
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalyst-derived hierarchy in 2D imine-based covalent organic frameworks†
Hao Guo a,
Joseph P. Clineb,
Ryan Thorpec,
Christopher J. Kielyabc,
Srinivas Rangarajana and
Mark A. Snyder*a
a,
Joseph P. Clineb,
Ryan Thorpec,
Christopher J. Kielyabc,
Srinivas Rangarajana and
Mark A. Snyder*a
aDept. of Chemical and Biomolecular Engineering, Lehigh University, Bethlehem, PA 18015, USA. E-mail: snyder@lehigh.edu
bDept. of Materials Science and Engineering, Lehigh University, Bethlehem, PA 18015, USA
cInstitute for Functional Materials and Devices, Lehigh University, Bethlehem, PA 18015, USA
First published on 17th January 2025
Abstract
Identifying facile strategies for hierarchically structuring crystalline porous materials is critical for realizing diffusion length scales suitable for broad applications. Here, we elucidate synthesis–structure–function relations governing how room temperature catalytic conditions can be exploited to tune covalent organic framework (COF) growth and thereby access unique hierarchical morphologies without the need to introduce secondary templates or structure directing molecules. Specifically, we demonstrate how scandium triflate, an efficient catalyst involved in the synthesis of imine-based COFs, can be exploited as an effective growth modifier capable of selectively titrating terminal amines on 2D COF layers to facilitate anisotropic crystal growth. We systematically map a compositional pseudo-phase space and uncover key mechanistic insights governing the catalyst-derived evolution of globular COFs with sub-micron diffusion length scales into unique rosette structures. Comprised of interconnected, high-aspect-ratio crystalline porous sheets of only several unit cells in thickness, the resulting COFs offer orders of magnitude reduction in diffusion length scales and several-fold increase in external surface area, enabling rapid uptake of bulky dyes. Generally, the resulting synthesis–structure–function relations hold promise for realizing unique control over COF mesostructure, morphology, and function.
Introduction
Covalent organic frameworks (COFs) are a burgeoning class of crystalline porous materials formed by the reticulation of building block molecules into two- (2D) or three-dimensional (3D) porous organic networks.1–3 Owing to the intrinsic stability4,5 and tunable functionality6 of the covalently bonded framework, and the ability to design building block structure and chemistry to tailor pore topology and size,1,7 COFs show promise in a wide range of applications, including in adsorption,8 catalysis,9 separations,10 and chemical sensing,11 among others.12 Such applications commonly rely on efficient molecular access to pore topologies.13–15 However, larger particle sizes, required for ease of processing in practical applications, offer characteristic diffusion length scales that stymie access to the intracrystalline pores. This is especially true in cases where diffusion becomes sluggish as pore diameters approach molecular dimensions.Efforts to overcome transport limits in crystalline porous materials have focused on strategies for introducing pore hierarchy to facilitate molecular access of the smallest pores deep within a particle. For example, strategies for the hierarchical structuring of zeolites,16 a class of crystalline microporous materials17,18 with pore topologies analogous to some COFs, have included post-synthetic processing (e.g., etching)19 and sacrificial templating20,21 resulting in embedment of mesopores within the microporous framework. Additional elegant strategies have employed designer surfactants and organic structure-directing agents22–24 or taken advantage of crystal twinning and defects25,26 as a means for growing intrinsically hierarchical zeolites.
2D COFs have a wider range of accessible pore sizes than zeolites, classifying them among microporous to mesoporous (ca. 5 Å to 5 nm) materials.27,28 Despite the wide range of nominal COF pore sizes, transport limits remain a persistent challenge due to the common micrometer particle sizes and concomitantly large characteristic diffusion length scales,29 pore restrictions deriving from the shifting of 2D COF sheets from eclipsed to staggered positions,30,31 and the growing interest in deploying COFs for processing of larger molecules (e.g., per- and polyfluoroalkyl substances – PFAS,32,33 other organic pollutants,34–36 bulky polysaccharides,37 etc.) that approach the size of COF pores. To circumvent transport limits in COFs, studies have employed judicious selection of building blocks to optimize pore size and chemistry32 and have focused on avoiding undesired pore restrictions by ensuring highly crystalline eclipsed stacking of 2D COF layers.38,39
In cases where the pore size is commensurate with molecular dimensions, strategies for hierarchically structuring COFs have been analogous to those used for zeolites.40–45 Sacrificial templating has led to COFs with meso to macroscale pores embedded within the COFs upon template removal,43,44 whereas in situ templating of macrostructured COFs has been achieved by adding foaming agents during COF crystallization.42 Separately, growth modifiers, building blocks with reduced functionality, have been introduced to control growth kinetics and realize structural hierarchy comprising thin interconnected COF sheets.46 Several studies have separately demonstrated how concentration-sensitive dimerization or temperature-dependent decomposition of specialized monomers can be employed for tuning COF morphology among globular, intrinsically hierarchical, or mesopore-etched structures.41,45
Dichtel and coworkers47–49 and others46,50 have extensively studied covalent linkage kinetics and synthetic catalysts with the goal of maximizing crystallinity by minimizing the defects of 2D imine-based COFs. Here, drawing inspiration from the elegant hierarchical structuring of zeolites, we explore the opposite scenario of how crystalline defects—induced by catalytic control of the covalent linkage kinetics—can be exploited for synthesizing intrinsically hierarchical COFs at room temperature and with facile and scalable strategies, namely without the need to introduce secondary templates or structure directing molecules. Specifically, we develop synthesis–structure–function relations linking catalyst type and composition to product morphology for representative 2D imine-based COFs, COF-LZU1 and TAPB-PDA. We show how scandium triflate, Sc(OTf)3, an alternative to acetic acid that is traditionally used in the synthesis of imine-based COFs, not only acts as an efficient catalyst but also tunes framework defects and appears to mediate anisotropic COF growth. The unique rosette-structured COFs that result bear interdigitated, planar features of only tens of nanometers in thickness, offering several fold higher external surface areas, orders of magnitude shorter characteristic diffusion lengths, and more accessible pores compared with globular forms of the same COFs synthesized under different catalytic conditions. The hierarchical structure and concomitant enhanced external surface area and expanded direct access to COF pores result in faster uptake of, and higher capacity for, bulky dyes.
Experimental
Chemicals
All chemicals were purchased from commercial vendors and used without further purification, including 1,3,5-Triformylbenzene (TFB, Thermo Scientific Chemicals, 98%), 1,4-Diaminobenzene (DAB, Sigma, 98%), 1,3,5-Tris(4-aminophenyl)benzene (TAPB, AmBeed, 97%), Terephthaldehyde (PDA, Sigma, 99%), Scandium triflate (Sc(OTf)3, Sigma, 99%), glacial acetic acid (Sigma, 99%), dioxane (Sigma, 99%), mesitylene (Sigma, 98%), methanol (Sigma, 99%), Rhodamine B (RdB, Sigma, <100%), and Methylene blue (MB, Sigma, <100%).COF-LZU1 synthesis
COF-LZU1 was synthesized from 1,3,5-Triformylbenzene (TFB) and 1,4-Diaminobenzene (DAB) using either acetic acid or Sc(OTf)3 as catalysts. In a typical synthesis 0.31 mmol of TFB and 0.46 mmol of DAB were added to a 20 mL scintillation vial under N2 protection and charged with 6.2 mL dioxane/mesitylene solution (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). The catalyst solution was prepared in a separate scintillation vial. In the case of acetic acid, a prescribed volume of 10.5 M aqueous acetic acid (0.44, 1.32, 3.09, 4.85, 6.17 mL) was added to 6.2 mL dioxane/mesitylene solution (4:1, v/v) to achieve 5, 15, 35, 55, and 70 eq. of acetic acid, respectively, relative to amine groups of DAB. Additionally, a water-free synthesis was carried out with a catalyst solution comprising 0.26 mL of glacial acetic acid in 6.2 mL dioxane/mesitylene solution (4:1, v/v), corresponding to 5 eq. acetic acid. In the case of Sc(OTf)3, prescribed amounts of the catalyst (0.9, 9.1, 18.2, and 41.0 mg) were dissolved in 6.2 mL dioxane/mesitylene solution (4:1, v/v) to achieve 0.002, 0.02, 0.04, and 0.09 eq. of Sc(OTf)3, respectively, relative to amine groups of DAB. The monomer solutions and catalyst solutions were sonicated separately at room temperature until the monomers and catalysts fully dissolved (i.e., 1–2 min). Catalyst solutions were added to the monomer solutions and vigorously stirred for 1 h at room temperature. Precipitates from the reaction were isolated by centrifugation at 10000 rpm for 10 min, followed by washing with reaction solvents (dioxane/mesitylene, 4:1, v/v) and vacuum filtration (Fisher Scientific, Q5 filter paper with 2–5 μm particle retention). The recovered wet powders were dried by rotary evaporation at 40 °C for 1 h.
:1, v/v). The catalyst solution was prepared in a separate scintillation vial. In the case of acetic acid, a prescribed volume of 10.5 M aqueous acetic acid (0.44, 1.32, 3.09, 4.85, 6.17 mL) was added to 6.2 mL dioxane/mesitylene solution (4:1, v/v) to achieve 5, 15, 35, 55, and 70 eq. of acetic acid, respectively, relative to amine groups of DAB. Additionally, a water-free synthesis was carried out with a catalyst solution comprising 0.26 mL of glacial acetic acid in 6.2 mL dioxane/mesitylene solution (4:1, v/v), corresponding to 5 eq. acetic acid. In the case of Sc(OTf)3, prescribed amounts of the catalyst (0.9, 9.1, 18.2, and 41.0 mg) were dissolved in 6.2 mL dioxane/mesitylene solution (4:1, v/v) to achieve 0.002, 0.02, 0.04, and 0.09 eq. of Sc(OTf)3, respectively, relative to amine groups of DAB. The monomer solutions and catalyst solutions were sonicated separately at room temperature until the monomers and catalysts fully dissolved (i.e., 1–2 min). Catalyst solutions were added to the monomer solutions and vigorously stirred for 1 h at room temperature. Precipitates from the reaction were isolated by centrifugation at 10000 rpm for 10 min, followed by washing with reaction solvents (dioxane/mesitylene, 4:1, v/v) and vacuum filtration (Fisher Scientific, Q5 filter paper with 2–5 μm particle retention). The recovered wet powders were dried by rotary evaporation at 40 °C for 1 h.
TAPB-PDA synthesis
TAPB-PDA was synthesized from 1,3,5-Tris(4-aminophenyl)benzene (TAPB) and Terephthalaldehyde (PDA) using either acetic acid or Sc(OTf)3 as catalysts. In a typical synthesis, 0.23 mmol of TAPB and 0.34 mmol of PDA were added to a 20 mL scintillation vial under N2 protection followed by addition of 4.6 mL dioxane/mesitylene solution (4:1, v/v). The catalyst solution was prepared in a separate scintillation vial. In the case of acetic acid, a prescribed volume of 10.5 M acetic acid in water (0.33, 1.3, 2.28, 3.58 mL) was added to 4.6 mL dioxane/mesitylene solution (4:1, v/v) to achieve 5, 20, 35, and 55 eq. of acetic acid, respectively, relative to amine groups of TAPB. Sc(OTf)3 solutions were prepared by addition of prescribed amounts of the catalyst (0.67, 6.7, 30.2, 50.4 mg) to 4.6 mL dioxane/mesitylene solution (4:1, v/v) to achieve 0.002, 0.02, 0.09, and 0.15 eq. of Sc(OTf)3, respectively to amine groups of TAPB. The monomer and catalyst solutions were sonicated and combined as described for COF-LZU1, but the reaction was carried out for 24 h before the same washing, recovery, and drying steps.
Activation of COF-LZU1
Activation of COF-LZU1 for dye adsorption requires the removal of residual solvents and Sc(OTf)3. Residual dioxane and mesitylene were removed from as-made dried powders of COF-LZU1 by dispersion of the powders (50 mg) in 45 mL of methanol, followed by sonication for 20 min, vacuum filtration, and rotary evaporation of the recovered wet powders at 40 °C for 30 min. The resulting powders are referred to as “methanol-washed COF-LZU1”. Sc(OTf)3 was fully removed from the methanol-washed COF-LZU1 powders by sonication in 45 mL DI water for 20 min followed by vacuum filtration. Finally, the resulting wet powders were dispersed by sonication for 1 min in water and soaked at room temperature for ca. 10 h before recovery by filtration and drying by rotary evaporation at 40 °C for 1 h. This water dispersion, soaking, and drying process was repeated once to obtain fully activated COF-LZU1 powders.Instrumentation and characterization
:1 (v/v) dioxane/mesitylene) onto an ultra-thin holey carbon film supported on a copper mesh (150 μm) TEM grid (Electron Microscopy Science, HC400-Cu-150) followed by overnight vacuum drying.000g for 5 min. Duplicate portions of the resulting supernatant, Vmeasure, were diluted with DI water into the calibration range, and UV-Vis absorbance was measured. The remaining solution was sonicated to redisperse the pelletized COF and returned to the uptake solution. Dye uptake at the time of the nth aliquot (Qtn [mmol g−1]) was calculated according to eqn (1),
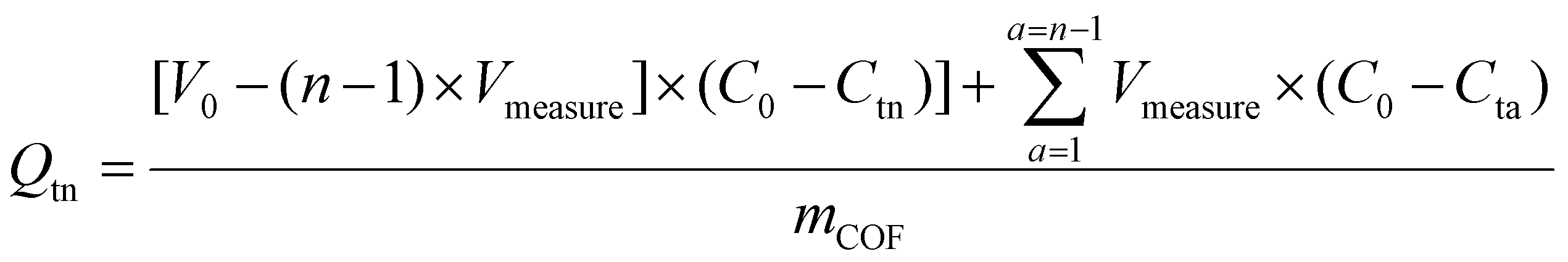 | (1) |
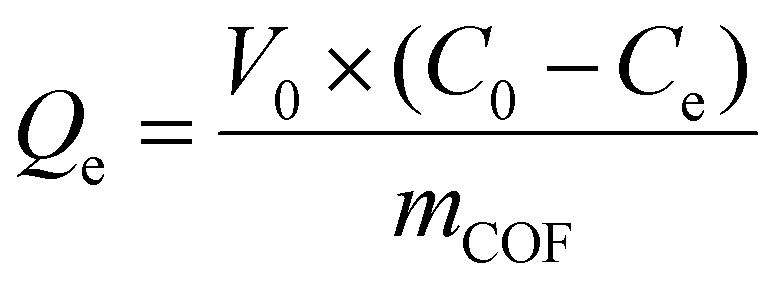 | (2) |
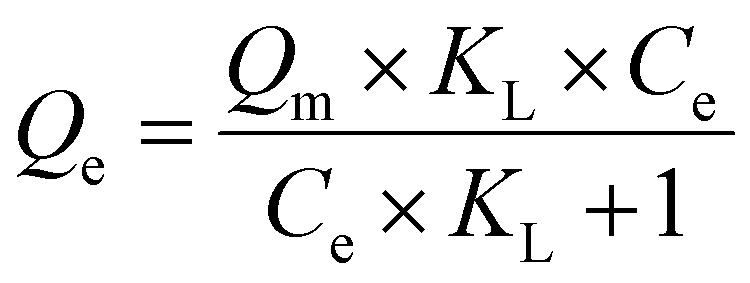 | (3) |
The maximum specific loading of a dye on the external surface area of a given COF, Lmax|EXT[mmol g−1], was calculated according to eqn (4),
| Lmax|EXT = Sexternal/Sdye | (4) |
:Sc as well as Yb and Zn) were carried out using identical settings in a box with at least 10 Å of vacuum in all directions. The binding energy (BE) of the triflate onto a site (physisorbed or chemisorbed) was generally calculated as:| BE = ECOF+M(OTf)x− ECOF− EM(OTf)x | (5) |
Results and discussion
We selected COF-LZU1 and TAPB-PDA from the array of established 2D imine-based structures to elucidate how synthetic catalysts might be exploited generally to tailor morphology, and, thereby, the characteristic diffusion length scale. These two imine-based COFs exhibit hexagonal pores and were chosen due to their room-temperature synthesis relative to the solvothermal processing common among other non-hexagonal COFs (e.g., triangular HPB-COF;57 rhombic TAPPy-PDA58 and Py-DHPh/Py-BPyPh;59 pentagonal TAPP-PBTD-COF;60 bi-porous PT-COF61). The comparable synthesis routes for COF-LZU1 and TAPB-PDA COFs are depicted in Scheme 1, wherein room temperature reticulation is achieved with distinct monomer combinations in conventional dioxane/mesitylene solvents for reaction times resulting in maximum crystallinity.47,62 With the dioxane/mesitylene solvent ratio held fixed throughout this work at the 4:1 (v/v) ratio employed broadly in the 2D imine-based COF literature, we studied two common synthesis catalysts, acetic acid and Sc(OTf)3, exploring sensitivities of COF chemistry, morphology, and functionality to catalyst concentration. We focused on Sc(OTf)3 as an alternative to acetic acid because of its identification from among a range of metal triflates (e.g., Sc, Eu, In, Yb, Y, Zn) as the most efficient triflate-based transimination catalyst leading to highly crystalline 2D imine-base COF products.47
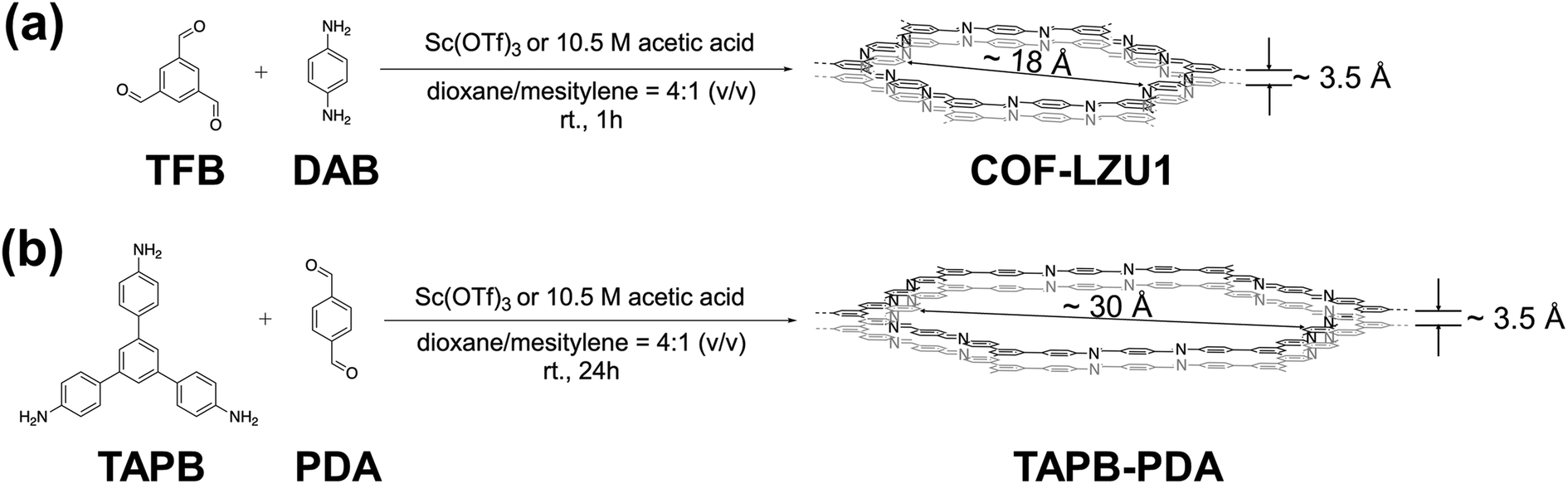 | ||
| Scheme 1 Synthesis routes to (a) COF-LZU1 and (b) TAPB-PDA using either acetic acid or Sc(OTf)3 as catalyst. | ||
Catalyst sensitivity of COF morphology
Fig. 1 shows scanning electron microscopy (SEM) and transmission electron microscopy (TEM) images of the COF-LZU1 and TAPB-PDA products resulting from acetic acid or Sc(OTf)3 catalytic conditions. Consistent with earlier literature reports,63 acetic acid-based synthesis of COF-LZU1 results in globular particles (Fig. 1a). Such globular morphology persists for syntheses at low to high acetic acid concentrations (Fig. S1†). The alternative use of a nominally low concentration (≤0.02 eq., Fig. 1b and S1†) of Sc(OTf)3 catalyst47 results in strikingly hierarchical rosette-like particles. Upon increasing the concentration of Sc(OTf)3, the rosette-like features become less pronounced, appearing as more subtle surface roughness (Fig. 1c), with final emergence of fully globular structures at the highest Sc(OTf)3 concentrations studied (Fig. 1d). SEM imaging of the product of triplicate syntheses (Fig. S2†) confirms the reproducibility of the COF-LZU1 morphological evolution as well as characteristic particle size with catalyst type and concentration. Additionally, while literature reports have suggested sensitivity of particle morphology to reaction time,64 time-series experiments reveal the robustness of the unique rosette-like COF-LZU1 morphology and characteristic particle size to reaction times spanning 1 h to 24 h (Fig. S3a†). | ||
| Fig. 1 SEM and TEM images of COF-LZU1 synthesized under (a) 15 eq. 10.5 M acetic acid and (b) 0.02, (c) 0.04, and (d) 0.09 eq. Sc(OTf)3; and TAPB-PDA synthesized under (e) 20 eq. 10.5 M acetic acid and (f) 0.02, (g) 0.09, and (h) 0.15 eq. Sc(OTf)3. | ||
We observe analogous particle morphologies relative to catalytic synthesis conditions for a different 2D imine COF, TAPB-PDA (Fig. 1e–h and Fig. S1†). Namely, TAPB-PDA products tend to be globular when synthesized with acetic acid as the catalyst, but distinctly rosette-like when low concentrations of Sc(OTf)3 are used. Similar to the morphological trends observed for COF-LZU1, increasing the concentration of Sc(OTf)3 leads to TAPB-PDA product morphologies that evolve toward globular structures (Fig. 1h). As with COF-LZU1, SEM imaging also confirms the reproducibility (Fig. S4†) and reaction-time invariability (Fig. S3b†) of TAPB-PDA COF prepared with the nominal catalytic conditions studied here. The similarities in morphological evolution point to potential generalizability of the effects of catalyst type and concentration on 2D imine COFs.
COF composition and crystallinity relative to catalytic conditions
Fig. 2 shows the FTIR spectra collected for as-made COF-LZU1 and TAPB-PDA COFs prepared under specified catalytic conditions, with band assignments concluded in some cases from reference spectra in Fig. S5.† The strong bands at 1620 cm−1 for COF-LZU1, and 1595 and 1621 cm−1 for TAPB-PDA COF indicate imine linkage formation.47,63 The weaker bands at 3200–3600 cm−1 and 1690 cm−1 are assigned, respectively, to amino and formyl groups comprising terminal functionality and/or framework defects (Fig. 2c).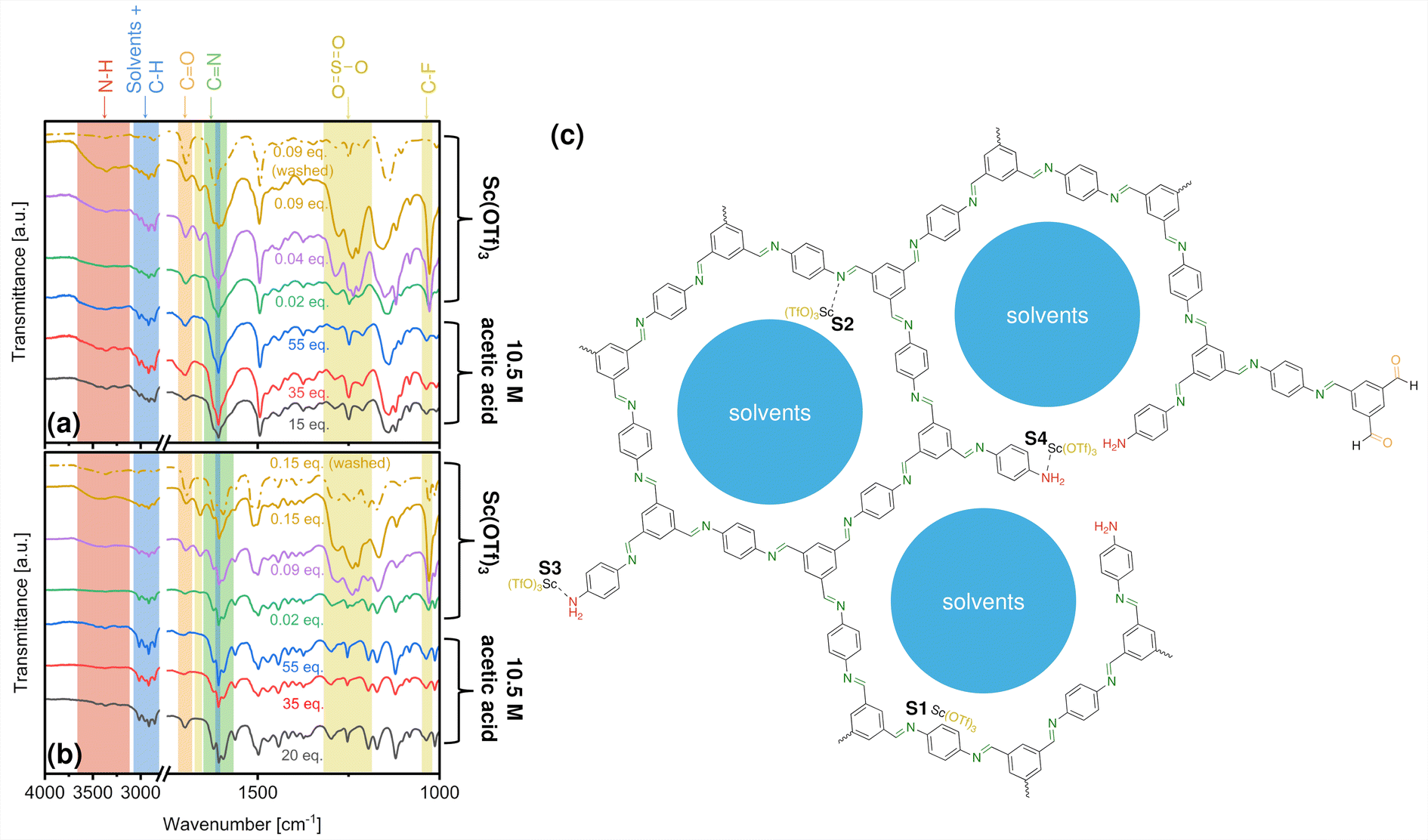 | ||
| Fig. 2 FTIR spectra of as-made (a) COF-LZU1 and (b) TAPB-PDA synthesized under different catalytic conditions, with (c) molecular representation of the pores (shown for COF-LZU1) as well as possible framework defects, terminal functional groups, and potential sitings of Sc(OTf)3 within the COF framework, including: (S1) physisorbed in the pores, coordinated with (S2) a framework N, (S3) a dangling terminal amine, or (S4) an amine N from a framework defect. | ||
Bands between 2750–3100 cm−1 are attributable to a combination of characteristic dioxane and mesitylene peaks overlaying C–H vibrations originating from the COF framework. Further, the band at 1617 cm−1 is a combination of characteristic mesitylene and COF-based C–C stretching vibrations. Taken together, these spectra indicate that reaction solvents remain despite rotary evaporation at 40 °C, owing to their high boiling points (101–165 °C).
In addition to solvent occlusion within the COF framework, FTIR bands at around 1660 cm−1, 1250 cm−1, and 1030 cm−1 indicate the presence of triflate ligands as well. Specifically, the band at 1250 cm−1 can be assigned to both SO3 stretches from triflate ligands and COF-based vibrations, whereas the band at 1030 cm−1 can be assigned to C–F stretching from the triflate ligands alone. The amount of triflate incorporated within the COFs trends qualitatively with the concentration of the catalyst used in the synthesis, as indicated by the corresponding increase in the intensity of (OTf)3-associated bands. Separately, EDS elemental mapping (Fig. S6†), carried out on a representative COF-LZU1 sample prepared with 0.02 eq. Sc(OTf)3 catalyst, reveals a uniform distribution of Sc throughout the COF-LZU1 particles, suggesting the incorporation of the full Sc(OTf)3 moiety within the COF structure.
Potential siting of Sc(OTf)3 in the COFs could conceivably occur via physisorption in the pores, or by coordination with framework imines or with amines from framework edges or defects, as depicted in Fig. 2c. Complementary XPS analysis offers critical insight into Sc incorporation during synthesis. Fig. 3 shows N 1s and Sc 2p spectra for COFs synthesized under different catalytic conditions (C 1s, S 2p, and F 1s spectra included in Fig. S7–S9†). The increasing intensity of the Sc 2p peaks (Fig. 3a and c) indicates increasing Sc incorporation with increasing catalyst concentrations in the reaction solution as quantified in Tables 1, S1, and S2.†
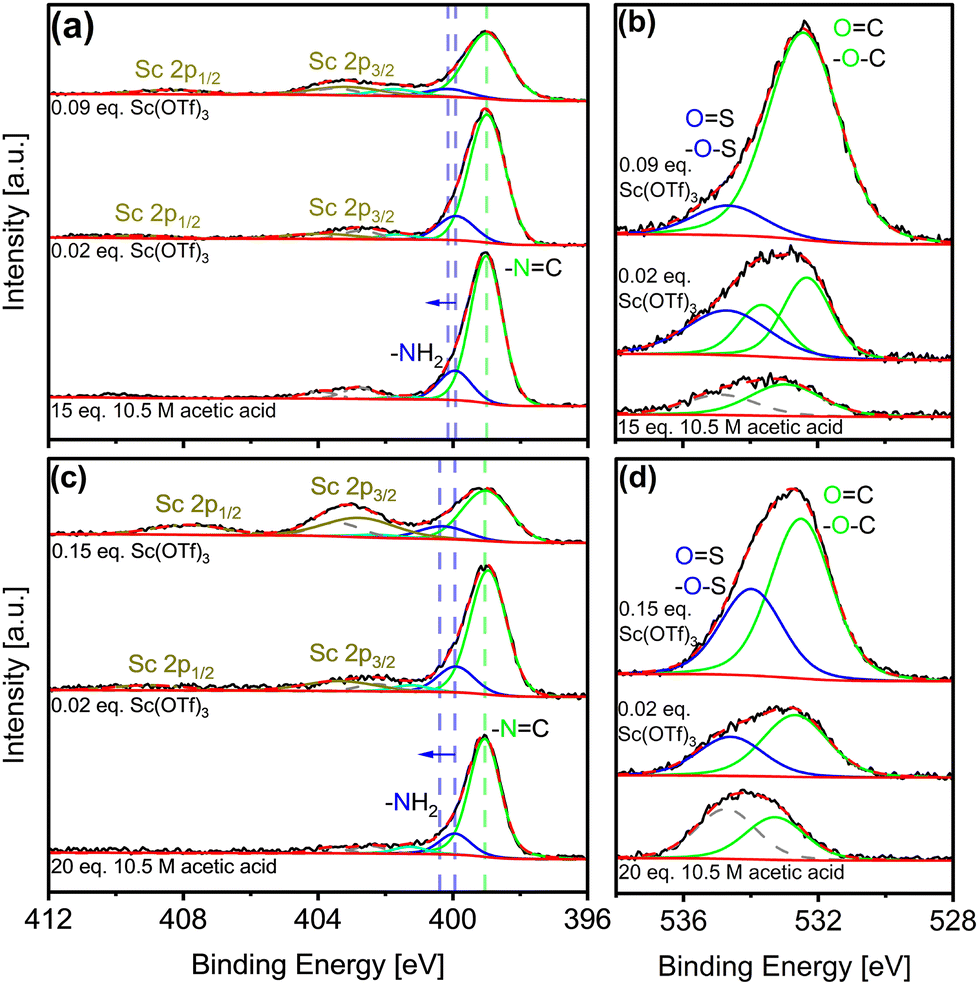 | ||
| Fig. 3 XPS of as-made (a and b) COF-LZU1 and (c and d) TAPB-PDA synthesized under different catalytic conditions; (a and c) N 1s and Sc 2p spectra, (b and d) O 1s XPS spectra. | ||
| Catalytic condition | COF-LZU1 | TAPB-PDA | ||
|---|---|---|---|---|
| Sc:N |
O % (pristine COFs) | Sc:N |
O % (pristine COFs) | |
| 15 or 20 eq. 10.5 M acetic acid | N/A | 2.74 | N/A | 5.40 |
| 0.02 eq. Sc(OTf)3 | 0.03:1 |
3.79 | 0.05:1 |
3.45 |
| 0.09 eq. Sc(OTf)3 | 0.07:1 |
11.59 | — | — |
| 0.15 eq. Sc(OTf)3 | — | — | 0.16:1 |
9.27 |
Deconvolution of each spectrum from 396 to 402 eV according to contributions from amino (-NH2) and imine (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) nitrogens reveals a slight shift of amino fitted peaks to a higher binding energy by ca. 0.5 eV with increasing Sc(OTf)3 concentration. Such a shift is expected upon coordination of the lone pair electron of the amino nitrogens with metals, as has been reported for Pd-imine COF coordination.65 Here, the observed shift is attributed to the increasing amount of Sc(OTf)3 coordinating with the N-containing dangling bonds associated with framework defects and/or the terminal edges of the COF framework, owing to the electron donation of lone pair electrons from amino nitrogens to Sc.66 Additional quantification of O content (Fig. 3b and d) offers insight into framework formyl groups comprising defects and terminal dangling bonds. Specifically, the increase in oxygen content (Table 1) with increasing Sc(OTf)3 catalyst concentration is indicative of commensurate increases of framework defects, likely resulting from the faster reaction kinetics during COF formation.67,68
N) nitrogens reveals a slight shift of amino fitted peaks to a higher binding energy by ca. 0.5 eV with increasing Sc(OTf)3 concentration. Such a shift is expected upon coordination of the lone pair electron of the amino nitrogens with metals, as has been reported for Pd-imine COF coordination.65 Here, the observed shift is attributed to the increasing amount of Sc(OTf)3 coordinating with the N-containing dangling bonds associated with framework defects and/or the terminal edges of the COF framework, owing to the electron donation of lone pair electrons from amino nitrogens to Sc.66 Additional quantification of O content (Fig. 3b and d) offers insight into framework formyl groups comprising defects and terminal dangling bonds. Specifically, the increase in oxygen content (Table 1) with increasing Sc(OTf)3 catalyst concentration is indicative of commensurate increases of framework defects, likely resulting from the faster reaction kinetics during COF formation.67,68
Such defects are borne out by the COF crystallinity as revealed by comparison of experimental diffraction patterns collected from materials synthesized under specific catalytic conditions for nominal reaction times leading to the maximum crystallinity (Fig. S10†), with simulated XRD patterns (Fig. 4). Namely, the attenuation of XRD reflections with increasing Sc(OTf)3 catalyst concentration is consistent with XPS evidence of concomitant increases in framework defects. Scherrer equation analysis of the (100) and (001) peaks reveals the presence of anisotropic crystallites with in-plane dimensions (ca. 21.7 nm) that average ca. 2.7–3 times the out-of-plane dimensions (Table S3†). Interlayer spacing, calculated from Bragg's law analysis of the (001) peak, was estimated to be ca. 3.5 Å under nearly all catalytic conditions considered.
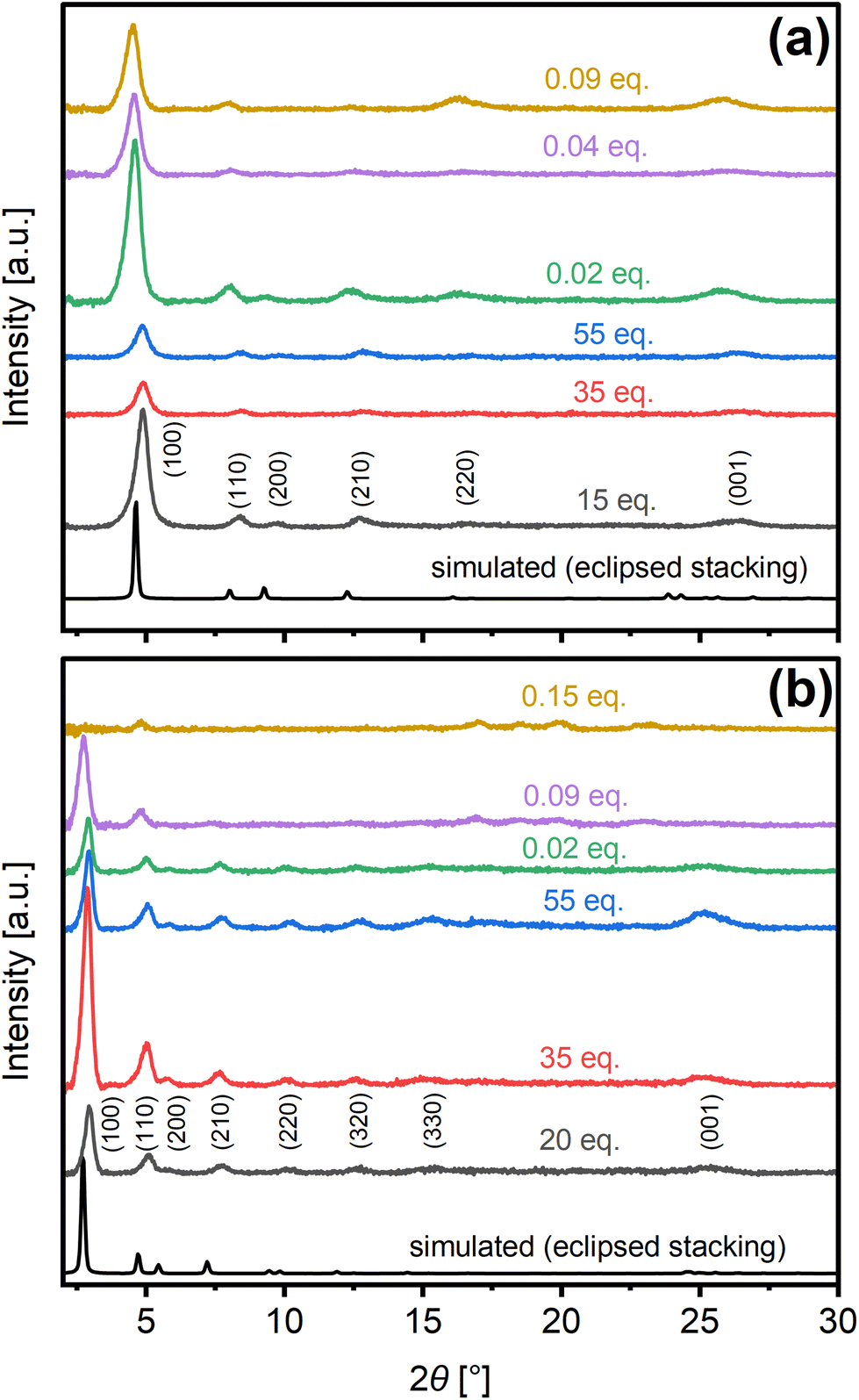 | ||
| Fig. 4 Experimental PXRD patterns of as-made (a) COF-LZU1 and (b) TAPB-PDA synthesized under different catalytic conditions compared to patterns simulated for eclipsed structures. | ||
Comparison of the 2θ angles of the (100) and (001) reflections as well as lattice parameters extracted from the Gaussian fittings of the peaks (Fig. S11†) and Pawley refinement of the diffraction patterns using Material Studio (Fig. S12†), confirms nearly eclipsed stacking in all cases.47,63 A uniform shift of the reflections relative to the theoretical patterns for perfectly eclipsed structures is observed as the synthesis catalyst is changed between acetic acid and scandium triflate. The subtle shifts to lower angles in the case of the Sc(OTf)3-catalyzed materials, indicate minor expansion of the unit cell that we attribute to sensitivity of the COF framework to metal coordination as reported in the literature.69,70 The shifts to higher angles in the case of acetic acid-derived COFs indicate a relative contraction of the unit cell/pores. This reproducible phenomenona, one that we are investigating further, is likely a manifestation bond rotation owing to the weakening of π-π stacking energies upon shifting of COF layers under room temperature synthesis.
The emergence of subtle higher angle reflections in the case of TAPB-PDA synthesized under 0.09 eq. and higher Sc(OTf)3 indicates the development of mixed crystalline phases. Hence, we will focus on the apparently phase-pure COF-LZU1 for the remainder of this work.
DFT calculations: Sc(III) siting and mechanistic insight into anisotropic crystal growth
Motivated by the XPS evidence of Sc(OTf)3 coordination with framework amines and imines (Fig. 3), we have carried out density functional theory (DFT) calculations to elucidate the strength of Sc(III) triflate coordination within the COF framework and to thereby assess where Sc(OTf)3 is most likely to site. Specifically, as shown in Fig. 5, we have computed the binding energies associated with various Sc(OTf)3-COF configurations enumerated in Fig. 2c, including the physical adsorption of Sc(OTf)3 in the pores (Fig. 5a) and the coordination of Sc(OTf)3 with various imine or amine functional groups, including coordination of Sc(OTf)3 with an imine group of a pristine COF layer surface (i.e., crystal 001 surface) (Fig. 5b), simultaneous coordination of Sc(OTf)3 with an amine comprising a framework defect and a framework imine within an adjacent COF layer (Fig. 5c), and co-coordination of Sc(OTf)3 with amine groups comprising adjacent framework defects (Fig. 5d).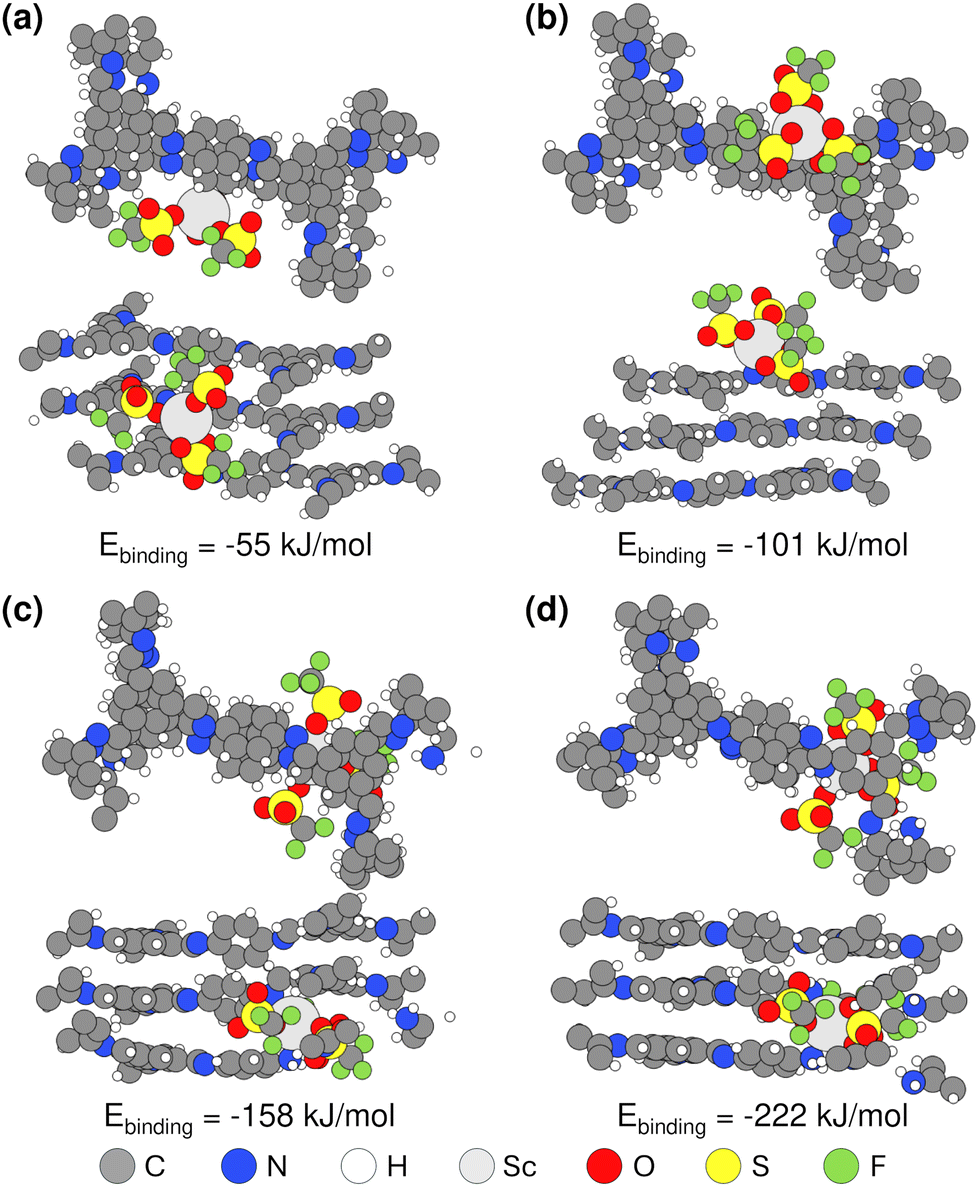 | ||
| Fig. 5 DFT simulation results of optimized structures (top and side view) and the calculated binding energies within COF-LZU1 trilayers for Sc(OTf)3 (a) physisorbed in the pore, (b) adsorbed on an imine bond of the top layer of the pristine COF-LZU1, (c) adsorbed on a single defect resulting in an imine-Sc(OTf)3-amine linkage, and (d) coordinated with two defects resulting in an amine-Sc(OTf)3-amine linkage. | ||
Sc(OTf)3 coordinates most strongly to adjacent amines comprising a double framework defect, a configuration that is also representative of Sc(OTf)3-mediated co-coordination between dangling amines terminating adjacent crystallites. This coordination state (BE: −222 kJ mol−1) is 60–160 kJ mol−1 more favored than Sc(OTf)3 coordinated with framework imines, and the strength of Sc(OTf)3 binding with all frameork N exceeds by ca. 84–300% the energy associated with Sc(OTf)3 physisorption in COF pores. The coordination of Sc(OTf)3 with amino nitrogen rather than absorption in COF pores was confirmed by the shift of the amino fitted XPS peaks with increasing Sc(OTf)3 concentrations (Fig. 3).
According to XPS analysis (Table S1†), amino nitrogen comprises at most ca. 15% of all N in the 0.02 eq. Sc(OTf)3-catalyzed COF-LZU1, and terminal amines make up a maximum of ca. 8% of all N according to approximate crystallite sizes estimated from XRD data (Table S3 and Scheme S1†) in the limit of all terminal groups being amines and not formyl groups. Thus, the XPS-measured coordination of Sc with just 3% of all amino N in the case of 0.02 eq. Sc(OTf)3-catalyzed COF-LZU1 (Table S1†), would be fully accomodated by terminal amines, the strongest and likely most accessible binding sites. Such preferential titration of terminal amines decorating the edge of 2D COF sheets at low Sc(OTf)3 concentrations could enable recruitment of, and co-coordination with, amine-functionalized monomers, oligomers, or precursor sheets or nanocrystals at the edge of COF sheets (Scheme 2a). At the same time, the lack of Sc(OTf)3 coordination with surface imines may suppress layer stacking and, thereby, result in anisotropic crystal growth consistent with the high-aspect-ratio features of the rosette structures formed at low Sc(OTf)3 concentrations.
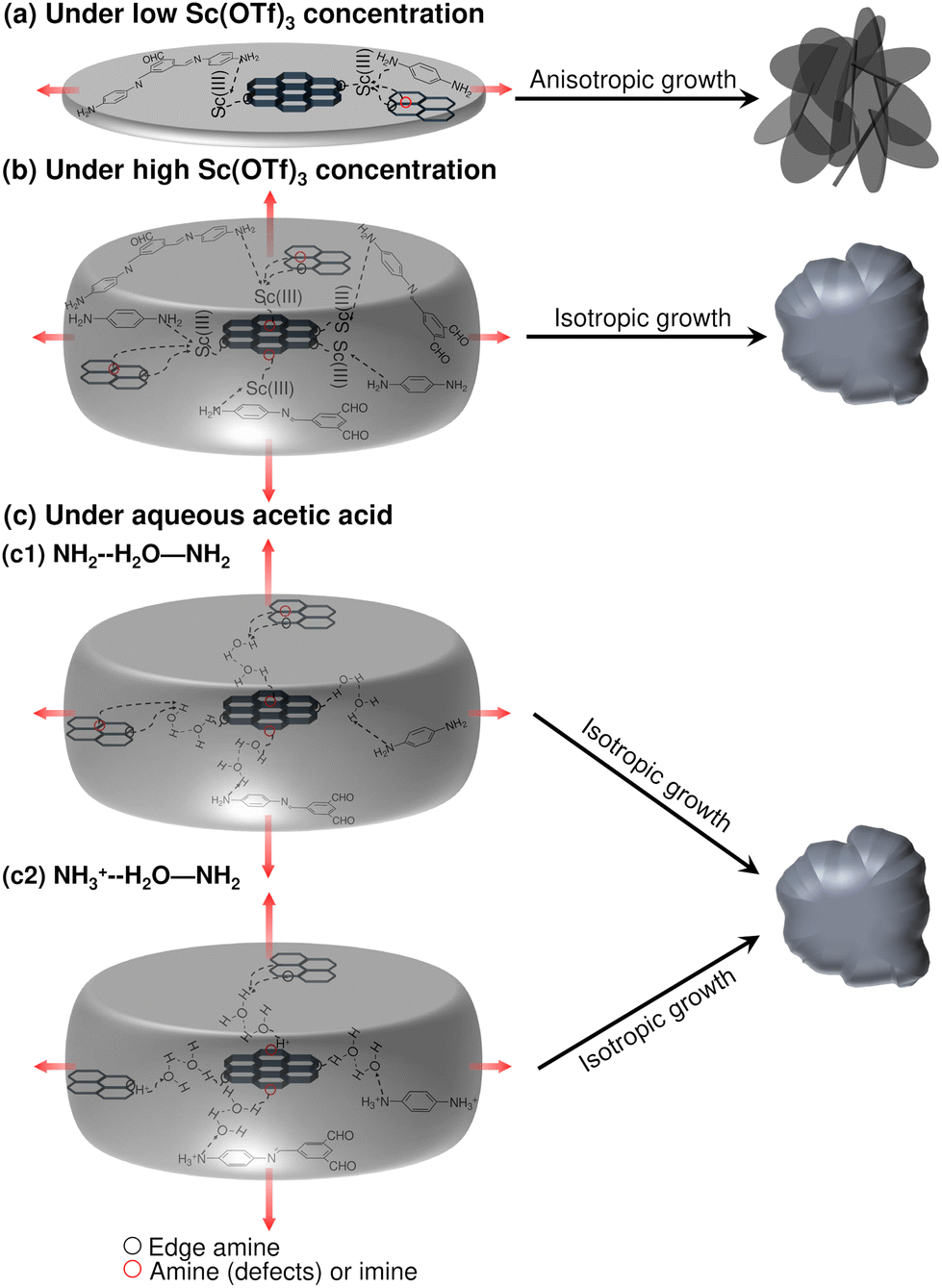 | ||
| Scheme 2 Proposed mechanism of anisotropic and isotropic COF particle growth under different catalytic conditions. | ||
The relative binding energies determined by DFT calculations (Fig. 5) suggest that Sc(OTf)3 would titrate amines/imines on the 2D COF layers only at higher concentrations of the catalyst. For the 0.09 eq. Sc(OTf)3-catalyzed COF-LZU1, XPS analysis shows an increase in Sc coordination to 7% of all N (Table 1), likely exceeding the number of terminal amino N (Table S3 and Scheme S1†) when accounting for the inevitable mixture of amine and formyl group terminations, and thereby increasing the likelihood of Sc(OTf)3 to also titrate more sterically hindered defect-derived framework amines and lower binding energy surface/framework imines. The concomitant coordinative bonding among amines/imines on different COF sheets supports the emergence of a sheet stacking mechanism simultaneously with continued 2D COF sheet growth. Such isotropic growth is consistent with the observed formation of globular COF-LZU1 structures at higher Sc(OTf)3 concentrations.
Complementary DFT calculations (Table S4†) show that the difference in binding energies between adjacent amine linkages and surface imines of ca. 120 kJ mol−1 is also preserved for other representative metal triflates (e.g., Yb, Zn), suggesting that triflates may more generally preferentially titrate double framework defects and edge amines over surface imines. Yet, the higher binding energies for the Yb- and Zn-based triflates make it less clear whether sequential titration of these sites would similarly encourage preferential lateral layer growth or if the higher binding strengths may instead explain reports by Dichtel and co-workers47 of the decreasing efficiency (Sc > Yb > Zn) of these catalysts to form crystalline 2D imine-based COF products.
When acetic acid is instead used as a catalyst, we conceive an isotropic growth mechanism like that which occurs at high Sc(OTf)3 concentrations is at play in the formation of only globular structures. Namely, Jesus and Redinha71 have demonstrated the hydrogen bonding of water with primary and/or protonated amines. Both types of amines likely populate COF framework edges and defects in this system, and hence serve as points for water- and/or acetic acid-mediated lateral growth of 2D COF layers and layer stacking as schematized in Scheme 2c. A third possibility for hydrogen bonding with amine radicals as reported in the literature,72 is not considered here given the unlikely formation of amine radicals under the synthesis conditions studied.
We have determined (not shown) that 5 eq. of acetic acid is sufficient to drive COF-LZU1 formation, far exceeding the amount of catalyst equivalence in the case of Sc(OTf)3-based COF-LZU1 synthesis (i.e., 0.002–0.15 eq.). Therefore, the high concentrations of hydrogen bonding mediators (i.e., acetic acid, water) under the minimal conditions sufficient for COF-LZU1 growth would enable simultaneous growth and stacking of COF layers akin to that observed for high concentrations of Sc(OTf)3. Indeed, by introducing controlled amounts of water (as high as 60 eq.), and thereby more hydrogen bonding mediators, to 0.02 eq. Sc(OTf)3 synthesis solutions, the COF-LZU1 morphology (Fig. S13†) evolves from its nominal rosette structure toward featureless globular structures bearing signature bonding and crystallinity as confirmed by FTIR (Fig. S14a†) and PXRD (Fig. S14b†), respectively.
Activation and textural analysis of a COF-LZU1
As-made COF-LZU1 products were activated by sequential solvent exhange. Initial methanol treatment removes residual solvents, as indicated by attenuation of FTIR bands at ca. 3000 cm−1 (dioxane, mesitylene) and 1617 cm−1 (mesitylene). Triflate ligands are also partially removed, as denoted by attenuation of peaks at 1660 cm−1, ca. 1200–1300 cm−1, and 1030 cm−1 (Fig. 6a). Low energy ion scattering (LEIS) (Fig. 6b) confirms clear attenuation of triflate-specific O, F, and S whereas the persistent Sc signal highlights the ineffectiveness of methanol for breaking coordination between Sc and framework N. Subsequent water washing is sufficient to break Sc–N coordination, leading to full removal of Sc(OTf)3 as confirmed by the vanishing FTIR bands (i.e., 1660 cm−1, 1030 cm−1) (Fig. 6a) and loss of F, S, and Sc LEIS signals (Fig. 6b). Powder XRD (Fig. 6c) and complementary SEM images (Fig. S15†) of activated COF-LZU1 samples synthesized under various catalytic conditions highlight the robustness of the crystallinity and morphology to full activation.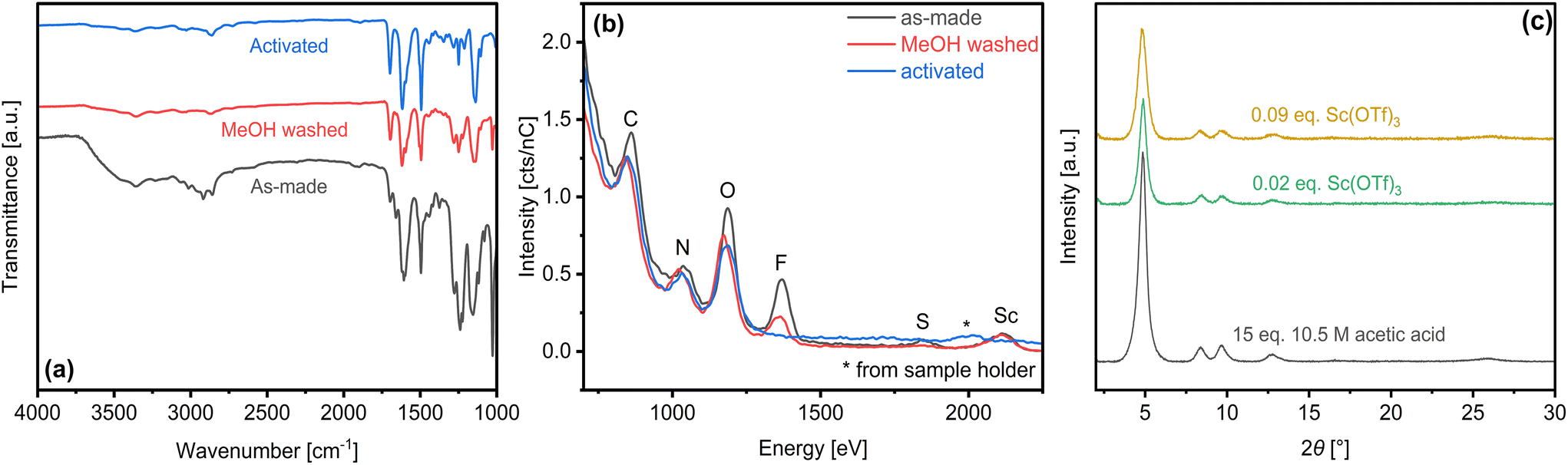 | ||
| Fig. 6 (a) FTIR spectra and (b) average LEIS spectra over 3nm sputtering depth for 0.09 eq. Sc(OTf)3-derived COF-LZU1 at points along the solvent activation process. (c) PXRD patterns of activated COF-LZU1 synthesized under different catalytic conditions. | ||
Nitrogen physisorption (Fig. 7) reveals Type 1(b) isotherms,73 characterized by precipitous micropore filling that gives way, at higher relative pressures, to adsorption in distributed but narrow mesopores. Gradual multi-layer adsorption in mesostructures at intermediate relative pressures and unrestricted monolayer-to-multilayer adsorption at the highest relative pressures is most distinct in the case of the rosette material (0.02 eq. Sc(OTf)3).
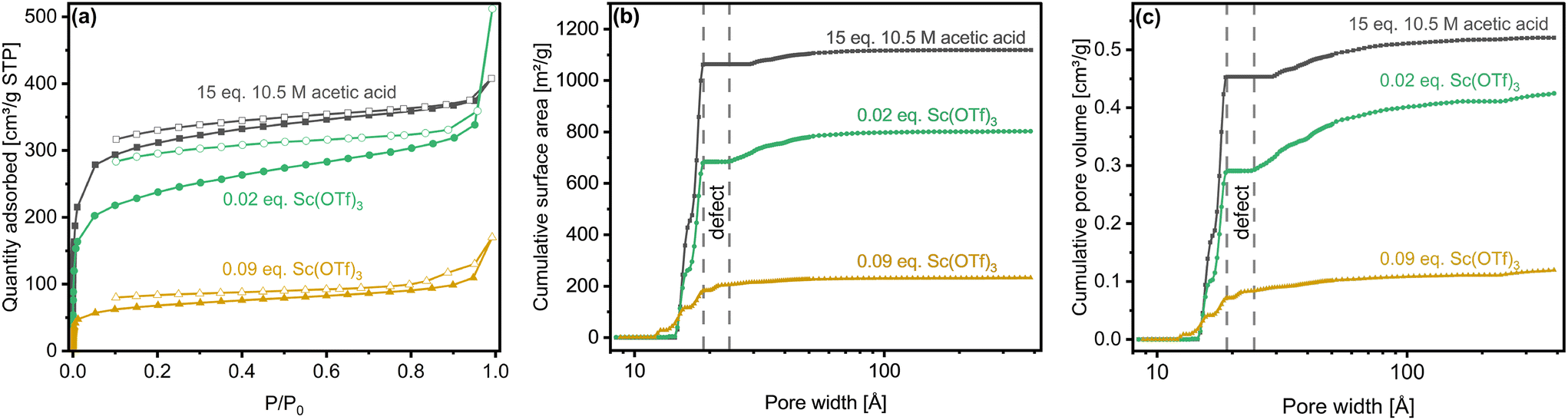 | ||
| Fig. 7 Representative (a) N2 adsorption and desorption isotherms (77K), (b) cumulative surface areas, and (c) cumulative pore volumes of activated COF-LZU1 synthesized under different catalytic conditions. | ||
Cumulative surface areas, cumulative pore volumes, and corresponding quantitative textural measures (Table 2) derived from adsorption branch NLDFT analysis of triplicate COF syntheses help differentiate COF-LZU1 synthesized under the various catalytic conditions studied here. The globular acetic-acid synthesized material has the largest BET surface area (Table 2), attributable to its high crystallinity (Fig. 6c), whereas the total specific surface areas for the rosette and 0.09 eq. Sc(OTf)3-synthesized globular structure were ca. 24% and 84% lower, respectively.
| Catalytic condition | Smicro [m2 g−1] | Sdefect [m2 g−1] | Sexternal [m2 g−1] | SBET [m2 g−1] | Vmicro [cm3 g−1] | Vdefect [cm3 g−1] | Vexternal [cm3 g−1] | Vtotal [cm3 g−1] |
|---|---|---|---|---|---|---|---|---|
| 15 eq. 10.5 M acetic acid | 1029 ± 44 | 0 | 52 ± 10 | 1149 ± 27 | 0.44 ± 0.01 | 0 | 0.06 ± 0.02 | 0.50 ± 0.02 |
| 0.02 eq. Sc(OTf)3 | 782 ± 93 | 0 | 95 ± 21 | 906 ± 40 | 0.33 ± 0.04 | 0 | 0.11 ± 0.02 | 0.44 ± 0.02 |
| 0.09 eq. Sc(OTf)3 | 163 ± 50 | 27 ± 3 | 30 ± 8 | 235 ± 40 | 0.06 ± 0.02 | 0.02 ± 0.01 | 0.05 ± 0.03 | 0.12 ± 0.003 |
Micropore surface areas and pore volumes (Fig. 7b, c and Table 2), classically associated with pores of 20 Å or less in diameter, also trend with material crystallinity and defects. Namely, the most crystalline and least defective material prepared under acetic acid conditions shows the most crystalline micropores and, hence, the largest cumulative micropore surface area and micropore volume. The reduced crystallinity of the 0.02 eq. Sc(OTf)3-derived material leads to ca. 25% reduction in both cumulative micropore surface area and micropore volume. The presence of significant framework defects, introduced when the highest amount (i.e., 0.09 eq.) of Sc(OTf)3 catalyst is used, further reduces the cumulative micropore surface area and micropore volume by ca. 82%. These defects are manifested by the corresponding distributions of features (‘defect’ in Fig. 7bc) in the ca. 19–23 Å size range that are absent in the case of the less-defected materials prepared with acetic acid and 0.02 eq. Sc(OTf)3. The characteristic size of the defect features is consistent with the likely pore opening that occurs with framework defects (Fig. S16†). We estimate a corresponding surface area and pore volume associated with such defects in Table 2.
The general trends in textural measures change when it comes to external surface areas and pore volumes. Namely, the rosette materials (0.02 eq. Sc(OTf)3) have the highest external surface area and pore volumes, nearly twice that of the acetic acid-derived materials and 2–3 times larger than the 0.09 eq. Sc(OTf)3-derived materials. This stark difference in external textural measures can be simply rationalized according to the distinct hierarchical rosette morphology, and hence larger external surface area, of the 0.02 eq. Sc(OTf)3-derived material.
Ultimately, the ability to realize hierarchically structured 2D COF-LZU1 with enhanced textural properties simply by multi-purposing the Sc(OTf)3 catalyst at room tempeature for 1 h, distinguishes this approach from others reported in the literature (Table S5†41–46) in terms of its simplicity, scalability, and sustainability. Specifically, alternative COF structuring strategies commonly introduce secondary templates (requiring separate synthesis) or growth modifiers, employ higher-temperature (sometimes solvothermal) conditions to achieve crystalline products, require extended reaction periods of up to 2–15 days, and rely on complicated, multi-step pre-, reaction, and post-processing conditions. Direct comparison of this work to literature reports of the impact of structuring on textural properties is confounded by differences in the specific COF frameworks reported. Yet, the ca. 286% enhancement of the BET surface area, achieved herein by structuring Sc(OTf)3-catalyzed COF-LZU1 simply by reducing the catalyst concentration from 0.09 eq. (globular) to 0.02 eq. (rosette), is ca. 2–12 times higher than textural enhancements achieved in other COF systems.
Dye adsorption on activated COF-LZU1
In order to assess how the catalyst-tuned particle morphology and textural properties can be leveraged for applications, we have studied transient uptake and equilibrium adsorption of two cationic dyes, Rhodamine B (RdB) and Methylene blue (MB). These dyes were chosen on the basis of distinct kinetic diameters of ca. 11.3 Å and 5.1 Å, respectively, with RdB approaching the characteristic pore size of COF-LZU1 (ca. 18 Å) and MB being significantly smaller.13 In addition to van der Waals interactions of the dyes with the COF framework—specfiically in cases of strong confinement in the pores,74 the positively charged N+ and S+ centers of RdB and MB, respectively, likely drive adsorption on the electron-rich imine and defect-derived amine sites within COFs (Fig. 8; equilibration times of 5 days (RdB) and 4 days (MB) established in Fig. S17†). Cationic dyes that were significantly smaller than (e.g., 2-Naphthol, ca. 4–5 Å) and commensurate with (e.g., Reactive Black 5, ca. 17.0 Å) the COF-LZU1 pore size were also studied, but both led to negligible uptake (not shown).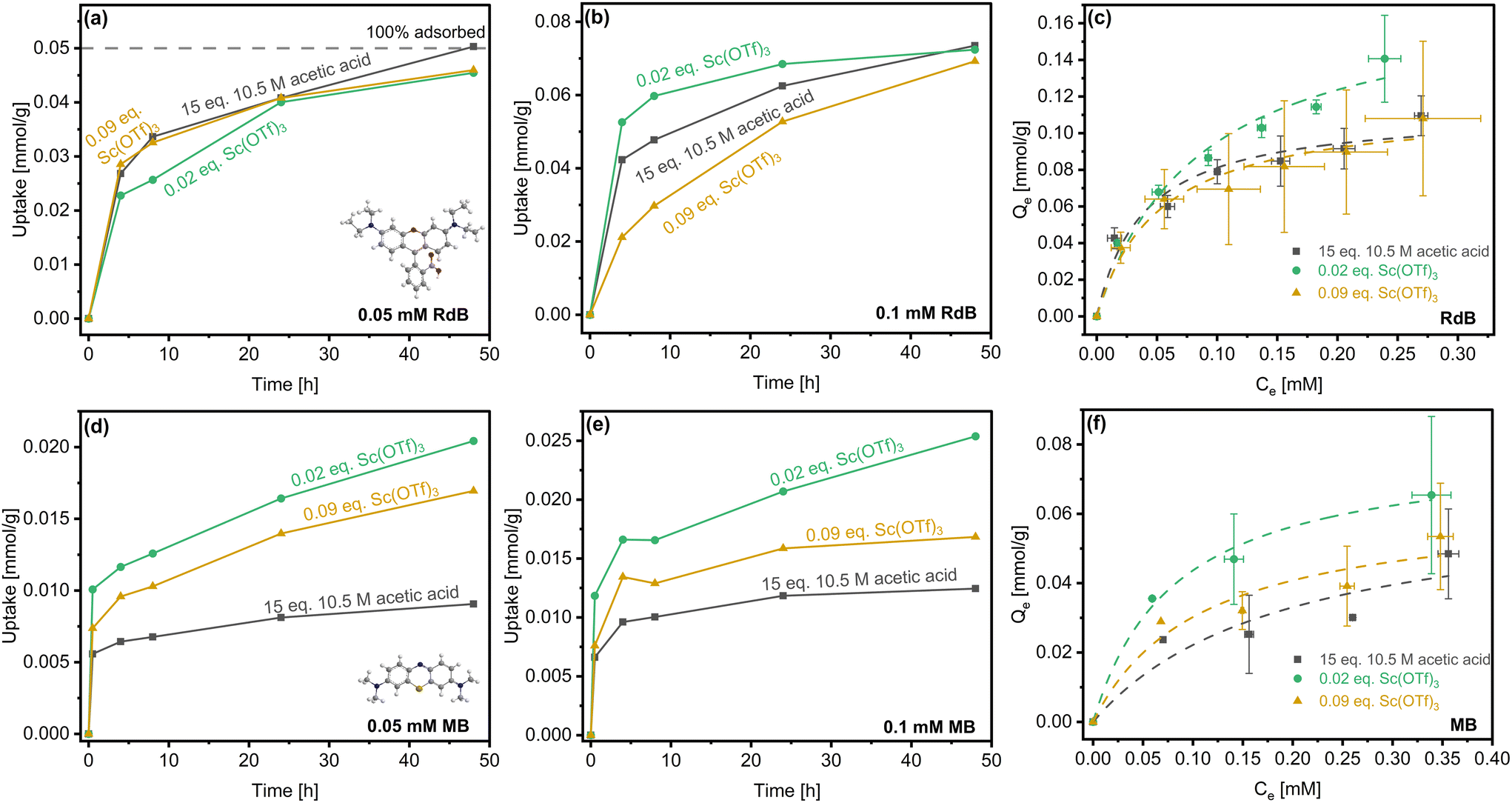 | ||
| Fig. 8 Room temperature uptake from 0.05 mM and 0.1 mM (a and b) RdB and (d and e) MB solutions, and (c and f) corresponding adsorption isotherms (dashed lines represent Langmuir fits) for activated COF-LZU1 synthesized under different catalytic conditions. | ||
Fig. 8a and b show RdB uptake from solutions of two sorbate concentrations over a period of 48 h by representative COF-LZU1 prepared with acetic acid and Sc(OTf)3 catalysts. In the case of the lower RdB concentration (0.05 mM, Fig. 8a), the uptake among the COF-LZU1 samples is effectively indistinguishable. Differentiation of the materials occurs at higher uptake, achieved upon doubling the RdB concentration (Fig. 8b). Specifically, the rate of uptake from 0.1 mM RdB solutions is fastest for the rosette-structured COF-LZU1 material (0.02 eq. Sc(OTf)3) and slower for the globular particles, with the most sluggish uptake measured for the 0.09 eq. Sc(OTf)3-derived material.
The sensitivity (or lack thereof) of the RdB uptake rates to adsorbents at the different sorbate concentrations can be understood in terms of the two modes of RdB adsorption on the accessible external surface and within the confining COF pores. While the textural properties vary among the three materials, the increasing rate of uptake at higher RdB concentrations (Fig. 8b) trends with increasing external surface area. Indeed, it is instructive to consider the maximum measured uptake relative to the estimated maximum loading of RdB that can be accomodated on the external surface of the three COF sorbents or directly within accesible pores, a measure we term Lmax|EXT (Table 3).
| Rhodamine B | Methylene blue | |||||
|---|---|---|---|---|---|---|
| Catalytic condition of activated COF-LZU1 | 15 eq. 10.5 M acetic acid | 0.02 eq. Sc(OTf)3 | 0.09 eq. Sc(OTf)3 | 15 eq. 10.5 M acetic acid | 0.02 eq. Sc(OTf)3 | 0.09 eq. Sc(OTf)3 |
| Lmax|EXT [mmol g−1] | 0.06 ± 0.01 | 0.12 ± 0.03 | 0.04 ± 0.01 | 0.12 ± 0.03 | 0.25 ± 0.06 | 0.08 ± 0.02 |
| KL [M−1] | 2.6 × 104 | 1.2 × 104 | 1.9 × 104 | 5.2 × 103 | 1.2 × 104 | 9.3 × 103 |
| Qm [mmol g−1] | 0.11 | 0.18 | 0.12 | 0.06 | 0.08 | 0.06 |
In the case of the lower RdB concentrations (Fig. 8a), the maximum specific uptake of ca. 0.045 mmol g−1 (48 h) can be nearly completely accommodated by adsorption on the accessible external surfaces of the three sorbents or directly within accessible pores without the slower diffusion into the COF pores impacting the uptake. Increasing the RdB concentration (Fig. 8b), however, results in higher RdB uptake (ca. 0.07 mmol g−1 at 48 h) that reaches or exceeds Lmax|EXT of the acetic acid- and 0.09 eq. Sc(OTf)3-synthesized materials, respectively. Specifically, with the lowest external surface area and hence fewest directly accessible COF pores, uptake by the 0.09 eq. Sc(OTf)3-synthesized sorbent is the first to be noticeably impacted by the sluggish rate-limiting diffusion of RdB in the COF pores. With further uptake, Lmax|EXT of the acetic acid-synthesized material is approached next, such that restricted RdB adsorption and diffusion in the associated COF pores slows the overall uptake. Ultimately, the total uptake is only approximately half the Lmax|EXT of the rosette-structured 0.02 eq. Sc(OTf)3-synthesized sorbent, with the most rapid RdB uptake resulting from adsorption on the accessible rosette surfaces and directly in accessible pores minimizing the impact of the slower diffusion in COF pores.
The impact of the accessible external surface area and thereby directly accessible pores of the rosette-structured sorbent over the globular sorbents is further indicated by the RdB isotherms collected at room temperature for the three materials (Fig. 8c). Langmuir fitting of the isotherms enabled calculations of the affinities (KL) and capacities (Qm) of dyes for the various activated COF-LZU1 samples (Table 3). The equilibrium isotherms are statistically indistinguishable at low RdB concentrations below the Lmax|EXT, and remain indistinguishable for the two globular materials even at higher sorbate concentrations despite the nearly sixfold higher total specific surface area of the acetic acid-derived COF-LZU1 relative to the 0.09 eq. Sc(OTf)3-synthesized material (Table 2). We hypothesize that the lower total specific surface area of the latter is offset by the larger number of framework defects (Table 1) comprising electron-rich terminal amines that serve as strong binding sites for RdB via its positively charged N+ center. Strong dispersion forces likely also contribute, in part, to this common equilibrium adsorption.
We attribute the divergence of the isotherm measured for the rosette sample at higher RdB concentrations to the contributions of enhanced sites for adsorption on the higher accessible external surface area. Such surface-dominated adsorption, for which dispersion forces are commonly weaker than for molecules under pore confinment, is further supported by the lower effective affinity, KL, for RdB on the rosette-structured materials compared to globular materials where we expect pore adsorption to play a bigger role at equilibrium.
Uptake of the smaller MB molecule (Fig. 8d and e) falls well below the Lmax|EXT (Table 3) for all three materials at both sorbate concentrations studied. Hence, the total uptake should be accomodated in all cases by adsorption on the external surface or directly in the accessible COF pores. The much smaller size of MB relative to the COF-LZU1 pore size likely limits the role of dispersion forces in MB adsorption and certainly eliminates any possible impact of diffusion limits on uptake. Instead, interaction of the S+ center of MB with electron-rich imine and defect-derived amine sites within COFs is expected to dominate adsorption, such that the differences in the uptake rates and equilibrium adsorption capacity can be attributed to differences in the number of accessible adsorption sites in these materials.
The most sluggish uptake and lowest equilibrium adsorption capacity occurs over the globular acetic acid-derived sample, owing to it being the least defective material. The larger number of defects (Table 1) in the 0.09 eq. Sc(OTf)3-catalyzed materials result in more rapid uptake, higher affinity, and similar capacity (Fig. 8f). The most rapid uptake and highest MB capacity are measured for the rosette-structured material (Fig. 8d–f and Table 3). While the specific defects are lower in this material than in the globular 0.09 eq. Sc(OTf)3-synthesized sample (Table 1), the hierarchical structure of the material (i.e., its ca. 4.5-fold higher external surface area) likely offers access to a larger specific number of those defects, resulting in the highest MB adsorption capacity.
Conclusions
The identification of facile strategies for tailoring structural hierarchy of porous materials is key to accessing diffusion length scales suitable for the efficient manipulation of molecules and ease of processing required for applications. Here, we have identified how catalytic conditions alone can be exploited as a powerful and scalable handle for tuning morphology of 2D imine-based COFs without the need to introduce complex structuring strategies, employ higher temperatures, require extended reaction periods, or rely on complicated multi-step reaction and processing steps. Through combined experimental and computational insights, this work offers mechanistic insight into the dual role of Sc(OTf)3 as an efficient synthetic catalyst and growth modifier. Rather than optimizing the system to minimize crystalline defects, we show how the titration of framework defects and terminal groups of different binding strengths can instead be exploited to tune crystal growth. The result is the ability to switch from isotropic crystal growth of globular porous materials with large diffusion length scales to anisotropic crystal growth of interconnected high-aspect-ratio hierarchical products. The sensitivity of this mechanism to triflate binding strength (e.g., stronger binding calculated for alternative metal triflates) and the influence of synthesis temperature (e.g., solvothermal processing) and pore topology (e.g., non-hexagonal topologies including triangular, rhombic, square, pentagonal) on the resulting morphology remain as potential additional design handles to be explored in the future. Yet, the rosette-structured COFs in this work offer unique textural properties with dramatically enhanced external surface area, highly accessible pores, and reduced diffusion length scales enabling their efficient interaction with bulky molecular sorbates. More generally, by exploiting rather than eliminating framework defects, this work offers a unique strategy for morphological design and COF synthesis that should extend beyond sorbents to impact anisotropic growth of COF thin films as a means for extending its impact.Author contributions
Hao Guo: investigation, methodology, and writing. Joseph P. Cline: formal analysis and reviewing. Ryan Thorpe: formal analysis and reviewing. Christopher J. Kiely: validation, writing, and reviewing. Srinivas Rangarajan: funding acquisition, formal analysis, methodology, visualization, writing, and reviewing. Mark A. Snyder: conceptualization, funding acquisition, methodology, project administration, supervision, validation, visualization, writing, and reviewing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge support for this work from the National Science Foundation, through the grant number CBET-1953245, and via a grant from the Commonwealth of Pennsylvania, Department of Community and Economic Development, through the Pennsylvania Infrastructure Technology Alliance (PITA). Portions of this research were conducted on Lehigh University's Research Computing infrastructure partially supported by NSF Award OAC-2019035. In addition, the authors gratefully acknowledge access to the Lehigh University Institute for Functional Materials and Devices Materials Characterization Facility (MCF).References
- A. P. Côté, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, Reticular Synthesis of Microporous and Mesoporous 2D Covalent Organic Frameworks, J. Am. Chem. Soc., 2007, 129(43), 12914–12915, DOI:10.1021/ja0751781.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Designed Synthesis of 3D Covalent Frameworks, Science, 2007, 316(5822), 268–272, DOI:10.1126/science.1139055.
- N. Huang, P. Wang and D. Jiang, Covalent Organic Frameworks: A Materials Platform for Structural and Functional Designs, Nat. Rev. Mater., 2016, 1, 16068, DOI:10.1038/natrevmats.2016.68.
- X. Li, S. Cai, B. Sun, C. Yang, J. Zhang and Y. Liu, Chemically Robust Covalent Organic Frameworks: Progress and Perspective, Matter, 2020, 3(5), 1507–1540, DOI:10.1016/j.matt.2020.09.007.
- L. Cusin, H. Peng, A. Ciesielski and P. Samorì, Chemical Conversion and Locking of the Imine Linkage: Enhancing the Functionality of Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2021, 60(26), 14236–14250, DOI:10.1002/anie.202016667.
- Y. Yusran, X. Guan, H. Li, Q. Fang and S. Qiu, Postsynthetic Functionalization of Covalent Organic Frameworks, Natl. Sci. Rev., 2020, 7(1), 170–190, DOI:10.1093/nsr/nwz122.
- C. S. Diercks and O. M. Yaghi, The Atom, the Molecule, and the Covalent Organic Framework, Science, 2017, 355(6328), eaal1585, DOI:10.1126/science.aal1585.
- W. Ji, L. Xiao, Y. Ling, C. Ching, M. Matsumoto, R. P. Bisbey, D. E. Helbling and W. R. Dichtel, Removal of GenX and Perfluorinated Alkyl Substances from Water by Amine-Functionalized Covalent Organic Frameworks, J. Am. Chem. Soc., 2018, 140(40), 12677–12681, DOI:10.1021/jacs.8b06958.
- Y. Peng, Z. Hu, Y. Gao, D. Yuan, Z. Kang, Y. Qian, N. Yan and D. Zhao, Synthesis of a Sulfonated Two-Dimensional Covalent Organic Framework as an Efficient Solid Acid Catalyst for Biobased Chemical Conversion, ChemSusChem, 2015, 8(19), 3208–3212, DOI:10.1002/cssc.201500755.
- H. Fan, M. Peng, I. Strauss, A. Mundstock, H. Meng and J. Caro, High-Flux Vertically Aligned 2D Covalent Organic Framework Membrane with Enhanced Hydrogen Separation, J. Am. Chem. Soc., 2020, 142(15), 6872–6877, DOI:10.1021/jacs.0c00927.
- Y. J. Hou, J. Deng, K. He, C. Chen and Y. Yang, Covalent Organic Frameworks-Based Solid-Phase Microextraction Probe for Rapid and Ultrasensitive Analysis of Trace Per- A Nd Polyfluoroalkyl Substances Using Mass Spectrometry, Anal. Chem., 2020, 92(15), 10213–10217, DOI:10.1021/acs.analchem.0c01829.
- A. G. Slater and A. I. Cooper, Function-Led Design of New Porous Materials, Science, 2015, 348(6238), aaa8075, DOI:10.1126/science.aaa8075.
- H. Wang, Y. Yang, X. Yuan, W. L. Teo, Y. Wu, L. Tang and Y. Zhao, Structure–Performance Correlation Guided Applications of Covalent Organic Frameworks, Mater. Today, 2022, 53, 106–133, DOI:10.1016/j.mattod.2022.02.001.
- Y. Tang, W. Li, Y. Muhammad, S. Jiang, M. Huang, H. Zhang, Z. Zhao and Z. Zhao, Fabrication of Hollow Covalent-Organic Framework Microspheres via Emulsion-Interfacial Strategy to Enhance Laccase Immobilization for Tetracycline Degradation, Chem. Eng. J., 2021, 421, 129743, DOI:10.1016/j.cej.2021.129743.
- S. Karak, K. Dey and R. Banerjee, Maneuvering Applications of Covalent Organic Frameworks via Framework-Morphology Modulation, Adv. Mater., 2022, 34(49), 2202751, DOI:10.1002/adma.202202751.
- D. P. Serrano, J. M. Escola and P. Pizarro, Synthesis Strategies in the Search for Hierarchical Zeolites, Chem. Soc. Rev., 2013, 42(9), 4004–4035, 10.1039/C2CS35330J.
- A. Primo and H. Garcia, Zeolites as Catalysts in Oil Refining, Chem. Soc. Rev., 2014, 43, 7548–7561, 10.1039/c3cs60394f.
- A. Corma, Inorganic Solid Acids and Their Use in Acid-Catalyzed Hydrocarbon Reactions, Chem. Rev., 1995, 95(3), 559–614, DOI:10.1021/cr00035a006.
- L. H. Chen, M. H. Sun, Z. Wang, W. Yang, Z. Xie and B. L. Su, Hierarchically Structured Zeolites: From Design to Application, Chem. Rev., 2020, 120(20), 11194–11294, DOI:10.1021/acs.chemrev.0c00016.
- W. Fan, M. A. Snyder, S. Kumar, P.-S. Lee, W. C. Yoo, A. V. McCormick, R. L. Penn, A. Stein and M. Tsapatsis, Hierarchical Nanofabrication of Microporous Crystals with Ordered Mesoporosity, Nat. Mater., 2008, 7(12), 984–991, DOI:10.1038/nmat2302.
- H. Chen, J. Wydra, X. Zhang, P.-S. Lee, Z. Wang, W. Fan and M. Tsapatsis, Hydrothermal Synthesis of Zeolites with Three-Dimensionally Ordered Mesoporous-Imprinted Structure, J. Am. Chem. Soc., 2011, 133(32), 12390–12393, DOI:10.1021/ja2046815.
- M. Choi, K. Na, J. Kim, Y. Sakamoto, O. Terasaki and R. Ryoo, Stable Single-Unit-Cell Nanosheets of Zeolite MFI as Active and Long-Lived Catalysts, Nature, 2009, 461(7261), 246–249, DOI:10.1038/nature08288.
- D. Xu, Y. Ma, Z. Jing, L. Han, B. Singh, J. Feng, X. Shen, F. Cao, P. Oleynikov, H. Sun, O. Terasaki and S. Che, π-π Interaction of Aromatic Groups in Amphiphilic Molecules Directing for Single-Crystalline Mesostructured Zeolite Nanosheets, Nat. Commun., 2014, 5, 4262, DOI:10.1038/ncomms5262.
- K. Na, C. Jo, J. Kim, K. Cho, J. Jung, Y. Seo, R. J. Messinger, B. F. Chmelka and R. Ryoo, Directing Zeolite Structures into Hierarchically Nanoporous Architectures, Science, 2011, 333, 328–332, DOI:10.1126/science.1205785.
- D. Xu, G. R. Swindlehurst, H. Wu, D. H. Olson, X. Zhang and M. Tsapatsis, On the Synthesis and Adsorption Properties of Single-Unit-Cell Hierarchical Zeolites Made by Rotational Intergrowths, Adv. Funct. Mater., 2014, 24(2), 201–208, DOI:10.1002/adfm.201301975.
- X. Zhang, D. Liu, D. Xu, S. Asahina, K. A. Cychosz, K. V. Agrawal, Y. Al Wahedi, A. Bhan, S. Al Hashimi, O. Terasaki, M. Thommes and M. Tsapatsis, Synthesis of Self-Pillared Zeolite Nanosheets by Repetitive Branching, Science, 2012, 336(6089), 1684–1687. DOI:10.1126/science.1221111.
- S. T. Emmerling, R. Schuldt, S. Bette, L. Yao, R. E. Dinnebier, J. Kästner and B. V. Lotsch, Interlayer Interactions as Design Tool for Large-Pore COFs, J. Am. Chem. Soc., 2021, 143(38), 15711–15722, DOI:10.1021/jacs.1c06518.
- Z. Qu, C. Lai, G. Zhao, A. Knebel, H. Fan and H. Meng, Pore Engineering in Covalent Organic Framework Membrane for Gas Separation, Adv. Membr., 2022, 2, 100037, DOI:10.1016/j.advmem.2022.100037.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Covalent Organic Frameworks: Design, Synthesis, and Functions, Chem. Rev., 2020, 120(16), 8814–8933, DOI:10.1021/acs.chemrev.9b00550.
- T. Sick, J. M. Rotter, S. Reuter, S. Kandambeth, N. N. Bach, M. Döblinger, J. Merz, T. Clark, T. B. Marder, T. Bein and D. D. Medina, Switching on and off Interlayer Correlations and Porosity in 2D Covalent Organic Frameworks, J. Am. Chem. Soc., 2019, 141(32), 12570–12581, DOI:10.1021/jacs.9b02800.
- A. M. Pütz, M. W. Terban, S. Bette, F. Haase, R. E. Dinnebier and B. V. Lotsch, Total Scattering Reveals the Hidden Stacking Disorder in a 2D Covalent Organic Framework, Chem. Sci., 2020, 11(47), 12647–12654, 10.1039/d0sc03048a.
- W. Wang, Y. Jia, S. Zhou and S. Deng, Removal of Typical PFAS from Water by Covalent Organic Frameworks with Different Pore Sizes, J. Hazard. Mater., 2023, 460, 132522, DOI:10.1016/j.jhazmat.2023.132522.
- N. Zhang, A. Ishag, Y. Li, H. Wang, H. Guo, P. Mei, Q. Meng and Y. Sun, Recent Investigations and Progress in Environmental Remediation by Using Covalent Organic Framework-Based Adsorption Method: A Review, J. Cleaner Prod., 2020, 277, 123360, DOI:10.1016/j.jclepro.2020.123360.
- X. Liu, H. Pang, X. Liu, Q. Li, N. Zhang, L. Mao, M. Qiu, B. Hu, H. Yang and X. Wang, Orderly Porous Covalent Organic Frameworks-Based Materials: Superior Adsorbents for Pollutants Removal from Aqueous Solutions, Innovation, 2021, 2(1), 100076, DOI:10.1016/j.xinn.2021.100076.
- V. D. da Silva, K. Zalewska, Z. Petrovski, C. D. Buarque, L. C. Branco and P. M. Esteves, Covalent Organic Frameworks as Promising Materials for the Removal of Metal and Organic Pollutants from Water, Mater. Today Sustainability, 2023, 270, 129523, DOI:10.1016/j.mtsust.2022.100279.
- A. R. Bagheri, N. Aramesh, F. Sher and M. Bilal, Covalent Organic Frameworks as Robust Materials for Mitigation of Environmental Pollutants, Chemosphere, 2021, 270, 129523, DOI:10.1016/j.chemosphere.2020.129523.
- L. Yang, L. Shao, Z. Wu, P. Zhan and L. Zhang, Design and Synthesis of Porous Organic Polymers: Promising Catalysts for Lignocellulose Conversion to 5-Hydroxymethylfurfural and Derivates, Polymers, 2023, 15(12), 2630, DOI:10.3390/polym15122630.
- C. H. Feriante, S. Jhulki, A. M. Evans, R. R. Dasari, K. Slicker, W. R. Dichtel and S. R. Marder, Rapid Synthesis of High Surface Area Imine-Linked 2D Covalent Organic Frameworks by Avoiding Pore Collapse During Isolation, Adv. Mater., 2020, 32(2), 1905776, DOI:10.1002/adma.201905776.
- M. Xue, J. Yang, F. Kang, X. Wang and Q. Zhang, Recent Progress in Single-Crystal Structures of Organic Polymers, J. Mater. Chem. C, 2022, 10, 17027–17047, 10.1039/d2tc02170f.
- X. Wang, Z. Mu, P. Shao and X. Feng, Hierarchically Porous Covalent Organic Frameworks: Synthesis Methods and Applications, Chem. – Eur. J., 2024, 30(14), e202303601, DOI:10.1002/chem.202303601.
- B. Garai, D. Shetty, T. Skorjanc, F. Gándara, N. Naleem, S. Varghese, S. K. Sharma, M. Baias, R. Jagannathan, M. A. Olson, S. Kirmizialtin and A. Trabolsi, Taming the Topology of Calix[4]Arene-Based 2D-Covalent Organic Frameworks: Interpenetrated vs Noninterpenetrated Frameworks and Their Selective Removal of Cationic Dyes, J. Am. Chem. Soc., 2021, 143(9), 3407–3415, DOI:10.1021/jacs.0c12125.
- S. Karak, K. Dey, A. Torris, A. Halder, S. Bera, F. Kanheerampockil and R. Banerjee, Inducing Disorder in Order: Hierarchically Porous Covalent Organic Framework Nanostructures for Rapid Removal of Persistent Organic Pollutants, J. Am. Chem. Soc., 2019, 141(18), 7572–7581, DOI:10.1021/jacs.9b02706.
- W. Li, H. X. Jiang, M. F. Cui, R. Wang, A. N. Tang and D. M. Kong, SiO2 Templates-Derived Hierarchical Porous COFs Sample Pretreatment Tool for Non-Targeted Analysis of Chemicals in Foods, J. Hazard. Mater., 2022, 432, 128705, DOI:10.1016/j.jhazmat.2022.128705.
- X. Zhao, P. Pachfule, S. Li, T. Langenhahn, M. Ye, C. Schlesiger, S. Praetz, J. Schmidt and A. Thomas, Macro/Microporous Covalent Organic Frameworks for Efficient Electrocatalysis, J. Am. Chem. Soc., 2019, 141(16), 6623–6630, DOI:10.1021/jacs.9b01226.
- Z. Mu, Y. Zhu, Y. Zhang, A. Dong, C. Xing, Z. Niu, B. Wang and X. Feng, Hierarchical Microtubular Covalent Organic Frameworks Achieved by COF-to-COF Transformation, Angew. Chem., Int. Ed., 2023, 62(17), e202300373, DOI:10.1002/anie.202300373.
- S. Wang, Y. Yang, H. Zhang, Z. Zhang, C. Zhang, X. Huang, D. Kozawa, P. Liu, B. G. Li and W. J. Wang, Toward Covalent Organic Framework Metastructures, J. Am. Chem. Soc., 2021, 143(13), 5003–5010, DOI:10.1021/jacs.0c13090.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, Rapid Low Temperature Formation of Imine-Linked Covalent Organic Frameworks Catalyzed by Metal Triflates, J. Am. Chem. Soc., 2017, 139(14), 4999–5002, DOI:10.1021/jacs.7b01240.
- C. Feriante, A. M. Evans, S. Jhulki, I. Castano, M. J. Strauss, S. Barlow, W. R. Dichtel and S. R. Marder, New Mechanistic Insights into the Formation of Imine-Linked Two-Dimensional Covalent Organic Frameworks, J. Am. Chem. Soc., 2020, 142(43), 18637–18644, DOI:10.1021/jacs.0c08390.
- B. J. Smith, A. C. Overholts, N. Hwang and W. R. Dichtel, Insight into the Crystallization of Amorphous Imine-Linked Polymer Networks to 2D Covalent Organic Frameworks, Chem. Commun., 2016, 52(18), 3690–3693, 10.1039/c5cc10221a.
- S. Wang, V. A. Reddy, M. C. Y. Ang, J. Cui, D. T. Khong, Y. Han, S. I. Loh, R. Cheerlavancha, G. P. Singh, S. Rajani and M. S. Strano, Single-Crystal 2D Covalent Organic Frameworks for Plant Biotechnology, J. Am. Chem. Soc., 2023, 145(22), 12155–12163, DOI:10.1021/jacs.3c01783.
- D. Ongari, A. V. Yakutovich, L. Talirz and B. Smit, Building a Consistent and Reproducible Database for Adsorption Evaluation in Covalent-Organic Frameworks, ACS Cent. Sci., 2019, 5(10), 1663–1675, DOI:10.1021/acscentsci.9b00619.
- G. Kresse and J. Furthmüller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, 1996, vol. 6 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, 1996 Search PubMed.
- S. Grimme, Accurate Description of van Der Waals Complexes by Density Functional Theory Including Empirical Corrections, J. Comput. Chem., 2004, 25(12), 1463–1473, DOI:10.1002/jcc.20078.
- S. Grimme, Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction, J. Comput. Chem., 2006, 27(15), 1787–1799, DOI:10.1002/jcc.20495.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104, DOI:10.1063/1.3382344.
- W. Chen, P. Chen, D. Chen, Y. Liu, G. Zhang, L. Wang and L. Chen, Triangular Topological 2D Covalent Organic Frameworks Constructed via Symmetric or Asymmetric “Two-in-One” Type Monomers, Adv. Sci., 2022, 9(19), 2105517, DOI:10.1002/advs.202105517.
- A. Natraj, W. Ji, J. Xin, I. Castano, D. W. Burke, A. M. Evans, M. J. Strauss, M. Ateia, L. S. Hamachi, N. C. Gianneschi, Z. A. Alothman, J. Sun, K. Yusuf and W. R. Dichtel, Single-Crystalline Imine-Linked Two-Dimensional Covalent Organic Frameworks Separate Benzene and Cyclohexane Efficiently, J. Am. Chem. Soc., 2022, 144(43), 6366–6378, DOI:10.1021/jacs.2c07166.
- C. Xiong, H. Ning, G. Jia, X. Hong, X. Fei and J. Donglin, Towards Covalent Organic Frameworks with Predesignable and Aligned Open Docking Sites, Chem. Commun., 2014, 50(46), 6161–6163, 10.1039/c4cc01825g.
- F. Wen, K. Xu, Y. Feng and N. Huang, Two-Dimensional Covalent Organic Frameworks with Pentagonal Pores, J. Am. Chem. Soc., 2024, 146(29), 19680–19685, DOI:10.1021/jacs.4c06438.
- T. Banerjee, F. Haase, S. Trenker, B. P. Biswal, G. Savasci, V. Duppel, I. Moudrakovski, C. Ochsenfeld and B. V. Lotsch, Sub-Stoichiometric 2D Covalent Organic Frameworks from Tri- and Tetratopic Linkers, Nat. Commun., 2019, 10(1), 2689, DOI:10.1038/s41467-019-10574-6.
- S. Y. Ding, X. H. Cui, J. Feng, G. Lu and W. Wang, Facile Synthesis of -C=N- Linked Covalent Organic Frameworks under Ambient Conditions, Chem. Commun., 2017, 53(87), 11956–11959, 10.1039/c7cc05779b.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, Construction of Covalent Organic Framework for Catalysis: Pd/COF-LZU1 in Suzuki-Miyaura Coupling Reaction, J. Am. Chem. Soc., 2011, 133(49), 19816–19822, DOI:10.1021/ja206846p.
- Y. He, N. An, C. Meng, K. Xie, X. Wang, X. Dong, D. Sun, Y. Yang and Z. Hu, High-Density Active Site COFs with a Flower-like Morphology for Energy Storage Applications, J. Mater. Chem. A, 2022, 10, 11030–11038, 10.1039/D2TA01065H.
- I. Romero-Muñiz, A. Mavrandonakis, P. Albacete, A. Vega, V. Briois, F. Zamora and A. E. Platero-Prats, Unveiling the Local Structure of Palladium Loaded into Imine-Linked Layered Covalent Organic Frameworks for Cross-Coupling Catalysis, Angew. Chem., Int. Ed., 2020, 59(31), 13013–13020, DOI:10.1002/anie.202004197.
- L. Wang, H. Xu, Y. Qiu, X. Liu, W. Huang, N. Yan and Z. Qu, Utilization of Ag Nanoparticles Anchored in Covalent Organic Frameworks for Mercury Removal from Acidic Waste Water, J. Hazard. Mater., 2020, 389, 121824, DOI:10.1016/j.jhazmat.2019.121824.
- L. Yang, Q. Guo, H. Kang, R. Chen, Y. Liu and D. Wei, Self-Controlled Growth of Covalent Organic Frameworks by Repolymerization, Chem. Mater., 2020, 32(13), 5634–5640, DOI:10.1021/acs.chemmater.0c01140.
- S. Daliran, M. Blanco, A. Dhakshinamoorthy, A. R. Oveisi, J. Alemán and H. García, Defects and Disorder in Covalent Organic Frameworks for Advanced Applications, Adv. Funct. Mater., 2023, 34(18), 2312912, DOI:10.1002/adfm.202312912.
- J. Dong, X. Han, Y. Liu, H. Li and Y. Cui, Metal–Covalent Organic Frameworks(MCOFs): A Bridge Between Metal–Organic Frameworks and Covalent Organic Frameworks, Angew. Chem., Int. Ed., 2020, 59(33), 13722–13733, DOI:10.1002/anie.202004796.
- A. Kuc, M. A. Springer, K. Batra, R. Juarez-Mosqueda, C. Wöll and T. Heine, Proximity Effect in Crystalline Framework Materials: Stacking-Induced Functionality in MOFs and COFs, Adv. Funct. Mater., 2020, 30(41), 1908004, DOI:10.1002/adfm.201908004.
- A. J. Lopes Jesus and J. S. Redinha, Molecular Insight into the Amine-Water Interaction: A Combined Vibrational, Energetic and NBO/NEDA Study, Comput. Theor. Chem., 2013, 1023, 74–82, DOI:10.1016/j.comptc.2013.09.006.
- J. Zhong, Y. Zhao, L. Li, H. Li, J. S. Francisco and X. C. Zeng, Interaction of the NH2 Radical with the Surface of a Water Droplet, J. Am. Chem. Soc., 2015, 137(37), 12070–12078, DOI:10.1021/jacs.5b07354.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report), Pure Appl. Chem., 2015, 87(9–10), 1051–1069, DOI:10.1515/pac-2014-1117.
- J. L. Fenton, D. W. Burke, D. Qian, M. O. D. L. Cruz and W. R. Dichtel, Polycrystalline Covalent Organic Framework Films Act as Adsorbents, Not Membranes, J. Am. Chem. Soc., 2021, 143(3), 1466–1473, DOI:10.1021/jacs.0c11159.
Footnote |
| † Electronic supplementary information (ESI) available: Morphology of COFs synthesized under different amounts of acetic acid and a low concentration of 0.002 eq. Sc(OTf)3; reproducibility of COFs synthesized under different catalytic conditions and different reaction times; FTIR comparisons of as-made COFs, monomers, solvents, and Sc(OTf)3 catalyst; Sc(OTf)3 dispersion in COF-LZU1 agglomerated particles; quantified elemental analysis of as-made COFs; crystallographic indexing of as-made COFs, reaction time sensitivity, and estimation of edge amines; DFT simulation of alternative metal triflate binding to COF framework; morphology, composition, and crystallinity of as-made COF-LZU1 under 0.02 eq. Sc(OTf)3 with added DI water; retention of morphology with activation; pore size changes with introduction of defects; hierarchical COF literature comparison; RdB and MB adsorption equilibrium time. See DOI: https://doi.org/10.1039/d4nr04717f |
| This journal is © The Royal Society of Chemistry 2025 |