
Correlating photochemical H2 production and excited state lifetimes of heterostructured and doped ZnCdS nanoparticles†
Mathew T.
Fortunato
a,
Joseph M.
O'Shea
 b,
Jie
Huang
a,
Hashini
Chandrasiri
b,
Eun Byoel
Kim
b,
Abdelqader M.
Jamhawi
b,
A. Jean-Luc
Ayitou
*b,
Preston T.
Snee
*b and
Claudia
Turro
*a
b,
Jie
Huang
a,
Hashini
Chandrasiri
b,
Eun Byoel
Kim
b,
Abdelqader M.
Jamhawi
b,
A. Jean-Luc
Ayitou
*b,
Preston T.
Snee
*b and
Claudia
Turro
*a
a4109 Newman & Wolfrom Laboratory, 100 W 18th Ave, Columbus, OH 43210, USA. E-mail: turro.1@osu.edu
bSES Building, 845 W. Taylor St., Chicago, Il 60607, USA. E-mail: aayitou@uic.edu; sneep@uic.edu
First published on 9th December 2024
Abstract
A variety of ZnCdS-based semiconductor nanoparticle heterostructures with extended exciton lifetimes were synthesized to enhance the efficacy of photocatalytic hydrogen production in water. Specifically, doped nanoparticles (NPs), as well as core/shell NPs with and without palladium and platinum co-catalysts, were solubilized into water using various methods to assess their efficacy for solar H2 fuel synthesis. The best results were obtained with low bandgap ZnCdS cores and ZnCdS/ZnS core/shell NPs with palladium co-catalysts. The results, augmented with DFT and tight binding electronic structure calculations, revealed the importance of exciton charge carrier separation via tunneling. While the systems studied here were photocatalytically active, they nonetheless lagged behind the quantum efficiency observed from “gold standard” CdSe/CdS·Pt dot-in-rod nanoparticles as evident from quantum efficiencies that were estimated to be 0.5 → 2%.
Introduction
Tuning the composition, size, and shape of nanomaterials affords the ability to maximize the properties for applications in several fields such as solar fuel production.1 Currently, one of the best-known systems for this purpose is composed of quantum-confined CdSe core nanoparticles (NPs), also called quantum dots, overcoated with rod-shaped CdS shells.2–5 Tipping the CdS shell with a platinum co-catalyst greatly enhances solar H2 production due to efficient separation of exciton charge carriers.6,7 While highly effective for this purpose, this system is complex both structurally and photochemically.8 Furthermore, multiple steps are required to synthesize CdSe/CdS dot-in-rod catalysts. Herein, we sought to prepare a simpler isotropic structure, ZnCdS, with and without a passivating shell and metal co-catalyst for solar H2 generation.This work was motivated by the fact that surface passivated ZnCdS/ZnS NPs were recently shown to be more effective reducing agents for organic substrates compared to the core ZnCdS alone.9 This is contrary to the expectation that the electronically insulating shell should reduce the redox activity towards substrates. The unexpected activity resulted from the 3-fold enhancement in the lifetime of the core–shell nanoparticles due to the shell's passivation of surface trap states, which, in turn, enhanced the likelihood of substrate reduction before the catalyst relaxed to the ground state.9 Additional efficacy may have been realized from stronger substrate binding to the ZnS shell and the fact that the ZnCdS/ZnS system is quasi-type II due to hole delocalization through the core and shell. The latter originates from the fact that sulphur p-orbitals comprise the hole states of both the core and shell. It is notable that suppression of trap states has been found to enhance the photocatalytic activity of CdS/ZnS and CdSe/CdS NPs for H2 synthesis.10,11
The present study further explores the ZnCdS material system to ascertain a correlation between photochemical dynamics and kinetics with the yield and activity of hydrogen evolution reaction.12–14 A successful demonstration would advance green energy applications as core/shell NPs have greater long-term (photo and thermal) stability compared to the core alone.15 This is especially true for zinc sulphide passivated materials that are more environmentally compatible compared to the cadmium alternative. Furthermore, the materials under study are simpler to prepare compared to CdSe/CdS·Pt dot-in-rod catalysts that are the current “gold standard”. Copper-doped CdS NPs were also synthesized and investigated for H2 catalytic competency, as copper dopants capture holes which greatly extends the excited state lifetimes.16 Along the same lines, ZnCdS/ZnS NPs with electron-sequestering palladium and platinum co-catalysts were examined.11,14,17 Aside from quantifying the effects of a co-catalyst, these materials provide a more meaningful comparison to results obtained from CdSe/CdS·Pt NPs. Theoretical results, from DFT on small quantum dots to tight binding calculations on large dot-in-rod heterostructures, augment the conclusions presented herein. Last, the competence of polymer encapsulated NPs for catalysis is demonstrated.
Results and discussion
Synthesis
Various samples of ZnCdS NPs and heterostructures were synthesized, starting with ∼450 nm bandgap ZnCdS core and ZnCdS/ZnS core–shell NPs using procedure adapted from prior reports.9,18,19 As these procedures do not afford substantial bandgap tunability, it was necessary to modify these protocols to create larger ZnCdS cores with a ∼485 nm bandgap using a less reactive tri-octylphosphine sulphide precursor. Metal tipping of the core/shell NPs with platinum and palladium was accomplished using the heat-up method developed by Amirav et al.6 Additionally, photochemical deposition of palladium onto ZnCdS/ZnS catalysts was performed using the method of Dukovic;7 however, the platinum salt precursor was found to be unreactive under light driven conditions. Copper- and zinc-doped CdS NPs were synthesized using Cu4(SPh)6 and Zn4Se4(SPh)16 nucleation catalysts via the cluster seed method.20–22 CdSe/CdS dot-in-rod materials were prepared using small CdSe core dots that were overcoated according to ref. 4, and were tipped with platinum via thermolysis, see ref. 6. Optical and TEM data for these samples are provided in Fig. S1 and S2.† Further, the samples were water solubilized with cap exchange using Zn-ligated 11-mercaptoundecanoic acid,23 as well as using an amphiphilic polymer for encapsulation.24,25Water solubilization and photochemical H2 synthesis
All samples were diluted to ∼0.4 OD at 410 nm, while slight differences were accounted for by normalizing the H2 data by the absorptivity at the excitation wavelength. Fresh sodium sulphide and sodium sulphite sacrificial reductants were added immediately before each measurement. Light-driven H2 production experiments were performed over 4 hours using a 410 nm light emitting diode (LED) excitation source.As-prepared semiconductor nanoparticles were coated with oleic acid as a stabilizer, which rendered the materials hydrophobic. To convert them into an aqueous colloid for photocatalytic H2 production, the NPs must either be encapsulated with amphiphilic polymers or undergo ligand (cap) exchange as illustrated in Fig. 1A.26 Previous research has employed the latter, as guided by the concept that cap exchange should enhance the exposure of the semiconductor surface to substrates and sacrificial reductants.6,11 However, cap exchanged colloidal NPs lack long term stability, beyond a few days, which impairs their use as an alternative energy resource.23 This prompted us to explore the photocatalytic activity of amphiphilic polymer encapsulated NPs, since such dispersions can be stable over a multi-year timescale because, in part, the native NP ligands are left intact.24 It should be noted that the stability comes at the expense of a larger hydrodynamic diameter. To this end CdSe/CdS dot-in-rod with Pt co-catalysts were prepared according to established methods (see Fig. 1B). Next, these materials were water solubilized with both 40% octylamine-modified poly(acrylic acid) encapsulation and zinc-ligated mercaptoundecanoic acid cap exchange. It is evident in the photocatalytic H2 production data shown in Fig. 1C that the activity of polymer encapsulated NPs is nearly equivalent to that of cap exchanged materials. As a result, we used polymer encapsulated NPs for the remaining studies; however, we note that precipitation was occasionally observed especially if a material was highly active.
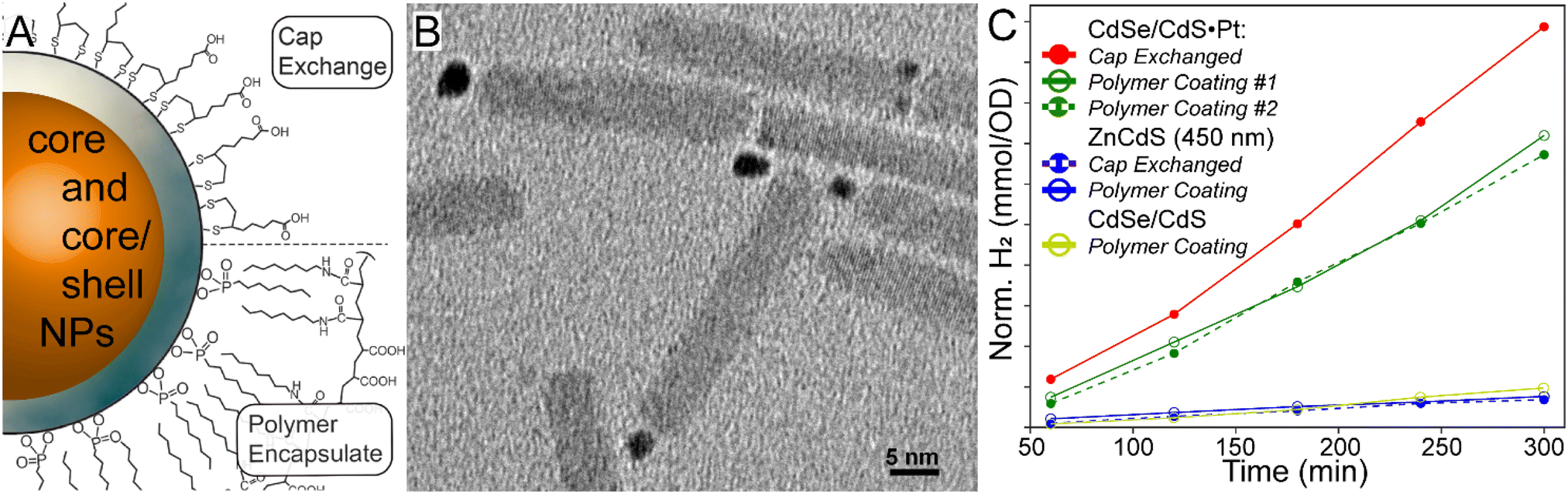 | ||
| Fig. 1 A. Scheme of nanoparticle cap exchange and polymer encapsulation for nanoparticle water solubility. B. TEM micrograph of dot-in-rod CdSe/CdS·Pt photocatalysts. C. H2 synthesis yields, normalized for catalyst absorption at 410 nm, over a 4-hour period of various samples in this study. | ||
ZnCdS core and related heterostructures were examined to potentially correlate the excited state lifetime with the photochemical activity. Before examining the details of the various structure–property relationships, it is instructive to first evaluate the performance of the ZnCdS and heterostructured materials against the “gold standard” CdSe/CdS·Pt system. As shown in Fig. 1C, the latter outperformed the ZnCdS-based materials prepared in this study; nonetheless, the results reveal the underlying photochemical mechanisms important in solar fuel synthesis that may be instructive for designing better catalysts.
Core vs. core/shell heterostructure
Interest in ZnCdS NPs was instigated by our previous demonstration of enhanced reduction efficiency of core/shell ZnCdS/ZnS vs. ZnCdS nanoparticles towards an organic substrate.9 The result was counterintuitive because a ZnS shell should hinder electron dislocation from the core NP. However, the shell also enhanced the excited state lifetime due to surface trap passivation, which resulted in better photoreducing effects. Furthermore, the degeneracy of the hole within the core/shell NP may have played a role, either by reducing the barrier to charge transfer or by enabling hole removal to “reset” the catalyst back into an active charge-neutral state. This study evaluated whether the same photodynamics may be applicable for light-driven hydrogen evolution reaction (HER) using core/shell ZnCdS-based NPs, which would benefit from the greater stability of core/shell heterostructures. The effects of other lifetime enhancing modifications were also interrogated for their potential to positively impact photochemical HER.Small (∼485 nm) and larger bandgap (∼450 nm) ZnCdS core nanomaterials were prepared and subsequently shelled with zinc sulphide. The absorption spectra of these ZnCdS-based materials are shown in Fig. 2; additional optical and electron microscopy characterizations are provided in Fig. S3 and S4.† All samples were water solubilized using polymer encapsulation. Fresh portions of the ∼450 nm bandgap core and core/shell NPs were interrogated with transient absorption spectroscopy. Kinetics of the parent bleach shown in Fig. 3A reveal a modest elongation (∼43%) of the excited state lifetime of the core/shell NPs (228 ± 4 ns) compared to the core (160 ± 6 ns), see Table S1 of the ESI.† As in our earlier study this is attributed to trap state passivation; furthermore, the average lifetimes are significantly greater than the typical ∼10's ns observed from as-prepared quantum dots. This is due to the presence of very long >100 ns components that comprise ∼50% of the decay kinetics for both the core and core/shell NPs. We attributed these behaviours to electrons bound to surface trap sites,27 mostly due to the correlation with the kinetics of a low energy absorption feature that decayed along a similarly long ∼100's of ns timescale as the parent bleach recovery (Fig. S4†). This was observed in previous studies and was attributed to trapped charge carriers.10,14,28 It is notable that the samples’ lifetimes are greater than previously reported,14 which is simply due to the wider range of time resolutions (femto- to microsecond) available using our current time-resolved laser system.
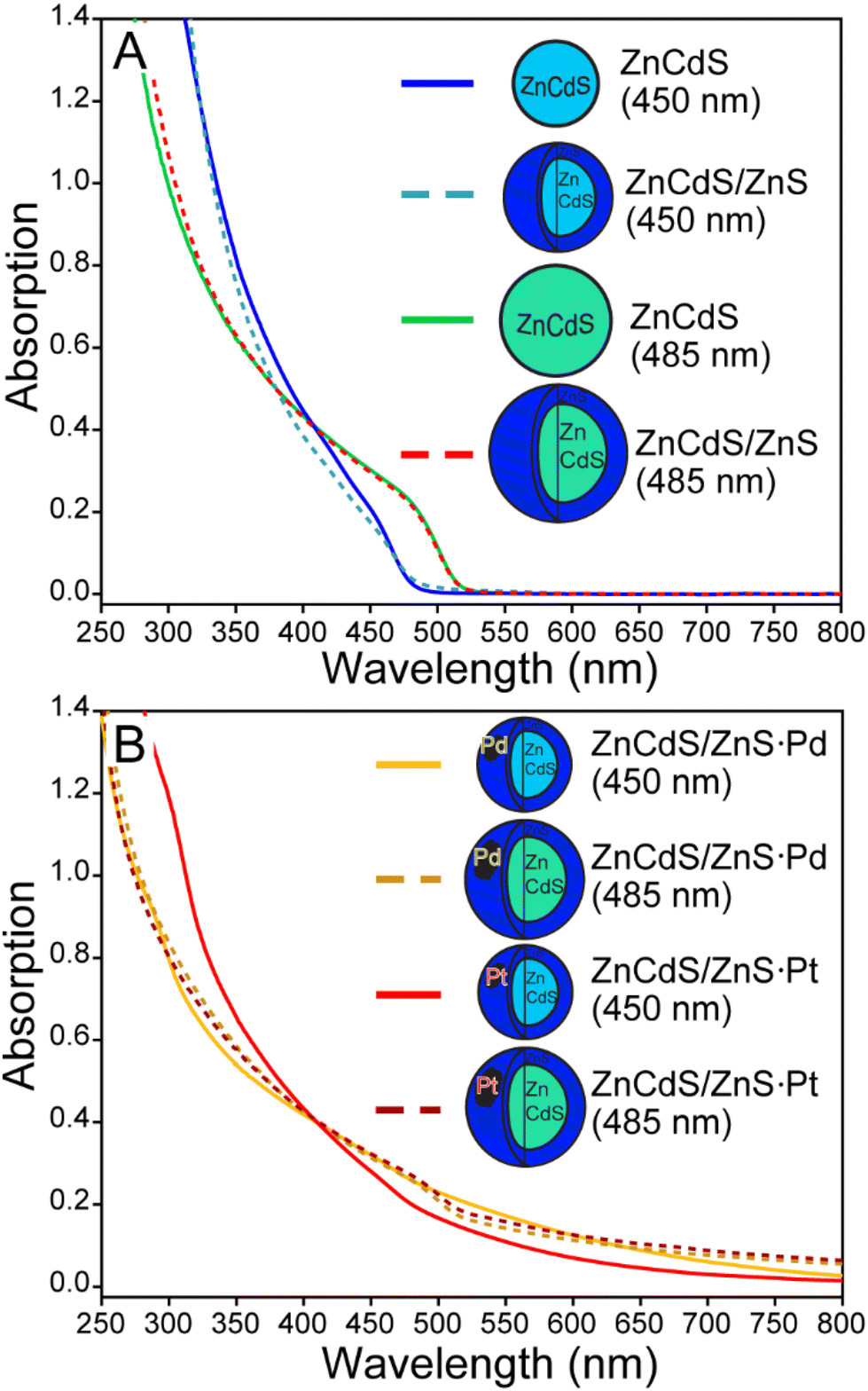 | ||
| Fig. 2 A. Absorption spectra of various ZnCdS and ZnCdS/ZnS nanoparticles prepared in this study. B. The absorption spectra of semiconductor-metal co-catalyst created using thermolysis. | ||
 | ||
| Fig. 3 A. Kinetics of the parent bleach of large bandgap core ZnCdS and ZnCdS/ZnS NPs. Inset: Heatmap of the same. B. H2 synthesis yields, normalized by absorption at the excitation wavelength, for various ZnCdS heterostructures prepared in this study. | ||
Having established the basic photophysical properties of the small and large ZnCdS and ZnCdS/ZnS NPs, the relative production of H2 from each system was compared as shown in Fig. 3B. It was found that the lower bandgap 485 nm ZnCdS NPs produced significantly more H2 over a 4-hour period (>10×) compared to the 450 nm ZnCdS particles. Concerning surface passivation, the characterizations revealed minimal-to-no beneficial effect to H2 productivity as a ∼5% increase in H2 was observed for the 450 nm core/shell compared to the core, which is consistent with a previous report on small sized CdS and CdS/ZnS NPs.11 In contrast, the lower bandgap core/shell NPs’ efficacy was impaired by zinc sulphide shelling by 7×. Overall, the sum of these results negates our hypothesis that the heterostructure's longer lifetime will correlate with greater H2 productivity. Most likely the findings are related to the complexity of the HER, including sacrificial reductants and aqueous solvent, and thus the shell served only to impair various redox processes in the case of the overcoated low bandgap ZnCdS.
Lifetime enhancement with hole-sequestering Cu dopants
Doping a semiconductor can significantly extend the excited state lifetime, on the order of 100's of ns, which is ostensibly attractive with an exciton splitting Cu(1+) guest. This is because a plethora of experimental and theoretical research has demonstrated exciton hole capture by a copper dopant embedded in a CdS matrix – see the density functional theory result in Fig. 4B, inset. Thus, the conduction band electron may remain free to reduce protons and/or water with a greater H2 yield realized due to suppressed electron–hole recombination.29–35 Copper-doped CdS NPs were prepared via the cluster seed method,21 whereby Cu4(SPh)62− organometallic clusters nucleate the formation of the nanoparticles. This procedure assures that each and every doped NP has four copper ions, thus the characterization data are not convoluted with results from pristine CdS semiconductors. Control Zn4:CdS NPs were prepared using Zn4(SPh)62− nucleation catalysts; spectra of the doped and control samples are shown in Fig. 4A (inset) and Fig. S5 of the ESI.† Both the doped and control NPs emit with significantly enhanced Stokes’ shifts, which calls into question whether the Cu4:CdS NPs’ electronic properties are due to the dopants or surface trap states, as is the case with Zn4:CdS NPs. Photoluminescence (PL) lifetime analysis shown in Fig. 4A was used to differentiate the results, which revealed that the Cu4:CdS NPs emit over 335 ± 15 ns whereas the control Zn4:CdS NPs have a 74 ± 4 ns lifetime (Table S2†). With this observation and other characterizations provided in ref. 21, we conclude that the photodynamics of water solubilized Cu4:CdS NPs are dominated by hole sequestration by the dopants.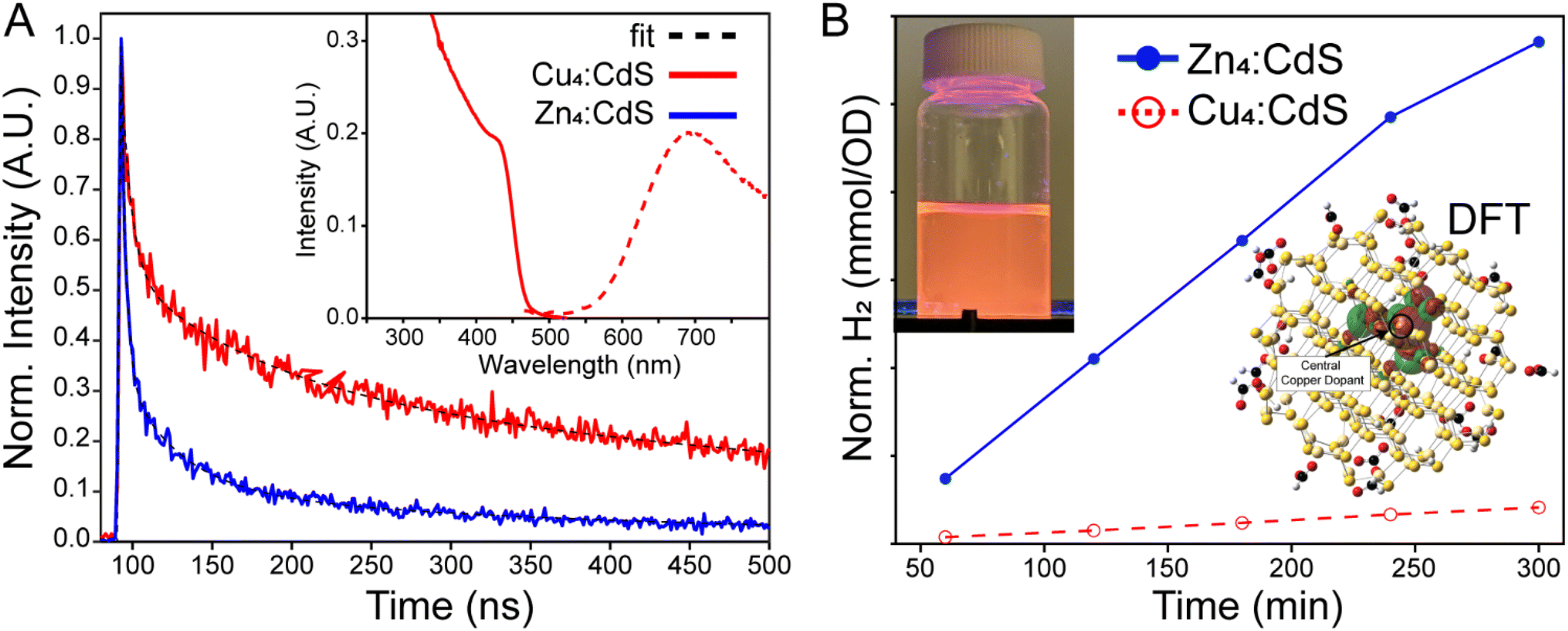 | ||
| Fig. 4 A. Time-resolved PL from Cu4:CdS and Zn4:CdS revealed significantly enhanced lifetime in the copper-doped materials, which distinguish them from the zinc-doped controls that emit from surface trap states. Inset: Absorption and emission of Cu4:CdS NPs. B. H2 produced over time using Cu4:CdS and control Zn4:CdS catalysts in water. Inset: The TDDFT/NTO orbital of the hole state is centred on copper in a (HCO2H)18CuCd113S114 cluster. Also, a photograph of the Cu4:CdS NPs emitting in water. | ||
The control Zn4:CdS NPs exhibit photocatalytic activity for HER as illustrated in Fig. 4B. However, the efficacy of the copper-doped nanoparticles, despite their enhanced lifetime, is nearly zero. These results could be explained using the trends observed in the photophysical mechanisms of H2 synthesis using CdSe/CdS NPs. Specifically, sacrificial reductants must “reset” the catalyst after H+ reduction, and it is this step that is rate limiting for CdSe/CdS.36 As it applies to Cu4:CdS NPs, it may be true that the efficacy of the sacrificial reductants is substantially mollified by hole sequestration by the copper, which, in turn, essentially leaves the photocatalyst in a charged “off” state after reducing a single substrate. At the same time, it has been noted that exciton splitting by electron-capturing nickel dopants in CdS hosts likewise suppresses H2 evolution,37 which implies that the use of dopants for exciton splitting is not an effective strategy due to a mechanism that is currently obscure to us.
Metal co-catalysts
As sequestration of the ZnCdS NP hole by copper dopants proved highly deleterious towards H2 generation, exciton separation via electron localization to a metal co-catalyst was explored to realize enhanced photocatalytic efficiency. This approach is highly successful when applied to a CdSe/CdS dot-in-rod heterostructure,6 whereby a Pt cluster is conjoined to the apex of the CdS shell. The CdSe/CdS·Pt system and similar variants have been the subject of extensive research, the consensus for which is that the exciton is divided between a CdSe core-bound hole and a Pt-sequestered electron that can effectively split water and/or reduce H+.11,35,38–40 To ascertain whether the same enhancement of photocatalytic efficacy could be observed on ZnCdS/ZnS NPs, palladium and platinum was deposited onto the core/shell semiconductor both photochemically and by thermal decomposition of metal precursors.6 The resulting optically clear semiconductor-metal dispersions were dark brown to black in colour. Furthermore, the emission of the core/shell NP was quenched, which is evidence of exciton dissociation, see the characterization data provided in Fig. S6–S11.† The catalysts were processed by addition of isopropanol/methanol to isolate the nanoparticulates that were subsequently water solubilized with polymer encapsulation. The spectra of these samples are shown in Fig. 2B and 5.Initial studies revealed modest alterations to the photochemical efficacy of hydrogen production due to the addition of a platinum co-catalyst. The larger bandgap (450 nm) ZnCdS/ZnS·Pt NPs actually produced −36% less H2 compared to the core/shell semiconductor alone, while the smaller bandgap (485 nm) NPs generated +63% more H2. The palladium co-catalysts had a minimal effect on H2 generation, as there was no alteration of the productivity of the larger bandgap (450 nm) ZnCdS/ZnS·Pd NPs, while a +28% increase in H2 was observed with the smaller bandgap (485 nm) NPs. These results pale in comparison to the substantial increase in activity of CdSe/CdS dot-in-rod NPs due to the addition of a metal co-catalyst, which prompted further experimental and theoretical investigations. To this end it was noted that excessive use of precursors and/or thermolysis at overly high temperatures resulted in free metal nanoparticles as quantified using size-selective precipitation (see Fig. S6 and S7†). While the thermal syntheses of ZnCdS/ZnS·Pt(Pd) NPs were optimized to minimize the nucleation of free metal particles, it is nonetheless probable that their presence mitigated the efficacy of photocatalysis.
An alternative method to synthesize semiconductor-metal co-catalyst NPs was explored to minimize the self-nucleation of the metal component. Following the general protocol of Dukovic et al.,7 a mixture of palladium acetate, triethylamine, and ZnCdS/ZnS NPs was continuously excited in a UV reactor system to reduce the metal salt onto the surface of the semiconductor (i.e. photodeposition).41 After ∼1/2 h the samples became light to dark brown; additional incubation in the reactor did not alter the colour further. It should be noted that the platinum precursor Pt(cod)2 was found to be unreactive under these conditions. As shown in Fig. 5, the photochemically prepared ZnCdS/ZnS·Pd NPs display less absorption by the metal co-catalyst compared to samples created by thermolysis. This may be due to a lack of free palladium particles, smaller Pd co-catalysts, or both. As shown in the inset of Fig. 5, 22- to 69-fold more H2 was generated by the 450 nm and 485 nm bandgap ZnCdS/ZnS·Pd NPs, respectively; note that the productivity nonetheless lags behind that observed with CdSe/CdS·Pt NPs. It is thus sensible to ascribe the enhancement to better contact between the host semiconductor and metal as has been cited previously as a significant factor for CdS·Pt catalysts.42 At the same time, prior research has also attributed variations in the photochemical efficacy among several CdS-metal systems to several factors including alloying the metal with sulphur,17,42 the presence of multiple metal oxidation states,43,44 the metal/semiconductor stoichiometry,17,45 and the metal particle size.44,46,47
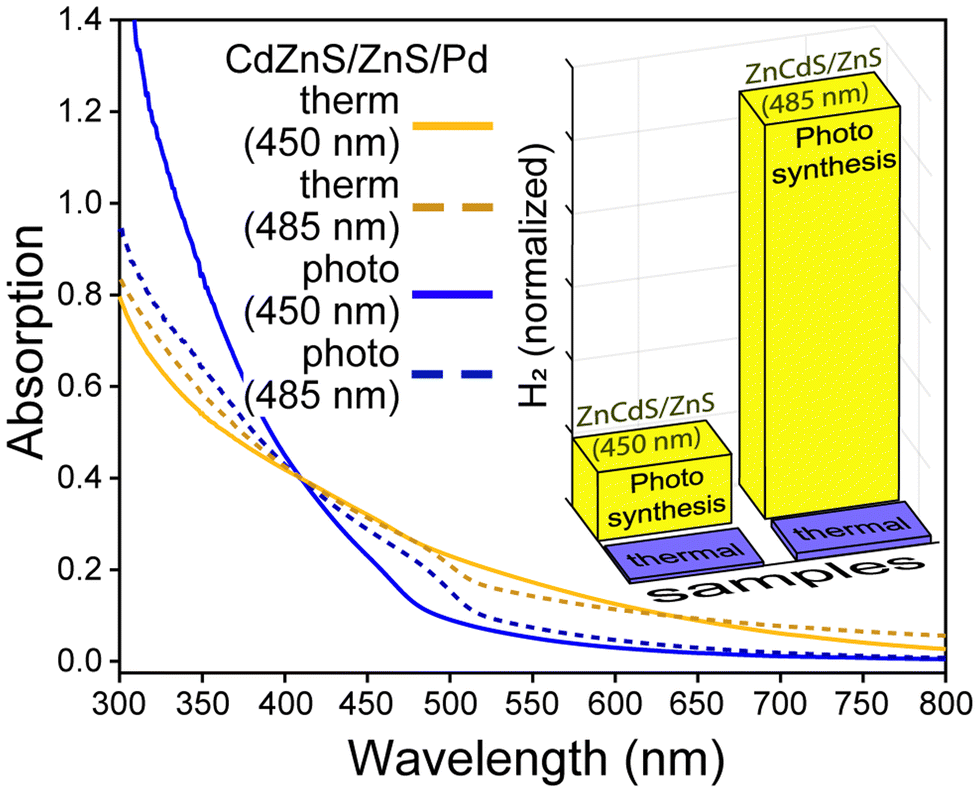 | ||
| Fig. 5 Absorption spectra of ZnCdS/ZnS·Pd NPs prepared by thermolysis and photodeposition of palladium acetate. Inset: H2 synthesis yields, normalized by absorption at the excitation wavelength for the same NPs. | ||
Electronic structure calculations
Theoretical characterizations were applied to gain insight into the exciton structure of ZnCdS·Pd(Pt) NPs and CdSe/CdS dot-in-rod heterostructures. First, a model of a 3 nm diameter CdSe quantum dot embedded in a 15 × 4 nm CdS rod was created using bulk structure parameters. Tight binding (TB) theory48 was used because the model size (∼2500 atoms) is too large for DFT analysis. The TB Hamiltonian was parameterized from various literature sources that were adjusted to reproduce the bulk CdSe–CdS band offsets; see Table S3 of the ESI† for further discussion. Diagonalization of the Hamiltonian and plotting of the various orbitals shown in Fig. 6 was performed using MATLAB. The HOMO–LUMO (electron–hole) orbitals are centred within the CdSe core, which is consistent with previous experimental and theoretical studies.49,50 The electron and hole orbitals of the CdS rod are 0.30 eV above (LUMO+26) and −0.32 eV below (HOMO−22) the core exciton states as shown in Fig. 6. Thus, the CdS shell creates a sizable ∼12× kBT barrier for the core-bound electron to reduce a surface metal co-catalyst. As a result, tunnelling is the only reasonable mechanism for splitting off the electron from the CdSe core.10,14,51 The electron and hole tunnelling transmission coefficients (Te,h) can be calculated by applying the tight binding results into Wentzel–Kramers–Brillouin theory as discussed in the ESI,† the results from which are Te = 0.53 and Th = 0.27 for an electron or hole tunnelling through a 10 nm CdS barrier. The transmission for the hole is reduced mostly due to the charge carrier's higher effective mass. These findings reveal the correlation of photocatalytic quantum efficiency to the heterostructure's length is the result of the differential in the electron–hole transmission coefficients.6 For example, the ratio Te/Th increases to ∼50× for a 60 nm rod and to ∼700× for a 100 nm CdS rod. | ||
| Fig. 6 Tight binding calculations of a 15 nm × 4 nm CdSe/CdS dot-in-rod NP reveal electron delocalization into the CdS rod at +0.3 eV above the conduction band. The CdS-centred hole state is similarly below the valence state. | ||
The electronic structure and excited states of ligated ZnCdS·M6 (M = Pd, Pt) were studied using DFT at the PBE1PBE/LANL2DZ level of theory, which is tractable given the smaller sizes of these systems. As shown in Fig. 7A and B, the density of states for a ∼2 nm diameter model (HCO2H)18Zn30Cd84S114·M6 as well as the orbitals visualized in Fig. 7C for the palladium system reveal metal-centred HOMO and semiconductor-centred LUMO states. While it is tempting to assume that these represent the exciton electron and hole, in fact characterization of the lowest triplet excited state using time-dependent density functional theory (TDDFT)/natural transition orbital analysis (NTO) also shown in Fig. 7C reveal that both the electron and hole are localized on the metal co-catalyst with an excitation energy that is lower than predicted from the DOS's bandgap. The same was observed for the optically allowed singlet state (Fig. S12†); similar results were found for the platinum system as shown in Fig. S13 of the ESI.† These differences are the result of significant electron redistribution instigated by the promotion of an electron into a higher lying orbital, which is why TDDFT/NTO analysis of these systems is necessary.
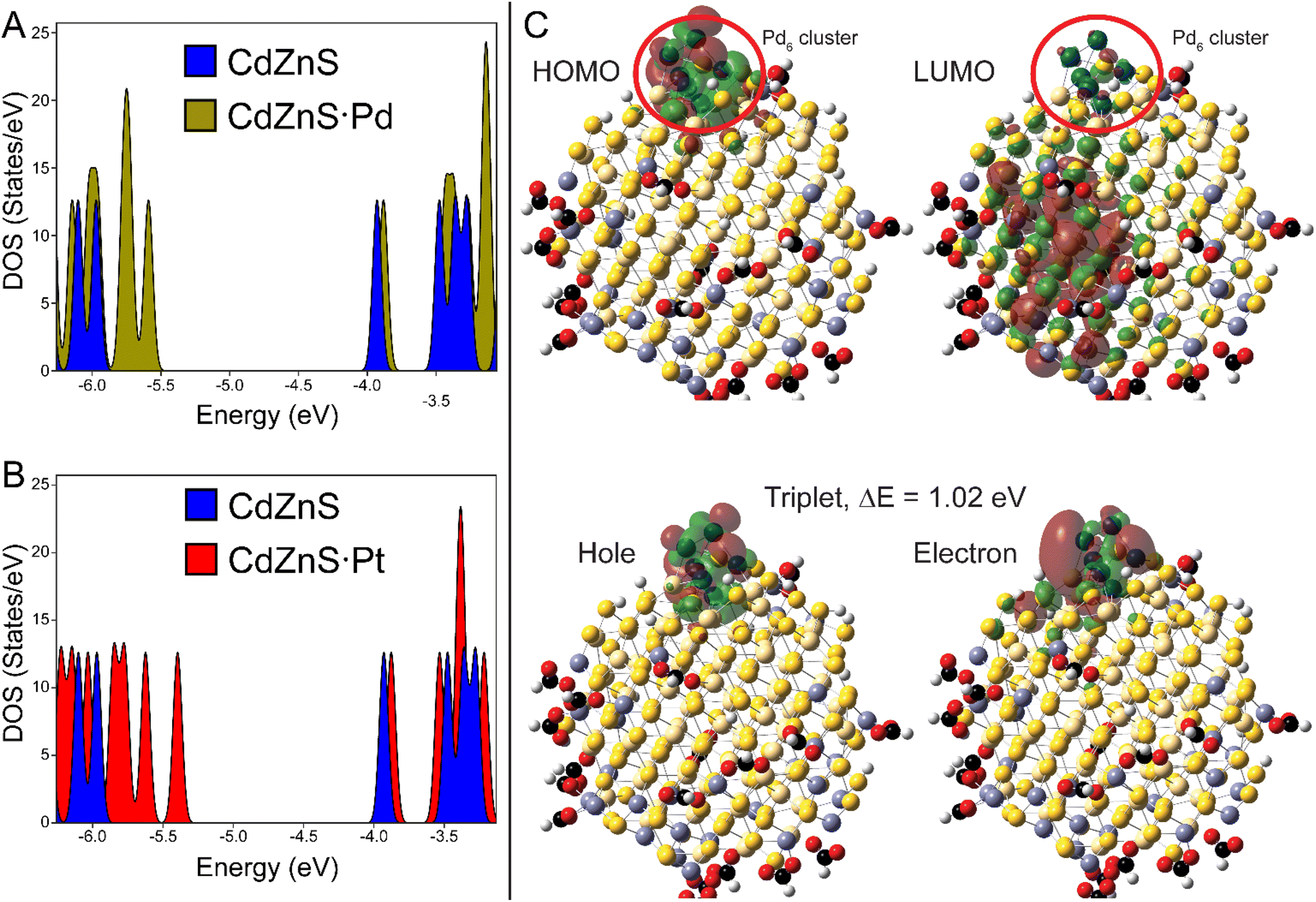 | ||
| Fig. 7 A. DOS of (HCO2H)18Zn30Cd84S114·Pd6 reveal a semiconductor LUMO and a metal centred HOMO. B. DOS of the platinum model is qualitatively similar to the palladium calculation. C. The HOMO-LUMO states and TDDFT/NTO orbitals of the lowest electronic (triplet) excited states of the palladium system. | ||
The co-localization of the exciton electron and hole on a metal cluster should result in thermalization of the excitation energy, assuming that the metal nanoparticle has a sufficient density of states to host free carriers. In this context the TDDFT/NTO results may explain why core/shell CdS/ZnS NPs tipped with Pt or PdS express significantly enhanced activity compared to core-metal particles.11 Furthermore it is interesting to note that the Pd6-centered electron has better overlap with the ZnCdS cluster, whereas the Pt6-bound electron is more insular (Fig. 7C and S13†). The Pt cluster has a 0.17 eV excitation energy, while the palladium system has a much larger bandgap (1.02 eV, Fig. 7C). Consequently, the small palladium clusters gain some semiconductor characteristics that may minimize energy loss by thermalization, which could explain the differences in photochemistry between these co-catalyst metals observed as by us and others.
Conclusions
The efficacy of ZnCdS and various heterostructures for solar hydrogen fuel production were studied. It was expected that the photochemical paradigms observed in the reduction of organic substrates by ZnCdS and ZnCdS/ZnS NPs in toluene would also apply to a more complex aqueous environment including sacrificial reductants. Specifically, that the longer lifetime ZnCdS/ZnS heterostructure would result in greater photocatalytic efficacy. However, this wasn't realized; we posit that the neutralization of trap states by the shell did not compensate for the fact that the shell presents a physical barrier for substrate reduction.Along the same lines, it was hypothesized that extending the exciton lifetime by copper doping CdS hosts would accentuate photochemical hydrogen yields; however, the actual result was a significant reduction in H2 production. Based on previous studies, this can be attributed to the fact that cluster seeding internalizes the Cu dopants, which mollified the efficacy of the sacrificial reductants. Enhanced H2 yields would likely have resulted had the copper been bound to the surface.52
As hole sequestration did not yield the intended effect, we explored electron localization on metallic platinum and palladium co-catalysts. To guide these efforts, DFT modelling was used to examine the structure/property relationships of semiconductor-metal co-catalyst, the results from which revealed localization of both the electron and hole on the metal cluster in the ground exciton state. This should facilitate charge carrier recombination over substrate reduction, a non-productive dynamic that is circumvented in dot-in-rod CdSe/CdS·Pt NPs by separating the core electron via tunnelling through the CdS rod. This mechanism is biased against the hole due to that charge carrier's greater effective mass. These results prompted the use of core/shell ZnCdS/ZnS NPs as substrates for metal deposition to prevent direct coupling of the metal and semiconductor core. In fact, a significant enhancement of H2 production for the palladium system prepared photochemically was observed, which has been noted in other studies.43 Coupled with the fact that other ZnCdS/ZnS-metal NPs examined here did not consistently express an enhancement of H2 production, we conclude that the photochemically prepared ZnCdS/ZnS·Pd had the best separation of exciton charge carriers due to the ZnS effectively insulating the semiconductor hole from the metal electron. While this is an exciting finding due to the greater simplicity of ZnCdS/ZnS·Pd compared to the dot-in-rod CdSe/CdS·Pt “gold-standard” photocatalysts, overall, the latter nonetheless outperformed this and all other systems characterized in this study.
Most reports apply the cap exchange method for aqueous phase transfer of colloidal semiconductor nanoparticles for photocatalysis.23,53,54 And while this would ostensibly expose the surface for chemical transformation, cap exchanged NPs are unstable and quickly precipitate if kept under ambient conditions.55 Encapsulation with amphiphilic polymers render enhanced stability on a multi-year timescale, although we posit that this approach has been avoided in photocatalytic research due to the likelihood that access to the surface may be limited by both the polymer and native caps that are left intact. As shown here, the polymer encapsulation method renders catalytically competent nanomaterials with comparable activity to cap exchanged NPs. This approach may afford additional catalytic tunability varying the surface ligand properties of the particles before encapsulation. We hope that this result will enhance the potential for colloidal nanomaterials to be applied for alternative energy generation.
Experimental
Materials and methods
Most chemicals were used as received, see the ESI.† Tri-n-octylphosphine oxide (TOPO, 90% Sigma-Aldrich) was distilled prior to use. Oleic acid was recrystallized from acetonitrile.56 40% octylamine-modified polyacrylic acid was prepared as per ref. 25 using 1800 MW polyacrylic acid from Sigma-Aldrich. The zinc and copper clusters used for seeding were prepared from ref. 21 for [Me4N]2Cu4(SPh)6 and ref. 57 for [Me4N]2Zn4(SPh)6. Diphenylphosphine selenide (DPPSe) was prepared according to ref. 58.Large bandgap (450 nm) ZnCdS core
To a 3-neck round-bottom flask was added 35 mg CdO (0.27 mmol), 36 mg ZnO (0.44 mmol), 1 g oleic acid (3.54 mmol) in 5 mL ODE. The conversion of the metal oxides to oleates was achieved by heating the solution to 280 °C (or higher if necessary) until optical clarity was achieved, upon which time the flask was cooled back to 80 °C for further vacuum degassing. An injection solution was prepared from 25 mg elemental sulphur (0.78 mmol) in 1 g dodecylamine in a small vial that was stirred and gently heated with a heat gun under vacuum. After the pressure equalized the vial was flushed with dry nitrogen, the flask was heated to 310 °C, and the sulphur solution was swiftly injected in. The NPs were allowed to grow at 300 °C for 30 min. The quantity of quantum dots per gram of growth stock (5.36 × 10−8 mol g−1) was estimated using a ZnCdS NP molar absorptivity of ε = 2.7 × 105 M−1 cm−1 at 450 nm.19 These samples are identical to those prepared in ref. 9, for which TEM characterization revealed a 6.1 ± 0.3 nm size distribution.Core/shell (450 nm bandgap) ZnCdS/ZnS
Core ZnCdS NPs (1.866 g) were precipitated and washed with isopropanol and were then transferred to a 4-neck round-bottom flask charged with degassed 4 g dodecylamine and 5 mL TOP. The solution was heated to 170 °C, upon which time 60 mg ZnEt2 in 3 mL TOP and 65 mg (TMS)2S in 3 mL TOP were added over the course of 3 hours using a syringe injector. During this time the temperature was raised to 220 °C. The final mass was weighed to estimate the number of nanoparticles per gram of growth solution (8.10 × 10−9 mol g−1) for further derivatization. These samples were prepared according to ref. 9, which revealed a 6.8 ± 0.7 nm size distribution.Low bandgap (485 nm) ZnCdS core
To a 3-neck round-bottom flask was added 75 mg Cd(acetate)2·2H2O (0.28 mmol), 102 mg Zn(acetate)2 (0.56 mmol), 1 g oleic acid (3.54 mmol), 1 g hexadecylamine in 5 mL ODE which was heated to 110 °C under vacuum for 1 h. A precursor solution was prepared of 25 mg elemental sulphur with 1.0 mL tri-n-octylphosphine, which was stirred until clear. The sulphur solution was injected into the flask at 310 °C and grown at 300 °C for 30 min. The molar absorptivity of these larger samples was estimated (ε ∼ 5.0 × 105 M−1 cm−1 at 485 nm) using a variety of literature sources. The quantity of quantum dots per gram of growth stock was estimated to be 3.14 × 10−8 mol g−1. The 6.5 ± 0.5 nm diameter particles were characterized with TEM as provided in Fig. S3A of the ESI.† Additional characterizations (X-ray photoelectron and diffraction, Raman, and optical) are provided in Fig. S8–S11.†Core/shell (485 nm bandgap) ZnCdS/ZnS
Core ZnCdS NPs (3.1903 g) were overcoated using the same procedure as the 450 nm bandgap cores. At the end of the process the sample was annealed at 280 °C briefly. The final mass was weighed to estimate the number of nanoparticles per gram of growth solution (7.81 × 10−9 mol g−1) for further derivatization. TEM characterization is provided in Fig. S3B of the ESI† reveal an 8.4 ± 1.0 nm size distribution; however, a loss of crystallinity impairs accurate quantitation of the diameter. Additional characterizations (X-ray diffraction, Raman and optical) are provided in Fig. S8–S10.†Pd, Pt co-catalyst preparation: thermolysis procedure. Precursor solution preparation
A platinum stock solution was prepared from 7.4 mg (1,5-cyclooctadiene)dimethylplatinum(II) (Pt(cod)2), dissolved in 5 mL toluene, while the palladium precursor was comprised of 10 mg palladium acetate (Pd(ace)2), in 10 mL toluene. Pt capping of ZnCdS/ZnS (450 nm): A 0.94 g portion of ZnCdS/ZnS stock (7.62 × 10−9 mol) was precipitated and washed with isopropanol with a small quantity of methanol. The NPs were mixed with 5 mL of ODE, 0.2 mL of oleic acid, 0.2 mL of oleylamine, and 1 mL Pt-toluene precursor solution (4.44 × 10−6 mol Pt) and was sonicated to homogeneity. The precursors were placed in 3-neck round-bottom flask and degassed at 50 °C for one hour. Afterwards, the solution was heated to 240 °C whereby the colour darkened, and the flask was immediately cooled to room temperature. Pd capping of ZnCdS/ZnS (450 nm): The same procedure for platinum tipping was employed with the addition of 1 mL of the Pd-toluene precursor (1.56 × 10−5 mol Pd). The solution was observed to darken upon degassing at 50 °C for a half hour. The colour intensified significantly when heating to 100 °C, and as such the flask was immediately cooled to room temperature afterwards. Pt capping of ZnCdS/ZnS (485 nm): A 0.973 g portion of ZnCdS/ZnS stock (7.60 × 10−9 mol) was processed following the same procedure applied for Pt tipping the smaller bandgap core/shell sample. The NP and platinum precursor solution was heated to 240 °C whereby the colour darkened, and the flask was immediately cooled to room temperature. Pd capping of ZnCdS/ZnS (485 nm): The same procedure for platinum tipping was employed with the addition of 1 mL Pd-toluene solution. These larger QDs had to be heated to 190 °C to observe significant colour darkening, upon which time the solution was cooled to room temperature.Pd co-catalyst preparation: photodeposition procedure. Precursor solution preparation
Semiconductor–palladium metal heterostructures were created using a light brown coloured solution of 0.010 g Pd(ace)2 (0.0441 mmol) and 0.78 g trioctylamine in 8.4469 g toluene. Pd capping of ZnCdS/ZnS: A 7.62 × 10−9 mol portion of either small or large bandgap ZnCdS/ZnS stock (was precipitated and subsequently washed with isopropanol with a small quantity of methanol). Next, 0.868 g of stock Pd solution (4.41 × 10−6 mol Pd) was added and mixed with sonication. Next, the solution was irradiated in a Rayonet photolysis cell for 40 min while stirring. Afterwards the solution was golden brown and non-fluorescent as shown in Fig. S10† for the case of the low bandgap ZnCdS sample. A Raman spectrum of the ZnCdS/ZnS·Pd sample is provided in Fig. S9.†CdSe/CdS dot-in-rods. CdSe cores
This procedure is a modification of that reported in ref. 4. Core CdSe NPs were prepared according to ref. 59, which is summarized here. Into a 50 mL 3-neck round-bottom flask was added 128 mg CdO, 2.4 mL oleic acid with 10 mL ODE. The solution was degassed under vacuum, and then heated to 280 °C to form Cd(oleate)2. A selenium precursor solution was prepared by addition of 161 mg DPPSe into 2.5 mL ODE in a septum capped vial, which was degassed under vacuum for 1 h. The Cd(oleate)2 solution was cooled to 265 °C, upon which 2 mL of the DPPSe solution was injected. The flask remained under heat for 4 min and was then allowed to cool to room temperature. CdS shell: A portion of core CdSe growth solution (7.36 × 10−8 mol) was processed using two cycles of precipitation and redispersion. A tri-n-octylphosphine sulphide precursor solution was prepared by first mixing 0.240 g elemental sulphur with ∼3.6 mL TOP (3.00 g) in a glove box and was stirred until clear and homogeneous. Into a 3-neck round-bottom flask was added 0.12 g CdO (0.0935 mmol), 0.16 g hexyl phosphonic acid (0.963 mmol), 0.58 g octadecyl phosphonic acid (1.734 mmol), and 6.0 g TOPO. The mixture was degassed under vacuum at 150 °C and was then heated to 300 °C under dry nitrogen gas; the growth solution was clear and colourless at this point. The processed CdSe cores were mixed with the TOPS in the glove box and were stirred until dissolution upon which time they were loaded into a 3 mL syringe. The core CdSe NPs were injected into the growth solution at 350 °C, which was held at that temperature for an additional 8 min. The sample was cooled to room temperature and stored under ambient conditions.Pt capping of CdSe/CdS: Pt stock solution
A stock solution of the platinum precursor was prepared by addition of 0.0073 g Pt(cod)2 (0.022 mmol) and 0.1115 g triethylamine (1.1 mmol) in 5 mL toluene. The solution was briefly sonicated to clarity.Pt capping of CdSe/CdS
This procedure is based on ref. 6. A portion of CdSe/CdS dot-in-rod growth solution (2.50 × 10−8 mol NPs) was precipitated with isopropanol to form a coloured phosphonic acid paste material. The nanoparticles were extracted and isolated with hexane, after which they were precipitated once more with isopropanol. The CdSe/CdS NPs were dissolved into a 10 mL of ODE, 0.2 mL oleic acid, 0.2 mL of oleylamine, and 17 mg of the Pt(cod)2 solution. This mixture was sonicated to homogeneity, after which it was placed into a 3-neck round-bottom flask and was degassed at 80 °C for one hour. The solution was quickly heated to 280 °C and then cooled back to room temperature. The platinum NPs started to form at ∼140 °C. A portion of the CdSe/CdS·Pt stock solution was precipitated, and then water solubilized with amphiphilic polymer to yield a 2.5 μM aqueous solution.Cu4:CdS
The procedure was replicated from ref. 21.Zn4:CdS
The procedure for Cu4:CdS NPs was employed with a substitution of Zn4(SPh)6 as nucleating cluster seeds.Water solubilization
Water solubilization via encapsulation was performed using 40% octylamine-modified poly(acrylic acid); see ref. 25. Water solubilization via cap exchange was performed using the zinc metathesis method, see ref. 23. Excess reagents (polymer, mercaptoacids etc.) and electrolytes were removed with dialysis with 300 kDa MWCO filter tubes overnight. Samples were prepared to create 3 mL of 2.1 → 2.5 μM NP catalyst solutions in DI water.Transient optical characterization
Details of all spectroscopic methods can be found in the ESI.†XRD characterization
X-Ray Diffraction (XRD) analysis was performed with a Bruker D8 Advance diffractometer with a Cu Kα radiation source (λ = 1.5418 Å) operating at 40 kV and 40 mA.Raman characterization
Raman spectra were collected using a Renishaw inVia Reflex microscope with a scattering red 633 nm/17.5 mW HeNe laser source. Data were background subtracted prior to analysis.Photochemical H2 generation
Hydrogen production experiments were performed using two light emitting diodes (725 mW @ 500 mA, LEDs, Luxeon Star) centred at 410 nm. The distance between the LEDs and the solution was kept at 2 cm for each trial. The photolysis solutions contained 0.07 M Na2S and 0.13 M Na2SO3 as sacrificial reductants; furthermore, their presence is known to reduce CdS photocorrosion.43 All samples were diluted to an absorption of 0.4 as measured at 410 nm in aqueous solution with ambient oxygen, which has been shown not to impede photochemical hydrogen production.60 For each measurement, 3 mL of sample was added to a 10 mL vial and sealed with a rubber septum. The amount of H2 product was detected each hour over 4 hours using a Shimadzu GC-2014 gas chromatograph, where 250 mL of gas from the headspace of the cell was injected into the column. A calibration curve was used to measure the H2. The total headspace volume of the cell was measured to calculate the amount of hydrogen produced. The abilities of the hydrogen production between different quantum dots samples were compared using the amount of hydrogen produced per absorbance at 410 nm, which is calculated by dividing the amount of hydrogen produced in the headspace by the absorbance of the solution at 410 nm. The external quantum efficiency can be estimated from the excitation flux, which was found to be 0.5% → 2% for the majority of ZnCdS core and core/shell samples of this study.Electronic structure calculations. DFT
Ligated semiconductor cluster models, (HCO2H)18Zn30Cd84S114·M6 (M = Pd,Pt) were studied using DFT at the PBE1PBE/LANL2DZ level of theory with Gaussian-16.61 The geometries were optimized, and then analysed with TDDFT followed by NTO characterization of the excited states. Visualization was performed with Gaussview.62 Previous studies on copper doping of CdS using a (HCO2H)18CuCd113S114 cluster model were reported in ref. 21.Tight binding
Large heterostructures cannot be modelled with DFT, and as a result tight binding was applied to a CdSe/CdS dot-in-rod model (2500 atoms) with an sp3s* basis set. Parameterization was accomplished starting with literature values for CdSe and CdS that were then subject to minor adjustment to impart electron/hole offsets at the Γ point of the bulk semiconductors as well as the correct bulk bandgaps. Cadmium 5s and Se 4pz orbitals were offset to mimic the crystal field splitting of a wurtzite structure, which is necessary as tight binding cannot distinguish between zinc blende and wurtzite in the nearest neighbour formalism. The model was comprised of a 4 nm diameter × 15 nm long rod shape atomistic model was created using bulk zinc blende structural parameters. A 3 nm diameter spherical cutout 4 nm from the end of the rod was designated as CdSe while the remaining atomic sites were assigned as CdS. Surface states were passivated using the method of ref. 63. MATLAB was used for tight binding calculations and visualization.Author contributions
PTS synthesized samples, performed theoretical calculations, and performed data analysis, MF, JH, and CT designed experiments and measured H2 generation, HC and EBK assisted with materials characterizations, and JMO, AMJ, and AJA measured time resolved optical data. AJA, CT, and PTS prepared the manuscript.Data availability
Data for this article, including XRD, XPS, and optical spectra, are available at UIC's Indigo Repository at https://indigo.uic.edu/.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Illinois Chicago for support of this research. Acknowledgements are also made to the donors of the American Petroleum Research Fund. We acknowledge Randall Meyer, Frank Osterloh, and Delmar Larsen for assistance with early investigations of ZnCdS nanoparticle photocatalysis. Also Fengyuan Shi of the UIC Research Resource Center for assistance with Raman and XPS data collection. The authors gratefully acknowledge the Advanced Cyberinfrastructure for Education and Research (ACER) group at the University of Illinois Chicago for providing computational resources and services needed to deliver research results delivered within this paper. https://acer.uic.edu. C.T. thanks the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (DE-SC0020243), for partial support of this work.References
- F. A. Frame, E. C. Carroll, D. S. Larsen, M. S. Sarahan, N. D. Browning and F. E. Osterloh, Chem. Commun., 2008, 2206–2208 RSC.
- D. V. Talapin, R. Koeppe, S. Götzinger, A. Kornowski, J. M. Lupton, A. L. Rogach, O. Benson, J. Feldmann and H. Weller, Nano Lett., 2003, 3, 1677–1681 CrossRef CAS.
- L. Carbone, C. Nobile, M. De Giorgi, F. D. Sala, G. Morello, P. Pompa, M. Hytch, E. Snoeck, A. Fiore, I. R. Franchini, M. Nadasan, A. F. Silvestre, L. Chiodo, S. Kudera, R. Cingolani, R. Krahne and L. Manna, Nano Lett., 2007, 7, 2942–2950 CrossRef CAS PubMed.
- T. Kodanek, H. M. Banbela, S. Naskar, P. Adel, N. C. Bigall and D. Dorfs, Nanoscale, 2015, 7, 19300–19309 RSC.
- D. V. Talapin, J. H. Nelson, E. V. Shevchenko, S. Aloni, B. Sadtler and A. P. Alivisatos, Nano Lett., 2007, 7, 2951–2959 CrossRef CAS PubMed.
- L. Amirav and A. P. Alivisatos, J. Phys. Chem. Lett., 2010, 1, 1051–1054 CrossRef CAS.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS.
- P. Kalisman, Y. Nakibli and L. Amirav, Nano Lett., 2016, 16, 1776–1781 CrossRef CAS PubMed.
- M. Pálmai, E. B. Kim, V. P. Schnee and P. T. Snee, Nanoscale, 2020, 12, 23052–23060 RSC.
- A. Thibert, F. A. Frame, E. Busby, M. A. Holmes, F. E. Osterloh and D. S. Larsen, J. Phys. Chem. Lett., 2011, 2, 2688–2694 CrossRef CAS.
- L. Huang, X. Wang, J. Yang, G. Liu, J. Han and L. Can, J. Phys. Chem. C, 2013, 117, 11584–11591 CrossRef CAS.
- F. E. Osterloh, Chem. Mater., 2008, 20, 35–54 CrossRef CAS.
- N. Kakuta, K. H. Park, M. F. Finlayson, A. Ueno, A. J. Bard, A. Campion, M. A. Fox, S. E. Webber and J. M. White, J. Phys. Chem., 1985, 89, 732–734 CrossRef CAS.
- E. Busby, A. Thibert, L. E. Page, A. M. Jawaid, P. T. Snee and D. S. Larsen, Chem. Phys. Lett., 2013, 573, 56–62 CrossRef CAS.
- B. O. Dabbousi, J. RodriguezViejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- J. Abdul Nasir, Z. ur Rehman, S. N. Ali Shah, A. Khan, I. S. Butler and C. R. A. Catlow, J. Mater. Chem. A, 2020, 8, 20752–20780 RSC.
- H. Yan, J. Yang, G. Ma, G. Wu, X. Zong, Z. Lei, J. Shi and C. Li, J. Catal., 2009, 266, 165–168 CrossRef CAS.
- X. H. Zhong, Y. Y. Feng, W. Knoll and M. Y. Han, J. Am. Chem. Soc., 2003, 125, 13559–13563 CrossRef CAS PubMed.
- L. E. Page, X. Zhang, C. M. Tyrakowski, C.-T. Ho and P. T. Snee, Analyst, 2016, 141, 6251–6258 RSC.
- A. M. Jawaid, S. Chattopadhyay, D. J. Wink, L. E. Page and P. T. Snee, ACS Nano, 2013, 7, 3190–3197 CrossRef CAS PubMed.
- A. Hassan, X. Zhang, X. Liu, C. E. Rowland, A. M. Jawaid, S. Chattopadhyay, A. Gulec, A. Shamirian, X. Zuo, R. F. Klie, R. D. Schaller and P. T. Snee, ACS Nano, 2017, 11, 10070–10076 CrossRef CAS PubMed.
- N. Pickett, US Pat., US7867556B2, 2006 Search PubMed.
- D. Liu and P. T. Snee, ACS Nano, 2011, 5, 546–550 CrossRef CAS PubMed.
- X. Y. Wu, H. J. Liu, J. Q. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. F. Ge, F. Peale and M. P. Bruchez, Nat. Biotechnol., 2003, 21, 41–46 CrossRef CAS PubMed.
- Y. Chen, R. Thakar and P. T. Snee, J. Am. Chem. Soc., 2008, 130, 3744–3745 CrossRef CAS PubMed.
- C. M. Tyrakowski and P. T. Snee, Phys. Chem. Chem. Phys., 2014, 16, 837–855 RSC.
- K. Wu, Y. Du, H. Tang, Z. Chen and T. Lian, J. Am. Chem. Soc., 2015, 137, 10224–10230 CrossRef CAS PubMed.
- P. Tyagi and P. Kambhampati, J. Chem. Phys., 2011, 134, 094706 CrossRef PubMed.
- K. E. Hughes, K. H. Hartstein and D. R. Gamelin, ACS Nano, 2018, 12, 718–728 CrossRef CAS PubMed.
- H. D. Nelson, X. S. Li and D. R. Gamelin, J. Phys. Chem. C, 2016, 120, 5714–5723 CrossRef CAS.
- K. E. Knowles, K. H. Hartstein, T. B. Kilburn, A. Marchioro, H. D. Nelson, P. J. Whitham and D. R. Gamelin, Chem. Rev., 2016, 116, 10820–10851 CrossRef CAS PubMed.
- G. K. Grandhi, R. Tomar and R. Viswanatha, ACS Nano, 2012, 6, 9751–9763 CrossRef CAS PubMed.
- B. B. Srivastava, S. Jana and N. Pradhan, J. Am. Chem. Soc., 2011, 133, 1007–1015 CrossRef CAS PubMed.
- S. Gul, J. K. Cooper, C. Corrado, B. Vollbrecht, F. Bridges, J. Guo and J. Z. Zhang, J. Phys. Chem. C, 2011, 115, 20864–20875 CrossRef CAS.
- M. Zhukovskyi, H. Yashan and M. Kuno, Res. Chem. Intermed., 2019, 45, 4249–4260 CrossRef CAS.
- K. Wu, Z. Chen, H. Lv, H. Zhu, C. L. Hill and T. Lian, J. Am. Chem. Soc., 2014, 136, 7708–7716 CrossRef CAS PubMed.
- J. Wang, T. Xia, L. Wang, X. Zheng, Z. Qi, C. Gao, J. Zhu, Z. Li, H. Xu and Y. Xiong, Angew. Chem., Int. Ed., 2018, 57, 16447–16451 CrossRef CAS PubMed.
- M. J. Berr, A. Vaneski, C. Mauser, S. Fischbach, A. S. Susha, A. L. Rogach, F. Jäckel and J. Feldmann, Small, 2012, 8, 291–297 CrossRef CAS PubMed.
- T. O'Connor, M. S. Panov, A. Mereshchenko, A. N. Tarnovsky, R. Lorek, D. Perera, G. Diederich, S. Lambright, P. Moroz and M. Zamkov, ACS Nano, 2012, 6, 8156–8165 CrossRef PubMed.
- K. Wu, H. Zhu, Z. Liu, W. Rodríguez-Córdoba and T. Lian, J. Am. Chem. Soc., 2012, 134, 10337–10340 CrossRef CAS PubMed.
- T. Sakata, T. Kawai and K. Hashimoto, Chem. Phys. Lett., 1982, 88, 50–54 CrossRef CAS.
- I. B. Rufus, B. Viswanathan, V. Ramakrishnan and J. C. Kuriacose, J. Photochem. Photobiol., A, 1995, 91, 63–66 CrossRef CAS.
- N. Buehler, K. Meier and J. F. Reber, J. Phys. Chem., 1984, 88, 3261–3268 CrossRef CAS.
- J. Jin, J. Yu, G. Liu and P. K. Wong, J. Mater. Chem. A, 2013, 1, 10927–10934 RSC.
- N. Bao, L. Shen, T. Takata and K. Domen, Chem. Mater., 2008, 20, 110–117 CrossRef CAS.
- I. Majeed, M. A. Nadeem, M. Al-Oufi, M. A. Nadeem, G. I. N. Waterhouse, A. Badshah, J. B. Metson and H. Idriss, Appl. Catal., B, 2016, 182, 266–276 CrossRef CAS.
- Y. Liu, W. Yang, Q. Chen, D. A. Cullen, Z. Xie and T. Lian, J. Am. Chem. Soc., 2022, 144, 2705–2715 CrossRef CAS PubMed.
- M. Korkusinski, O. Voznyy and P. Hawrylak, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 245304 CrossRef.
- A. Sitt, F. D. Sala, G. Menagen and U. Banin, Nano Lett., 2009, 9, 3470–3476 CrossRef CAS PubMed.
- W. Sukkabot, Chin. J. Phys., 2021, 72, 240–247 CrossRef CAS.
- L. Dworak, V. V. Matylitsky, V. V. Breus, M. Braun, T. Basché and J. Wachtveitl, J. Phys. Chem. C, 2011, 115, 3949–3955 CrossRef CAS.
- L. Borrell, S. Cervera-March, J. Giménez, R. Simarro and J. M. Andújar, Sol. Energy Mater. Sol. Cells, 1992, 25, 25–39 CrossRef CAS.
- W. C. W. Chan and S. M. Nie, Science, 1998, 281, 2016–2018 CrossRef CAS PubMed.
- H. Mattoussi, J. M. Mauro, E. R. Goldman, G. P. Anderson, V. C. Sundar, F. V. Mikulec and M. G. Bawendi, J. Am. Chem. Soc., 2000, 122, 12142–12150 CrossRef CAS.
- J. Aldana, Y. A. Wang and X. G. Peng, J. Am. Chem. Soc., 2001, 123, 8844–8850 CrossRef CAS PubMed.
- R. L. Arudi, M. W. Sutherland and B. H. Bielski, J. Lipid Res., 1983, 24, 485–488 CrossRef CAS.
- I. G. Dance, A. Choy and M. L. Scudder, J. Am. Chem. Soc., 1984, 106, 6285–6295 CrossRef CAS.
- C. M. Evans, M. E. Evans and T. D. Krauss, J. Am. Chem. Soc., 2010, 132, 10973–10975 CrossRef CAS PubMed.
- K. Welsher, S. A. McManus, C.-H. Hsia, S. Yin and H. Yang, J. Am. Chem. Soc., 2015, 137, 580–583 CrossRef CAS PubMed.
- J. R. Darwent and G. Porter, J. Chem. Soc., Chem. Commun., 1981, 145–146 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. Caricato, V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- S. Lee, F. Oyafuso, P. von Allmen and G. Klimeck, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 045316 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04427d |
| This journal is © The Royal Society of Chemistry 2025 |