
Sulphur-decorated Ti3C2 MXene structures as high-capacity electrode for Zn-ion batteries: a DFT study
Sunita
Saharan
,
Umesh
Ghanekar
and
Shweta
Meena
 *
*
Department of Electronics and Communication Engineering, National Institute of Technology Kurukshetra, Kurukshetra 136119, Haryana, India. E-mail: mail2shwetameena@nitkkr.ac.in
First published on 20th February 2025
Abstract
MXene-based structures have gained tremendous attention in energy storage applications, especially in ion batteries due to their promising electrical conductivity and high energy storage properties. Herein, we studied sulphur-decorated Ti3C2 MXene structures for Zn-ion batteries with augmented storage capacity (462.5 mAh g−1). Moreover, we systematically investigated the adsorption energy, structural stability, electronic band structure, open-circuit voltage and diffusion barrier of the sulphur-decorated Ti3C2 MXene using first-principles calculations. Our findings reveal that the studied MXene structures retain metallic characteristics with high open-circuit voltage (1.13 V) and charge transfer of 1.30 |q|. Hence, the designed sulphur-modified Ti3C2 MXene structures could be considered as promising cathode materials in Zn-ion batteries.
1 Introduction
As global energy demand increases, there is a pressing need for more sustainable and environmentally friendly energy storage solutions. Currently, lithium-ion batteries (LIBs) dominate the commercial battery market due to their high energy densities and extended cycle life.1 Market projection indicates a significant expansion from $30 billion to $100 billion by 2025.2 However, despite their advantages, LIBs face notable challenges such as diminishing lithium metal resources, high production costs, capacity degradation due to corrosion and dendrite formation, and safety concerns associated with flammable organic electrolytes and overcharging incidents.2–4 Beyond these concerns, the rapid expansion of markets for electronic devices, electric vehicles, and renewable energy storage systems demands batteries with higher energy densities, longer life spans, quicker charging times, and lower costs. Consequently, there is a growing imperative to explore alternative battery technologies.Several potential replacements for lithium, including sodium, potassium, magnesium, calcium, aluminium, and zinc, have been identified.2 Among these, zinc-ion batteries (ZIBs) have emerged as promising contenders due to their non toxicity and lower production costs facilitated by abundant zinc resources, making them viable replacements for traditional lithium-ion batteries.5,6 Additionally, ZIBs utilize safer and less complex electrolytes, offering advantages over LIBs in terms of electrolyte composition.2 ZIBs have relatively low redox potential (approximately −0.76 V against SHE) compared to other metal anodes used in aqueous solutions.7,8 Moreover, their capability to undergo a two-electron transfer mechanism during charging/discharging contributes to their high energy density. Also, ZIBs possess longer service lives, improved environmental sustainability, and enhanced stability in aqueous electrolytes.2,9 Yet, the advancement of ZIBs remains in the early stages due to challenges such as the formation of dendrites during charging and discharging, the absence of appropriate cathode materials for zinc, and unwanted reactions between zinc and the electrolyte.10,11
There is a critical need for novel cathode materials in ZIBs that offer stable structures and high ion-diffusion rates. Developing cathodes with layered structures could significantly enhance zinc-ion migration and increase surface activity, ultimately improving battery performance.12 This focus on alternative battery technologies underscores a shift towards more sustainable and efficient energy storage solutions to meet the future demands of technology and renewable energy. The majority of reported cathode materials for ZIBs include Prussian blue analogues,1 manganese oxides,13,14 compounds based on vanadium,14–17 and conductive polymers.18,19 While manganese oxide-based cathodes have potential zinc storage capability, the majority of these compounds have poor capacity, which leads to low energy density.20 Furthermore, manganese-based materials will disintegrate during the electrochemical reaction process, resulting in low cycling stability and impeding further growth.20 The poor specific capacitance of Prussian blue counterparts prevents them from being developed further, despite their potential for high voltage and high energy density applications.21
Significantly, vanadium-based electrode materials have a large theoretical capacity, a variety of crystal shapes, and multiple valence states. They may also facilitate multi-electron transfer and facilitate the rapid intercalation and deintercalation of Zn2+ ions. V2O5, a typical example of a layered vanadium-based cathode, features a high theoretical capacity of 589 mAh g−1. However, the capacity of vanadium oxides to store divalent zinc-ions in the specified vanadium oxide cathodes is significantly restricted by their intrinsic electrochemical constraints. These include the dissolution of vanadium in acidic or neutral electrolytes, resulting in the degradation of electrode structure, contamination of electrolytes, and corrosion of the zinc anode.22,23 To tackle these issues, researchers are looking into using other 2D materials as cathodes for zinc-ion batteries.24,25
MXenes, a class of novel 2D materials consisting of transition metal nitrides and carbides have been extensively researched and acknowledged for energy storage.26–28 They exhibit superior electrical conductivity, thermodynamic stability, tunable surface features and layered structures. They have emerged as promising materials for zinc-ion batteries due to their multifunctional roles as coating layers, structural reinforcements, and active electrode components. As a protective coating, MXenes help stabilize electrodes by suppressing dendrite growth and mitigating side reactions, thereby enhancing battery lifespan.29–31 When employed as reinforcement materials, they improve mechanical durability and facilitate efficient ion transport, contributing to better electrochemical performance. MXenes, known for their excellent electrical conductivity and rich surface functional groups, have been employed to enhance the performance of these cathode materials. These diverse functionalities position MXenes as key candidates for advancing zinc-ion battery technology, necessitating further exploration for their practical deployment.28,32
Liu et al. synthesized a V2O5/MXene heterostructure as a cathode material for ZIBs to overcome the vanadium dissolution problem. In addition, density functional theory (DFT) was used to validate the adsorption, ion diffusion performance and electron interaction of the V2O5/MXene heterostructure. The reported storage capacity and diffusion barrier of the V2O5/MXene heterostructure was 243.6 mAh g−1 and 0.3 eV, respectively.24 In another study, Yang et al.25 developed a Mn2+-doped MoS2/Ti3C2 heterostructure to improve the conductivity and electrochemical energy storage performance of the cathode for ZIBs. The reported capacity for the studied heterostructure was 191.7 mAh g−1 at 0.1 Ag−1. Additionally, with faster Zn2+-ion diffusion, improved conductivity and lower activation energy were also observed.
Qi et al.33 developed a flexible cathode for ZIBs by using carbon cloth as a substrate in MnO2 followed by coating of Ti3C2Tx using electrodeposition. By repeating the process, different samples of CC@MnO2@Ti3C2Tx MXene composites were developed. The studied composite demonstrated a storage capacity of 517 mAh g−1 at 0.1 Ag−1. The Ti3C2Tx MXene was responsible for the improved electron and ion transfer, and the MXene layer also contributed additional Zn2+ storage sites, enhancing the overall capacity of the electrode. Liu and colleagues34 introduced a (Na0.55Mn2O4·1.5H2O)/MXene hybrid consisting of pre-inserted Na+ ions for high-performance zinc-ion batteries. The pre-intercalation of Na+ in (Na0.55Mn2O4·1.5H2O) nanosheets enhanced structural stability and the presence of MXene facilitated fast electron transfer. The reported MXene hybrid cathode exhibited a high rate capacity of 180 mAh g−1 at 10 C and demonstrated prolonged cycling stability, lasting over 2500 cycles at 5 C. Feng et al.35 developed the VS2/MXene cathode to improve the performance of ZIBs. The combination of the large layer spacing and hydrophobic characteristics of VS2, along with the high electrical conductivity of Ti3C2Tx MXene, enhanced ionic transfer and diffusion kinetics, leading to superior electrochemical performance. The VS2/MXene cathodes exhibited a high capacity of 213.4 mAh g−1 at 0.2 Ag−1.
Despite recent advancements in ZIBs through the development of novel cathode materials, such as vanadium-based compounds, MXene heterostructures and advanced electrolyte engineering, several challenges remain, including limited storage capacity, sluggish ion diffusion, and low charge transfer capabilities. To fully unlock the potential of ZIBs, there is a pressing need to design and develop next-generation cathode materials that not only enhance key performance metrics like energy density and cycle life, but also improve overall electrochemical performance.
In the present study, we employed first-principles calculations to investigate the potential of sulphur-decorated monolayer Ti3C2 (m Ti3C2-S2) and sulphur-decorated bilayer Ti3C2 (b Ti3C2-S2) MXenes as electrode materials for ZIBs. We systematically evaluated their feasibility as cathode materials by analysing adsorption energy, structural stability through ab initio molecular dynamics simulations (AIMD), electronic band structure, projected density of states (PDOS), Bader charge analysis, open circuit voltage (OCV), storage capacity, and diffusion barriers. The electronic band structure and PDOS analyses revealed that both m Ti3C2-S2 and b Ti3C2-S2 MXenes maintain their metallic characteristics after Zn-ion adsorption and intercalation, respectively. We identified the optimal adsorption sites (top sites) for Zn ions and calculated the corresponding adsorption energies. This is the first report on sulphur-functionalized Ti3C2 MXene structures (monolayer and bilayer) as cathode materials for ZIBs.
2 Computational details
All the first-principles calculations were performed using the density functional theory (DFT)-based materials modeling tool, atomistic tool kit (ATK). The atomic structure of the unit cell of Ti3C2-S2 MXenes was initially optimized using the DFT module in the ATK. Generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE) functional was applied to describe the exchange–correlation effects.36,37 Additionally, the Grimme DFT-D3 method was utilized to accurately account for van der Waals interactions and interatomic forces.38 A density mesh cut-off of 2000 eV with 4 × 4 × 1 Brillouin-zone sampling points and a temperature of 500 K was used for all the computations. A 3 × 3 × 1 supercell of m Ti3C2-S2 and b Ti3C2-S2 was constructed and optimized using the ATK. The optimized structure of the m Ti3C2-S2 MXene and the b Ti3C2-S2 MXene is depicted in Fig. 1 and 2, respectively.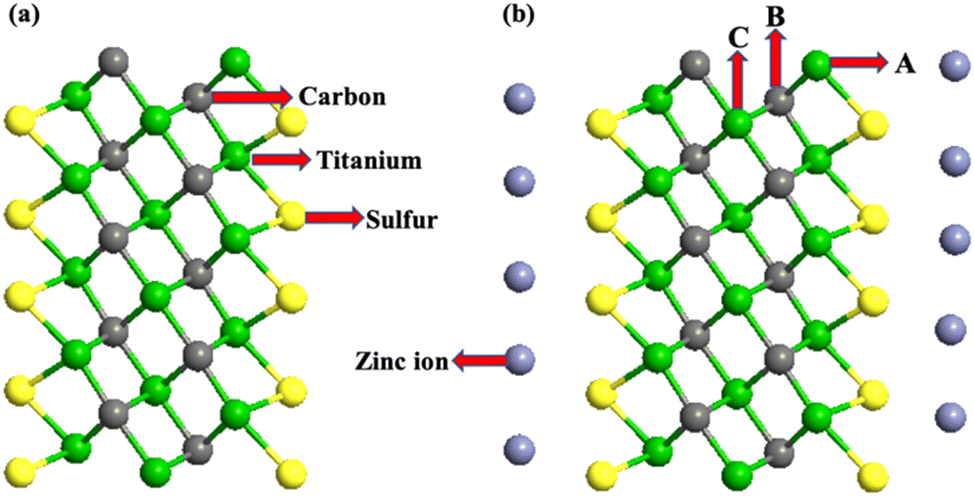 | ||
| Fig. 1 Side view of (a) the optimised structure of the m Ti3C2-S2 MXene before Zn-ion adsorption and (b) the optimised structure of the m Ti3C2-S2 MXene after Zn-ion adsorption (A, B and C correspond to the top adsorption sites for Zn ions). | ||
 | ||
| Fig. 2 Side view of (a) the optimized structure of the b Ti3C2-S2 MXene before Zn-ion intercalation and (b) the optimized structure of the b Ti3C2-S2 MXene after Zn-ion intercalation. | ||
The limited-memory Broyden–Fletcher–Goldfarb–Shanno (LBFGS) optimizer method was employed for geometry optimization. All the atoms were fully relaxed until the forces on each atom was less than 0.05 eV Å−1. A vacuum distance of 30 Å and an interlayer distance of 9.5 Å were used between Ti3C2-S2 layers to minimize interactions between the neighbouring atoms. The ion migration and diffusion barriers of Zn ions within Ti3C2-S2 were investigated using the climbing image nudged elastic band (CI-NEB) method.39 The electronic band structures and PDOS of m Ti3C2-S2 and b Ti3C2-S2 MXenes, before and after Zn-ion adsorption and intercalation, respectively, were calculated using DFT computations. Band structure calculations were conducted using a 7 × 7 × 1 k-point grid, with a density mesh cut-off of 2000 eV at a temperature of 500 K. The meta-GGA exchange–correlation method, including spin polarization and self-consistent computations, was used to calculate the electronic band structure accurately.
3 Results and discussion
3.1 Structural and adsorption characteristics
A unit cell of Ti3C2 MXene was designed and optimized using the ATK, where the structure consists of alternating layers of titanium (Ti) and carbon (C) atoms. To enhance the surface chemistry, sulphur (S) atoms were introduced on the surface of the Ti3C2 MXene, resulting in the formation of Ti3C2-S2. We selected sulphur (S) for MXene decoration because of its ability to form strong and stable bonds with surface titanium atoms in Ti3C2. This bond stability enhances the structural integrity of the material and strengthens interactions with Zn ions during electrochemical processes. Sulphur's higher reactivity, attributed to its atomic number (16) and tendency to complete its octet, makes it particularly suitable for improving the surface chemistry of MXenes. These properties collectively contribute to better electrochemical performance, making sulphur an optimal choice for modifying MXenes for Zn-ion battery applications.40A 3 × 3 × 1 supercell of a sulphur-decorated monolayer Ti3C2, named m Ti3C2-S2, was created, along with a bilayer structure, named b Ti3C2-S2. The final optimized structures are shown in Fig. 1 and 2, respectively. To check the structural stability of the considered MXene structures, we have computed the formation energy and performed the energy analysis using AIMD. To better understand which site is most stable for Zn-ion adsorption/intercalation, the adsorption energies of Zn ions have been calculated for different positions on both the m Ti3C2-S2 and b Ti3C2-S2 MXene structures. This computational analysis helps identify where Zn ions are most likely to bind securely. A negative adsorption energy indicates that the intercalation process is stable and will occur spontaneously. The adsorption energy (Ead) of Zn ions on the surfaces of m Ti3C2-S2 and b Ti3C2-S2 MXene structures is characterized as follows:
| Ead = Etot − Ehost − EZn | (1) |
It is observed that the structural stability of m Ti3C2-S2 and b Ti3C2-S2 MXenes intercalated with Zn ions remained preserved, with no evidence of cluster formation observed during the structure optimization process (see Fig. 1 and 2). In the present study, Zn ions are intercalated at three distinct positions (top sites) on the Ti3C2-S2 structure: adjacent to the outer titanium atoms, at the sites occupied by carbon atoms and near the inner titanium atoms (represented by A, B and C in Fig. 1). The hollow and bridge sites are unfavourable for Zn-ion intercalation, as indicated by their positive adsorption energies (+2.17 eV for the bridge site and +1.57 eV for the hollow site). The adsorption energies of Zn ions are calculated to be −0.754 eV at site A, −0.617 eV at site B, and −0.541 eV at site C. These values indicate that site A, located on the top of the outer titanium atoms, is the most stable adsorption site for Zn ions, suggesting a stronger and more favourable interaction at site A compared to site B and site C. This finding underscores the significance of the outer titanium positions in facilitating stable Zn-ion adsorption on the m Ti3C2-S2 MXene and Zn-ion intercalation on the b Ti3C2-S2 MXene. We incrementally increased the number of intercalated Zn ions on the Ti3C2-S2 structure, calculating the adsorption energy for each addition up to a total of 12 ions on m Ti3C2-S2 and 25 ions on b Ti3C2-S2. After intercalating the first 15 Zn ions, we proceeded to intercalate the 16th to 20th Zn ions on the left surface and the 21st to 25th Zn ions on the right surface of the b Ti3C2-S2 MXene. Notably, we observed a significant drop in the OCV after the intercalation of the 15th Zn ion. This abrupt change prompted us to cease further Zn-ion intercalation at this juncture. Subsequently, we concentrated on detailed OCV measurements post-15th ion intercalation to gain insights into the electrochemical stability and the potential limits of the b Ti3C2-S2 structure when utilized as an electrode material in a zinc-ion battery. It is clearly observed from Fig. 3 that the adsorption energy increases linearly with increasing Zn-ion concentration in both m Ti3C2-S2 and b Ti3C2-S2 MXenes. These calculated adsorption energies are detailed in Table 1.
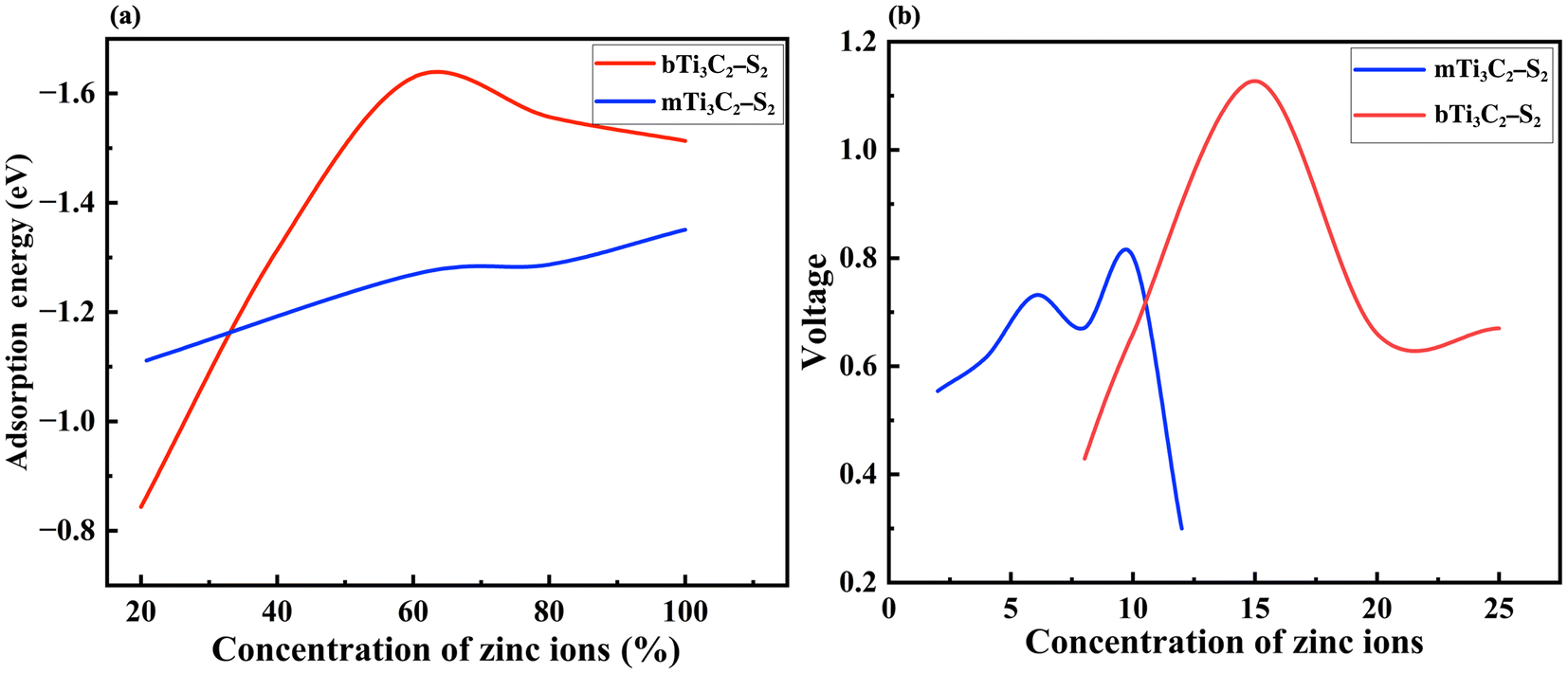 | ||
| Fig. 3 Representations of (a) the adsorption energy versus Zn-ion concentration curve, showing the variation in adsorption energy with Zn-ion concentration for m Ti3C2-S2 and b Ti3C2-S2 MXene structures and (b) the voltage profile of m Ti3C2-S2 and b Ti3C2-S2 MXene structures with respect to Zn-ion concentration. | ||
| Material | Structure + Zn concentration | Adsorption energy (eV) | OCV (V) |
|---|---|---|---|
| m Ti3C2-S2 | m Ti3C2-S2 + 2Zn | −1.1081 | 0.554 |
| m Ti3C2-S2 + 4Zn | −1.1713 | 0.617 | |
| m Ti3C2-S2 + 6Zn | −1.2686 | 0.732 | |
| m Ti3C2-S2 + 8Zn | −1.2868 | 0.671 | |
| m Ti3C2-S2 + 10Zn | −1.3509 | 0.803 | |
| m Ti3C2-S2 + 12Zn | −1.2154 | 0.376 | |
| b Ti3C2-S2 | b Ti3C2-S2 + 5Zn | −0.9882 | 0.421 |
| b Ti3C2-S2 + 10Zn | −1.2434 | 0.891 | |
| b Ti3C2-S2 + 15Zn | −1.3134 | 1.130 | |
| b Ti3C2-S2 + 20Zn | −1.4101 | 0.670 | |
| b Ti3C2-S2 + 25Zn | −1.6293 | 0.663 | |
Similarly, for the m Ti3C2-S2 MXene, we incrementally adsorbed Zn ions, calculating the adsorption energies for each addition, up to a total of 12 Zn ions. Notably, upon the adsorption of the 12th Zn ion, we observed a significant decrease in the OCV. This observation led us to halt further adsorption at this point. Consequently, a total of 10 Zn ions were successfully adsorbed into the m Ti3C2-S2 MXene structure. The average adsorption energy recorded after the adsorption of the 10th Zn ion was found to be −1.3509 eV. A negative adsorption energy indicates that the adsorption process is exothermic and energetically favourable, suggesting proper adsorption. Fig. 3 shows the relationship between Zn-ion concentration and adsorption energies. It is evident from Fig. 3 that as the number of adsorbed ions increases, the adsorption energy becomes more negative, indicating a more favourable adsorption process. This trend signifies that the interactions between the Zn ions and the considered MXene structures are becoming stronger, enhancing the material's capability for efficient ion storage.
3.2 Electronic characteristics
The electronic band structure and projected density of states for the m Ti3C2-S2 and b Ti3C2-S2 MXene structures have also been examined both before and after Zn-ion intercalation. Fig. 4 represents the band structure and PDOS of the m Ti3C2-S2 MXene before and after adsorption, clearly illustrating a zero bandgap in both states. Similarly, Fig. 5 represents the bandgap and PDOS of the b Ti3C2-S2 before and after intercalation, further confirming the material's metallic characteristics. The results indicate that the bandgap is zero in both cases, signifying that the m Ti3C2-S2 and b Ti3C2-S2 MXene structures exhibit metallic behaviour. The absence of a bandgap implies that there are free electrons available for conduction, which is a hallmark of a material's metallic nature and is crucial for high electrical conductivity. The intercalation process, which involves inserting Zn ions between the layers of the MXene, does not alter its metallic nature, further confirming the robustness of its electronic properties. The PDOS analysis supports these findings, showing continuous electronic states at the Fermi level. These insights into the electronic structure of m Ti3C2-S2 and b Ti3C2-S2 MXene structures are vital for their potential use as cathode materials in zinc-ion batteries, where high conductivity and efficient electron transport are essential for optimal battery performance. The metallic nature of both these MXenes ensures rapid electron transfer, contributing to the overall efficiency and rate capability of the zinc-ion battery. | ||
| Fig. 4 Representations of (a) the band structure of the m Ti3C2-S2 MXene before Zn-ion adsorption; (b) PDOS of the m Ti3C2-S2 MXene before Zn-ion adsorption; (c) the band structure of the m Ti3C2-S2 MXene after Zn-ion adsorption; and (d) PDOS of the m Ti3C2-S2 MXene after Zn-ion adsorption. | ||
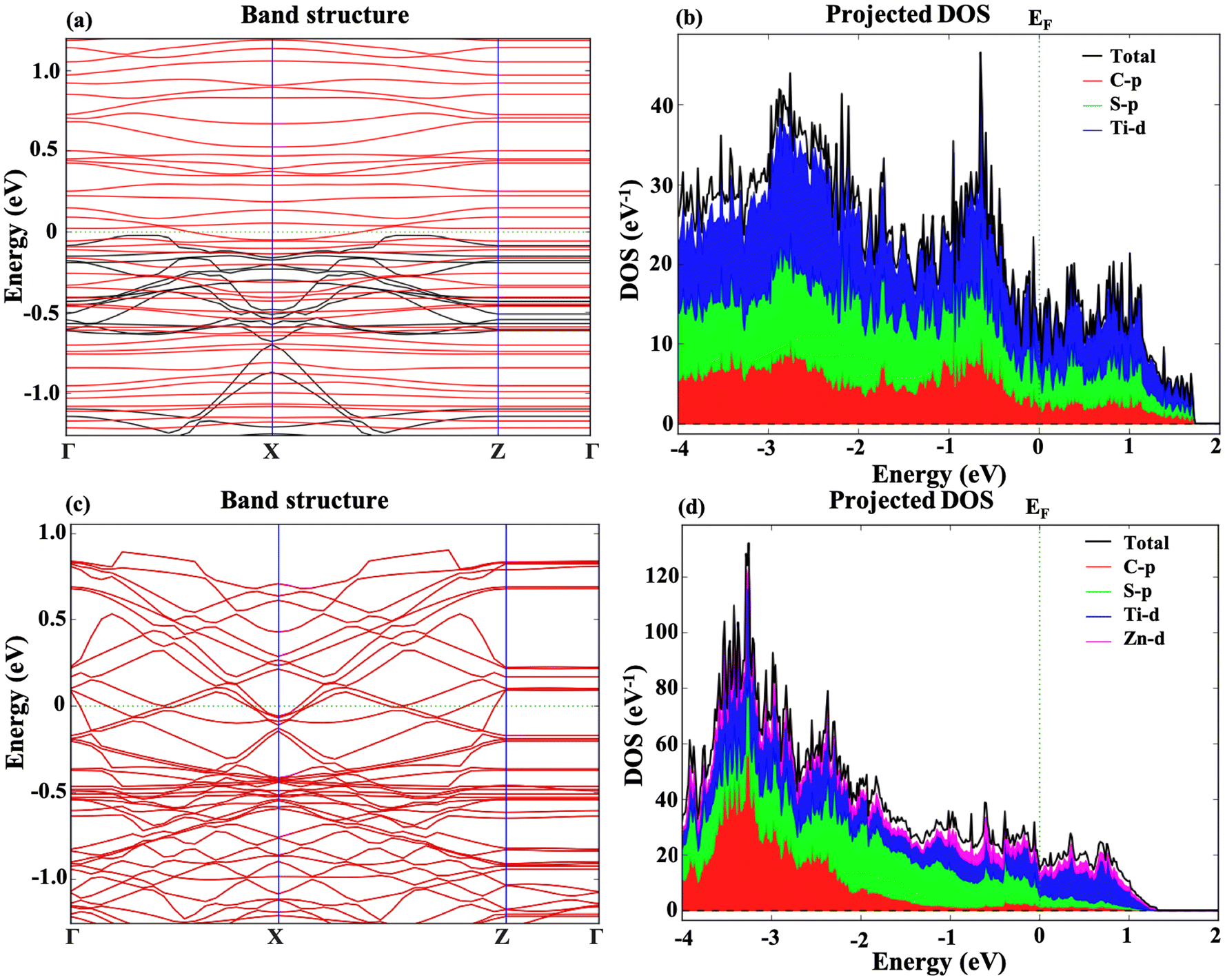 | ||
| Fig. 5 Representations of (a) the band structure of the b Ti3C2-S2 MXene before Zn-ion intercalation; (b) PDOS of the b Ti3C2-S2 MXene before Zn-ion intercalation; (c) band structure of the b Ti3C2-S2 MXene after Zn-ion intercalation; and (d) PDOS of the b Ti3C2-S2 MXene after Zn-ion intercalation. | ||
3.3 Open circuit voltage and theoretical capacity
Here, the open circuit voltage and storage capacity are determined to further assess the electrochemical performance of m Ti3C2-S2 and b Ti3C2-S2 MXene structures for ZIBs. For a more comprehensive understanding of a material's functioning as an electrode, the OCV stands out as an essential parameter for determining whether the system serves as a cathode or anode material. The OCV of an electrode depends on the change in Gibbs free energy (ΔGf) during the ion adsorption process, expressed by the equation OCV = (−ΔGf/2xe), where x denotes the number of ions adsorbed/intercalated on the MXene and e represents the value of the transferred charge. The factor “2” represents the valency of Zn ions. The ΔGf value, defined as ΔGf = ΔEf + PΔV − TΔS, includes changes in energy (ΔEf), volume (ΔV), and entropy (ΔS). By ignoring both volume and entropy effects, the OCV simplifies to OCV = −(ΔEf/2xe), as calculated using the following equation:41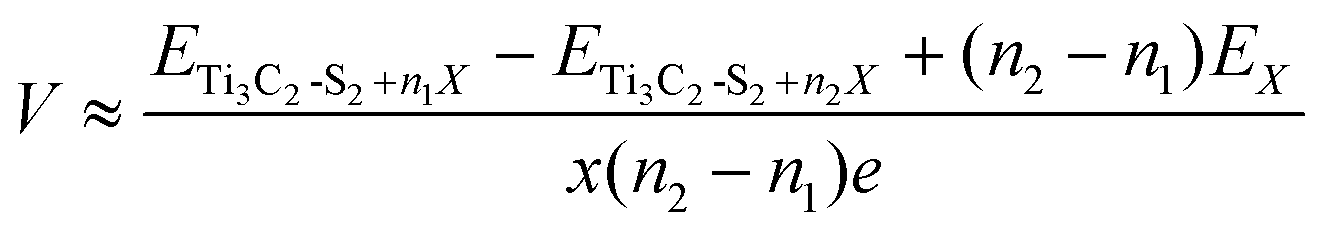 | (2) |
We explored the adsorption of Zn ions into the m Ti3C2-S2 and intercalation on the b Ti3C2-S2 MXene structures, meticulously recording the OCV after Zn-ion adsorption and intercalation on m Ti3C2-S2 and b Ti3C2-S2, respectively. The OCV for the b Ti3C2-S2 MXene was measured at several key points: after the intercalation of 5 ions, the OCV was 0.43 V; after 10 ions, it increased to 0.88 V; and after 15 ions, it reached 1.13 V. The results depicted in Fig. 3(b), demonstrate a consistent increase in OCV correlating with the rising concentration of intercalated Zn ions. This increase is primarily attributed to the enhanced electrochemical potential created by the accumulation of positively charged ions within the b Ti3C2-S2 MXene structure, which elevates the energy state of the system. A significant observation was made upon the intercalation of the 16th Zn ion, where there was an unexpected decrease in OCV from 1.13 V to 0.6 V. The observed sudden drop in OCV, despite the continued favourable adsorption energy, can be attributed to a combination of electronic, electrochemical, and structural factors. Bader charge analysis reveals that beyond the 15th Zn-ion intercalation, charge transfer from the Zn ion to the MXene surface decreases significantly, reducing the effective potential difference and thereby lowering the OCV. This indicates the saturation of active sites, meaning the Zn ion is no longer interacting directly with the MXene, leading to a drop in OCV despite the thermodynamically favourable adsorption energy. However, we have not observed any lattice distortions in the considered MXene structures after optimization beyond the 15th Zn-ion intercalation. Overall, these results demonstrate that OCV behaviour in the b-MXene is not solely influenced by adsorption energy, but is also determined by charge redistribution and other electrochemical factors.
Similarly, for the m Ti3C2-S2 MXene, the computed OCV values are as follows: 0.55 V after the second Zn-ion adsorption, 0.61 V after the fourth, 0.73 V after the sixth, 0.67 V after the eighth, 0.80 V after the 10th, and 0.37 V after the 12th Zn-ion adsorption. These findings indicate that the OCV increases with the concentration of Zn ions up to the 10th adsorption. However, after the 12th Zn-ion adsorption, there is a sudden drop in OCV from 0.80 V to 0.37 V, as shown in Fig. 3(b). This significant decrease suggests that the adsorption process should be halted after the 10th Zn-ion adsorption to avoid electrochemical instability, structural changes or side reactions within the material. Furthermore, the relationship between the OCV and the adsorption energy curve provides deeper insights (refer to Fig. 3(a) and (b)). Initially, the increase in OCV aligns with increasing adsorption energies, indicating efficient ion accommodation within m Ti3C2-S2 and b Ti3C2-S2 MXene layers. This detailed analysis of the OCV in relation to ion adsorption concentration in MXene structures highlights the delicate balance between ion capacity and electrochemical performance. It underscores the importance of optimizing ion adsorption processes to enhance the efficiency and stability of MXene-based energy storage devices, particularly when operating near or at full ion capacity. Understanding these dynamics is crucial for advancing the design and application of next-generation energy storage materials.
It is clearly observed from Fig. 3(a) and (b) that the OCV follows the trend of adsorption energy. Specifically, the OCV increases with increasing adsorption energy with respect to ion intercalation. The adsorption energy becomes more negative with Zn-ion intercalation, indicating a stronger affinity of the material for Zn ions. This stronger binding enhances the stability of the intercalated ions and leads to an increase in OCV. From eqn (2), it is evident that the OCV is directly related to the change in energy. The change in energy resulting from ion binding (i.e., adsorption energy) influences the total Gibbs free energy of the system. As the adsorption energy becomes more negative (indicating stronger binding of the Zn ions), the Gibbs free energy decreases. This reduction in Gibbs free energy contributes to an increase in OCV, with a more negative adsorption energy leading to a more favourable energy state and, consequently, a higher OCV. However, both the OCV and adsorption energy start to decrease after the 15th ion intercalation in the b Ti3C2-S2 MXene structure and after the 12th ion adsorption in the m Ti3C2-S2 MXene structure. The OCV values clearly suggest that both of these MXene structures are suitable for use as cathode materials in ZIBs.
The following equation can be used to calculate the maximum capacity:42
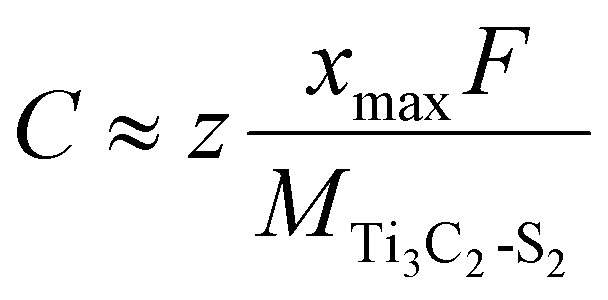 | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801 mAh mol−1), z is the valence number and xmax is the corresponding maximum Zn-ion concentration. MTi3C2-S2 represents the relative molecular mass of the Ti3C2-S2 MXene.
801 mAh mol−1), z is the valence number and xmax is the corresponding maximum Zn-ion concentration. MTi3C2-S2 represents the relative molecular mass of the Ti3C2-S2 MXene.
The maximum theoretical capacity of Zn-intercalated m Ti3C2-S2 and b Ti3C2-S2 MXene structures are 462.5 mAh g−1 and 346.93 mAh g−1, respectively. Our findings reveal that the Zn-ion storage capacities of Ti3C2-S2 MXenes surpass those of the other MXene-based structures documented in the literature (see Table 2).43–48 This suggests a superior performance of Ti3C2-S2 MXenes as electrode materials for Zn-based batteries.
| Ref. | Material structure | Reported capacity (mAh g−1) | OCV (V) | Diffusion barrier (eV) | Bader charge (|q|) |
|---|---|---|---|---|---|
| 52 | V2CTx | 508 | 0.1–1.85 | — | — |
| 43 | RGO@V2O5 | 254.9 | ≈1.5 | — | — |
| 44 | 1T-MoS2/Ti3C2 | 284.3 | — | — | — |
| 45 | VO2/MXene | 445 | ≈1.6 | — | — |
| 46 | H2V3O8/MXene | 346 | ≈1.6 | — | — |
| 47 | 3D porous carbon@VO2 | 388 | ≈1.6 | — | — |
| 48 | Porous carbons@MnFe2O4 | 168 | — | — | — |
| Present work | m Ti3C2-S2 | 462.5 | 0.8 | 0.057 | 0.45 |
| Present work | b Ti3C2-S2 | 346.93 | 1.13 | 0.41 | 1.30 |
Additionally, the Bader charge analysis revealed significant charge transfer values of 0.45|q| for m Ti3C2-S2 and 1.30 |q| for b Ti3C2-S2, which are higher than those in previously reported studies.28,49 This indicates strong interactions between Zn ions and the surface of these MXenes, further supporting their enhanced storage capabilities and confirming their potential as high-performance electrode materials in energy storage applications.50,51
3.4 Diffusion barrier
A promising electrode material should exhibit a low diffusion barrier and high mobility, as these factors significantly influence the charge and discharge rate performance of a battery. In the present study, we have investigated the diffusion barrier for Zn ions on m Ti3C2-S2 and b Ti3C2-S2 MXene structures by using the CI-NEB approach. We defined the initial and final positions (see Fig. 6) of the Zn ions and generated a series of intermediate images between these states. Each of these images was optimized to find the diffusion barrier. In the CI-NEB approach, the image with the highest energy (the transition state) is further optimized to ensure it accurately represents the saddle point, namely the highest energy point along the minimum energy path as shown in Fig. 7. The diffusion barrier values obtained are 0.057 eV for m Ti3C2-S2 and 0.41 eV for b Ti3C2-S2 (see Fig. 7). The lower barrier for m Ti3C2-S2 indicates it is a more promising candidate for efficient Zn-ion diffusion compared to b Ti3C2-S2. Additionally, we observed multiple transition states between the initial and final states in m Ti3C2-S2, suggesting the presence of multiple energy barriers. Furthermore, the end-point energy for both MXenes is not zero. For m Ti3C2-S2, the end-point energy is approximately 0.057 eV, while for b Ti3C2-S2 it is around 0.4 eV. This indicates that the system absorbs some energy from its surroundings, which is reflected in these non-zero end-point energies. The reported diffusion barrier value for m Ti3C2-S2 is much lower than the values in previously reported studies,50,51,53,54 highlighting its superior potential for Zn-ion diffusion. | ||
| Fig. 6 Schematic representation of the top view of the energetically optimized migration pathways of Zn ions on the m Ti3C2-S2 MXene and the b Ti3C2-S2 MXene. | ||
 | ||
| Fig. 7 Diffusion energy profiles for Zn ions in (a) the m Ti3C2-S2 MXene and (b) the b Ti3C2-S2 MXene. | ||
3.5 Ab initio molecular dynamics simulations
To evaluate the structural stability of the m Ti3C2-S2 and b Ti3C2-S2 MXenes, ab initio molecular dynamics (AIMD) simulations were conducted at 500 K for 20 picoseconds and 8000 picoseconds, respectively. The energy analysis from these simulations is illustrated in Fig. 8, which shows the potential energy and total energy of both the considered MXene structures. The results demonstrate that both the potential energy and total energy for the m Ti3C2-S2 and b Ti3C2-S2 systems remain negative and constant throughout the AIMD simulations. This stability in energy levels indicates that the structures do not undergo any significant changes or degradation under the simulated conditions. Specifically, the consistent behaviour of the potential energy and total energy suggests that the atomic configurations of the considered MXene structures are preserved, affirming their structural stability. Moreover, these findings align with the binding energy and formation energy analyses conducted separately in this study. The negative and stable energies observed throughout the simulations further confirm that the considered MXene structures maintain their stability and do not experience destabilization. These insights are crucial for understanding the resilience of m Ti3C2-S2 and b Ti3C2-S2 in practical applications, where structural stability is a key factor in their performance as electrode materials or in other energy storage devices.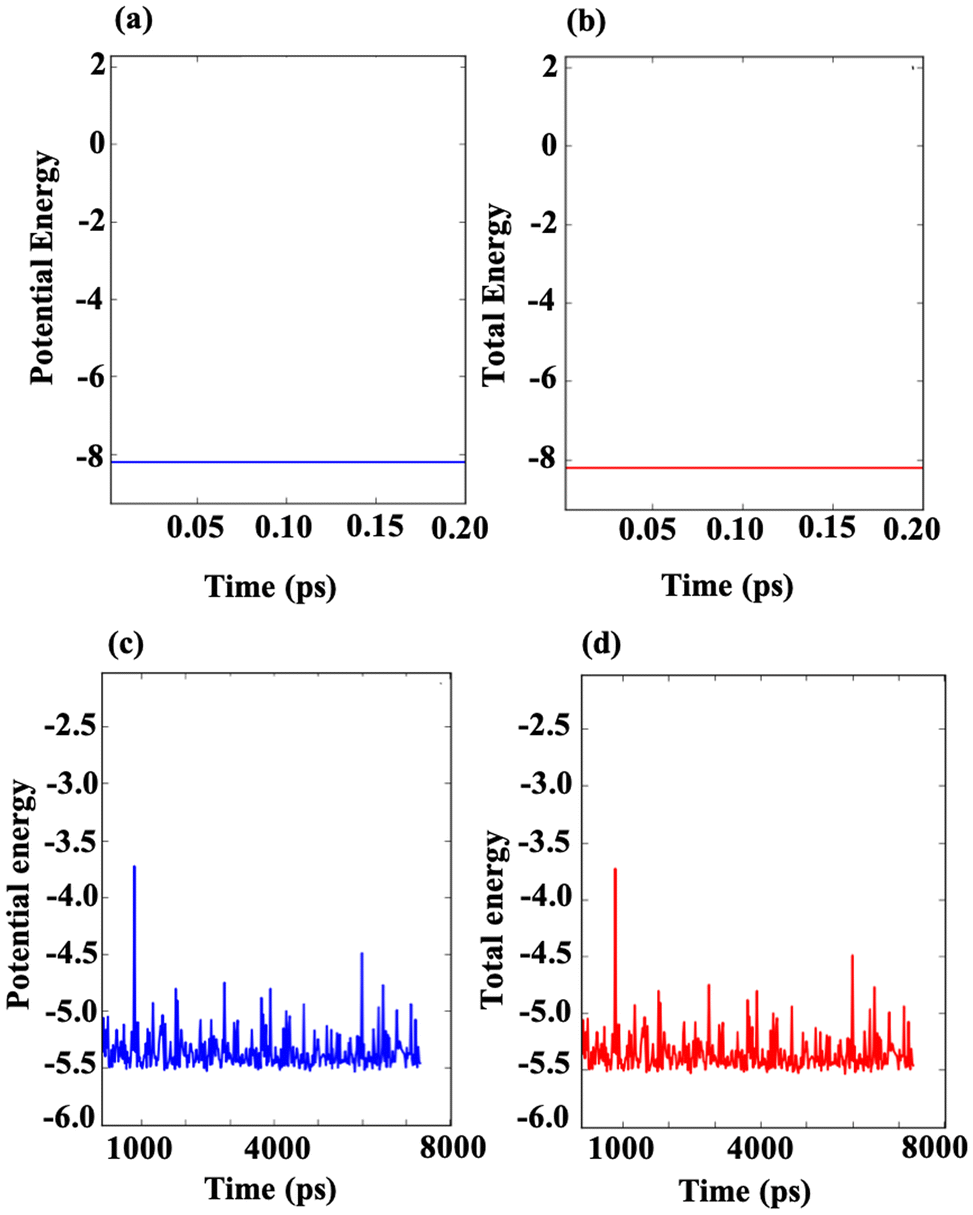 | ||
| Fig. 8 Illustrations of the AIMD energy profiles for the m Ti3C2-S2 MXene and the b Ti3C2-S2 MXene after simulations conducted for 20 picoseconds (a and b) and 8000 picoseconds (c) and (d) at 500 K. | ||
4 Conclusions
In summary, this study explores the electronic and electrochemical energy storage properties of m Ti3C2-S2 and b Ti3C2-S2 MXenes through first-principles calculations. Our findings reveal that both m Ti3C2-S2 and b Ti3C2-S2 MXenes exhibit robust dynamic stability. The electronic structure analysis, including PDOS and band structure calculations, shows that both the considered MXene structures possess strong metallic character, contributing to their excellent conductivity. The theoretical storage capacities are notable, reaching 462.5 mAh g−1 for m Ti3C2-S2 and 346.93 mAh g−1 for b Ti3C2-S2. Additionally, the computed open circuit voltages and Bader charges for m Ti3C2-S2 and b Ti3C2-S2 are 1.13 V and 0.8 V, 0.45 |q| and 1.30 |q|, respectively. Low diffusion barrier values of 0.057 eV and 0.41 eV for m Ti3C2-S2 and b Ti3C2-S2 MXene structures, respectively, indicate rapid and efficient Zn-ion transport. Their structural stability, confirmed through ab initio molecular dynamics simulations, further supports their potential for long-term use in practical applications. Overall, these Ti3C2-S2 MXenes, with their favourable OCVs, high storage capacities, low diffusion barriers, high charge transfer and robust stability, represent highly promising cathode materials for zinc-ion batteries. This study not only underscores their potential in high-performance energy storage, but also lays the groundwork for future experimental and theoretical investigations into 2D MXenes.Author contributions
Shweta Meena and Sunita Saharan conceptualized the idea and wrote the original draft. Shweta Meena supervised the project. Sunita Saharan conducted all the simulations. Umesh Ghanekar reviewed and edited the manuscript.Data availability
Data will be made available on reasonable request to the corresponding author.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to the Department of Electronics and Communication Engineering, NIT Kurukshetra, India, for providing the computational resources essential for this work.References
- G. Fang, J. Zhou, A. Pan and S. Liang, ACS Energy Lett., 2018, 3, 2480–2501 CrossRef CAS.
- S. Ponnada, M. S. Kiai, R. Krishnapriya, R. Singhal and R. K. Sharma, Energy Fuels, 2022, 36, 6013–6026 CrossRef CAS.
- W. Lu, C. Zhang, H. Zhang and X. Li, ACS Energy Lett., 2021, 6, 2765–2785 CrossRef CAS.
- T. Wang, C. Li, X. Xie, B. Lu, Z. He, S. Liang and J. Zhou, ACS Nano, 2020, 14, 16321–16347 CrossRef CAS PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Sci. Adv., 2020, 6, eaba4098 CrossRef CAS PubMed.
- Y. Liang, C.-Z. Zhao, H. Yuan, Y. Chen, W. Zhang, J.-Q. Huang, D. Yu, Y. Liu, M.-M. Titirici and Y.-L. Chueh, et al. , InfoMat, 2019, 1, 6–32 CrossRef CAS.
- Y. Mao, B. Zhao, J. Bai, P. Wang, X. Zhu and Y. Sun, Nanoscale, 2024, 16, 5042–5059 RSC.
- L. Zhang, J. Shi, K. Niu, P. Jia, Y. Gao and G. Gao, ACS Appl. Nano Mater., 2023, 6, 20812–20822 CrossRef CAS.
- N. Wang, H. Wan, J. Duan, X. Wang, L. Tao, J. Zhang and H. Wang, Mater. Today Adv., 2021, 11, 100149 CrossRef CAS.
- C. Li, X. Xie, S. Liang and J. Zhou, Energy Environ. Mater., 2020, 3, 146–159 CrossRef CAS.
- X. Zhang, Y. Tang, P. He, Z. Zhang and T. Chen, Carbon, 2021, 172, 207–213 CrossRef CAS.
- A. Konarov, N. Voronina, J. H. Jo, Z. Bakenov, Y.-K. Sun and S.-T. Myung, ACS Energy Lett., 2018, 3, 2620–2640 CrossRef CAS.
- G. Yang and H. Wan, Front. Chem., 2022, 10, 1000337 CrossRef CAS PubMed.
- F. Jing, J. Pei, Y. Zhou, Y. Shang, S. Yao, S. Liu and G. Chen, J. Colloid Interface Sci., 2022, 609, 557–565 CrossRef CAS PubMed.
- D. Batyrbekuly, S. Cajoly, B. Laïk, J.-P. Pereira-Ramos, N. Emery, Z. Bakenov and R. Baddour-Hadjean, ChemSusChem, 2020, 13, 724–731 CrossRef CAS PubMed.
- W. Li, X. Jing, K. Jiang and D. Wang, ACS Appl. Energy Mater., 2021, 4, 2797–2807 CrossRef CAS.
- N. An, J. Xin, W. Li, Z. Guo, L. Shang, Y. He, L. Lv, D. Sun, Y. Zhang and Z. Hu, Appl. Surf. Sci., 2022, 599, 153881 CrossRef CAS.
- X. Li, X. Xie, R. Lv, B. Na, B. Wang and Y. He, Energy Technol., 2019, 7, 1801092 CrossRef.
- F. Guo, S. Gao, C. Ji, H. Mi, H. Li, W. Zhang and H. Pang, Solid State Ionics, 2021, 364, 115612 CrossRef CAS.
- F. Tang, J. Gao, Q. Ruan, X. Wu, X. Wu, T. Zhang, Z. Liu, Y. Xiang, Z. He and X. Wu, Electrochim. Acta, 2020, 353, 136570 CrossRef CAS.
- M. Xia, X. Zhang, T. Liu, H. Yu, S. Chen, N. Peng, R. Zheng, J. Zhang and J. Shu, Chem. Eng. J., 2020, 394, 124923 CrossRef CAS.
- Q. Zong, Y. Zhuang, C. Liu, Q. Kang, Y. Wu, J. Zhang, J. Wang, D. Tao, Q. Zhang and G. Cao, Adv. Energy Mater., 2023, 13, 2301480 CrossRef CAS.
- Y. Tong, S. Su, X. Li, B. Liang, J. Peng, J. Hou and M. Luo, J. Power Sources, 2022, 528, 231226 CrossRef CAS.
- H. Liu, L. Jiang, B. Cao, H. Du, H. Lu, Y. Ma, H. Wang, H. Guo, Q. Huang and B. Xu, et al. , ACS Nano, 2022, 16, 14539–14548 CrossRef CAS PubMed.
- W. Yang, L. Mou, B. Xiao, J. Chen, D. Wang, S. Peng and J. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 51231–51240 CrossRef CAS PubMed.
- S. Saharan, U. Ghanekar and S. Meena, ChemistrySelect, 2022, 7, e202203288 CrossRef CAS.
- P. P. Dinda and S. Meena, J. Phys.: Condens. Matter, 2021, 33, 175001 CrossRef CAS PubMed.
- S. Saharan, U. Ghanekar, B. R. Shivankar and S. Meena, J. Phys. Chem. C, 2024, 128, 12840–12848 CrossRef CAS.
- Y. Zhang, Z. Cao, S. Liu, Z. Du, Y. Cui, J. Gu, Y. Shi, B. Li and S. Yang, Adv. Energy Mater., 2022, 12, 2103979 CrossRef CAS.
- Z. Yu, X. Wang, X. He, R. Li, A. Zhang and J. Zhang, ACS Appl. Mater. Interfaces, 2024, 16, 69187–69198 CrossRef CAS PubMed.
- L. Tan, C. Wei, Y. Zhang, Y. An, S. Xiong and J. Feng, Chem. Eng. J., 2022, 431, 134277 CrossRef CAS.
- J. Li, C. Wang, Z. Yu, Y. Chen and L. Wei, Small, 2024, 20, 2304543 CrossRef CAS PubMed.
- M. Qi, F. Li, Z. Zhang, Q. Lai, Y. Liu, J. Gu and L. Wang, J. Colloid Interface Sci., 2022, 615, 151–162 CrossRef CAS PubMed.
- Y. Liu, Y. Mei, W. Xu, M. Zhang, J. Hou and Y. Dong, J. Alloys Compd., 2023, 957, 170397 CrossRef CAS.
- Y. Feng, Y. Feng, Y. Zhang, L. Sun, X. Li, M. Meng, Y. Zhu and K. Liu, J. Power Sources, 2022, 545, 231944 CrossRef CAS.
- M. Naguib, J. Halim, J. Lu, K. M. Cook, L. Hultman, Y. Gogotsi and M. W. Barsoum, J. Am. Chem. Soc., 2013, 135, 15966–15969 CrossRef CAS PubMed.
- J. Hu, B. Xu, S. A. Yang, S. Guan, C. Ouyang and Y. Yao, ACS Appl. Mater. Interfaces, 2015, 7, 24016–24022 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- K. A. Papadopoulou, A. Chroneos and S.-R. G. Christopoulos, J. Phys. Chem. Solids, 2022, 166, 110713 CrossRef CAS.
- P. Gong, X. Zhang, F. Liu, K. Yao and S. Zhu, Surf. Interfaces, 2022, 32, 102091 CrossRef CAS.
- X. Zhang, J. Hu, Y. Cheng, H. Y. Yang, Y. Yao and S. A. Yang, Nanoscale, 2016, 8, 15340–15347 RSC.
- X. Zhong, Z. Kong, Q. Liu, C. Yang, Y. Chen, J. Qiu and L. Zang, ACS Appl. Mater. Interfaces, 2023, 15, 58333–58344 CrossRef CAS PubMed.
- F. Long, Q. Zhang, J. Shi, L. Wen, Y. Wu, Z. Ren, Z. Liu, Y. Hou, K. Mao and K. Niu, et al. , Chem. Eng. J., 2023, 455, 140539 CrossRef CAS.
- W. Kou, L. Yu, Q. Wang, Y. Yang, T. Yang, H. Geng, X. Miao, B. Gao and G. Yang, J. Power Sources, 2022, 520, 230872 CrossRef CAS.
- P. Liang, K. Zhu, Y. Rao, H. Zheng, Z. Kong, J. Chen, J. Liu, K. Yan and J. Wang, J. Electroanal. Chem., 2023, 951, 117931 CrossRef CAS.
- C. Wang, M. Wang, L. Liu and Y. Huang, ACS Appl. Energy Mater., 2021, 4, 1833–1839 CrossRef CAS.
- X. Liu, X. Shen, T. Chen and Q. Xu, J. Alloys Compd., 2022, 904, 164002 CrossRef CAS.
- Y. Zhang, J. Xu, C. Liu, H. Cheng, X. Cai, D. Jia and H. Lin, J. Power Sources, 2022, 549, 232075 CrossRef CAS.
- J. Zhang, X. Lu, J. Zhang, H. Li, B. Huang, B. Chen, J. Zhou and S. Jing, Front. Chem., 2021, 9, 677620 CrossRef CAS PubMed.
- Y. Ding, Q. Deng, C. You, Y. Xu, J. Li and B. Xiao, Phys. Chem. Chem. Phys., 2020, 22, 21208–21221 RSC.
- X. Li, M. Li, Q. Yang, H. Li, H. Xu, Z. Chai, K. Chen, Z. Liu, Z. Tang, L. Ma, Z. Huang, B. Dong, X. Yin, Q. Huang and C. Zhi, ACS Nano, 2020, 14, 541–551 CrossRef CAS PubMed.
- S. Tang, C. Wang, X. Pu, X. Gu and Z. Chen, Acta Phys.-Chim. Sin., 2023, 39, 2212037 Search PubMed.
- L. Zhang, J. Shi, K. Niu, P. Jia, Y. Gao and G. Gao, ACS Appl. Nano Mater., 2023, 6, 20812–20822 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |