
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eco-friendly synthesis of a porous reduced graphene oxide–polypyrrole–gold nanoparticle hybrid nanocomposite for electrochemical detection of methotrexate using a strip sensor†
Reshmi A.
Sukumaran
a,
Kavitha
Lakavath
a,
V. V. N.
Phani Kumar
b,
Sampath
Karingula
a,
Kuldeep
Mahato
 c and
Yugender Goud
Kotagiri
*a
c and
Yugender Goud
Kotagiri
*a
aDepartment of Chemistry, Indian Institute of Technology Palakkad, Palakkad, Kerala 678 557, India. E-mail: yugenderkotagiri@iitpkd.ac.in
bCentre for Automotive Energy Materials, International Advanced Research Centre for Powder Metallurgy and New Materials (ARCI), Chennai 600113, Tamil Nadu, India
cDepartment of Nanoengineering, University of California San Diego, 9500 Gilman Drive, La Jolla, California 92093, USA
First published on 26th December 2024
Abstract
Chemotherapy is a crucial cancer treatment, but its effectiveness requires precise monitoring of drug concentrations in patients. This study introduces an innovative electrochemical strip sensor design to detect and continuously monitor methotrexate (MTX), a key chemotherapeutic drug. The sensor is crafted through an eco-friendly synthesis process that produces porous reduced graphene oxide (PrGO), which is then integrated with gold nanocomposites and polypyrrole (PPy) to boost the performance of a screen-printed carbon electrode (SPCE). Advanced techniques were employed for detailed characterization of the nanocomposites such as X-ray diffraction (XRD), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), field emission scanning electron microscopy (FE-SEM), and BET analysis. The enhanced sensor exhibited a notable increase in the electrochemical oxidation signals of MTX, attributed to the improved electron transfer at the SPCE/PrGO–PPy–Au electrode interface. Superior electrochemical interfacial properties were well characterized with the techniques of cyclic voltammetry, electrochemical impedance spectroscopy, and square wave voltammetry. The sensor demonstrates an efficient electrochemical response toward the detection of MTX with a broad detection range from 130 nM to 1 μM, an impressively low detection limit of 0.4 nM in human serum, and a sensitivity of 24.1 μA μM−1. This combination highlights its exceptional performance in detecting analytes with high precision and sensitivity. The sensor exhibited a long-term continuous monitoring stability response to monitor the MTX drug in human serum for 4 hours. The sensor's high sensitivity, selectivity, reproducibility, and stability over time emphasize its potential as a valuable tool for the swift on-site testing of anticancer drugs in clinical and environmental settings.
1. Introduction
In the fight against cancer, chemotherapy stands as a leading treatment method. Often combined with surgery, radiation, or hormonal therapies, it employs potent drugs in high doses to target and eradicate fast-growing cancer cells.1,2 This aggressive approach underscores its crucial role in modern cancer care, offering a powerful tool against the disease's progression.3 Methotrexate (MTX), an antifolate compound (2,4-diamino-N10-methylpteroyl-glutamic acid), structurally mimics folic acid. In its structure, the hydroxyl group at the C4 position of the pyrimidine ring is replaced by an amino group. Highly effective as a chemotherapy drug, it is used to treat a range of cancers, such as osteosarcoma, leukemia, breast cancer and lymphoma.4 Methotrexate is metabolized in the liver and eliminated through the kidneys. MTX closely resembles folic acid (FA) in structure, leading to the inhibition of dihydrofolate reductase. While it effectively fights cancers, it can cause side effects. Ensuring the right dosage is crucial to avoid under-treatment or harmful side effects, making accurate blood level tracking essential for patient safety.5,6 Beyond healthcare, MTX's environmental impact through pharmaceutical waste poses risks to ecosystems, especially aquatic life.7 Onsite rapid detection offers a compassionate solution to monitor MTX in medical settings and for environmental protection.8 These efforts aim to strike a balance between delivering effective cancer treatment and safeguarding our environment from potential contamination. Analytical and electroanalytical methods are essential in medical and environmental analyses, providing accurate and sensitive means to detect various substances.9 In medicine, these techniques are crucial for monitoring therapeutic drugs, metabolites, and biomarkers, enabling personalized medicine and disease diagnosis.10 Specifically, in therapeutic drug monitoring (TDM), electroanalytical methods such as voltammetry and amperometry offer rapid, cost-effective, and highly sensitive tools for real-time drug quantification.11 These methods ensure optimal drug dosages, reducing toxicity risks and enhancing treatment efficacy.Several methods, such as HPLC,12 fluorescence,13 UV–visible spectrophotometry,14 enzymatic assays,15 fluorimetric assays16 and capillary electrophoresis,17 have been developed to detect MTX with high sensitivity. However, these techniques are problematic due to their time-consuming nature, complex procedures requiring technical expertise, and expensive equipment. Electroanalytical methods like pulse voltammetry and electrochemical impedance analysis offer a promising solution to address these challenges. Electrochemical sensors, in particular, offer high sensitivity, portability, and real-time analysis, making them ideal for detecting low concentrations in complex samples.18,19
In this study, we aim to detect methotrexate at much lower concentrations than previously reported.20–22 Our approach aims to push the detection limit from micromolar levels to nanomolar, surpassing previously established benchmarks; moreover, we also add a new methodology strategy for continuous monitoring of MTX under real-time conditions.
Nanomaterials like nano graphite,23 reduced graphene oxide,24 graphene,25 carbon nanotubes,26 metal nanoparticles,27 and core–shell nanoparticles28 have revolutionized biosensor technology. These materials have been instrumental in enhancing the sensor performance across various applications. Graphene, renowned for its impressive specific surface area and high electrical conductivity, has become a focal point for electrochemical applications. Yet, these ideal properties are only seen in single-layer graphene with perfect crystallinity.29 Graphene oxide (GO) offers better mass production and dispersibility in polar solvents but suffers from poor conductivity and numerous defects.30,31 The reduction process of GO partially restores the sp2 carbon network, resulting in enhanced electron transport and thermal conductivity, and improves GO's electrical properties and solvent compatibility, but still has limitations due to oxygen functional groups and defects causing restacking.32 To overcome these issues, active rGO was developed with a porous structure that significantly enhances conductivity. Integrating graphene into porous structures revolutionizes their electrochemical performance by creating highly conductive pathways.33,34 In this study, we synthesized a porous reduced graphene oxide material through an eco-friendly method.
Traditional methods to produce rGO often use hazardous chemicals and energy-intensive processes, leading to environmental and health risks.35–37 To address these concerns, green synthesis methods for rGO have gained significant attention.38 One promising approach uses ascorbic acid, a naturally occurring and environmentally friendly reducing agent, to produce porous rGO (PrGO). Notably, nanocomposites that integrate two or more distinct components are engineered to amplify the properties of each element, paving the way for the creation of advanced electrochemical sensors. Conductive polymers, like inorganic semiconductors and metals, exhibit distinctive electrical and optical properties due to their conjugated carbon chains containing alternating single and double bonds, which create electron-dense π bonds.39 Notable conductive polymers include polypyrrole (PPy), polyindole, polythiophene (PTH), polyacetylene (PA), poly(para-phenylene) (PPP), poly(3,4-ethylenedioxythiophene) (PEDOT), polyaniline (PANI), poly(phenylenevinylene) (PPV) and polyfuran (PF). With their significant technological potential, these materials have become integral to various applications.40 Among them, PPy is especially noteworthy for its remarkable stability and conductivity, ease of forming homopolymers and composites, fast charge/discharge processes, simple synthesis, flexibility, high energy density, and low cost.41 The PrGO–PPy–Au hybrid electrode boasts superior electrochemical properties due to its smart design. Introducing polypyrrole to porous reduced graphene oxide significantly enhances its properties, making it ideal for conducting applications.42 PPy, known for its low cost, flexibility, high conductivity, eco-friendliness, and excellent redox stability, forms π–π stacking interactions with PrGO, boosting its conductivity.43 This synergy improves electrochemical cycling stability and overall performance, making the PPy–PrGO composite highly effective for various sensing applications.44 In this study, PPy was employed both as a conductive polymer and as an efficient linker to aid in the attachment of rGO and nano Au to the electrode surface via electrostatic and π–π interactions. This method improves mechanical stability and ensures effective conjugation within the nanohybrid composite. Incorporating gold nanoparticles on the surface of PrGO has been found to further improve the conductivity of the composite.45 A particularly exciting advancement lies in the creation of a nanocomposite where gold nanoparticles are intricately layered on the surface of rGO, which has a PPy matrix. This innovative design not only improves conductivity, but also renders it exceptionally effective for numerous sensor applications.46
In this study, we are utilizing a modified screen-printed carbon electrode (SPCE). SPCEs are highly preferred in electrochemical sensors due to their large surface area, portability, reproducibility, and ease of mass production.47 They allow for experiments with small solution volumes (30–40 μL), which is crucial for analyzing biological samples. SPCEs are well-suited for point-of-care applications because they can be incorporated into compact, portable devices, enhancing convenience and accessibility in various environments.48 This research is dedicated to developing a cutting-edge, portable wireless electrochemical sensor strip for continuous monitoring and smartphone-based point-of-care detection of methotrexate, a vital anticancer drug. By emphasizing an eco-friendly synthesis method for the PrGO–PPy–Au biocomposite, this study aims to innovate the way we track methotrexate levels. The novel composite is employed as an advanced electroactive surface modifier, enabling precise and real-time detection of methotrexate. This continuous monitoring capability is crucial, as it ensures timely and accurate drug level assessments, directly impacting the effectiveness of cancer treatment and patient outcomes. PrGO–PPy–Au ink was prepared by dissolving in a chitosan solution and drop-casting onto a screen-printed electrode; this approach streamlines characterization and testing of the composite's electrochemical sensing abilities. The modified sensor underwent characterization using advanced electroanalytical techniques. To assess the sensor's analytical performance for methotrexate detection, square-wave voltammetry (SWV) and chronoamperometry (CA) methods were employed. Additionally, the sensor's real-time application was demonstrated through interference studies and the analysis of real samples.
2. Experimental section
Chemicals and materials are listed in section 1 and details on the preparation of graphene oxide are provided in section 2 of the ESI.†2.1 Synthesis of the PrGO–PPy–Au nanocomposite
The process for preparing graphene oxide is covered in section 1 of the ESI.†3. Results and discussion
3.1. Design and approach for modified screen-printed electrodes
The effectiveness of electrochemical sensing largely hinges on the electrode surface. In this work, we devised a simple method for synthesizing the PrGO–PPy–Au nanocomposite, which was utilized as a coating for methotrexate detection. The nanocomposite was applied to a clean SPCE and allowed to dry overnight. To prevent interference, a Nafion layer was subsequently added. As shown in Fig. 1A, the nanocomposite was synthesized by mixing PrGO and PPy with HAuCl4 solutions, leading the formation of Au nanoparticles and PPy chains on the rGO surface. In the preparation of the PrGO–PPy–Au ink, a 1% chitosan solution was used. 1 mL of chitosan solution was used to dissolve 1 mg of the PrGO–PPy–Au composite.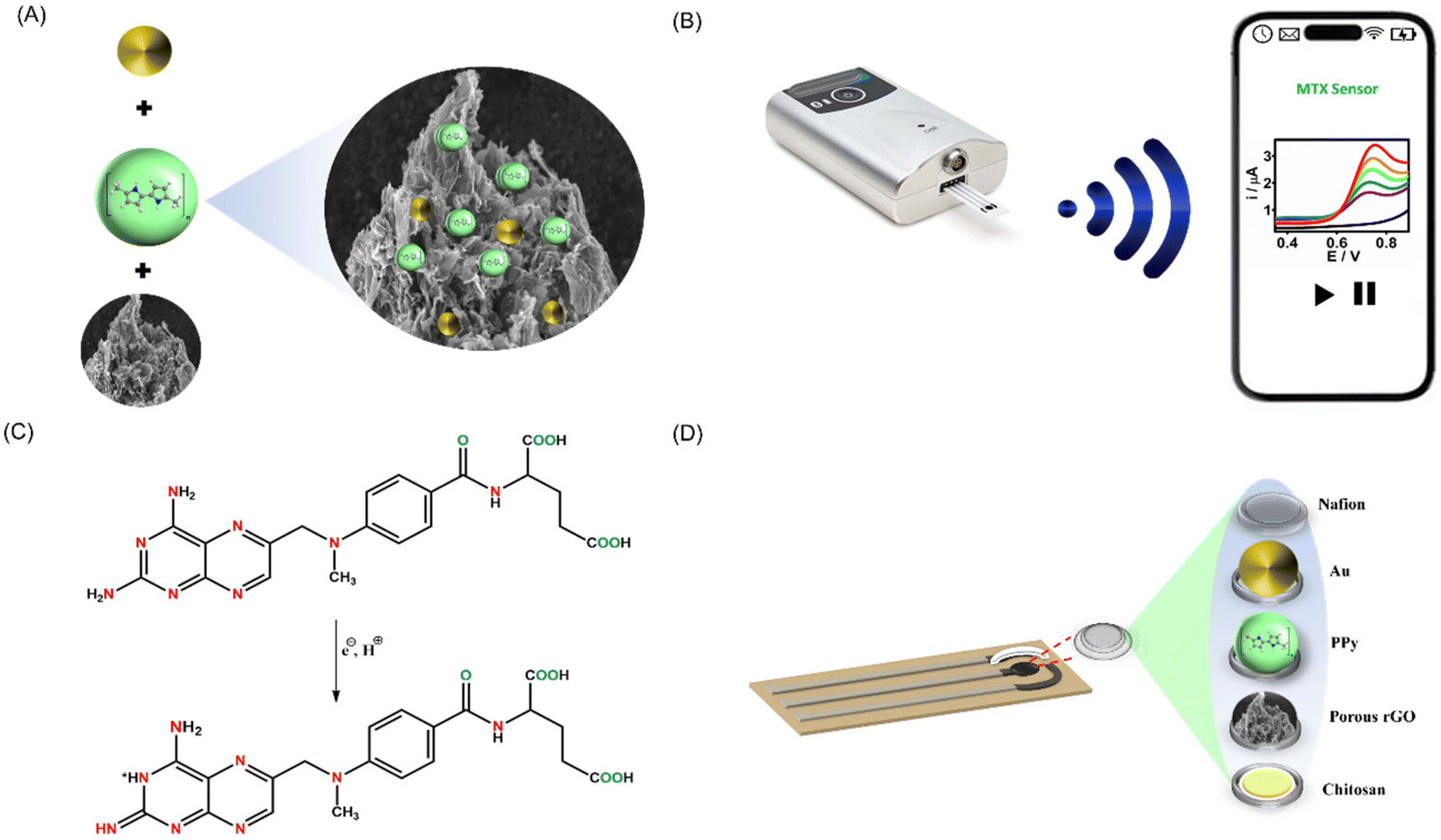 | ||
| Fig. 1 Diagrams showing the synthesis and sensing process. (A) The PrGO–PPy–Au nanocomposite. (B) Recording voltammograms of methotrexate using a bluetooth-enabled handheld potentiostat and a mobile device. (C) Electrochemical oxidation behaviour of methotrexate. (D) Diagram depicting the modified SPCE sensor's layered architecture incorporating a PrGO–PPy–Au composite within a chitosan matrix and protected by a Nafion layer. | ||
The prepared ink was drop-cast onto a clean SPCE, followed by electrochemical measurements conducted with a portable potentiostat linked to a smartphone, as illustrated in Fig. 1B. Methotrexate oxidation is a complex process governed by pH and controlled by diffusion. It involves the transfer of two electrons and two protons, leading to the formation of the electroactive compound 7-hydroxymethotrexate, which participates in a reversible redox reaction, as depicted in Fig. 1C. Fig. 1D depicts the layered structure on the SPCE. After dissolving the nanohybrid composite in chitosan, it was applied to the SPCE and then covered with a Nafion protective layer.
3.2 Structural and morphological analysis of the PrGO–PPy–Au nanocomposite
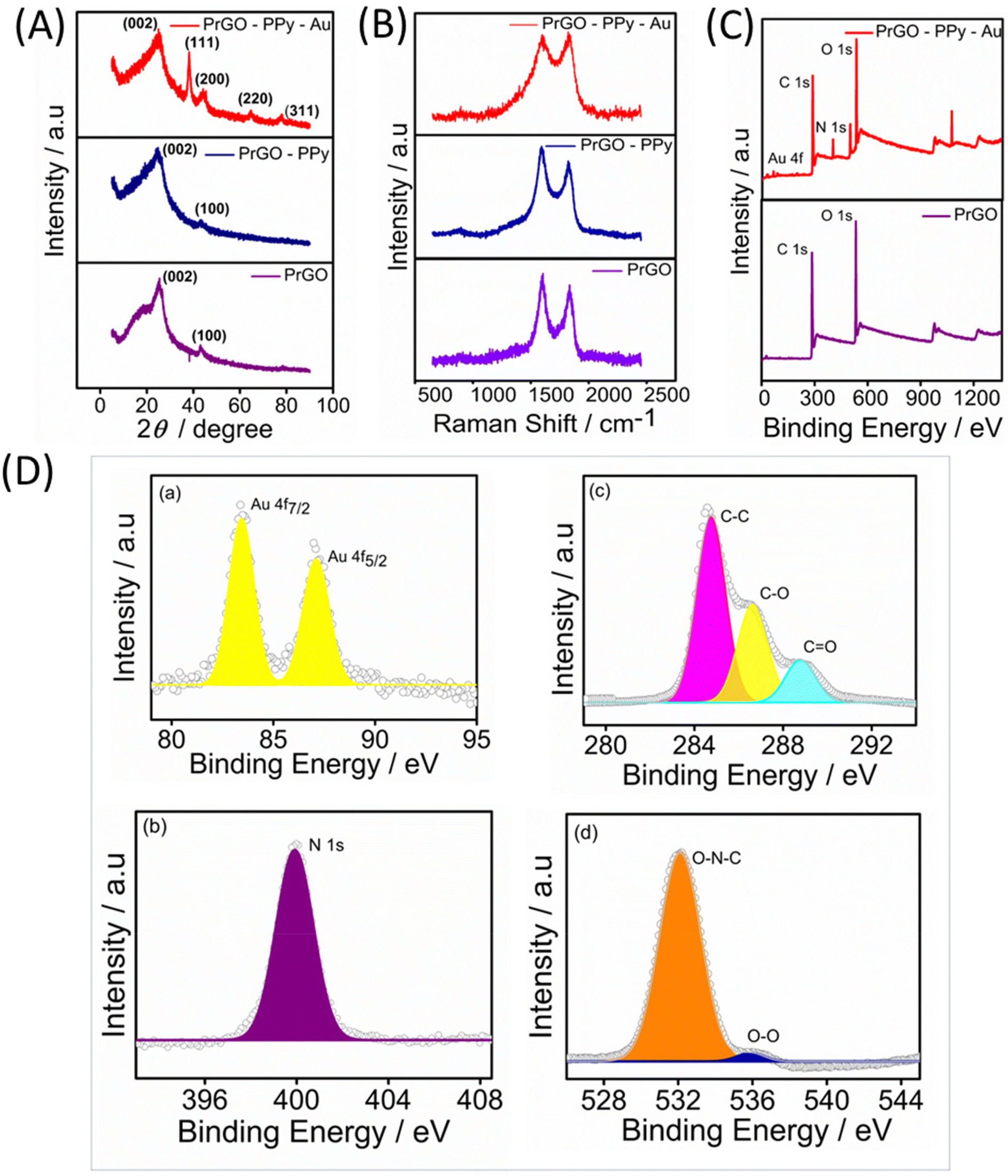 | ||
| Fig. 2 (A) Powder XRD patterns of PrGO, PrGO–PPy, and PrGO–PPy–Au. (B) Raman shifts of PrGO, PrGO–PPy, and PrGO–PPy–Au. (C) XPS survey spectra of PrGO and PrGO–PPy–Au. (D) High-resolution XPS spectra of Au (a), nitrogen (b), carbon (c), and oxygen (d). | ||
The Raman spectra for the rGO, rGO–PPy, and rGO–PPy–Au nanocomposites were recorded, with the results displayed in Fig. 2B. Two characteristic peaks were observed in the Raman spectra of the samples. The peak near 1352 cm−1 (D-band) is linked to defects and disorder in the hexagonal lattice, indicating the presence of vacancies or imperfections in the carbon structure, as well as the presence of sp3-hybridized carbons, while the peak at 1582 cm−1 (G-band) is associated with the vibration of sp2-bonded carbon atoms in the 2D hexagonal lattice.51 The D/G intensity ratios (ID/IG) indicate the atomic ratio of sp2 to sp3 carbons, reflecting the degree of disorder in the graphite structure.52 For rGO, the calculated ID/IG ratio is 0.85. In the case of rGO–PPy, the D-band appears at around 1346 cm−1 and the G-band appears at 1575 cm−1. In comparison to pure rGO, the minor blue shifts observed in the D-band and G-band of the PPy nanosphere/rGO composites suggest charge redistribution and interactions between the PPy nanospheres and rGO.53 Furthermore, the ID/IG ratios of PPy nanosphere/rGO composites are lower than those of pure rGO, as the physical defects such as wrinkles in pure rGO contribute to a higher ID/IG value.54 As noted, rGO frequently experiences significant bending and aggregation into a partially graphitic structure due to intense π–π re-stacking. This leads to high conductivity and problematic impedance mismatching. The D-band and G-band of are shifted to 1348 cm−1 and 1588 cm−1 for PrGO–PPy–Au, indicating that the immobilized Au nanoparticles effectively reduce defects on the rGO sheets.55 The heightened relative intensity of the D-band signifies a strong interaction between the PPy monomer, rGO, and Au. The observed shifts are also attributed to π–π interactions between PPy and rGO, along with significant interactions with AuNPs.56,57
The composition of PrGO–PPy–Au was analyzed using X-ray photoelectron spectroscopy (XPS). Fig. 2C presents the XPS survey spectra of PrGO and PrGO–PPy–Au. In the PrGO sample, the presence of C 1s and O 1s peaks is observed at approximately 288 eV and 548 eV, respectively, with the O 1s peak showing higher intensity than the C 1s peak. The PrGO–PPy–Au composite, in addition to carbon and oxygen, exhibits characteristic peaks corresponding to Au 4f and N 1s. In Fig. 2D(a), the Au 4f peak is clearly observed, indicating the reduction of Au3+ to metallic Au0 using tri sodium citrate during the synthesis,58 confirming the formation of AuNPs on the surface of PrGO–PPy nanocomposites. High-resolution XPS analysis further confirms the nitrogen, carbon, and oxygen binding energies of the PrGO–PPy–Au nanocomposites. Fig. 2D(b) shows the N 1s spectrum with a characteristic peak at 399.8 eV, corresponding to the graphitic N signal.59 The high-resolution C 1s spectra (Fig. 2D(c)) show three main components: C–C and C–O at ∼284.56 eV and ∼286.61 eV, respectively, and a minor component from C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O at 288.1 eV. The peaks at 286.7 and 288.4 can be attributed to various C–O bonding configurations resulting from the intense oxidation and disruption of the sp2 atomic structure of graphite.54 Similarly, Fig. 2D(d) shows O 1s peaks at 535.2 eV and 532 eV, corresponding to the O–O and O–N–C bonds.
O at 288.1 eV. The peaks at 286.7 and 288.4 can be attributed to various C–O bonding configurations resulting from the intense oxidation and disruption of the sp2 atomic structure of graphite.54 Similarly, Fig. 2D(d) shows O 1s peaks at 535.2 eV and 532 eV, corresponding to the O–O and O–N–C bonds.
Thermogravimetric analysis (TGA) was conducted to determine the degassing temperature of the PrGO material prior to performing the BET analysis. The thermogravimetric analysis (TGA) of the PrGO nanomaterial, illustrated in Fig. S1,† reveals an initial 11% weight loss between 30 and 120 °C, primarily due to adsorbed water and inherent moisture. As the temperature rises, PrGO undergoes two distinct degradation stages. The first stage, occurring between 225 and 400 °C, results in a significant 50% weight loss, driven by the decomposition of epoxy, hydroxyl groups, and residual water molecules. The second stage, a change in the slope is again observed from 450 to 600 °C, involves the breakdown of more stable oxygen-containing functional groups and the carbon skeleton, marking the complete pyrolysis process forming CO and CO2. Notably, PrGO remains thermally stable up to approximately 200 °C, beyond which significant decomposition and pyrolysis occur.49,60,61
3.3. Morphological characterization
The surface morphologies of the PrGO-, PrGO–PPy-, and PrGO–PPy–Au-modified electrodes were analyzed using FE-SEM, with the representative results depicted in Fig. 3A. The FE-SEM images of the rGO composite reveal a highly porous and wrinkled surface with mesopores distributed across the surface, as illustrated in Fig. 3A(a and d). Ascorbic acid helps in the reduction process of graphene oxide(GO) functional groups while simultaneously and creating a porous structure within the rGO sheets. During the reduction, functional groups on the GO surface are removed, generating small molecules like CO2 and H2O as by-products. The release of these gases disrupts the stacking of rGO sheets, leading to the formation of a unique porous structure. Fig. S2† presents the SEM images of GO and PrGO at different resolutions. The images clearly reveal that GO exhibits a non-porous, sheet-like structure, while PrGO distinctly displays a porous morphology.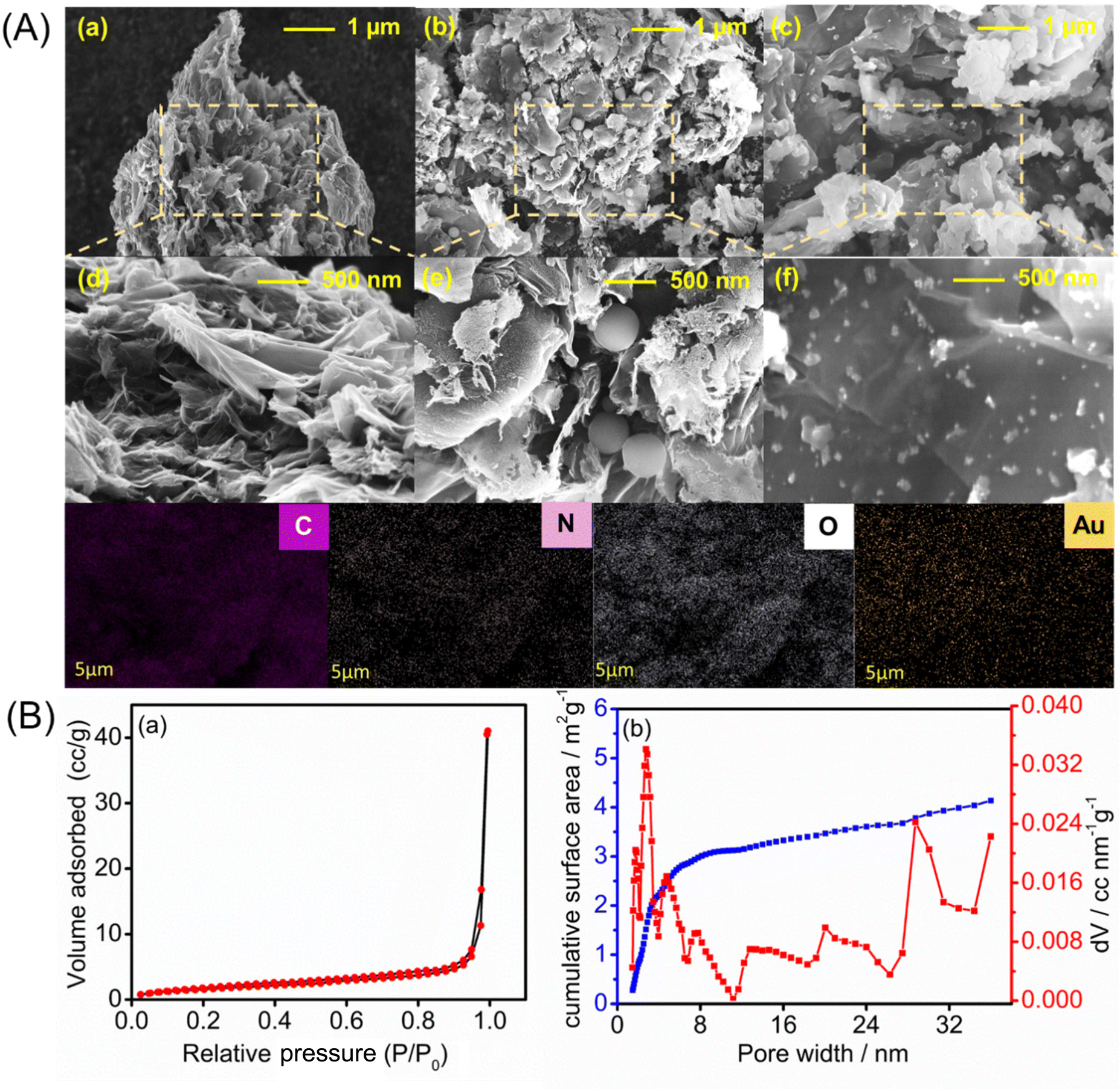 | ||
| Fig. 3 (A) FE-SEM images of PrGO at 1 μm (a) and 500 nm (d), PrGO–PPy at 1 μm (b) and 500 nm (e), and PrGO–PPy–Au at 1 μm (c) and 500 nm (f). (B) Specific surface area and pore size characterization: (a) adsorption/desorption isotherm and (b) DFT pore distribution. | ||
The porous and wrinkled structure of rGO aids in preventing the aggregation of GO and preserving a high surface area. The observed imperfections and irregularities in rGO are due to the presence of oxygen-containing functional groups on the surfaces and edges of the rGO nanosheets.62 On the surface of the porous rGO, a layer of PPy forms, with round-shaped PPy particles penetrating into the pores, which is clearly visible in the SEM images in Fig. 3A(b and e), while the rGO–PPy composite is wrapped on the surface of porous rGO. As shown in Fig. S2,† PPy nanospheres have smooth spherical surfaces. Some are loosely dispersed and hidden under rGO, while others cluster on its surface. The PPy nanospheres are wrapped in rGO, connecting through it as a medium. Unlike pure rGO, which shows severe crimping and aggregation due to strong π–π restacking after removing surface functional groups, PrGO–PPy displays less crimping and aggregation. This suggests that PPy nanospheres act as barriers, preventing the restacking of rGO.63 FE-SEM images of the PrGO–PPy–AuNPs nanocomposite are shown in Fig. 3A(c) and A(f). These results suggest the uniform dispersion and formation of AuNPs on the PrGO–PPy sheets, providing clear evidence of their interaction. The FE-SEM images of the NCs show that spherical AuNPs are homogeneously immobilized on the surface of PrGO–PPy. Moreover, dense Au nanoparticles were uniformly distributed across the PrGO–PPy networks. Moreover, energy-dispersive X-ray spectroscopy (EDX) reveals the presence of C, N, O, and Au elements on the modified electrode, confirming the successful creation of PrGO–PPy–Au nanocomposites (Fig. S3†).
Conformational and compositional changes in rGO and rGO–PPy are confirmed by UV–Vis spectroscopy. The π–π* transitions of the aromatic CC bonds in rGO are observed at 252 nm.64 The spectrum of PPy shows an absorption peak at 415 nm, corresponding to the π–π* transition in the conjugated polymer backbone or bipolaronic state. In contrast, rGO–PPy exhibits a broad absorption band in the range of 300–500 nm,65 indicating noncovalent interactions between PPy and the rGO sheets. This is evidenced by the shift of the characteristic PPy peak from 415 nm to a longer wavelength,66 suggesting the formation of the rGO–PPy composite52 Due to noncovalent interactions within the composite, the characteristic peak of PPy (415 nm) shifts to a longer wavelength (Fig. S4†).
The FTIR spectrum of porous rGO reveals several characteristic peaks: a broad, intense peak at 3400 cm−1 corresponding to O–H stretching, a peak at 1545 cm−1 for CC stretching, and another at 1190 cm−1 for C–C stretching. Additionally, the spectrum shows peaks at 1735 cm−1 and 1092 cm−1, attributed to the CO stretching of carbonyl groups and the C–O stretching of epoxy groups, respectively. In the PrGO–PPy–Au nanocomposite, a notable peak at 1574 cm−1 is observed, likely due to the contribution from aromatic CC stretching.67 Weak peaks at 1710 cm−1 and 1385 cm−1 in rGO are indicative of red-shifted carbonyl (CO) and carboxyl (C–O) stretching vibrations resulting from π–π interactions between rGO sheets and PPy nanoparticles52 (Fig. S5†).
The porous nature of the rGO materials was characterized using the BET nitrogen adsorption–desorption isotherm, as shown in Fig. 3B(a). The analysis revealed a BET surface area of approximately 6.067 m2 g−1. Furthermore, the non-local density functional theory (NLDFT) equilibrium model for N2 adsorption at 77 K, conducted over a relative pressure range of 0.0000–1.0000, demonstrated that the rGO materials exhibit a significant degree of porosity (Fig. 3B(b)). The average pore volume was found to be 0.018 cc g−1 with the pore structure predominantly within the mesoporous range, having an average pore width of 20.9 nm. Notably, around 90% of the surface area comprises pores with widths ≤25 nm. These results underscore the rGO's well-defined mesoporous structure,68 enhancing its suitability for various applications due to its uniform and controlled pore sizes.
From the DFT study, we concluded that most of the pores are mesoporous. Therefore, we conducted a BJH study (Fig. S6†), which is specifically suited for analysing mesoporous materials. The BJH results indicated that most pores fall within the 0–20 nm range. In contrast, DFT analysis revealed a broader pore size distribution, extending beyond 20 nm. This discrepancy suggests that the rGO material possesses a more complex pore structure, with a significant proportion of larger mesopores that the BJH method may not fully capture. The complementary use of both DFT and BJH analyses provides a complete view of the pore distribution, enhancing our understanding of the material's potential applications. The porous characteristics of the PrGO–PPy–Au composite were analyzed using BET nitrogen adsorption–desorption isotherms, as illustrated in Fig. S7(a).† The material exhibited a BET surface area of approximately 14.135 m2 g−1, confirming the significant porosity of the PrGO–PPy–Au composite, as shown in Fig. S7(b).† The composite displayed an average pore volume of 0.00258 cc g−1 and predominantly microporous characteristics with an average pore width of 3.19 nm. Notably, about 90% of the surface area comprised pores with widths ≤15 nm. These findings highlight the composite's well-defined micro–mesoporous structure with uniform and controlled pore sizes, enhancing its potential for various advanced applications.
DFT analysis further revealed that most of the pores in the final composite were in the microporous range. A comparison of the porous rGO and the final PrGO–PPy–Au composite indicated that the final composite exhibited a higher surface area but a noticeable shift in pore distribution from mesoporous to microporous. This transition suggests that the incorporation of polypyrrole into the PrGO structure filled most of the mesopores, leading to reduced mesoporosity in the final material.
3.4. Optimization of parameters
Optimizing parameters such as the amount of the transducer layer, solvents, and solution pH is crucial for developing a sensitive sensing method. The transducer layer was refined by applying different volumes of PrGO–PPy–Au to the working electrode. 1 mg mL−1 solution of PrGO–PPy–Au in chitosan was drop-cast onto an SPCE, which had been pretreated with acidic and basic solutions to eliminate impurities and introduce functional groups on the carbon edges. The amino groups in the PPy polymers and graphene layers interacted with the electrode surface, enhancing stability. To create a single layer, 1 μL of the composite solution was drop-cast onto the working electrode. For comparison purposes, varying numbers of layers double, triple, and quadruple of PrGO–PPy–Au were applied. The triple layer showed the highest current response for detecting MTX (Fig. S11A†). For solvent optimization, we used chitosan, ethanol, and water. The composite showed high current in the chitosan solution (Fig. S11B†).Attaining the optimal pH is essential for effective analytical performance. We assessed the sensor's performance across a range of pH levels from 3.0 to 9.0, with the resulting voltammograms presented in Fig. S12A† and the peak current versus pH data depicted in Fig. S12B.† These results indicate that the electrooxidation peaks of MTX were influenced by the buffer's pH. At higher pH values, MTX exhibited a slightly higher current response at lower potential, whereas at lower pH values, the oxidation peak potential shifted towards a more positive potential. Specifically, pH levels 3, 4, 5, and 6 showed slightly higher potential values compared to pH 7, 8, and 9. Building on this, the dual potential shifts observed in the pH study of methotrexate further highlight the role of protonation and deprotonation in modulating its electrochemical behaviour. Under acidic conditions, the predominant protonation of methotrexate increases its positive charge density, thereby facilitating electron transfer at lower applied potential and causing a shift in the reduction or oxidation peak towards lower potential. In contrast, under basic conditions, deprotonation reduces the positive charge density, necessitating higher energy for electron transfer, which shifts the peaks to higher potential. This behavior aligns with the Nernst equation, linking potential to the concentration of protonated and deprotonated species, and may also depends on pH. Notably, pH 7 exhibited a comparatively higher current response at a lower oxidation potential than other pH levels.66,67 Considering both the current and potential, pH 7 8 and 9 yielded the best results. Considering its physiological significance and the lower potential required, pH 7.4 was chosen as the optimal condition for subsequent electrochemical analysis.
3.5. Electrode and electrolyte interface characterization
Metal nanoparticle composites, especially those combining polymers with carbon-based materials, are gaining attention for their exceptional properties. Gold nanoparticle composites, with their superior conductivity, biocompatibility, and mechanical strength, enhance electrochemical sensors by promoting electron transfer and redox reactions.The interfacial properties between the electrode and the electrolyte were investigated using CV, SWV, and EIS techniques. CV experiments were conducted on various transducer electrodes in a K3[Fe(CN)6] solution at a scan rate of 100 mV s−1, with the resulting voltammograms presented in Fig. 4A. The results indicated that anodic and cathodic peaks were observed in both the modified and unmodified transducer electrodes. Notably, the PrGO–PPy–Au-modified electrode showed a stronger peak current response and a narrower peak potential separation than the bare SPCE electrode. This suggests that the integration of Au nanoparticles and PPy onto the transducer surface enhances the electron transfer rate between the electrode and the electrolyte. This behaviour was additionally verified using EIS analysis of the electrode–electrolyte interface. The modified electrode demonstrates a distinct advantage over the bare electrode, exhibiting an oxidation peak at a significantly lower potential range in the CV analysis.
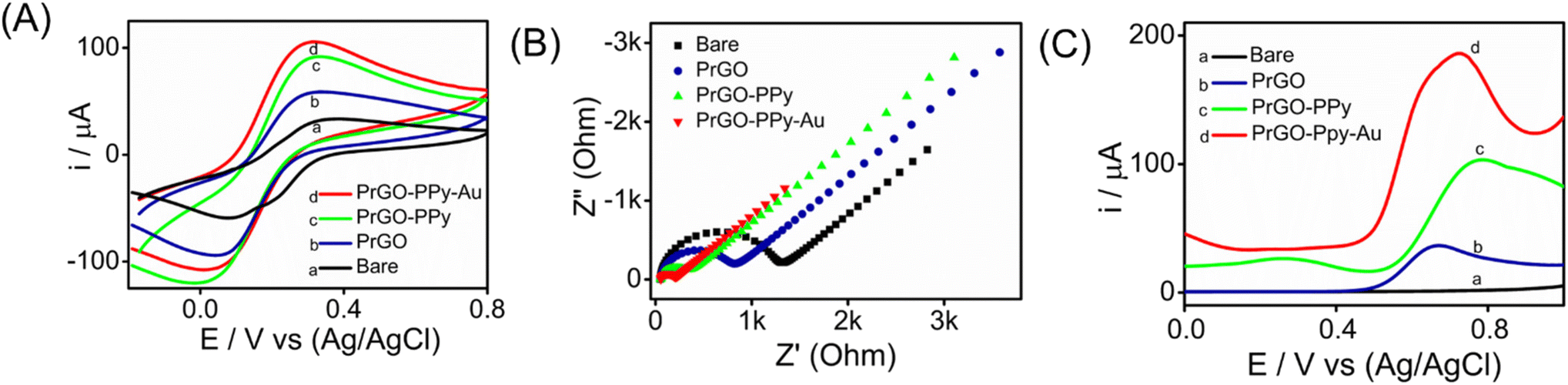 | ||
| Fig. 4 (A) CV responses of different modified electrodes: (a) bare, (b) PrGO, (c) PrGO–PPy, and (d) PrGO–PPy–Au obtained in a solution containing 2 mM K3[Fe(CN)6] and 0.1 M KCl. The potential was applied between −0.1 V and 0.8 V at a scan rate of 100 mV s−1. (B) Nyquist plots obtained for the SPCE and various modified electrodes in an aqueous solution of 2 mM K3[Fe(CN)6] and 0.1 M KCl. (C) SWV responses of various electrodes: (a) bare, (b) PrGO, (c) PrGO–PPy, and (d) PrGO–PPy–Au recorded at a methotrexate (MTX) concentration of 25 μM. | ||
The experiments were conducted in a 2 mM K3[Fe(CN)6] solution with 0.1 M KCl buffer at the open-circuit potential, spanning a frequency range from 100 kHz to 10 MHz for both the modified and unmodified transducer electrode surfaces. The Nyquist plots obtained, as shown in Fig. 4B, reveal semicircular patterns for both electrodes and were analyzed using a basic Randles equivalent circuit (Fig. S8†), with the Rct values listed in Table S1.† The data reveal that the modified sensor (SPCE/PrGO–PPy–Au) exhibited a significantly lower Rct value (132 Ω) compared to the unmodified electrode (bare SPCE) with an Rct value of 1133 Ω. These findings emphasize the excellent conductivity and catalytic properties of the hybrid nanocomposite transducer made from graphene, gold nanoparticles, and a conducting polymer. Additionally, the PrGO and PrGO–PPy electrodes showed Rct values of 650 Ω and 244 Ω, respectively, indicating less resistance compared to the bare electrode. Fig. 4C illustrates the SWV responses of the modified electrode in comparison to different working electrodes for detecting 25 μM methotrexate. The PrGO–PPy–Au electrode demonstrated efficient and higher current with a lower potential input, meaning that less potential was required for the oxidation of methotrexate.
CV analysis was performed at various scan rates (10–100 mV s−1) for SPCE/PrGO–PPy–Au in a 2 mM ferri–ferrocyanide solution over a potential range of −0.2 to 0.9 V. The results showed well-defined oxidation (ipa) and reduction (ipc) peak currents, both of which increased proportionally with the scan rate. The CV curve is depicted in Fig. S10A of the ESI.† The calibration graph displaying peak current against the square root of the scan rate, shown in Fig. S10B,† exhibited a linear correlation with an R2 value of 0.9985 for ipa, indicating that the electrochemical oxidation of ferricyanide is diffusion controlled. The electroactive surface area of the modified electrode was calculated using the Randles–Sevcik equation, as described in the ESI.†
3.6. Electroanalytical capabilities of the SPCE/PrGO–PPy–Au sensor for MTX detection in PBS
The analytical performance of the developed SPCE/PrGO–PPy–Au sensor for detecting MTX was assessed by recording cyclic voltammetry (CA) and square wave voltammetry (SWV) in a phosphate-buffered saline (PBS) solution at pH 7.4.Fig. 5 illustrates the MTX measurements in the buffer at this pH using SWV and CA across different MTX concentration ranges. SWV (Fig. 5A) effectively detects MTX within a range of 165 nM–1.41 μM, achieving a strong correlation (R2 = 0.994) with a lowest detection limit of 1.8 nM. CA (Fig. 5B) detects MTX in the range of 130 nM–950 nM, with an R2 = 0.9761 having an LOD of 25 nM. These voltammograms clearly show that the PrGO–PPy–Au film enhances the current response to MTX. To demonstrate the sensor's continuous monitoring capability for industrial quality control applications, long-term stability studies were conducted for 1.3 μM MTX detection using SWV (Fig. 5C) and 1.3 μM MTX using CA (Fig. 5D) at room temperature over 4 hours, with readings taken every 10 minutes. Throughout these stability studies, the modified SPCE/PrGO–PPy–Au electrode maintained stable current responses.
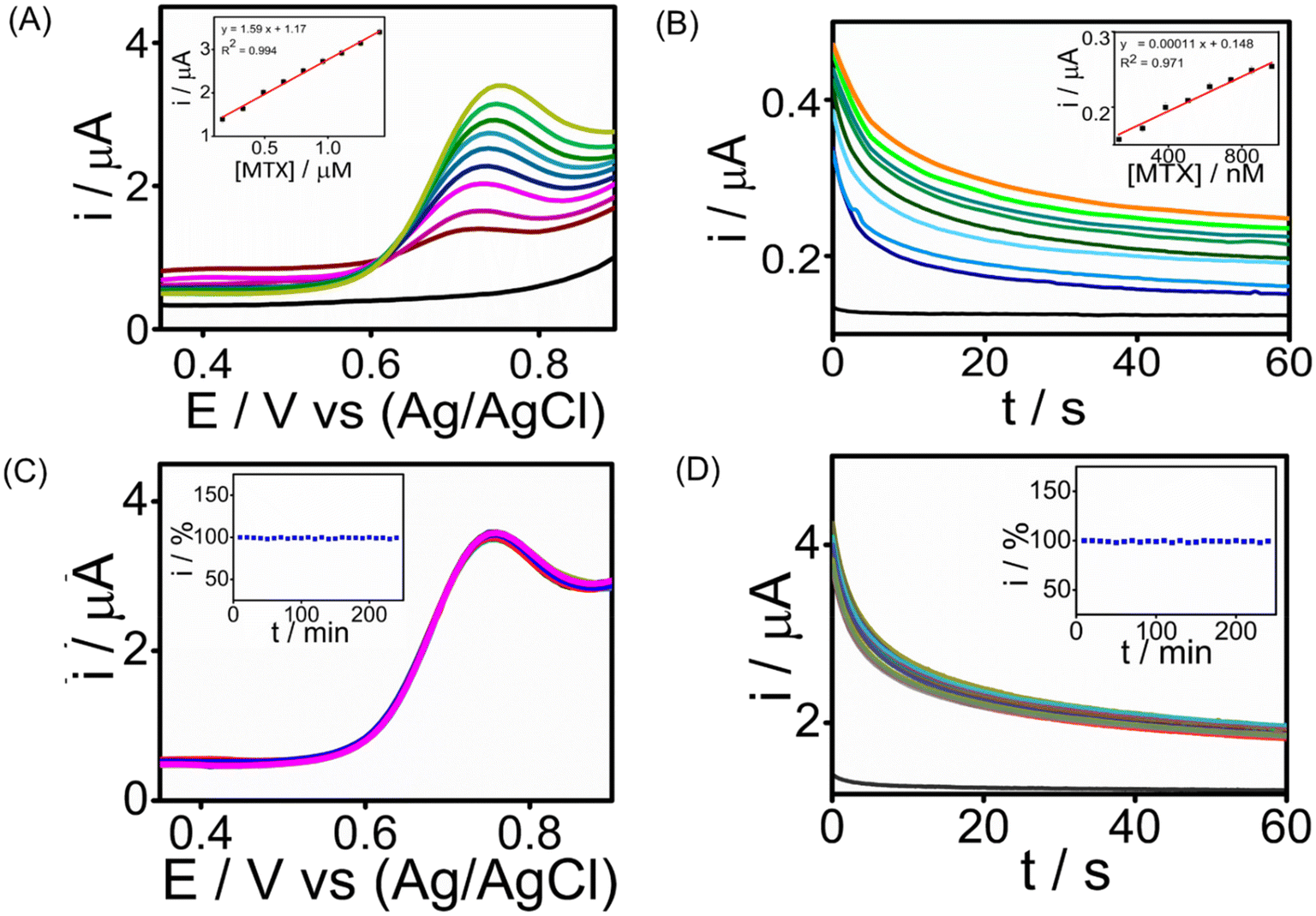 | ||
| Fig. 5 Electroanalytical performance of the SPCE/PrGO–PPy–Au sensor in PBS. (A) SWV responses recorded in PBS (pH 7.4) for MTX detection spanning a concentration range of 165 nM–1.41 μM. (B) Chronoamperometric measurements of MTX in PBS (pH 7.4) performed over a concentration range of 130 nM–950 nM. (C) Stability of MTX with a concentration of 1.3 μM in PBS (pH 7.4) evaluated for 4 hours using SWV with measurements taken at 10-minute intervals. (D) Additional stability test for 1.3 μM MTX in PBS (pH 7.4) conducted for 4 hours with chronoamperometric measurements recorded every 10 minutes (all inset graphs show the corresponding calibration curves). | ||
3.7. Determination of MTX in human serum
To evaluate the practicality of our electrochemical sensor, we assessed the ability of the SPCE/PrGO–PPy–Au electrode to detect MTX in human serum. In this experiment, the electrochemical sensor was evaluated using 98% human serum purchased from Sigma-Aldrich, which was diluted with phosphate-buffered saline (PBS) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. This dilution was made to minimize potential interference from the serum's components while maintaining the physiological relevance of the sample for testing the sensor's performance. We conducted chronoamperometry and square wave voltammetry tests for MTX detection, and the results depicted in Fig. 6A & B show MTX readings in human serum using SWV (R2 = 0.991) and CA (R2 = 0.995), respectively, achieving the lowest detection limit of 0.4 nM in SWV and 1 nM in CA. The sensitivity of the sensor was found to be 24.1 μA μM−1 cm−2 for SWV and 11 μA μM−1 cm−2 for CA, respectively (ESI†). The peak current consistently increased with higher MTX concentrations, underscoring the potential of the graphene–Au-integrated conducting polymer screen-printed sensor strip for point-of-care MTX analysis in healthcare settings. Long-term stability tests were performed for detecting 1.3 μM MTX using chronoamperometry (CA) and square wave voltammetry (SWV) over a 4-hour period at room temperature with measurements taken every 10 minutes (Fig. 6C and D). During these tests, the modified SPCE/PrGO–PPy–Au electrode consistently delivered stable current responses. When compared with recent studies (Table S2 in the ESI†), our sensor demonstrated a comparable electroanalytical performance, featuring a lower detection limit than other reported methods. Importantly, our sensor transducer showed exceptional stability over 4 hours in real serum samples, a characteristic not previously reported in the literature.
:1 ratio. This dilution was made to minimize potential interference from the serum's components while maintaining the physiological relevance of the sample for testing the sensor's performance. We conducted chronoamperometry and square wave voltammetry tests for MTX detection, and the results depicted in Fig. 6A & B show MTX readings in human serum using SWV (R2 = 0.991) and CA (R2 = 0.995), respectively, achieving the lowest detection limit of 0.4 nM in SWV and 1 nM in CA. The sensitivity of the sensor was found to be 24.1 μA μM−1 cm−2 for SWV and 11 μA μM−1 cm−2 for CA, respectively (ESI†). The peak current consistently increased with higher MTX concentrations, underscoring the potential of the graphene–Au-integrated conducting polymer screen-printed sensor strip for point-of-care MTX analysis in healthcare settings. Long-term stability tests were performed for detecting 1.3 μM MTX using chronoamperometry (CA) and square wave voltammetry (SWV) over a 4-hour period at room temperature with measurements taken every 10 minutes (Fig. 6C and D). During these tests, the modified SPCE/PrGO–PPy–Au electrode consistently delivered stable current responses. When compared with recent studies (Table S2 in the ESI†), our sensor demonstrated a comparable electroanalytical performance, featuring a lower detection limit than other reported methods. Importantly, our sensor transducer showed exceptional stability over 4 hours in real serum samples, a characteristic not previously reported in the literature.
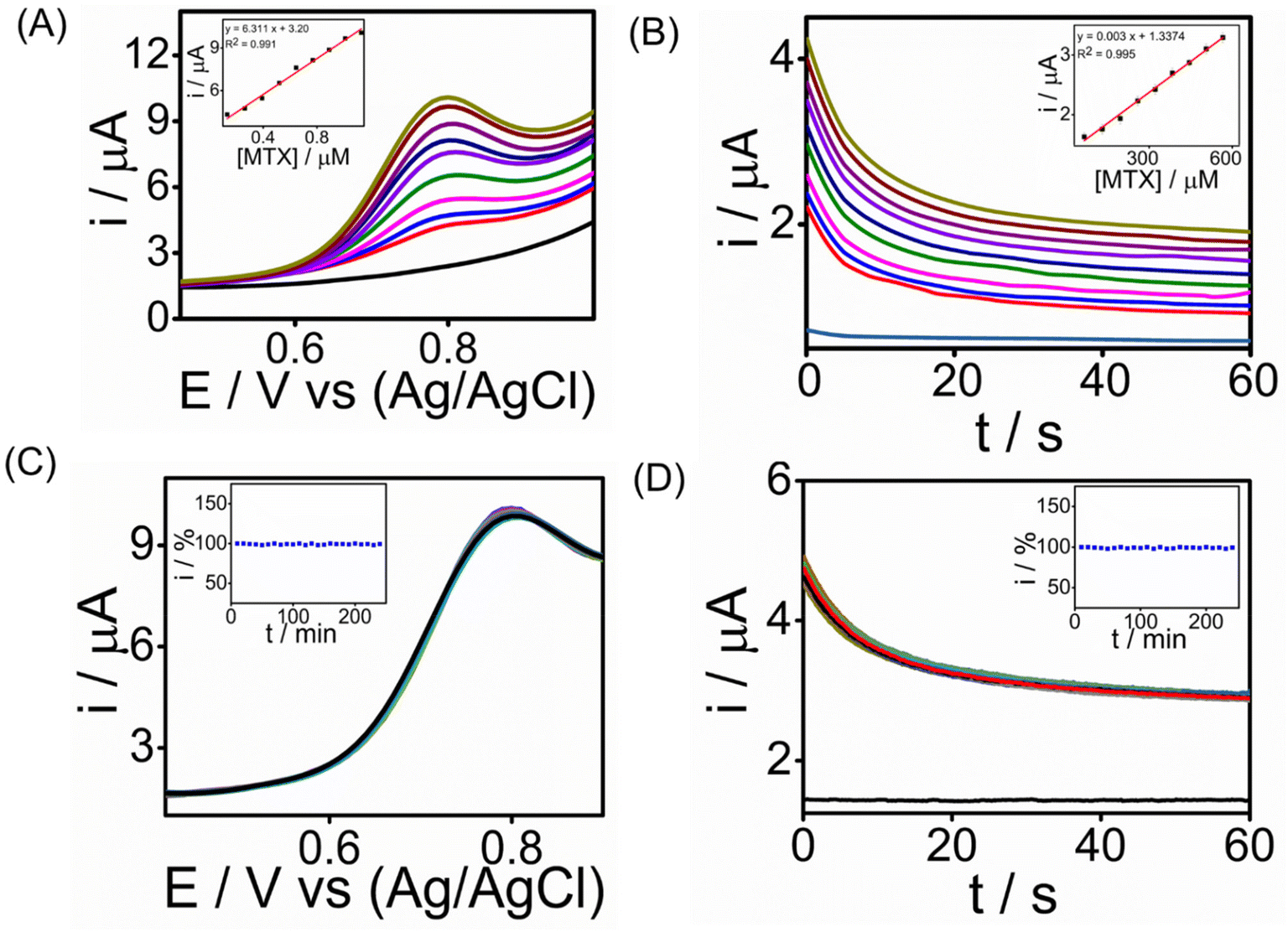 | ||
| Fig. 6 Electroanalytical performance of the SPCE/PrGO–PPy–Au sensor in serum. (A) SWV analysis of MTX in serum (pH 7.4) conducted over a concentration range of 130 nM–1 μM. (B) Chronoamperometric measurements of MTX in serum (pH 7.4) recorded for a concentration range of 65 nM–565 nM. (C) Stability studies of MTX with a concentration of 1.3 μM in serum (pH 7.4) conducted for 4 hours with measurements taken every 10 minutes. (D) Stability studies of MTX with a concentration of 1.3 μM in serum (pH 7.4) for 4 hours with readings taken every 10 minutes. (The corresponding calibration curves are displayed in all the inset graphs.) | ||
3.8. Selectivity, reproducibility, and long-term storage stability of the sensor
Assessing the selectivity of an electrochemical sensor is crucial for accurate target analyte detection without interference. To evaluate the selectivity of the SPCE/PrGO–PPy–Au sensor for MTX detection, SWV measurements were performed in the presence of 5 μM dopamine (DA) (Fig. 7A). The findings revealed that methotrexate exhibited a strong SWV response across different concentrations (0, 1.0, 2.0, and 3.0 μM) even in the presence of dopamine. Furthermore, CA (Fig. 7B) and SWV (Fig. S13†) measurements were conducted with common biological interferents, including uric acid (UA), ascorbic acid (AA), folic acid (FA), fluorouracil (FU), and dopamine (DA), each at a concentration of 10 μM. In this study, methotrexate, a folic acid analogue, was tested for its potential as an interfering molecule. Fluorouracil, another anticancer drug, was also included to assess its interference effects (Fig. 7B). The concentration of methotrexate used in this study was 2 μM. Fig. 7C and D present the CA responses of the MTX sensor at 1 μM in the presence of 5 μM interfering molecules in serum, along with the corresponding bar graph. The chronoamperometry analysis demonstrated that these substances did not interfere with methotrexate detection. Reproducibility and long-term storage stability are vital for enhancing electrochemical sensor performance. We evaluated the sensor's reproducibility by independent electrodes for MTX detection at different concentrations (100 and 500 nM) using CA (Fig. 7E). The results demonstrate that the sensors exhibited consistent responses with a standard deviation of 1%. In addition, the long-term stability of the sensor was evaluated by storing it at room temperature (25 °C) for three weeks, followed by testing its ability to detect 200 nM MTX. We have achieved the regeneration capability after performing 20 scans of the cyclic voltammograms in PBS buffer solution. This indicates the good regeneration capability of the sensor interface. Fig. 7F illustrates the long-term stability of the sensor, showing that it maintained stable performance over a prolonged storage period of 4 weeks at 5 °C. The sensors exhibited consistent responses throughout the evaluation, with only a slight decrease observed in the CA signals, indicating minimal performance degradation and reliable stability under refrigerated conditions.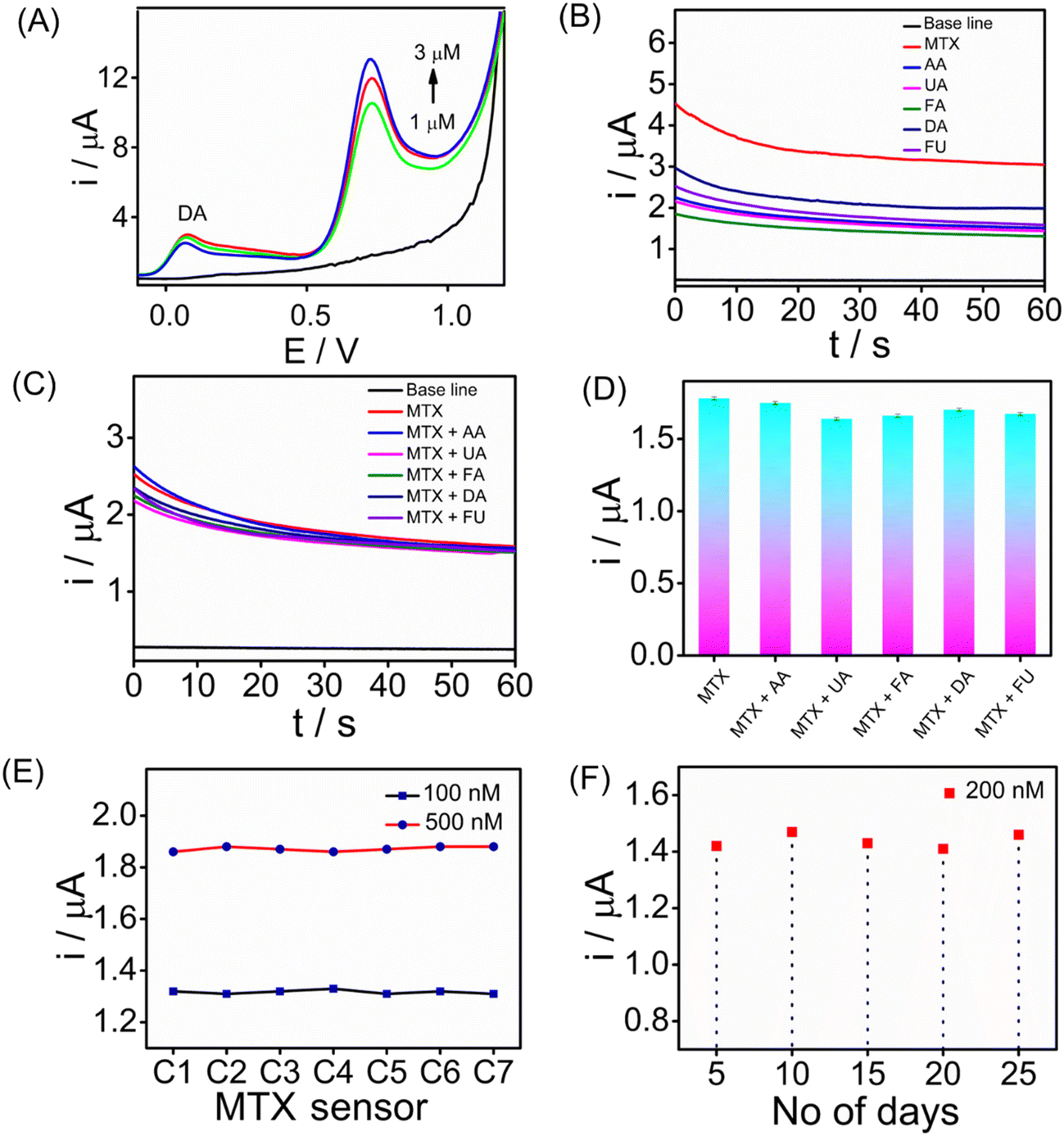 | ||
| Fig. 7 (A) SWV analysis of human serum with varying concentrations of methotrexate (0, 1.0, 2.0, and 3.0 μM) in the presence of DA (5 μM) measured against an Ag/AgCl reference electrode. (B) Chronoamperometric study of MTX (2 μM) and an interfering molecule with five times the concentration of MTX (10 μM). (C) Chronoamperometric responses of the MTX (1 μM) sensor in serum containing 5 μM of each interfering molecule. (D) Corresponding bar chart showing the results. (E) Chronoamperometric responses of seven independent SPCE/PrGO–PPy–Au sensors for MTX detection at concentrations of 100 nM and 500 nM. (F) Chronoamperometric responses of the MTX sensor for 25 days at room temperature for detecting 200 nM MTX. | ||
4 Conclusions
Electrochemical methods for detecting methotrexate are favoured due to its structure, which contains amino and carboxylic groups that can undergo oxidation or reduction. Modifying the electrode with specialized materials can significantly boost the sensor's selectivity and sensitivity to methotrexate. In this study, we successfully improved disposable SPCEs by incorporating PrGO–PPy–Au through a green synthesis and chemical process. The modified electrode was then assessed for its capability to electrochemically detect the anticancer drug MTX. The PrGO–PPy–Au hybrid nanocomposite ink was created by dispersing the synthesized black powder in a freshly prepared chitosan solution. This mixture was then drop-cast onto thoroughly cleaned SPCEs in varying volumes (1, 2, 3, and 4 μL) to optimize the transducer surface. The sensor's interfacial properties were characterized using CV, EIS, and SWV techniques. Additionally, the modified SPCE/PrGO–PPy–Au sensor was evaluated for MTX detection using SWV (covering a broad linear range from 165 nM to 1.41 μM) and CA (covering a broad linear range from 130 nM to 950 nM), achieving an LOD of 1.8 nM in PBS solution via SWV. The sensor's performance was further validated using human serum samples, achieving an LOD of 0.4 nM in SWV analysis with a broad linear range from 130 nM to 1 μM. The sensor's sensitivity was determined using SWV and CA techniques, yielding values of 24.1 μA μM−1 for SWV and 11 μA μM−1 for CA in serum. Even in the presence of biological interfering molecules, the modified sensor successfully detected MTX at nanomolar concentrations, showing high sensitivity, excellent reproducibility, and stability over the modified electrode. The PrGO–PPy–Au sensor, with its straightforward design and impressive electrochemical properties, holds significant promise for continuous MTX monitoring.Author contributions
The manuscript was written with contributions from all authors. All authors have given approval to the final version of the manuscript.Data availability
Data files can be shared on reasonable request.Conflicts of interest
The authors state that there are no financial interests or personal relationships that could have influenced the work presented in this paper.Acknowledgements
Yugender Goud Kotagiri acknowledges the financial support from the Department of Science and Technology, Science and Engineering Research Board (DST-SERB/RJF/2021/000113, Ramanujan Board), and MNRE project no.: 350/2/2018NT. The authors thank the Indian Institute of Technology Palakkad for financial support and research facilities. The authors acknowledge the central instrumentation facility at Palakkad for chemical and materials characterizations.References
- H. Chen, Y. Li, H. Li, X. Chen, H. Fu, D. Mao, W. Chen, L. Lan, C. Wang, K. Hu, J. Li, C. Zhu, I. Evans, E. Cheung, D. Lu, Y. He, A. Behrens, D. Yin and C. Zhang, Nature, 2024, 631, 663–669 CrossRef PubMed.
- B. Liu, H. Zhou, L. Tan, K. T. H. Siu and X.-Y. Guan, Signal Transduction Targeted Ther., 2024, 9, 175 CrossRef.
- U. Anand, A. Dey, A. K. S. Chandel, R. Sanyal, A. Mishra, D. K. Pandey, V. De Falco, A. Upadhyay, R. Kandimalla, A. Chaudhary, J. K. Dhanjal, S. Dewanjee, J. Vallamkondu and J. M. Pérez De La Lastra, Genes Dis., 2023, 10, 1367–1401 CrossRef PubMed.
- S. Wang, Z. Qi, H. Huang and H. Ding, Anal. Lett., 2012, 45, 1658–1669 CrossRef.
- K. M. Hamed, I. M. Dighriri, A. F. Baomar, B. T. Alharthy, F. E. Alenazi, G. H. Alali, R. H. Alenazy, N. T. Alhumaidi, D. H. Alhulayfi, Y. B. Alotaibi, S. S. Alhumaidan, Z. A. Alhaddad, A. A. Humadi, S. A. Alzahrani and R. H. Alobaid, Cureus, 2022, 14, 29518 Search PubMed.
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef PubMed.
- F. Karami, S. Ranjbar, Y. Ghasemi and M. Negahdaripour, J. Pharm. Anal., 2019, 9, 373–391 CrossRef.
- P. Jariwala, V. Kumar, K. Kothari, S. Thakkar and D. D. Umrigar, Case Rep. Dermatol. Med., 2014, 2014, 1–3 Search PubMed.
- H. Alessa, J. Umm Al-Qura Univ. Appl. Sci., 2023, 9, 378–390 CrossRef.
- S. Qiu, Y. Cai, H. Yao, C. Lin, Y. Xie, S. Tang and A. Zhang, Signal Transduction Targeted Ther., 2023, 8, 132 CrossRef.
- D. R. Cunha, M. A. Segundo and M. B. Quinaz, Biosens. Bioelectron., 2024, 116988 Search PubMed.
- H. Aboleneen, J. Simpson and D. Backes, J. Chromatogr. B: Biomed. Sci. Appl., 1996, 681, 317–322 CrossRef PubMed.
- A. Mohammadzadeh, A. Jouyban, M. Hasanzadeh, V. Shafiei-Irannejad and J. Soleymani, Anal. Methods, 2021, 13, 4280–4289 RSC.
- N. Q. Febrianti, M. G. R. Tunggeng, I. D. Ramadhany, R. M. Asri, Y. Y. Djabir and A. D. Permana, Spectrochim. Acta, Part A, 2024, 315, 124258 CrossRef PubMed.
- L. C. Falk, D. R. Clark, S. M. Kalman and T. F. Long, Clin. Chem., 1976, 22, 785–788 CrossRef.
- S. Chen, Z. Zhang, D. He, Y. Hu, H. Zheng and C. He, Luminescence, 2007, 22, 338–342 CrossRef PubMed.
- C.-Y. Kuo, H.-L. Wu, H.-S. Kou, S.-S. Chiou, D.-C. Wu and S.-M. Wu, J. Chromatogr., A, 2003, 1014, 93–101 CrossRef PubMed.
- F. Gao, C. Liu, L. Zhang, T. Liu, Z. Wang, Z. Song, H. Cai, Z. Fang, J. Chen, J. Wang, M. Han, J. Wang, K. Lin, R. Wang, M. Li, Q. Mei, X. Ma, S. Liang, G. Gou and N. Xue, Microsyst. Nanoeng., 2023, 9, 1 CrossRef PubMed.
- F. A. Castelli, G. Rosati, C. Moguet, C. Fuentes, J. Marrugo-Ramírez, T. Lefebvre, H. Volland, A. Merkoçi, S. Simon, F. Fenaille and C. Junot, Anal. Bioanal. Chem., 2022, 414, 759–789 CrossRef PubMed.
- E. Jara-Cornejo, S. Khan, J. Vega-Chacón, A. Wong, L. C. Da Silva Neres, G. Picasso and M. D. P. T. Sotomayor, Biomimetics, 2023, 8, 77 CrossRef PubMed.
- A. Mir, M. Shabani-Nooshabadi and N. Ziaie, Chemosphere, 2023, 338, 139427 CrossRef PubMed.
- M. Zamani, N. Tavakkoli and N. Soltani, Diamond Relat. Mater., 2023, 136, 109954 CrossRef.
- V. Marchianò, A. Tricase, M. Caputo, E. Farinini, R. Leardi, A. Imbriano, D. Leech, R. Kidayaveettil, L. Gentile, L. Torsi, E. Macchia and P. Bollella, Chem. Mater., 2024, 36, 358–370 CrossRef.
- M. Hosseine, S. M. Naghib and A. Khodadadi, Sci. Rep., 2024, 14, 11928 CrossRef PubMed.
- E. B. Bahadır and M. K. Sezgintürk, TrAC, Trends Anal. Chem., 2016, 76, 1–14 CrossRef.
- S. M. Monavari, F. Marsusi, N. Memarian and M. Qasemnazhand, Sci. Rep., 2023, 13, 3118 CrossRef PubMed.
- K. Białas, D. Moschou, F. Marken and P. Estrela, Microchim. Acta, 2022, 189, 172 CrossRef PubMed.
- S. Bilge, B. Dogan-Topal, M. M. Gürbüz, S. A. Ozkan and A. Sınağ, Microchim. Acta, 2024, 191, 240 CrossRef PubMed.
- K. Rana, H. Kaur, N. Singh, T. Sithole and S. S. Siwal, Next Mater., 2024, 3, 100107 CrossRef.
- P. Kumar, M. Šilhavík, Z. A. Zafar and J. Červenka, J. Phys. Chem. C, 2023, 127, 10599–10608 CrossRef PubMed.
- A. Iakunkov and A. V. Talyzin, Nanoscale, 2020, 12, 21060–21093 RSC.
- S. Lin, J. Tang, K. Zhang, Y. Chen, R. Gao, H. Yin and L.-C. Qin, Nanoscale Adv., 2023, 5, 1163–1171 RSC.
- A. Devendran and A. Nagai, Mater. Adv., 2023, 4, 2524–2543 RSC.
- D. K. Padhi, T. K. Panigrahi, K. Parida, S. K. Singh and P. M. Mishra, ACS Sustainable Chem. Eng., 2017, 5, 10551–10562 CrossRef.
- B. Anegbe, I. H. Ifijen, M. Maliki, I. E. Uwidia and A. I. Aigbodion, Environ. Sci. Eur., 2024, 36, 15 CrossRef.
- H. Lin, T. Buerki-Thurnherr, J. Kaur, P. Wick, M. Pelin, A. Tubaro, F. C. Carniel, M. Tretiach, E. Flahaut, D. Iglesias, E. Vázquez, G. Cellot, L. Ballerini, V. Castagnola, F. Benfenati, A. Armirotti, A. Sallustrau, F. Taran, M. Keck, C. Bussy, S. Vranic, K. Kostarelos, M. Connolly, J. M. Navas, F. Mouchet, L. Gauthier, J. Baker, B. Suarez-Merino, T. Kanerva, M. Prato, B. Fadeel and A. Bianco, ACS Nano, 2024, 18, 6038–6094 CrossRef PubMed.
- S. Lam, F. Wang, S. P. Yeap, K. H. Teng, A. Shaw, V. Karunamoothei, V. Khosravi and C. Liu, ChemSusChem, 2024, e202400845 CrossRef.
- M. Gao, X. Li, D. Qi and J. Lin, ACS Omega, 2020, 5, 32706–32714 CrossRef.
- J. Mahmood, N. Arsalani, S. Naghash-Hamed, Z. Hanif and K. E. Geckeler, Sci. Rep., 2024, 14, 11653 CrossRef PubMed.
- K. Namsheer and C. S. Rout, RSC Adv., 2021, 11, 5659–5697 RSC.
- S. Lee, Sci. Rep., 2024, 14, 3883 CrossRef PubMed.
- Z. Liu, Z. Gao, L. Xu and F. Hu, RSC Adv., 2020, 10, 17524–17533 RSC.
- N. Ratsameetammajak, T. Autthawong, K. Khunpakdee, M. Haruta, T. Chairuangsri and T. Sarakonsri, Polymers, 2023, 15, 4638 CrossRef.
- J. Xue, Q. Sun, Y. Zhang, W. Mao, F. Li and C. Yin, ACS Omega, 2020, 5, 10995–11004 CrossRef.
- B. Wu, N. Zhao, S. Hou and C. Zhang, Nanomaterials, 2016, 6, 220 CrossRef.
- N. Kavitha, M. Elavarasan, R. Ramachandran, S. Uthayakumar, A. Chandramohan and K. Dinakaran, Curr. Res. Green Sustainable Chem., 2022, 5, 100316 CrossRef.
- M. Pavličková, L. Lorencová, M. Hatala, M. Kováč, J. Tkáč and P. Gemeiner, Sci. Rep., 2022, 12, 11900 CrossRef.
- G. Adedokun, M. Alipanah and Z. H. Fan, Lab Chip, 2024, 24, 3626–3650 RSC.
- G. B. Mahendran, S. J. Ramalingam, J. B. B. Rayappan, S. Kesavan, T. Periathambi and N. Nesakumar, J. Mater. Sci.: Mater. Electron., 2020, 31, 14345–14356 CrossRef.
- B. Demirkan, S. Bozkurt, K. Cellat, K. Arıkan, M. Yılmaz, A. Şavk, M. H. Çalımlı, M. S. Nas, M. N. Atalar, M. H. Alma and F. Sen, Sci. Rep., 2020, 10, 2946 CrossRef PubMed.
- S. P. Lim, N. M. Huang and H. N. Lim, Ceram. Int., 2013, 39, 6647–6655 CrossRef.
- S. P. Lim, A. Pandikumar, Y. S. Lim, N. M. Huang and H. N. Lim, Sci. Rep., 2014, 4, 5305 CrossRef PubMed.
- Y. Wang, Y. Du, B. Wu, B. Han, S. Dong, X. Han and P. Xu, Polymers, 2018, 10, 998 CrossRef PubMed.
- S. Some, Y. Kim, Y. Yoon, H. Yoo, S. Lee, Y. Park and H. Lee, Sci. Rep., 2013, 3, 1929 CrossRef PubMed.
- F. Yousefimehr, S. Jafarirad, R. Salehi and M. S. Zakerhamidi, Sci. Rep., 2021, 11, 11900 CrossRef PubMed.
- D. Mathivanan, K. S. Shalini Devi, G. Sathiyan, A. Tyagi, V. A. O. P. Da Silva, B. C. Janegitz, J. Prakash and R. K. Gupta, Sens. Actuators, A, 2021, 328, 112769 CrossRef.
- S. Xing and G. Zhao, Mater. Lett., 2007, 61, 2040–2044 CrossRef.
- S. V. Otari, M. Kumar, M. Z. Anwar, N. D. Thorat, S. K. S. Patel, D. Lee, J. H. Lee, J.-K. Lee, Y. C. Kang and L. Zhang, Sci. Rep., 2017, 7, 10980 CrossRef PubMed.
- X.-Y. Wang, X.-X. Yan, Y.-P. Wu, X.-Q. Wu, Y.-M. Yin, S. Li, Q. Zhang, B. Liu and D.-S. Li, Inorg. Chem., 2023, 62, 10256–10262 CrossRef CAS PubMed.
- S. Abdolhosseinzadeh, H. Asgharzadeh and H. Seop Kim, Sci. Rep., 2015, 5, 10160 CrossRef CAS PubMed.
- R. Chuah, S. C. B. Gopinath, P. Anbu, M. N. Salimi, A. R. W. Yaakub and T. Lakshmipriya, 3 Biotech, 2020, 10, 364 CrossRef.
- F. A. Bezza, S. A. Iwarere, H. G. Brink and E. M. N. Chirwa, Sci. Rep., 2024, 14, 13793 CrossRef CAS.
- X. Chen, J. Chen, F. Meng, L. Shan, M. Jiang, X. Xu, J. Lu, Y. Wang and Z. Zhou, Compos. Sci. Technol., 2016, 127, 71–78 CrossRef CAS.
- S. Yang, W. Yue, D. Huang, C. Chen, H. Lin and X. Yang, RSC Adv., 2012, 2, 8827 RSC.
- H. Yu, T. Wang, B. Wen, M. Lu, Z. Xu, C. Zhu, Y. Chen, X. Xue, C. Sun and M. Cao, J. Mater. Chem., 2012, 22, 21679 RSC.
- S. Park, J. An, I. Jung, R. D. Piner, S. J. An, X. Li, A. Velamakanni and R. S. Ruoff, Nano Lett., 2009, 9, 1593–1597 CrossRef CAS PubMed.
- N. Atikah Md Jani, M. Aidil Ibrahim, T. Ishak Tunku Kudin, A. Malik Marwan Ali, H. Osman and O. Hasdinor Hassan, Mater. Today: Proc., 2017, 4, 5138–5145 Search PubMed.
- S. Karingula, S. K. Venishetty, Y. G. Kotagiri and K. V. Gobi, J. Energy Storage, 2024, 75, 109641 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04010d. Fabrication of the SPCE; TGA; EDX; UV; BJH analysis; EIS; Randles equivalent circuit; different scan rates; transducer concentration optimization; pH optimization; a comparison table. |
| This journal is © The Royal Society of Chemistry 2025 |