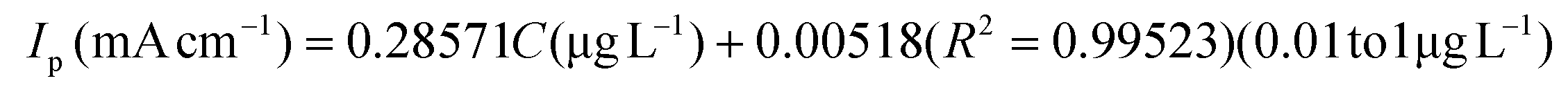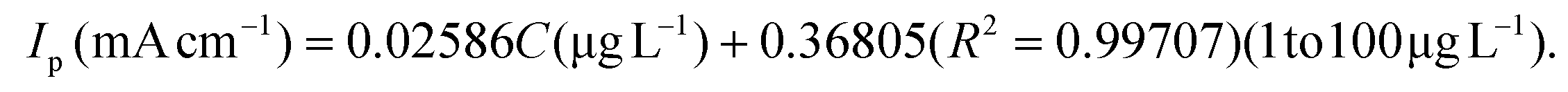DOI:
10.1039/D4NR05021E
(Paper)
Nanoscale, 2025,
17, 7229-7243
A modified MXene composite sensor with sulphur impurities for electrochemical detection of lead in the aqueous system†
Received
29th November 2024
, Accepted 14th February 2025
First published on 17th February 2025
Abstract
The detection of heavy metal ions, particularly Pb2+, is critical due to their severe environmental and health impacts. This study introduces a novel sensor based on an alkali-modified Ti-MXene decorated with bismuth and sulphur (S–Bi@TiMX), designed for the ultra-sensitive electrochemical detection of Pb2+. Synthesized via hydrothermal deposition, the composite material exhibits a distinctive marigold-like morphology, enhanced active sites, and robust synergistic interactions between its constituents. These features mitigate issues like MXene sheet restacking and promote superior electron transfer kinetics. The composite was drop-cast onto a low-cost, disposable plastic chip electrode (PCE), achieving remarkable sensitivity and an impressive detection limit of 0.0002 μg L−1 Pb2+, well below the WHO safety threshold. The sensor demonstrated good repeatability (RSD < 2.75%), excellent reproducibility (RSD < 2.8%), minimal interference from competing ions, and high recovery rates (99.2%–99.6%) in real water samples. Optimized parameters like pH, preconcentration potential, and time ensured precise, robust performance across a broad linear range (0.01–100 μg L−1). This work highlights the potential of the S–Bi@TiMX/PCE sensor for environmental monitoring and offers a scalable, cost-effective solution for detecting ultra-trace Pb2+ in diverse aqueous systems.
1. Introduction
Heavy metal ions and dyes are the main sources of water pollution. Most heavy metal ions are persistent pollutants and carcinogens that seriously harm both ecosystems and human health. The presence of heavy metals in water bodies is increasing day by day with industrial growth. Lead is a heavy metal in water, particularly dangerous because it tends to accumulate in living organisms and strongly binds to biological molecules, disrupting essential processes within cells.1 When lead enters the human body, it can mimic important metals like calcium, iron, and zinc, which are crucial for various biochemical reactions. By replacing these essential metals, lead interferes with vital functions, which can damage organs and systems, especially affecting the brain, kidneys, and blood.2–4 Lead toxicity stems from its chemical behaviour in the body, where it forms stable compounds with proteins and enzymes, distorting their shape and interfering with processes like nerve signalling and cell repair.5 Unlike some other pollutants, lead cannot be broken down or metabolized by organisms, which means that it builds up over time in both the environment and biological tissues. Therefore, even low levels of lead pose long-term health risks, making it essential to detect it and control its exposure early.6 Sources of lead exposure include mining, smelting, leaded gasoline, and lead-based paints.7 Lead can enter the body through hand-to-mouth contact or by consuming contaminated food or water. Detecting trace amounts of lead in water, soil, and other environmental sources is crucial to preventing harmful accumulation and protecting public health, highlighting the need for sensitive, reliable sensors and monitoring tools. The World Health Organization (WHO) has set the safe limit for lead in drinking water at 10 ppb (48 nM).8 Various methods are employed to detect trace levels of lead in contaminated water, with techniques like inductively coupled plasma–mass spectrometry (ICP-MS) and atomic absorption spectroscopy (AAS) being particularly effective. These methods offer high accuracy and can detect even trace amounts of lead within complex biomolecular samples.9 However, their widespread application is limited by the complexity of their instrumentation and the time-intensive nature of their operation, which restricts their use for rapid ion detection in the field. To address these limitations, researchers are increasingly exploring electrochemical approaches to detect heavy metals in real samples, working toward the development of ion-selective electrodes (ISEs) tailored for specific toxic ions. While ISEs are commonly available for ions like sodium, calcium and can be further designed for lead, the standard commercial Pb2+ ISEs, such as the HI4112 lead/sulfate combination and HI4012 lead/sulfate half-cell electrodes, are limited in their sensitivity.10–12 These electrodes are generally not capable of reliably measuring lead concentrations in the low part-per-billion (ppb) range, making them more suitable for industrial applications, such as plant monitoring, rather than environmental testing at ultra-low levels.11 Additionally, when using ISEs to detect lead in water, potential interference from other ions such as Fe2+, Na+, Ca2+, Mg2+, and Cu2+ must be carefully managed. Anodic stripping voltammetry (ASV) is the most commonly used electrochemical technique for detecting heavy metal ions, offering a detection limit as low as 10−10 M, making it particularly suitable for analyzing trace levels of these elements.13 Numerous modifications and advancements have been reported in electrode materials to enable the detection of metal ions at extremely low concentrations. Traditional electrode substrates such as glassy carbon electrodes (GCE),14 indium tin oxide (ITO),15 aluminium,16 stainless steel,17 and gold18 have been engineered with various coatings to enhance their sensitivity. These modifications include coatings of metals like bismuth,19 antimony,20 and mercury,21 carbon-based materials such as reduced graphene oxide and carbon nanotubes (CNTs),22–24 boron-doped diamond,25 and conducting polymers (polyaniline, polypyrrole).17,24,26 However, each of these substrates presents challenges, including environmental toxicity (e.g., Hg), high cost, or complex fabrication processes that can hinder their application for ultra-trace detection.
In response to these limitations, this work presents the design of an innovative electrode using a plastic chip substrate modified with MXenes and bismuth. Comprising poly(methyl methacrylate) (PMMA) and conductive graphite. The plastic chip electrode (PCE) offers enhanced electron transfer kinetics and reduced capacitive behaviour over conventional materials.27 Its scalability and low cost make it ideal for miniaturized, disposable sensing devices, enabling ultra-sensitive detection down to parts per trillion (ppt) while providing a practical alternative to traditional electrodes.28 MXenes, discovered in 2011, come from a family of 2D metal carbides and nitrides and are being used in a wide realm of electrochemical applications due to their impressive features like high electrical conductivity, hydrophilicity, and tuneable surface properties.29 Synthesized by etching the A-layers from layered MAX phases (Mn+1AXn, where M is a transition metal, A is a group IIIA/IVA element, and X is C/N), MXene surfaces are naturally terminated with groups like –OH, –O, and –F.30 These functional groups enhance their reactivity and tunability, allowing for tailored interactions with ions and molecules. MXenes typically exhibit a laminar, sheet-like morphology, which can lead to restacking issues, thereby limiting ion diffusion pathways and reducing performance. Unmodified Ti3C2Tx MXenes have demonstrated remarkable sensitivity and selectivity for Pb2+ detection.31 However, the incorporation of Pb2+-sensitive materials offers an opportunity to further enhance their performance. To achieve this, researchers have explored various modifications, including functionalization and composite formation. Recent advancements include functionalization with amino and thiol groups, such as NH2-functionalized MXenes (NH2-Ti3C2Tx/SPCE) and NH2/SH-functionalized MXenes, achieving detection limits of 0.31 μg L−1 (ref. 32) and 2.30 μg L−1,33 respectively, for Pb2+. Beyond functionalization, MXene composites with carbon-based materials and metal oxides have been extensively investigated for improved Pb2+ detection due to their enhanced selectivity and lower limits of detection (LOD). For instance, a melamine-doped rGO/MXene aerogel/SPCE demonstrated sensitive detection of Pb2+ with a detection limit of 0.29 μg L−1.34 Similarly, a Ti3C2/Fe3O4/g-C3N4/GCE nanocomposite exhibited an impressive LOD of 0.02 μg L−1 for Pb2+.35 Other notable examples include the MXene aerogel-CuO/CC electrochemical sensor with an LOD of 0.2 μg L−1 for Pb2+![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 and the UiO-66-NH2-doped MXene@rGO composite aerogel, which achieved an LOD of 0.40 μg L−1 for Pb2+.37 Bismuth film electrodes are frequently noted for their high activity and as practical, mercury-free alternatives in electrochemical sensing, though they often require a complex, sensitive electrodeposition process with various drawbacks. So, instead of Bi films, Bi nanostructures are expected to be more effective as they provide added active sites. And the incorporation of these nanostructures onto the MXene laminar structure eventually solves the problem of restacking of the MXene sheets and agglomeration of Bi nanostructures.38
36 and the UiO-66-NH2-doped MXene@rGO composite aerogel, which achieved an LOD of 0.40 μg L−1 for Pb2+.37 Bismuth film electrodes are frequently noted for their high activity and as practical, mercury-free alternatives in electrochemical sensing, though they often require a complex, sensitive electrodeposition process with various drawbacks. So, instead of Bi films, Bi nanostructures are expected to be more effective as they provide added active sites. And the incorporation of these nanostructures onto the MXene laminar structure eventually solves the problem of restacking of the MXene sheets and agglomeration of Bi nanostructures.38
In this work, we design a composite material using the hydrothermal method to integrate Bi nanostructures onto the Ti-MXene surface, followed by uneven dispersion of sulphur. The morphology of the MXene is changed from conventional sheet like to flower like which subsequently offers a greater number of active sites. Sulphur's thiol groups tend to oxidatively dimerize, forming disulphides that are expected to enhance the binding affinity with Pb2+ ions and increase the active surface area. The synthesised composite material is drop cast onto in-house prepared PCE electrodes which overall offers a novel combination of a biodegradable and scalable sensor for lead detection. This configuration merges cost-efficiency with heightened sensitivity, allowing metal lead detection down to ppt levels.
2. Methodology
2.1 Materials
For the synthesis of PCE, the graphite powder was obtained from Qualikems Pvt. Ltd, and Otto Pvt Ltd provided us with poly(methyl methacrylate) (PMMA). For the preparation of the composite material, bismuth nitrate pentahydrate (Bi (NO3)3·5H2O, 99%) and precursor MAX phase Ti3AlC2 were procured from Sigma Aldrich and from Nano Research Elements (Haryana, India), respectively. Thiourea was used to impart sulphur impurities and was purchased from Loba Chemicals. For the preparation of test solutions, lead chloride (PbCl2) and perchloric acid were acquired from Merck for use in the experimental procedures.
2.2 Fabrication of the plastic chip electrode (PCE)
The PCE was prepared by adapting a previously established method.27 The comprehensive procedure is provided in Section SI-1 of the ESI.†
2.3 Synthesis of the composite material
2.3.1 Synthesis of alkali-modified Ti-MXene.
Alkali-modified Ti-MXene (Ti3C2) was prepared following the previously reported method by our group.39 To summarize, 500 mg Ti3AlC2 and 500 g of lithium fluoride (LiF) were weighed and submerged in 50 mL of 9 M HCl at 60 °C for 48 hours. This method is an in situ HF generation method for MXene synthesis. After this etching, the solution was centrifuged at 7000 rpm for 10 minutes, and the black precipitate was separated following the removal of the supernatant. This black precipitate was then treated with 40 mL of 49% HF at 25 °C for 24 hours to eliminate any remaining aluminium moieties. The final black powder was thoroughly rinsed until the pH became neutral and dried overnight in a vacuum oven at 60 °C. It was further treated with alkali and 100 mg of the synthesized Ti3C2 was dispersed in 40 mL of 1 M KOH with 0.7 mL of 30% H2O2. This mixture was placed in a Teflon-lined autoclave and heated to 150 °C for 12 hours. Afterward, the product was extensively washed with deionized water, and the top suspension was separated by centrifugation and subsequently dried under vacuum at 60 °C. The obtained product was used as a test sample and termed TiMX.
2.3.2 Synthesis of the sulphur decorated Bi-MXene composite.
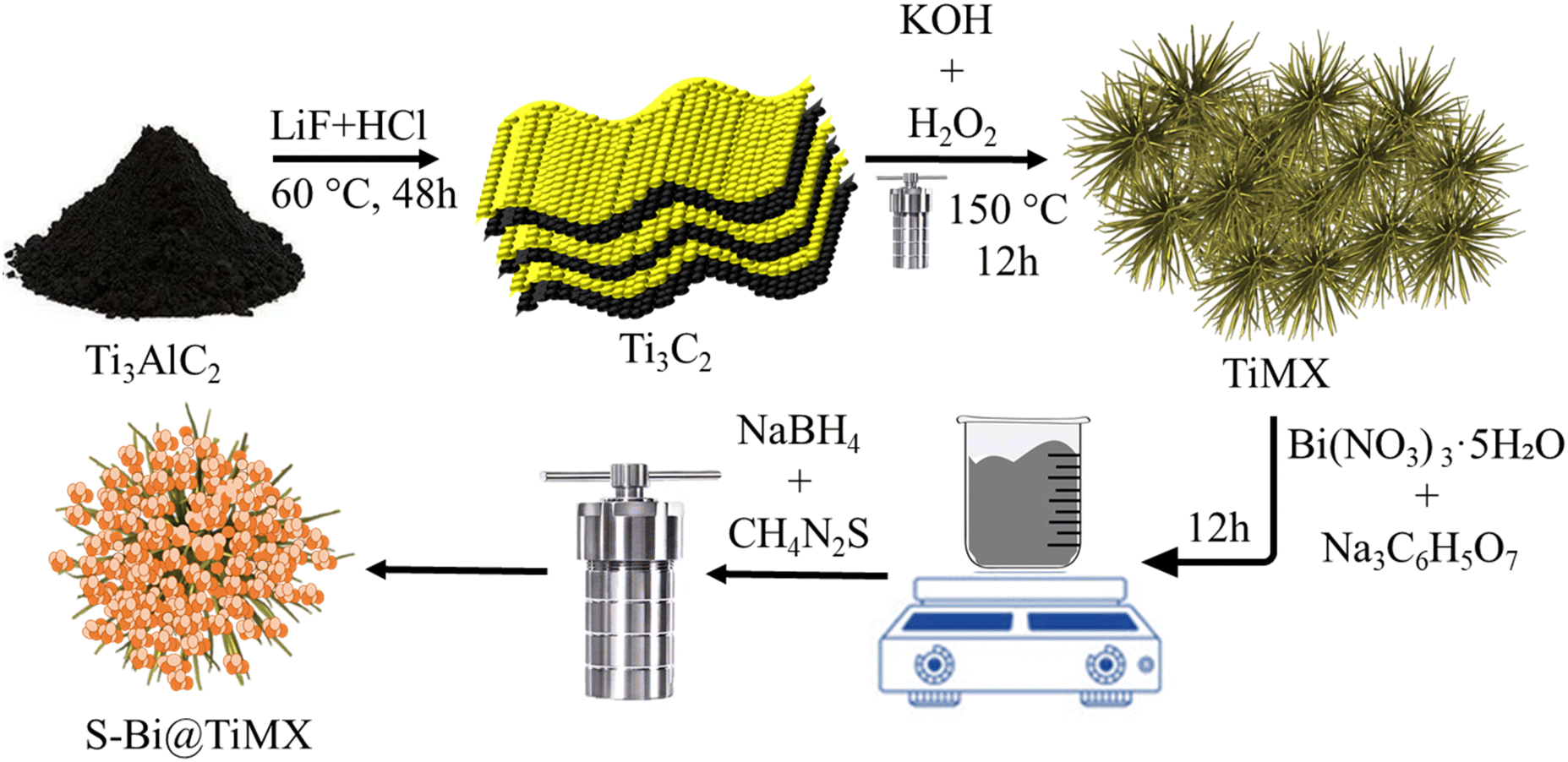
The final composite material was synthesized using a one-step hydrothermal method. First, 40 mg of TiMX was dissolved in 100 mL of ethanol and stirred for 45 minutes. Subsequently, 100 mg of sodium citrate and 430 mg of Bi (NO3)3·5H2O were added, and the solution was stirred continuously for 12 hours to achieve thorough homogenization. Following this, 1 mL of 0.1 M NaBH4 solution was introduced, and the mixture was stirred for an additional hour at room temperature. To incorporate sulphur moieties, 20 mg of thiourea was added to the solution. This mixture was then transferred to a custom-prepared Teflon-lined 50 mL autoclave and treated in a muffle furnace at 180 °C for 3 hours. The resulting product was centrifuged with water and subsequently dried, yielding the final composite material, designated as S–Bi@TiMX. To assess the impact of TiMX, a control sample was prepared following the same procedure without adding TiMX and was labelled as S–Bi. The synthesis scheme for preparation of TiMX and S-Bi@TiMX has been shown in (Fig. 1).
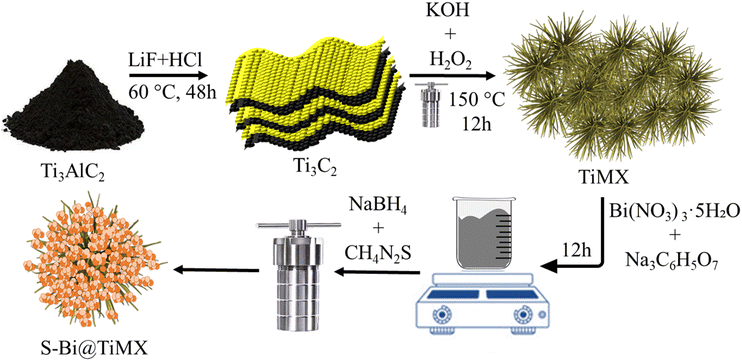 |
| | Fig. 1 The synthetic route followed for the preparation of S–Bi@TiMX. | |
2.4 Fabrication of the composite electrode
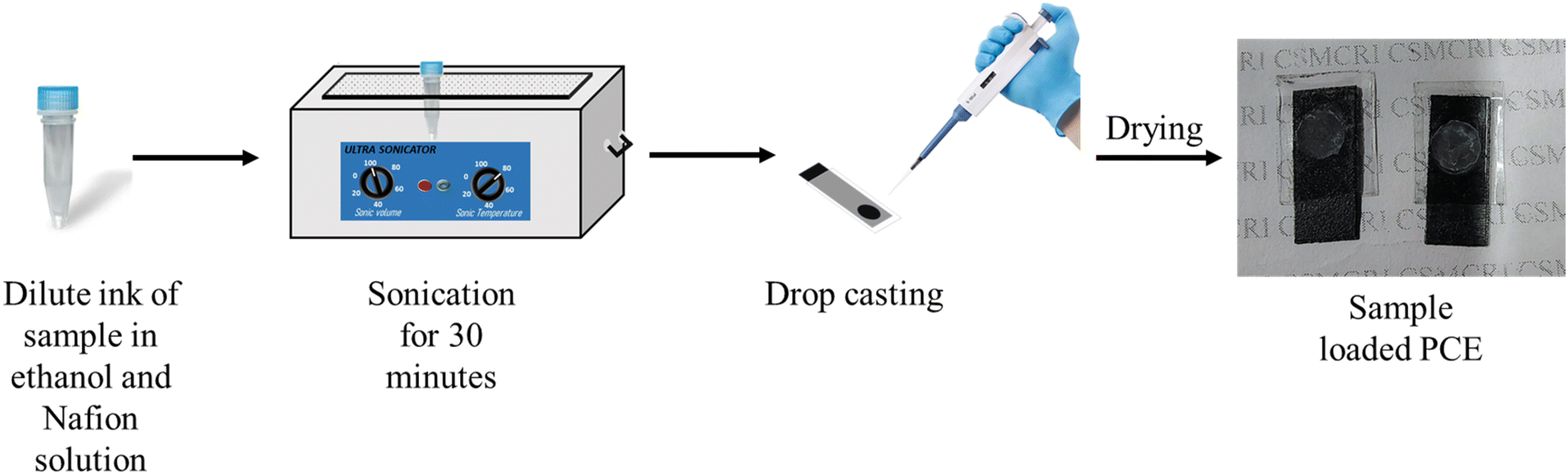
To prepare the composite electrode, an ink of the S–Bi@TiMX sample was first formulated as represented in (Fig. 2). Specifically, 3 mg of S–Bi@TiMX was dispersed in 500 μL of ethanol, to which 10 μL of Nafion solution was added as a binder. The ink was ultrasonicated and then drop-cast onto the exposed active area of the PCE. The electrode was allowed to dry for 1 hour before being used in all subsequent electrochemical experiments.
 |
| | Fig. 2 The procedure followed to prepare sample loaded PCEs. | |
3. Characterization
3.1 Compositional and morphological analysis
The structure of the composite and the presence of all the elements’ bonding nature were confirmed using powder X-ray diffraction (PXRD). Fourier transform infrared spectroscopy (FT-IR) was used to characterize surface functional groups using the PerkinElmer Spectrum GX 2000 FT-IR spectrometer. Using X-ray photoelectron spectroscopy (XPS), which was made possible by the Thermo Scientific Nexsa G2 Surface Analysis System (voltage: 220–240 V; frequency: 50/60 Hz) fitted with Mg Kα X-ray sources, the oxidation states of elements included in the sample were measured. Field Emission Scanning Electron Microscopy (FE-SEM) (JEOL JSM 7100F) was used to identify the morphology of all test materials at an acceleration voltage of 200 kV.
3.2 Electrochemical analysis and detection of lead ions
An Autolab PGSTAT M204 Potentiostat running Nova 2.1.6 software was used for all electrochemical assays. The analysis was conducted in a three electrode set-up in which Ag/AgCl and platinum wire mimiced as the reference and counter electrode, respectively whereas, modified PCEs were employed as working electrode. The interfacial properties and redox activity of the electrodes were investigated using cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS). CV was performed in a 5.0 mM ferricyanide solution with 0.1 M KCl throughout a potential range of −0.2 V to +0.7 V. Performance of different prepared electrodes was evaluated with 0.08 M HClO4 containing 10 mg L−1 Pb2+ at a scan rate of 0.05 V s−1. Interfacial properties of the electrodes were determined by EIS experiments in 0.08 M HClO4 electrolyte with 10 mg L−1 Pb2+ throughout the frequency range of 10 Hz to 1 MHz at Eocp. The square wave anodic stripping voltammetry (SW-ASV) method was applied for the detection of Pb2+. Pb2+ ions were preconcentrated on the electrode interface for 480 seconds at −1.8 V, followed by a stripping step with a potential step of 2 mV, pulse amplitude of 50 mV, and frequency of 20 Hz over a potential range of −1.2 V to −0.2 V.
4. Results and discussion
4.1 Structure and compositional profiling
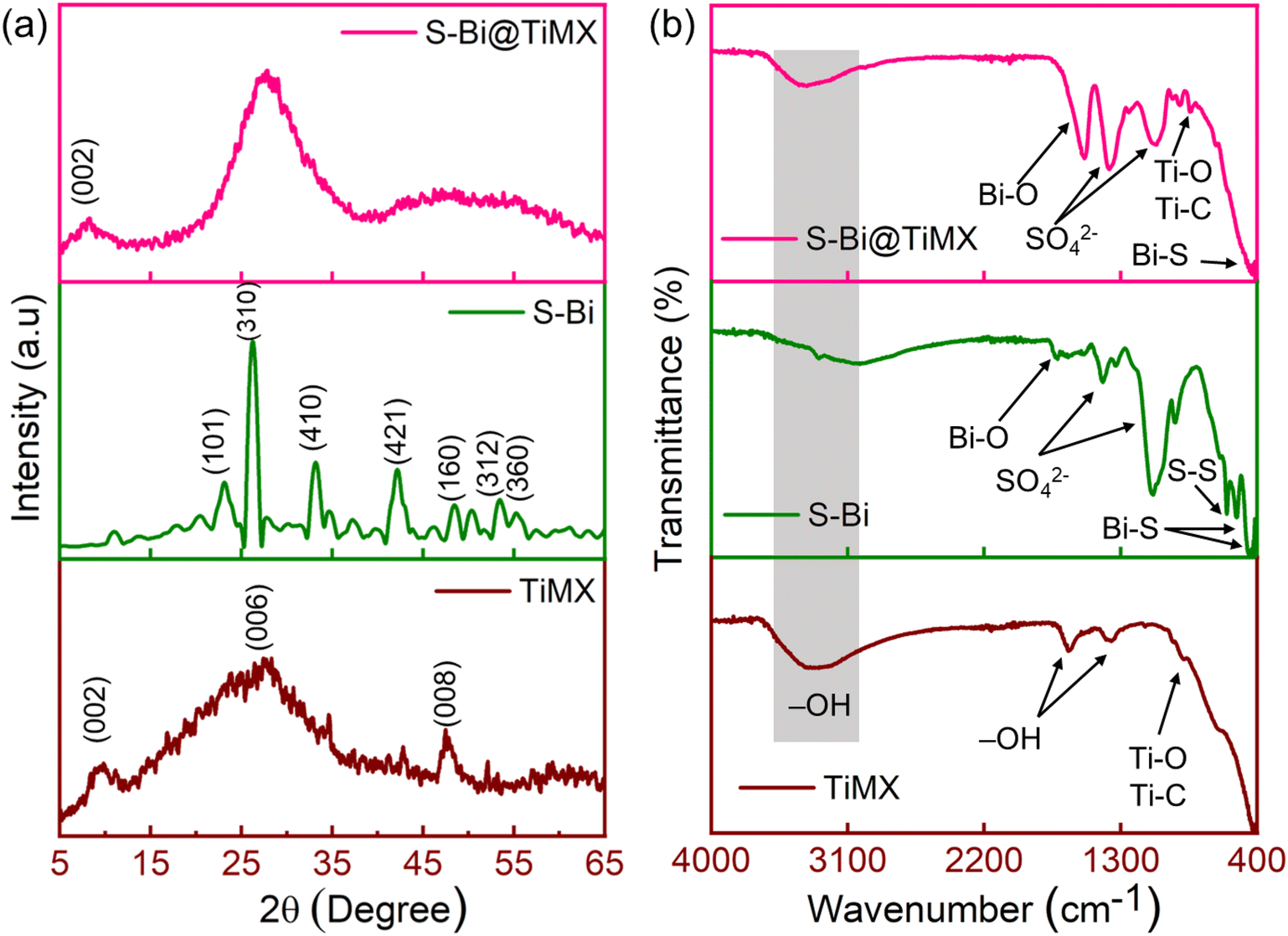
XRD analysis was done to examine the crystallographic structure and phase composition of synthesized samples, as depicted in Fig. 3(a). The diffraction peak observed at 2θ = 9.4° corresponds to the (002) plane of Ti-MXene (Ti3C2).40 The absence of any peaks attributable to aluminium suggests the successful removal of aluminium from the MAX phase during the etching process. Additional broad peaks at 2θ = 29.1° and 49.5° are indexed to the (006) and (008) planes of Ti3C2. TiMX sample exhibits a significant amorphous character, likely due to the morphological transformation of the MXene from sheet-like to a ‘marigold flake’ like structure upon treatment with KOH. This altered morphology enhances the active surface area and minimizes sheet agglomeration, thus favouring its functional attributes. For the S–Bi sample, the diffraction peaks are consistent with literature reports41 and can be indexed to the (101), (310), (410), (421), (160), (312), and (360) planes of orthorhombic Bi2S3 (JCPDS no. 17-0320), appearing at 2θ values of 23.7°, 25.2°, 33.1°, 42.6°, 49.01°, 53.04°, and 54.4°, respectively. S–Bi@TiMX composite reveals a predominantly amorphous structure, as evidenced by the presence of a broad hump in the 2θ range of 16°–38°, which exhibits higher intensity compared to the individual TiMX and S–Bi samples. A small broad peak at (002), inherited from the TiMX phase, is also observed. The incorporation of S–Bi moieties into the modified MXene structure induces a reduction in crystallinity and an increase in structural disorder. This enhanced disorder augments the overall exposure of active sites, thereby facilitating efficient Pb2+ ion adsorption and improving the electrochemical sensitivity of the composite. FT-IR spectroscopy was conducted on all samples to identify surface functional groups and to elucidate the bonding interactions formed during the synthesis process. Fig. 3(b) presents the FT-IR spectrum of TiMX, which exhibits a broad peak at 3321 cm−1, attributed to the surface –OH groups, reflecting the hydrophilic nature of MXene. Additional –OH associated peaks are observed at 1638 cm−1 and 1365 cm−1. A minor peak at 894 cm−1 corresponds to characteristic Ti–O and Ti–C bonds typical of MXene structures.42 Similarly, the S–Bi sample displays a broad peak indicating O–H stretching vibrations from surface-adsorbed water. Prominent peaks at 1087 cm−1 and 1389 cm−1 are assigned to symmetric vibrations of the SO42− group. Peaks at 454 cm−1, 540 cm−1, and 931 cm−1 are attributed to Bi–S resonances, while a weak peak at 602 cm−1 corresponds to S–S bond vibrations.43,44 In the composite sample, S–Bi@TiMX, charge localization effects are evident, as peaks observed at 1087 cm−1 and 1389 cm−1 in S–Bi are shifted to lower wavelengths. The O–H stretching band persists but appears less intense or slightly shifted, suggesting reduced water adsorption or alterations in surface chemistry due to S–Bi incorporation.45 Bi–S vibrations from the S–Bi sample are retained but exhibit minor shifts, indicating interactions between S–Bi and TiMX. Peaks corresponding to Ti–O and Ti–C vibrations (from TiMX) broaden or overlap, signifying strong interfacial interactions within the composite. These spectral observations confirm that the S–Bi@TiMX composite integrates the functional characteristics of both TiMX and S–Bi, demonstrating interactions that likely influence its electronic and chemical properties.
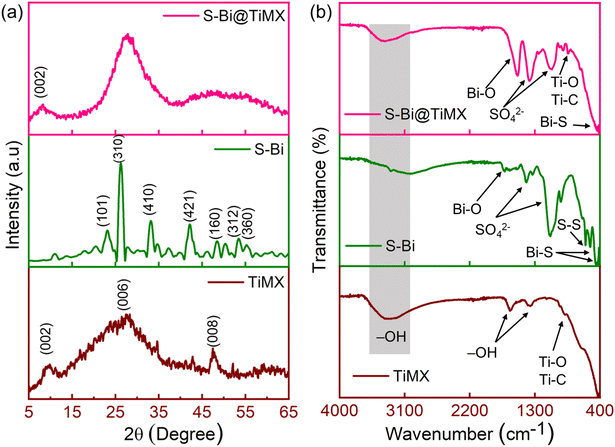 |
| | Fig. 3 (a) Results for XRD and (b) FT-IR of TiMX, S–Bi and S–Bi@TiMX. | |
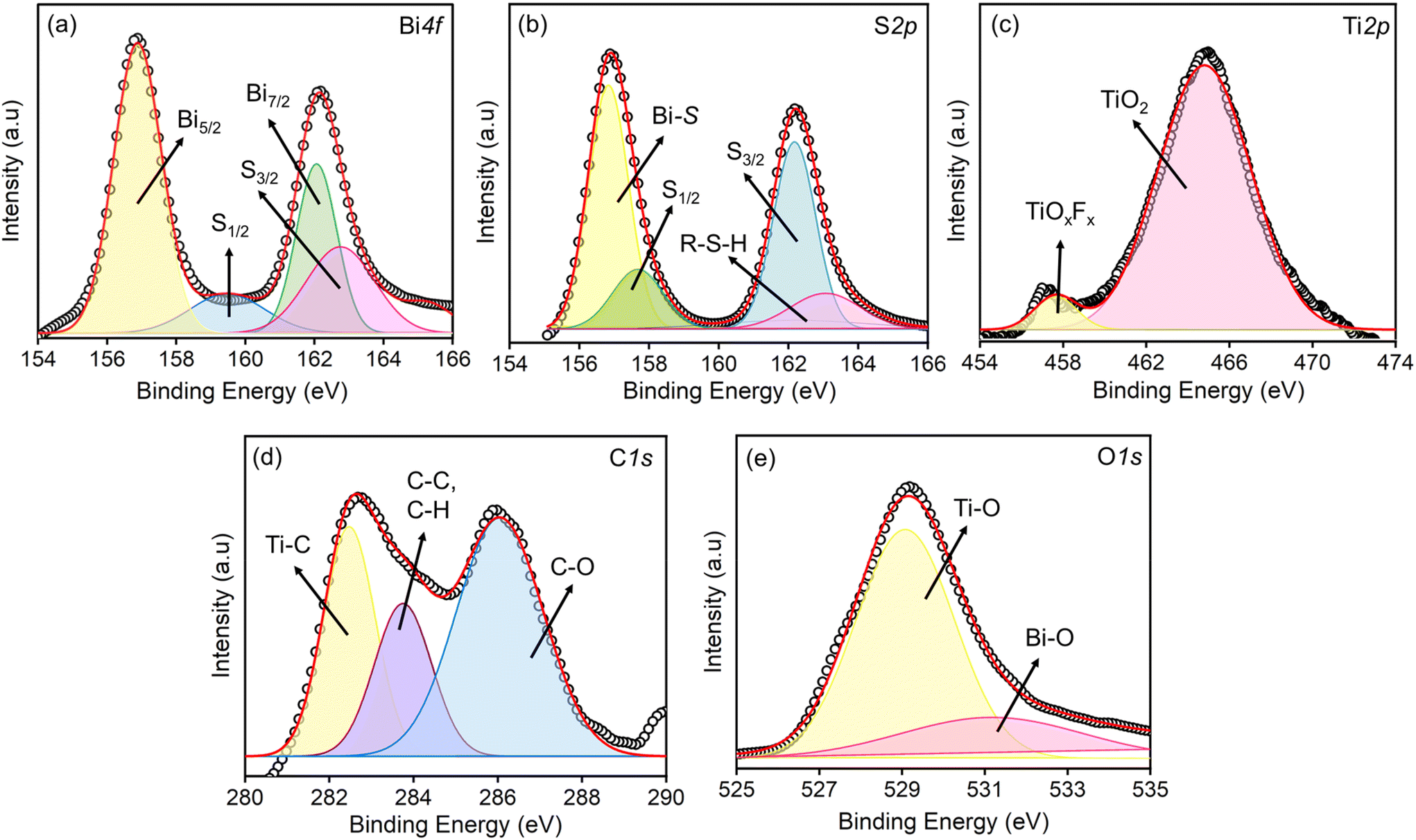
XPS analysis was carried out to determine the oxidation states of elements in the synthesised composite. Fig. 4(a) presents the core-level spectra of Bi 4f, displaying two high-intensity peaks at binding energies (BE) of 163.0 eV and 156.7 eV, corresponding to Bi 4f7/2 and Bi 4f5/2, respectively. These peaks confirm the presence of Bi in the Bi3+ oxidation state. Additionally, two significant peaks at BE = 160.1 eV and 162.0 eV are attributed to the low binding energy contributions of S 2p1/2 and S 2p3/2, indicating the formation of Bi–S bonds.46Fig. 4(b) shows two distinct peaks at BE = 162.1 eV and 159.4 eV, corresponding to S 2p3/2 and S 2p1/2, which confirm the S2− state of sulphur. A high-intensity peak at 156.1 eV is attributed to Bi–S bonding, while a smaller peak at 163.1 eV is linked to contributions from thiol groups (R–S–H bonds).47,48Fig. 4(c) depicts the Ti 2p spectra, where the components are deconvoluted into a prominent peak at 464.9 eV and a smaller peak at 457.0 eV. These peaks are assigned to Ti ions with a formal valence of +4, corresponding to TiO2 and TiOxFx species formed due to spontaneous surface oxidation. In the C 1s spectrum, Fig. 4(d), a peak at BE = 282.2 eV is assigned to the carbide component (Ti–C) and C–C/C–H bonding respectively, while another peak at 285.1 eV arises from C–O bonds, likely formed during the selective dissolution of Ti during chemical etching. The O 1s spectrum (Fig. 4(e)) displays a broad peak at 531 eV, attributed to Bi–O, lattice oxygen suggesting enhanced interaction between surface oxygen functional groups and Bi moieties. A sharp peak at 529 eV is observed due to the formation of TiO2 through MXene surface oxidation.49
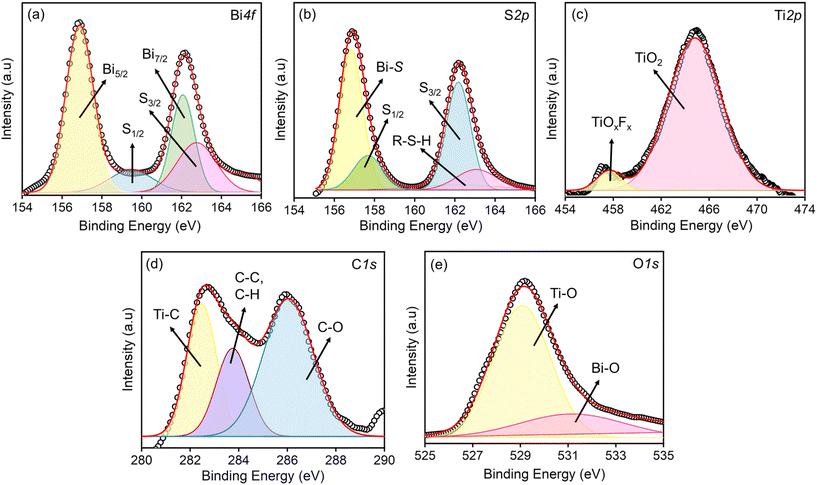 |
| | Fig. 4 XPS core level spectra of the elements present in the S–Bi@TiMX composite (a) Bi, (b) S, (c) Ti, (d) C and (e) O. | |
4.2 Morphological profiling
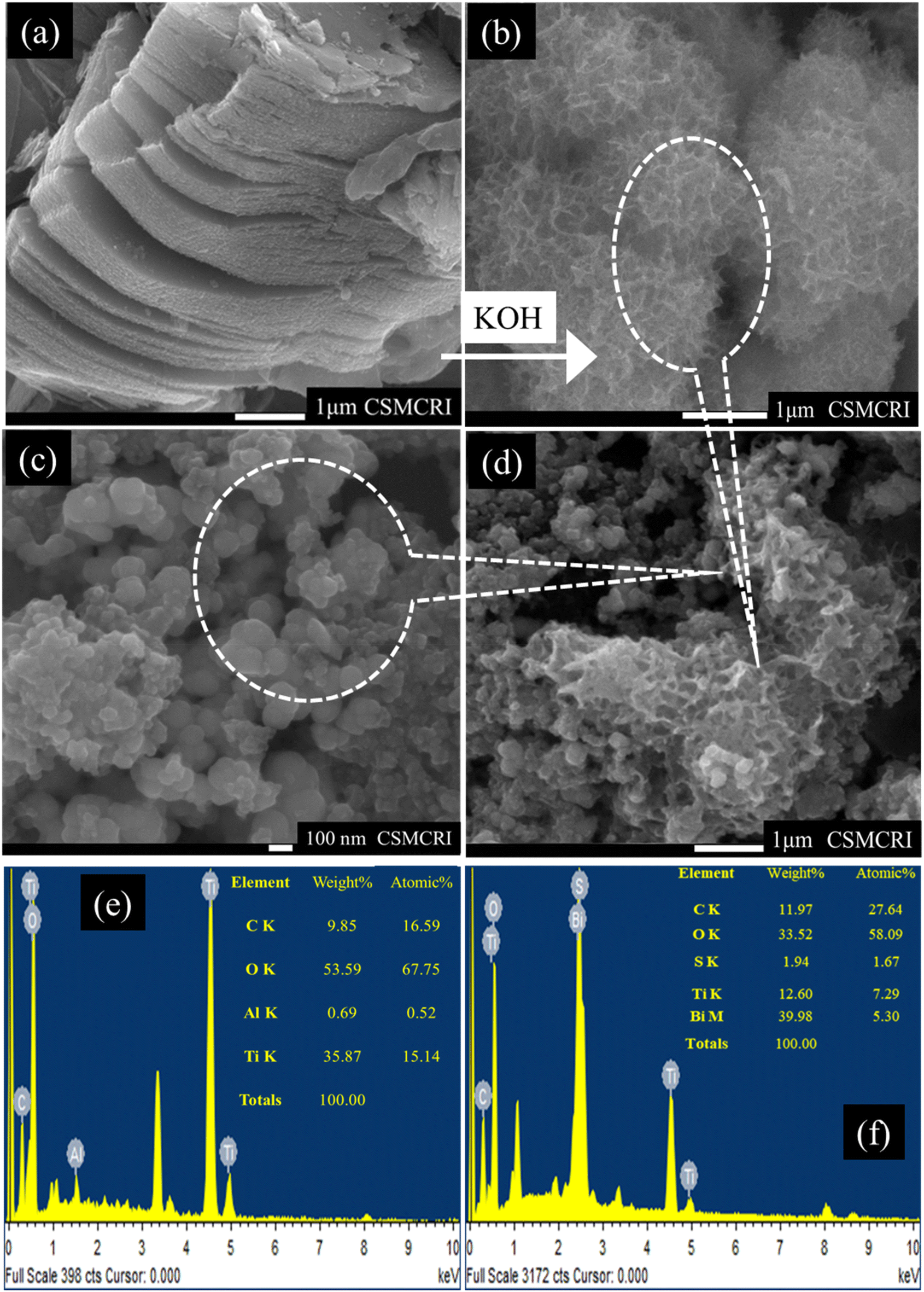
To gain deeper insights into the morphological characteristics of the S–Bi@TiMX composite, FE-SEM analysis was performed. Fig. 5(a) illustrates the conventional sheet-like morphology of Ti-MXene, which was synthesized through LiF and HCl etching. Notably, minimum traces of aluminium are present in Ti-MXene, confirmed by the EDX analysis, as shown in Fig. 5(e), confirming the successful etching process of the parent MAX phase. To enhance the catalytic activity and increase the surface area, an alkalization treatment was applied to Ti-MXene. This modification induced a significant transformation in morphology from sheet-like structures to distinctive “marigold”-like architectures, as shown in Fig. 5(b). Such morphological changes are indicative of increased active sites and improved surface properties. The SEM image in Fig. 5(c) reveals the formation of spherical clusters in the S–Bi sample. These spheres are observed to be uniformly deposited onto the alkali-modified Ti-MXene framework, as evidenced in Fig. 5(d), thereby confirming the successful synthesis of the S–Bi@TiMX composite. Furthermore, the EDX analysis confirmed the presence of all the elements in the composite matching with the obtained XPS results shown in Fig. 5(f). This unique structural configuration is anticipated to result in a pronounced synergistic interaction between S–Bi and TiMX, significantly enhancing the material's performance in lead detection applications.
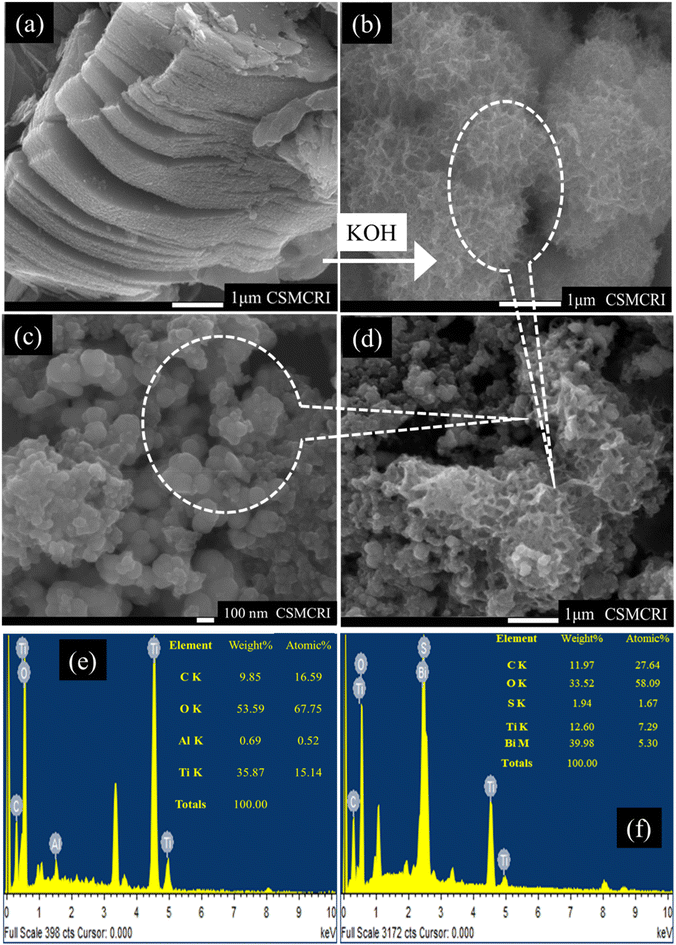 |
| | Fig. 5 SEM images of (a) laminar Ti-MXene, (b) alkali modified Ti-MXene, (c) S–Bi and (d) S–Bi@TiMX. EDX analysis of (e) TiMX and (f) S–Bi@TiMX/PCE. | |
4.3 Electrochemical behaviour of bare PCE and PCE composite electrodes
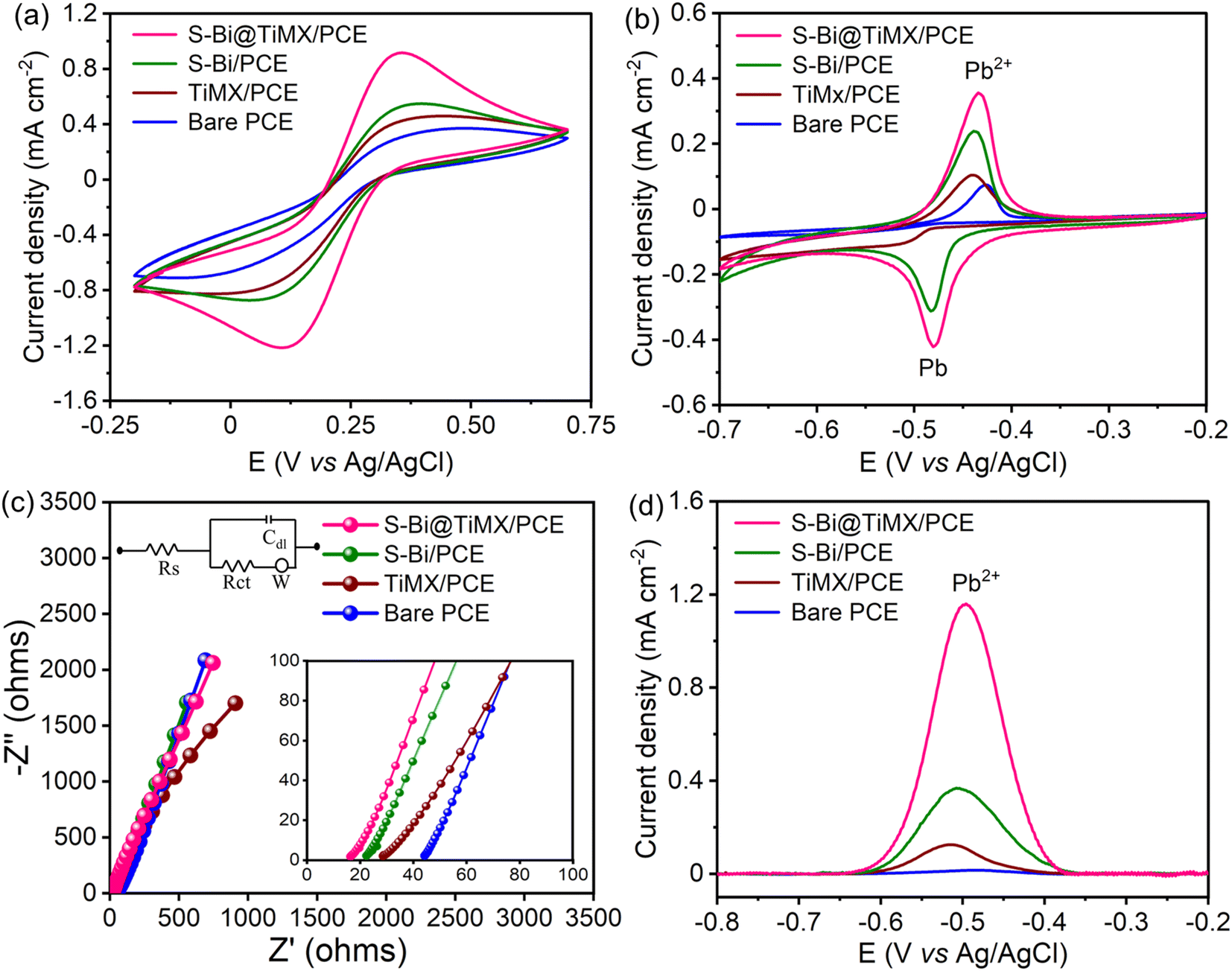
CV of bare PCE, TiMX/PCE, S–Bi/PCE, and S–Bi@TiMX/PCE electrodes was performed using a 5 mM K4[Fe (CN)6] solution containing 0.1 M KCl as the supporting electrolyte and 0.08 M HClO4 with 10 mg L−1 Pb2+, to evaluate their electrochemical activity in the voltage range of −0.2 V to 0.7 V with 50 mV s−1 scan rate as illustrated in Fig. 6(a and b). The results demonstrated a gradual increase in both anodic and cathodic peak currents across the series of electrodes. Bare PCE exhibited the lowest peak currents due to its limited electroactive surface area and intrinsic conductivity. Incorporating MXene significantly enhanced the current response, attributed to its high electrical conductivity, modified flower-like structure, and increased active sites for electron transfer. S–Bi led to a noticeable improvement in peak currents, suggesting enhanced electron transfer kinetics due to the synergistic effect of semiconducting properties in S–Bi and its interaction with the redox species. The S–Bi@TiMX composite demonstrated the highest anodic and cathodic peak currents, indicating superior electrochemical performance. This enhancement can be ascribed to the combined effect of the conductive network of TiMX and active sites of S–Bi, which collectively facilitate efficient charge transfer and increased electroactive surface area. Additionally, CV measurements were performed at varying scan rates (10–200 mV s−1) to investigate the reaction mechanism, as shown in Fig. S1.† A strong linear relationship was observed between the peak currents and the square root of the scan rate, confirming that redox processes at all electrodes are diffusion-controlled. The S–Bi@TiMX/PCE composite exhibited the highest slope in these plots, further emphasizing its superior diffusion characteristics and charge transfer efficiency. The slopes of the corresponding calibration curves were used to evaluate the active surface area of all the samples using the Randles–Sevcik equation (eqn S(i)†).50 The active surface area was calculated as 0.009, 0.012, 0.015, and 0.028 cm2 for bare PCE, TiMX, S–Bi, and S–Bi@TiMX/PCE, respectively. To calculate the resistance offered at the electrode–electrolyte interface, all test samples were subjected to EIS, and the findings are illustrated in Fig. 6(c). The Nyquist plot reveals a straight line for all three samples, indicating that the charge transfer process is not dominant in the given acidic medium. Instead, the impedance is primarily governed by diffusive processes. The Rs values were calculated by magnifying the obtained plot (inset Fig. 6c), where S–Bi@TiMX/PCE, S–Bi, TiMX, and bare PCE show Rs values of 16.27, 22.43, 28.14, and 43.43 Ω, respectively. These results support the faster kinetics of S–Bi@TiMX/PCE electrodes, and these findings collectively establish S–Bi@TiMX/PCE as a highly efficient material for electrochemical sensing.
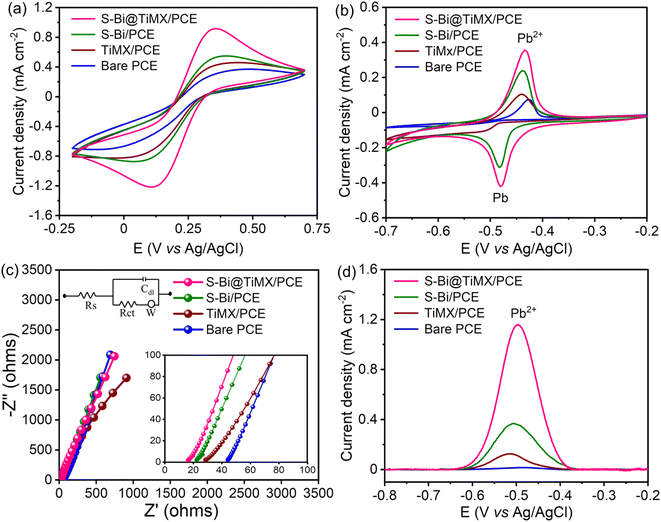 |
| | Fig. 6 CVs of bare PCE, TiMX/PCE, S–Bi/PCE, and S–Bi@TiMX/PCE at 50 mV s−1 in (a) 5.0 mM (K4[Fe (CN)6]) solution with 0.1 M KCl, (b) 0.08 M HClO4 containing 10 mg L−1 Pb2+, (c) EIS, and (d) SWASV responses of bare PCE, TiMX/PCE, S–Bi/PCE, and S–Bi@TiMX/PCE in 0.08 M HClO4 solution containing 50 μg L−1 Pb2+. | |
SW-ASV was conducted using all synthesised electrodes to evaluate their performance for detecting 50 μg L−1 Pb2+. The results demonstrate a continuous increase in stripping peak currents in the order bare PCE, TiMX/PCE, S–Bi/PCE, and S–Bi@TiMX/PCE, as shown in Fig. 6(d). Bare PCE exhibited the lowest peak current, reflecting its limited sensitivity and lack of active sites for Pb2+ adsorption. TiMX showed improved peak currents compared to PCE, indicating enhanced electrochemical activity. S–Bi further increased the peak currents due to its semiconducting properties and affinity of electronegative sulphur for Pb2+. Finally, the S–Bi@TiMX composite displayed the highest peak currents, attributed to the synergistic effect of abundant active sites of S–Bi and the excellent conductivity of TiMX due to the large surface area. These findings highlight the superior detection capability of the S–Bi@TiMX composite for Pb2+, making it a promising material for sensitive electrochemical applications.
4.4 Optimization of S–Bi@TiMX/PCE operational parameters for detection of Pb2+
Operational parameters, such as solution pH, preconcentration potential, and preconcentration time, play a crucial role in augmenting the electrochemical performance of the developed electrode and the detection of Pb2+. For an enhanced electrochemical signal, a key requirement is achieving an efficient and complete reduction of Pb2+ at active sites of the S–Bi@TiMX/PCE surface. When Pb2+ undergoes full reduction at the electrode, a measurable current is generated, which correlates with the concentration of Pb2+. The complete reduction of present Pb2+ maximizes response intensity, thereby improving the sensitivity of detection. Optimization ensures that each parameter is balanced to maximize the electrode's performance, allowing for precise quantification of ions even in complex sample matrices. Essentially, parameter optimization enhances the reliability, reproducibility, repeatability, robustness, and sharpness of peaks of Pb2+ in electrochemical detection, which makes it essential for applications like environmental monitoring and medical diagnostics where precision is crucial. All SW-ASV experiments were performed in a 20 ml solution of 0.08 M HClO4 electrolyte containing 50 μg L−1 Pb2+.
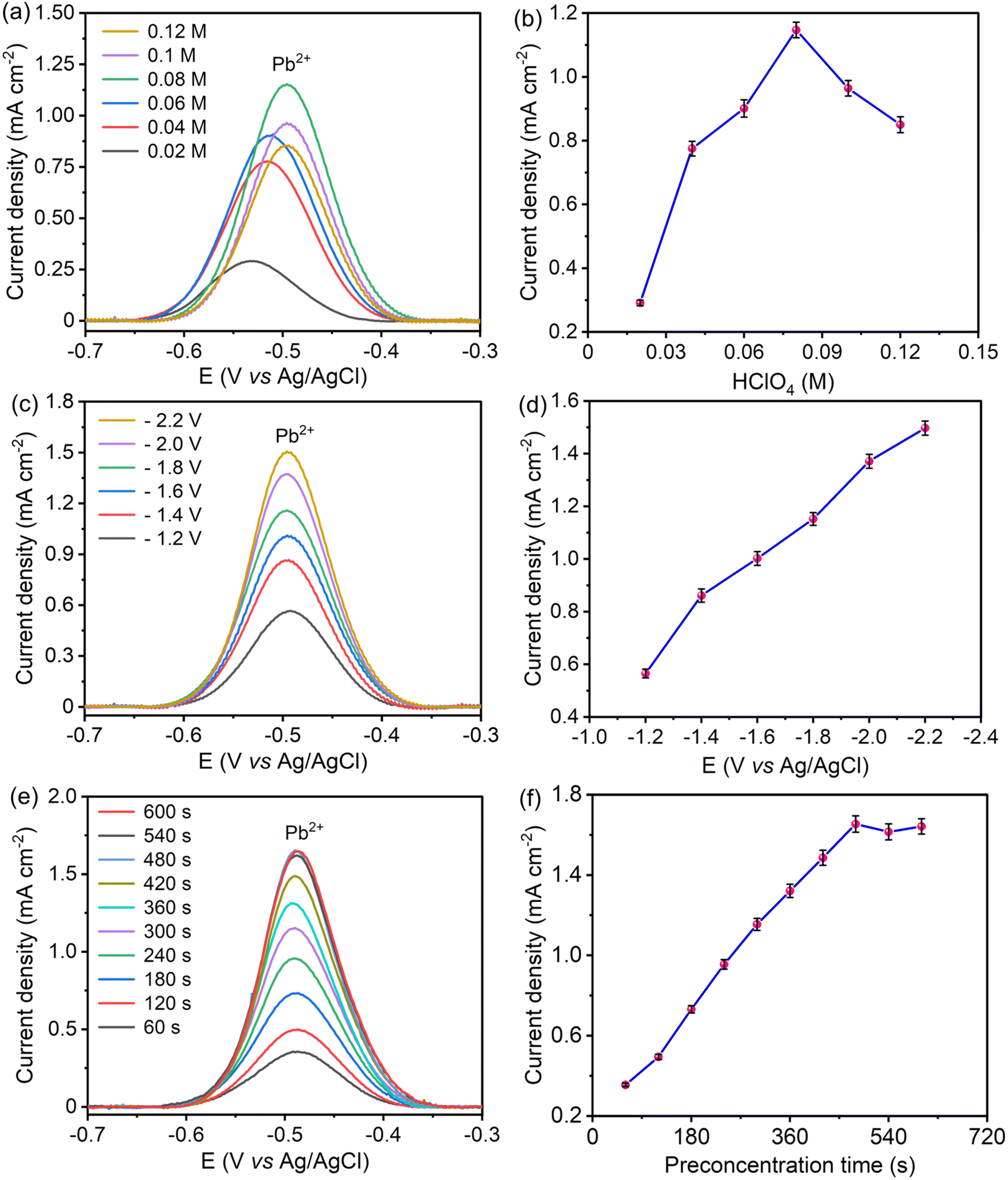
The solution pH influences the chemical environment around the electrode, impacting the stability, charge, and availability of target ions. At too high and too low solution pH, ions may either precipitate or undergo other reactions, leading to inconsistent detection. Therefore, optimal pH improves sensitivity by maximizing ion concentration at the electrode surface and selectivity by minimizing interference from other species. For optimization of solution pH, the concentration of perchloric acid varied from 0.02 M to 0.12 M (pH value of 1.7 to 0.9), as displayed in Fig. 7(a and b). At pH values higher than 1.1, Pb2+ hydrolyzed to form the hydroxide precipitate, which reduced the availability of Pb2+ at the electrode surface and resulted in gradual decay in the stripping peak current.51 At very low pH values, the presence of more hydrogen ions promotes the hydrogen evolution reaction, and the generated hydrogen bubbles lead to the blocking of active centers of the electrode surface. Additionally, the hydrogen evolution reaction lowers the enrichment of Pb2+ at the electrode surface by competing for active centers, resulting in a decrease in the stripping peak current of Pb2+ at low pH.52 Hence, 0.08 M HClO4 (pH 1.1) was chosen as the optimal pH for further experiments.
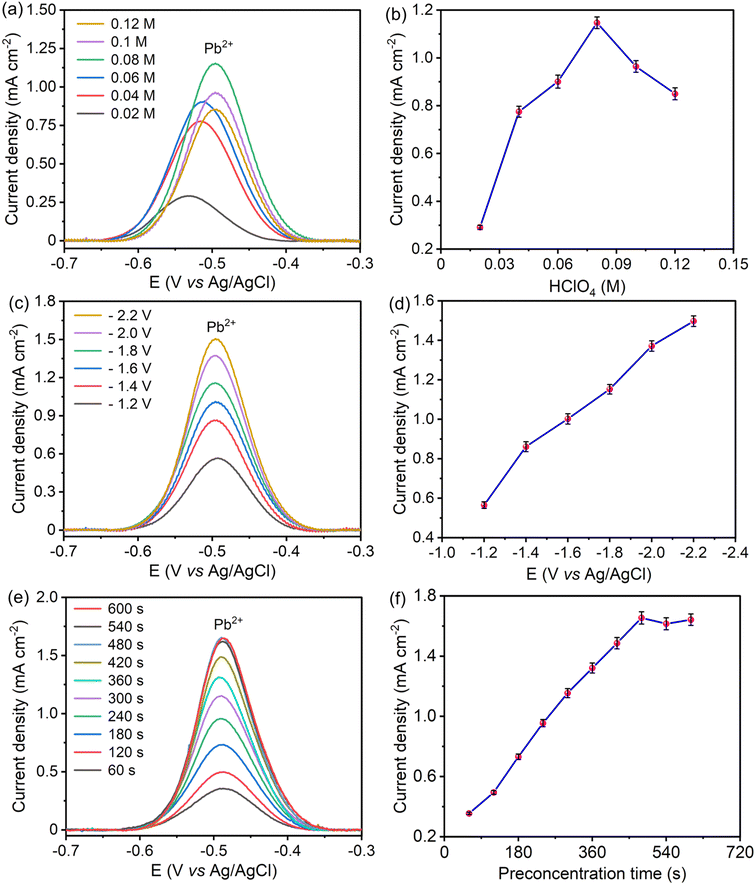 |
| | Fig. 7 SWASV responses and corresponding stripping peak currents of S–Bi@TiMX/PCE showing the impact of (a) and (b) solution pH, (c) and (d) preconcentration potential, and (e) and (f) preconcentration time in 0.08 M HClO4 solution containing 50 μg L−1 Pb2+. | |
Preconcentration potential also plays a significant role in the sensitive electrochemical detection of heavy metal ions. A significant negative potential attracts Pb2+ electrostatically, which maximizes the reduction of Pb2+ at the electrode surface, resulting in an enhanced stripping peak current of Pb2+. To find out the optimal potential, SW-ASV was performed at potentials ranging from −1.2 V to −2.2 V vs. Ag/AgCl as shown in Fig. 7(c and d). For Pb2+ detection, the stripping peak current increased with increasing applied negative potential. At lower negative potential, low peak responses indicate insufficient enrichment of Pb2+ at the electrode surface. As deposition potential increased towards a negative direction, peak responses increased due to significant enrichment of Pb2+ at the electrode surface.53 Additionally, at a higher potential than −1.8 V, hydrogen bubbles generated from hydrogen evolution affect the reproducibility and repeatability of the signal. Therefore, to achieve sensitive, reproducible, and precise detection of Pb2+ at the electrode, −1.8 V was the favorable preconcentration potential.
Optimal preconcentration time balances sufficient ion accumulation for a strong current signal with minimal noise or interference. This ensures that the electrode provides a stable, reproducible response that accurately reflects the Pb2+ concentration in the contaminated sample. For assessment of optimal preconcentration time, reduction of Pb2+ at the electrode surface was performed over a period from 60 seconds to 600 seconds. With a longer preconcentration time, more Pb2+ accumulates on the electrode surface, resulting in an enhanced signal and improved sensitivity, as shown in Fig. 7(e and f). This heightened accumulation helps to detect lower concentrations of Pb2+, as it maximizes the amount of analyte available for the electrochemical reaction. However, after 480 seconds, the stripping peak current becomes saturated for further increment in deposition duration. At prolonged preconcentration time, active centers of the electrode surface get saturated due to the overabundance of Pb2+ at the electrode surface.54 Hence, 480 seconds was found to be the optimal time for a stable and reproducible current signal.
4.5 Electrochemical detection of Pb2+ by the S–Bi@TiMX/PCE sensor
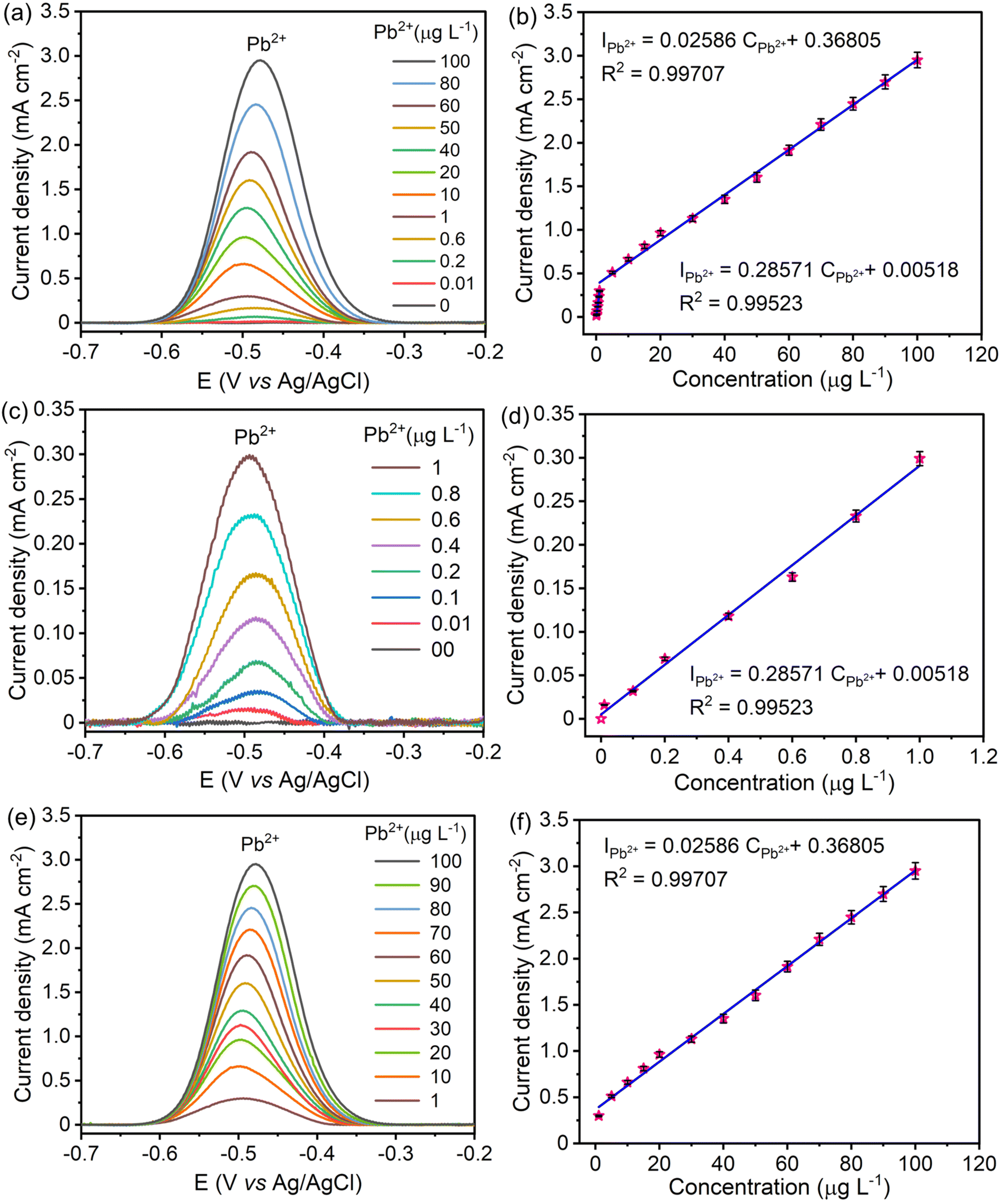
The linear range and sensitivity of the developed S–Bi@TiMX/PCE sensor for Pb2+ detection were evaluated under optimized conditions. SW-ASV was performed using the S–Bi@TiMX/PCE sensor across varying Pb2+ concentrations, revealing a gradual increase in stripping peak current with rising Pb2+ levels, as shown in Fig. 8(a and b). The sensor exhibited a dual-linearity response, effectively adapting to detect Pb2+ across distinct concentration ranges. Fig. 8(c and d) show that at ultra-trace levels (0.01 μg L−1 to 1 μg L−1), the sensor demonstrated a strong linear behaviour with exceptional sensitivity, described by linear regression eqn (i). Beyond this range, a second linear response was observed for the concentration of Pb2+ from 1 μg L−1 to 100 μg L−1 with regression eqn (ii), as depicted in Fig. 8(e and f). The corresponding regression equations from calibration plots are as follows:| | 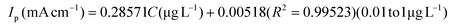 | (i) |
| | 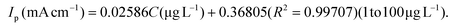 | (ii) |
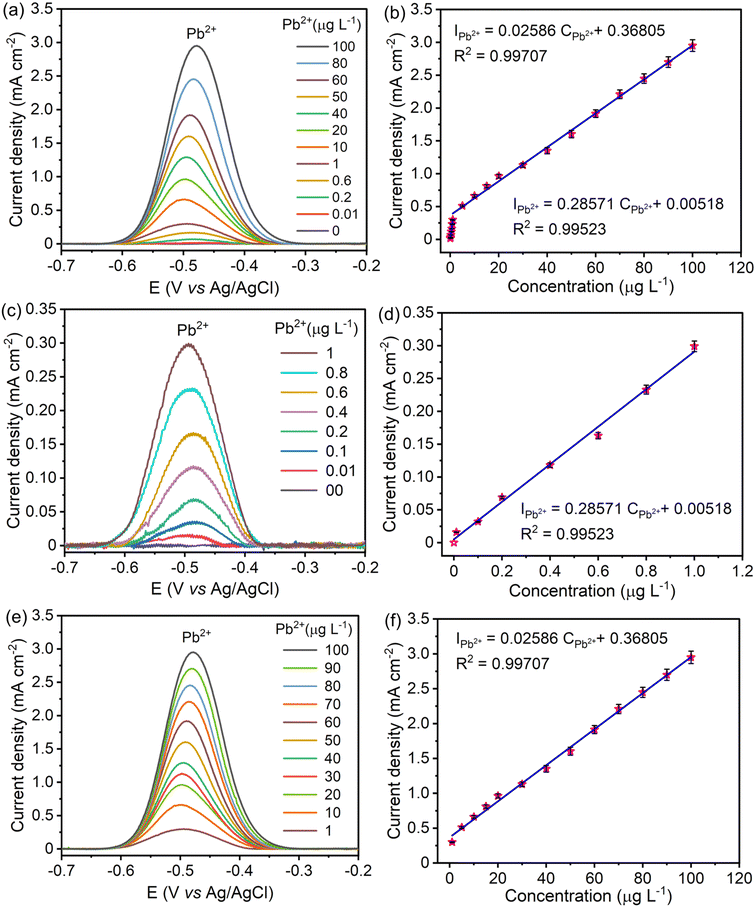 |
| | Fig. 8 SWASV responses of S–Bi@TiMX/PCE and the corresponding linear calibration plots of Pb2+ in 0.08 M HClO4 solution, (a) and (b) 0.01 μg L−1 to 100 μg L−1, (c) and (d) 0.01 μg L−1 to 1 μg L−1, and (e) and (f) 1 μg L−1 to 100 μg L−1 Pb2+. | |
Using the first calibration equation, the limit of detection (LOD) was calculated to be 0.0002 μg L−1 (LOD = 3SD/S, where SD is the standard deviation of the blank signal, and S is the slope of the calibration curve).55 The sensitivities of the S–Bi@TiMX/PCE sensor were found to be 285.71 μA L cm−2 μg−1 (0.01 μg L−1 to 1 μg L−1) and 25.86 μA L cm−2 μg−1 (1 μg L−1 to 100 μg L−1), respectively. Notably, the LOD is well below the WHO's stipulated drinking water threshold of 10 μg L−1, underscoring the capability of the developed sensor for real-world applications. The observed dual linear behaviour of S–Bi@TiMX/PCE can be attributed to surface heterogeneity. At ultra-low concentrations of Pb2+, sulphur present on the composite's surface likely reacts efficiently. However, at higher concentrations, the bulk species of the composite become active, enhancing overall surface diffusibility and interacting differently with Pb2+. This shift in interaction dynamics due to surface heterogeneity explains the dual linear response exhibited by the electrode across varying concentrations.56 The dual-linear behavior of the sensor highlights its high sensitivity and wide dynamic range, making it a robust and reliable platform for Pb2+ detection, from ultra-trace monitoring to high-level quantifications in diverse environmental and analytical scenarios.
A comparison of the electrochemical performance of the developed S–Bi@TiMX/PCE with previously reported bismuth and MXene-based electrodes for Pb2+ detection was performed (Table 1). According to this study, fabricated S–Bi@TiMX/PCE has the least LOD values, broad linear detectable ranges, and, most importantly, the best sensitivity for the Pb2+ ions under investigation.
Table 1 Comparative data of analytical performance of S–Bi@TiMX/PCE with previously reported bismuth and MXene-based electrodes for the detection of Pb2+
| Working electrode |
Detection method |
Linearity range (μg L−1) |
LOD (μg L−1) |
Ref. |
|
MGMA: 3D melamine-doped rGO/MXene aerogel; Alk: alkalization; H-C3N4: protonated carbon nitride; Bi@d-Ti3C2: delaminated MXene-bismuth; MSA: microelectrode assembly.
|
| NH2-Ti3C2Tx/SPCE |
DPASV |
5–500 |
0.31 |
32
|
| 3D MGMAa/SPCE |
DPASV |
3–900 |
0.29 |
34
|
| Ti3C2/Fe3O4/g-C3N4/GCE |
DPASV |
1–100 |
0.02 |
35
|
| UiO-66-NH2-Ti3C2Tx@rGO/GCE |
DPASV |
1–600 |
0.40 |
37
|
| Alka-Ti3C2/GCE |
SWASV |
20.7–310 |
8.5 |
38
|
| BiNPs/Ti3C2Tx/GCE |
SWASV |
12.4–124.3 |
2.24 |
57
|
| Ti3C2@N–C/GCE |
SWASV |
10.4–414 |
0.228 |
58
|
| H-C3N4a/Ti3C2Tx/GCE |
SWASV |
2–310 |
0.124 |
59
|
| Bi@d-Ti3C2a/MSAa |
SWASV |
1–20 |
0.2 |
60
|
| Bi-MOF-SH/Mx/Bi-MOF-SH/GCE |
DPASV |
0.03–20 |
0.012 |
61
|
| S–Bi/TiMX/PCE |
SWASV |
0.01–100 |
0.0002 |
This work |
4.6 Repeatability, reproducibility, and selectivity of the S–Bi@TiMX/PCE sensor
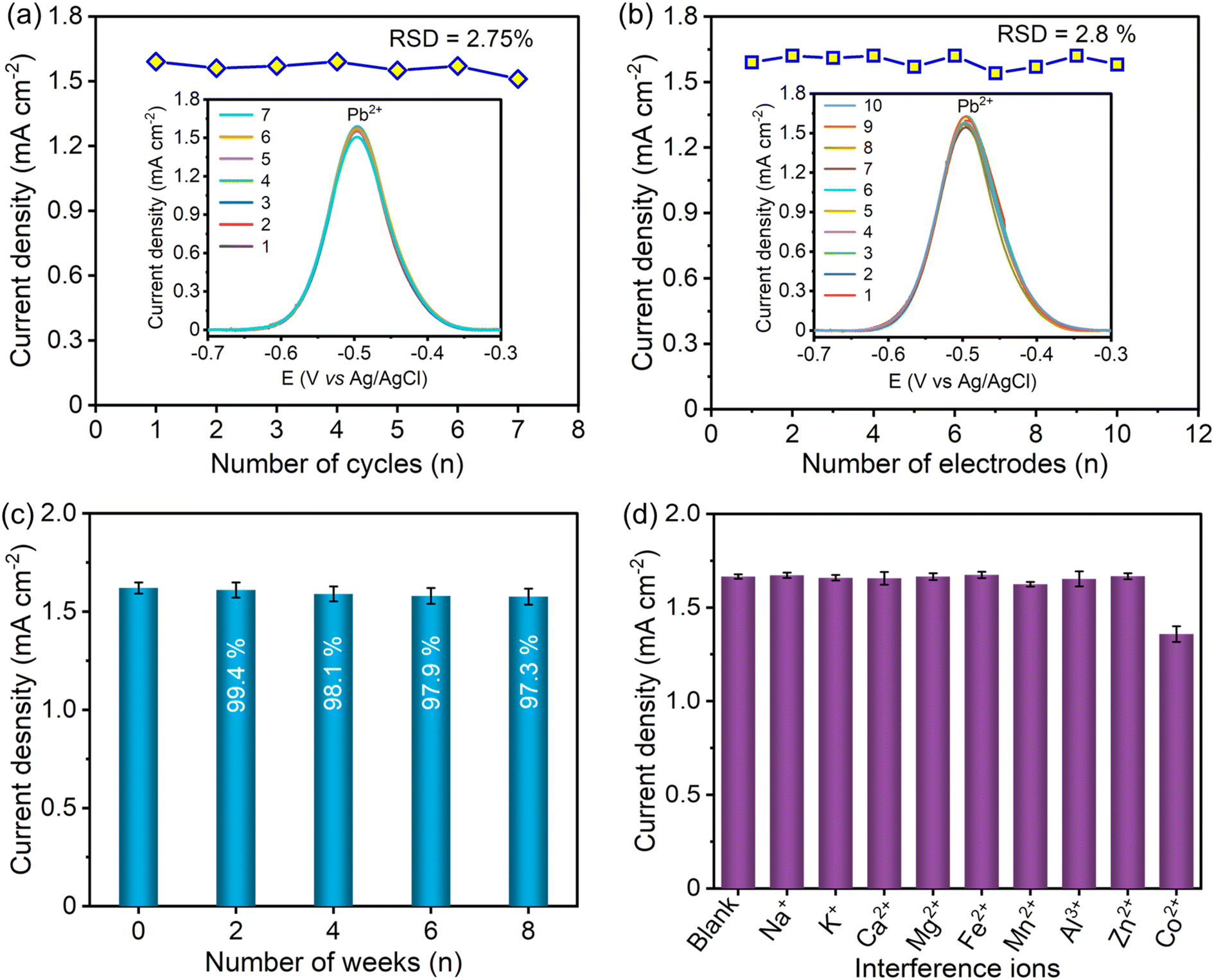
The repeatability of the S–Bi@TiMX/PCE sensor was evaluated by performing multiple consecutive cycles. The S–Bi@TiMX/PCE sensor attained considerably good repeatability up to 7 consecutive cycles with a relative standard deviation (RSD) of 2.75% for the detection of 50 μg L−1 Pb2+, as shown in Fig. 9(a). The reproducibility of the S–Bi@TiMX/PCE sensor was inspected for 10 identically fabricated electrodes by operating SW-ASV measurements under similar working conditions as presented in Fig. 9(b), which shows excellent reproducibility with an RSD value of 2.8%. For stability assessment of the developed sensor, S–Bi@TiMX/PCE was stored under ambient conditions for up to 8 weeks. SW-ASV measurements for Pb2+ were recorded every 2 weeks, revealing minimal degradation over time. As illustrated in Fig. 9(c), the sensor maintained stability throughout the 8-week period under these conditions.
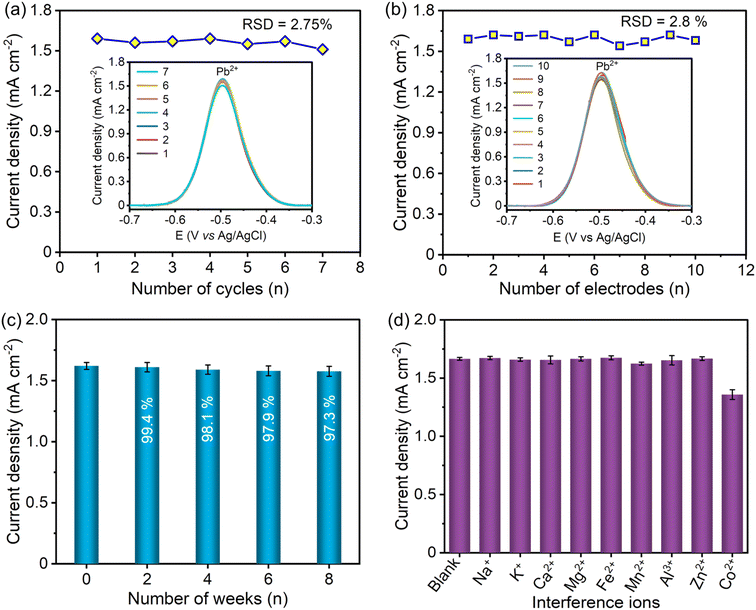 |
| | Fig. 9 (a) SWASV responses of S–Bi@TiMX/PCE for (a) 7 successive measurements and (b) 10 freshly prepared electrodes, (c) stability over time, and (d) SWASV responses of S–Bi@TiMX/PCE in 0.08 M HClO4 solution containing 50 μg L−1 Pb2+ in the presence of interfering ions (Na+, K+, Ca2+, Mg2+, Fe2+, Mn2+, Al3+, Zn2+, and Co2+). | |
Apart from high sensitivity and rapid detection of target ions, the effectiveness of the developed sensor depends on its ability to resist interference from potentially interfering ions. Environmental water samples are complex, containing a wide range of coexisting ions at varying concentrations. Some ions, such as Na+, K+, Ca2+, and Mg2+, are commonly present at high concentrations, reaching up to hundreds of ppm, while others, including Fe2+, Mn2+, Al3+, Zn2+, and Co2+, are found at lower levels, typically up to tens of ppm. To simulate realistic conditions, Na+, K+, Ca2+, and Mg2+ (each in 10 mg L−1 concentration, which is 200-fold higher than Pb2+ concentration) were added to a 0.08 M HClO4 solution containing 50 μg L−1 Pb2+. Additionally, Fe2+, Mn2+, Al3+, Zn2+, and Co2+ (each in 1 mg L−1 concentration, which is 20-fold higher than the Pb2+ concentration) were introduced into the test solution. The minimal interference due to Na+, K+, Ca2+, Mg2+, Fe2+, Mn2+, Al3+, and Zn2+ was observed, as shown in Fig. 9(d). The addition of Co2+ results in a decreased response, with the stripping current retaining only 81.6% of the initial signal. This reduction in response for Pb2+ in the presence of Co2+ may be attributed to competition for active sites.62 The results showed that the S–Bi@TiMX/PCE sensor possessed appreciable resistance to other metal matrices. These outcomes confirm the excellent reproducibility, good repeatability, and selectivity of the developed S–Bi@TiMX/PCE sensor for the detection of Pb2+ in a complex matrix.
4.7 Real sample analysis with the S–Bi@TiMX/PCE sensor
Real sample testing was observed to establish the practical applicability of the S–Bi@TiMX/PCE sensor for the detection of Pb2+ in tap water, which was first diluted 4-fold to reduce matrix effects. Then, known concentrations of Pb2+ (30, 40, and 60 μg L−1) were added into 0.08 M HClO4 solution of diluted samples, and detection was carried out using SW-ASV under optimized conditions. The measured concentrations were in good agreement with the spiked values, demonstrating the accuracy of the method. The calculated recovery percentages ranged between 99.2% and 99.6%, indicating minimal interference from the sample matrix. Furthermore, the RSD values for three replicate measurements were below 3%, confirming the reliability and reproducibility of the results. The detailed recoveries and RSD values for each concentration are briefed in Table 2.
Table 2 Analytical performance of the S–Bi@TiMX/PCE sensor in tap water samples
| Samples |
Spiked (μg L−1) |
Found (μg L−1) |
Recovery (%) |
RSD (%) |
| Tap water |
30 |
29.78 |
99.3 |
1.73 |
| 40 |
39.67 |
99.2 |
2.05 |
| 60 |
59.80 |
99.6 |
2.56 |
5 Conclusion
This study demonstrates the successful synthesis of an alkali-modified Ti-MXene doped with bismuth and sulphur moieties tailored for the effective electrochemical sensing of Pb2+ in both laboratory-prepared and tap water samples. The alkali-modified MXene exhibits a distinctive “marigold-like” morphology, which facilitates the enhanced deposition of bismuth species. The strong electronegativity of sulphur imparts a high affinity for Pb2+, thereby significantly enhancing the electrode's activity. The synthesized composite material was drop-cast onto an in-house prepared PCE, offering multiple advantages such as disposability, economic feasibility, and excellent stability. The distinctive network architecture of the S–Bi@TiMX/PCE composite significantly improves the electron transfer kinetics of the electrode, offers plentiful binding sites for Pb2+, and promotes effective accumulation on the sensing interface. The developed sensor displayed exceptional sensitivity (25.9 μA L cm−2 μg−1), a remarkable detection threshold of 0.0002 μg L−1, and a broad dynamic detection ability from 0.01 μg L−1 to 100 μg L−1, making it highly suitable for detecting ultra-low concentrations of Pb2+ in aqueous systems with outstanding accuracy. Furthermore, repeatability and reproducibility tests performed across seven cycles and ten different electrodes, respectively, yielded low RSD values of 2.75% and 2.8%. The sensor showed good resistance to the presence of potential interfering ions (e.g., Na+, K+, Ca2+, Mg2+, Fe2+, Mn2+, Al3+, Zn2+, and Co2+) on the stripping peak current of Pb2+. Real water testing for different concentrations of Pb2+ with acceptable recoveries (99.2%–99.6%) proved that S–Bi@TiMX/PCE is a potential alternative for onsite monitoring. This work underscores the potential of alkali-modified MXene composites in advancing electrochemical sensing technologies, particularly for environmental monitoring of heavy metals. The sensor's remarkable sensitivity and stability highlight its applicability in real-world scenarios, contributing to sustainable and cost-effective solutions for water quality assessment.
Data availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.
Conflicts of interest
Authors declare no conflict of interest.
Acknowledgements
The author, KS, sincerely acknowledges the Ministry of Education, New Delhi, for providing financial support for this research. Gratitude is also extended to the Central Instrumentation Facility of CSIR-CSMCRI for access to the instrumental resources essential for this work.
References
- K. Steenland and P. Boffetta, Am. J. Ind. Med., 2000, 38, 295–299 CrossRef CAS.
- G. Flora, D. Gupta and A. Tiwari, Interdiscip. Toxicol., 2012, 5, 47–58 CAS.
- A. Carocci, A. Catalano, G. Lauria, M. S. Sinicropi and G. Genchi, Rev. Environ. Contam. Toxicol., 2016, 45–67 CAS.
- K. S. Sangeetha and S. Umamaheswari, J. Clin. Diagn. Res., 2020, 14(12) DOI:10.7860/JCDR/2020/45615.14345.
- A. H. Sani and M. Amanabo, GSC Biol. Pharm. Sci., 2021, 15, 055–062 CrossRef CAS.
- M. A. Assi, M. N. M. Hezmee, A. W. Haron, M. Y. Sabri and M. A. Rajion, Vet. World, 2016, 9, 660–671 CrossRef CAS PubMed.
- W. Kang, X. Pei, C. A. Rusinek, A. Bange, E. N. Haynes, W. R. Heineman and I. Papautsky, Anal. Chem., 2017, 89, 3345–3352 CrossRef CAS PubMed.
-
World Health Organization. Guidelines for Drinking-Water Quality, WHO Press, Geneva, Switzerland, 4th edn, 2011 Search PubMed.
- M. Arjomandi and H. Shirkhanloo, Anal. Methods Environ. Chem. J., 2019, 2, 97–126 CrossRef CAS.
-
https://www.edt.co.uk/Lead-Combination-Ion-Selective-Electrode-3231
.
-
https://www.nico2000.net/analytical/lead.htm
.
-
https://hannainst.com/hi4112-lead-sulfate-combination-ion-selective-electrode.html
.
- H. Gunasingham and R. R. Dalangin, Anal. Chim. Acta, 1991, 246, 309–313 CrossRef CAS.
- A. Mourya, S. K. Sinha and B. Mazumdar, Microchem. J., 2019, 147, 707–716 CrossRef CAS.
- M. A. Deshmukh, M. Gicevicius, A. Ramanaviciene, M. D. Shirsat, R. Viter and A. Ramanavicius, Sens. Actuators, B, 2017, 248, 527–535 CrossRef CAS.
- A. Eftekhari, Sens. Actuators, B, 2003, 88, 234–238 CrossRef CAS.
- M. A. Deshmukh, R. Celiesiute, A. Ramanaviciene, M. D. Shirsat and A. Ramanavicius, Electrochim. Acta, 2018, 259, 930–938 CrossRef CAS.
- Y. Bonfil, M. Brand and E. Kirowa-Eisner, Anal. Chim. Acta, 2000, 424, 65–76 CrossRef CAS.
- G. Kefala, A. Economou, A. Voulgaropoulos and M. Sofoniou, Talanta, 2003, 61(5), 603–610 CrossRef CAS PubMed.
- S. B. Hocevar, I. Švancara, B. Ogorevc and K. Vytřas, Anal. Chem., 2007, 79, 8639–8643 CrossRef CAS PubMed.
- X. Zhu, C. Gao, J.-W. Choi, P. L. Bishop and C. H. Ahn, Lab Chip, 2005, 5, 212 RSC.
- N. Wang and X. Dong, Anal. Lett., 2008, 41, 1267–1278 CrossRef CAS.
- R. Pauliukaitė and C. M. A. Brett, Electroanalysis, 2005, 17, 1354–1359 CrossRef.
- M. A. Deshmukh, M. D. Shirsat, A. Ramanaviciene and A. Ramanavicius, Crit. Rev. Anal. Chem., 2018, 48(4), 293–304 CrossRef CAS PubMed.
- K. E. Toghill, G. G. Wildgoose, A. Moshar, C. Mulcahy and R. G. Compton, Electroanalysis, 2008, 20, 1731–1737 CrossRef CAS.
- M. A. Deshmukh, G. A. Bodkhe, S. Shirsat, A. Ramanavicius and M. D. Shirsat, Front. Chem., 2018, 6, 451 CrossRef CAS PubMed.
- M. Perween, D. B. Parmar, G. R. Bhadu and D. N. Srivastava, Analyst, 2014, 139, 5919–5926 RSC.
- S. Gupta, R. Singh, M. D. Anoop, V. Kulshrestha, D. N. Srivastava, K. Ray, S. L. Kothari, K. Awasthi and M. Kumar, Appl. Phys. A, 2018, 124, 737 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- B. Anasori, Y. Xie, M. Beidaghi, J. Lu, B. C. Hosler, L. Hultman, P. R. C. Kent, Y. Gogotsi and M. W. Barsoum, ACS Nano, 2015, 9, 9507–9516 CrossRef CAS PubMed.
- S. Zukauskas, A. Rucinskiene, S. Ramanavicius, A. Popov, G. Niaura, I. Baginskiy and A. Ramanavicius, Sci. Total Environ., 2024, 950, 175190 CrossRef CAS PubMed.
- Y. Chen, P. Zhao, Z. Hu, Y. Liang, H. Han, M. Yang and D. Huo, Food Chem., 2023, 402, 134269 CrossRef CAS PubMed.
- S. Bagheri, R. Chilcott, S. Luo and A. Sinitskii, Langmuir, 2022, 38(42), 12924–12934 CrossRef CAS PubMed.
- Y. Chen, P. Zhao, Y. Liang, Y. Ma, Y. Liu, J. Zhao and D. Huo, Talanta, 2023, 256, 124294 CrossRef CAS PubMed.
- M. Akhtar, M. Sohail, M. F. Warsi, M. M. Ibrahim, M. A. Amin and M. Shahid, FlatChem, 2023, 41, 100537 CrossRef CAS.
- L. Wen, J. Dong, H. Yang, J. Zhao, Z. Hu, H. Han and D. Huo, Sci. Total Environ., 2022, 851, 158325 CrossRef CAS PubMed.
- J. Dong, X. Li, L. Wen, Y. Ma, J. Xu, H. Luo and D. Huo, Food Chem., 2024, 437, 137835 CrossRef CAS PubMed.
- Y. He, L. Ma, L. Zhou, G. Liu, Y. Jiang and J. Gao, Nanomaterials, 2020, 10, 866 CrossRef CAS PubMed.
- S. K. Raj, V. Sharma, S. Mishra and V. Kulshrestha, RSC Appl. Interfaces, 2024, 1, 1057–1068 RSC.
- S. K. Raj, K. B. Patel, V. Sharma, D. N. Srivastava and V. Kulshrestha, Energy Fuels, 2023, 37, 16856–16865 CrossRef CAS.
- A. K. Dutta, S. K. Maji, K. Mitra, A. Sarkar, N. Saha, A. B. Ghosh and B. Adhikary, Sens. Actuators, B, 2014, 192, 578–585 CrossRef CAS.
- B. Scheibe, K. Tadyszak, M. Jarek, N. Michalak, M. Kempiński, M. Lewandowski, B. Peplińska and K. Chybczyńska, Appl. Surf. Sci., 2019, 479, 216–224 CrossRef CAS.
- Y. Han, J. Tao, A. Khan, R. Ullah, N. Ali, N. Ali, S. Malik, C. Yu, Y. Yang and M. Bilal, Environ. Res., 2022, 215, 113978 CrossRef CAS PubMed.
- S. Subramanian, P. Chithra Lekha and D. Pathinettam Padiyan, Curr. Appl. Phys., 2009, 9, 1140–1145 CrossRef.
- K. Sanjeev Kumar, K. Giribabu, R. Suresh, R. Manigandan, S. Praveen Kumar and V. Narayanan, Mater. Lett., 2021, 283, 128804 CrossRef CAS.
- T. Bashir, Q. Mao, W. Zhu, J. Yang and L. Gao, ACS Appl. Energy Mater., 2024, 7, 2487–2495 CrossRef CAS.
- F. P. Ramanery, A. A. P. Mansur, H. S. Mansur, S. M. Carvalho and M. C. Fonseca, Nanoscale Res. Lett., 2016, 11, 187 CrossRef PubMed.
- S. V. P. Vattikuti, P. C. Nagajyothi and J. Shim, J. Mater. Sci.: Mater. Electron., 2019, 30, 5681–5690 CrossRef CAS.
- H. Zhou, S. J. Han, H. Lee, D. Zhang, M. Anayee, S. H. Jo, Y. Gogotsi and T. Lee, Adv. Mater., 2022, 34(41), 2206377 CrossRef CAS PubMed.
- E. A. Shalaby, A. M. Beltagi, A. A. Hathoot and M. A. Azzem, RSC Adv., 2023, 13, 7118–7128 RSC.
- S. Lee, S.-K. Park, E. Choi and Y. Piao, J. Electroanal. Chem., 2016, 766, 120–127 CrossRef CAS.
- J. Liu, J. Shi, M. Zhong, Y. Wang, X. Zhang, W. Wang, Z. Chen, Y. Tan, D. Xu, S. Yang and L. Li, Anal. Methods, 2024, 16, 244–252 RSC.
- Q. Yang, X. Sun, Y. Sun, X. Shen and Y. Pang, ACS Appl. Nano Mater., 2023, 6, 7901–7909 CrossRef CAS.
- M. Kim, J. Park, H. Park, W. Jo, W. Lee and J. Park, ACS Sustainable Chem. Eng., 2023, 11, 6844–6855 CrossRef CAS.
- W. Liu, J. Guan, B. Kong, H. Lu, Y. Wu, X. Qin, H. Jiang and X. Liu, J. Solid State Electrochem., 2023, 27, 3393–3404 CrossRef CAS.
- K. M. Zeinu, H. Hou, B. Liu, X. Yuan, L. Huang, X. Zhu, J. Hu, J. Yang, S. Liang and X. Wu, J. Mater. Chem. A, 2016, 4, 13967–13979 RSC.
- X. Zhu, B. Liu, H. Hou, Z. Huang, K. M. Zeinu, L. Huang, X. Yuan, D. Guo, J. Hu and J. Yang, Electrochim. Acta, 2017, 248, 46–57 CrossRef CAS.
- X. Zhang, D. An, Z. Bi, W. Shan, B. Zhu, L. Zhou, L. Yu, H. Zhang, S. Xia and M. Qiu, J. Electroanal. Chem., 2022, 911, 116239 CrossRef CAS.
- X. Lv, F. Pei, S. Feng, Y. Wu, S.-M. Chen, Q. Hao and W. Lei, J. Electrochem. Soc., 2020, 167, 067509 CrossRef CAS.
- X. Zhu, B. Liu, L. Li, L. Wu, S. Chen, L. Huang, J. Yang, S. Liang, K. Xiao, J. Hu and H. Hou, Microchim. Acta, 2019, 186, 776 CrossRef CAS PubMed.
- S. U. Saleem, Y. Liu, J. Sui, G. I. N. Waterhouse, Z. Zhang and L. Yu, J. Environ. Chem. Eng., 2024, 12, 113603 CrossRef CAS.
- Y. Beyene, Z. Bitew and F. Fekade, Mater. Adv., 2022, 3, 5882–5892 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *bc
*bc