
Built-in electric field in the Mn/C60 heterojunction promotes electrocatalytic nitrogen reduction to ammonia†
Hao
Xue
ab,
Kaiheng
Zhao
c,
Denglei
Gao
d,
Fangying
Duan
e,
Zijian
Gao
f,
Wenjia
Yu
g,
Sha
Li
 *h,
Menglei
Yuan
*be and
Zongjing
Lu
*a
*h,
Menglei
Yuan
*be and
Zongjing
Lu
*a
aInstitute of Photochemistry and Photofunctional Materials, University of Shanghai for Science and Technology, Shanghai 200093, China. E-mail: zongjinglu@usst.edu.cn
bQueen Mary University of London Engineering School, Northwestern Polytechnical University, Xi'an 710129, China
cKey Laboratory of Photochemistry, Institute of Chemistry, Chinese Academy of Sciences, University of Chinese Academy of Sciences, Beijing 100190, China
dSchool of Chemical Engineering, Shandong Institute of Petroleum and Chemical Technology, Dongying 257061, China
eState Key Laboratory of Solidification Processing and School of Materials Science and Engineering, Northwestern Polytechnical University, Xi'an 710072, China. E-mail: mlyuan@nwpu.edu.cn
fCAS Key Laboratory of Green Process Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
gYantai Research Institute of Harbin Engineering University, Yantai 264006, China
hChemistry and Chemical Engineering Guangdong Laboratory, Shantou 515021, China. E-mail: lisha@ccelab.com.cn
First published on 8th January 2025
Abstract
The electrochemical nitrogen reduction reaction (NRR) has been regarded as a green and promising alternative to the traditional Haber–Bosch process. However, the high bond energy (940.95 kJ mol−1) of the N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond hinders the adsorption and activation of N2 molecules, which is a critical factor restricting the catalytic performance of catalysts and their large-scale applications. Herein, an Mn/C60 heterostructure is constructed via a simple grinding and calcination process and achieves an extraordinary faradaic efficiency of 42.18% and an NH3 yield rate of 14.52 μg h−1 mgcat−1 at −0.4 V vs. RHE in 0.08 M Na2HPO4. Our experimental and theoretical results solidly confirm that the spontaneous charge transfer at the Mn/C60 heterointerface promotes the formation of a built-in electric field, which facilitates the electron transfer from Mn towards C60 and creates localized electrophilic and nucleophilic regions. The formation of the space-charge region effectively optimized the adsorption energy of the key intermediate *NH–*NH2 and also reduced the free energy barrier for the hydrogenation step of *NH–*NH to *NH–*NH2. Furthermore, the calculated lower limiting potential (UL(NRR)) in Mn/C60 relative to the HER (UL(HER)) demonstrates its enhanced selectivity toward the NRR. This work provided new insights into enhancing the activity and performance of electrocatalysts for the NRR by constructing heterojunctions to improve nitrogen adsorption.
N triple bond hinders the adsorption and activation of N2 molecules, which is a critical factor restricting the catalytic performance of catalysts and their large-scale applications. Herein, an Mn/C60 heterostructure is constructed via a simple grinding and calcination process and achieves an extraordinary faradaic efficiency of 42.18% and an NH3 yield rate of 14.52 μg h−1 mgcat−1 at −0.4 V vs. RHE in 0.08 M Na2HPO4. Our experimental and theoretical results solidly confirm that the spontaneous charge transfer at the Mn/C60 heterointerface promotes the formation of a built-in electric field, which facilitates the electron transfer from Mn towards C60 and creates localized electrophilic and nucleophilic regions. The formation of the space-charge region effectively optimized the adsorption energy of the key intermediate *NH–*NH2 and also reduced the free energy barrier for the hydrogenation step of *NH–*NH to *NH–*NH2. Furthermore, the calculated lower limiting potential (UL(NRR)) in Mn/C60 relative to the HER (UL(HER)) demonstrates its enhanced selectivity toward the NRR. This work provided new insights into enhancing the activity and performance of electrocatalysts for the NRR by constructing heterojunctions to improve nitrogen adsorption.
 Menglei Yuan | Menglei Yuan is an associate professor in the School of Materials Science and Engineering, Northwestern Polytechnical University. He received his bachelor's degree in engineering from the North University of China in 2017. In the same year, he went to the Institute of Process Engineering, Chinese Academy of Sciences, for direct doctoral training. He joined the Northwestern Polytechnical University in 2022. Currently, he focuses on electrocatalytic small molecule activation and organic electrosynthesis reactions for the preparation of high-value chemicals such as ammonia, urea, amides, olefins, etc. He mainly focuses on two aspects, namely electrocatalyst design and regulation and electrolyte coupling enhancement. |
 Zongjing Lu | Zongjing Lu received his Ph.D. in applied chemistry from Tianjin University, China, under the supervision of Prof. Jiannian Yao. He subsequently joined the University of Shanghai for Science and Technology in November 2023 as an assistant professor and established his research group. His current research interests mainly focus on the development of new catalytic materials for catalysis-related applications, such as electrolytic nitrogen/nitrate reduction, plastic decomposition, biomass conversion, and the in-depth understanding of catalytic mechanisms. |
Introduction
Ammonia (NH3) is indispensable in agriculture and industry, enabling large-scale fertilizer production and supporting various chemical processes.1–3 Its high hydrogen concentration and carbon-free nature position it as a promising energy carrier for sustainable energy solutions and climate mitigation.4,5 The large-scale synthesis of NH3, initiated by the Haber–Bosch process in the early 20th century, is thermodynamically exothermic and spontaneous (N2(g) + 3H2(g) → 2NH3(g), ΔrHmθ = −92.22 kJ mol−1, and ΔrGmθ = −32.90 kJ mol−1). This reaction requires high temperature (350 °C–450 °C) to accelerate the reaction rate due to the strong bond energies of the NN triple bond (940.95 kJ mol−1), and the substantial energy barrier (410 kJ mol−1) in the first step of NN triple bond dissociation and high pressure (100–200 bar) to ensure a forward reaction according to Le Chatelier's principle.5–8 However, the Haber–Bosch process not only leads to significant energy costs but also generates large amounts of greenhouse gases (mainly CO2) during the steam methane reformation (SMR) process to produce H2.9 Recently, the electrocatalytic nitrogen reduction reaction (NRR) powered by renewable energy sources, such as wind and solar, in aqueous electrolytes presents a promising alternative to the Haber–Bosch process for efficient NH3 synthesis.10–13 Nevertheless, the performance of NRR electrocatalysts remains insufficient to meet the requirements for practical applications.14,15
Encouragingly, designing a heterogeneous interface to construct a built-in electric field is considered an effective strategy for enhancing electrocatalytic activity, which accelerates the reaction kinetics by promoting electron transfer and reactant diffusion.16 In general, to construct the built-in electric field, at least one of the two heterojunction components should be a semiconductor. The work function difference between the two materials of a heterojunction induces interfacial polarization, generating a potential difference that promotes the spontaneous movement of electrons near the interface until the Fermi level on two sides is balanced, which ultimately forms localized electron-rich and electron-deficient regions, creating a charge distribution gradient and establishing a built-in electric field.16,17 This built-in electric field facilitates electron transfer and modulates the charge density around active sites, thereby enhancing the intrinsic activity of the electrocatalyst. According to the types of the two materials, the heterojunction could be categorized as: (i) p–n heterojunction, (ii) p–p heterojunction, (iii) n–n heterojunction, and (iv) Mott–Schottky heterojunction. The Mott–Schottky heterojunction is a metal–semiconductor interface formed when a metal comes into close contact with a semiconductor that possesses a different Fermi level than that of the metal. For example, if a metal is in contact with a p-type semiconductor (which has electron holes as majority carriers), the electrons in the metal region will flow towards the p-type semiconductor due to the work function difference, which ultimately constructs a built-in electric field from the metal to the semiconductor. A variety of transition metals (TMs), such as Fe,18,19 Ru,20,21 and Mo,22 have demonstrated excellent NRR activity due to their favorable combination of occupied and unoccupied d orbitals, which possesses suitable energy and symmetry for σ donation from the occupied σ orbital of N2 to the empty orbital of the metal atom and π back-donation from the occupied orbital of the metal atom to the empty π* orbital of N2.23,24 Ludden et al. indicated that Mn element is capable of improving the catalytic activity of nitrogenases extracted from the photosynthetic bacterium Rhodospirillum rubrum.25 Mn has 3d orbitals with appropriate energy and symmetry, enabling it to achieve the aforementioned donation and back-donation effects, which are beneficial for the adsorption and activation of N2 molecules.2,26–28 However, the unoccupied d orbitals in the metal may lead to the formation of strong metal–H bonds, which kinetically enhance the unfavorable competitive hydrogen evolution reaction (HER), causing a relatively poor selectivity towards the NRR.23,29 Recent studies have demonstrated that tuning the electronic structure around Mn can effectively enhance its NRR activity and selectivity.26,28,30,31 Du and colleagues proposed a band structure regulation strategy through alloying, which helps to mitigate the HER issue of Mn.32 The constructed MnP alloy showed a reduced HOMO energy level compared to Mn and suppressed HER behavior. Buckminsterfullerene (C60) has a closed cage-like structure composed of hexagons with alternating single and double bonds and pentagons connected by single bonds,33 which not only provides a curved surface but also exhibits unique electron-withdrawing properties to work as an excellent electron acceptor.34,35 Its narrow bandgap (approximately 1.6–1.9 eV) makes it an excellent semiconductor material for the construction of Mott–Schottky heterojunctions.36–38 Moreover, a series of theoretical and experimental studies have demonstrated the pronounced electron-withdrawing capability of C60, underscoring its potential to effectively mitigate the competitive HER at metal active sites while facilitating the NRR.39–41 Therefore, exploring the strategy of constructing a Mn/C60 Mott–Schottky heterojunction for the NRR holds great promise. However, no such studies have been reported to date.
In this work, a simple grinding and calcination process was utilized to synthesise and construct an Mn/C60 heterostructure with the purpose of forming a built-in electric field, which was advantageous for accelerating charge transfer and optimizing the Gibbs free energy for intermediate adsorption. The built-in electric field could promote electron transfer and tune the charge density around active Mn sites to suppress the competitive HER and reduce the energy barriers of the NRR. Therefore, the fabricated Mn/C60 catalyst achieved a remarkable faradaic efficiency of 42.18% and an NH3 yield rate of 14.52 μg h−1 mgcat−1 at −0.4 V vs. RHE. This work aimed to achieve efficient electrochemical ammonia synthesis at ambient temperature and pressure through exploring the strategy of constructing an Mn/C60 heterostructure.
Results and discussion
The Mn/C60 catalyst was synthesized via a simple grinding and low-temperature (300 °C) annealing process. As shown in Fig. 1a, the X-ray diffraction (XRD) patterns of the prepared Mn/C60 catalyst exhibit pronounced characteristic C60 peaks and recognizable peaks of Mn. The peaks at 10.8°, 17.7°, 20.8°, 21.7°, 27.4°, 28.2°, 30.9°, 32.8°,42.2° and 48.1° were indexed to the (111), (220), (311), (222), (331), (420), (422), and (511) planes of C60 and the (101) and (110) planes of Mn, respectively. The other peaks in the XRD graph may be attributed to MnOx generated from the oxidation of Mn element.42 The corresponding morphology of the catalyst, as revealed by the scanning electron microscopy (SEM) image (Fig. 1b and c), demonstrated that an abundant amount of nanoparticles with small particle sizes (an average diameter of approximately 0.18 μm) (Fig. S1†) were highly dispersed on the substrate. Furthermore, the high resolution transmission electron microscopy (HRTEM) image of Mn/C60 further validated that Mn nanoparticles were well dispersed on the C60 substrate (Fig. 1d and e). In the HRTEM image of Mn/C60 (Fig. 1f), the well-resolved lattice fringes of 2.14 nm corresponded to the (101) lattice of Mn. Similarly, the HRTEM image of the substrate exhibited interplanar distances of 2.72 nm and 3.16 nm, which could be ascribed to the (511) and (420) planes of C60, indicating the successful preparation of Mn/C60, consistent with the SEM observations. Due to the low concentration of Mn, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) (Fig. 1g) and the corresponding energy dispersive spectroscopy (EDS) were employed to precisely identify the spatial distribution and elemental composition of Mn on the C60 substrate. The EDS elemental mapping images of C (Fig. 1h) and Mn (Fig. 1i) elements in the Mn/C60 catalyst show that Mn was uniformly dispersed across the C60 substrate in the form of nanoparticles. Combined with the weak Mn characteristic peaks in the XRD pattern, the high dispersion of Mn on the C60 substrate was further confirmed, establishing a critical foundation for the strong electronic interactions between Mn and C60. Moreover, the well-defined lattice fringe distribution was indicative of the heterogeneous structure formed between Mn and C60, facilitating the formation of an intrinsic built-in electric field. | ||
| Fig. 1 (a) Experimental XRD patterns of the as-prepared Mn/C60; (b and c) SEM image of the Mn/C60 catalyst; (d–f) high-magnification TEM image of Mn/C60; (g–i) the corresponding energy-dispersive X-ray spectroscopy (EDS) elemental mapping of Mn/C60. | ||
Based on the results obtained from the aforementioned characterization techniques, we successfully demonstrated the construction of the Mn/C60 heterostructure. Subsequently, it was imperative to confirm the presence of the built-in electric field and examine the charge distribution around Mn and C60 through a combination of theoretical calculations and experimental characterization. First, based on the crystallographic information observed from XRD and TEM, we modeled the Mn (101) crystal plane and C60 and computed their work functions, respectively. The work function of the Mn (101) crystal plane was found to be 3.75 eV, while that of C60 was 5.52 eV, confirming that the two materials met the necessary conditions for the formation of a built-in electric field, specifically, an appropriate work function difference (ΔΦ = 1.77 eV) (Table S1†). The work function difference between the two materials drove directional electron transfer at the interface, aligning the Fermi levels on both sides. Further differential charge density and Bader charge analysis (Fig. 2a and b) confirmed the spontaneous electron transfer from Mn to C60 (1.10 e−), which was consistent with the results observed in X-ray photoelectron spectroscopy (XPS). The corresponding charge accumulation and depletion generated locally electrophilic Mn sites and nucleophilic C60 sites, optimizing the adsorption of the reactant/intermediate during the NRR through electrostatic interactions.
 | ||
| Fig. 2 (a) Charge density difference of the Mn/C60 heterojunction; yellow and cyan indicate electron accumulation and depletion, respectively, with isosurface values of 0.009 e Å−3. (b) Bader charge analysis for Mn/C60. Colour code: pink, C; blue, Mn. | ||
X-ray photoelectron spectroscopy (XPS) was performed to further elucidate the chemical composition and variations in the elemental valence state of the synthesized Mn/C60 catalyst. The significant signals for C, O, and Mn could be observed in the survey spectra (Fig. 3a). More specifically, in the high-resolution C 1s spectra (Fig. 3b), the highest characteristic peak at 284.75 eV corresponds to the C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond and the other three weak peaks at 286.49 eV, 288.67 eV and 290.54 eV could be attributed to the C–O bond, CO bond and π–π* satellite, respectively. Meanwhile, in the high-resolution O 1s spectra (Fig. 3c), the three peaks at 530.05 eV, 533.16 eV and 531.58 eV could be attributed to the O–Mn, O–H, and O–C bonds, stemming from inevitable metal surface oxidation, surface-adsorbed water and oxygen, respectively.43 Intriguingly, in the high-resolution Mn 2p spectra, the Mn0 2p3/2 peaks depicted a shift towards higher binding energies (Fig. 3d) compared with the characteristic peak of pristine Mn nanoparticles (639.0 eV),43,44 indicating strong electronic interactions between Mn and C60. More concretely, the positive shift of Mn0 2p3/2 peaks implied an increased oxidation state of Mn, which could be ascribed to the charge transfer from Mn to C60. The aforementioned results indicated the successful construction of the Mn/C60 heterostructure and effective charge transfer between Mn and C60, resulting in the formation of localized electrophilic and nucleophilic regions.17,45 This local charge redistribution established a built-in electric field that optimized the adsorption of the reactant/intermediate, thereby improving the performance of the electrocatalytic NRR.
C bond and the other three weak peaks at 286.49 eV, 288.67 eV and 290.54 eV could be attributed to the C–O bond, CO bond and π–π* satellite, respectively. Meanwhile, in the high-resolution O 1s spectra (Fig. 3c), the three peaks at 530.05 eV, 533.16 eV and 531.58 eV could be attributed to the O–Mn, O–H, and O–C bonds, stemming from inevitable metal surface oxidation, surface-adsorbed water and oxygen, respectively.43 Intriguingly, in the high-resolution Mn 2p spectra, the Mn0 2p3/2 peaks depicted a shift towards higher binding energies (Fig. 3d) compared with the characteristic peak of pristine Mn nanoparticles (639.0 eV),43,44 indicating strong electronic interactions between Mn and C60. More concretely, the positive shift of Mn0 2p3/2 peaks implied an increased oxidation state of Mn, which could be ascribed to the charge transfer from Mn to C60. The aforementioned results indicated the successful construction of the Mn/C60 heterostructure and effective charge transfer between Mn and C60, resulting in the formation of localized electrophilic and nucleophilic regions.17,45 This local charge redistribution established a built-in electric field that optimized the adsorption of the reactant/intermediate, thereby improving the performance of the electrocatalytic NRR.
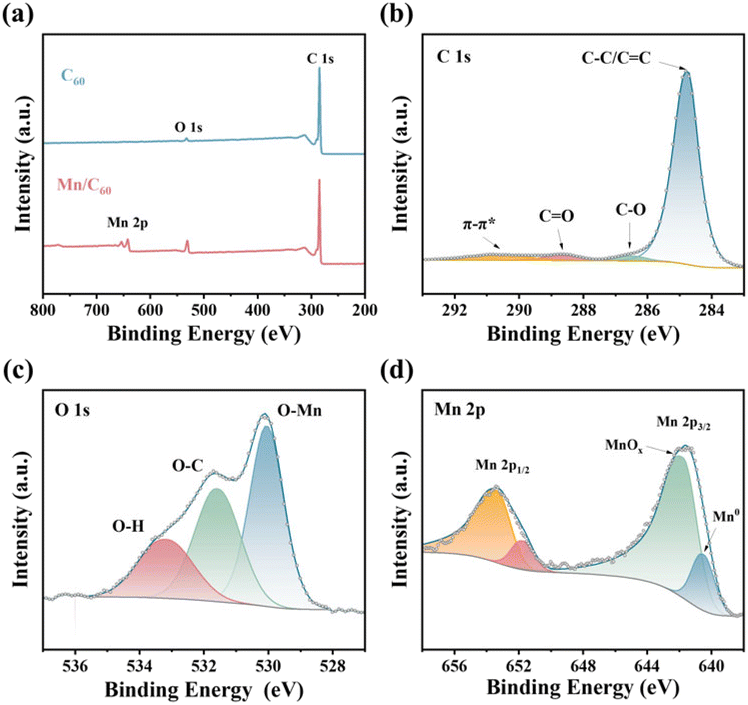 | ||
| Fig. 3 (a) XPS survey spectrum of Mn/C60, and high-resolution XPS spectra of (b) C 1s, (c) O 1s, and (d) Mn 2p. | ||
The NRR measurements were carried out with a typical three-electrode system in an N2-saturated 0.08 M Na2HPO4 solution utilizing a single-chamber electrolytic cell. A graphite rod, an Ag/AgCl electrode (filled with saturated KCl solution), and a piece of pretreated carbon cloth (1 × 1 cm2) (with a catalyst loading of 0.3 mg) were used as the counter electrode, reference electrode, and working electrode, respectively. Note that all potentials were reported on the reversible hydrogen electrode (RHE) scale in this work. The linear sweep voltammetry (LSV) curves were first recorded in Ar-saturated and N2-saturated solutions to indicate the electrocatalytic synthesis of ammonia. As shown in Fig. 4a, the current intensity in the Ar-saturated solution was smaller than that in the N2-saturated solution, exhibiting an extra current density contribution from the NRR. The time-dependent chronoamperometry (CA) test was subsequently performed within the potential range of −0.2 V to −1.0 V vs. RHE to further quantitatively assess the electrocatalytic NRR performance of the Mn/C60 catalyst. Fig. 4b displays all the chronoamperometric curves recorded for 1 h at various potentials in N2-saturated solution with stable current densities. Obviously, the current density of Mn/C60 increased with the applied potential. The employed ultraviolet-visible (UV-vis) absorption spectra of electrolytes based on the indophenol blue method demonstrated the largest absorption intensity (Fig. 4c) at −0.4 V vs. RHE after electrolysis for 1 h. Quantitatively, the NH3 yields and associated FEs from the NRR were evaluated utilizing a concentration–absorbance calibration curve constructed via the indophenol blue method (Fig. 4d and e). The corresponding highest NH3 yield rate achieved was 14.52 μg h−1 mgcat−1 with an FE of 42.18% at −0.4 V (Fig. 4f), outperforming most of the recently reported catalysts, including Pd/C,46 Mn3O4 nanocube47 and C-doped TiO2 nanoparticles48 (Fig. 4g). However, the electrocatalytic NRR performance decreased when the potential becomes more negative than −0.4 V, which can be ascribed to the enhancement of the competitive HER.49–51
 | ||
| Fig. 4 (a) Linear sweep voltammetry (LSV) of Mn/C60 in N2-saturated (red line) and Ar-saturated (blue line) 0.08 M Na2HPO4 electrolytes at 10 mV s−1. (b) the chronoamperometric curves of Mn/C60 in the N2-saturated 0.08 M Na2HPO4 electrolyte at various potentials (−0.2 V, −0.4 V, −0.6 V, −0.8 V and −1.0 V) for 1 h. (c) UV-vis absorption spectra of the electrolytes colored with the indophenol indicator for 2 h. (d and e) Concentration–absorbance of NH4Cl solution with a series of standard concentrations (0–1.0 μg mL−1) in 0.08 M Na2HPO4. (f) The corresponding NH3 yield rate and faradaic efficiency at each given potential. (g) Comparison of the NH3 yield rate and faradaic efficiency of Mn/C60 with selected reported NRR electrocatalysts. (h) LSV curves under different conditions. (i) 1H NMR spectra (700 MHz) for 14NH4+ and 15NH4+ produced from N2 electrochemical reduction using N2 and 15N2 as the feeding gas, respectively. (j) Electrochemical performance of Mn/C60, Mn and C60. | ||
In order to show that the detected NH3 was obtained from N2 by the electrocatalytic NRR on the Mn/C60 catalyst, a series of control experiments were carried out under the following conditions: (i) Ar as the feed gas and C60/Mn as the NRR catalyst, (ii) N2 as the feed gas and C60 as the NRR catalyst, (iii) N2 as the feed gas and Mn as the NRR catalyst, and (iv) N2 as the feed gas and Mn/C60 as the NRR catalyst (Fig. 4h). In addition, 15N2 isotope labeling experiments were carried out to further exclude possible interference from any contaminants (such as adventitious NH3 and NOX). As shown in Fig. 4i, the triplet coupling of 14NH4+ and the doublet coupling of 15NH4+ of the standard samples can be detected in 1H NMR. Only 15NH4+ was detected when utilizing 15N2 as the feed gas for the NRR, which illustrated that NH3 was produced by the electroreduction of N2. All of these aforementioned results further confirmed that the synthesized NH3 here originated from the directly supplied nitrogen during the electrocatalytic NRR instead of from other contaminants or decomposition of catalysts. Subsequently, the corresponding NH3 yield and FE after electrolysis for 1 h at −0.4 V vs. RHE utilizing N2 as the feed gas and Mn/C60 as the NRR catalyst were much higher than those obtained utilizing C60 or Mn as the NRR catalyst, respectively (Fig. 4j), corroborating that the electrocatalytic NRR performance was enhanced by the local charge redistribution in the Mn/C60 heterojunction.
To further explore the intrinsic reasons behind the enhanced NRR activity of Mn/C60, DFT calculations were first performed to investigate the adsorption of *N2 on pristine C60, Mn, and Mn/C60. For pristine C60, a relatively high positive free energy barrier (0.49 eV) for *N2 adsorption was observed (Fig. 5a), suggesting that nitrogen molecules encountered significant difficulty in adsorbing onto C60, which aligned with the suboptimal NRR performance of pristine C60, as shown in Fig. 4j. In contrast, as anticipated, pristine Mn exhibited excellent N2 adsorption capability with a thermodynamically spontaneous *N2 adsorption process (Fig. 5b). This property was also observed in Mn/C60, presenting a spontaneously downhill step with a negative energy barrier for *N2 adsorption (Fig. 5c). Additionally, an ideal NRR catalyst must carefully consider the competing HER, involving both the adsorption energies and the limiting potentials of the HER and NRR, the latter of which is discussed further in the following sections. Regarding the discussion of adsorption energies, a lower *N2 adsorption energy is typically required compared to that of *H. DFT calculations revealed the adsorption energies of *N2 and *H on Mn/C60 to be −1.23 and −0.60 eV, respectively, indicating that the constructed Mn/C60 heterostructure was energetically favored to adsorb N2 rather than competitively adsorb *H (Fig. 5e). However, pristine Mn also exhibited good selectivity in the competitive adsorption of *N2 and *H (ΔG*N2 − ΔG*H = −0.95 eV < 0) (Fig. 5b and d). Therefore, it was necessary to further investigate the reasons behind the enhanced NRR performance of Mn/C60 compared to Mn. Subsequently, to further elucidate the impact of introducing C60 on the NRR activity of Mn, we performed electrochemical impedance spectroscopy (EIS) measurements to investigate the influence of C60 on the charge transfer kinetics of the Mn catalyst. The charge-transfer resistance (Rct) of the catalyst was investigated by EIS. Nyquist plots for Mn/C60 in Fig. 5f demonstrated a smaller semicircle than those of pristine Mn and C60, suggesting that Mn/C60 exhibited smaller charge transfer impedance than pristine Mn and C60, revealing that the strong electronic interactions between Mn and C60 stemming from the built-in electric field can effectively facilitate effective charge transfer and thereby create localized electrophilic and nucleophilic regions. The created localized nucleophilic and electrophilic regions facilitated the adsorption of reactants/intermediates, thereby altering the reaction kinetics and ultimately enhancing NRR activity.52
 | ||
| Fig. 5 (a) The free energy of N2 → *N2H over C60. (b and c) The model and results of N2 adsorption energy calculations for (b) pristine Mn (101) and (c) Mn(101)/C60 with different molecular configurations. (d) The calculated free-energy diagram of the HER for pristine C60, pristine Mn, and Mn/C60. (e) The computed adsorption energy of *N2 and *H on Mn/C60. (f) Nyquist plots of electrochemical impedance spectra (EIS) of Mn/C60, C60, and Mn. (g) Three different reaction pathways for the NRR including the distal-associative pathway, alternating-associative pathway, and enzymatic pathway. (h and i) DFT-calculated reaction pathways and the corresponding energy barriers for the NRR on pristine Mn and Mn/C60. (j) Maximum free energies required for activating the NRR and HER on pristine Mn and Mn/C60, where the green and blue regions indicate NRR and HER dominance, respectively. | ||
So far, we have validated the role of the rationally designed built-in electric field in facilitating the adsorption of inert N2, suppressing competitive *H adsorption and accelerating the reaction kinetics. However, the previously mentioned (i) limiting potentials of the HER and NRR, and (ii) how the built-in electric field reduced the energy barrier of the NRR, had not yet been discussed. To be more specific, both of them were closely related to the reaction process and pathway of the NRR. Therefore, further DFT calculations were performed to elaborate on the reaction mechanism by analyzing the variation of intermediates and the energy barriers throughout the reaction process. Both the NRR and the HER are electrochemical processes that involve the transfer of both protons and electrons, a mechanism known as proton-coupled electron transfer (PCET).53 In the absence of Li mediation and no N atoms on the catalyst surface, the NRR mechanism can be classified into the following pathways: distal-associative pathway, alternating-associative pathway, dissociative pathway, and enzymatic pathway.6,23,54 Among these, the dissociative pathway was difficult to achieve under mild conditions, as it required the direct cleavage of the NN triple bond to obtain nitrogen atoms for protonation and hydrogenation. Therefore, the feasible reaction pathways were limited to the (i) distal-associative pathway, (ii) alternating-associative pathway, and (iii) enzymatic pathway (Fig. 5g). Generally, the optimal reaction pathway could be determined by the adsorption configuration of N2 and the hydrogenation order of N atoms.
As aforementioned, the adsorption of N2 molecules onto the pristine C60 surface was highly unfavorable due to the relatively high free energy barrier (Fig. 5a). Therefore, the reaction pathways for the NRR were only discussed for Mn and Mn/C60. Fig. 5b demonstrates that, compared to the end-on configuration, N2 adsorbed in a side-on configuration on pristine Mn sites exhibited a lower adsorption energy (end-on: −0.46 eV and side-on: −1.50 eV), indicating that the adsorption behavior of N2 on Mn was primarily dominated by side-on adsorption. Similarly, after calculating the adsorption energies of the two N2 adsorption configurations on Mn/C60, it was found that N2 was more favored to adsorb in a side-on configuration at the Mn sites on Mn/C60, and geometrically, it tended to adopt a more energetically stable configuration, as demonstrated in Fig. 5c (ΔG*N2: configuration 1, −1.23 eV; configuration 2, −0.57 eV). Therefore, the enzymatic pathway was considered the primary reaction pathway for the NRR on Mn/C60.
The relevant reaction intermediates and NRR free energy diagrams obtained through DFT calculations are presented in Fig. 5h and i, with detailed calculation methods in the ESI.† Typically, in the enzymatic pathway, the hydrogenation steps proceeded alternately, with hydrogen atoms binding to nitrogen atoms in a sequential manner.55 As shown in Fig. 5h and i, on both Mn and Mn/C60, the intermediate *NH–*NH formed in the second hydrogenation step exhibited a lower free energy compared to *N–*NH2 (Mn: *NH–*NH, −1.21 eV; *N–*NH2, −0.66 eV; Mn/C60: *NH–*NH, −1.01 eV; *N–*NH2, −0.56 eV). Therefore, the reaction mechanism followed an alternating hydrogenation strategy, i.e., the enzymatic pathway. For pristine Mn, the third hydrogenation step (*NH–*NH + H+ + e− → *NH–*NH2) was the potential-determining step (PDS) of the NRR, with an energy barrier of 0.96 eV. However, for Mn/C60, the built-in electric field in the Mn/C60 heterojunction effectively lowered the Gibbs free energy of the key intermediate *NH–*NH2 from −0.25 eV to −0.86 eV and thereby reduced the free energy barrier for the potential-determining step in pristine Mn from 0.96 eV to 0.15 eV. The potential-determining step for the NRR on Mn/C60 was confirmed to be the hydrogenation reduction of *NH2–*NH2 to *NH2 and NH3, for which a Gibbs free energy of only 0.55 eV was required to overcome the free energy barrier, demonstrating a lower energy barrier for the potential-determining step. The thermodynamic limiting potential (UL = −ΔGPDS/e) was defined as the highest potential at which none of the reaction steps were uphill in free energy.56,57 It is noteworthy that the thermodynamic limiting potential for the NRR on pristine Mn was considerably lower than that for the HER (UL(NRR): −0.96 V; UL(HER): −0.55 V). In contrast, the thermodynamic limiting potential for the NRR on Mn/C60 (UL(NRR): −0.55 V) was higher than that for the HER (UL(HER): −0.60 V), suggesting enhanced NRR selectivity (Fig. 5j).
Combining the aforementioned experimental and theoretical calculation results, the electrochemical NRR process catalyzed by the Mn/C60 heterostructure could be summarized in the following steps: (i) the spontaneous charge transfer at the heterointerfaces generated a built-in electric field, which then (ii) facilitated the formation of electrophilic/nucleophilic regions by electrostatic interaction. (iii) The space-charge regions enhanced the adsorption of the key intermediate *NH–*NH2, lowered the energy barrier of the NRR to −0.55 eV for the potential-determining step, improved the limiting potential of the NRR and thereby enhanced NRR activity and selectivity.
Conclusions
In summary, we have demonstrated that the Mn/C60 heterostructure obtained by a simple grinding and low-temperature (300 °C) annealing process showed excellent electrocatalyst activity for the electrocatalytic conversion of N2 to NH3 under ambient conditions. The fabricated Mn/C60 achieved a high FE of 42.18% and a high NH3 yield rate of 14.52 μg h−1 mgcat−1 in 0.08 M Na2HPO4 solution at −0.4 V vs. RHE. A series of characterization techniques, electrochemical tests, and theoretical calculations suggested that the built-in electric field at the Mn/C60 heterojunction promoted local charge redistribution, thereby endowing the Mn/C60 surface with distinct nucleophilic and electrophilic sites that can effectively optimize the adsorption energy of the key intermediate *NH–*NH2 from −0.25 eV to −0.86 eV and reduce the free energy barrier to 0.15 eV for the third hydrogenation step (*NH–*NH + H+ + e− → *NH–*NH2). Additionally, the NRR on Mn/C60 exhibited a lower limiting potential (−0.55 V) compared to the HER (−0.60 V), indicating an enhancement in NRR selectivity. This study indicates that designing electrocatalysts through the design and construction of heterostructures is a promising strategy for the electrocatalytic NRR.Author contributions
Hao Xue: conceptualization, methodology, software, validation, formal analysis, investigation, resources, data curation, writing – original draft, writing – review & editing, and visualization. Kaiheng Zhao: XRD and SEM investigations, formal analysis, investigation, and data curation. Denglei Gao: DFT calculations, methodology, software, formal analysis, investigation, resources, data curation, and writing – review & editing. Fangying Duan: methodology, electrochemical testing, software, validation, formal analysis, investigation, resources, and data curation. Zijian Gao: methodology, electrochemical testing, software, validation, formal analysis, investigation, resources, and data curation. Wenjia Yu: methodology, electrochemical testing, software, validation, formal analysis, investigation, resources, and data curation. Sha Li: conceptualization, validation, formal analysis, investigation, resources, data curation, writing – review & editing, project administration and funding acquisition. Menglei Yuan: conceptualization, validation, resources, data curation, writing – original draft, writing – review & editing, visualization, supervision, project administration and funding acquisition. Zongjing Lu: conceptualization, validation, formal analysis, investigation, resources, data curation, writing – review & editing, supervision, project administration and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (52302310), the QinChuangYuan Cites High-level Innovation and Entrepreneurship Talent Programs (QCYRCXM-2022-335), the Shanghai 2023 “Science and Technology Innovation Action Plan” Social Development Science and Technology Research Project (23DZ1203004), the Guangdong Basic and Applied Basic Research Foundation (2023A1515012816), the Fundamental Research Funds for the Central Universities (G2022KY05111), and the Open Project Program of Anhui Province International Research Center on Advanced Building Materials (JZCL2303KF).References
- V. Rosca, M. Duca, M. T. de Groot and M. T. M. Koper, Chem. Rev., 2009, 109, 2209–2244 CrossRef CAS.
- Z. Wang, F. Gong, L. Zhang, R. Wang, L. Ji, Q. Liu, Y. Luo, H. Guo, Y. Li, P. Gao, X. Shi, B. Li, B. Tang and X. Sun, Adv. Sci., 2019, 6, 1801182 CrossRef.
- X. Fu, V. A. Niemann, Y. Zhou, S. Li, K. Zhang, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. Enemark-Rasmussen, P. Benedek, A. Xu, N. H. Deissler, J. B. V. Mygind, A. C. Nielander, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Nat. Mater., 2024, 23, 101–107 CrossRef CAS PubMed.
- S. Li, Y. Zhou, X. Fu, J. B. Pedersen, M. Saccoccio, S. Z. Andersen, K. Enemark-Rasmussen, P. J. Kempen, C. D. Damsgaard, A. Xu, R. Sažinas, J. B. V. Mygind, N. H. Deissler, J. Kibsgaard, P. C. K. Vesborg, J. K. Nørskov and I. Chorkendorff, Nature, 2024, 629, 92–97 CrossRef CAS PubMed.
- X. Fu, J. Zhang and Y. Kang, Chem Catal., 2022, 2, 2590–2613 CrossRef CAS.
- G. Duan, Y. Chen, Y. Tang, K. A. M. Gasem, P. Wan, D. Ding and M. Fan, Prog. Energy Combust. Sci., 2020, 81, 100860 CrossRef.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Shi, S. Li, X. Wang and G. Zhang, Chem. Sci., 2021, 12, 6048–6058 RSC.
- J. Mu, X.-W. Gao, T. Yu, L.-K. Zhao, W.-B. Luo, H. Yang, Z.-M. Liu, Z. Sun, Q.-F. Gu and F. Li, Adv. Sci., 2024, 11, 2308979 CrossRef CAS PubMed.
- N. G. Mohan and K. Ramanujam, Curr. Opin. Electrochem., 2024, 45, 101520 CrossRef CAS.
- H. Wang, C. Zhang, B. Liu, W. Li, C. Jiang, Z. Ke, D. He and X. Xiao, Adv. Mater., 2024, 36, 2401032 CrossRef CAS.
- Y. Wan, Z. Wang, J. Li and R. Lv, ACS Nano, 2022, 16, 643–654 CrossRef CAS PubMed.
- H. Xue, Z.-H. Zhao, M. Yuan and G. Zhang, Green Energy Environ., 2024 DOI:10.1016/j.gee.2024.06.006.
- M. Qiao, J. Xie and D. Zhu, Nanoscale, 2024, 16, 3676–3684 RSC.
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- Z. Lu, H. Wang, Y. Tao, S. Zhu, W. Hao, X. Liu, Y. Min and J. Fan, Nanoscale, 2023, 15, 14847–14857 RSC.
- X. Zhao, M. Liu, Y. Wang, Y. Xiong, P. Yang, J. Qin, X. Xiong and Y. Lei, ACS Nano, 2022, 16, 19959–19979 CrossRef CAS.
- M. Yuan, J. Chen, Y. Bai, Z. Liu, J. Zhang, T. Zhao, Q. Wang, S. Li, H. He and G. Zhang, Angew. Chem., Int. Ed., 2021, 60, 10910–10918 CrossRef CAS PubMed.
- M. Wang, S. Liu, T. Qian, J. Liu, J. Zhou, H. Ji, J. Xiong, J. Zhong and C. Yan, Nat. Commun., 2019, 10, 341 CrossRef CAS.
- S. Zhang, M. Jin, T. Shi, M. Han, Q. Sun, Y. Lin, Z. Ding, L. R. Zheng, G. Wang, Y. Zhang, H. Zhang and H. Zhao, Angew. Chem., Int. Ed., 2020, 59, 13423–13429 CrossRef CAS PubMed.
- H. Tao, C. Choi, L.-X. Ding, Z. Jiang, Z. Han, M. Jia, Q. Fan, Y. Gao, H. Wang, A. W. Robertson, S. Hong, Y. Jung, S. Liu and Z. Sun, Chem, 2019, 5, 204–214 CAS.
- Z. Geng, Y. Liu, X. Kong, P. Li, K. Li, Z. Liu, J. Du, M. Shu, R. Si and J. Zeng, Adv. Mater., 2018, 30, 1803498 CrossRef.
- L. Han, X. Liu, J. Chen, R. Lin, H. Liu, F. Lü, S. Bak, Z. Liang, S. Zhao, E. Stavitski, J. Luo, R. R. Adzic and H. L. Xin, Angew. Chem., Int. Ed., 2019, 58, 2321–2325 CrossRef CAS.
- S. Qiang, F. Wu, J. Yu, Y.-T. Liu and B. Ding, Angew. Chem., Int. Ed., 2023, 62, e202217265 CrossRef CAS.
- M.-A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef.
- P. W. Ludden and R. H. Burris, Science, 1976, 194, 424–426 CrossRef CAS PubMed.
- L. Zhang, X.-Y. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC.
- L. Han, M. Hou, P. Ou, H. Cheng, Z. Ren, Z. Liang, J. A. Boscoboinik, A. Hunt, I. Waluyo, S. Zhang, L. Zhuo, J. Song, X. Liu, J. Luo and H. L. Xin, ACS Catal., 2021, 11, 509–516 CrossRef CAS.
- Y. Ma, Y. Lu, C. Li, L. Hu, H. Zhang and J. Feng, Catal. Lett., 2024, 154, 5830–5837 CrossRef CAS.
- L. Zhang, L.-X. Ding, G.-F. Chen, X. Yang and H. Wang, Angew. Chem., Int. Ed., 2019, 58, 2612–2616 CrossRef CAS PubMed.
- K. Chu, Y.-p. Liu, Y.-b. Li, Y.-l. Guo, Y. Tian and H. Zhang, Appl. Catal., B, 2020, 264, 118525 CrossRef CAS.
- X. Wang, D. Wu, S. Liu, J. Zhang, X.-Z. Fu and J.-L. Luo, Nano-Micro Lett., 2021, 13, 125 CrossRef CAS.
- W. Lu, T. Zheng, X. Zhang, T. He, Y. Sun, S. Li, B. Guan, D. Zhang, Z. Wei, H. Jiang, H. J. Fan and F. Du, Angew. Chem., Int. Ed., 2024, e202417171 Search PubMed.
- S. Singla, S. Sharma, S. Basu, N. P. Shetti and T. M. Aminabhavi, Int. J. Hydrogen Energy, 2021, 46, 33696–33717 CrossRef CAS.
- R. Zhang, Y. Li, X. Zhou, A. Yu, Q. Huang, T. Xu, L. Zhu, P. Peng, S. Song, L. Echegoyen and F.-F. Li, Nat. Commun., 2023, 14, 2460 CrossRef CAS.
- B. Zhao, F. Chen, C. Cheng, L. Li, C. Liu and B. Zhang, Adv. Energy Mater., 2023, 13, 2204346 CrossRef CAS.
- Y. Pan, X. Liu, W. Zhang, Z. Liu, G. Zeng, B. Shao, Q. Liang, Q. He, X. Yuan, D. Huang and M. Chen, Appl. Catal., B, 2020, 265, 118579 CrossRef CAS.
- V. Martínez-Agramunt and E. Peris, Inorg. Chem., 2019, 58, 11836–11842 CrossRef.
- M. Grandcolas, J. Ye and K. Miyazawa, Ceram. Int., 2014, 40, 1297–1302 CrossRef CAS.
- T. He, G. Gao, L. Kou, G. Will and A. Du, J. Catal., 2017, 354, 231–235 CrossRef CAS.
- A. R. Puente Santiago, T. He, O. Eraso, M. A. Ahsan, A. N. Nair, V. S. N. Chava, T. Zheng, S. Pilla, O. Fernandez-Delgado, A. Du, S. T. Sreenivasan and L. Echegoyen, J. Am. Chem. Soc., 2020, 142, 17923–17927 CrossRef CAS PubMed.
- M. A. Ahsan, T. He, K. Eid, A. M. Abdullah, M. L. Curry, A. Du, A. R. Puente Santiago, L. Echegoyen and J. C. Noveron, J. Am. Chem. Soc., 2021, 143, 1203–1215 CrossRef CAS PubMed.
- H. Chen, W. Zhou, D. Zhu, Z. Liu, Z. Feng, J. Li and Y. Chen, J. Alloys Compd., 2020, 813, 151812 CrossRef CAS.
- Q. Huang, W. Yang, Y. Yan, S. Xie, A. Yu, T. Xu, Y. Zhao, P. Peng and F.-F. Li, Adv. Funct. Mater., 2024, 2409406 CrossRef CAS.
- A. Chourasia and D. Chopra, Surf. Sci. Spectra, 1994, 3, 74–81 CrossRef.
- J. Hu, A. Al-Salihy, J. Wang, X. Li, Y. Fu, Z. Li, X. Han, B. Song and P. Xu, Adv. Sci., 2021, 8, 2103314 CrossRef CAS.
- J. Wang, L. Yu, L. Hu, G. Chen, H. Xin and X. Feng, Nat. Commun., 2018, 9, 1795 CrossRef.
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Small, 2018, 14, 1803111 CrossRef.
- K. Jia, Y. Wang, Q. Pan, B. Zhong, Y. Luo, G. Cui, X. Guo and X. Sun, Nanoscale Adv., 2019, 1, 961–964 RSC.
- Z. Xue, C. Sun, M. Zhao, Y. Cui, Y. Qu, H. Ma, Z. Wang and Q. Jiang, ACS Appl. Mater. Interfaces, 2021, 13, 59834–59842 CrossRef CAS.
- M. Liu, S. Yin, T. Ren, Y. Xu, Z. Wang, X. Li, L. Wang and H. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 47458–47464 CrossRef CAS PubMed.
- J. Wang, H. Jang, G. Li, M. G. Kim, Z. Wu, X. Liu and J. Cho, Nanoscale, 2020, 12, 1478–1483 RSC.
- M. Yuan, S. Dipazir, M. Wang, Y. Sun, D. Gao, Y. Bai, M. Zhang, P. Lu, H. He, X. Zhu, S. Li, Z. Liu, Z. Luo and G. Zhang, J. Mater. Chem. A, 2019, 7, 3317–3326 RSC.
- L. Hu, Z. Xing and X. Feng, ACS Energy Lett., 2020, 5, 430–436 CrossRef CAS.
- T. He, S. K. Matta and A. Du, Phys. Chem. Chem. Phys., 2019, 21, 1546–1551 RSC.
- F. Lai, W. Zong, G. He, Y. Xu, H. Huang, B. Weng, D. Rao, J. A. Martens, J. Hofkens, I. P. Parkin and T. Liu, Angew. Chem., Int. Ed., 2020, 59, 13320–13327 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- Y. Zhang, J. Huang and M. Eikerling, Electrochim. Acta, 2021, 400, 139413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr04496g |
| This journal is © The Royal Society of Chemistry 2025 |