
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The fate of nanoparticle surface chemistry during reductive electrosynthesis in aprotic media†
Xenia V. Medvedeva ,
Jury J. Medvedev,
Xingya Zhao,
Elena Smith and
Anna Klinkova*
,
Jury J. Medvedev,
Xingya Zhao,
Elena Smith and
Anna Klinkova*
Department of Chemistry and Waterloo Institute for Nanotechnology University of Waterloo, Waterloo, Ontario N2L 3G1, Canada. E-mail: aklinkova@uwaterloo.ca
First published on 14th February 2025
Abstract
Reductive electrochemical coupling of carbon dioxide with organic molecules (electrocarboxylation, EC) represents a green route towards value-added carboxylic acids and serves as a promising strategy for carbon footprint mitigation. Despite the industrial prospects of this synthetic process, little has been done towards the optimization of cathode materials at the nanoscale. Herein, we pave the way towards the use of metal nanoparticles (NPs) as electrocatalysts in EC by demonstrating the effects of NP surface chemistry on electroorganic transformations and the evolution of surface functionalization in the course of reductive electrosynthesis in aprotic media. Using spherical Au NPs capped with citrate or cetylpyridinium chloride (CPC) as our study subjects, we examined the effect of Au NP surface chemistry on the selectivity of EC of benzyl bromide in acetonitrile and determined the fate of the surface adsorbates of Au NPs in the course of the reaction using Raman spectroscopy and X-ray photoelectron spectroscopy. We show that the CPC-stabilized Au NPs outperform the citrate-stabilized NPs at a low applied potential of −1.5 V vs. Ag/Ag+ with the former showing an almost two-fold increase in the faradaic efficiency towards phenylacetic acid. This higher selectivity is attributed to the reaction on the liberated Au surface stemming from the stripping of CPC molecules. In contrast to the CPC-functionalized NPs, the citrate-stabilized Au NPs retain their adsorbates during the reaction, which undergo electrochemical transformations during EC.
Introduction
Electroorganic synthesis has attracted a lot of attention in the last few decades as a powerful tool for carbon–carbon bond formation under mild conditions.1–4 The electrochemical approach offers multiple advantages over traditional synthetic methods, including high atom economy, unique selectivity, and the avoidance of toxic redox agents as electron sources.5 Despite the great potential of electrochemical methods, just a few processes have found industrial application,6,7 mainly due to the existing barriers to adopting electrochemical techniques in the organic synthesis field, including engineering difficulties for scale-up, degraded selectivity at industrially relevant current densities, and the absence of effective strategies for precise control over the reaction stereoselectivity.3 Now, with the declining prices of renewable electricity and significant advances in electrochemical technologies, the development of sustainable methods for the electrosynthesis of valuable chemicals is on the rise.Electrochemical coupling of carbon dioxide with a wide variety of organic compounds (electrocarboxylation, EC) has proved to be a promising strategy for the direct synthesis of carboxylic acids.8–12 Recently, multiple studies have suggested routes towards the efficient EC of organohalides,13–20 pseudohalides,21–23 ketones,24–28 imines,29–33 and unsaturated organic compounds.34–38 The increased interest in EC has been further fueled by global initiatives to mitigate climate change as EC represents a promising CO2 utilization technology.
Historically, EC in aprotic media was widely studied on non-catalytic carbon-based electrodes, such as glassy carbon.39 Despite the good yields of target carboxylic acids, the reaction on inert electrodes suffered from slow kinetics and low current densities as a result of the outer sphere electron transfer mechanism.40,41 To facilitate EC and to tune the reaction selectivity, an electrocatalyst can be employed. Notably, the nature of the metal used as an electrocatalyst can significantly affect both the process rate and selectivity as demonstrated in the example of the EC of organohalides and imines.29,40
Further advances in the catalyst design for EC can be achieved by employing nanoscale electrocatalysis by analogy with catalyst improvement strategies for electrocatalytic reactions in aqueous media.42–45 Notably, the electrochemical performance is also affected by the exposed crystallographic facets that depend on nanoparticle (NP) shape.46–50 However, besides the facets, a change in the NP morphology is often associated with a difference in surface chemistry that is dependent on the selected production method.51–54 Recently, it has been established that the presence of organic molecules on the electrocatalyst surface significantly improves the material performance in CO2 reduction in aqueous media.55,56 At the same time, little is known about the surface chemistry effects for electroorganic transformations such as EC that require an aprotic medium.57
Moreover, disentangling the effects of surface atomic structures (i.e., exposed facets, undercoordinated sites, etc.) and surface adsorbates (i.e., ligands originating from the NP synthesis) is essential for rational catalyst design that would synergistically combine both the metal nanoscale and the atomic surface structure with molecular surface functionalization to achieve the desired activity and selectivity in EC. A deeper understanding of the behavior of surface adsorbates on metal NPs in the course of electrocatalysis can also provide insights into long-term ligand effects in atomically precise metal nanoclusters that are now being explored in various electrocatalytic reactions.58,59
Here we investigate the effects of the surface chemistry of a nanoscale electrocatalyst on its catalytic properties in cathodic electroorganic synthesis. Specifically, we study the EC of benzyl bromide using Au NPs with different surface functionalization as a cathodic electrocatalyst. We demonstrate that variation in NP surface chemistry leads to dramatic changes in the selectivity of EC. To obtain insights into the structure–performance relationship, we conducted a detailed study of the surface chemistry changes occurring in the course of electrolysis using ex situ Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). Based on the extensive spectroscopic data, we determined how the reaction medium and applied potential change the surface chemistry of the Au NPs. Finally, we suggest the mechanism of observed surface transformations and explain how these changes affect the catalyst performance. The results of this work highlight the importance of material surface chemistry consideration for the rational design of nanostructured catalysts for electroorganic transformations.
Results and discussion
Catalyst material synthesis
To explore the ligand-specific behavior of electrocatalysts for EC, we selected gold as a cathodic material due to its great performance in the EC of organohalides based on the studies of bulk metal electrocatalysts40 and its reasonable stability in this reaction. We produced near-spherical Au NPs (Fig. 1) using two of the most common wet synthesis routes: the Turkevich method60 resulting in citrate-capped Au NPs (Au-citrate) and seed-mediated synthesis in aqueous solution of a cationic surfactant,61 cetylpyridinium chloride (CPC), leading to Au NPs stabilized with micellar double layers (Au-CPC). Both approaches were carefully adjusted to yield comparable NP sizes, resulting in Au-citrate with a size of 53 ± 9 nm and Au-CPC with a diameter of 52 ± 1.5 nm. The synthesized nanoparticles were concentrated and deposited on a 0.5 × 0.5 cm2 Ti foil substrate using drop-casting with an even coverage and a mass loading of about 1 mg cm−2 (Fig. S1†).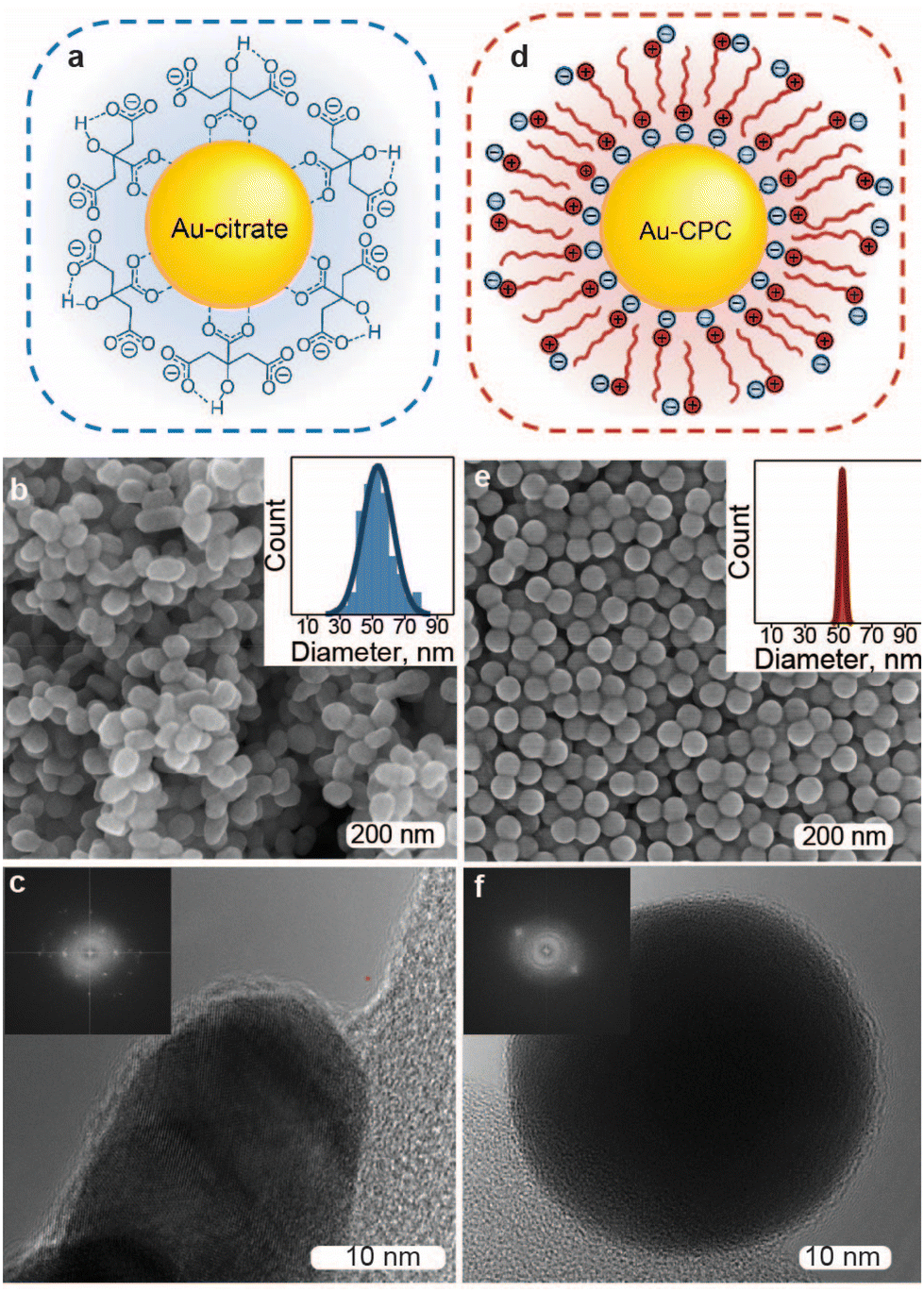 | ||
| Fig. 1 Au nanoparticles chosen as objects for this study. Schematics representing Au NP stabilization for Au-citrate (a) and Au-CPC (d) with blue balls showing chloride anions and red balls with lines showing cetylpyridinium cations. SEM images of Au-citrate (b) and Au-CPC (e) with insets showing NP size distributions, which were determined via ≥150 measurements for Au-citrate and via ≥100 measurements for Au-CPC. High-Resolution Transmission Electron Microscopy (HRTEM) images of Au-citrate (c) and Au-CPC (f) with insets showing the corresponding Fast Fourier Transformations (FFTs). | ||
While Au-CPC is single crystalline due to the slow growth kinetics of CPC-based Au NP synthesis62 (Fig. 1f and Fig S7a†), the Au-citrate obtained through the Turkevich method is polycrystalline63 (Fig. 1c and Fig S7b†) and includes grain boundaries that potentially can affect its electrocatalytic properties.64 To disambiguate the surface adsorbate effects from the Au metal surface structure, we included a control sample, where CPC in the as-synthesized Au-CPC was replaced with citrate. Specifically, we employed a direct ligand exchange of CPC with citrate on the Ti-supported Au-CPC particles using 100 mM sodium citrate solution (see Fig. S1† for details). The surface adsorbates of the resulting ligand-exchanged Au NP electrode (Au-LE) consisted of both CPC and citrate molecules, which was confirmed by Raman spectroscopy and XPS (see Fig. S2† for details).
To ensure that the prepared electrodes had similar surface areas of exposed gold for electrochemical reactions, we employed a common surface characterization method for Au-based electrodes: Pb underpotential deposition (PbUPD, Fig. S3†).65 Based on the Pb stripping curves, all 3 samples showed similar electrochemically active surface areas and their surfaces comprised mostly mixed low index facets.65
Electrochemical performance
To probe the effect of Au NP electrocatalyst surface functionalization on the outcome of an electrochemical process in aprotic medium, we studied these materials in the EC of benzyl bromide. To understand which reaction steps can be inhibited or facilitated by the catalyst surface chemistry, all possible reaction pathways need to be considered. Recently, our group demonstrated that the formation of carboxylate from organohalides (R–Hal) via EC can be described as R–Hal + CO2+2e− → RCO2− + Hal−, which proceeds either through the direct reduction of R–Hal (Fig. 2a, path 1) or through the activation of CO2 (Fig. 2a, path 2).40,57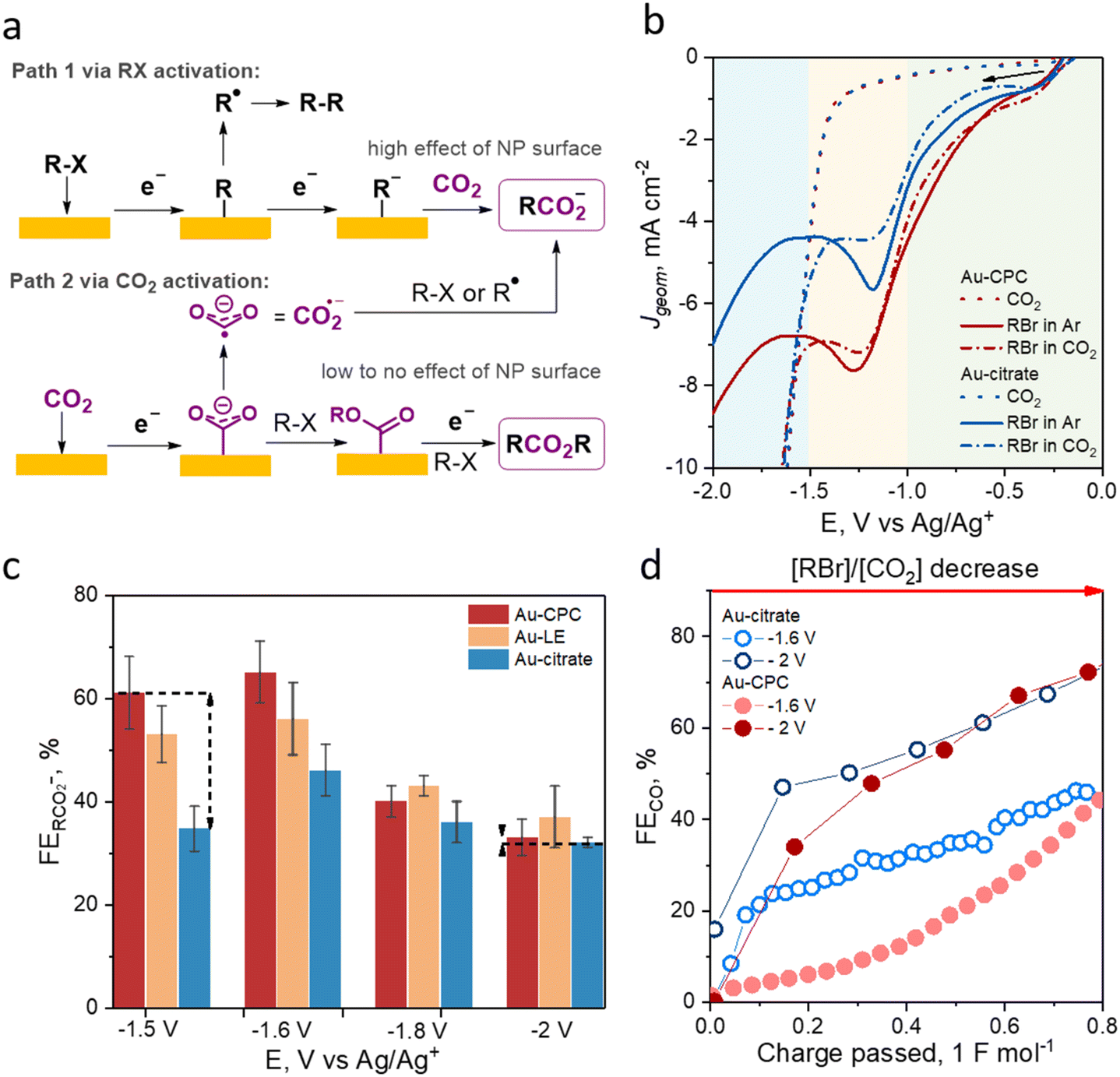 | ||
| Fig. 2 Effect of Au nanoparticle surface chemistry on the EC outcome. (a) Mechanism of benzyl halide (RHal) electrocarboxylation. (b) LSV curves recorded in 0.1 M TEABF4 in CH3CN at 50 mV s−1 in CO2 saturated solution (dotted trace), 25 mM benzyl bromide (RBr) in Ar-saturated solution (solid lines), and 25 mM benzyl bromide in CO2-saturated solution (dash dotted lines) for the Au-CPC electrode (red traces) and the Au-citrate electrode (blue traces). Potential zones are colored as follows: the adsorption of benzyl bromide and the first electron transfer is green, the second electron transfer is yellow, and CO2 activation is blue. (c) FE of carboxylate after passing 1 F mol−1 under a constant applied potential in CO2-saturated 25 mM benzyl bromide in TBABr in CH3CN. (d) Representative changes in the FEs of CO during EC at different applied potentials. | ||
The direct reduction of R–Hal starts with the adsorption of the organohalide on the cathode surface followed by the first electron transfer that results in R–Hal bond cleavage and the formation of R˙.66 The radical R˙ either undergoes radical coupling at less negative applied potentials (E ≥ −1 V vs. Ag/Ag+) or a second electron transfer at more negative applied potentials (E ≤ −1 V vs. Ag/Ag+) with the formation of R−.10,16 This anion in turn attacks CO2 in a nucleophilic manner with the formation of the target carboxylate RCO2−.10,16 The alternative pathway (Fig. 2a, path 2) starts with the adsorption of a CO2 molecule and proceeds through its electrochemical activation with the formation of CO2˙− that further reacts with R–Hal or R˙, yielding the target carboxylate.16,67 Possible side reactions include radical and anion transformations in path 1, leading to dimers, oligomers, etc. or direct CO2 reduction to CO in path 2.
To determine potential zones corresponding to paths 1 and 2, we performed linear sweep voltammetry (LSV) studies (Fig. 2b). As EC via R–Hal activation requires a lower applied potential than that via CO2 activation, we determined the potential range where CO2 activation does not occur by performing LSV in CO2− acetonitrile. The onset potential for CO2 reduction for all saturated solution of an inert electrolyte (0.1 M TEABF4) in the studied Au NPs was about −1.38 V vs. Ag/Ag+ and the potential at 5 mA cm−2 was −1.51 V vs. Ag/Ag+. To determine the electrochemical behavior of benzyl bromide (RBr) on Au-CPC and Au-citrate, we carried out LSV studies of the Ar-saturated solution of 0.1 M TEABF4 in acetonitrile with the addition of 25 mM benzyl bromide. Notably, the peaks corresponding to the first and second electron transfers to benzyl bromide were not resolved and only one broad peak was present, which means that the two reduction steps described above are co-occurring at these potentials.40 We observed an earlier onset on Au-CPC for benzyl bromide reduction by about 70 mV and a higher current density of the two-electron transfer peak by 35% compared to that of Au-citrate. The same trends were observed for the CO2-saturated solution. The similar behavior of the curves with and without CO2 can be attributed to similar initial electrochemical steps (RBr → R˙, R−), which do not involve CO2. The difference in activation energy for RBr at the Au-CPC and Au-citrate surfaces is due to the difference in the interactions of RBr and the reaction intermediates with the catalytic surface. While the Au-citrate surface is hydrophilic and has poor interactions with organic halide molecules, the hydrophobic Au-CPC surface has better affinity to RBr, resulting in earlier onset potential. At the same time, no significant difference in LSV curve behaviour was observed for the reduction of small non-polar CO2 molecules at both surfaces. It is worth noting that Au-citrate showed a noticeable decrease in the benzyl bromide reduction activity in the CO2 saturated solution, suggesting CO2 adsorption on the surface at low potentials that competes with that of benzyl bromide. These observations indicate that Au-citrate has a higher affinity to CO2 compared to that of Au-CPC, which, in turn, shows higher activity towards the electrochemical transformations of benzyl bromide.
Based on the determined boundary of approximately −1.5 V vs. Ag/Ag+ between R–Hal activation and competing activation of both R–Hal and CO2, we selected the potentials of interest for further chronoamperometry (CA) studies with current densities above 5 mA cm−2. To study the potentiostatic behavior of EC when either R–Hal or CO2 activation pathways are dominant, we chose potentials of −1.5 and −2 V vs. Ag/Ag+, respectively. Additionally, to gradually assess the change of the reaction selectivity in the potential window where both pathways are enabled, we studied potentials of −1.6 and −1.8 V vs. Ag/Ag+.
The outcome of the potentiostatic electrolysis at chosen potentials was determined by product distribution analysis both in solution and in the gas phase using 1H nuclear magnetic resonance (NMR) and in-line gas chromatography (GC), respectively. We performed CAs on freshly prepared electrodes to eliminate the possibility of surface chemistry fluctuations. All reactions were carried out under a constant delivery of CO2 at a rate of 14 sccm in a solution of 0.1 M TBABr in acetonitrile with a starting benzyl bromide concentration of 25 mM. The faradaic efficiencies (FEs) of carboxylate after passing 1 F mol−1 equivalent of electrons are shown in Fig. 2c.
Potentiostatic electrolysis at −1.5 V vs. Ag/Ag+ showed a drastic difference between Au-CPC, Au-LE, and Au-citrate cathodes with respective carboxylate FEs of 61%, 53%, and 34%. These results show that the NP surface chemistry significantly affects the preferential adsorption and the energy barriers required for R–Hal activation with citrate being less favorable than CPC for this process.
For the more negative applied potential of −1.6 V vs. Ag/Ag+, we expected a similar dependence of the reaction outcome on the Au NP surface chemistry considering low CO2 activity at this potential and the trend of EC FEs for the R–Hal activation pathway. Indeed, electrolysis at −1.6 V vs. Ag/Ag+ showed the highest EC FE for Au-CPC followed by Au-LE and Au-citrate. Overall, a slight decrease in the applied potential resulted in slightly higher FEs of carboxylate (by 5–12%) for all tested Au NP cathodes due to the decreased contribution of the R˙-driven side reactions associated with the facilitated second electron transfer at higher energies.16,40
At more negative applied potentials of −1.8 and −2 V vs. Ag/Ag+, where the impact of the CO2 activation pathway was expected to be more prominent, only a small difference in the EC FEs for all Au NP cathodes was observed. For CAs at −1.8 and −2 V vs. Ag/Ag+, the respective EC FEs for Au-CPC were 40% and 33%, which were comparable to those of 36% and 32% observed for Au-citrate. The obtained FE data suggest that Au NP surface chemistry has little to no effect on the CO2 activation pathway of EC.
The effect of surface chemistry on CO2 adsorption and activation can also be assessed based on the rate of direct CO2 reduction to CO during EC. We monitored the FE of CO as a function of the passed charge during the EC of benzyl bromide using in-line gas chromatography. The experiments were conducted for Au-citrate and Au-CPC at −2 V vs. Ag/Ag+, representing a high CO2 reactivity region as well as at −1.6 V vs. Ag/Ag+ where the CO2 reduction rate is minimal. At −2 V, no significant difference was observed after passing 10C (the first 20 minutes of the reaction). At −1.6 V vs. Ag/Ag+, the FE of CO for Au-citrate was high from the beginning of the electrolysis and increased from 20% to 46% in the course of the reaction, while the FE of CO for Au-CPC was negligible in the beginning and increased exponentially with benzyl bromide depletion. The CO FE trends observed using GC indicate that at all studied potentials of Au-citrate have either an enhanced affinity to CO2 or a reduced one to R–Hal, compared to that to the Au-CPC surface.
By summarizing both carboxylate and CO FE trends, the observed potentiostatic behavior can be explained from the surface chemistry perspective. The higher polarity of Au-citrate surface species compared to that of Au-CPC creates a more polar environment in the vicinity of the electrode surface. Thus, the adsorption of not very polar R–Hal molecules is less favorable on Au-citrate as R–Hal will need to penetrate the polar shell to undergo direct electron transfer. The suppressed R–Hal adsorption on the electrode surface in turn enables more pronounced competing CO2 adsorption. To validate this hypothesis, further evaluation of the chemical environment in the vicinity of the electrode needs to be performed by determining the surface species stability in the course of electrolysis.
To evaluate surface chemistry stability in the course of EC, we first performed three 1 hour consecutive electrolysis experiments under EC conditions at −1.5 V vs. Ag/Ag+ (Fig. S4†). Even though the CA curves were somewhat similar for all runs for both Au-CPC and Au-citrate, in the case of Au-CPC, the FE of carboxylate increased by a factor of 1.8 for the second electrolysis run and 1.87 for the third electrolysis run compared to the FE of the first short term CA. This change in the EC outcome signifies a change in the Au-CPC cathode surface chemistry associated with the applied current. Interestingly, Au-citrate showed a stable FE of carboxylate regardless of the number of runs.
Catalyst surface characterization
To further understand the evolution of the NP surface chemistry in the aprotic media and under the EC conditions, we studied the Au-CPC and Au-citrate electrodes using ex situ Raman spectroscopy and XPS and determined general changes after the materials were exposed to acetonitrile under the EC conditions at −1.5 V vs. Ag/Ag+ for 1 hour (Fig. 3).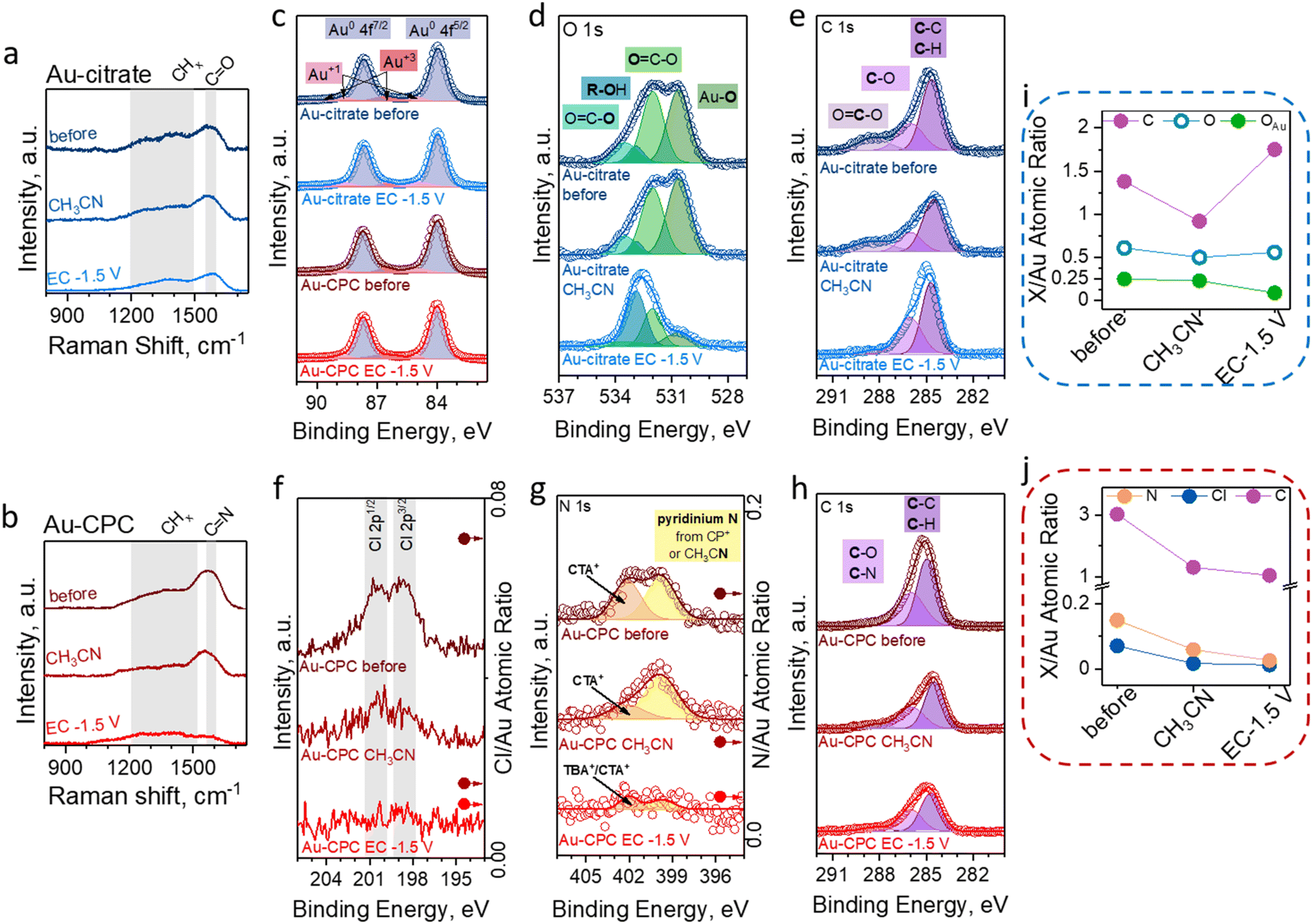 | ||
| Fig. 3 Spectroscopic assessment of the electrode surface chemistry. Raman spectra of the (a) Au-citrate and (b) Au-CPC electrodes as-prepared (before), after extensive washing with acetonitrile (CH3CN), and after EC CA at −1.5 V vs. Ag/Ag+ for 1 hour followed by a thorough washing with CH3CN. XPS evaluation of the surfaces of the Au-citrate electrode as-prepared (dark blue traces), after washing with acetonitrile (blue traces), and after EC CA at −1.5 V for 1 hour (light-blue traces) and the Au-CPC electrode as-prepared (very deep red traces), after washing with acetonitrile (dark red traces), and after EC CA at −1.5 V for 1 hour (red traces). (c) Au 4f peaks before and after electrochemical treatment. (d) O 1s fitted data for the Au-citrate electrode. C 1s fitted data for (e) Au-citrate and (h) Au-CPC. (f) Cl 2p data for the Au-CPC electrodes with the corresponding Cl/Au atomic ratios calculated from XPS. (g) N 1s data for the Au-CPC electrodes with the corresponding N/Au atomic ratios calculated from XPS. For all fitted XPS datasets, raw data are depicted as hollow circles, fitted peaks are coloured, and fitted data are shown as a bold trace. (i) C/Au (violet trace), O/Au (blue trace with hollow circles), and OAu/Au (green trace) ratios calculated from XPS of the Au-citrate electrode as-prepared, after washing with acetonitrile, and after EC at −1.5 V vs. Ag/Ag+ for 1 hour. (j) C/Au (violet trace), Cl/Au (deep blue trace), N/Au (orange trace), and OAu/Au (green trace) ratios calculated from XPS of the Au-CPC electrode as-prepared, after washing with acetonitrile, and after EC at −1.5 V vs. Ag/Ag+ for 1 hour. | ||
First, spectroscopic studies were performed for the Au-citrate electrode. The surface of the freshly prepared electrode was evaluated by Raman spectroscopy (Fig. 3a), showing a band at around 1600 cm−1 which was attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of citrate carboxylic groups.68,69 The detailed examination of the surface using XPS revealed peaks characteristic of adsorbed citrate molecules.70 Specifically, the XPS spectrum of O 1s (Fig. 3d) showed four peaks at 530.6 eV, 532 eV, 532.9 eV, and 533.5 eV that correspond to Au-bound oxygen (OAu), the CO bond of the carboxylic group, oxygen of the alcohol group, and the C–O bond of the carboxylic group, respectively.71 The XPS spectrum of C 1s (Fig. 3e) showed three peaks at 284.5 eV, 286 eV, and 288.5 eV that were assigned to the C–C/C–H and C–O bonds of the alcohol group and the carbon of the carboxylic group, respectively.71 Notably, the XPS signal for Au 4f (Fig. 3c) showed that Au-citrate comprised Au0 with some oxidized Au that behaves as adsorption sites for citrate.71–73
O stretching of citrate carboxylic groups.68,69 The detailed examination of the surface using XPS revealed peaks characteristic of adsorbed citrate molecules.70 Specifically, the XPS spectrum of O 1s (Fig. 3d) showed four peaks at 530.6 eV, 532 eV, 532.9 eV, and 533.5 eV that correspond to Au-bound oxygen (OAu), the CO bond of the carboxylic group, oxygen of the alcohol group, and the C–O bond of the carboxylic group, respectively.71 The XPS spectrum of C 1s (Fig. 3e) showed three peaks at 284.5 eV, 286 eV, and 288.5 eV that were assigned to the C–C/C–H and C–O bonds of the alcohol group and the carbon of the carboxylic group, respectively.71 Notably, the XPS signal for Au 4f (Fig. 3c) showed that Au-citrate comprised Au0 with some oxidized Au that behaves as adsorption sites for citrate.71–73
After the Au-citrate electrode was extensively washed with acetonitrile, we did not observe any qualitative changes in both the Raman spectra and the XPS spectra. However, the quantitative analysis of XPS data showed an approximately 30% decrease in C/Au and O/Au ratios (Fig. 3i), hinting at the loss of some citrate molecules from the surface. Interestingly, the OAu/Au ratio remained the same, suggesting that only not surface-bound citrate molecules were removed from the surface by washing with acetonitrile.
Next, we examined the Au-citrate electrode that was exposed to the EC conditions at −1.5 V vs. Ag/Ag+ for 1 hour. We noticed a decrease in the CO signal in the Raman spectrum. Additionally, the XPS data indicated dramatic changes in the organic species present on the surface. Specifically, by investigating both the XPS spectra of O 1s and C 1s, we noticed a decrease in the OAu and CO2− signals and an increase in the peaks related to the alcohol group (R–OH and C–O).71 This result suggests partial electroreduction of carboxylic groups to alcohols in the adsorbed citrate molecules. Another noticeable change was the 2-fold increase in carbon content, specifically in the C–C/C–H peak. As no nitrogen was detected by XPS, this carbon cannot originate from the electrolyte (TBA+) or solvent, thus we hypothesize that this increase in C content is related to the presence of the products and intermediates of the electroorganic reaction on the Au-citrate surface even after extensive washing with acetonitrile. Overall, citrate is strongly bound to the Au surface and only undergoes chemical transformations under the applied potential, notably without any significant effect on the electrocatalytic properties of the electrocatalyst.
Second, we studied the behavior of the Au-CPC electrode. The as-prepared electrode showed a characteristic band at 1560 cm−1 in the Raman spectrum attributed to CN stretching of the pyridinium group (Fig. 3b).69 Further evaluation of the surface chemistry composition was performed by XPS, showing the presence of Cl (Fig. 3f) and Br (Fig. S5†).71 Additionally, the XPS spectrum of N 1s (Fig. 3g) showed the signals of quaternary ammonium (402 eV) and pyridinium (399.8 eV) nitrogens71,74 that respectively correspond to CTAB and CPC – cationic surfactants used during the synthesis of Au-CPC.
After the Au-CPC electrode was washed with acetonitrile, we did not observe the disappearance of the CN stretching band in the Raman spectrum. The analysis of XPS data showed a noticeable decrease in the Cl 2p, N 1s, and C 1s (Fig. 3h) peaks along with an approximately 3-fold decrease in the Cl/Au, N/Au, and C/Au ratios (Fig. 3j), indicating that the micellar double layer was destroyed and any surfactant molecules that were not bound to Au were removed.
Lastly, we evaluated the surface chemistry of the Au-CPC electrode that was exposed to the EC conditions at −1.5 V vs. Ag/Ag+ for 1 h. Raman spectroscopy showed the disappearance of the CN signal. Further investigation by XPS also confirmed the decrease in N and Cl content on the Au surface to trace amounts; however, Br 3d peaks were still present (Fig. S5†) and the Br/Au ratio increased by 20% due to the remaining surface-bound Br− from the initial surface functionalization and additional adsorption of Br− from the electrolyte and benzyl bromide during EC. Interestingly, while the N/Au ratio decreased by more than a factor of 2, the C/Au ratio decreased by only 20%, indicating that cationic surfactants are not the main source of C on the surface after EC. In analogy with Au-citrate, we believe that the remaining carbon is related to the products and intermediates of the electrochemical reaction. Notably, the presence of a small amount of quaternary ammonium N in the N 1s XPS spectrum suggests that some electrolyte may be adsorbed on the surface. Overall, in the case of Au-CPC, the double layer of micelles is destroyed once the material is introduced into a polar organic solvent, leaving only a monolayer of adsorbed surfactant molecules on the Au surface. Furthermore, this adsorbed surfactant is being stripped from the Au surface during EC, leaving only Br− and reaction-related molecules on the surface, as discussed below. The removal of the surfactant layer plays a crucial role in the electrocatalytic behavior of Au-CPC in EC. The proposed evolution of Au NP surface chemistry is summarized in Fig. 4.
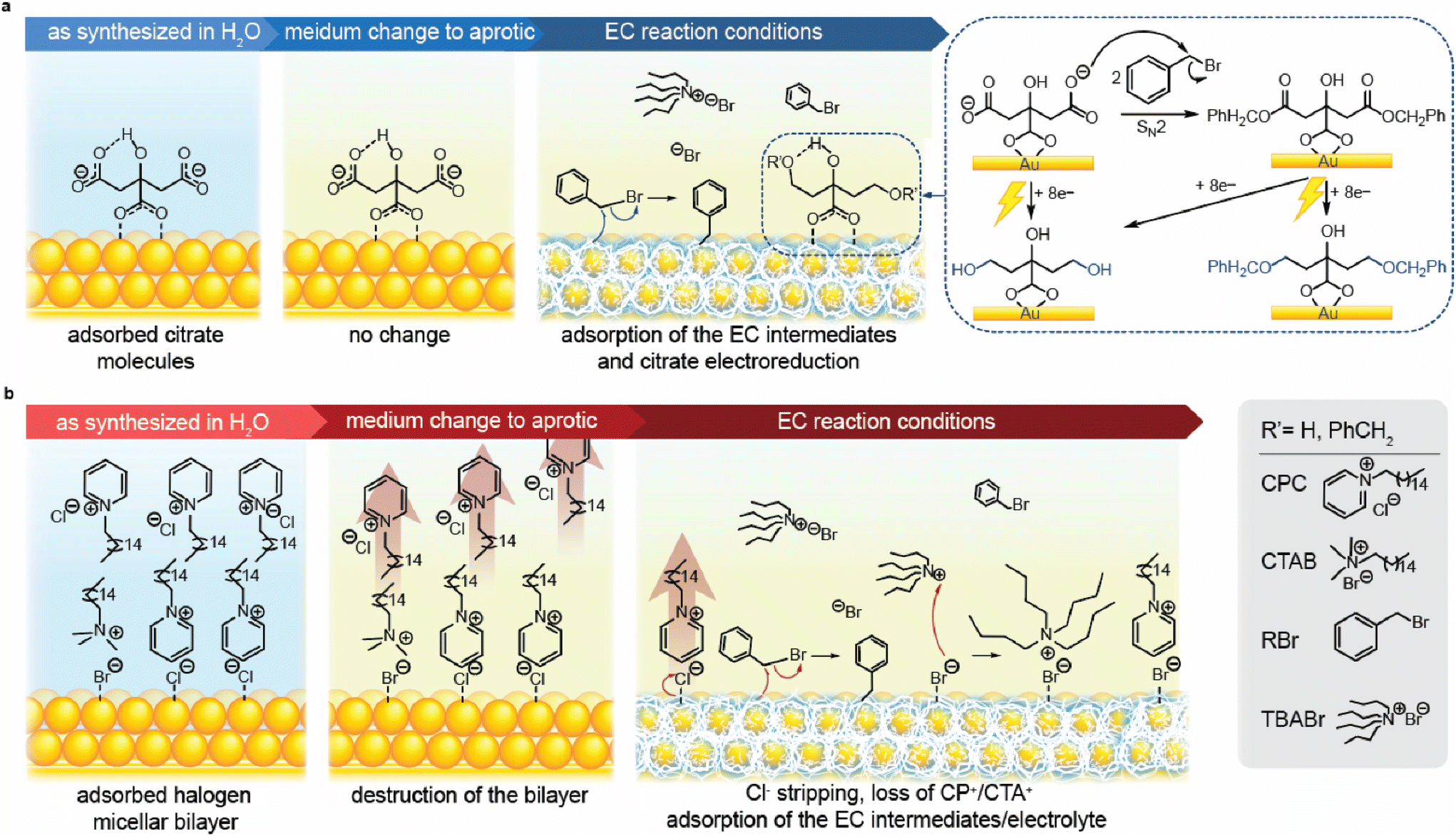 | ||
| Fig. 4 Proposed stepwise evolution of the Au NP surface chemistry driven by medium nature change and electrolysis under the EC conditions. (a) From left to right: citrate-capped Au electrode surface as-prepared in aqueous medium; after extensive washing with acetonitrile showing no change; after CA at −1.5 V vs. Ag/Ag+ under the EC conditions with the schematic inset showing the process of citrate reduction as well as citrate esterification (SN2 substitution) using benzyl bromide and the reduction of the product thereof. (b) From left to right: CPC-capped Au electrode surface as-prepared in aqueous medium; after extensive washing with acetonitrile showing double layer destruction; after CA at −1.5 V vs. Ag/Ag+ under the EC conditions showing surfactant desorption as well as adsorption of the electrolyte and reaction intermediates. The large red transparent arrows in (b) depict the departure of molecules from the Au surface vicinity into the bulk solution. | ||
Proposed surface chemistry evolution mechanism
Based on the obtained spectroscopic data, we reconstructed the surface chemistry evolution for both Au-citrate and Au-CPC. For the former, the surface of the as-synthesised NPs comprised Au atoms with citrate molecules chemisorbed through O–Au bonds. This adsorption can occur through the coordination of one or two carboxylic groups to the metal surface (Fig. 4a).73 Upon immersion of the substrate-supported citrate-stabilized NPs into acetonitrile, surface-bound citrate molecules remain adsorbed on the Au surface without any change. When a negative potential is applied under the EC conditions, the adsorbed citrate molecules undergo reductive transformations. Specifically, the carboxylic groups of citrate can be reduced with the formation of alcohol groups. Moreover, citrate carboxylates can chemically react with the EC precursor, PhCH2Br, through SN2 substitution to produce a corresponding ester. This ester can in turn be reduced to an alcohol or ether. Additionally, the benzyl bromide transformations on the Au surface in the course of EC result in the surface-bound reaction intermediates, including radicals and anions.In the case of Au-CPC, stabilization occurs by the chemisorbed halide ions originating from the employed surfactants and an outer micellar bilayer (Fig. 4b). This bilayer predominantly consists of CPC with some CTAB molecules present as remnants of the NP seed synthesis. The surfactant molecules in the bilayer are arranged with the charged groups interacting with the NP surface electrostatically in the first layer and the aqueous solution in the second layer rendering NP colloidal stability, while the hydrophobic tails of both layers are tucked in the middle. Upon immersion of the substrate-supported CPC-stabilized NPs in acetonitrile, the outer layer of CPC is removed from the double layer rendering the new outer layer hydrophobic. The bilayer to monolayer transition is forced by the medium change from a polar protic solvent to a polar aprotic solvent. This switch in the solvent nature resulted in inefficient stabilization of the charged micellar heads and increased solubility of the hydrophobic tails. The disruption in stabilization leads to the diffusion of the outer layer of CPC into the bulk solution, leaving Au NPs with a monolayer of CPC on the surface. When a negative potential is applied under the EC conditions, most halide ions (Hal) and CP+ are stripped from the Au surface. This stripping occurs through an electron transfer-assisted Au–Hal bond cleavage75,76 followed by desorption of the whole surfactant molecule. Residual Br− remains on the surface due to having stronger Au–Hal interactions compared to Cl−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ,77 resulting in a minimal presence of TBA+ and CP+ on the surface. Similarly to the Au-citrate case, surface-bound reaction intermediates remaining from the EC of RBr are also present based on the relative ratios of C/Au and N/Au found by XPS (Fig. 3j, see Fig. S6† for details).
,77 resulting in a minimal presence of TBA+ and CP+ on the surface. Similarly to the Au-citrate case, surface-bound reaction intermediates remaining from the EC of RBr are also present based on the relative ratios of C/Au and N/Au found by XPS (Fig. 3j, see Fig. S6† for details).
The short exposure of Au-CPC to the reductive electrochemical conditions in organic medium results in a Au surface that is mostly “clean” of the surface adsorbates other than the EC reaction intermediates. In contrast, the Au-citrate surface retains reduced citrate species that occupy some of the catalytic sites and inevitably affect EC in organic media.
Experimental section
Materials
Cetyltrimetylammonium bromide (CTAB, ≥99.0%, BioUltra, for molecular biology), cetylpyridinium chloride (CPC, USP grade), sodium citrate (for molecular biology, ≥99%), iron(III) chloride (FeCl3, reagent grade, 97%), L-ascorbic acid (AA, ≥99%, anhydrous, ACS grade), sodium borohydride (NaBH4, ≥98.0%), gold(III) chloride trihydrate (HAuCl4·3H2O, ≥99.9%), gold(III) chloride solution (HAuCl4, 99.99%, 30 wt% solution in dilute HCl), sodium hydroxide (NaOH, ≥97%, ACS grade), acetonitrile (CH3CN, ACN, MeCN; anhydrous, 99.8%) and benzyl bromide (98%) were purchased from Sigma Aldrich. Tetrabutylammonium bromide (TBABr, C16H36BrN, >98%) was purchased from TCI. Ti foil (>99%) was purchased from VWR. Ar (99.998%) and CO2 (99.999%) were purchased from Praxair. All chemicals were used as received without further purification. All solutions were freshly prepared before use. Ultrapure water (Millipore, 18.2 MΩ cm) was used for all aqueous solutions. The solutions of organic electrolytes were kept over molecular sieves prior to use.Nanoparticle synthesis
000g and redispersed in 50 mL of water. (iii) The entire volume of Au octahedra was placed in a 500 mL Erlenmeyer flask and diluted to 200 mL. Then 32 mL of 0.1 M CPC solution was added and the solution was heated in a 30 °C water bath for 5 minutes under mild stirring. Then the solution was removed from the water bath and 5 mL of 15 mM HAuCl4 solution was added, followed by 16 mL of 0.1 M AA solution. The resulting solution was left undisturbed for 8 hours. (iv) The NPs formed in (iii) were washed for 15 min at 27 °C at 5000g and redispersed in the same volume. Then, the resulting solution was mixed with 0.1 M FeCl3 solution in a 1:1 volume ratio and left for 2 hours under occasional stirring. The etched Au spherical NPs were washed for 15 min at 5000g, topped up with 50 mM CPC, washed with water again, and concentrated. The resulting concentrated solution was further used for imaging and electrode preparation.Materials characterization
Electrochemical experiments
Product analysis
where ni is the number of electrons transferred, F is the Faraday constant, Φi is the volume fraction of the gas product being quantified (calculated by calibrating the GC data using a diluted mixture of gases of known concentrations), I is the current value at the beginning of the measurement, and Fm is the gas flow rate of molar CO2.
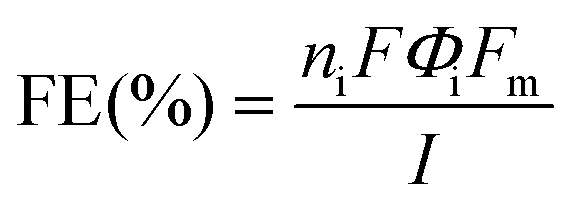
Procedure for quantification. After the electrolysis, 0.7 ml of the catholyte was taken as a sample, and then the solvent was evaporated to obtain a colorless or bright yellow oil (with the presence of colorless crystals). This mixture was dissolved in 0.8 ml of a 33.3 mM solution of 1,2,4,5-tetrachlorobenzene in CDCl3 and analyzed using a Bruker 300 MHz NMR spectrometer. For every sample, 64 scans were recorded. The peak areas of products were compared to the peak area of the standard (2H singlet at 7.55 ppm) that was chosen as it does not interfere with the peak from the electrolyte, the starting bromide and the products.
Since NMR signals are shifted from the standard values due to the electrolyte present in the mixture, 1H-NMR spectra of the reaction mixtures without the electrolyte were also analyzed and compared with the literature data.
Procedure for removing the electrolyte (A). Acetonitrile was removed from the reaction mixture after the electrolysis followed by the addition of diethyl ether. We found that all organic products are soluble in Et2O while the organic electrolyte is completely insoluble.
Procedure for removing the electrolyte (B). After the electrolysis, the solvent was evaporated under reduced pressure and the residual material was acidified with 3 ml of concentrated HCl; after 10 minutes, an additional 3 ml of water was added followed by the extraction of the products with diethyl ether (3 × 20 ml). The extracts were washed with water and dried over anhydrous MgSO4.
After the evaporation of diethyl ether form the solution obtained by method A or B, the residue was analyzed by 1H-NMR with the internal standard.
Conclusions
In conclusion, we found that Au NPs stabilized by cationic surfactants outperform Au NPs stabilized by citrate ligands in the EC reaction. Specifically, at a low applied potential of −1.5 V vs. Ag/Ag+ Au-CPC showed almost two-fold higher FE for the EC of benzyl bromide compared to Au-citrate. This phenomenon is associated with the electrochemically induced surface liberation from the adsorbed molecules in the case of cationic surfactants, resulting in a “clean” hydrophobic surface. In contrast, in the course of the EC reaction on Au-citrate, surface-bound citrate molecules remained on the surface and underwent reductive electrochemical transformations, resulting in a functionalized hydrophilic surface that is not favourable for the EC precursor and intermediate adsorption and facilitates direct CO2 reduction instead. Fundamentally, our findings provide important insights into the rational design of nanostructured electrocatalysts for EC and electroorganic transformations in general. In particular, our results highlight the pivotal role of the surface chemistry of NPs, and thus the choice of their manufacturing method, in their electrocatalytic behavior in aprotic medium.Author contributions
X. Z. and E. S. conducted the experiments; J. M. conducted the experiments, performed formal analysis, and edited the paper; X. M. designed and conducted the experiments, performed formal analysis and visualization as well as wrote the paper; A. K. acquired funds, performed conceptualization and supervision and reviewed and edited the paper.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Waterloo, the National Science and Engineering Research Council of Canada, the Canada Foundation for Innovation, and the Ontario Research Fund for the financial support of this work and the Rodney Smith group for their help with Raman spectroscopy.References
- J. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- C. Schotten, T. P. Nicholls, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Green Chem., 2020, 22, 3358–3375 RSC.
- N. Tanbouza, T. Ollevier and K. Lam, iScience, 2020, 23, 101720 CrossRef CAS PubMed.
- C. A. C. Sequeira and D. M. F. Santos, J. Braz. Chem. Soc., 2009, 20, 387–406 CrossRef CAS.
- X. V. Medvedeva, J. J. Medvedev and A. Klinkova, Adv. Energy Sustainability Res., 2021, 2, 2100001 CrossRef CAS.
- X.-F. Liu, K. Zhang, L. Tao, X.-B. Lu and W.-Z. Zhang, Green Chem. Eng., 2022, 3, 125–137 CrossRef.
- R. Matthessen, J. Fransaer, K. Binnemans and D. E. De Vos, Beilstein J. Org. Chem., 2014, 10, 2484–2500 CrossRef PubMed.
- J. Mao, Y. Wang, B. Zhang, Y. Lou, C. Pan, Y. Zhu and Y. Zhang, Green Carbon, 2024, 2, 45–56 CrossRef.
- S. Wang, T. Feng, Y. Wang and Y. Qiu, Chem.– Asian J., 2022, 17, e202200543 CrossRef CAS PubMed.
- Y. Cao, D. Li, C. Ding, S. Ye, X. Zhang, H. Chi, L. Liu, Y. Liu, J. Xiao and C. Li, ACS Catal., 2023, 13, 11902–11909 CrossRef CAS.
- A. A. Isse and A. Gennaro, Indian J. Chem., Sect. A, 2003, 42, 751–757 Search PubMed.
- D. Yimin, N. Lanli, L. Hui, Z. Jiaqi, Y. Linping and F. Qiuju, Int. J. Electrochem. Sci., 2018, 13, 1084–1095 CrossRef.
- J. J. Medvedev, X. V. Medvedeva, F. Li, T. A. Zienchuk and A. Klinkova, ACS Sustainable Chem. Eng., 2019, 7, 19631–19639 CrossRef CAS.
- V. Rajagopal, P. Manivel, N. Nesakumar, M. Kathiresan, D. Velayutham and V. Suryanarayanan, ACS Omega, 2018, 3, 17125–17134 CrossRef CAS PubMed.
- D. F. Niu, L. P. Xiao, A. J. Zhang, G. R. Zhang, Q. Y. Tan and J. X. Lu, Tetrahedron, 2008, 64, 10517–10520 CrossRef CAS.
- S. Bazzi, G. Le Duc, E. Schulz, C. Gosmini and M. Mellah, Org. Biomol. Chem., 2019, 17, 8546–8550 RSC.
- Y.-C. Lan, H. Wang, L.-X. Wu, S.-F. Zhao, Y.-Q. Gu and J.-X. Lu, J. Electroanal. Chem., 2012, 664, 33–38 CrossRef CAS.
- D.-T. Yang, M. Zhu, Z. J. Schiffer, K. Williams, X. Song, X. Liu and K. Manthiram, ACS Catal., 2019, 9, 4699–4705 CrossRef CAS.
- B. Batanero and F. Barba, Tetrahedron Lett., 2006, 47, 8201–8203 CrossRef CAS.
- H. Senboku, K. Sakai, A. Fukui, Y. Sato and Y. Yamauchi, ChemElectroChem, 2019, 6, 4158–4164 CrossRef CAS.
- B.-L. Chen, Z.-Y. Tu, H.-W. Zhu, W.-W. Sun, H. Wang and J.-X. Lu, Electrochim. Acta, 2014, 116, 475–483 CrossRef CAS.
- S.-F. Zhao, H. Wang, Y.-C. Lan, X. Liu, J.-X. Lu and J. Zhang, J. Electroanal. Chem., 2012, 664, 105–110 CrossRef CAS PubMed.
- A. S. C. Chan, T. T. Huang, J. H. Wagenknecht and R. E. Miller, J. Org. Chem., 1995, 60, 742–744 CrossRef CAS.
- O. Scialdone, A. Galia, A. A. Isse, A. Gennaro, M. A. Sabatino, R. Leone and G. Filardo, J. Electroanal. Chem., 2007, 609, 8–16 CrossRef CAS.
- S. Khaja Lateef, R. Ramesh Raju, S. Krishna Mohan and S. Jayarama Reddy, Synth. Commun., 2006, 36, 31–36 CrossRef.
- A. Dmitrieva, J. J. Medvedev, X. V. Medvedeva, E. Krivoshapkina and A. Klinkova, J. Electrochem. Soc., 2023, 170, 075501 CrossRef CAS.
- Y. Qu, C. Tsuneishi, H. Tateno, Y. Matsumura and M. Atobe, React. Chem. Eng., 2017, 2, 871–875 RSC.
- C.-H. Li, X.-Z. Song, L.-M. Tao, Q.-G. Li, J.-Q. Xie, M.-N. Peng, L. Pan, C. Jiang, Z.-Y. Peng and M.-F. Xu, Tetrahedron, 2014, 70, 1855–1860 CrossRef CAS.
- V. G. Koshechko, V. E. Titov, V. N. Bondarenko and V. D. Pokhodenko, J. Fluorine Chem., 2008, 129, 701–706 CrossRef CAS.
- V. E. Titov, V. N. Bondarenko, V. G. Koshechko and V. D. Pokhodenko, Theor. Exp. Chem., 2008, 44, 271–277 CrossRef CAS.
- G.-Q. Yuan, H.-F. Jiang, C. Lin and S.-J. Liao, Electrochim. Acta, 2008, 53, 2170–2176 CrossRef CAS.
- H. Senboku, H. Komatsu, Y. Fujimura and M. Tokuda, Synlett, 2001, 0418–0420 CrossRef CAS.
- D. A. Tyssee and M. M. Baizer, J. Org. Chem., 1974, 39, 2819–2823 CrossRef CAS.
- C. Li, G. Yuan and H. Jiang, Chin. J. Chem., 2010, 28, 1685–1689 CrossRef CAS.
- F. Köster, E. Dinjus and E. Duñach, Eur. J. Org. Chem., 2001, 2001, 2507–2511 CrossRef.
- J. A. Cleary, M. S. Mubarak, K. L. Vieira, M. R. Anderson and D. G. Peters, J. Electroanal. Chem. Interfacial Electrochem., 1986, 198, 107–124 CrossRef CAS.
- J. J. Medvedev, X. V. Medvedeva, H. Engelhardt and A. Klinkova, Electrochim. Acta, 2021, 387, 138528 CrossRef CAS.
- D. M. Heard and A. J. J. Lennox, Angew. Chem., Int. Ed., 2020, 59, 18866–18884 CrossRef CAS PubMed.
- W. Choi, D. H. Won and Y. J. Hwang, J. Mater. Chem. A, 2020, 8, 15341–15357 RSC.
- J. Linnemann, K. Kanokkanchana and K. Tschulik, ACS Catal., 2021, 11, 5318–5346 CrossRef CAS.
- L. Fan, C. Xia, F. Yang, J. Wang, H. Wang and Y. Lu, Sci. Adv., 2020, 6, eaay3111 CrossRef CAS PubMed.
- A. A. Feidenhans'l, Y. N. Regmi, C. Wei, D. Xia, J. Kibsgaard and L. A. King, Chem. Rev., 2024, 124, 5617–5667 CrossRef PubMed.
- C. Wang, Q. Zhang, B. Yan, B. You, J. Zheng, L. Feng, C. Zhang, S. Jiang, W. Chen and S. He, Nano-Micro Lett., 2023, 15, 52 CrossRef CAS PubMed.
- C. Xiao, B.-A. Lu, P. Xue, N. Tian, Z.-Y. Zhou, X. Lin, W.-F. Lin and S.-G. Sun, Joule, 2020, 4, 2562–2598 CrossRef CAS.
- A. Klinkova, P. De Luna, C.-T. Dinh, O. Voznyy, E. M. Larin, E. Kumacheva and E. H. Sargent, ACS Catal., 2016, 6, 8115–8120 CrossRef CAS.
- L. Han, C. Wang, S. Luo, Y. Zhou, B. Li and M. Liu, Green Energy Environ., 2024, 9, 1314–1320 CrossRef CAS.
- I. Terekhina, J. White, A. Cornell and M. Johnsson, ACS Appl. Nano Mater., 2023, 6, 11211–11220 CrossRef CAS.
- J. Mosquera, D. Wang, S. Bals and L. M. Liz-Marzán, Acc. Chem. Res., 2023, 56, 1204–1212 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- A. Klinkova, E. M. Larin, E. Prince, E. H. Sargent and E. Kumacheva, Chem. Mater., 2016, 28, 3196–3202 CrossRef CAS.
- N. Baig, I. Kammakakam and W. Falath, Mater. Adv., 2021, 2, 1821–1871 RSC.
- Z. Cao, D. Kim, D. Hong, Y. Yu, J. Xu, S. Lin, X. Wen, E. M. Nichols, K. Jeong, J. A. Reimer, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2016, 138, 8120–8125 CrossRef CAS PubMed.
- Q. Zhu, C. J. Murphy and L. R. Baker, J. Am. Chem. Soc., 2022, 144, 2829–2840 CrossRef CAS PubMed.
- J. J. Medvedev, X. V. Medvedeva, F. Li, T. A. Zienchuk and A. Klinkova, ACS Sustainable Chem. Eng., 2019, 7, 19631–19639 CrossRef CAS.
- X.-K. Wan, J.-Q. Wang, Z.-A. Nan and Q.-M. Wang, Sci. Adv., 2017, 3, e1701823 CrossRef PubMed.
- Q. Zhu, X. Huang, Y. Zeng, K. Sun, L. Zhou, Y. Liu, L. Luo, S. Tian and X. Sun, Nanoscale Adv., 2021, 3, 6330–6341 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55 RSC.
- E. Galati, M. Tebbe, A. Querejeta-Fernández, H. L. Xin, O. Gang, E. B. Zhulina and E. Kumacheva, ACS Nano, 2017, 11, 4995–5002 CrossRef CAS PubMed.
- W. Niu, S. Zheng, D. Wang, X. Liu, H. Li, S. Han, J. Chen, Z. Tang and G. Xu, J. Am. Chem. Soc., 2009, 131, 697–703 CrossRef CAS PubMed.
- J. Kimling, M. Maier, B. Okenve, V. Kotaidis, H. Ballot and A. Plech, J. Phys. Chem. B, 2006, 110, 15700–15707 CrossRef CAS PubMed.
- S. Mondal, S. K. De, R. Jana, A. Roy, M. Mukherjee, A. Datta, B. Satpati and D. Senapati, ACS Appl. Energy Mater., 2021, 4, 3017–3032 CrossRef CAS.
- X. Min, Y. Chen and M. W. Kanan, Phys. Chem. Chem. Phys., 2014, 16, 13601–13604 RSC.
- Y.-F. Huang, D.-Y. Wu, A. Wang, B. Ren, S. Rondinini, Z.-Q. Tian and C. Amatore, J. Am. Chem. Soc., 2010, 132, 17199–17210 CrossRef CAS PubMed.
- J. A. Rosso, S. G. Bertolotti, A. M. Braun, D. O. Mártire and M. C. Gonzalez, J. Phys. Org. Chem., 2001, 14, 300–309 CrossRef CAS.
- C. Farcau, N. M. Sangeetha, N. Decorde, S. Astilean and L. Ressier, Nanoscale, 2012, 4, 7870–7877 RSC.
- D. Lin-Vein, N. Colthup, W. Fateley and J. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, 1991 Search PubMed.
- L. F. Gorup, B. Perlatti, A. Kuznetsov, P. A. de P. Nascente, E. P. Wendler, A. A. Dos Santos, W. R. Padilha Barros, T. Sequinel, I. de M. Tomitao, A. M. Kubo, E. Longo and E. R. Camargo, RSC Adv., 2020, 10, 6259–6270 RSC.
- X-ray Photoelectron Spectroscopy (XPS) reference pages. https://www.xpsfitting.com/.
- J. M. Gisbert-González, W. Cheuquepán, A. Ferre-Vilaplana, E. Herrero and J. M. Feliu, J. Electroanal. Chem., 2020, 875, 114015 CrossRef.
- D.-B. Grys, B. de Nijs, A. R. Salmon, J. Huang, W. Wang, W.-H. Chen, O. A. Scherman and J. J. Baumberg, ACS Nano, 2020, 14, 8689–8696 CrossRef CAS PubMed.
- R. J. J. Jansen and H. van Bekkum, Carbon, 1995, 33, 1021–1027 CrossRef CAS.
- R. C. Salvarezza and P. Carro, J. Electroanal. Chem., 2018, 819, 234–239 CrossRef CAS.
- K. Kwan Li, C.-Y. Wu, T.-H. Yang, D. Qin and Y. Xia, Acc. Chem. Res., 2023, 56, 1517–1527 CrossRef CAS PubMed.
- S. K. Meena, S. Celiksoy, P. Schäfer, A. Henkel, C. Sönnichsen and M. Sulpizi, Phys. Chem. Chem. Phys., 2016, 18, 13246–13254 RSC.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional electrode characterization for Au-LE and electrochemical electrode stability data. See DOI: https://doi.org/10.1039/d4nr04135f |
| This journal is © The Royal Society of Chemistry 2025 |