
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning structures and properties of self-healing silsesquioxane films using block copolymers†
Yoshiaki
Miyamoto
a,
Takamichi
Matsuno
 abc and
Atsushi
Shimojima
*abc
abc and
Atsushi
Shimojima
*abc
aDepartment of Applied Chemistry, Faculty of Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan. E-mail: shimojima@waseda.jp
bWaseda Research Institute for Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
cKagami Memorial Research Institute for Materials Science and Technology, Waseda University, 2-8-26 Nishiwaseda, Shinjuku-ku, Tokyo 169-0051, Japan
First published on 10th February 2025
Abstract
Designing siloxane-based materials with self-healing properties is crucial for various applications. In this study, we report the preparation of organosiloxane-based thin films with a crack-healing capability, using 1,2-bis(triethoxysilyl)ethane ((EtO)3Si–C2H4–Si(OEt)3) and poly(ethylene oxide)–poly(propylene oxide)–poly(ethylene oxide)-type amphiphilic block copolymers. A lamellar mesostructure with alternating polymer and silsesquioxane layers (O1.5Si–C2H4–SiO1.5), featuring a periodicity of 9.56 nm, was formed through evaporation-induced self-assembly. The film exhibited reversible swelling behavior in response to changes in humidity. The cracks on the film disappeared immediately under humid conditions (90% relative humidity at room temperature). The nanoindentation hardness and elastic modulus of the film were higher than those of conventional lamellar silsesquioxane thin films prepared using a quaternary ammonium-type surfactant, which was attributable to the formation of thicker siloxane layers. Improved solvent resistance of the film was also confirmed by the fact that the mesostructure and crack-healing abilities were maintained even after immersion in ethanol. Thus the use of polymeric surfactants enhanced the mechanical properties and chemical stability of the mesostructured composites. These findings will lead to the development of self-healing siloxane-based coatings for various applications.
Introduction
Siloxane-based materials are widely used in fields such as coatings, optics, electronics, and aerospace because of their transparency, excellent insulation, weather resistance, and high thermal stability.1–4 Precise control over the structure and composition of siloxane-based materials, along with their complexation with other components, gives rise to unique properties and functions.1,2,5–8 The development of self-healing properties is highly sought after to enhance the longevity and durability of siloxane-based materials. A reliable method involves incorporating microcapsules filled with healing agents,9,10 which has been successfully used for the extrinsic healing of polydimethylsiloxane (PDMS) elastomers.11,12 However, it is difficult to repair the same area multiple times or fully restore the physical properties. On the other hand, intrinsic healing is based on reversible bonds,13,14 allowing molecular-level healing through the rearrangement of networks around the damaged area. Partially cross-linked PDMS elastomers with silanolate (Si–O−) ends have self-healing capabilities.4,15,16 Their healing mechanism relies on the flexibility of the PDMS chains, and the room-temperature healing requires a low cross-linking density.15 Stiff self-healing epoxy vitrimers with excellent mechanical properties have been prepared by employing oligosiloxane chains with Si–O− end groups.17 However, the healing of such siloxane-based vitrimers generally requires heating above glass transition temperatures. Therefore, achieving spontaneous self-healing under ambient conditions while simultaneously tuning other properties remains a significant issue.Recently, we reported a new class of self-healing siloxane-based materials with a spontaneous crack-healing ability. Silica thin films with lamellar mesostructures, prepared through the evaporation-induced self-assembly of hydrolyzed alkoxysilanes and surfactants, exhibit crack-healing behavior under high-humidity conditions.18 This healing mechanism, based on the swelling of the lamellar structure,18 is markedly different from those of conventional self-healing siloxane-based materials. Furthermore, lamellar bridged silsesquioxane [(O1.5Si–R–SiO1.5)n, R = C2H4 and C6H4] films demonstrated superior swelling and healing abilities, effectively repairing cracks of greater widths and more varied shapes that could not be healed by lamellar silica films.19 These siloxane-based lamellar films were prepared using double-chained cationic surfactants, didodecyldimethylammonium bromide (DDAB). Understanding the effects of the thickness of the siloxane and surfactant layers, and nature of the inorganic–organic interface on the crack-healing ability is of fundamental and practical interest. It is also important to establish design guidelines for improving the mechanical properties and chemical stability as smart coatings.
In addition to alkylammonium-type surfactants, nonionic polymer surfactants are commonly used as structure-directing agents in the preparation of mesostructured siloxane-based materials.1,5–7 Amphiphilic block copolymers composed of poly(ethylene oxide) (PEO) and poly(propylene oxide) (PPO) blocks are widely used in industry, primarily due to their low cost and minimal toxicity.20,21 Such polymer surfactants can form silica mesostructures with considerably larger periodicities than those prepared using low-molecular-weight surfactants, mainly because of the larger sizes of the micellar assemblies.1,6,22 The nature of the siloxane–organic interface also varies because of the difference in the interactions between the siloxane moieties and surfactant. The PEO blocks interact with multiple SiOH/OH2+ sites and can be partially integrated into the thick siloxane layers,23,24 which is expected to enhance the mechanical properties and chemical stability of the mesostructured composites. Therefore, the use of block copolymers as structure directing agents is of great interest for the design of siloxane-based materials with crack-healing abilities. However, there are only a few studies on the preparation of lamellar structures using PEO–PPO–PEO-type block copolymers. For example, Wu et al. reported the preparation of silica-based lamellar thin films using a triblock copolymer, Pluronic P123 (PEO20PPO70PEO20).25 We have recently reported the preparation of lamellar structured films using P123 and PEO–PDMS–PEO-type block copolymers.26 However, to the best of our knowledge, there are no reports on the preparation of lamellar silsesquioxane films using only PEO–PPO–PEO-type block copolymers and their crack-healing behaviors induced by swelling.
Herein, we report the preparation of silsesquioxane-based lamellar thin films with a crack-healing ability using 1,2-bis(triethoxysilyl)ethane ((EtO)3Si–C2H4–Si(OEt)3, hereafter denoted as BTSE) and P123. The films were prepared on substrates though evaporation-induced self-assembly process and their mesostructure was controlled by adjusting the P123/BTSE molar ratios. The crack-healing behaviors under humid conditions were evaluated by inducing artificial cracks with an indenter, and the effects of the micro- and meso-structures on the self-healing properties were investigated. Furthermore, the structural features, mechanical properties (nanoindentation hardness and elastic modulus), and chemical stability of the lamellar silsesquioxane–P123 films were compared with those of conventional lamellar thin films prepared using alkylammonium-type surfactant (DDAB). The obtained results showed that the use of PEO–PPO–PEO-type block copolymers can contribute to the enhancement of various properties of the self-healing lamellar thin films.
Experimental
Materials
1,2-Bis(triethoxysilyl)ethane (BTSE, >96.0%) was purchased from Sigma-Aldrich; tetraethoxysilane (TEOS, >98.0%) and didodecyldimethylammonium bromide (DDAB, >98.0%) were purchased from Tokyo Chemical Industry Co., Ltd.; ethanol (>99.5%) was purchased from Junsei Chemical Co., Ltd.; 6 N HCl aq. and 0.1 N HCl aq. were purchased from FUJIFILM Wako Pure Chemical Corp.; Pluronic® P123 (average Mn = 5800; PEO20PPO70PEO20) was purchased from Sigma-Aldrich. These chemicals were used without further purification. Si (100) substrates were purchased from Silicon Technology.Preparation of mesostructured silsesquioxane thin films using BTSE and P123
Mesostructured thin films were prepared using BTSE as a silsesquioxane precursor and P123 as a structure directing agent. The precursor solutions were prepared by stirring a mixture of BTSE, P123, EtOH and HCl aq. for 1 h at room temperature in a 15 mL sample bottle, with the molar composition of BTSE/P123/EtOH/H2O/HCl = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :x:15:20:0.008, where x = 0.02, 0.025, 0.03, and 0.04 (P123/Si ratios of 0.01, 0.0125, 0.015, and 0.02, respectively). The corresponding weight ratios of P123 to BTSE were 0.327, 0.409, 0.490, and 0.654, respectively. Under these conditions, the SiOEt groups of BTSE were almost completely hydrolyzed (Fig. S1, ESI†). The precursor solutions (350 μL) were spin-coated (3000 rpm for 30 s) on Si substrates (2 × 2 cm) at 25 °C and 40% relative humidity (RH). The Si substrates were ultrasonically cleaned in Semico Clean 23 (Furuuchi Chemical Co.) for 15 min and then in deionized water for 15 min before spin-coating. The prepared thin films were air-dried at room temperature for 1 d. The film was also prepared on a soft elastic PDMS substrate (2 mm × 20 mm × 20 mm) under otherwise identical conditions. Before spin-coating, the PDMS substrate was plasma-treated for 15 min (using PIB-10, Vacuum Device Inc.) to make the surface hydrophilic.
:x:15:20:0.008, where x = 0.02, 0.025, 0.03, and 0.04 (P123/Si ratios of 0.01, 0.0125, 0.015, and 0.02, respectively). The corresponding weight ratios of P123 to BTSE were 0.327, 0.409, 0.490, and 0.654, respectively. Under these conditions, the SiOEt groups of BTSE were almost completely hydrolyzed (Fig. S1, ESI†). The precursor solutions (350 μL) were spin-coated (3000 rpm for 30 s) on Si substrates (2 × 2 cm) at 25 °C and 40% relative humidity (RH). The Si substrates were ultrasonically cleaned in Semico Clean 23 (Furuuchi Chemical Co.) for 15 min and then in deionized water for 15 min before spin-coating. The prepared thin films were air-dried at room temperature for 1 d. The film was also prepared on a soft elastic PDMS substrate (2 mm × 20 mm × 20 mm) under otherwise identical conditions. Before spin-coating, the PDMS substrate was plasma-treated for 15 min (using PIB-10, Vacuum Device Inc.) to make the surface hydrophilic.
Investigation of crack healing behavior
The crack healing behavior of the mesostructured films were investigated by observing artificial cracks made by pressing a micro indenter. A Shimadzu HMV-2000 Knoop indenter was used to form cracks on the surface of the films, and the optical microscope attached to the indenter was used to observe the cracks. The film on the Si substrate was set on the stage and indented for 5 s with a load of 147 mN using a Knoop indenter. Subsequently, water vapor (room temperature, 90% RH) was blown on the cracked films and the change of the cracks was observed.Analysis
Liquid-state 1H NMR spectrum was obtained using a JEOL JNM-ECZ 500 spectrometer with a resonance frequency of 500.16 MHz at ambient temperature. The chemical shift was referenced to internal tetramethylsilane at 0 ppm. Ethanol-d6 was used to obtain lock signals. θ–2θ X-ray diffraction (XRD) patterns were collected in Bragg–Brentano geometry using Ultima IV diffractometer (Rigaku) with Fe Kα radiation (40 kV and 30 mA). Grazing-incidence small-angle X-ray scattering (GI-SAXS) patterns were collected in reflection mode using a NANO-Viewer (Rigaku) with Cu Kα radiation (40 kV and 30 mA) and a hybrid photon counting detector (Pilatus, Dectris) at 0.2° angle of incidence. Fourier transform infrared spectroscopy (FT-IR) spectra were obtained using a JASCO FT/IR-6100 spectrometer. Attenuated total reflection infrared spectroscopy (ATR-IR) spectra were obtained using a JASCO FT/IR-6100 spectrometer with attachment of a diamond prism. Solid-state 29Si magic-angle spinning (MAS) nuclear magnetic resonance (NMR) spectra were measured using a JEOL JNM-ECX-400 spectrometer at a resonance frequency of 79.4 MHz with a 90° pulse and a recycle delay of 100 s. The samples were set in a 4 mm zirconia rotor and the chemical shifts were referenced to the signal of polydimethylsilane at −33.8 ppm. Scanning electron microscopy (SEM) images were taken using a Hitachi S5500 microscope at an accelerating voltage of 1 kV. Cross-sectional transmission electron microscopy (TEM) and high-angle annular dark field (HAADF) scanning TEM (STEM) images were obtained using a JEOL JEM-2100F microscope with an accelerating voltage of 200 kV. The distributions of Si, O, C in the STEM images were recorded by energy-dispersive X-ray spectroscopy (EDS) using a Si (Li) detector. The specimen was prepared using focused ion beam (FIB) milling (JEOL JIB-4000) to reduce the thickness to <100 nm after carbon-coating to protect the surface. The surface topographies of the film samples were observed using atomic force microscopy (AFM) (Jupiter-XR) at a scanning rate of 1.00 Hz. The images were collected by scanning a 10 × 10 μm2 area. The hardness of the film samples was calculated using a NEC San-ei Instruments MH4000 mechanical property tester attached with a triangular pyramidal indenter with an 80° face angle. The indentation speed was 2.7 nm s−1 and each sample was tested 10 times at different points. Thermogravimetry-differential thermal analysis (TG-DTA) was conducted using a Rigaku Thermo plus EVO2 TG8121 under air flow (200 mL min−1) at a temperature increase rate of 10 °C min−1. α-Al2O3 was used as the standard sample, and an Pt pan was used as the sample container.Results & discussion
Structural analysis of mesostructured thin films
GI-SAXS patterns of the thin films prepared using P123 and BTSE at various P123/Si molar ratios are shown in Fig. 1a. At relatively low P123/Si ratios of 0.01 and 0.0125, two-dimensional (2D)-hexagonal structures with diffraction spots corresponding to the (10) and (01) planes were formed. In contrast, the films prepared at higher P123/Si ratios of 0.015 and 0.02 exhibited patterns with intense spots that arose from the structural periodicity perpendicular to the substrate, indicating the formation of lamellar structures. The XRD pattern of the thin film prepared at P123/Si = 0.02 (hereafter named BTSE–P123) showed an intense peak with a d-spacing of 9.56 nm, along with second- and third-order diffractions (Fig. 1b).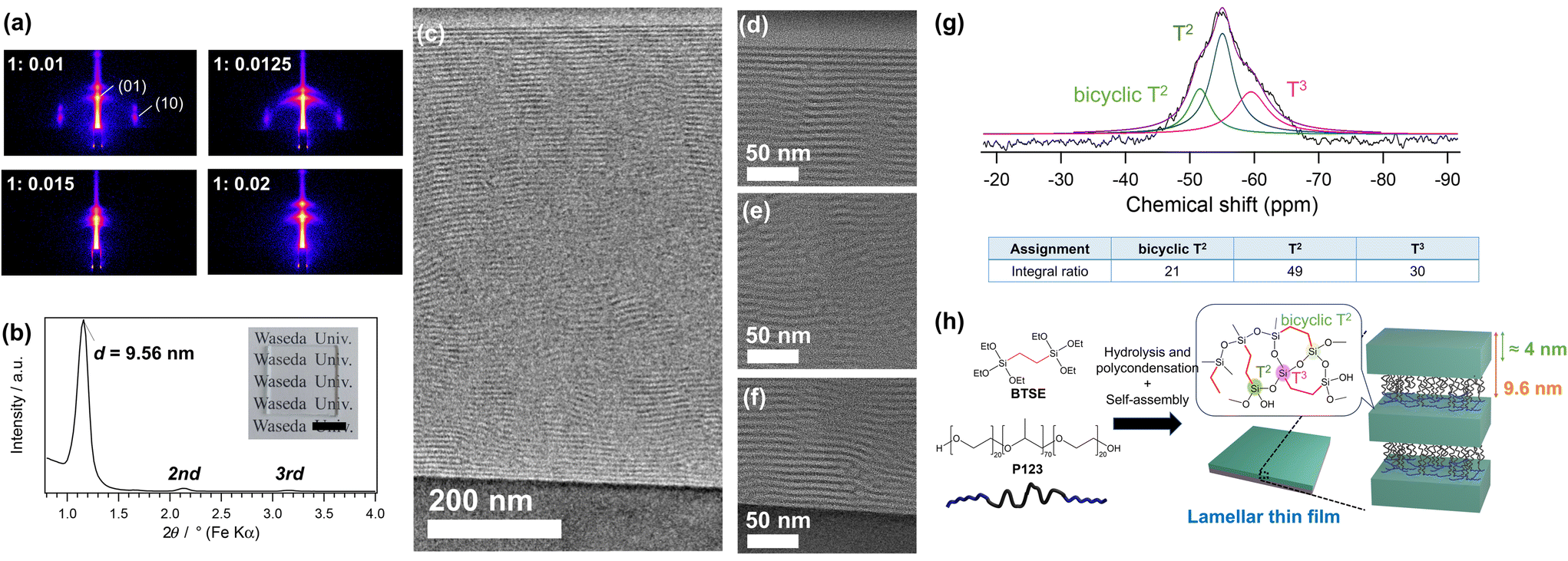 | ||
| Fig. 1 (a) GI-SAXS patterns of the mesostructured films prepared using 1,2-bis(triethoxysilyl)ethane (BTSE) and P123 at different P123/Si ratios of 0.01, 0.0125, 0.015, and 0.02. (b) XRD pattern and (c–f) cross-sectional TEM images of the film prepared at P123/Si = 0.02 (BTSE–P123). The inset in panel (b) shows the photograph of the lamellar film prepared on a glass substrate (scale bar: 1 cm). The images in panels (d), (e), and (f) are the magnified images near the film surface, in the middle of the film, and near the substrate, respectively. The film was cut using FIB milling for the TEM observation. (g) Solid-state 29Si MAS NMR spectrum and (h) a proposed structural model of BTSE–P123. | ||
The mesophase behavior of the BTSE–P123 system differed from those of conventional TEOS–P123 systems.25 At a P123/Si molar ratio of 0.01, a silica-based thin film with a lamellar structure (d = 9.09 nm) was formed using TEOS, as previously reported (Fig. S2, ESI†).25 This contrasts with the formation of a 2D-hexagonal mesostructure using BTSE at the same P123/Si ratio of 0.01 (Fig. 1a). Higher P123/Si ratios were required to form lamellar structures using BTSE, which was probably due to the larger volumes of the hydrolyzed monomers and oligomers derived from BTSE compared with those derived from TEOS because of the presence of ethylene bridging groups in BTSE. Previous studies on the preparation of mesostructured materials using block copolymers have shown that the mesostructure is governed by the hydrophobic/hydrophilic volume ratios of the polymers and siloxane–polymer composites.1,27,28 Mesostructures with lower surface curvature, such as lamellar structures, are favored when the hydrophobic/hydrophilic volume ratio is lower.1
The lamellar thin film prepared using BTSE and P123 at a P123/Si ratio of 0.02 (BTSE–P123) was further characterized in detail. The thickness of the film was approximately 800 nm, as confirmed by SEM observation of the fracture surface (Fig. S3a, ESI†). The surface of the film is locally uneven, though the cause remains unclear (Fig. S3b, ESI†).
The cross-sectional TEM images of the BTSE–P123 film cut vertically using FIB-milling showed layered stripes with a periodicity of approximately 8.6 nm (Fig. 1c–f). The layers were well-oriented near the film–air and film–substrate interfaces and were relatively ill-oriented in the middle region of the film. It was reasonable to assume that these stripes corresponded to the stacked P123 and silsesquioxane layers. The slightly smaller periodicity of the stripes (≈8.6 nm) compared with the d-spacing (9.56 nm) attributed to the shrinkage of the film during FIB processing. In fact, the film thickness observed by TEM is about 700 nm, which is smaller than the thickness of 800 nm observed by SEM. These results confirmed the formation of a partially oriented lamellar structure.
The FT-IR spectrum of BTSE–P123 (Fig. S4, ESI†) showed a peak attributed to the Si–O–Si stretching vibration at 1000–1100 cm−1,19,29 indicating the formation of siloxane frameworks through hydrolysis and condensation of BTSE. A peak attributed to C–O–C stretching vibration at 1100–1200 cm−1 indicated the presence of P123 in the film.30,31 Solid-state 29Si MAS NMR analysis was conducted to obtain detailed structural information. Thick lamellar films were prepared by drop-casting the precursor solution onto Si substrates, peeling it off the substrates, and pulverizing it for measurements. The 29Si MAS NMR spectrum (Fig. 1g) showed the largest signal at −55 ppm with a shoulder signal at −60 ppm, corresponding to typical T2 and T3 units, respectively [Tn; CSi(OSi)n(OH)3−n]. The small signal at −51 ppm, which was also observed for the previously reported lamellar film prepared using BTSE and DDAB (hereafter denoted as BTSE–DDAB),19 corresponded to the T2 units of bicyclic structures that were shifted to lower magnetic fields compared with those of typical unstrained T2 units.19 The average numbers of the Si–O–Si bonds per Si, calculated from the relative intensities of the T2 and T3 signals, was 2.31 for BTSE–P123, which was higher than that for BTSE–DDAB (2.01, Fig. S5, ESI†). This may be partly due to the greater thickness of the siloxane layers compared with those of BTSE–DDAB (discussed later). Note that the T2 signal at −55 ppm might overlap with the bicyclic T3 signals, which were shifted to lower magnetic fields compared with typical T3 signals.19,32 These data indicated the formation of silsesquioxane layers with well cross-linked siloxane networks (Fig. 1h).
The TG–DTA curves of BTSE–P123 (Fig. S6a, ESI†) showed the weight loss and endothermic peak below 150 °C due to the loss of physically adsorbed water molecules. A large weight loss of approximately 68% in total was observed between 150 and 600 °C, which was attributed to the thermal decomposition of P123 and bridging ethylene groups of silsesquioxane. The TG–DTA curves of P123 showed combustion in the 150–250 °C range (Fig. S6b, ESI†). It has also been reported that in mesostructured materials prepared using TEOS and P123, the decomposition of P123 begins at 150 °C.33 Therefore, the large exothermic peak observed in the DTA curve of BTSE–P123 from 150 to 270 °C was attributed to the decomposition of P123 (weight loss of 57%). After heating to 900 °C, 30% of the total weight remained as SiO2, appearing as a white powder. From these values, the molar ratio of P123 to SiO2 was calculated to be 0.02, which was consistent with the molar ratio of P123 to BTSE (0.04) in the precursor solution. The smaller exothermic peak from 270 to 500 °C was mainly attributed to the combustion of the bridging ethylene groups in the silsesquioxane layers.34
In general, amphiphilic block copolymers form mesostructured silica-based materials with thick siloxane layers.1 In the case of mesoporous silicas SBA-15 and MCM-41, prepared using P123 and alkylammonium-type surfactants, respectively, SBA-15 exhibits a larger wall thickness (typically 3–5 nm) and pore size.1,22,35 Therefore, BTSE–P123 likely has a thicker siloxane layer compared with that of BTSE–DDAB. The d-spacing of BTSE–P123 (9.56 nm) is considerably larger than that of BTSE–DDAB (≈3 nm).19 To obtain information about the thickness of the siloxane layers, P123 was removed from BTSE–P123 by solvent extraction (Fig. S7a, ESI†). The d-spacing decreased to 4.23 nm after P123 removal (Fig. S7b and S7c, ESI†). The thickness of the siloxane layer obtained from the TEM images of BTSE–P123 (Fig. 1d–f) was about 4.2 nm. From these results, the thickness of the siloxane layers was estimated to be approximately 4 nm (Fig. 1h), which was larger than the thickness of the siloxane layers in BTSE–DDAB (≈1.0 nm, assuming the thickness of the DDAB bilayers to be 2.4 nm (ref. 36 and 37)).
Properties of the lamellar film (BTSE–P123)
The thickening of the siloxane layers is expected to improve the mechanical properties of lamellar thin films. Nanoindentation tests revealed that the hardness of BTSE–P123 was 235 MPa, which was approximately 2 times harder than that of BTSE–DDAB (102 MPa) (Fig. S8, ESI†). The elastic modulus measured by AFM was also higher for BTSE–P123 (146 MPa) than for BTSE–DDAB (115 MPa) (Fig. S9, ESI†).In previously reported lamellar thin films prepared using DDAB, including BTSE–DDAB,18,19 the d-spacing increased instantaneously when the film was exposed to water vapor. The same behavior was observed for BTSE–P123. The d-spacing at 40% RH was 9.56 nm, whereas it increased to 9.90 nm at 90% RH (Fig. 2a), indicating a swelling of approximately 4%. The lateral swelling of the film could be a driving force for the closure of small cracks.18,19 An indentation with cracks made on BTSE–P123 was targeted for healing. The cracks closed quickly upon exposure of the films to water vapor at 90% RH (Fig. 2b and Movie S1, ESI†). For further analysis, the top surface and cross-section of the healed film were observed by electron microscopy and AFM. The surface SEM image clearly shows that the indentation and cracks were closed, with no observable gaps between the attached crack surfaces (Fig. 2c). The AFM image of the top surface also showed that the crack was closed with only a slight depression remained on the surface (Fig. S10, ESI†). Cross-sectional TEM and STEM observations of the healed indentation showed that healing did not involve reorganization of the lamellar structure. The fractured edges of the siloxane layers on the fracture surface curved and overlapped to fill the gaps (Fig. 2d and e). STEM-EDS mapping of the healed area (Fig. 2e) revealed a uniform distribution of siloxane and organic moieties at the nanometer level, indicating that healing was not caused by filling the gap with P123 eluted from the surroundings. Considering that reopening of the closed crack did not occur even after drying, the closed crack surfaces are likely chemically bonded to each other, probably through siloxane bonds reformed by condensation of silanol groups18,38 and/or by non-covalent interactions between the block copolymers and silsesquioxane layers.
 | ||
| Fig. 2 (a) In situ XRD patterns of the lamellar thin film (BTSE–P123) at different humidities. Immediately after the measurement at 40% RH, the humidity was increased to 90% RH by exposing the film with water vapor. After the measurement at 90% RH, the exposing of water vapor was stopped and the measurement at 40% RH was performed again. (b) Optical microscope images of the cracks on BTSE–P123 before and after exposure to water vapor flow (90% RH at room temperature). (c) SEM image of the closed cracks after the exposure to water vapor flow. (d) cross-sectional TEM image and (e) STEM-EDS mapping of the cracked area after exposure to water vapor flow (Scale bars: 200 nm). | ||
A detailed observation of the healed area clearly showed depression of the substrate and disordering of the orientation of the lamellae by the indenter (Fig. 2d). In previously reported silica-based lamellar thin films, a random orientation of the lamellar structures was more favorable for crack closure.18 In the case of BTSE–P123, healing was achieved even though the layers were mostly oriented parallel to the substrate. This may be due to the orientation change of the lamellae around the indentation (Fig. S11, ESI†). We speculated that the swelling of these disordered lamellae induced expansion of the film in the direction of the crack closure.
We confirmed that the cracks formed on BTSE–P123 by bending the substrate can be repeatedly healed at 90% RH (Fig. S12, ESI†). The film was prepared on a flexible PDMS substrate. The GI-SAXS pattern exhibited a ring pattern with a relatively intense spot (Fig. S12b, ESI†), indicating an oriented lamellar structure with some randomly oriented regions. Many cracks formed on this film by bending disappeared when exposed to humid air at 90% RH. Some of these cracks reformed when the substrate was bent again, but they disappeared at 90% RH (Fig. S12a and S12c, ESI†).
To clarify the effect of the mesostructure on healing, a silsesquioxane-based thin film with a 2D-hexagonal mesostructure, prepared at P123/Si = 0.01, was tested. This film underwent almost no change in the d-spacing with increasing humidity (Fig. S13a, ESI†). The optical microscopy image of the cracked film surface after exposure to water vapor showed that the cracks closed to some extent (Fig. S13b, ESI†). However, cross-sectional observations using TEM and STEM (Fig. S13c and S13d, ESI†) confirmed that the fracture surfaces were not reattached at the nanometer scale. These results indicated that swelling of the lamellar mesostructure played a crucial role in healing.
The effects of the composition of the siloxane layers on the crack healing were also investigated. Our previous study showed that BTSE–DDAB had a higher crack-healing ability than lamellar silica-based films prepared using TEOS and DDAB (Fig. S14a, ESI†).19 Therefore, we compared the crack healing abilities of BTSE–P123 with those of the lamellar silica-based film prepared using TEOS and P123.25 As a result, in contrast to BTSE–P123, no crack closure was observed for the silica-based lamellar film (Fig. S14b, ESI†). This may be due to the rigidity of the silica frameworks compared with those of the silsesquioxane frameworks. The nanoindentation hardness of the silica-based lamellar film was 1.12 GPa, which was approximately 5 times higher than that of BTSE–DDAB.
The effects of heating on the mechanical properties and crack healing ability were examined. BTSE–P123 was heated in air at 80 °C for 1 d. FT-IR spectrum showed a slight increase in the relative intensity of the Si–O–Si band compared to the Si–OH band (Fig. S15a, ESI†), confirming the progression of polycondensation. The nanoindentation hardness of the film increased from 235 MPa to 372 MPa by heating (Fig. S8, ESI†), which can be attributed to the densification of the silsesquioxane layers. Crack healing at room temperature and 90% RH was still observed (Fig. S15b, ESI†). Thus, an improvement in mechanical properties was achieved while maintaining self-healing capability.
The stability of the lamellar films against surfactant elution and delamination of the siloxane layers in an organic solvent was investigated. BTSE–P123 remained intact even after 10 min of immersion in ethanol (Fig. 3a). The XRD pattern showed diffraction peaks assignable to the lamellar structure (Fig. S16a, ESI†). Although the slight decrease in the d-spacing implied that P123 was partially removed from the interlayers, the FT-IR analysis confirmed that the majority of P123 still remained (Fig. S16b, ESI†). Most importantly, even after immersion, the film exhibited a crack-healing ability (Movie S2, ESI†). In contrast, BTSE–DDAB disappeared after 1 min of immersion in ethanol (Fig. 3b), suggesting that the film peeled off from the substrate and/or that the layers were delaminated via the intercalation of ethanol.
 | ||
| Fig. 3 Photographs of (a) BTSE–P123 and (b) BTSE–DDAB thin films before and after immersion in ethanol for 10 min (scale bars: 1 cm). ATR-IR spectra of the powder samples obtained by pulverizing the thick films prepared by casting the precursor solutions of (c) BTSE–P123 and (d) BTSE–DDAB before and after washing with ethanol. The samples were washed twice with ethanol, collected by centrifugation, and dried overnight under a reduced pressure. | ||
Powdered samples of BTSE–P123 and BTSE–DDAB prepared by drop-casting followed by pulverization were analyzed before and after washing with ethanol. FT-IR analysis revealed that the C–H stretching vibration bands (2800–3000 cm−1) significantly decreased for BTSE–DDAB after washing (Fig. 3d), whereas the decrease of the bands in BTSE–P123 was negligible (Fig. 3c). In addition, the TG–DTA curves (Fig. S17, ESI†) showed nearly no weight loss derived from DDAB (from 150 to 350 °C). In contrast, BTSE–P123, after washing with ethanol, still showed weight loss with an exothermic peak attributed to P123 decomposition. These results indicated that DDAB was leached out from the interlayers of BTSE–DDAB upon immersion in ethanol and that BTSE–P123 had a considerably higher solvent resistance compared with that of BTSE–DDAB. The hydrophilic PEO blocks of the block copolymer can partially penetrate the siloxane networks23,24 and interact with silanol groups at multiple points, which probably suppressed elution of P123 from the interlayers of BTSE–P123. These results showed that the solvent resistance was significantly improved by using the block copolymer P123.
The lamellar silsesquioxane films with crack-healing ability are promising candidates for protective coatings, as they can repair various types of cracks under mild conditions. However, further improvement of their mechanical and thermal properties remains a challenge for practical applications. The design of block copolymers offers an effective approach. For example, introducing terminal Si(OEt)3 groups into P123 enables covalent bonding with the siloxane layers,39 which is expected to lead to a significant enhancement of stability and mechanical properties. The use of other block copolymers with more thermostable blocks is also of interest.
Conclusions
We demonstrated the enhancement of the mechanical and chemical properties of the lamellar thin films using block copolymers, while preserving their self-healing capabilities. The lamellar films prepared with BTSE and P123 had thicker silsesquioxane layers than those prepared with conventional alkylammonium-type surfactants. The increase in the thickness of the siloxane layer achieved an increase in the hardness. In addition, the prepared film exhibited a high resistance to the alcohol solvent and retained its self-healing ability even after immersion in the solvent. Thus, nanostructural and compositional control was proven to be important for the design of siloxane-based self-healing materials. These findings contribute to the material design for application in smart coatings.Author contributions
Yoshiaki Miyamoto: conceptualization, all experimental data collection, writing– original draft and editing. Takamichi Matsuno: writing– review & editing. Atsushi Shimojima: writing– review & editing, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by JSPS KAKENHI (grant no. 20H02826). This work was the result of using research equipment (JEOL JNM-ECX-400 (C1025), JEOL JNM-ECZ500R (C1028), Oxford Instruments Jupiter-XR (C1047), Rigaku NANO-Viewer (G1008), JEOL JEM-2100F (G1026), Hitachi S-5500 (G1028), JEOL JIB-4000 (G1033), Rigaku RINT Ultima IV (G1036), and Shimadzu HMV-2000 (G1064)) shared in the MEXT Project for promoting public utilization of advanced research infrastructure (Program for supporting construction of core facilities) Grant Number JPMXS0440500024. Y. M. is grateful to JST SPRING (grant no. JPMJSP2128). The authors are grateful to Dr N. Hanzawa and Mr T. Akanuma (Kagami Memorial Research Institute for Materials Science and Technology) for the STEM observation, and Mr T. Goto (Material Characterization Central Laboratory, Waseda University) for the AFM observation. We gratefully acknowledge Dr T. Hayashi, Dr M. Yatomi, Mr T. Hikino, Ms R. Wakino and Ms M. Suzuki (Waseda University) for fruitful discussions.References
- Y. Wan and D. Zhao, Chem. Rev., 2007, 107, 2821–2860 CrossRef CAS PubMed.
- K. Kuroda, A. Shimojima, K. Kawahara, R. Wakabayashi, Y. Tamura, Y. Asakura and M. Kitahara, Chem. Mater., 2014, 26(1), 211–220 CrossRef CAS.
- K. Glosz, A. Stolarczyk and T. Jarosz, Int. J. Mol. Sci., 2020, 21, 6387 CrossRef CAS PubMed.
- B. Yi, S. Wang, C. Hou, X. Huang, J. Cui and X. Yao, Chem. Eng. J., 2021, 405, 127023 CrossRef CAS.
- A. Mehdi, C. Reye and R. Corriu, Chem. Soc. Rev., 2011, 40, 563–574 RSC.
- Q. Lei, J. Guo, A. Noureddine, A. Wang, S. Wuttke, C. J. Brinker and W. Zhu, Adv. Funct. Mater., 2020, 30, 1909539 CrossRef CAS.
- Y. Liang, Nanoscale Adv., 2021, 3, 6827–6868 RSC.
- S. Porrang, S. Davaran, N. Rahemi, S. Allahyari and E. Mostafavi, Int. J. Nanomed., 2022, 17, 1803–1827 CrossRef PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- A. Ouarga, N. Lebaz, M. Tarhini, H. Noukrati, A. Barroug, A. Elaissaric and H. B. Youcef, J. Mol. Liq., 2022, 354, 118862 CrossRef CAS.
- M. W. Keller, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2007, 17, 2399–2404 CrossRef CAS.
- Z. Yin, J. Guo, J. Qiao and X. Chen, Colloid Polym. Sci., 2020, 298, 67–77 CrossRef CAS.
- A. Kowalewska and K. Majewska-Smolarek, Polymers, 2024, 16, 487 CrossRef CAS PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, 34–59 CrossRef CAS.
- W. Schmolke, N. Perner and S. Seiffert, Macromolecules, 2015, 48, 8781–8788 CrossRef CAS.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027 CrossRef CAS PubMed.
- X. Wu, X. Yang, X. Zhao, Y. Zhang and W. Huang, J. Mater. Chem. A, 2018, 6, 10184–10188 RSC.
- S. Itoh, S. Kodama, M. Kobayashi, S. Hara, H. Wada, K. Kuroda and A. Shimojima, ACS Nano, 2017, 11, 10289–10294 CrossRef CAS PubMed.
- S. Kodama, Y. Miyamoto, S. Itoh, T. Miyata, H. Wada, K. Kuroda and A. Shimojima, ACS Appl. Polym. Mater., 2021, 3, 4118–4126 CrossRef CAS.
- N. A. D. Spirito, N. Grizzuti and R. Pasquino, Phys. Fluids, 2024, 36, 111302 CrossRef.
- K. Kuperkar, D. Patel, L. I. Atanase and P. Bahadur, Polymers, 2022, 14, 4702 CrossRef CAS PubMed.
- M. Ide, M. El-Roz, E. D. Canck, A. Vicente, T. Planckaert, T. Bogaerts, I. V. Driessche, F. Lynen, V. V. Speybroeck, F. Thybault-Starzykb and P. V. D. Voort, Phys. Chem. Chem. Phys., 2013, 15, 642–650 RSC.
- A. Galarneau, H. Cambon, F. D. Renzo and F. Fajula, Langmuir, 2001, 17, 8328–8335 CrossRef CAS.
- M. A. Smith and R. F. Lobo, Microporous Mesoporous Mater., 2010, 131, 204–209 CrossRef CAS.
- C.-W. Wu, J.-B. Pang and M. Kuwabara, Chem. Lett., 2002, 31, 974–975 CrossRef.
- Y. Miyamoto, T. Matsuno and A. Shimojima, Chem. Commun., 2025, 61, 3319–3322 RSC.
- F. R. D. Fernandes, F. G. H. S. Pinto, E. L. F. Lima, L. D. Souza, V. P. S. Caldeira and A. G. D. Santos, Appl. Sci., 2018, 8, 725 CrossRef.
- G. Zhou, Y. Chen, J. Yang and S. Yang, J. Mater. Chem., 2007, 17, 2839–2844 RSC.
- Y. Hattori, T. Hayashi, T. Hikino, R. Miwa, Y. Oka, K. Fujino, N. Sato, T. Matsuno, H. Wada, K. Kuroda and A. Shimojima, J. Sol-Gel Sci. Technol., 2023, 108, 392–400 CrossRef CAS.
- L. S. Vitorino, T. C. dos Santos, I. A. A. Bessa, E. C. S. Santos, B. R. F. Verçoza, L. A. S. de Oliveira, J. C. F. Rodrigues and C. M. Ronconi, Data Brief, 2022, 41, 107841 CrossRef CAS PubMed.
- M. K. Oo, B. Alallam, A. A. Doolaanea, A. Khatib, F. Mohamed and B. Chatterjee, ACS Omega, 2022, 7, 27126–27134 CrossRef CAS PubMed.
- K. Yamamoto, J. Ohshita, T. Mizumo and T. Tsuru, J. Sol-Gel Sci. Technol., 2014, 71, 24–30 CrossRef CAS.
- A. S. Araujo, S. A. Quintella and A. C. S. L. S. Coutinho, Adsorption, 2009, 15, 306–311 CrossRef CAS.
- X. Yu, H. Nagasawa, M. Kanezashi and T. Tsuru, J. Mater. Chem. A, 2018, 6, 23378–23387 RSC.
- J. F. Bardeau, A. Gourbil, M. Dutreilh-Colas, S. Dourdain, A. Mehdi and A. Gibaud, Thin Solid Films, 2006, 495, 191–196 CrossRef CAS.
- M. Dubois and T. Zemb, Langmuir, 1991, 7, 1352–1360 CrossRef CAS.
- G. Tsagkaropoulou, F. J. Allen, S. M. Clarke and P. J. Camp, Soft Matter, 2019, 15, 8402–8411 RSC.
- R. Girard, A. Faivre and F. Despetis, J. Am. Ceram. Soc., 2011, 94, 2402–2407 CrossRef CAS.
- C. Urata, Y. Tamura, Y. Yamauchi and K. Kuroda, J. Mater. Chem., 2011, 21, 3711–3717 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04435e |
| This journal is © The Royal Society of Chemistry 2025 |