators. A key strategy for achieving STC conversion without redox mediators involves developing single particulate PCs capable of driving both reduction and oxidation reactions. However, to date, there have been no demonstrations of BHJ PCs that perform oxidation reactions other than sacrificial oxidation, although a few PCs based on single conjugated polymers for overall solar water splitting have been demonstrated.86 Most existing BHJ PC systems rely on the HER using sacrificial agents to facilitate charge separation. However, these sacrificial agents are not sustainable and add complexity to the system. Furthermore, the reliance on sacrificial electron or hole acceptors has often obscured a clear understanding of the underlying physics since the Gibbs free energy for the given reaction becomes close to zero or even negative.124 A notable example is black (defective) TiO2, which generates substantial amounts of H2 under illumination, but only in the presence of fast sacrificial hole acceptors.125 Thus, the development of sacrificial agent-free BHJ PC systems for overall reactions is a prerequisite for practical STC conversions. Achieving this will require the design of novel BHJ blends, the simultaneous deposition of co-catalysts for reduction and oxidation reactions, and interface engineering within BHJ PCs.
(5) Exploration of PEC cells and PCs beyond solar water splitting: to mitigate charge accumulation caused by the kinetically slow water oxidation, coupling a BHJ photocathode for solar H2 production with a BHJ photoanode for alternative oxidation reactions, or developing PCs capable of producing H2 while driving alternative oxidation reactions, presents a promising strategy. This approach not only addresses the issue of slow oxygen evolution but also enables the production of valuable chemicals beyond dispensable O2. Indeed, a PEC tandem cell that split HI into H2 and I3− through photoreduction and photooxidation reactions, respectively, has been recently reported based on an OS-based photocathode and an organic/inorganic hybrid photoanode.88 However, the performance of BHJ PEC tandem cells for alternative oxidation reactions remains extremely low, and no BHJ PCs capable of simultaneously performing solar-driven HER and alternative oxidation have been demonstrated to date. Therefore, continued research efforts must be dedicated to enhancing the overall performance of BHJ-based tandem cells and developing novel BHJ PCs designed for such dual functions. In parallel, expanding the range of potential products from solar-driven alternative oxidation reactions could amplify the overall impact of these technologies. Furthermore, careful attention must be given to the design of device architecture, PC reactors, and electrolyte composition to ensure the simultaneous execution of both oxidation and reduction reactions.
The notable examples discussed in this report provide guidance for further enhancements and illustrate that BHJ-based PEC cells and PCs hold promising potential for STC conversions. With continued substantial research efforts, a new energy paradigm can be achieved in which solar-driven fuels and feedstocks generated by BHJ-based energy conversion systems become prospective alternatives to fossil fuels and petrochemicals.
Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H.-H. Cho thanks the National Research Foundation of Korea (NRF) for grants funded by the Korean Government (Ministry of Science and ICT, MSIT) (no. RS-2023-00211341 and RS-2023-00217968). T. H. Lee thanks the National Research Foundation of Korea (NRF) grant funded by the Korean Government (MSIT) (no. RS-2023-00247635), and the New Faculty Research Grant of Pusan National University, 2023.References
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- B. A. Pinaud, J. D. Benck, L. C. Seitz, A. J. Forman, Z. Chen, T. G. Deutsch, B. D. James, K. N. Baum, G. N. Baum, S. Ardo, H. Wang, E. Miller and T. F. Jaramillo, Energy Environ. Sci., 2013, 6, 1983–2002 RSC.
- H. Nishiyama, T. Yamada, M. Nakabayashi, Y. Maehara, M. Yamaguchi, Y. Kuromiya, Y. Nagatsuma, H. Tokudome, S. Akiyama, T. Watanabe, R. Narushima, S. Okunaka, N. Shibata, T. Takata, T. Hisatomi and K. Domen, Nature, 2021, 598, 304–307 CrossRef PubMed.
- E. A. Gibson, Nat. Catal., 2021, 4, 740–741 CrossRef.
- X. Liu, J. Iocozzia, Y. Wang, X. Cui, Y. Chen, S. Zhao, Z. Li and Z. Lin, Energy Environ. Sci., 2017, 10, 402–434 RSC.
- M. Ye, J. Gong, Y. Lai, C. Lin and Z. Lin, J. Am. Chem. Soc., 2012, 134, 15720–15723 CrossRef.
- Y. Bai, K. Hippalgaonkar and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 16222–16232 RSC.
- C. Zhao, Z. Chen, R. Shi, X. Yang and T. Zhang, Adv. Mater., 2020, 32, 1907296 CrossRef.
- Y. Guo, Q. Zhou, B. Zhu, C. Y. Tang and Y. Zhu, EES Catal., 2023, 1, 333–352 RSC.
- Z. Wu, X. Li, Y. Zhao, Y. Li, K. Wei, H. Shi, T. Zhang, H. Huang, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 60561–60570 CrossRef.
- L. Yao, A. Rahmanudin, N. Guijarro and K. Sivula, Adv. Energy Mater., 2018, 8, 1802585 CrossRef.
- M. V. Pavliuk, S. Wrede, A. Liu, A. Brnovic, S. Wang, M. Axelsson and H. Tian, Chem. Soc. Rev., 2022, 51, 6909–6935 RSC.
- Z. Fu, A. Vogel, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 4291–4296 RSC.
- K. Oka, O. Tsujimura, T. Suga, H. Nishide and B. Winther-Jensen, Energy Environ. Sci., 2018, 11, 1335–1342 RSC.
- H. C. Rojas, S. Bellani, F. Fumagalli, G. Tullii, S. Leonardi, M. T. Mayer, M. Schreier, M. Grätzel, G. Lanzani, F. D. Fonzo and M. R. Antognazza, Energy Environ. Sci., 2016, 9, 3710–3723 RSC.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, R. Cornut, V. Artero and B. Jousselme, ACS Appl. Mater. Interfaces, 2015, 7, 16395–16403 CrossRef.
- T. Abe, K. Nagai, S. Kabutomori, M. Kaneko, A. Tajiri and T. Norimatsu, Angew. Chem., Int. Ed., 2006, 45, 2778–2781 CrossRef PubMed.
- L. Wang, D. Yan, D. W. Shaffer, X. Ye, B. H. Layne, J. J. Concepcion, M. Liu and C.-Y. Nam, Chem. Mater., 2018, 30, 324–335 CrossRef.
- P. Bornoz, M. S. Prévot, X. Yu, N. Guijarro and K. Sivula, J. Am. Chem. Soc., 2015, 137, 15338–15341 CrossRef.
- S. Yanagida, A. Kabumoto, K. Mizumoto, C. Pac and K. Yoshino, J. Chem. Soc., Chem. Commun., 1985, 474–475 RSC.
- R. J. Lyons and R. S. Sprick, Mater. Horiz., 2024, 11, 3764–3791 RSC.
- L. Liu, M. A. Kochman, Y. Xu, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 17242–17248 RSC.
- Y. Bai, K. Nakagawa, A. J. Cowan, C. M. Aitchison, Y. Yamaguchi, M. A. Zwijnenburg, A. Kudo, R. S. Sprick and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 16283–16290 RSC.
- R. S. Sprick, K. J. Cheetham, Y. Bai, J. A. Fernandes, M. Barnes, J. W. Bradley and A. I. Cooper, J. Mater. Chem. A, 2020, 8, 7125–7129 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef PubMed.
- R. S. Sprick, Y. Bai, A. A. Y. Guilbert, M. Zbiri, C. M. Aitchison, L. Wilbraham, Y. Yan, D. J. Woods, M. A. Zwijnenburg and A. I. Cooper, Chem. Mater., 2019, 31, 305–313 CrossRef.
- Y. Bai, L. Wilbraham, H. Gao, R. Clowes, H. Yang, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, J. Mater. Chem. A, 2021, 9, 19958–19964 RSC.
- L. Liu, M. A. Kochman, W. Zhao, M. A. Zwijnenburg and R. S. Sprick, Chem. Commun., 2022, 58, 10639–10642 RSC.
- W.-C. Lin, J. Jayakumar, C.-L. Chang, L.-Y. Ting, M. H. Elsayed, M. Abdellah, K. Zheng, A. M. Elewa, Y.-T. Lin, J.-J. Liu, W.-S. Wang, C.-Y. Lu and H.-H. Chou, Appl. Catal., B, 2021, 298, 120577 CrossRef.
- R. S. Sprick, J.-X. Jiang, B. Bonillo, S. Ren, T. Ratvijitvech, P. Guiglion, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, J. Am. Chem. Soc., 2015, 137, 3265–3270 CrossRef PubMed.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. G. Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Nat. Mater., 2020, 19, 559–565 CrossRef PubMed.
- D. Zhang, H. Cho, J. Yum, M. Mensi and K. Sivula, Adv. Energy Mater., 2022, 12, 2202363 CrossRef.
- T. H. Lee, R. R. Rao, R. A. Pacalaj, A. A. Wilson and J. R. Durrant, Adv. Energy Mater., 2022, 12, 2103698 CrossRef.
- H.-H. Cho, L. Yao, J.-H. Yum, Y. Liu, F. Boudoire, R. A. Wells, N. Guijarro, A. Sekar and K. Sivula, Nat. Catal., 2021, 4, 431–438 CrossRef.
- L. Yao, N. Guijarro, F. Boudoire, Y. Liu, A. Rahmanudin, R. A. Wells, A. Sekar, H.-H. Cho, J.-H. Yum, F. Le Formal and K. Sivula, J. Am. Chem. Soc., 2020, 142, 7795–7802 CrossRef PubMed.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef PubMed.
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C.-C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef PubMed.
- Z. Li, S. Fang, H. Sun, R. Chung, X. Fang and J. He, Adv. Energy Mater., 2023, 13, 2203019 CrossRef.
- A. Wadsworth, Z. Hamid, J. Kosco, N. Gasparini and I. McCulloch, Adv. Mater., 2020, 32, 2001763 CrossRef PubMed.
- J. M. Yu and J.-W. Jang, Catalysts, 2023, 13, 814 CrossRef.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K.-Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 1–19 CrossRef.
- T. Bourgeteau, D. Tondelier, B. Geffroy, R. Brisse, C. Laberty-Robert, S. Campidelli, R. de Bettignies, V. Artero, S. Palacin and B. Jousselme, Energy Environ. Sci., 2013, 6, 2706–2713 RSC.
- M. Haro, C. Solis, G. Molina, L. Otero, J. Bisquert, S. Gimenez and A. Guerrero, J. Phys. Chem. C, 2015, 119, 6488–6494 CrossRef CAS.
- A. Mezzetti, F. Fumagalli, A. Alfano, D. Iadicicco, M. R. Antognazza and F. di Fonzo, Faraday Discuss., 2017, 198, 433–448 RSC.
- F. Fumagalli, S. Bellani, M. Schreier, S. Leonardi, H. C. Rojas, A. Ghadirzadeh, G. Tullii, A. Savoini, G. Marra, L. Meda, M. Grätzel, G. Lanzani, M. T. Mayer, M. R. Antognazza and F. D. Fonzo, J. Mater. Chem. A, 2016, 4, 2178–2187 RSC.
- W. Shi, W. Yu, D. Li, D. Zhang, W. Fan, J. Shi and C. Li, Chem. Mater., 2019, 31, 1928–1935 CrossRef.
- T. Kim, R. Younts, W. Lee, S. Lee, K. Gundogdu and B. J. Kim, J. Mater. Chem. A, 2017, 5, 22170–22179 RSC.
- W. Shi, D. Li, D. Tu, D. Li, W. Yu, J. Shi and C. Li, Adv. Funct. Mater., 2023, 33, 2209211 CrossRef.
- Y. Wu, D. Liu, H. Zhuang, J. Le and Y. Kuang, Chin. Chem. Lett., 2023, 34, 107480 CrossRef.
- W. Shi, D. Li, W. Fan, J. Ma, C. Li, W. Yu, J. Shi and C. Li, Adv. Funct. Mater., 2020, 30, 2003399 CrossRef.
- D. Shao, X. Zhao, T. Chen, M. Lin, H. Wang and L. Li, Catal. Lett., 2024, 154, 610–618 CrossRef.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. Tang, C. Sun, Z. Luo, K. Chang, Z. Zhang, J. Yuan, T. Li, W. Tang, E. Zhou, Z. Xiao, L. Ding, Y. Zou, X. Zhan, C. Yang, Z. Li, C. J. Brabec, Y. Li and J. Min, J. Mater. Chem. A, 2019, 7, 25088–25101 RSC.
- M. Salvador, N. Gasparini, J. D. Perea, S. H. Paleti, A. Distler, L. N. Inasaridze, P. A. Troshin, L. Lüer, H.-J. Egelhaaf and C. Brabec, Energy Environ. Sci., 2017, 10, 2005–2016 RSC.
- R. Zhang, X. Sun, L. Zheng, L. Diao, F. Chen, Y. Li, S. Wang, Y. Wang, W. Wang, F. Lu, H. Dong, H. Liu and Y. Cheng, Chem. – Eur. J., 2022, 28, e202103495 CrossRef PubMed.
- L. Li, Z. Cai, Q. Wu, W.-Y. Lo, N. Zhang, L. X. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7681–7686 CrossRef PubMed.
- Y. Wu, D. Liu, J. Le, H. Zhuang and Y. Kuang, Small, 2023, 19, 2206763 CrossRef PubMed.
- H. C. Rojas, S. Bellani, E. A. Sarduy, F. Fumagalli, M. T. Mayer, M. Schreier, M. Grätzel, F. Di Fonzo and M. R. Antognazza, ACS Omega, 2017, 2, 3424–3431 CrossRef PubMed.
- F. Fumagalli, S. Bellani, M. Schreier, S. Leonardi, H. C. Rojas, A. Ghadirzadeh, G. Tullii, A. Savoini, G. Marra, L. Meda, M. Grätzel, G. Lanzani, M. T. Mayer, M. R. Antognazza and F. D. Fonzo, J. Mater. Chem. A, 2016, 4, 2178–2187 RSC.
- B. Seger, T. Pedersen, A. B. Laursen, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, J. Am. Chem. Soc., 2013, 135, 1057–1064 CrossRef PubMed.
- A. J. Cowan and J. R. Durrant, Chem. Soc. Rev., 2013, 42, 2281–2293 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef.
- S. R. Pendlebury, A. J. Cowan, M. Barroso, K. Sivula, J. Ye, M. Grätzel, D. R. Klug, J. Tang and J. R. Durrant, Energy Environ. Sci., 2012, 5, 6304–6312 RSC.
- A. Sekar, J. M. Moreno-Naranjo, Y. Liu, J.-H. Yum, B. P. Darwich, H.-H. Cho, N. Guijarro, L. Yao and K. Sivula, ACS Appl. Mater. Interfaces, 2022, 14, 8191–8198 CrossRef.
- C. Xiao, W. Jiang, X. Li, L. Hao, C. Liu and Z. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 18098–18103 CrossRef.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- P. Chao, H. Chen, Y. Zhu, N. Zheng, H. Meng and F. He, Macromolecules, 2020, 53, 165–173 CrossRef.
- P. Chao, Z. Mu, H. Wang, D. Mo, H. Chen, H. Meng, W. Chen and F. He, ACS Appl. Energy Mater., 2018, 1, 2365–2372 CrossRef.
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef PubMed.
- M.-H. Lee, Sol. RRL, 2023, 7, 2300533 CrossRef.
- R. Godin, A. Kafizas and J. R. Durrant, Curr. Opin. Electrochem., 2017, 2, 136–143 CrossRef.
- R. Godin and J. R. Durrant, Chem. Soc. Rev., 2021, 50, 13372–13409 RSC.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef.
- W. Zhu, Y. Yuan and Y. Ma, Sustainable Energy Fuels, 2024, 8, 1077–1084 RSC.
- Z. Ai, Y. Shao, B. Chang, L. Zhang, J. Shen, Y. Wu, B. Huang and X. Hao, Appl. Catal., B, 2019, 259, 118077 CrossRef CAS.
- Q. Shi and H. Duan, Chem. Catal., 2022, 2, 3471–3496 CrossRef.
- T. H. Lee, S. A. J. Hillman, S. Gonzalez-Carrero, A. Difilippo and J. R. Durrant, Adv. Energy Mater., 2023, 13, 2300400 CrossRef.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Nat. Energy, 2019, 4, 746–760 CrossRef.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C.-L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- J. Peña-Bahamonde, C. Wu, S. K. Fanourakis, S. M. Louie, J. Bao and D. F. Rodrigues, J. Catal., 2020, 381, 508–519 CrossRef.
- S. Goetz, S. Edinger, C. Linke, E. Franzke, J. Winkler, M. Valtiner and T. Dimopoulos, J. Mater. Chem. C, 2023, 11, 4899–4906 RSC.
- H.-H. Cho and K. Sivula, Trends Chem., 2022, 4, 93–95 CrossRef.
- K. Sivula and R. van de Krol, Nat. Rev. Mater., 2016, 1, 1–16 Search PubMed.
- M. Liras, M. Barawi and V. A. de la Peña O'Shea, Chem. Soc. Rev., 2019, 48, 5454–5487 RSC.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, M. Bahri, H. Yang, A. Gardner, M. A. Zwijnenburg, N. D. Browning, A. J. Cowan, A. Kudo, A. I. Cooper and R. S. Sprick, Angew. Chem., Int. Ed., 2022, 61, e202201299 CrossRef PubMed.
- S. Ye, W. Shi, Y. Liu, D. Li, H. Yin, H. Chi, Y. Luo, N. Ta, F. Fan, X. Wang and C. Li, J. Am. Chem. Soc., 2021, 143, 12499–12508 CrossRef PubMed.
- L. Yao, Y. Liu, H.-H. Cho, M. Xia, A. Sekar, B. P. Darwich, R. A. Wells, J.-H. Yum, D. Ren, M. Grätzel, N. Guijarro and K. Sivula, Energy Environ. Sci., 2021, 14, 3141–3151 RSC.
- S. A. J. Hillman, R. S. Sprick, D. Pearce, D. J. Woods, W.-Y. Sit, X. Shi, A. I. Cooper, J. R. Durrant and J. Nelson, J. Am. Chem. Soc., 2022, 144, 19382–19395 CrossRef PubMed.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef PubMed.
- S. An, Z. Wu, H. Jeong, J. Lee, S. Y. Jeong, W. Lee, S. Kim, J. W. Han, J. Lim, H. Cha, H. Y. Woo and D. S. Chung, Small, 2023, 19, 2204905 CrossRef PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- C. Yang, B. C. Ma, L. Zhang, S. Lin, S. Ghasimi, K. Landfester, K. A. I. Zhang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 9202–9206 CrossRef PubMed.
- C. Cheng, X. Wang, Y. Lin, L. He, J.-X. Jiang, Y. Xu and F. Wang, Polym. Chem., 2018, 9, 4468–4475 RSC.
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Adv. Energy Mater., 2020, 10, 2001935 CrossRef.
- H. Yang, X. Li, R. S. Sprick and A. I. Cooper, Chem. Commun., 2020, 56, 6790–6793 RSC.
- A. Liu, L. Gedda, M. Axelsson, M. Pavliuk, K. Edwards, L. Hammarström and H. Tian, J. Am. Chem. Soc., 2021, 143, 2875–2885 CrossRef PubMed.
- Y. Yang, D. Li, P. Wang, X. Zhang, H. Zhang, B. Du, C. Guo, T. Wang and D. Liu, Polymer, 2022, 244, 124667 CrossRef.
- A. Liu, S. Wang, H. Song, Y. Liu, L. Gedda, K. Edwards, L. Hammarström and H. Tian, Phys. Chem. Chem. Phys., 2023, 25, 2935–2945 RSC.
- A. Dolan, X. Pan, M. J. Griffith, A. Sharma, J. M. de la Perrelle, D. Baran, G. F. Metha, D. M. Huang, T. W. Kee and M. R. Andersson, Adv. Mater., 2024, 36, 2309672 CrossRef PubMed.
- B. Meng, J. Liu and L. Wang, Polym. Chem., 2020, 11, 1261–1270 RSC.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, W. Zhang, M. Moser, R. Sheelamanthula, L. Zhao, B. Willner, T. C. Hidalgo, H. Faber, B. Purushothaman, M. Sachs, H. Cha, R. Sougrat, T. D. Anthopoulos, S. Inal, J. R. Durrant and I. McCulloch, Adv. Mater., 2022, 34, 2105007 CrossRef.
- J. Brebels, E. Douvogianni, D. Devisscher, R. T. Eachambadi, J. Manca, L. Lutsen, D. Vanderzande, J. C. Hummelen and W. Maes, J. Mater. Chem. C, 2018, 6, 500–511 RSC.
- X. Liu, Y. Zhao, Y. Ni, F. Shi, X. Guo and C. Li, Energy Environ. Sci., 2023, 16, 4065–4072 RSC.
- E. Reisner, D. J. Powell, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 18457–18466 CrossRef.
- B. L. Greene, C. A. Joseph, M. J. Maroney and R. B. Dyer, J. Am. Chem. Soc., 2012, 134, 11108–11111 CrossRef PubMed.
- K. A. Brown, S. Dayal, X. Ai, G. Rumbles and P. W. King, J. Am. Chem. Soc., 2010, 132, 9672–9680 CrossRef CAS.
- G. A. M. Hutton, B. Reuillard, B. C. M. Martindale, C. A. Caputo, C. W. J. Lockwood, J. N. Butt and E. Reisner, J. Am. Chem. Soc., 2016, 138, 16722–16730 CrossRef CAS.
- K. Holá, M. V. Pavliuk, B. Németh, P. Huang, L. Zdražil, H. Land, G. Berggren and H. Tian, ACS Catal., 2020, 10, 9943–9952 CrossRef.
- D. Chen, I.-C. Wu, Z. Liu, Y. Tang, H. Chen, J. Yu, C. Wu and D. T. Chiu, Chem. Sci., 2017, 8, 3390–3398 RSC.
- M. V. Pavliuk, M. Lorenzi, D. R. Morado, L. Gedda, S. Wrede, S. H. Mejias, A. Liu, M. Senger, S. Glover, K. Edwards, G. Berggren and H. Tian, J. Am. Chem. Soc., 2022, 144, 13600–13611 CrossRef.
- S. Wang, G. Kumar and H. Tian, Sol. RRL, 2022, 6, 2200755 CrossRef.
- H. G. Cha and K.-S. Choi, Nat. Chem., 2015, 7, 328–333 CrossRef PubMed.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef.
- S. Wang, B. Cai and H. Tian, Angew. Chem., Int. Ed., 2022, 61, e202202733 CrossRef.
- C. Jiang, T. Zhao, P. Yuan, N. Gao, Y. Pan, Z. Guan, N. Zhou and Q.-H. Xu, ACS Appl. Mater. Interfaces, 2013, 5, 4972–4977 CrossRef PubMed.
- S. Wang, M. V. Pavliuk, X. Zou, P. Huang, B. Cai, O. M. Svensson and H. Tian, Nat. Commun., 2024, 15, 6765 CrossRef.
- K. M. Mancini, Y. Khatib, L. Shahine and G. D. O'Neil, Langmuir, 2024, 40, 17536–17546 CrossRef PubMed.
- S. Casalini, C. A. Bortolotti, F. Leonardi and F. Biscarini, Chem. Soc. Rev., 2017, 46, 40–71 RSC.
- M. S. Prévot and K. Sivula, J. Phys. Chem. C, 2013, 117, 17879–17893 CrossRef.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef.
- W.-H. Cheng, M. H. Richter, M. M. May, J. Ohlmann, D. Lackner, F. Dimroth, T. Hannappel, H. A. Atwater and H.-J. Lewerenz, ACS Energy Lett., 2018, 3, 1795–1800 CrossRef.
- X. Shi, K. Zhang, K. Shin, M. Ma, J. Kwon, I. T. Choi, J. K. Kim, H. K. Kim, D. H. Wang and J. H. Park, Nano Energy, 2015, 13, 182–191 CrossRef.
- A. J. Kaufman, A. C. Nielander, G. J. Meyer, S. Maldonado, S. Ardo and S. W. Boettcher, Nat. Catal., 2024, 7, 615–623 CrossRef.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |