
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Lauryl-NrTP6 lipopeptide self-assembled nanorods for nuclear-targeted delivery of doxorubicin†
Amanda
Phungula‡
 abc,
Sofia
Zuffi‡
cd,
Sunisa
Thongsom‡
c,
Paolo
Di Gianvincenzo
c,
Santiago Gimenez
Reyes
ce,
Ana Beatriz
Caribé dos Santos Valle
cf,
Frederico
Pittella
f,
Fernando
Albericio
ag,
Beatriz G.
de la Torre
*b and
Sergio E.
Moya
*c
abc,
Sofia
Zuffi‡
cd,
Sunisa
Thongsom‡
c,
Paolo
Di Gianvincenzo
c,
Santiago Gimenez
Reyes
ce,
Ana Beatriz
Caribé dos Santos Valle
cf,
Frederico
Pittella
f,
Fernando
Albericio
ag,
Beatriz G.
de la Torre
*b and
Sergio E.
Moya
*c
aPeptide Science Laboratory, School of Chemistry and Physics, University of KwaZulu-Natal, Durban 4001, South Africa
bSchool of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban 4001, South Africa. E-mail: garciadelatorreb@ukzn.ac.za
cSoft Matter Nanotechnology, Center for Cooperative Research in Biomaterials (CIC biomaGUNE), Basque Research and Technology Alliance (BRTA), Paseo de Miramon 194, 20014, Donostia-San Sebastián, Spain. E-mail: smoya@cicbiomagune.es
dMolecular Oncology Laboratory, IIS BioGipuzkoa, P° Dr Beguiristain s/n, 20014 San Sebastián, Gipuzkoa, Spain
eInstituto de Fisica del Sur (IFISUR-CONICET), Av. Alem 1253, Bahia Blanca 8000, Argentina
fLaboratório de Desenvolvimento de Sistemas Nanoestruturados, Faculdade de Farmácia, Universidade Federal de Juiz de Fora, Rua José Lourenço Kelmer, 36036-900, Juiz de Fora, MG, Brasil
gCIBER-BBN and Department of Organic Chemistry, University of Barcelona, Barcelona 08001, Spain
First published on 15th January 2025
Abstract
Targeted delivery offers solutions for more efficient therapies with fewer side effects. Here, lipopeptides (LPs) prepared by conjugation of the nuclear-targeting peptide analogue H-YKQSHKKGGKKGSG-NH2 (NrTP6) and two lauric acid chains are used to encapsulate the chemotherapeutic agent doxorubicin (DX) through a solvent-exchange protocol. LPs spontaneously form nanosized rod-like assemblies in phosphate buffer. DX is trapped in the peptide regions of the assemblies. Confocal laser scanning microscopy shows that the peptide assemblies translocate into the nucleus. Cytotoxicity studies over 72 h in A549 and HeLa cancer cell lines show less toxicity for the LP encapsulated DX than for free DX. In contrast, subtoxic doses of encapsulated DX are more effective than free DX in avoiding colony formation over 14 days, with a complete absence of colonies for the LP-encapsulated DX. The results show a more efficient and slow delivery of DX to the nucleus through LP encapsulation, paving the way for the use of lower DX doses as a chemotherapeutic agent.
1. Introduction
Nucleolar targeting peptides (NrTP), capable of targeting the nucleoli in the cells, were prepared through minimization of crotamine, one protein component of the venom toxin derived from the South American rattlesnake Crotalus durissus terrificus. NrTP consists of the residues (1–9)–(38–42) of crotamine that correspond to the N- and C-termini, respectively.1 The analogue H-YKQSHKKGGKKGSG-NH2 (NrTP6), in which Cys 4 was replaced by a Ser residue, resulted in a cell-penetrating peptide (CPP) with low cytotoxicity.1–3 In a previous work,4 we have described the synthesis and the self-assembly behaviour of lipopeptides produced by anchoring 1, 2, and 4 lauric acid chains, a linear twelve-carbon chain saturated fatty acid, to NrTP6.5,6 The lipopeptides resulting from the conjugation of NrTP6 and lauric acid form spherical, fibre-like, or rod-like structures depending on the number of lipid chains in the molecule, ionic strength, and the presence of phosphate ions.7,8 The nanoscale structures formed by the self-assembled lipopeptides show hydrophilic and hydrophobic regions, corresponding to the peptide chains and the lipid domains, respectively. The peptide is more voluminous than the lipid and the structures formed are multilamellar. The presence of lipid and peptide domains in the assemblies allows for the encapsulation of both hydrophobic and hydrophilic drugs. The targeting properties of NrTP6 make these assemblies particularly appealing for the delivery of drugs that need to be directed to the nucleus for therapeutic action.In this work, we explore the use of NrTP6 peptides with two lauric acid chains (from now on LP, lipopeptide) for the delivery of doxorubicin (DX), a well-known chemotherapeutic agent belonging to the anthracycline family. DX intercalates between adjacent base pairs of DNA, interfering with both cell translation and replication and triggering cell death. DX has high water solubility and can usually be administered without a carrier.9,10 Moreover, free DX easily enters cells, crossing cell membranes and translocating into the nucleus. However, the intravenous administration of non-encapsulated DX has several drawbacks, as the drug can enter any cell, not just the tumour site, and in addition, DX is cardiotoxic. Therefore, there is a need for carriers that direct DX towards the tumour and that ensure efficient nuclear delivery, which will help reduce the amount of DX required for therapy and undesired side effects.11–14 We will show here the cell uptake and intracellular localization of LP assemblies, demonstrating that they retain the nuclear-targeting properties of the unconjugated peptides. Then, we will show DX encapsulation in the lipopeptides and conduct cell proliferation studies in selected cancer cell lines after DX administration. The long-term action of the formulation on cancer cells will be assessed by its capacity to prevent cell colony formation. We will demonstrate that sub-lethal doses of encapsulated DX are more efficient than free DX in decreasing colony formation because of the targeted delivery of DX when encapsulated in the assemblies.
2. Experimental
2.1. Materials
Doxorubicin, sulforhodamine B, and trichloroacetic acid were purchased from Merck, Darmstadt, Germany. Paraformaldehyde and nuclear staining Hoechst 33342 were purchased from Thermo Fisher Scientific, Waltham, MA, USA. Three cell lines were used: A549 (lung carcinoma) purchased from Merck Darmstadt, Germany; HeLa (cervical adenocarcinoma) and MRC-5 (human lung fibroblasts) purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). All three cell lines were cultured in Dulbecco's Modified Eagle's medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% foetal bovine serum, streptomycin (100 mg mL−1) and penicillin (100 UI mL−1), at 37 °C and 5% CO2.2.2. Lipopeptide synthesis
The peptide NrTP6 is synthesized by the Solid Phase Peptide Synthesis (SPPS) technique through the Fmoc/tBu strategy. The peptide is synthesized using a microwave peptide synthesizer (CEM). NrTP6 has 14 amino acids with the sequence YQSHKKGGKKGSG. Fmoc rink amide resin (390 mg, 0.64 mmol g−1 loading) is used as the solid support. A scale of 0.1 mmol is used. First, the resin is swelled in DMF; then, Fmoc is removed using 20% piperidine/DMF. A 0.2 M concentration of Fmoc amino acid is coupled for 2 min, using 0.5 M DIC/Oxyma as the coupling reagents. After each coupling, Fmoc is removed with 20% piperidine/DMF. Following the coupling of all amino acids, the resin is removed from the synthesizer and the peptide continues manually to couple the fatty acids. Dodecanoic acid was used as the fatty acid in all the lipopeptides. To incorporate two lauric chains, one Fmoc-Lys(Fmoc)-OH is used for branching, and Fmoc is removed and the lauric acid is coupled to give FA2-NrTP6.2.3. Doxorubicin encapsulation in LP
To encapsulate DX in LP, both LP and DX were dissolved in ethanol and mixed at a molar ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively. Then, ethanol was allowed to evaporate at 37 °C, and the resulting powder was dissolved in 2 mM phosphate buffer (PB) to achieve a final DX concentration of 1 mg ml−1. The suspension was stirred for 24 h. Further dilutions were performed in water. In each step of complexation and experimental handling, DX was carefully protected from environmental light.
:1, respectively. Then, ethanol was allowed to evaporate at 37 °C, and the resulting powder was dissolved in 2 mM phosphate buffer (PB) to achieve a final DX concentration of 1 mg ml−1. The suspension was stirred for 24 h. Further dilutions were performed in water. In each step of complexation and experimental handling, DX was carefully protected from environmental light.
2.4 Fluorescence spectroscopy
The drug encapsulation was monitored by fluorescence spectroscopy using the following parameters: wavelength range of 500–700 nm, excitation wavelength of 470 nm, emission and excitation slit of 2.5 nm, scan speed of 200 nm min−1 and 10 accumulations. The emission spectra of DX were recorded before and after encapsulation in LP. The ratio of the peaks of the fluorescence intensity spectra of DX at 560 and 590 nm was calculated to assess the polarity of the DX environment.2.5 1H and DOSY NMR
All samples were dissolved in D2O and placed in 5 mm NMR tubes. All NMR data were collected on a Bruker AVANCE III NMR spectrometer (11.7 T) equipped with a 5 mm1H/19F BBI probe with an actively shielded z-gradient that was used in combination with a Bruker gradient amplifier providing a maximum current of 10 A, which results in a 65 G cm−1 gradient. 1H diffusion NMR measurements15 were performed without sample spinning at 298 K, using a stimulated echo with bipolar gradient pulses from Bruker's sequence library (ledbpgp2s) with the following parameters: 16 k acquisition complex points, SW 14 ppm, NS 240, DS 16, D1 2 s, D2O (little delta) 0.6 s, P30 (big Delta/2) 1.8 ms, and 32 equally spaced gradient strengths from 5 to 95%.2.6 Transmission electron microscopy (TEM)
TEM analysis was performed with a JEOL JEM-2100F UHR microscope operating at an acceleration voltage of 200 kV. Carbon films (Aname, CF400-CU) were pre-treated in a glow discharge chamber Emitek K100X (2 min, 35 mA). LP-DX solution (0.5 μL) was deposited on an activated carbon grid for 1 min. Staining was carried out with (NH4)2MoO4 (2%, 2 μL) for 1 minute. Excess solution was removed by blotting using filter paper. The sample was washed once with 5 μL of Milli-Q water.2.7 Dynamic light scattering (DLS)
Samples were analyzed for size distribution and zeta potential using a Malvern ZetaSizer Nano ZSP with dynamic light scattering with a DTS1070 cell at 25 °C with backscatter. The measurements were done in triplicate.2.8 DX release in vitro
The in vitro release of DX from LP-DX was assessed in 2 mM PB with pH 7.4 using a dialysis method. LP-DX samples equivalent to 0.1 mg of DX were dissolved in 1 mL of PB, placed in Spectra-Por® Float-A-Lyzer® G2 (5 mL, MWCO 3.5–5 kDa) dialysis tubes and dipped in 15 mL of 2 mM PB. The release was allowed to proceed under stirring (200 rpm) at 37 °C for 96 h. The outer membrane buffer was completely replaced with fresh pre-warmed buffer at different times (1 h, 2 h, 4 h, 6 h, 8 h, 24 h, 48 h, and 96 h), freeze dried, dissolved in 1 mL of Milli-Q H2O and analysed. The amount of DX in the release buffer was measured by fluorescence spectroscopy at 560 nm and the cumulative release data were obtained using a calibration curve for DX. Data were obtained as the average of two independent experiments.2.9 Flow cytometry
To study DX uptake via flow cytometry, A549 cells were seeded at a density of 5 × 104 cells per well in 24-well plates. The following day, the cells were treated with free or encapsulated DX at concentrations of 0.1 and 1 μg mL−1. At 4, 24 or 48 h, the medium was removed, and the cells were rinsed with phosphate-buffered saline (PBS) and detached, rinsed with cycles of centrifugation (300 g, 5 min, and room temperature) and resuspension in PBS. Then, the cells were resuspended in 4% paraformaldehyde (PFA) for 10 min to fix them and then rinsed with additional cycles of centrifugation and resuspension in PBS. Finally, the cells were resuspended in 200 μl prior to analysis. Flow cytometry studies were performed on a MACSQuant10 flow cytometer (Miltenyi Biotec GmbH). DX was excited using a 488 nm laser and the signal was acquired using a 525/50 band-pass filter. Data were analysed using MACSQuant Software: the cell population was gated according to morphology, singlets and DX fluorescence. The data are expressed as the percentage of DX-positive cells relative to the parental population.2.10 Confocal laser scanning microscopy (CLSM)
To study DX uptake via CLSM, A549 cells were seeded at a density of 2 × 104 cells per well in 8-well-Ibidi plates. The following day, the cells were treated with free or encapsulated DX at concentrations of 1 and 5 μg mL−1. At 4, 24 or 48 h, the cells were rinsed with PBS and fixed using 4% PFA for 10 min. The cells were rinsed with PBS and nuclei were stained with Hoechst 33342 (Invitrogen, Waltham, MA, USA) following company instructions. Images were acquired on a Zeiss LSM 880 confocal microscope (Carl Zeiss Microscopy, Jena, Germany). For the visualization of Hoechst 33342, fluorescein-labelled LPs and DX, 405 nm, 488 nm, and 561 nm were chosen as the excitation wavelengths, respectively. The excitation at 561 nm for DX was chosen to avoid the excitation of fluorescein and to be sure that all fluorescence at this wavelength comes from DX. All CLSM experiments were performed with the same settings. For colocalization analysis of confocal images, several regions of interest (ROIs) were selected in each acquisition. The Pearson's coefficient r for colocalization was calculated using ImageJ software. Each event was then represented in the plot and the mean and SD are represented. The statistical analysis was performed as described above.2.11 Cell viability assay
For cytotoxicity evaluation of DX and LP-DX on cancer cell lines HeLa and A549, and non-cancerous lung fibroblast cell line MRC-5, the cells were seeded in a 96-well plate at a density of 5–7.5 × 103 cells per well for 24 h and then treated with free DX or LP-DX at increasing concentrations (0–10 μg mL−1) in complete media. After 24 h, the supernatant was discarded, and the sulforhodamine B (SRB) assay was performed as described previously.16 The absorbance was measured at 570 nm using a Synergy H1 Multi-Mode Reader (BioTek Industries, Inc., VT, USA). As compared to the untreated control, the percentage of cell viability in treated wells was calculated using the following equation: cell viability (%) = (absorbance of treated cells/absorbance of control) × 100. The viability inhibition concentrations at 50% (IC50) were then calculated using GraphPad Prism 8.0 (San Diego, CA, USA) by plotting absorbance data against the concentrations tested for each sample.For the cell proliferation assay, HeLa and A549 cells were seeded at 5 × 103 cells per well for 24 h and then treated with free DX or LP-DX at concentrations of 0, 0.04, 0.016, and 0.625 μg mL−1 for 24, 48, and 72 h. After the indicated times, cell viability was determined by the SRB assay as described above.
2.12 Colony formation assay
HeLa or A549 cells were seeded into 6-well plates with 500 cells per well and treated with a non-toxic dose of DX and LP-DX for 48 h, after which the medium was replaced with the normal medium and further cultured for 7 or 14 days. Blank lipopeptides were used as a control. Colonies were fixed with fixation solution (acetic acid:ethanol; 1:3) and stained with 0.1% crystal violet solution (Sigma). Colonies were imaged using a digital camera and were counted with ImageJ software (National Institutes of Health (NIH), Bethesda, MD, USA).
2.13 Statistical analysis
All the data were expressed as mean ± standard deviation (SD). All the statistical analyses (Student's t-test/one-way ANOVA) were performed using GraphPad Prism 8.0 software (San Diego, CA, USA). A statistical difference of p < 0.05 was considered significant.3. Results and discussion
The chemical formula of NrTP6 with two lauric chains (LP, lipopeptide) is shown in Scheme 1 together with the formula of DX. DX was encapsulated in the LPs as explained in the Experimental section. Both DX and the lipids were dissolved in ethanol. The ethanol solutions of DX and LPs were mixed, and the solvent was evaporated. The solid residue was resuspended in PB. 1H NMR spectra of DX, LP and LP-DX were recorded (Fig. S1†). NMR peaks corresponding to both DX and LP were found in the LP-DX spectra, but the signals were broader than in the spectra of individual molecules, suggesting the formation of a supramolecular structure and the complexation of DX with the LPs. In parallel, DOSY NMR experiments showed that the encapsulation of DX was complete (Fig. 1A). Diffusion times of free DX (red points, D = 1.99 × 10−6 cm2 s−1) could not be detected in the LP-DX formulation (green, D = 3.49 × 10−7 cm2 s−1), which showed a single diffusion time, meaning that no free DX is present and all DX is encapsulated in the LPs.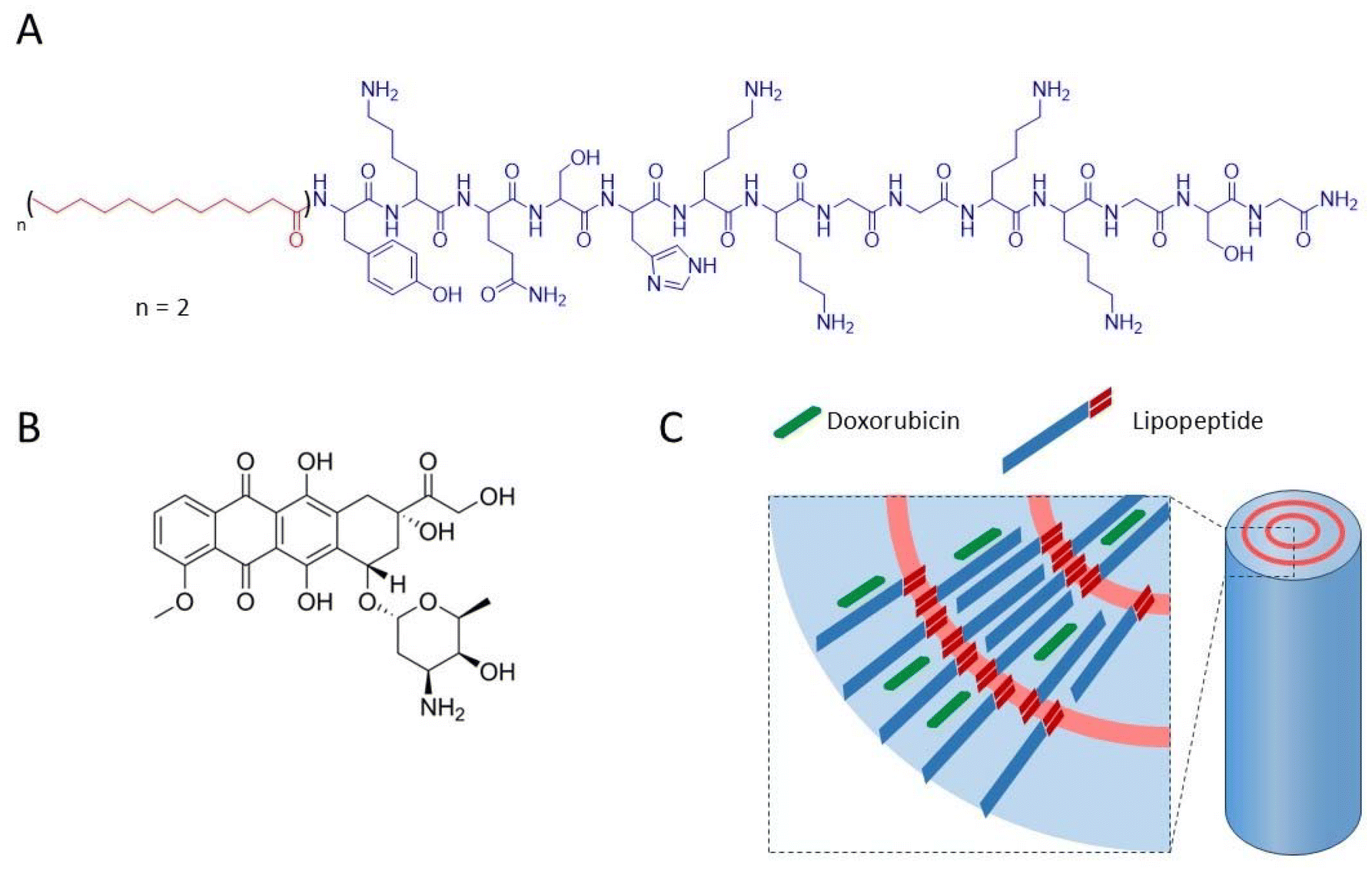 | ||
| Scheme 1 Chemical structure of the NrTP6 lipopeptide (LP) with two lauric acid chains (A) and doxorubicin (DX) (B), and sketch of the nanorod complex formed by DX and LP (C). | ||
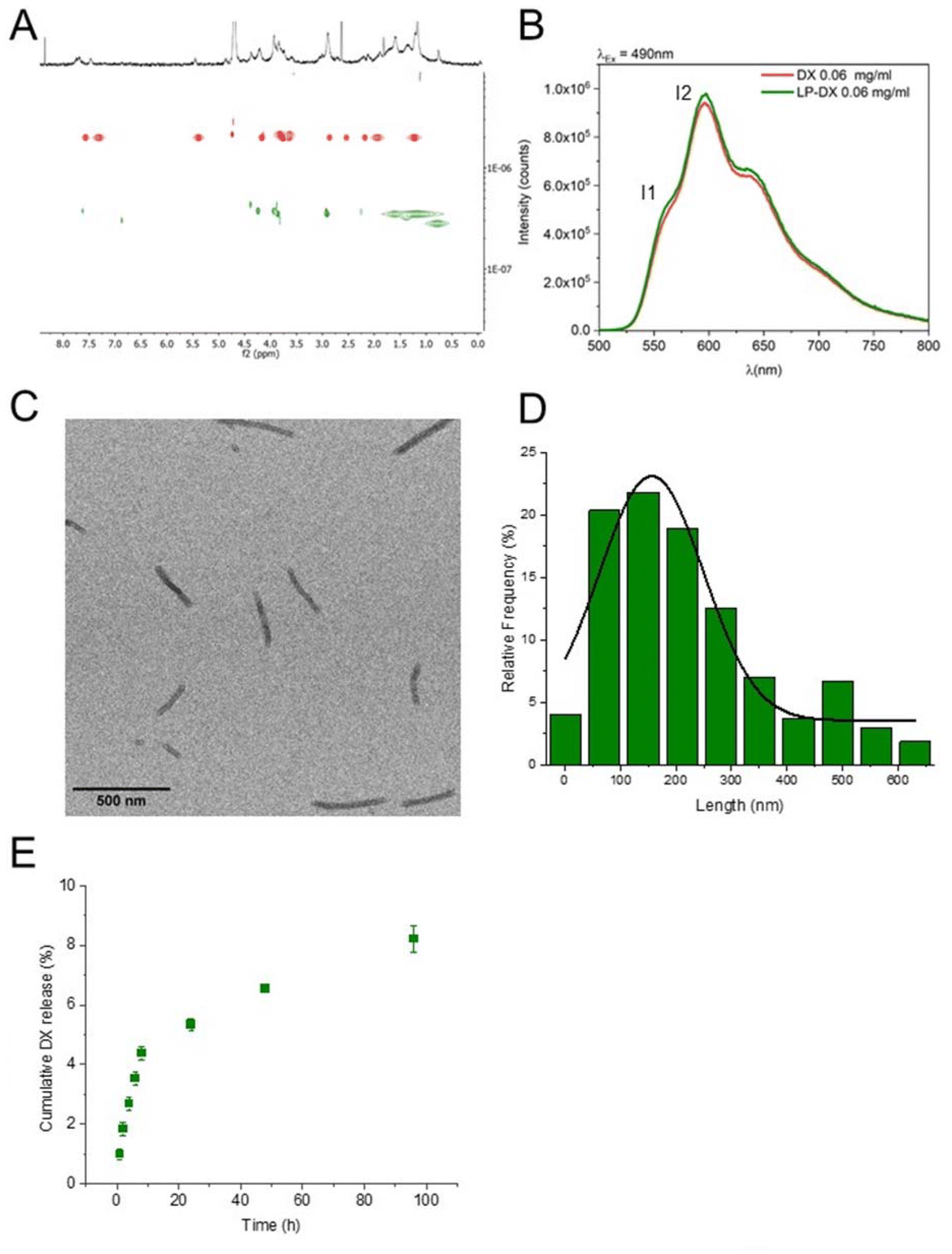 | ||
| Fig. 1 LP-DX physico-chemical characterization. (A) Independent DOSY NMR experiments in D2O: free DX in red and LP-DX in green. (B) Fluorescent emission of free DX (red curve) and LP-DX (green curve), excitation at 490 nm and DX concentration: 0.06 mg ml−1. (C) TEM image of LP-DX and scale bar: 500 nm. (D) Length distribution of LP-DX nanorods measured by TEM. (E) Kinetic study of the release of DX from LP-DX in 2 mM PB (96 h). Values are expressed as the mean of two independent experiments. | ||
DX was dissolved in ethanol in its protonated form, DX·HCl, and it can be expected to remain protonated when resuspended in water. Anthracyclines like DX are fluorescent, and their fluorescence spectra are very sensitive to the polarity of the environment. The fluorescence spectrum of encapsulated DX is shown in Fig. 1B (DX in red and LP-DX in green). The ratio of the intensity of the first and second emission bands of DX (I1, 560 nm and I2, 590 nm, respectively), I1/I2, used as a measure of polarity, had a value of 0.55, very close to the I1/I2 of DX in water (0.52). If DX were in the hydrophobic regions of the lipopeptide, the I1 intensity would increase and I1/I2 would be larger than 1.11 Since we know from DOSY experiments that DX was all encapsulated, we can conclude that the molecule is located in the hydrophilic regions of the assembly, probably trapped by hydrogen bonding interactions between the peptides and DX. TEM images (Fig. 1C) show that the assemblies with DX form nanorod structures with lengths around 150 nm (Fig. 1D) and diameters of 20 nm. From the size distribution analysis, we can conclude that most of the rods have sizes between 100 and 200 nm but larger rods, up to 600 nm, can also be found. The observed diameter indicates that the assemblies are multilamellar structures as observed for the lipopeptides without encapsulated drugs in a previous paper. The assemblies showed a zeta potential of +25.5 mV, indicating positive charges.4 The in vitro release of DX from DX-LP was performed in 2 mM PB (pH 7.4) at 37 °C, simulating physiological conditions. The cumulative release of the drug was plotted against time (Fig. 1E) in order to obtain the drug-delivery profile and analyse the release mechanism. The release of DX is slow with a total amount of DX released after 96 h corresponding to 8% of the initial amount. Physicochemical characterization of the assemblies leads us to conclude that these are nanorods and that DX is located in the peptide regions of the LP assemblies as sketched in Scheme 1.
Following the physicochemical characterization of the lipopeptide formulation with DX and having proved that DX was successfully encapsulated, we proceeded to study the cell uptake and DX delivery with the LPs by CLSM and flow cytometry and by means of cell proliferation and colony assays.
Fig. 2A shows the CLSM images of A549 cells exposed to LP with encapsulated DX and to free DX as a control. The cell nucleus was stained with Hoechst 33342 for visualization. The experiments were carried out at a concentration of 1 μg mL−1. Free DX can be visualized in the nucleus after just 4 h. At 4 and 24 h, the DX intensity increases but at 48 h it largely decreases, meaning also that the DX uptake is fast, being at maximum before 48 h.
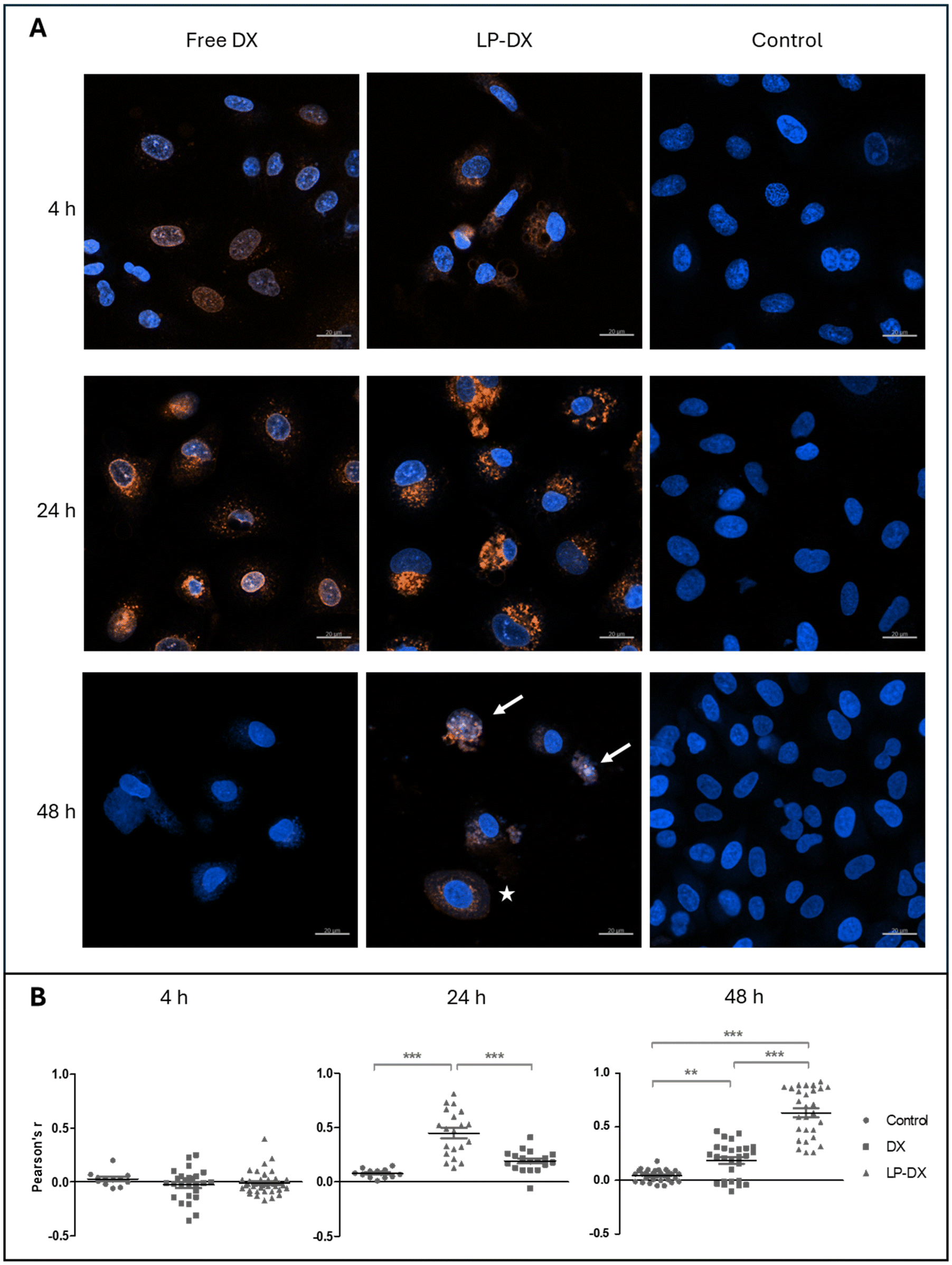 | ||
| Fig. 2 Confocal laser scanning microscopy of free and encapsulated DX. (A) CLSM images of A549 cells treated with 1 μg ml−1 of DX (free DX, left), encapsulated DX (LP-DX, center) and mock-treated cells (control, right) at 4, 24, and 48 h. Reference bars equal to 20 μm. Blue: nuclei; red: DX; pink: colocalization. White arrows point to nuclei with ongoing changes in morphology. The white star marks a cell with its nucleus surrounded by a halo of DX. (B) Pearson's correlation coefficient, r, at. 4, 24, and 48 h calculated to assess colocalization of nuclei staining (Hoechst 33342) and DX. Each event reported for each condition is represented by a symbol, with the mean and SD. **p < 0.01, ***p < 0.001. | ||
DX encapsulated in the LP can be barely seen in the nucleus at 4 h but is present in the cytoplasm. Over longer times, fluorescence around the nucleus increases, while at 48 h the fluorescence intensity starts decreasing. However, at 48 h some LP-treated cells show colocalization of DX and nuclear staining (blue and red light appear as pink), and the nuclear morphology strongly suggests ongoing toxic action (white arrows in Fig. 2), while other cells show a perinuclear localization of DX, almost forming a halo around the nucleus (white star in Fig. 2). This is indicative of a more progressive uptake of the encapsulated DX by the cells. Colocalization of DX and the nuclei was quantified via Pearson's coefficient (Fig. 2B) and confirmed this behaviour: at 4 h no clear correlation is observed for free and encapsulated DX, possibly because, as Fig. 2A shows, only some cells have already taken up DX in the cells. At 24 hours, DX colocalizes with nuclear staining (Pearson's coefficient r 0.45 ± 0.19, indicating a moderate correlation), while LP-DX does not show any significant correlation at this time point. However, at 48 hours, the encapsulated DX shows moderate colocalization (Pearson's coefficient r 0.63 ± 0.22), while the colocalization of free DX and nuclei decreases (Pearson's coefficient r 0.19 ± 0.16). These data suggest a slower internalization and targeting of the nucleus, by the encapsulated DX.
We also performed uptake experiments with LPs labelled with fluorescein, which allows us to trace the location of the lipopeptides in the cell. Fluorescein and DX fluorescence were resolved without spectral overlap after laser adjustment. Confocal images of the labelled LPs are shown in Fig. 3. LP and DX fluorescence colocalize in the cytoplasm at 4 and 24 h, and later at 48 h in the nucleus, and this suggests that DX is reaching the nucleus encapsulated and it is liberated there, rather than being released in endosomes. In the time frame of our measurements, 48 h, we are unable to visualize DX fluorescence not colocalizing with LP fluorescence, which suggests that DX is retained in the LPs or if liberated remains close to the LPs, and it is not possible to discriminate free and encapsulated DX with the resolution of confocal microscopy. However, flow cytometry data and proliferation studies suggest that DX is released slowly from the LPs.
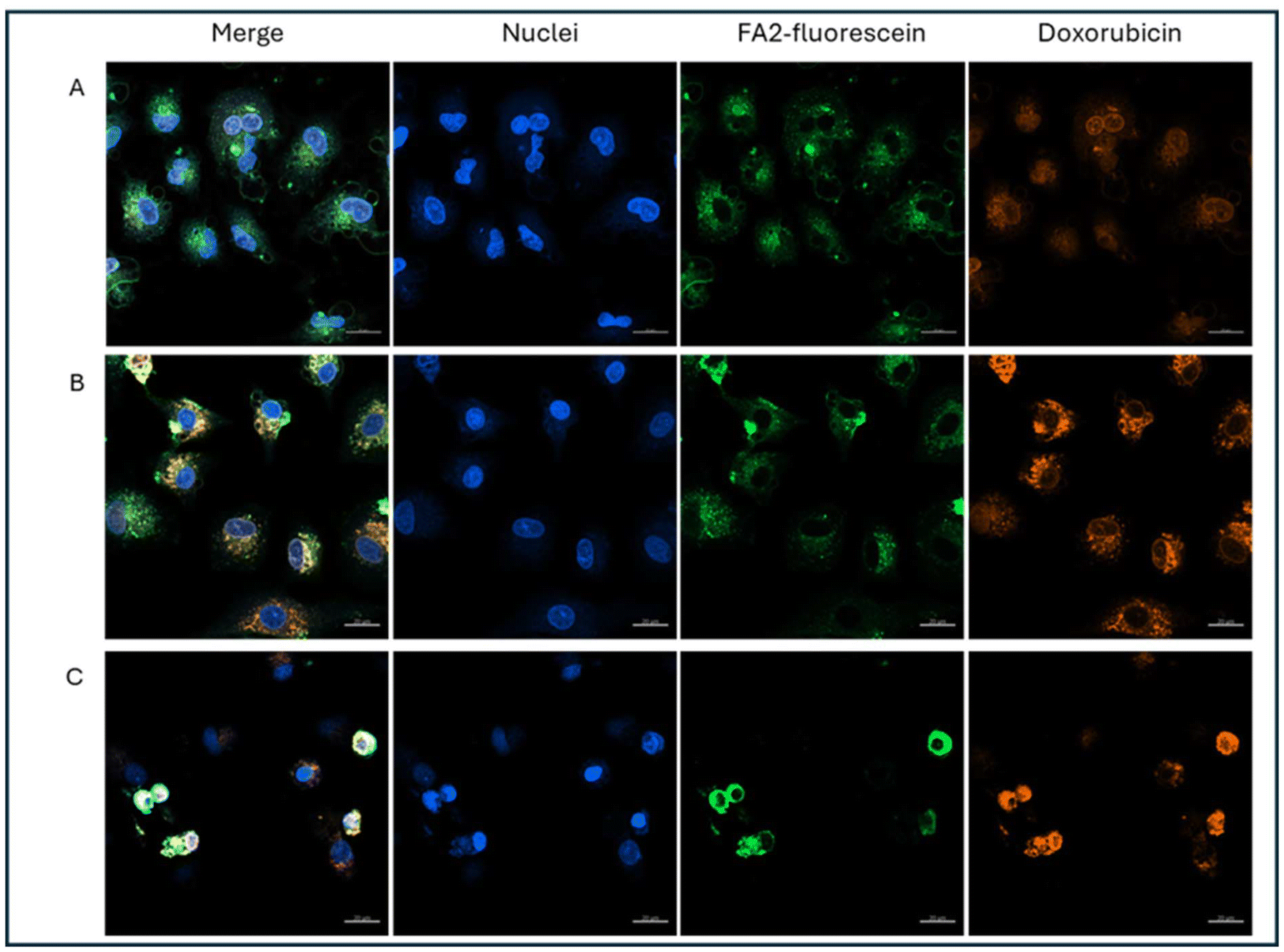 | ||
| Fig. 3 Uptake of fluorescein labeled LPs with encapsulated DX (1 μg mL−1) in A549 cells via CLSM. (A) 4 h, (B) 24 h, and (C) 48 h after treatment. Reference bars represent 20 μm. Blue: nuclei:Hoechst 33342; green: fluorescein-labelled FA2NRP6; red: DX. | ||
We performed flow cytometry measurements to quantify the uptake of LP-DX in A549 cells, assessing the fluorescence intensity, coming from DX, at the cell level. The uptake studies are shown in Fig. 4 and correspond to free and encapsulated DX in A549 at 0.1 μg mL−1 DX. At 4 h, a small percentage of cells already started the uptake of DX. Free DX uptake is slightly faster than that of encapsulated DX (15.5% and 12% of DX-positive cells, respectively). However, at 24 and 48 h, the LP-encapsulated DX leads to higher percentages of internalization compared to free DX, with a difference between encapsulated and free DX of more than 20% at each time point. At 1 μg mL−1, virtually all cells resulted in a positive DX signal at 24 and 48 h, while at 4 h the percentage of DX-positive cells was higher for encapsulated DX (data not shown). For the lowest DX concentration used, 0.1 μg mL−1, the uptake of DX correlates with the time of exposure, peaking at 48 h with encapsulated DX. The difference in uptake at equal concentrations of free and encapsulated DX is due to the different mechanisms of uptake involved. Free DX diffuses through the cell membrane, while encapsulated DX enters the cell through endocytic processes.
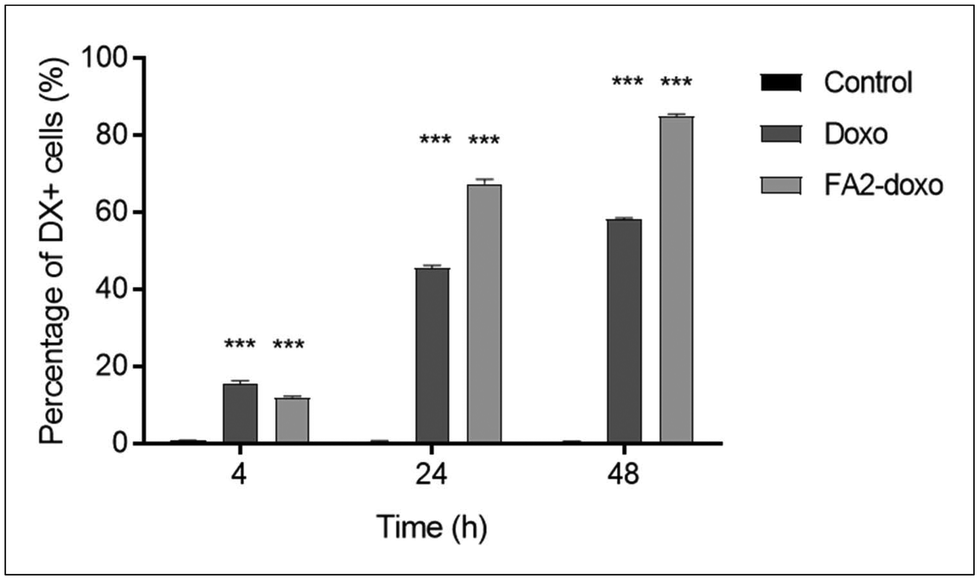 | ||
| Fig. 4 Percentage of DX positive A549 cells following exposure to 0.1 μg mL−1 of free or encapsulated DX (LP-DX), at 4, 24, and 48 h, as measured by flow cytometry. Error bars represent the standard deviation of duplicate samples. Each treatment condition is compared to the untreated control at the corresponding time point, and significance is calculated using an ANOVA test: ***p < 0.001. | ||
However, DX uptake and uptake kinetics may not correlate with the cell toxicity as DX must reach the nucleus and intercalate with the nucleic acids to induce toxicity. We have seen from confocal images that there is a delay in observing DX fluorescence in the nucleus when it is encapsulated compared with free DX and that DX may remain encapsulated in the LP assemblies up to 48 h. Cell proliferation assays were performed in two cancer cell lines, A549, lung carcinoma, and HeLa cells, cervical cancer, and in a non-cancerous cell line: MRC-5. HeLa cells are known to be more DX sensitive than A549 cells. Experiments were performed at DX concentrations from 0 to 10 μg mL−1 for 24 h. The impact of free DX, encapsulated DX (LP-DX), and blank LP on cell viability was determined using the SRB assay. The cytotoxicity of free DX and LP-DX on cancer cell lines HeLa and A549 and non-cancerous lung fibroblast MRC-5 cells is shown in Fig. 5A. Both DX and LP-DX are less toxic to non-cancerous cells but show cytotoxicity in cancer cells. Free DX and LP-DX showed a dose-dependent cytotoxic effect in both cancer cell lines, while the blank LP has no dose-dependent effect. The IC50 values of free DX, LP-DX, and LP against HeLa and A549 cells are shown in Fig. 5B. For HeLa cells, IC50 values for free DX and LP-DX were 0.05 μg mL−1 and 0.25 μg mL−1, respectively. For A549 cells, IC50 values for free DX and LP-DX were 0.41 μg mL−1 and 1.08 μg mL−1, respectively. The results revealed that LP-DX exhibited lower cytotoxicity on HeLa and A549 cells compared to free DX, with IC50 values that increased by 5 and 2.6 fold, respectively.
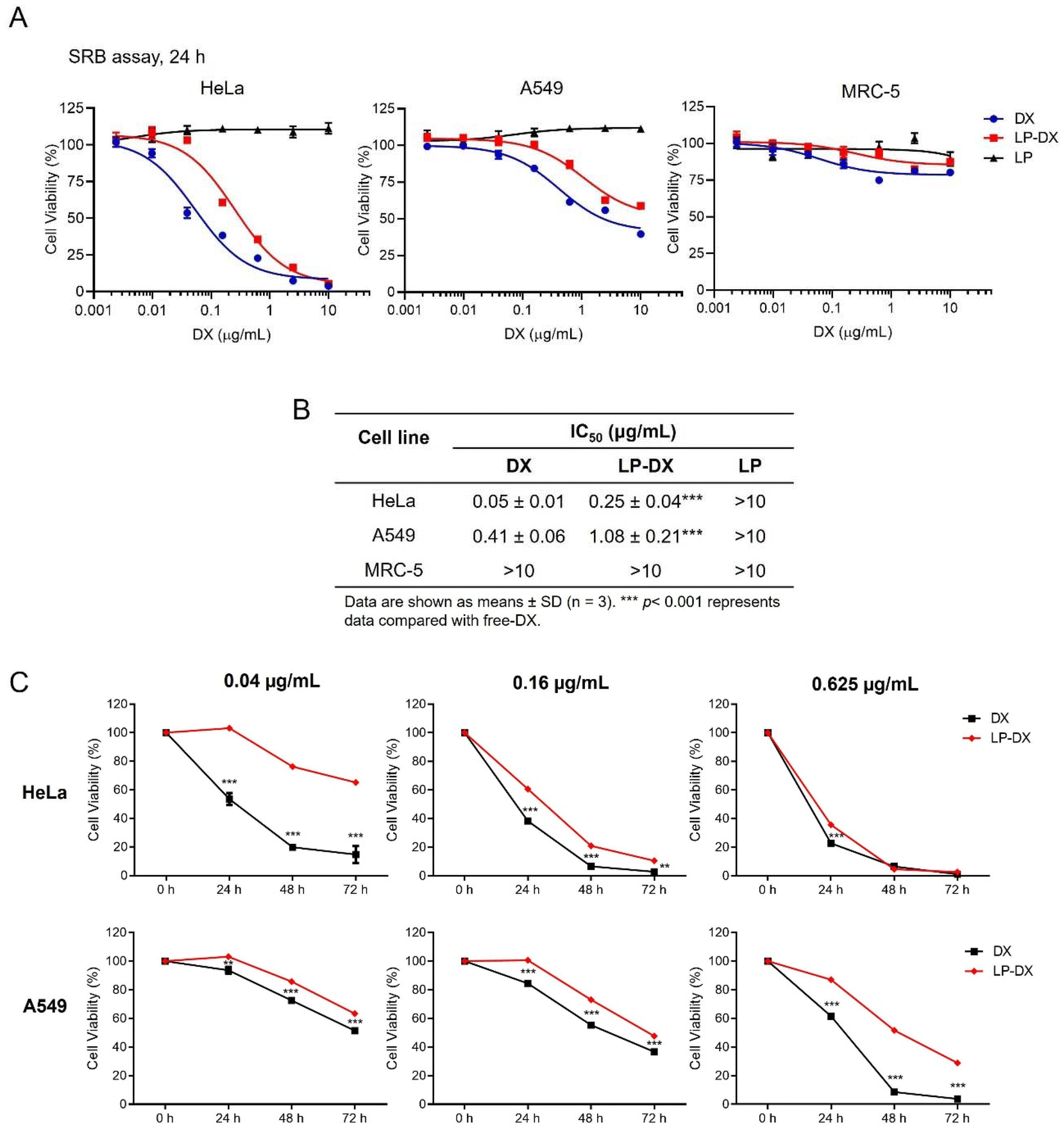 | ||
| Fig. 5 Cytotoxicity and antiproliferative activity of DX and LP-DX on cancer cell lines HeLa and A549 cells and in the non-cancerous cell line MRC-5. (A) Cytotoxicity of DX, LP-DX, and LP on cancer cell lines HeLa and A549 and non-cancerous cell line MRC-5. (B) IC50 values derived from the SRB cytotoxicity assay of DX, LP-DX, and LP in HeLa, A549 and MRC-5 cells. (C) The effect of DX and LP-DX on cell proliferation in HeLa and A549 cells. Cell viability was determined using the SRB assay. Data are expressed as the percentage of cell viability vs. untreated control cells. Each value is the mean (± SD) from triplicate experiments. **p < 0.01 and ***p < 0.001 vs. free DX. | ||
We further compared the antiproliferative activity of free DX and LP-DX on HeLa and A549 cells at various time points (24 h, 48 h, and 72 h) (Fig. 5C). At the lowest DX concentrations, free DX has a stronger effect on cell proliferation than encapsulated DX. The decrease in cell proliferation is more significant for the free DX at all time points considered. As the DX concentration increases, the differences in proliferation between the cell lines diminish. For HeLa cells at the highest concentration tested, the decrease in proliferation for encapsulated and free DX is practically the same, while for the more resistant A549, free DX continues to be more toxic. We know from flow cytometry that the uptake of encapsulated DX is always higher than for free DX, for the same administered dose. Therefore, we can safely conclude that the amount of DX interacting with the nucleic acids in the nucleus is smaller for encapsulated DX, since toxicity depends on the amount of DX in the nucleus. However, we observed that the fluorescence intensity of DX is higher around the nucleus at longer time points when DX is encapsulated, which corroborates that DX is trapped in the lipopeptide assemblies and not released.
Since release may take place at a slow pace even if the encapsulated DX is in close vicinity to the nucleus we decided to assess the impact of DX over longer periods and we evaluated the action of DX lipopeptide formulations on colony formation.17 For this, cancer cell lines HeLa and A549 were seeded at 500 cells per well in 6-well plates and then exposed to free and encapsulated DX in subtoxic doses (corresponding to a cell viability of more than 80%) for 48 h (Fig. 6A) and allowed to grow and form colonies for 7–14 days. Subtoxic doses were confirmed with no cytotoxic effects on non-cancerous MRC-5 cells even when exposed for 96 h (ESI Fig. S2†). The ability of colony formation was assessed at 7 days and 14 days for HeLa cells and A549 cells, respectively. Both HeLa and A549 not exposed to DX form large colonies, as shown in Fig. 6B and C. Free DX reduces colonies partially while encapsulated DX reduces colony formation completely as can be observed in the light microscopy image. The suppression of colony formation at longer time points than in the proliferation studies suggests that the encapsulated DX is acting on the cells even if the dose of DX is below toxicity and that the encapsulated DX may be released very slowly in the cells. Indeed, we observe that more DX enters the cells when encapsulated and confocal microscopy hints that DX reaches the nucleus with the lipopeptide assemblies. The fact that the lipopeptide carrier reaches the nucleus may enhance the efficiency of DX action but also lead to a marked delay in toxicity and DX action on cell metabolism as the DX liberation is slow.
 | ||
| Fig. 6 Colony formation assay corresponding to HeLa and A549 cells exposed to DX or encapsulated DX (LP-DX). (A) Non-toxic doses of DX or LP-DX on HeLa and A549 cells were determined after cell treatment for 48 h. (B and C) Colony formation assay of HeLa and A549 after treatment with subtoxic doses of DX or LP-DX for 48 h and allowed to grow to form visible colonies. The right panel of B and C shows the quantitative analysis performed using ImageJ software. Each value is the mean (± SD) from triplicate experiments. **p < 0.01 and ***p < 0.001 vs. untreated control; ##p < 0.01 and ###p < 0.001 vs. DX. | ||
4. Conclusions
We have loaded LPs with DX by mixing lipopeptides and DX in ethanol, evaporating the solvent and resuspending in PB. LP-DX self-assembles into nanorods. The loading of DX in the lipopeptides is complete and DX is retained in the hydrophilic regions of the assemblies, being released slowly. DX uptake in cells is more pronounced when DX is encapsulated in LPs than when free DX is used. CLSM images show that the lipopeptides target the nucleus and that DX largely remains encapsulated in the lipopeptide assemblies around the nucleus at 48 h. Despite the higher uptake of encapsulated DX than free DX, the slow release of DX from the lipopeptides inside the cells results in lower cell toxicity for the encapsulated DX. However, over the long term encapsulated DX at subtoxic concentrations is more effective than free DX in preventing colony formation. We conclude that this is because encapsulated DX is more effective in reaching the nucleus than free DX, but the slow release of DX results in observable effects at longer time periods. These results open novel possibilities for targeted nuclear delivery using lipopeptides. The capacity of the lipopeptide assemblies to reach the nucleus allows for a more effective delivery of chemotherapeutic drugs with nuclear action and foresees the use of lower doses of the drugs, which will reduce the potential side effects of the chemotherapeutics.Author contributions
A.P.: research; S.Z.: research and draft revision; S.T.: research and methodology, P.d.G.: research, methodology, and writing – original draft; S.G.R.: research; B.V.: research; F.A.: supervision, funding acquisition, and draft revision; F.P.: supervision; B.T.: supervision, funding acquisition, and draft revision; S.E.M.: experimental design, supervision, funding, and writing – original draft.Data availability
Data for this article, including dynamic light scattering readouts, fluorescence readouts, NMR spectra, UV readouts, flow cytometry readouts, transmission electron microscopy micrographs, and confocal laser fluorescence scanning micrographs, are available at Zenodo at https://doi.org/10.5281/zenodo.13880040.Conflicts of interest
The authors report no conflict of interest.Acknowledgements
The authors acknowledge support from the H2020 MSCA-RISE 2020 Project SUPRO-GEN, Grant Agreement no. 101008072. S. E. M. thanks the PID2020-114356RB-I00 project from the Ministry of Science and Innovation of the Government of Spain and the Maria de Maeztu Units of Excellence Program from the Spanish State Research Agency – Grant no. MDM-2017-0720.References
- G. Rádis-Baptista, B. G. de la Torre and D. Andreu, A Novel Cell-Penetrating Peptide Sequence Derived by Structural Minimization of a Snake Toxin Exhibits Preferential Nucleolar Localization, J. Med. Chem., 2008, 51(22), 7041–7044 CrossRef PubMed.
- M. Rodrigues, D. Andreu and N. C. Santos, Uptake and cellular distribution of nucleolar targeting peptides (NrTPs) in different cell types, Biopolymers, 2015, 104(2), 101–109 CrossRef CAS PubMed.
- M. Lindgren, M. Hällbrink, A. Prochiantz and Ü. Langel, Cell-penetrating peptides, Trends Pharmacol. Sci., 2000, 21(3), 99–103 CrossRef CAS PubMed . Available from: https://www.sciencedirect.com/science/article/pii/S0165614700014474.
- A. Phungula, A. Y. Waddad, M. D. Fernandez Leyes, P. Di Gianvincenzo, B. Espuche and S. Zuffi, et al., Self-assembly of NrTP6 cell-penetrating lipo-peptide with variable number of lipid chains: Impact of phosphate ions on lipid association, J. Colloid Interface Sci., 2024, 654, 124–133 CrossRef CAS PubMed.
- K. R. Meena and S. S. Kanwar, Lipopeptides as the antifungal and antibacterial agents: Applications in food safety and therapeutics, BioMed Res. Int., 2015, 2015, 473050 Search PubMed.
- E. Wiman, E. Zattarin, D. Aili, T. Bengtsson, R. Selegård and H. Khalaf, Development of novel broad-spectrum antimicrobial lipopeptides derived from plantaricin NC8 β, Sci. Rep., 2023, 13, 4104 CrossRef CAS PubMed.
- P. Andreozzi, C. Ricci, J. E. M. Porcel, P. Moretti, D. Di Silvio and H. Amenitsch, et al., Mechanistic study of the nucleation and conformational changes of polyamines in presence of phosphate ions, J. Colloid Interface Sci., 2019, 543, 335–342 CrossRef CAS PubMed.
- P. Andreozzi, E. Diamanti, K. R. Py-Daniel, P. R. Cáceres-Vélez, C. Martinelli and N. Politakos, et al., Exploring the pH Sensitivity of Poly(allylamine) Phosphate Supramolecular Nanocarriers for Intracellular siRNA Delivery, ACS Appl. Mater. Interfaces, 2017, 9(44), 38242–38254 CrossRef CAS PubMed.
- O. Tacar, P. Sriamornsak and C. R. Dass, Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems, J. Pharm. Pharmacol., 2013, 65(2), 157–170, DOI:10.1111/j.2042-7158.2012.01567.x.
- C. Pérez-Arnaiz, N. Busto, J. M. Leal and B. García, New insights into the mechanism of the DNA/doxorubicin interaction, J. Phys. Chem. B, 2014, 118(5), 1288–1295 CrossRef PubMed.
- L. Desiderio, N. S. Gjerde, E. Tasca, L. Galantini, I. Llarena and P. Di Gianvincenzo, et al., Determination of the optimal pH for doxorubicin encapsulation in polymeric micelles, J. Colloid Interface Sci., 2024, 664, 972–979 CrossRef CAS PubMed.
- E. R. Gillies and J. M. J. Fréchet, pH-responsive copolymer assemblies for controlled release of doxorubicin, Bioconjugate Chem., 2005, 16(2), 361–368 CrossRef CAS PubMed.
- E. Tasca, A. Del Giudice, L. Galantini, K. Schillén, A. M. Giuliani and M. Giustini, A fluorescence study of the loading and time stability of doxorubicin in sodium cholate/PEO-PPO-PEO triblock copolymer mixed micelles, J. Colloid Interface Sci., 2019, 540, 593–601 CrossRef CAS PubMed.
- E. Tasca, P. Andreozzi, A. Del Giudice, L. Galantini, K. Schillén, A. M. Giuliani, M. D. L. A. Ramirez, S. E. Moya and M. Giustini, Poloxamer/sodium cholate co-formulation for micellar encapsulation of doxorubicin with high efficiency for intracellular delivery: An in vitro bioavailability study, J. Colloid Interface Sci., 2020, 579, 551–561 CrossRef CAS PubMed.
- P. Groves, Diffusion ordered spectroscopy (DOSY) as applied to polymers, Polym. Chem., 2017, 8(44), 6700–6708, 10.1039/C7PY01577A.
- S. Thongsom, W. Suginta, K. J. Lee, H. Choe and C. Talabnin, Piperlongumine induces G2/M phase arrest and apoptosis in cholangiocarcinoma cells through the ROS-JNK-ERK signaling pathway, Apoptosis, 2017, 22(11), 1473–1484 CrossRef CAS PubMed.
- N. A. P. Franken, H. M. Rodermond, J. Stap, J. Haveman and C. van Bree, Clonogenic assay of cells in vitro, Nat. Protoc., 2006, 1(5), 2315–2319, DOI:10.1038/nprot.2006.339.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04068f |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |