
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProper aggregation of Pt is beneficial for the epoxidation of styrene by O2 over Ptx/γ-Al2O3 catalysts†
Fengfeng
Li‡
,
Chenyang
Shen‡
,
Yu
He
,
Haoyu
Lu
,
Rongtian
Gu
 ,
Jun
Yao
,
Zhewei
Zhang
,
Feifei
Mei
,
Taotao
Zhao
,
Xiangke
Guo
,
Nianhua
Xue
and
Weiping
Ding
*
,
Jun
Yao
,
Zhewei
Zhang
,
Feifei
Mei
,
Taotao
Zhao
,
Xiangke
Guo
,
Nianhua
Xue
and
Weiping
Ding
*
Key Lab of Mesoscopic Chemistry, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China. E-mail: dingwp@nju.edu.cn
First published on 7th February 2025
Abstract
The dispersion of metal catalysts has multiple effects on catalytic performance, and higher dispersions do not necessarily imply better performance. Herein, we report the epoxidation reaction of styrene over supported platinum catalysts as an example. Compared with the Pt1/γ-Al2O3 catalyst, the Ptn/γ-Al2O3 catalyst with a larger Pt cluster size showed a much better performance. Combining the results of various characterizations and density functional theory calculations, Ptn/γ-Al2O3 was found to be more favorable for oxygen adsorption and activation to generate singlet oxygen species, further promoting the styrene oxidation reaction to styrene oxide in terms of kinetics. In contrast the metallic center of Pt1 in Pt1/γ-Al2O3 was too small to efficiently activate the diatomic oxygen molecule. These insights provide valuable guidance for designing high-performance metal catalysts.
1 Introduction
Supported precious metal catalysts offer a broad spectrum of applications in the modern chemical industry, environmental protection, and energy technology. Their effectiveness is closely tied to structural features including particle size, composition, morphology, and metal–support interactions.1 Particle size, in particular, can fundamentally influence the catalytic performance and availability of active sites within these catalysts.2,3 Recent developments have highlighted the potential of single-atom catalysts (SACs) for their nearly complete surface exposure of active atoms, which can directly facilitate catalytic reactions.4–6 However, despite their advantages, the instability and structural variability of SACs limit their practical applications. In response, further research is needed to determine whether single-atom catalysts, nanoparticles or cluster catalysts are more effective for a particular reaction.7 By carefully adjusting the size,8,9 shape,10,11 composition,12,13 and surface modifications,14,15 researchers can significantly alter the electronic properties and surface activity of the catalysts. Such enhancements would not only capitalize on the benefits of extreme dispersion but also improve stability and customization, fostering synergistic interactions at the interface between the active sites and reactants.16,17 However, achieving uniformity and predictability in the distribution of active sites of supported precious metal catalysts remains challenging despite the advancements made in nanoparticle- and cluster-catalysts in the past decades. Nevertheless, addressing this issue is crucial for maximizing the catalytic efficiency and selectivity in practical applications.Epoxidation reactions, such as the transformation of styrene into styrene oxide, are crucial for producing essential industrial chemicals used in products like surfactants and resins.18 The catalytic oxidation of styrene through molecular oxygen presents an environmentally friendly and economically feasible alternative to traditional peroxide-based methods. In this process, molecular oxygen is activated and then converted to reactive oxygen species (ROS), and this procedure is vital for supported heterogeneous catalysts to effectively catalyze the epoxidation of styrene under aerobic conditions.19 Although many advancements have improved ROS generation, these reactions typically require harsh conditions, such as high temperature or photothermal synergy.20–22 Multiple mechanisms have been proposed for molecular oxygen activation. One involves the absorption of molecular oxygen at the oxygen vacancies (Ov) on defective oxide supports.23–25 Alternatively, the adsorption-dissociation process primarily occurs on noble metal surfaces.26–28 In this context, platinum (Pt) catalysts are particularly effective owing to their outstanding catalytic activity and efficiency in promoting these activation mechanisms. In general, the particle size of platinum is critical to its catalytic activity since the particle size dictates the amount of active sites on the catalytic surface, thereby influencing the adsorption and activation efficiency of oxygen molecules. Recent studies have underscored the importance of particle size in catalysis, highlighting how its variations can influence the electronic properties and chemical reactivity of the catalyst.29–31 These studies emphasize the significance of the catalyst size in the activation of molecular oxygen, which is essential for enhancing the epoxidation of styrene and a critical step in this reaction.
Herein, we adopted a facile and effective impregnation strategy to construct catalysts with different Pt sizes to explore the species that can promote the generation of more singlet oxygen species and have a higher selectivity of styrene epoxidation. These Pt catalysts are composed of atomically dispersed Pt at the γ-Al2O3 support (labelled as Pt1/γ-Al2O3) and nanocluster Pt at the γ-Al2O3 support (denoted as Ptn/γ-Al2O3). The latter catalyst in styrene oxidation, in which the dispersion and interaction of Pt with the support is modified by the presence of oleic acid during preparation, shows higher selectivity and activity with reduced metal–support interaction, achieving 81.8% styrene conversion ratio and 70.1% styrene oxide (SO) selectivity, which is more stable compared with the Pt1/γ-Al2O3 catalyst. According to the characterizations using X-ray absorption spectroscopy (XAS), aberration-corrected transmission electron microscopy (AC-TEM), as well as density functional theory (DFT) calculations, Pt in Pt1/γ-Al2O3 or Ptn/γ-Al2O3 catalysts is atomically dispersed or in nanoclusters, respectively. The oxygen activation mechanism over the two catalysts was analyzed through oxygen temperature-programmed desorption (TPD) and in situ electron paramagnetic resonance (EPR) measurements, revealing that Pt particle size can influence oxygen activation and overall catalytic performance.
2 Results and discussion
Ptx/γ-Al2O3 catalyst texture
Both Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts were prepared using the incipient wetness impregnation method.32 Generally, precisely controlling the volume of the precursor solution ensures a thorough coating of the support. Following impregnation, the materials are subjected to drying and calcinating to achieve the final catalyst. For the synthesis of Ptn/γ-Al2O3, an additional step of surface modification is necessary, which alters the interactions between platinum and the support (Fig. S1, ESI†).33 As shown in Table S1,† the practical loadings of platinum are 0.77 and 0.82 wt% for Pt1/γ-Al2O3 and Ptn/γ-Al2O3, respectively, analyzed by ICP, which are close to the theoretical amount (0.80 wt%). Also, compared with bare γ-Al2O3, sufficient pore volume (∼0.50 cm3 g−1) with a high surface area (∼130 m2 g−1) is exhibited by both catalysts in Fig. S3.† Based on the XRD patterns in Fig. S2,† all diffraction peaks of the two catalysts can be indexed to γ-Al2O3 (PDF # 10-0425). The absence of a discernible phase of the platinum-related species is due to the low content and high dispersion of Pt species. HR-TEM images and the corresponding EDS elemental mappings of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 are shown in Fig. 1. Both the catalysts show similar morphology as the γ-Al2O3 support used. For the Pt1/γ-Al2O3 catalyst (in Fig. 1a), no apparent trace of any Pt species can be identified, but the Ptn/γ-Al2O3 catalyst (in Fig. 1b) gives evident aggregation of Pt as nanoparticles (NPs) loaded on alumina. The EDS elemental mappings (in Fig. 1c and d) confirm the different dispersion states of the two catalysts. | ||
| Fig. 1 (a) HR-TEM images of Pt1/γ-Al2O3 and (b) Ptn/γ-Al2O3; EDX elemental map distributions of Pt, Al and O of (c) Pt1/γ-Al2O3 and (d) Ptn/γ-Al2O3. | ||
Due to the intrinsic interactions between the Pt and γ-Al2O3, the Pt species disperse atomically on the surface of the support (Fig. 2a and b and Fig. S4†). For Ptn/γ-Al2O3, Pt NPs can be observed at low magnification in Fig. 2d, revealing that the weakened metal–support interactions induced the aggregation of Pt atoms. Subsequent statistical analysis also confirmed that the average size of Pt NCs is ca. 3.5 nm. Furthermore, the lattice fringes of 0.22 nm are observed in the metal particle in Ptn/γ-Al2O3 (Fig. 1b and 2e), which corresponds to the {111} plane of PtO2. The images confirm that Pt species in the Pt1/γ-Al2O3 catalyst is atomically dispersed on the γ-Al2O3 support surface, while in the Ptn/γ-Al2O3 catalyst, Pt aggregates into nanoparticles and are evenly dispersed on the support.
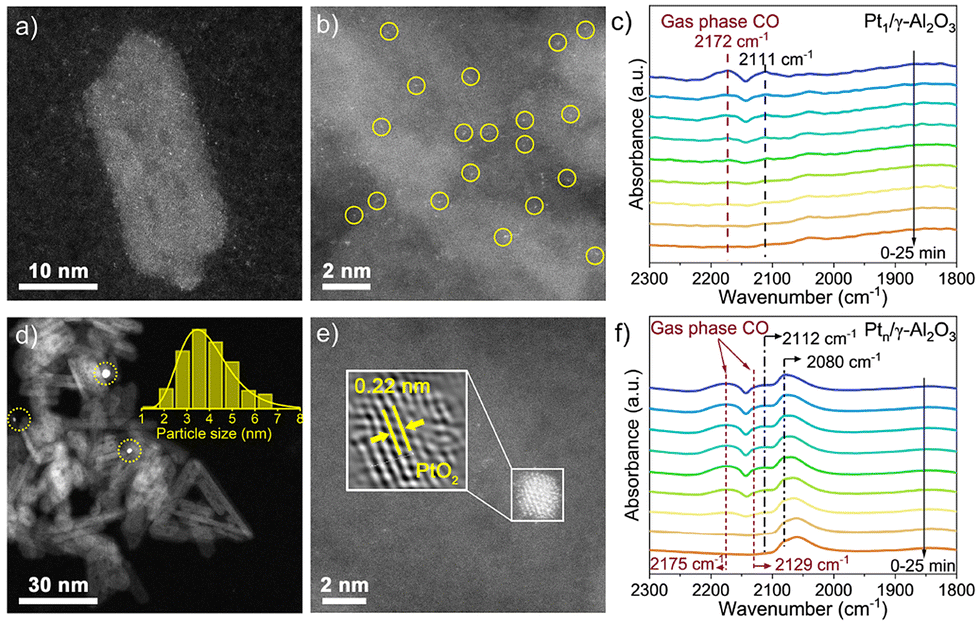 | ||
| Fig. 2 AC-HAADF-STEM images of Ptx/γ-Al2O3 samples. (a and b) Pt1/γ-Al2O3; (d and e) Ptn/γ-Al2O3; in situ CO DRIFTS spectra of Ptx/γ-Al2O3 samples. (c) Pt1/γ-Al2O3 and (f) Ptn/γ-Al2O3. | ||
In situ CO diffuse reflectance infrared Fourier transform spectroscopy (CO-DRIFTS) was used to characterize the electronic properties of the two catalysts. The CO band position can be influenced by the particle sizes, surface coverage, and interaction with the support. Fig. 2 shows the CO-DRIFTS spectra of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts. Two evident distinctive peaks at 2172 and 2175 cm−1 in the two catalysts are attributed to gaseous CO.34 For the Pt1/γ-Al2O3 catalyst, discernible peaks within the range of 1700 to 2300 cm−1 distinctly signify the vibrational and stretching modes associated with the adsorption of CO by Pt sites (Fig. 2c).35 Moreover, the singular and faint adsorption band manifested at a wavenumber of 2111 cm−1 implies that CO is linearly bonded to the atomically dispersed Pt sites with a positive charge.36,37 The CO-DRIFTS spectrum of Ptn/γ-Al2O3 catalysts shown in Fig. 2f, except for CO gas adsorption, manifests a novel band positioned at 2080 cm−1, indicating CO adsorption on the positively charged Ptδ+ species, which is a discernible state exclusively presented at the Pt–O-metal interfacial site.38–40 Throughout the whole purging process, the intensity of these peaks remains consistently stable, revealing the enduring stability of the CO adsorption state on the Pt nanocluster surface. In addition, the CO-DRIFTS results of both the Pt catalysts show the oxidation state of Pt with positive valence.
Further insights into the structure of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts were acquired by X-ray absorption fine structure (XAFS) experiments. Pt L3-edge X-ray absorption near-edge structure (XANES) spectra shown in Fig. 3a were used to determine the valence state of the Pt species. The corresponding reference spectra of Pt1/γ-Al2O3, Ptn/γ-Al2O3, as well as PtO2 and Pt foil, are plotted. Thereinto, the Ptx/γ-Al2O3 peak (white line) is located between the PtO2 and Pt foil peaks, which indicates that both atomically dispersed Pt atoms and NPs species are in the form of oxidized platinum. The intensity of the white line for each Pt catalyst is very similar to that of PtO2, with a consistent oscillation shape.41 However, the white line intensity of the Pt nanocluster is higher than that of the Pt SAC, which is shown in an enlarged view in the inset of Fig. 3a, indicating that Pt nanoparticles have a higher valence state, which is consistent with the XPS results. Owing to the overlapping of the Pt 4f peaks with Al 2p,42 the Pt 4d binding energies were used for the analysis (Fig. S5†). In the two Pt catalysts, Pt species are detected with Pt2+ and Pt4+ valence states, while the ratio of Pt4+/Pt2+ in Pt1/γ-Al2O3 is significantly higher than that in Ptn/γ-Al2O3. Notably, the oxidized Ptδ+ peak in the Pt 4d binding energy of Pt1/γ-Al2O3 distinctly shifts approximately 0.3 eV lower than that of Ptn/γ-Al2O3. This shift implies electron transfer from the Pt atoms to alumina, attributed to their strong interatomic interactions. It is also proven by the Al 2p peak shown in Fig. S5a.†
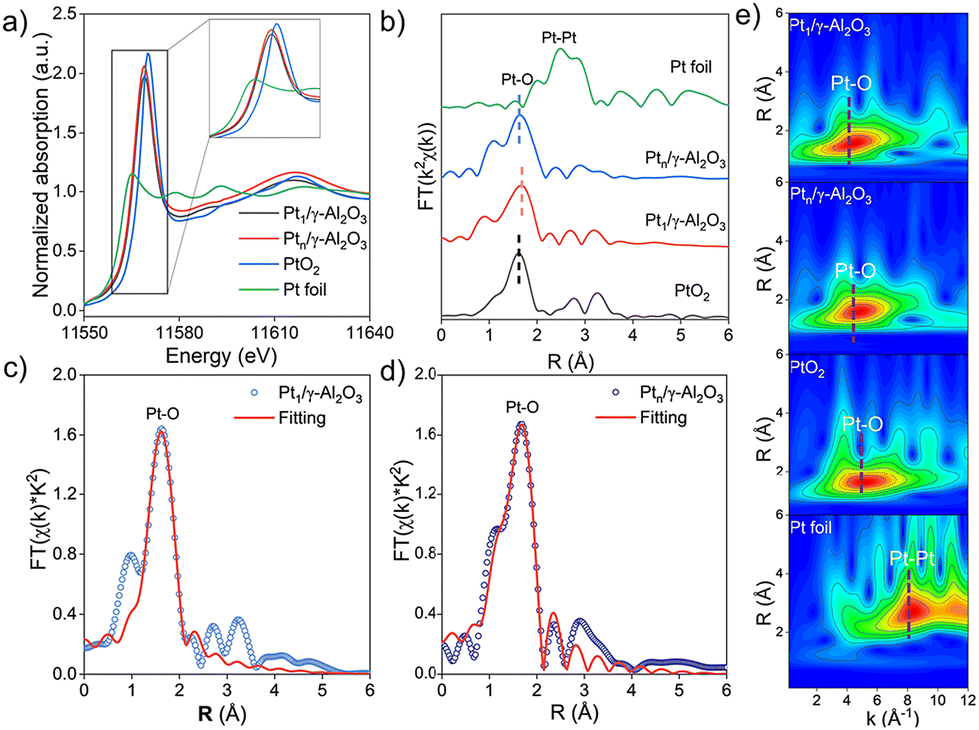 | ||
| Fig. 3 (a) Pt L3-edge X-ray absorption near-edge structure; (b) Fourier-transform EXAFS spectra of Pt1/γ-Al2O3, Ptn/γ-Al2O3, and the corresponding references; (c and d) Fourier transform EXAFS fitting spectrum of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 at R space; (e). Wavelet transform EXAFS of Pt1/γ-Al2O3, Ptn/γ-Al2O3, PtO2 and Pt foil. | ||
Further, we used Fourier-transformed (FT) k2-weighted extended X-ray absorption fine structure (EXAFS) spectra to analyze the bond lengths and coordination number of Pt species (Fig. 3b). In both Pt1/γ-Al2O3 and Ptn/γ-Al2O3, sharp peaks corresponding to the Pt–O layer of coordination at 1.64 Å and 1.68 Å, respectively, are observed, similar to the PtO2 reference.43 The least squares method was used to fit the EXAFS spectra to quantify the coordination number of the Pt species (Fig. 2d, e and Table S2†). In both Pt catalysts, there is no Pt–Pt bond, but the Pt–O bond exists, while the Pt–O coordination number is ∼5.3 at a distance of 2 Å for Pt1/γ-Al2O3 or ∼6 at a distance of 1.93 Å for Ptn/γ-Al2O3, respectively. The wavelet transform (WT) analysis is a powerful technique to discern the scattering atoms and provide resolutions in both R-space and k-space. In Fig. 3e, the WT-EXAFS spectrum of Pt1/γ-Al2O3 exhibited a peak at 1.5 Å in the R space and 4.4 Å−1 in the k-space, similar to the Ptn/γ-Al2O3 catalyst, which showed a maximum at 1.6 Å in the R space and 4.3 Å−1 in the k-space, attributable to Pt–O bonds.44 In contrast, the WT-EXAFS spectrum of the Pt foil displayed a peak at 2.7 Å inthe R space and 8.3 Å−1 in the k-space in the WT-EXAFS spectrum, corresponding to Pt–Pt bonds.45 This indicates that Pt atoms were only bonded to oxygen in both catalysts.
Combined with the above systematic characterization analyses, it is discerned that the Pt atoms in both catalysts are in the oxidized state. Notably, the Pt1/γ-Al2O3 catalyst was highly dispersed in the form of a single atom, whereas the Ptn/γ-Al2O3 catalyst was a nanocluster-type.
Catalytic performance
The catalytic properties of the Pt1/γ-Al2O3 and Ptn/γ-Al2O3 samples were explored using styrene epoxidation using O2 as the oxidant. Initial optimization revealed that Pt-based catalysts oxidize solvents and reactants simultaneously due to their strong oxidation capabilities. N,N-Dimethylformamide (DMF), reported to be an effective solvent for achieving the epoxidation of styrene,46,47 was utilized to assess the oxidation performance at different temperatures (Fig. S7a and S7b†). However, its strong interaction with the Pt center caused catalyst passivation and low styrene conversion, indicating its unsuitability. Subsequently, other solvents were tested, and it showed that 1,4-dioxane was the best solvent for the reaction (Fig. S7c†). Mannitol was added to prevent the oxidation of 1,4-dioxane at elevated temperatures.19 To exclude the effect of mass transfer on the reaction kinetics,48,49 we conducted a series of experiments at different stirring speeds (ranging from 100 rpm to 500 rpm) to assess the effect on catalytic performance. As shown in Fig. S6,† the styrene conversion rate first increased with the stirring speed, reaching a maximum at 300 rpm, and then decreased at higher stirring. Nevertheless, the selectivity of SO mainly decreased with the increase in stirring speed, indicating a shift in the reaction pathway at stirring speeds higher than 300 rpm, which could be due to the disturbance in oxygen transfer and adsorption. Therefore, it was speculated that the reaction kinetics are not significantly affected by mass transfer at 300 rpm, and this stirring speed was chosen for further optimization of the reaction conditions. Fig. 4a and b illustrate that the reaction temperature can influence the catalytic performance for styrene epoxidation after 10 h. An increase in both styrene conversion ratio and SO selectivity was observed as the reaction temperature rose. Notably, the performance of the Ptn/γ-Al2O3 catalyst consistently surpassed that of the Pt1/γ-Al2O3 catalyst. As shown in Fig. 4c and d, reaction time can also obviously influence the styrene conversion ratio and SO selectivity, and in this procedure, Ptn/γ-Al2O3 maintains superior catalytic activity over the Pt1/γ-Al2O3 catalyst. Moreover, as the reaction time extended under 100 °C, the optimum temperature, the reaction rate of both Pt catalysts showed an increasing trend. After 10 hours of the reaction, the Ptn/γ-Al2O3 catalyst shows the best catalytic activity, achieving 81.8% styrene conversion and 70.1% selectivity for SO, surpassing the Pt1/γ-Al2O3 catalyst, which reached a 65.3% of styrene conversion ratio and 51.5% of SO selectivity. The turnover frequency value for Ptn/γ-Al2O3 was confirmed to be 155 h−1, which is much higher than that of Pt1/γ-Al2O3 (99 h−1) in a 3 h reaction (Fig. 4e). The reusability tests showed that Ptn/γ-Al2O3 maintained high selectivity and activity over five cycles (Fig. 4f). X-ray diffraction (XRD) patterns of the used catalysts showed no significant changes compared with fresh catalysts, confirming the structural integrity (Fig. S8a†). Furthermore, the size of the Pt nanoparticles remained consistent, as evidenced by the high-resolution transmission electron microscopy (HR-TEM) images (Fig. S8b†). | ||
| Fig. 4 Catalytic performance of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 as a function of (a and b) temperature and (c and d) time; (e) calculated TOF values of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 for the oxygen of styrene; (f) cyclic stability of Ptn/γ-Al2O3. Reaction conditions: 4.33 mmol styrene, 100 mg catalysts, 20 mL 1,4-dioxane, 1 atm oxygen. | ||
Kinetics and mechanism
A kinetic study was performed to clarify the kinetic behaviors and Arrhenius plots for styrene epoxidation on Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts. According to the various reaction data obtained by the mass transfer and controlling the reaction temperature, the kinetics analysis for the Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts were processed, which can be linearly plotted against reaction time, as illustrated in Fig. 5a and b. Here, the application of pseudo-first-order kinetics is suitable for assessing the catalytic rate.50,53 The kinetic equation of this epoxidation reaction may be formulated as follows (eqn (1)):51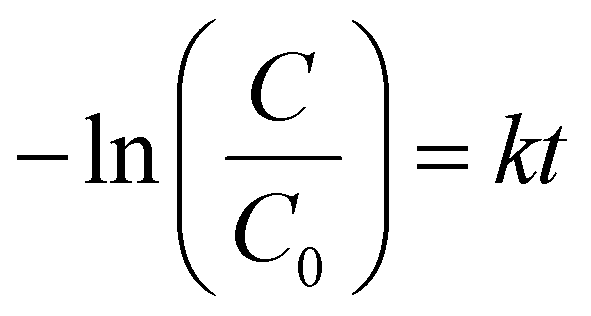 | (1) |
 | ||
| Fig. 5 Kinetic study of styrene epoxidation by Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts. (a and b) ln(C/C0) versus reaction time; (c) Arrhenius plots and the calculated apparent activation barriers of styrene epoxidation for Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts. | ||
Based on the results of kinetic analysis, the apparent reaction constants (k) were used to calculate the apparent activation energy (Ea) in the Arrhenius equation. Ea can be determined based on the Arrhenius equation (eqn (2)):53
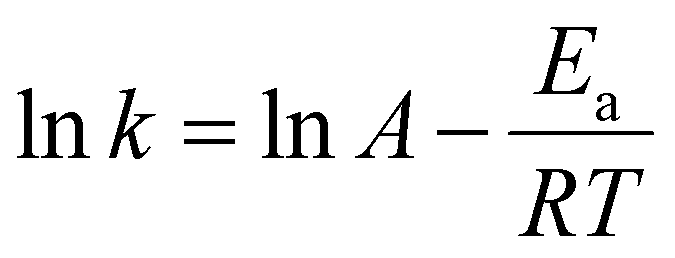 | (2) |
Compared with Pt1/γ-Al2O3, Ptn/γ-Al2O3 exhibits a higher reaction rate and lower activation energy, as shown in Table S3.† This result further demonstrates that the Ptn/γ-Al2O3 catalyst significantly enhances the reaction rate, which is conducive to accelerating the oxidation process of styrene.
The O2-TPD results are depicted in Fig. S9.† Generally, there are three regions of oxygen desorption, respectively, corresponding to the reactive oxygen species (O2−/O−, 100–300 °C), surface lattice oxygen species (Olatt2−, 350–500 °C), and bulk lattice oxygen species (Olatt2−, 500–700 °C).52 Ptn/γ-Al2O3 shows two peaks at 376 and 475 °C, assigned to the adsorbed and lattice oxygen, respectively, while Pt1/γ-Al2O3 has only one peak at 447 °C, attributed to the activated lattice oxygen (Olatt2−). The Ptn/γ-Al2O3 catalyst exhibits a lower temperature of oxygen desorption and a larger peak area compared to the Pt1/γ-Al2O3 catalyst. With the same loading of Pt, the Ptn/γ-Al2O3 sample is more active for oxidation.
Fig. 6a shows the catalytic performances of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 with various quenchers and experimental results for investigating the mechanism. Firstly, negligible styrene conversion was observed under a nitrogen atmosphere. When 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was introduced into the system, the reaction was inhibited completely, confirming the ROS-mediated route over the two catalysts. When 1,4-benzoquinone, an efficient O2− scavenger, was added into the system, the reaction was also shut down, indicative that O2− is not the main factor affecting SO production. It is interesting that the addition of mannitol (a hydroxyl radical ˙OH trap) cannot change the conversion of styrene and selectivity to SO, which suggests the ˙OH species do not influence styrene epoxidation.19 However, with the addition of 2,2,6,6-tetramethylpiperidine (TMP, an effective scavenger of 1O2)53 to the reaction system, the styrene conversion was reduced and SO selectivity was completely inhibited, revealing the singlet oxygen species 1O2 is responsible for the styrene epoxidation. In situ electron paramagnetic resonance (EPR) spectra were used to monitor the 1O2 species over Pt1/γ-Al2O3 and Ptn/γ-Al2O3 catalysts from 25 °C to 100 °C. As shown in Fig. 6b, a triplet peak with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 was attributed to TEMP-1O2. The intensity of the 1O2 radical for the Pt1/γ-Al2O3 catalyst is very weak, irrespective of the temperature at 25 °C or 100 °C. For the Ptn/γ-Al2O3 catalyst, however, an intense EPR signal was observed at 100 °C, though it was weak under 25 °C, indicating that the Ptn/γ-Al2O3 catalyst is more conducive to the generation of 1O2 driven by heat. These results demonstrate the higher catalytic activity of Ptn/γ-Al2O3 compared to Pt1/γ-Al2O3.
:1:1 was attributed to TEMP-1O2. The intensity of the 1O2 radical for the Pt1/γ-Al2O3 catalyst is very weak, irrespective of the temperature at 25 °C or 100 °C. For the Ptn/γ-Al2O3 catalyst, however, an intense EPR signal was observed at 100 °C, though it was weak under 25 °C, indicating that the Ptn/γ-Al2O3 catalyst is more conducive to the generation of 1O2 driven by heat. These results demonstrate the higher catalytic activity of Ptn/γ-Al2O3 compared to Pt1/γ-Al2O3.
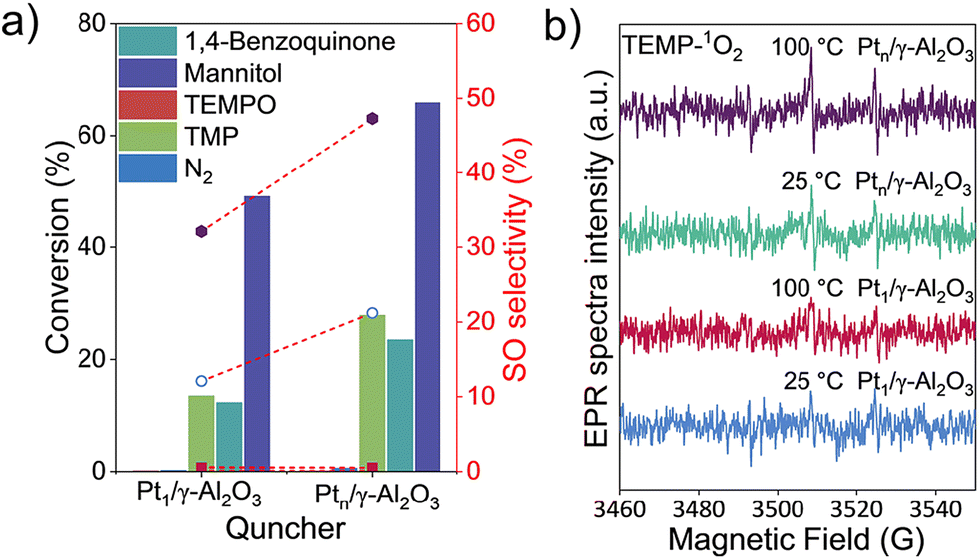 | ||
| Fig. 6 (a) Catalytic performance of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 with various quenchers, the red short dash represents the SO selectivity and column represents the styrene conversion; (b) in situ EPR spectra of Pt1/γ-Al2O3 and Ptn/γ-Al2O3 with TEMP-1O2 at 25 °C and 100 °C. | ||
Mechanism elucidated by DFT calculations
We have conducted density functional theory (DFT) calculations to thoroughly understand the mechanism of Pt catalysts with varying sizes for the epoxidation of styrene.The DFT analyses explored the interaction dynamics between the Pt species and oxygen molecules on γ-Al2O3 surfaces. The optimized configurations of Pt1O2/γ-Al2O3 and Pt11O18/γ-Al2O3 (Fig. 7a and Fig. S10†) revealed significant differences in electronic properties. According to the Bader charge analysis, both Pt catalysts are positively charged. The isolated platinum atom in Pt1O2/γ-Al2O3 is positively charged with +0.78 |e|, while more electrons were accumulated on Pt11O18, as shown in Fig. 7b, revealing the platinum atoms in Pt11O18/γ-Al2O3 with higher valence, agreeing well with the XPS analysis. In addition, the d-band center upward shift of Pt11O18 concerning the Fermi level was observed (Fig. S11†), which may strengthen the bond with oxygen molecules according to the d-band center theory9 and also benefit the adsorption and activation of molecular oxygen to a certain degree.
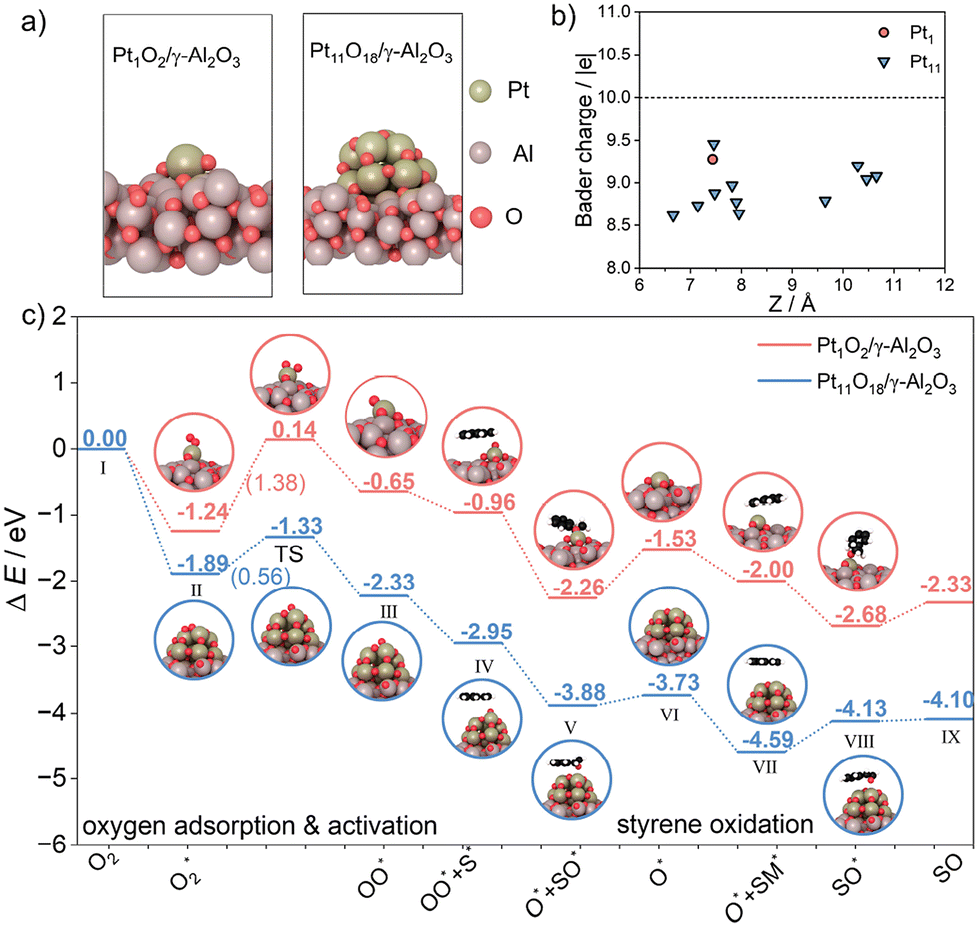 | ||
| Fig. 7 DFT calculations. (a) Optimized configurations of Pt1O2/γ-Al2O3 and Pt11O18/γ-Al2O3 models; (b) Bader charge analysis of the platinum atoms in both models. The black dotted line represents the number of valence electrons in platinum; (c) energy profiles of the styrene epoxidation reaction over Pt1O2/γ-Al2O3 and Pt11O18/γ-Al2O3. Color code: Pt (dark golden), Al (brown), O (red), C (black) and H (white). | ||
Based on the kinetic results, O2 activation is the first step within the consecutive styrene oxidation. According to the calculations, the adsorption of oxygen molecules over oxidized platinum NPs (−1.24 eV) was much stronger than the oxidized platinum single atom (−1.89 eV) (Fig. S12†). In Fig. 7c, the activation barrier of the adsorbed oxygen (O2*) and subsequent dissociation over Pt1O2/γ-Al2O3 was 1.38 eV (II to TS), which is kinetically difficult. By contrast, the reaction occurring on the Pt11O18/γ-Al2O3 surface (ΔE = −0.44 eV) is thermodynamically favorable with a barrier of 0.56 eV (II to TS), which is easy to overcome. This result indicated that the formation of the O* species with Pt NPs was far more facile, which is consistent with the EPR results (Fig. 6b). Configurations involved in the initial, transitional and final states are shown in Fig. S13 and S14.†
Along the reaction path, molecular oxygen is firstly adsorbed and activated to the O* species over both Pt1O2/γ-Al2O3 and Pt11O18/γ-Al2O3 and the latter has low exothermic values. Subsequently, a styrene molecule was activated (IV) and transformed to the epoxidation product SO (V), with an energy barrier of −0.93 eV on Pt11O18/γ-Al2O3 and −1.30 eV on Pt1O2/γ-Al2O3 (IV to V), which confirmed the Pt11O18/γ-Al2O3 catalyst with low energy barrier for the epoxidation progress. By overcoming relatively low energy barriers (V to VI), the remaining oxygen is transformed to new one-coordinated oxygen atoms. In this process, Pt11O18/γ-Al2O3 can more easily overcome the energy barrier of 0.15 eV compared to Pt1O2/γ-Al2O3, with a higher energy barrier of 0.73 eV. Subsequently, another new styrene molecule was activated and converted to SO (VII to IX). The energy barrier of Pt11O18/γ-Al2O3 is just 0.03 eV; however, the Pt1O2/γ-Al2O3 energy barrier was 0.35 eV, which demonstrated that the SO product is hardly desorbed from the Pt1O2/γ-Al2O3 catalyst surface. Hence, the DFT calculation results further proved that for the activation of styrene substrates and O2, Ptn/γ-Al2O3 is the optimum catalyst, while the desorption of SO products is also easier compared to that in Pt1/γ-Al2O3, consistent with the experimental results.
3 Conclusions
By precisely tuning the metal–support interaction, the single-atom Pt1/γ-Al2O3 and nanocluster-dispersed Ptn/γ-Al2O3 catalysts were prepared. Compared with Pt1/γ-Al2O3, the Ptn/γ-Al2O3 catalyst exhibited superior catalytic performance for styrene epoxidation. It was confirmed by experimental characterizations and theoretical calculations that Ptn/γ-Al2O3 was more active for the activation of O2 to reactive singlet oxygen species. In addition, owing to its larger size, it offers more sites simultaneously for styrene adsorption and epoxidation at the Pt clusters. Over the Pt1/γ-Al2O3 catalyst, however, both the O2 activation and the desorption energy of styrene oxide show a higher energy barrier. The results provide a comprehensive understanding of the impact of Pt size on the epoxidation of styrene and offer insights into regulation by the metal support for controlling the dispersion and size distribution of platinum on alumina.4 Experimental
Catalyst preparation
Reagents used for the catalyst preparation and detailed procedures are described in ESI.†Catalyst characterizations
Catalyst characterization and instruments used as well as specific test methods are given in the ESI.†Catalytic test
The styrene oxidation was conducted in a three-necked glass flask equipped with a reflux condenser. In a typical experiment, styrene, catalysts, and solvent were mixed and added to a 100 mL three-necked glass flask. 1 atm oxygen was then introduced into the system using a balloon. The reaction vessel was conducted at 100 °C in a silicon oil bath with continuous stirring. Once the reactor was cooled to room temperature, the liquid mixture was collected by filtration using a filter membrane (0.22 μm) and then analyzed with a gas chromatography system (GC 9560) connected to a mass detector, in which a known amount of nitrobenzene was introduced as an internal standard. The turnover frequency (TOF) was determined using the following equation (eqn (3)):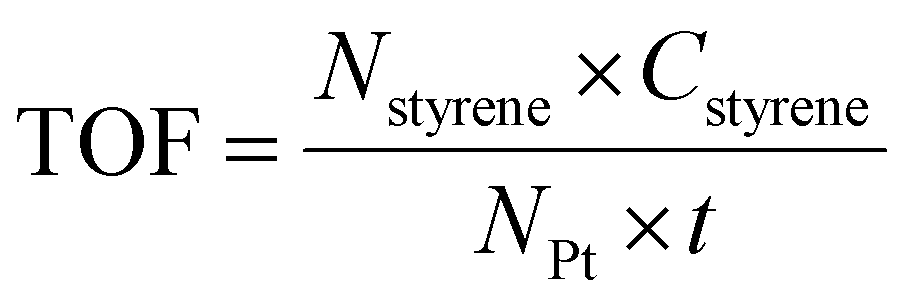 | (3) |
The styrene conversion (eqn (4)) and SO selectivity (eqn (5)) was calculated by the following equation:
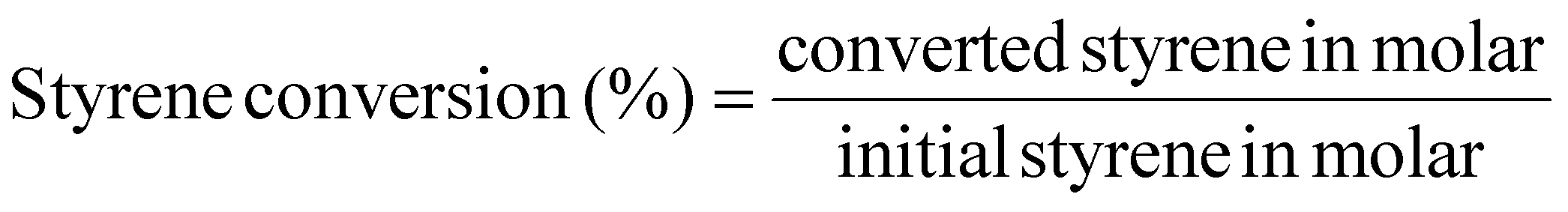 | (4) |
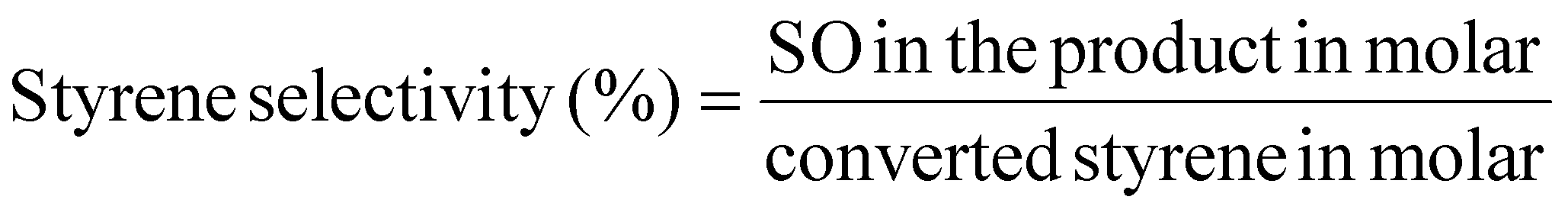 | (5) |
DFT calculations
All spin-polarized DFT calculations were carried out using the Vienna Ab initio Simulation Package (VASP 5.4.4),54,55 a periodic DFT code with projector augmented wave (PAW) potentials. The exchange–correlation energy was described using the generalized gradient approximation (GGA) formulated by the Perdew–Burker–Ernzerhof (PBE) functional.56 The plane-wave basis sets were converged by a kinetic energy cutoff of 400 eV and a Monkhorst–Pack (3 × 3 × 1) k-point was applied for the sampling of the Brillouin Zone. The convergence criterion of energy and gradient was set as 10−5 and 0.03 eV Å−1, respectively. The reaction energy (ΔE) and activation barrier (Ea) were defined as the energy difference between the initial state (IS) and final state (FS) and the energy difference between the initial state and transition state (TS), respectively. The transition states were located via the climbing-image nudged elastic band (CI-NEB) method57 together with the improved dimer method (IDM),58 and finally confirmed by the frequency vibrations. Atomic charges were computed through the Bader charge analysis.59The pristine γ-Al2O3(111) surface was modelled by a periodic (1 × 1) supercell slab containing three Al–O–Al atomic layers. A vacuum height of 15 Å along the Z direction was set between the slab surfaces to eliminate unphysical interactions. During the structural optimization, the atoms in the bottom layer were frozen in the bulk positions while the remaining atoms were fully relaxed. Based on the structural characterizations, two different models were taken into account. As for modelling the single-atom structured platinum, an isolated Pt1O2 was supported above the γ-Al2O3(111) surface and named Pt1O2/γ-Al2O3. Moreover, platinum oxides with bigger sizes were represented by substituting the Pt1O2 with an optimized Pt11O18 cluster, which was named as Pt11O18/γ-Al2O3.
Author contributions
F. F. L.: investigation, methodology, data curation, software, writing – original draft. C. Y. S.: software, validation, writing – review & editing. Y. H.: validation. H. Y. L: validation. R. T. G.: methodology. J. Y.: software, validation. Z. W. Z.: validation, data curation. F. F. M.: writing – review & editing. T. T. Z.: data curation. X. K. G. and N. H. X. review. W. P. D.: conceptualization, funding acquisition, writing – review & editing, supervision.Data availability
The authors state that the data included in the current paper are available on request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21932004, 91963206, 22172072, and 22072090) and the Ministry of Science and Technology of China (2021YFA1500301). The support from the NJU-HUACHANG Joint Institute of Meso Catalysis is also appreciated.References
- M. Macino, A. J. Barnes, S. M. Althahban, R. Qu, E. K. Gibson, D. J. Morgan, S. J. Freakley, N. Dimitratos, C. J. Kiely, X. Gao, A. M. Beale, D. Bethell, Q. He, M. Sankar and G. J. Hutchings, Nat. Catal., 2019, 2, 873–881 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS PubMed.
- A. Okrut, R. C. Runnebaum, X. Ouyang, J. Lu, C. Aydin, S.-J. Hwang, S. Zhang, O. A. Olatunji-Ojo, K. A. Durkin, D. A. Dixon, B. C. Gates and A. Katz, Nat. Nanotechnol., 2014, 9, 459–465 CrossRef CAS PubMed.
- L. Wang, H. Liu, J. Zhuang and D. Wang, Small Sci., 2022, 2, 2200036 CrossRef CAS.
- Y. Yao, Q. Dong, A. Brozena, J. Luo, J. Miao, M. Chi, C. Wang, I. G. Kevrekidis, Z. J. Ren, J. Greeley, G. Wang, A. Anapolsky and L. Hu, Science, 2022, 376, eabn3103 CrossRef CAS PubMed.
- W. Jing, H. Shen, R. Qin, Q. Wu, K. Liu and N. Zheng, Chem. Rev., 2022, 123, 5948–6002 CrossRef PubMed.
- M. Nasrollahzadeh, M. Sajjadi, S. Iravani and R. S. Varma, J. Hazard. Mater., 2021, 401, 123401 CrossRef CAS PubMed.
- J. Hui, H. Chu, W. Zhang, Y. Shen, W. Chen, Y. Hu, W. Liu, C. Gao, S. Guo, G. Xiao, S. Li, Y. Fu, D. Fan, W. Zhang and F. Huo, Nanoscale, 2018, 10, 8772–8778 RSC.
- C. Yang, Z. Dao, S. Pu, W. Yan, Y. Wang, D. Tian, C. Min, J. Zhao and C. Zhuang, ACS Appl. Nano Mater., 2024, 7, 6429–6441 CrossRef CAS.
- F. Xue, Q. Li, M. Lv, Y. Song, T. Yang, X. Wang, T. Li, Y. Ren, K. Ohara, Y. He, D. Li, Q. Li, X. Chen, K. Lin and X. Xing, J. Am. Chem. Soc., 2023, 145, 26728–26735 CrossRef CAS PubMed.
- J. Dou, X. Li, B. Xi, X. Yang, Y. Liu, C. Jin and J. Zhang, Catal. Sci. Technol., 2023, 13, 5592–5598 RSC.
- X. Li, J. Zhang, C. Jin, B. Yan, J. Cai, M. Li, X. Peng and Y. Wang, ACS Sustainable Chem. Eng., 2021, 9, 11062–11069 CrossRef CAS.
- H. Yang, Y. Wu, Z. Zhuang, Y. Li and C. Chen, Chin. J. Chem., 2021, 40, 515–523 CrossRef.
- A. Xu, R. Chen, W. Zhao, J. Lin, R. Hu, A. Khan, W. Li, X. Li, S. Zhao and Y. Qiu, Chem. Eng. J., 2023, 473, 145343 CrossRef CAS.
- W. Zhao, M. Adeel, P. Zhang, P. Zhou, L. Huang, Y. Zhao, M. A. Ahmad, N. Shakoor, B. Lou, Y. Jiang, I. Lynch and Y. Rui, Environ. Sci.: Nano, 2022, 9, 61–80 RSC.
- Y. Yu, Q. Zhang, Q. Yao, J. Xie and J. Y. Lee, Acc. Chem. Res., 2014, 47, 3530–3540 CrossRef CAS PubMed.
- H. Lv, W. Guo, M. Chen, H. Zhou and Y. Wu, Chin. J. Catal., 2022, 43, 71–91 CrossRef CAS.
- A. S. Sharma, V. S. Sharma, H. Kaur and R. S. Varma, Green Chem., 2020, 22, 5902–5936 RSC.
- M. Li, P. Wang, Z. Ji, Z. Zhou, Y. Xia, Y. Li and S. Zhan, Appl. Catal., B, 2021, 289, 120020 CrossRef CAS.
- M. Li, L. Ma, L. Luo, Y. Liu, M. Xu, H. Zhou, Y. Wang, Z. Li, X. Kong and H. Duan, Appl. Catal., B, 2022, 309, 121268 CrossRef CAS.
- F. Li, J. Tang, Q. Ke, Y. Guo, M. N. Ha, C. Wan, Z. Lei, J. Gu, Q. Ling, V. N. Nguyen and W. Zhan, ACS Catal., 2021, 11, 11855–11866 CrossRef CAS.
- S. Dissanayake, N. Vora, L. Achola, Y. Dang, J. He, Z. Tobin, X. Lu, A. Mirich, P.-X. Gao and S. L. Suib, Appl. Catal., B, 2021, 282, 119573 CrossRef CAS.
- S. Zhao, Y. Yang, F. Bi, Y. Chen, M. Wu, X. Zhang and G. Wang, Chem. Eng. J., 2023, 454, 140376 CrossRef CAS.
- S. Zhan, H. Zhang, X. Mi, Y. Zhao, C. Hu and L. Lyu, Environ. Sci. Technol., 2020, 54, 8333–8343 CrossRef CAS PubMed.
- L. Hao, H. Huang, Y. Zhang and T. Ma, Adv. Funct. Mater., 2021, 31, 2100919 CrossRef CAS.
- U. J. Etim, P. Bai, O. M. Gazit and Z. Zhong, Catal. Rev., 2021, 65, 239–425 CrossRef.
- G. Gao, G. Zhu, X. Chen, Z. Sun and A. Cabot, ACS Nano, 2023, 17, 20804–20824 CrossRef PubMed.
- S. Zhang, Z. Tian, Y. Ma and Y. Qu, ACS Catal., 2023, 13, 4629–4645 CrossRef CAS.
- T. Gan, J. Yang, D. Morris, X. Chu, P. Zhang, W. Zhang, Y. Zou, W. Yan, S.-H. Wei and G. Liu, Nat. Commun., 2021, 12, 2741 CrossRef CAS PubMed.
- Y. Zhuo, X. Guo, W. Cai, T. Shao, D. Xia, C. Li and S. Liu, Appl. Catal., B, 2023, 333, 122789 CrossRef CAS.
- B. Niu, Y. Wang, T. Zhao, X. Duan, W. Xu, Z. Zhao, Z. Yang, G. Li, J. Li, J. Cheng and Z. Hao, Environ. Sci. Technol., 2024, 58, 4428–4437 CrossRef CAS PubMed.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- R. Gu, D. Meng, M. She, Y. Wang, H. Yang, X. Guo, N. Xue and W. Ding, Chem. Commun., 2022, 58, 7630–7633 RSC.
- K. Yuan, Y. Guo, Q.-L. Lin, L. Huang, J.-T. Ren, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, J. Catal., 2021, 394, 121–130 CrossRef CAS.
- M. Kottwitz, Y. Li, R. M. Palomino, Z. Liu, G. Wang, Q. Wu, J. Huang, J. Timoshenko, S. D. Senanayake, M. Balasubramanian, D. Lu, R. G. Nuzzo and A. I. Frenkel, ACS Catal., 2019, 9, 8738–8748 CrossRef CAS.
- S. Rojas-Buzo, B. Bohigues, D. Salusso, A. Corma, M. Moliner and S. Bordiga, ACS Catal., 2023, 13, 9171–9180 CrossRef CAS PubMed.
- J. Xiong, Y. Wei, Y. Zhang, P. Zhang, Q. Yu, X. Mei, X. Liu, Z. Zhao and J. Liu, ACS Catal., 2020, 10, 7123–7135 CrossRef CAS.
- V. Matsouka, M. Konsolakis, R. M. Lambert and I. V. Yentekakis, Appl. Catal., B, 2008, 84, 715–722 CrossRef CAS.
- F. Morfin, C. Dessal, A. Sangnier, C. Chizallet and L. Piccolo, ACS Catal., 2024, 14, 9628–9639 CrossRef CAS.
- C. Dessal, T. Len, F. Morfin, J.-L. Rousset, M. Aouine, P. Afanasiev and L. Piccolo, ACS Catal., 2019, 9, 5752–5759 CrossRef CAS.
- S. Tian, B. Wang, W. Gong, Z. He, Q. Xu, W. Chen, Q. Zhang, Y. Zhu, J. Yang, Q. Fu, C. Chen, Y. Bu, L. Gu, X. Sun, H. Zhao, D. Wang and Y. Li, Nat. Commun., 2021, 12, 3181 CrossRef CAS PubMed.
- B. A. Riguetto, S. Damyanova, G. Gouliev, C. M. P. Marques, L. Petrov and J. M. C. Bueno, J. Phys. Chem. B, 2004, 108, 5349–5358 CrossRef CAS.
- Y. Li, Y. Li, Y. Ding, J. Ma, P. Das, B. Zhang, Z.-S. Wu and X. Bao, Chem. Catal., 2023, 3, 100658 CrossRef CAS.
- H. Liu, S. Qiang, F. Wu, X.-D. Zhu, X. Liu, J. Yu, Y.-T. Liu and B. Ding, ACS Nano, 2023, 17, 19431–19440 CrossRef CAS PubMed.
- L. Shi, Y. Zhou, S. Qi, K. J. Smith, X. Tan, J. Yan and C. Yi, ACS Catal., 2020, 10, 10661–10671 CrossRef CAS.
- C. Weerakkody, S. Biswas, W. Song, J. He, N. Wasalathanthri, S. Dissanayake, D. A. Kriz, B. Dutta and S. L. Suib, Appl. Catal., B, 2018, 221, 681–690 CrossRef CAS.
- Y. Xiong, W. Sun, P. Xin, W. Chen, X. Zheng, W. Yan, L. Zheng, J. Dong, J. Zhang, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2000896 CrossRef CAS PubMed.
- W. Chen, G. Qian, Y. Wan, D. Chen, X. Zhou, W. Yuan and X. Duan, Acc. Chem. Res., 2022, 55, 3230–3241 CrossRef CAS PubMed.
- W. Chen, W. Fu, X. Duan, B. Chen, G. Qian, R. Si, X. Zhou, W. Yuan and D. Chen, Engineering, 2022, 14, 124–133 CrossRef CAS.
- J.-H. Noh, R. Patala and R. Meijboom, Appl. Catal., A, 2016, 514, 253–266 CrossRef CAS.
- J. Liu, R. Meng, J. Li, P. Jian, L. Wang and R. Jian, Appl. Catal., B, 2019, 254, 124–222 Search PubMed.
- Q. Zhao, Y. Gu, H. Fu, X. Qu, Z. Xu, B. Chefetz, S. Zheng and D. Zhu, Environ. Sci. Technol., 2024, 58, 3483–3494 CAS.
- M. Neamţu, C. Nădejde, V. D. Hodoroaba, R. J. Schneider and U. Panne, Appl. Catal., B, 2018, 232, 553–561 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 1999, 111, 7010–7022 CrossRef CAS.
- R. F. Bader, Chemt. Rev., 1991, 91, 893–928 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Description about catalyst synthesis and characterization method. Results of XRD patterns, nitrogen sorption isotherms, XPS, solvent effects, EXAFS fitting parameters, O2-TPD, projected density of state, optimized configurations of O2 adsorption on the Pt1O2/γ-Al2O3 and Pt11O18/γ-Al2O3 model surfaces and the initial, transitional and final states of O2 dissociation. See DOI: https://doi.org/10.1039/d4nr05256k |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |