
Thin films based on nanocomposites of crumpled graphene fully decorated with Prussian blue: a new material for aqueous battery systems†
Vitor H. N.
Martins
 a,
Monize M.
Silva
ab,
Maria K.
Ramos
c,
Maria H.
Verdan
a,
Eduardo G. C.
Neiva
d,
Aldo J. G.
Zarbin
c and
Victor H. R.
Souza
*a
a,
Monize M.
Silva
ab,
Maria K.
Ramos
c,
Maria H.
Verdan
a,
Eduardo G. C.
Neiva
d,
Aldo J. G.
Zarbin
c and
Victor H. R.
Souza
*a
aFaculty of Exact Science and Technology, Universidade Federal da Grande Dourados (UFGD), Dourados, MS, Brazil. E-mail: victorsouza@ufgd.edu.br
bUniversidade do Estado do Amapá (UEAP), Macapá, AP, Brazil
cDepartment of Chemistry, Universidade Federal do Paraná (UFPR), Curitiba, PR, Brazil
dDepartment of Chemistry, Universidade Regional de Blumenau (FURB), Blumenau, SC, Brazil
First published on 13th March 2025
Abstract
This study involves synthesizing thin films through an interfacial method, which relies on composites of Prussian blue nanoparticles and nanostructures derived from graphene, known as crumpled graphene. The resulting compounds were subjected to evaluation for potential applications in aqueous battery-type energy storage systems. Considering the importance of structure–property relationships and applications, the carbon nanostructures were previously processed to assess their morphological characteristics and electrochemical performance for the growth of Prussian blue nanocubes. To this end, the spray-pyrolysis method was employed, resulting in crumpled graphene infused with β-FeOOH and Fe2O3 (β-iron(III) oxyhydroxide and iron (III) oxide) species. Composites of crumpled graphene and Prussian blue were synthesized through the electrodeposition method via cyclic voltammetry, which formed Prussian blue nanocubes on the surface of crumpled graphene with sizes ranging from 48 to 153 nm depending on the number of cycles. Specific capacity values varied based on the compound structure, with the highest recorded value of 50.4 mA h g−1 at a rate of 500 mA g−1 for the PB_10 composite achieved in an aqueous electrolyte of 0.1 mol L−1 KCl vs. Ag|AgCl (3.0 mol L−1 KCl). The PB_10 electrode was further studied using different electrochemical techniques and employed in a coin cell battery system, demonstrating a discharge capacity of 25.0 mA h g−1 at 250 mA g−1. Additionally, the device retained 97% of its capacity after cycling at various current densities, highlighting its stability.
 Victor H. R. Souza | Victor H. R. Souza received his PhD in Chemistry (2015) from the Universidade Federal do Paraná (Curitiba/Brazil). During his PhD, he undertook an Improvement Internship at Centre de Recherche Paul Pascal, CNRS, Bordeaux, France in 2014. He was a postdoctoral fellow at Northwestern University, USA from 2015 to 2016. He is currently an Associate Professor at the Universidade Federal da Grande Dourados, Dourados, Brazil. His current research interests are focused on designing new carbon-based nanocomposites for further application in energy storage devices and electrochemical sensors. His passion for football makes his research on paper ball-like crumpled graphene structures even more rewarding. |
1. Introduction
The current globalized world system, faced with increasing energy demand and the need for actions to mitigate climate change and the risks we are exposed to, has caused rapid awareness and accelerated the transition from fossil fuels to a clean and renewable energy production system.1 The growing environmental concern regarding carbon emissions, river pollution, and climate change brings the need for increasingly safe, efficient, and – from an ecological point of view – more friendly energy storage and conversion technologies. Energy production through renewable sources, such as wind and solar, is an alternative that is considered sustainable.2 However, intermittent energy sources require the advancement of innovative energy storage and conversion technologies to ensure reliable and efficient power supply. Batteries are up-and-coming devices for the efficient storage of electrical energy.3–5Among devices, current lithium-based batteries (LIBs) have stood out since their advent in the late 1990s due to their characteristics such as high energy density, stability over several use cycles, and energy efficiency. However, in contrast to the high demand and scarcity of lithium, the abundant resources of sodium and potassium in nature make them strong candidates for developing low-cost and sustainable technologies for aqueous battery-type devices. Due to their low cost and abundant resources, developing nanomaterial-based cathodes offers a promising future for sodium and potassium batteries. The use of conventional organic electrolytes is also a factor to consider since most are toxic, flammable, and environmentally unfriendly substances. These characteristics affect safety and limit their application in specific cases.6,7 Compared to organic electrolytes, aqueous electrolytes offer a good alternative to overcome the challenges of developing environmentally friendly products. In addition to being low-cost, this class of electrolytes is also safer and easy to make, offering exceptional characteristics such as high ionic conductivity. Therefore, finding materials suitable for aqueous potassium and sodium batteries has become an aim in materials science and the industrial sector.8–10 In addition, there are a growing number of studies involving the development of cathodes from nanocomposites that could intercalate these ions, as in the case of hexacyanometallates (HCMs).
Prussian blue (PB – Fe3+4[Fe2+(CN)6]3·nH2O), also known as iron(III) hexacyanoferrate(II), is the most well-known material in the HCM class due to its electrochemical properties. Specifically, PB is composed of high-spin Fe3+ atoms and low-spin Fe2+ atoms coordinated to cyano ligand units so that the ferrous species coordinate to carbon and the ferric species coordinate to nitrogen.11–15 Because of its structure, PB is recognized as a typical blue pigment. Its characteristics, such as high surface area and biocompatibility, make it an attractive material for applications in energy storage and electrochemical sensors.15–17 One of its most notable properties is the ability to act as a reversible electron mediator, allowing this material to be applied in various ways. Its electrochemical properties, reported by Neff in 1978,18 brought notoriety, and this material became the target of numerous studies, being studied mainly as an electrochemical sensor of hydrogen peroxide (H2O2). Its porous and small-sized structure, with interstitial sites with a diameter of approximately 3.2 Å,19 allows the insertion and removal of small cations and molecules. Its reversible redox behavior makes PB a target of studies for the most varied applications, including batteries,11 electrochromic devices,12 and sensors,15 among others. PB's main characteristic is different oxidation states. Its most reduced form is known as Everitt's salt or Prussian white (PW), and it only has sites with ferrous species, which are colourless. In its most oxidized form, the material has a greenish coloration known as Berlin green (BG). This colour variation can be explained by the possible internal transitions of the metal–ligand–metal type between the Fe2+ and Fe3+ species and the cyano bridge ligands.11,12,15 Despite all the potential, HCMs have several limitations, including low electrochemical activity and their semiconductor characteristics. On the other hand, if limited to the isolated use of HCMs, studies show that the formation of composites with nanostructured carbon materials such as carbon nanotubes11,15 and graphene derivatives (GO and rGO)12 reduce the electrochemical instability and increase the conductivity. In general, electrical energy storage systems based on composites of nanostructured carbon materials and Prussian blue nanoparticles offer a promising platform, mainly for aqueous systems of potassium ions. The processing of graphene and its derivatives can also become a challenge when considering the stacking effects of graphene layers, which result in a series of obstacles for manufacture and application. This occurs due to the interactions between graphene layers, which tend to stack in a disordered manner because of van der Waals forces.20 This effect causes loss of the material's active area and the formation of agglomerates above the nanometer scale.
New strategies capable of circumventing the stacking effects of graphene-based materials have emerged; among these new strategies is the development of composites21 and the modification of the two-dimensional structure.20 A new strategy consists of a three-dimensional material with a morphology similar to crumpled paper balls, preserving the chemical structure of graphene and its properties. This new material is called crumpled graphene (CG).20,22–27 Due to its unique properties, crumpled graphene has gained interest in new technology development. Combining CG and other materials allows the production of new materials with synergism between the characteristics of each component, being one of the strategies adopted for tuning properties. Transition metal oxides are good examples of materials with electrochemical activity that can contribute to the formation of composites with improved electrochemical response. Recently, composites of CG and transition metal species have been reported in the literature and demonstrated fine-tuning of the CG properties, enabling the application of this carbon nanostructure as an electrochemical sensor22,23,25–27 and in electrical energy storage.22 However, the ability to process materials is as important as the capacity to produce them. In recent years, the production of thin films of various materials using the LLIR (liquid–liquid interfacial route) has increased, where thin, transparent, and multifunctional films based on nanomaterials can be stabilized at the interface of immiscible liquids.11,12,14 Specifically, some of us have taken advantage of the LLIR to prepare nanocomposite thin films based on carbon nanostructures (nanotubes or graphene), Prussian blue and Prussian blue analogues with excellent performance as cathodes for aqueous potassium or sodium ion batteries.11,12,14,28–31 Husmann et al. presented a strategy based on obtaining carbon nanotubes filled with iron oxide and subsequent electrodeposition of PB nanoparticles.15 Herein, we demonstrate an innovative LLIR-based strategy to produce thin films based on crumpled graphene decorated with Prussian blue nanoparticles and the application of this novel nanocomposite as a cathode for aqueous potassium batteries.
2. Experimental
2.1. Synthesis of crumpled graphene (CG)/Fe nanocomposites
We synthesized new composites based on crumpled graphene fully decorated with iron-based nanoparticles using the methodology detailed in the previous literature.22Scheme 1 shows the method applied to prepare the composite of CG and iron-based nanoparticles. Briefly, graphene oxide (1 mg mL−1) and iron III chloride (FeCl3) were dispersed in ultrapure water. Then, 100 mL of the dispersion was nebulized into a quartz tube heated at 400 °C, with a purge flow of 1 L min−1 of nitrogen. At the end of the quartz tube, all the crumpled graphene already decorated with nanoparticles in a single step was collected using a polytetrafluoroethylene (PTFE) filter system. Finally, the material was washed with acetone and dried in an oven at 60 °C (CG/Fe). | ||
| Scheme 1 Synthetic scheme of the CG/Fe composite. | ||
2.2. Interfacial thin film electrodes based on crumpled graphene and Prussian blue
The CG/Fe films were prepared using the LLIR, as described elsewhere for films based on carbon nanotubes and graphene.32,33Scheme 2 shows this process. First, 5 mL of CG/Fe in toluene (0.5 mg mL−1) was dispersed for 3 minutes using an ultrasonic bath. The resulting dispersion was transferred to a round-bottom flask (25 mL capacity) containing 5 mL of ultrapure water. The system was maintained under magnetic stirring (1500 rpm) for 120 min at room temperature. Subsequently, the magnetic stirring was stopped. The thin film was stabilized at the organic/aqueous interface. Both phases were replaced three times with pure solvents (water and toluene). Finally, the interfacial film was transferred to a similar biphasic system in a beaker containing an indium tin oxide-coated glass (ITO) substrate (9 mm × 25 mm) fixed on a copper hast. The electrode was then produced by lifting the ITO substrate toward the CG/Fe film. The electrode with the stabilized film was immediately dried in an oven at 60 °C for 60 min.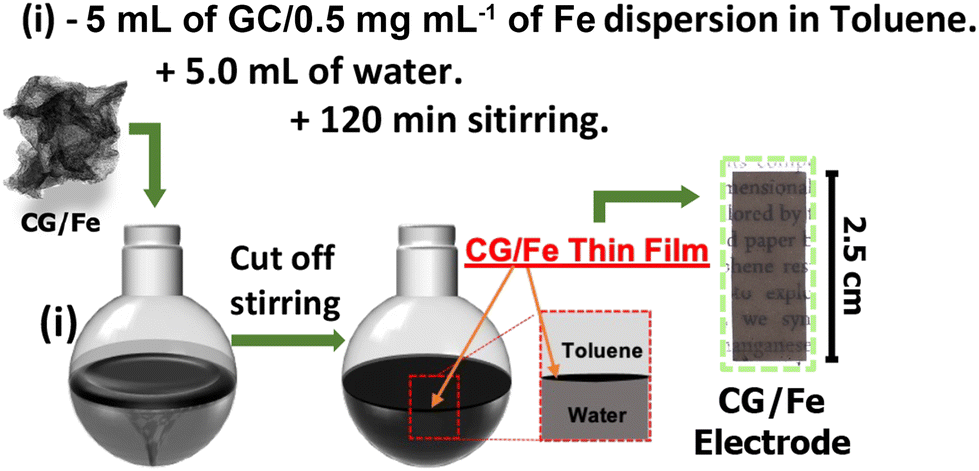 | ||
| Scheme 2 Experimental step to prepare CG/Fe films at the liquid–liquid interface. | ||
Afterwards, the area of the film deposited on the ITO substrate was adjusted to 1.8 cm2. The electrochemical deposition of Prussian blue nanocubes on CG/Fe thin films was carried out using an interfacial film electrode as the working electrode (WE), Ag|AgCl (3 mol L−1 KCl) as the reference electrode (RE), and a platinum plate as the counter electrode (CE). Cyclic voltammetry (CV) with a sweep potential from −0.3 to 1.4 V at a sweep rate of 50 mV s−1 in 0.1 mol L−1 KCl electrolyte and 1 mmol L−1 K3[Fe(CN)6] was performed for electrodeposition. Different electrodeposition cycles were adopted to produce materials with different characteristics, namely PB_2, PB_5, PB_10, PB_25, and PB_50, for films with 2, 5, 10, 25, and 50 voltammetric cycles, respectively. Fig. S1† shows the photographs of all the thin and transparent films synthesized herein.
2.3. Materials characterization
The crystalline structure of the nanocomposites was determined using a Shimadzu XRD-6000 instrument with Cu Kα radiation (λ = 1.5418 Å) and an incident angle of 0.1°. A SEM-FEG/TESCAN (MIRA-3) was used for morphology investigation at an accelerating voltage of 10.0 kV and a working distance of approximately 5 mm. The energy-dispersive X-ray spectroscopy (EDS) spectrum was recorded using a SEM-FEG. The thin films over the ITO substrate were used for SEM analysis. Transmission electron microscopy (TEM) images were obtained using a JEOL JEM-1200 microscope with a voltage of 120 kV. Dispersions of pure crumpled graphene and crumpled graphene decorated with iron-based nanoparticles in isopropanol were deposited by drop-casting on copper grids. Raman spectroscopy analyses were carried out using WiTec equipment (Oxford Instruments Alpha300 R) with a 633 nm laser and an accumulation time of 10 seconds, with 20 accumulations per spectrum in a spectral range from 250 to 3100 cm−1. Fourier transform infrared spectroscopy analysis was performed using an Invenio-R (Bruker) in transmittance mode in the range of 380–4000 cm−1 with a resolution of 2 cm−1 and 256 accumulations. X-ray photoelectron spectroscopy analysis was performed using a K-alpha XPS (Thermo Scientific) with an Al monochromatic source (1486 eV). Data acquisition of CG/Fe and CG decorated with the PB nanoparticle samples was carried out using their powder and their thin films on ITO substrates, respectively.2.4. Electrochemical measurements
All experimental procedures were conducted within a glass electrochemical cell (30.0 mL) employing a three-electrode system. An ITO decorated with the composite's substrate with a well-defined geometric surface area of 1.8 cm2 was used as the WE, a platinum plate (surface area of 2.5 cm2) was used as the CE and Ag|AgCl (3.0 mol L−1 KCl) was used as the RE. The modified electrode was covered with 10 μL of Nafion®–isopropanol at a concentration of 10.00% (w/v). Electrochemical measurements were performed using a portable μ-Autolab Type III potentiostat/galvanostat (Metrohm Autolab) employing Nova 2.1 software. Electrochemical impedance spectroscopy (EIS) measurements were performed using a portable EmStat 4s potentiostat/galvanostat (Palm Sens) in the frequency range of 104–10−2 Hz and an amplitude of 10 mV in an open circuit potential.2.5. Battery testing
The PB_10 sample was investigated by assembling CR2032 coin cells. A schematic representation is presented in Fig. S2.† The CG/Fe films were deposited on 16 mm gold-coated current collector discs. Similarly, the reference and counter electrodes based on the bare_CG sample were deposited on 16 mm gold-coated current collector discs. An aqueous potassium chloride solution (0.5 mol L−1) was used as the electrolyte. Glass fiber (TOB – China) in 19 mm diameter discs was used as the solid separator. The results were obtained considering the total mass of the electrodes.3. Results and discussion
3.1. Morphological and structural characterization
Designing materials as films by fully adjusting their chemical and physical properties is not easy. Among different sophisticated methods, the liquid–liquid interfacial route (LLIR) stands out for its quality and fine adjustment of the properties of materials as thin and transparent films. In addition, the production of electrodes based on thin films goes beyond the deposition of the same on substrates with good capacity to conduct electrons, as in the case of ITO.34,35 The aspects of the interface active material/electrolyte are important. The search for materials with potential for application in electrical energy storage systems, such as batteries, which replace traditional organic solvents and have reduced dependence on elements such as lithium is a recent subject of great environmental and economic importance. Here, we describe the use of thin films based on crumpled graphene decorated with iron-based nanoparticles, which will be used as a precursor for the electrodeposition of Prussian blue, and the investigation of the resulting CG/PB materials as electrodes in potassium-aqueous coin-cell battery devices.The synthesis of CG by the spray pyrolysis of the mixture of GO and the metal precursor dispersion into a heated quartz tube at 400 °C is an effective method for producing three-dimensional nanocomposites of crumpled graphene and metal-based nanoparticles. This synthesis method incorporates the simultaneous formation of metal-based species within the crumpled graphene structure by isotropic compression of confined GO sheets in water droplets.28 CG structures decorated with iron-based nanoparticles (CG/Fe) are shown in Fig. 1(a), and the new structure is a three-dimensional material from CG. Fig. S3(a and b)† shows the classic morphology of the bare crumpled graphene. This process results in a three-dimensional graphene-based material (CG) with structural modification that provides well-defined ridges and vertices to support the iron-based nanoparticles, as shown in Fig. 1(b-i). At higher magnifications, the SEM micrographs showed the composite CG/Fe morphology with iron-based nanoparticles on CG's surface. Similarly, the iron-based nanoparticles were observed in the backscattered electron (BSE) micrograph (Fig. 1(b-ii)). The distribution of iron species all over CG showed the elemental mapping obtained from EDS in Fig. 1(c).
 | ||
| Fig. 1 SEM micrographs of the CG/Fe nanocomposite: secondary electrons (a), (b-i), and backscattered electrons (b-ii); (c) EDS mapping of the CG/Fe composite with the presence of carbon (C), oxygen (O), and (Fe) iron species. | ||
The oxygen species in the elemental mapping of the nanocomposite suggests (i) functional groups remaining from GO, (ii) iron oxide-based nanoparticles, or (iii) a combination of these two factors. In the first case, the temperature adopted in the tubular furnace may be insufficient to fully reduce GO to CG. Secondly, the formation of iron hydroxides and oxyhydroxides is common through acid hydrolysis of the metal. Finally, this observation suggests that the material has functional groups from GO and iron oxide-based nanoparticles. The method developed here produces the CG/Fe composite and based on thermogravimetric analysis (TGA) (Fig. S3(d)†), percentages of 17.5% (100–350 °C) for functional groups and 30.8% of iron species from the residue were observed.
Among several methodologies for synthesizing PB, electrochemical deposition through the heterogeneous reaction between iron species confined in carbon-based materials (carbon nanotubes and graphene) can fine-tune the size and amount of PB and its electrochemical response. This approach is based on the gradual release of iron ions (or metals such as cobalt, nickel and copper for the case of Prussian blue analogues (PBA)) throughout the voltammetric cycles, allowing the electrodeposition of nanoparticles from the coordination between the released metal and the [Fe(CN)6]3− species in solution.15,29,36
Fig. 2(a) shows the electrodeposition profile of PB over the CG surface for 50 cyclic voltammetric (CV) cycles at 50 mV s−1 in 1 mmol L−1 K3[Fe(CN)6]/0.1 mol L−1 KCl aqueous solution. The voltammogram displays two redox pairs ranging from −0.10/0.50 V to 0.55/1.10 V (vs. Ag| AgCl, 3 mol L−1 KCl), corresponding to different redox processes. The first process is related to the conversion of Fe3+/Fe2+ species in solution.36 At higher potentials, the second redox pair is noticeable. This pair is the KFeIII[FeII(CN)6]/FeIII[FeIII(CN)6] redox pair in the PB/GB structure.37 For this, the iron species confined into the crumpled graphene act as a precursor of PB nanocubes. First, the iron species leached from the CG structure are reduced and the [Fe(CN)6]3− species react with Fe2+ to form the nanocubes of PB. With consecutive CV cycles, more PB was formed and deposited onto the surface of the carbon nanostructure. The deposition of nanocubes was measured by the increase of the cathodic peak current between 0.90 and 0.55 V, as shown in Fig. 2(b).
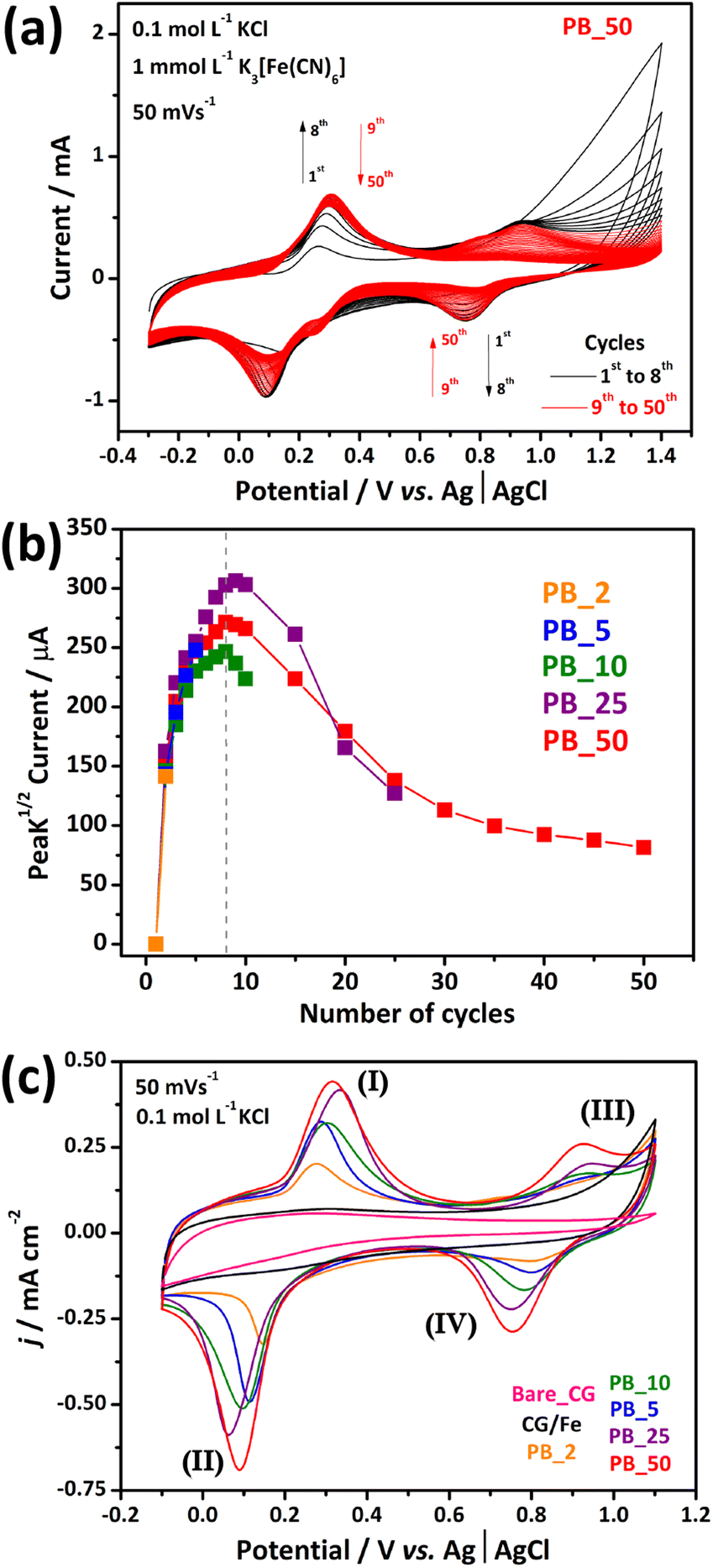 | ||
| Fig. 2 Electrodeposition of PB nanocubes performed in fifty cycles at a scan rate of 50 mV s−1 in 0.1 mol L−1 KCl electrolyte and 0.1 mmol L−1 K3[Fe(CN)6] for the PB_50 sample (a). Cathodic peak (between 0.90 and 0.55 V) current intensity as a function of the voltammetric cycles (b). Cyclic voltammograms for the modified electrodes: CG/Fe (black line) and pure CG (pink line) films before electrodeposition (c). | ||
The 50-cycle scan (red line in Fig. 2(b)) showed an increase in peak current from the second to about the eighth consecutive cycle, and then it slightly decreased. Knowing this, composites with different numbers of PB electrodeposition cycles were developed to investigate the growth of PB and the influence of the number of voltammetric cycles on the electrochemical performance, structure, and morphology of the nanocomposites. Fig. S4† shows the electrodeposition voltammograms of the PB_2, PB_5, PB_10, PB_25, and PB_50 electrodes modified with 2, 5, 10, 25, and 50 CV electrodeposition cycles of PB, respectively. The behavior of the composite curves presents a common profile in the first electrodeposition cycles. A pronounced current increase between the first two cycles of about 150 μA is common for all composites. This may indicate that the heterogeneous reaction for the formation of Prussian blue occurs, facilitated in the first two voltammetric cycles. Subsequently, a less intense increase in current was recorded for the following cycles for all electrodes until around the eighth cycle, when the loss of current for the pair began. The electrochemical profiles presented in Fig. 2(c) show capacitive characteristics for the CG/Fe and bare CG film electrodes, with only one discrete redox process being observed for the CG/Fe film in the range between 0.0 and 0.4 V. This corresponds to the oxidation–reduction of the iron species confined in the CG/Fe sample. On the other hand, when observing the profiles of the composites, all present two redox processes typical of Prussian blue. The first process between 0.0 and 0.4 V, corresponding to the high-spin iron species coordinated to nitrogen, is related to the transition between PB and PW ((I) KFeIII[FeII(CN)6]/ (II) K2FeII[FeII(CN)6]), and a second process between 0.6 and 1.0 V, corresponding to the low-spin iron species coordinated to carbon, is related to the conversion between GB and PB ((III) FeIII[FeIII(CN)6]/ (IV) KFeIII[FeII(CN)6]).36,37 The respective pairs are represented in the following equations:
 | (1) |
 | (2) |
In addition, a rise in current was observed according to an increase in electrodeposition cycles, indicating that as more voltammetric cycles occur, a higher amount of Prussian blue was deposited on the film. This behavior is accompanied by a potential shift generating greater hysteresis in systems with more electrochemically deposited PB.
3.2. Structural and vibrational characterization using XRD, XPS, and Raman analysis
The structural and vibrational characterization (XRD, XPS, and Raman analysis) was performed on CG/Fe films before and after the electrodeposition of PB. XRD analysis of the CG/Fe sample (Fig. 3(a)) shows specific peaks for the β-FeOOH species as the 11.6° (110), 16.6° (200), 26.7° (310), 34.1° (400), 35.2° (211), 39.3° (301), and 46.3° (411) planes (PDF 34-1266, β-FeOOH). This reveals that the synthesis of CG with the addition of the iron precursor (as described in section 3) led to the formation of β-FeOOH nanoparticles decorating the carbon nanostructure. The formation mechanism for the β-FeOOH species can proceed through the hydrolysis of an acidic FeCl3 solution, where the needed conditions for β-FeOOH are favored.38 The proposed mechanism in Scheme 3 represents the formation of β-FeOOH nanoparticles. Fig. 3(a) also confirms the presence of PB nanoparticles. The peaks of the CG/PB nanocomposites and the peak characteristics of PB (PDF 73-0687, Fe4[Fe(CN)6]3) are shown at 17.3° (200), 24.6° (220), and 35.3° (400) for the samples after electrodeposition. The peaks for the (110) and (310) planes are not clearly visible after the electrochemical modification of the electrodes, suggesting a release of the β-FeOOH species during the electrodeposition process of PB.11,12,36 | ||
| Fig. 3 X-ray diffractograms (a) and Raman spectra (λ = 633 nm) (b) of the CG/Fe (black line), PB_2 (orange line), PB_5 (blue line), PB_10 (green line), PB_25 (purple line), and PB_50 (red line) samples. XPS spectra of the CG/Fe and PB_10 samples (c). | ||
 | ||
| Scheme 3 Formation of β-FeOOH nanoparticles by hydrolysis of an acidic FeCl3 solution. | ||
The Raman spectra obtained from the film samples (Fig. 3(b)) show characteristic profiles for graphitic materials such as CG. The D band at 1335 cm−1 is associated with the breaking of symmetry between carbons in the structure of the rGO sheets composing CG. This occurs due to portions with different sp2 hybridization and also the “defects” located at the edges. In addition, the G band at 1597 cm−1 is characteristic of the stretching modes of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C; D′ at 1620 cm−1 and G′ at 2677 cm−1 are attributed to graphitic materials and are related to the organization of the structure; and the G + D band at 2937 cm−1 is present in the spectrum of the CG/Fe sample (black line).36,37,39
C; D′ at 1620 cm−1 and G′ at 2677 cm−1 are attributed to graphitic materials and are related to the organization of the structure; and the G + D band at 2937 cm−1 is present in the spectrum of the CG/Fe sample (black line).36,37,39
In addition to these, a set of bands that are characteristic vibrational modes of HCMs in 2090 and 2153 cm−1 are attributed to the stretching of the CN units of ligands.
Another band characteristic of HCMs appears at a lower wavelength, 530 cm−1, and is attributed to the vibrational modes of the Fe–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N portion.36,37 Fig. S5† presents complementary characterization studies by Fourier-transform infrared spectroscopy (FTIR), followed by the detailed assignment of all bands. Bands corresponding to the epoxide, carboxylic acid, and hydroxyl groups of CG were observed at 1230, 1724, and 3320 cm−1, respectively. Besides, the CN band at 2088 cm−1 is observed for the CG/PB samples. The CG samples do not exhibit any apparent change in the region from 1600 to 1640 cm−1, typical of interstitial water, except for the PB_50 sample, which showed a discrete alteration in the profile. Thus, the interaction between PB nanoparticles and crumpled graphene may directly influence the performance of the CG/PB composites rather than the presence of interstitial water.
N portion.36,37 Fig. S5† presents complementary characterization studies by Fourier-transform infrared spectroscopy (FTIR), followed by the detailed assignment of all bands. Bands corresponding to the epoxide, carboxylic acid, and hydroxyl groups of CG were observed at 1230, 1724, and 3320 cm−1, respectively. Besides, the CN band at 2088 cm−1 is observed for the CG/PB samples. The CG samples do not exhibit any apparent change in the region from 1600 to 1640 cm−1, typical of interstitial water, except for the PB_50 sample, which showed a discrete alteration in the profile. Thus, the interaction between PB nanoparticles and crumpled graphene may directly influence the performance of the CG/PB composites rather than the presence of interstitial water.
Fig. 3(c) exhibits the XPS survey spectra of the CG/Fe (black line) and PB_10 (green line) samples and Table S1† depicts the atomic percentages, peak positions, and the elements identified in both samples. Peaks related to carbon (C 1s), oxygen (O 1s), and iron (Fe 2p) were identified in the two samples. The peak corresponding to chlorine (Cl 2p) is only observed in CG/Fe, which may be attributed to the chloride residue from the iron precursor. Instead, a peak corresponding to nitrogen (N 1s) is noticed in the PB_10 sample, which indicates effectiveness in the electrodeposition of Prussian blue over crumpled graphene. All peaks assigned with an asterisk are related to indium (In) and tin (Sn) coming from the ITO/glass substrate.
The insets in Fig. 3(c) show the high-resolution XPS spectra of the carbon and iron regions, which provide a better understanding of the chemical environment before and after the PB electrodeposition. The C 1s high-resolution spectrum of the CG/Fe sample shows a peak corresponding to CC at 285.0 eV. This indicates an effective restoration of the aromatic sp2 bonds from GO to crumpled graphene throughout the spray-pyrolysis process. Peaks related to C–O, CO, and O–CO at 286.6, 288.3, and 289.5 eV, respectively, denote that the graphene-based material is not completely reduced. Still, in the C 1s region, it is worth noting that the PB_10 spectrum shows a small contribution of FeII–CN at 285.4 eV, indicating the presence of Prussian blue in the composite.40 The Fe 2p high-resolution spectrum of CG/Fe shows a peak at 712.3 eV, confirming the presence of FeOOH on crumpled graphene, as observed from the XRD results. Moreover, a contribution of 710.8 eV is attributed to the Fe2O3 species, although this crystalline structure was not observed in XRD. This is a reasonable result since XPS is a surface-sensitive technique and the co-existence of iron oxide species with similar crystallographic planes to those of FeOOH is feasible since the formation of Fe2O3 is the final step of an acidic FeCl3 solution hydrolysis. Finally, the Fe 2p high-resolution spectrum of PB_10 exhibits a peak at 708.7 eV related to the FeII σ-bonded to carbon in the Prussian blue structure, confirming again the electrodeposition. A higher contribution of a FeII species is a consequence of the electrochemical process since the cyclic voltammetry finishes at a negative potential.41
The morphology of the thin films prepared showed an evolution of the morphology as a function of the number of cycles of PB electrodeposition. For the PB_2 sample (Fig. 4(a)), many CG structures are observed over the surface of the ITO electrode. However, at higher magnifications (Fig. 4(b)), the PB_2 film contains PB nanocubes distributed only on the surface of CG. The formation of cubes on the CG structure indicates the composite formation of CG and PB nanocubes through the electrochemical deposition technique.
 | ||
| Fig. 4 SEM micrographs of the (a and b) PB_2, (c) PB_5, (d) PB_10, (e) PB_25, and (f) PB_50 samples. | ||
These results corroborate with the results of previous XRD, Raman, and XPS analyses. Furthermore, increasing the number and size of the PB cubes toward more CV cycles is straightforward, accompanied by the cube agglomeration tendency, especially in the PB_25 and PB_50 samples.
For the 2-cycle sample in Fig. 4(a), a rough film morphology was observed, with CG structures homogeneously distributed over most of the ITO substrate. This feature is also evident for the PB_5, and PB_10 films presented in Fig. 4(c and d), respectively. However, for the PB_25 and PB_50 samples shown in Fig. 4(e and f), the formation of agglomerates and an increase in empty regions (film defects) were observed, exposing the ITO substrate completely. In addition, cubic nanostructures are present in these defects, which were not previously observed for the PB_2, PB_5, and PB_10 samples. Although not evenly distributed over the film at higher magnifications, the presence of cubic structures with an average size of 48 ± 16 nm on the surface of the PB_2 film is evident. Similarly, the surface of the PB_5 film (Fig. 4(c)) presents cubic structures with a particle size around 70 ± 30 nm in addition to a higher quantity and better distribution of nanoparticles. It is observed that in both composites, no electrodeposited nanoparticles are present in the defective regions of the film. This can indicate that during the initial electrodeposition cycles, growth occurs predominantly on the surface of the CG film.
The histogram of the particle size of the PB cube is shown in Fig. S6.† The PB_10 film (Fig. 4(d)) presents a homogeneous distribution of nanocubes with a size of 82 ± 31 nm on the CG surface. For the PB_25 and PB_50 films, as shown in Fig. 4(e) and (f), respectively, an increase in the number of nanoparticles and their size was recorded, 121 ± 58 nm for the PB_25 film and 153 ± 62 nm for the PB_50 film. Generally, an increase in the number of voltammetric cycles contributes to morphological and structural changes in the film, resulting in different sizes of electrodeposited PB nanoparticles and for the PB_25 and PB_50 samples, the emergence of agglomerates. Electrochemical deposition proved to be efficient and a tool capable of controlling the morphology of the composite. Furthermore, the electrochemical results and morphological images indicate that the nucleation of nanoparticles occurs preferentially on the surface of the CG film between the second and fifth voltammetric cycles. Scheme 4 depicts the mechanism for the electrodeposition of PB nanocubes over the CG surface.
 | ||
| Scheme 4 Representative scheme of PB electrodeposition on thin films of crumpled graphene decorated with iron species. | ||
The electrosynthesis kinetics of PB nanoparticles from metallic species encapsulated in materials such as carbon nanotubes (CNT) and freestanding rGO/PAni films is a process that involves three crucial steps: (i) diffusion of the stabilized species in the carbon nanostructure, followed by the steps of (ii) nucleation and (iii) growth. The anodic direction of the voltammogram initiates the diffusion of the Fe3+ species originating from the β-FeOOH and Fe2O3 nanoparticles of the CG/Fe sample, setting the stage for the subsequent steps.15,36,37 As the working electrode undergoes inversion polarization in the second step, reduction of the iron species released occurs at the electrode/electrolyte interface to Fe2+, creating nucleation points for PB nanoparticles, due to the abundance of [Fe(CN)6]2− species in solution. This establishes the ground for the growth of pre-existent PB nanoparticles and the formation of new nucleation points until the end of the electrodeposition experiment. It is driven by the high concentration of [Fe(CN)6]3− in solution and the constant release of iron from the β-FeOOH and the Fe2O3 species of the CG/Fe sample.
3.3. Electrochemical behavior in different electrolytes and galvanostatic charge/discharge measurements
The intercalation of cations in the HCM structure is a critical aspect of the redox processes within these materials. It arises from the inherent properties of the metallic centers, playing a crucial role in maintaining the chemical structure's charge balance. When examining the intercalation process, it is essential to carefully consider factors such as the hydrated radius of the cation. The diffusion of these charge-carrying species directly and significantly impacts the electrochemical properties, particularly the stability of the HCM and electrochemical activity. Introducing cations with a large hydrated radius, such as Li+, into inadequately sized interstices can reduce electrochemical activity. Additionally, during the oxidation of the PB structure (and analogues), minor deformations can hinder or prevent the cations from leaving the HCM structure.42 Therefore, the electrochemical performance of PB relies heavily on the nature of the cation and structural characteristics, such as the coordination of water molecules and defects in the structures. For these reasons, a comprehensive analysis of various cations is imperative when considering the use of PB in electrochemically active materials for energy storage devices, like aqueous-based batteries.21Thus, the voltammetric stability of the PB_10 electrode was evaluated in 0.1 mol L−1 KCl, 0.1 mol L−1 NaCl, and 0.1 mol L−1 LiCl over 25 CV cycles at pH 5.0 in a similar system to that used for the electrodeposition of PB. For Li+, Na+, and K+ ions, the material showed a current drop below 70% of the anodic peak current intensity (IpA) and the cathodic peak current intensity (IpC) at the end of the 25th voltammetric cycle, as shown in Fig. 5(a). The voltammetric profiles can be found in Fig. S7.† It is possible to observe similar behavior for Li+ and Na+ ions. Both present a drop in IpA and IpC in the initial cycles (2nd cycle for Li+ and 6th cycle for Na+). This electrochemical behavior is directly related to the PB cell and the size of the hydrated radius of the species. Having a smaller unit cell size than those of its analogues, for example, the ruthenium analogue – known as Prussian purple – with 10.42 Å, Prussian blue (10.16 Å) has a certain limitation for electrolytes, with potassium electrolytes generally being used given the small hydrated radius (1.25 Å) when compared to Na+ (1.83 Å) and Li+ (2.37 Å).42 Thus, the insertion of Na+ and Li+ cations causes the breakage of the interstitial sites of PB, leading to the loss of the electrochemical activity of the films. For the K+ ion electrolyte, an increase in the intensities of IpA and IpC of around 135% was observed in the first cycle. There was some stability at this step until the 10th voltammetric cycle. This behavior, contrary to the others, is strong evidence of the compatibility of the K+ ions with the PB structure, followed by an accommodation event of the structure. Fig. 5(b) shows the electrochemical profile of each electrolyte. For the K+ electrolyte, the redox pair of Fe2+/Fe3+ (typical of the PW/PB redox transitions) was observed between 0.0 and 0.4 V and the redox pair for PB/GB between 0.6 and 1.0 V. The intensities and definition of pairs are other important evidence of the affinity of PB nanocubes with the potassium electrolyte.
 | ||
| Fig. 5 Stability of the peak current as a function of CV over 25 CV cycles (a) and voltammetric profiles in 0.1 mol L−1 KCl (black line), 0.1 mol L−1 NaCl (red line), and 0.1 mol L−1 LiCl (purple line) (b) – measurements were taken at 50 mV s−1; galvanostatic charge–discharge curves at 500 mA g−1 in 0.1 mol L−1 KCl (c); and specific capacities of the electrodes at different current densities in 0.1 mol L−1 KCl (d). | ||
Furthermore, the stability of the films was evaluated at different hydrogen potential values of the 0.1 mol L−1 KCl electrolytic solution in CV. As shown in Fig. S8,† an interesting behavior of the PB_10 composite was observed between the pH range of 4.0 and 7.0. For this purpose, films of the PB_10 sample were subjected to 50 voltammetric cycles in a potential range of −0.1 to 1.1 V at 50 mV s−1 by varying the pH of the potassium electrolyte. The relative current intensity between the 2nd and 50th voltammetric cycles was evaluated for the cathodic peak between 0.0 and 0.2 V, corresponding to the redox process of the Fe2+/Fe3+ species of PB. Different behaviors were observed when scanning at pH values of 4.0, 5.0, 6.0, 7.0, and 8.7. The composite of CG and PB showed high stability in acidic electrolytes with a peak intensity loss of current less than 10%, indicating a small loss of electroactive PB. In contrast, the scanning at pH 8.7 showed a significant decrease (∼75% of current loss), indicating the breakdown of the PB structure.
Charge/discharge measurements were performed for all materials for a preliminary study of application in potassium-aqueous battery systems. The presence of plateaus in the charge and discharge profiles for all composites of CG and PB can be observed in Fig. 5(c). Although not a symmetrical profile, there is a clear difference in the electrochemical performance of the composite samples and a typical pseudocapacitive electrochemical behavior for the bare_CG and CG/Fe films in Fig. S9.† Notably, the presence of plateaus is evidence of the battery-type mechanism, which is characteristic of potassium ion diffusion processes in the PB structure.43 The charge/discharge profiles for all composites at a current density of 500 mA g−1 and the potential range between 0.05 and 0.65 V showed different specific capacities among themselves, 23.1, 36.3, 50.4, 41.8, and 39.6 mA h g−1 for the PB_2, PB_5, PB_10, PB_25, and PB_50 composites, respectively. All electrodes were evaluated at different current densities and the specific capacity values are shown in Fig. 5(d) and Table S2.† An increase in the specific capacity of the composites was observed as they were manufactured with a greater number of CV cycles when comparing the materials PB_2, PB_5, and PB_10, reflecting the influence of large amounts of PB and the size of these nanocubes. However, lower specific capacity values were observed for the PB_25 and PB_50 composites, which may reflect the negative effects of nanoparticle aggregation observed in the micrographs of the samples in Fig. 4(e and f). The specific capacity results obtained in this work (Fig. 5(d)), although presenting lower values when compared to other electrodes based on different carbon nanostructures such as rGO and CNTs, reveal great potential given the ease of processing the CG/PB-based composites, without the need for dispersing agents and binders, among others. It should be noted that our study is the pioneering work in developing a nanocomposite based on crumpled graphene and the HCM class, specifically PB. Table 1 presents a short comparison of the PB_5 and PB_10 electrodes with other works.
The PB_10 composite was analyzed for its stability. First, we performed a study at different current densities, as shown in Fig. 6(a), through consecutive cycles of charge/discharge per current density. A significant and linear decrease in discharge capacity was observed when the current density increased to 2000 mA g−1, explained by the internal resistance of the composite that regulates the speed of the K+ ion diffusion process in the PB structure. Values of 43.1, 36.1, 29.6, 24.8, 21.5, and 19.5 mA h g−1 were obtained at rates of 250, 350, 500, 750, 1000, and 2000 mA g−1, respectively. By expanding the range of replicates analyzed, we performed a stability analysis of the material by subjecting it to 500 consecutive charge/discharge cycles at a current density of 2000 mA g−1. The stability of specific capacity is remarkable until around the 200th cycle, where the capacity remains at 40.0 mA h g−1. It was followed by a subtle and constant drop until the 500th cycle, as shown in Fig. 6(b and c), where the electrode showed a specific capacity of about 25.0 mA h g−1. On the other hand, the coulombic efficiency of the composite remains above 90% for the entire period, demonstrating the high reversibility of the redox process between PW and PB. Commonly, at high rates, such as 2000 mA g−1, the intercalation process in the structure of PB nanoparticles occurs less efficiently due to factors such as diffusional limitation and irreversible structural damage.11
 | ||
| Fig. 6 Measurements of the PB_10 sample. (a) Discharge profiles at different rate, (b) stability test at 2000 mA g−1, and (c) charge/discharge stability test profiles. | ||
EIS was performed to understand the reaction kinetics of the CG/Fe and PB_10 electrodes. The Nyquist diagram in Fig. S10† shows different behaviors for both materials. For the interpretation and fitting of the EIS curves, two equivalent circuits were adopted to analyze three main surface phenomena: (i) the charge transfer resistance (CTR) in the high-frequency region; (ii) the diffusion of species in the electric double layer in the intermediary frequency region; and (iii) a second diffusion resistance at low frequency related to the diffusion of species throughout the pores of the material. For the CG/Fe sample, a high resistance (2.2 kΩ) was observed in the low-frequency region, indicating low diffusion kinetics in the regions where the electrolyte accesses the cavities of the CG structure. Contrary to this, an easy electronic transport performed by the surface of the CG/Fe is observed, with a CTR in the high-frequency region of 6.2 Ω. For the PB_10 electrode, an increase in the CTR at a high frequency (29.9 Ω) compared to CG/Fe was recorded due to the electrochemical deposition of PB nanoparticles over CG. Moreover, the resistance in the low-frequency region increased (3.3 kΩ) as an extra diffusion resistance imposed by the PB nanoparticles. These results suggest a significant change in the electrochemical interface of the CG/Fe-based electrodes after modification with Prussian blue nanoparticles.
3.4. Coin-cell battery-type system
We demonstrate the application of thin films based on crumpled graphene and Prussian blue as cathodes for potassium-aqueous electrolyte batteries. As a proof of concept, the optimized PB_10 sample (cathode) was integrated into a coin-cell system with bare_CG as an anode and 0.5 mol L−1 KCl as an electrolyte. Fig. 7(a) shows the cyclic voltammograms recorded at 0.05 mV s−1 in the range from 0.2 to 1.0 V to stabilize the system. In the 2nd cycle, only one redox pair is observed between 0.5 and 0.9 V, corresponding to the typical redox process of PB previously discussed. The 10th voltammetric cycle presents a drop in the anodic process and a sudden rise in current during the cathodic process. The 12th cycle presents some stability for the redox pair. This behavior is mainly associated with activating the electrochemically active area of the electrodes. Before the charge and discharge studies, the cell was cycled three times at a density of 100 mA g−1 for stabilization. Fig. 7(b) shows the discharge profiles, where the cell presented a capacity of 25 mA h g−1 at a current density of 250 mA g−1. The high coulombic efficiency reflects the non-optimized anode based on the bare_CG sample. | ||
| Fig. 7 Measurements of the coin cell battery device using the PB_10 sample as a cathode and bare_CG as an anode. (a) Cyclic voltammetry profiles at 50 mV s−1, (b) charge/discharge profiles, and (c) rate handling performance. | ||
In Fig. 7(c), the cell exhibits stable behavior with a gradual decrease in specific capacity as the current density increases. However, reasonable performance and coulombic efficiency of almost 100% are observed for rates above 750 mA g−1, showing a good performance of the porous cathode based on CG/PB. Coulombic efficiency above 100% is a phenomenon commonly observed in the HCM class and recent studies suggest a direct relationship with the presence of [Fe(CN)6]3− moieties that are not part of the PB structure and come from the synthesis.46 It is important to note that after scanning at different current densities, when cycled again at 250 mA g−1, the device showed 97% capacity recovery, reflecting its stability.
4. Conclusions
This study successfully synthesized films based on composites of crumpled graphene (CG) and Prussian blue (PB) nanoparticles using a simple processing methodology. This approach involved the liquid–liquid interface route (LLIR) for film fabrication and cyclic voltammetry for the electrodeposition of PB nanoparticles, providing a composite with tuneable properties suitable for use as a cathode in aqueous potassium-ion battery energy storage systems.The fine-tuning of composite properties resulted in significant differences in the electrochemical activity of the synthesized materials, with specific capacity values varying according to the amount and size of PB nanoparticles formed. This highlights the direct relationship between the quantity and size of PB nanoparticles and the energy storage performance of the composites.
Moreover, the CG/PB nanocomposite demonstrated considerable promise in real coin-cell test systems. It combined the excellent processability of crumpled graphene with the favourable electrochemical properties of PB, as evidenced in coin-cell systems using aqueous K+-ion electrolytes. These characteristics present exceptional advantages, showcasing the ease of electrode fabrication for rechargeable battery systems and a lower environmental impact due to the use of aqueous electrolytes.
Author contributions
Vitor H. N. Martins: conceptualization, validation, investigation, formal analysis, and writing – original draft. Monize M. Silva: investigation, validation, and formal analysis. Maria K. Ramos: formal analysis and validation. Maria H. Verdan: writing – original draft and writing – review & editing. Eduardo G. C. Neiva: formal analysis and validation. Aldo J. G. Zarbin: writing – original draft, writing – review & editing, supervision, and funding acquisition. Victor H. R. Souza: conceptualization, validation, formal analysis, writing – original draft, writing – review & editing, supervision, project administration, and funding acquisition.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from CNPq, CAPES, National Institute of Science and Technology of Nanomaterials for Life (INCT NanoLife – Brazil, Grant #406079/2022-6), the INCT of Carbon Nanomaterials, Fundação de Apoio ao Desenvolvimento do Ensino, Ciência e Tecnologia do Estado de Mato Grosso do Sul (FUNDECT, #83/013.569/2023 and #71/055.441/2022), the National Council for Scientific and Technological Development (Grant #423918/2018-4, and 403197/2021-0) and Financiadora de Estudos e Projetos (FINEP – Brazil, Grant # 04.13.0448.00/2013). V. H. N. M. acknowledges the Coordination of Superior Level Staff Improvement (CAPES-Brazil). The authors thank the Brazilian Nanotechnology National Laboratory (LNNano) for the XPS measurements, the Quality Control and Thermal Analysis Laboratory (Labcat) at the Federal University of Grande Dourados for the TG analysis, and the Materials Chemistry Group at the Federal University of Paraná (GQM-UFPR) for the SEM-FEG, XRD, and Raman measurements.References
- S. Fawzy, A. I. Osman, J. Doran and D. W. Rooney, Environ. Chem. Lett., 2020, 18, 2069 Search PubMed.
- S. Christopher, M. P. Vikram, C. Bakli, A. K. Thakur, Y. Ma, Z. Ma, H. Cu, P. M. Cuce, E. Cuce and P. Singh, J. Cleaner Prod., 2023, 405, 136942 Search PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- D. Larcher and J.-M. Tarascon, Nat. Chem., 2014, 7, 19 Search PubMed.
- C. Schubert, Nature, 2011, 474, 531–533 Search PubMed.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- A. Amato, A. Becci, M. Villen-Guzman, C. Vereda-Alonso and F. Beolchini, J. Cleaner Prod., 2021, 300, 126954 Search PubMed.
- D. Chao, D. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S. Qiao, Sci. Adv., 2020, 6, eaba4090 Search PubMed.
- F. Wan, X. Zhou, Y. Lu, Z. Niu and J. Chen, ACS Energy Lett., 2020, 5, 3569 CrossRef CAS.
- G. Liang, F. Mo, X. Ji and C. Zhi, Nat. Rev. Mater., 2020, 6, 109 CrossRef.
- S. Husmann, A. J. G. Zarbin and R. A. W. Dryfe, Electrochim. Acta, 2020, 349, 136243 CrossRef CAS.
- L. C. Lopes, S. Husmann and A. J. G. Zarbin, Electrochim. Acta, 2020, 345, 136199 CrossRef CAS.
- B. Bornamehr, V. Presser, A. J. G. Zarbin, Y. Yamauchi and S. Husmann, J. Mater. Chem. A, 2023, 11, 10473 RSC.
- S. Husmann and A. J. G. Zarbin, Chem. - Eur. J., 2016, 22, 6643 CrossRef CAS PubMed.
- S. Husmann, E. Nossol and A. J. G. Zarbin, Sens. Actuators, B, 2014, 192, 782 CrossRef CAS.
- C. D. Wessells, S. V. Peddada, M. T. McDowell, R. A. Huggins and Y. Cui, J. Electrochem. Soc., 2012, 159, A98 CrossRef CAS.
- L. Chen, L. Zhang, X. Zhou and Z. Liu, ChemSusChem, 2024, 7, 2295 CrossRef PubMed.
- V. D. Neff, J. Electrochem. Soc., 1978, 125, 886 CrossRef CAS.
- D. Moscone, D. D'Ottavi, D. Compagnone, G. Palleschi and A. Amine, Anal. Chem., 2001, 73, 2529 CrossRef CAS PubMed.
- J. Luo, H. D. Jang, T. Sun, L. Xiao, Z. He, A. P. Katsoulidis, M. G. Kanatzidis, J. M. Gibson and J. Huang, ACS Nano, 2011, 5, 8943 CrossRef CAS PubMed.
- B. Konkena and S. Vasudevan, J. Phys. Chem. Lett., 2012, 3, 867 CrossRef CAS PubMed.
- L. H. Nonaka, T. S. D. Almeida, C. B. Aquino, S. H. Domingues, R. V. Salvatierra and V. H. R. Souza, ACS Appl. Nano Mater., 2020, 3, 4859 CrossRef CAS.
- L. M. Alencar, A. W. B. N. Silva, M. A. G. Trindade, R. V. Salvatierra, C. A. Martins and V. H. R. Souza, Sens. Actuators, B, 2022, 360, 131649 CrossRef CAS.
- X. Dou, A. R. Koltonow, X. He, H. D. Jang, Q. Wang, Y.-W. Chung and J. Huang, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1528 CrossRef CAS PubMed.
- D. A. Gonçalves, L. M. Alencar, J. P. B. Anjos, E. S. Orth and V. H. R. Souza, Mater. Today Commun., 2023, 36, 106746 CrossRef.
- A. E. Silva, V. H. R. D. Souza and E. G. C. Neiva, Appl. Surf. Sci., 2023, 622, 156967 CrossRef CAS.
- D. A. Gonçalves, V. H. N. Martins, D. D. Reis, M. M. Silva and V. H. R. Souza, RSC Adv., 2024, 14, 29134 RSC.
- S. Husmann, M. K. Ramos and A. J. G. Zarbin, Electrochim. Acta, 2022, 422, 140548 CrossRef CAS.
- E. G. C. Neiva and A. J. G. Zarbin, New J. Chem., 2022, 46, 11118 Search PubMed.
- S. Husmann and A. J. G. Zarbin, Electrochim. Acta, 2018, 283, 1339 Search PubMed.
- E. Nossol, V. H. R. Souza and A. J. G. Zarbin, J. Colloid Interface Sci., 2016, 478, 107 Search PubMed.
- R. V. Salvatierra, M. M. Oliveira and A. J. G. Zarbin, Chem. Mater., 2010, 22, 5222 Search PubMed.
- A. Schmidt, M. K. Ramos, C. S. Pinto, A. F. Pereira, V. H. R. Souza and A. J. G. Zarbin, Electrochem. Commun., 2022, 134, 107183 CrossRef CAS.
- V. H. R. Souza, E. Flahaut and A. J. G. Zarbin, J. Colloid Interface Sci., 2017, 502, 146 CrossRef CAS PubMed.
- C. S. Pinto, V. H. R. Souza, A. Schmidt and A. J. G. Zarbin, Synth. Met., 2023, 293, 117259 Search PubMed.
- V. H. N. Martins, M. M. D. Silva, D. A. Gonçalves, V. Presser, S. Husmann and V. H. R. Souza, ACS Omega, 2024, 9, 31569 CrossRef CAS PubMed.
- E. Nossol and A. J. G. Zarbin, Adv. Funct. Mater., 2009, 19, 3980 CrossRef CAS.
- A. Šarić, K. Nomura, S. Popović, N. Ljubešić and S. Musić, Mater. Chem. Phys., 1998, 52, 214 Search PubMed.
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang and K. S. Novoselov, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS PubMed.
- J. B. Martins, S. Husmann, A. G. Veiga, A. J. G. Zarbin and M. L. M. Rocco, ChemPhysChem, 2024, 25, e202300590 CrossRef CAS PubMed.
- Y. Xue, Y. Chen, X. Shen, A. Zhong, Z. Ji, J. Cheng, L. Kong and A. Yuan, J. Colloid Interface Sci., 2022, 609, 297 CrossRef CAS PubMed.
- A. L. Crumbliss, P. S. Lugg and N. Morosoff, Inorg. Chem., 1984, 23, 4701 Search PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- Y. Wei, H. Wang, J. Wang, C. Gao, H. Zhang, F. Yuan, J. Dong, D. Zhai and F. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 54079 CrossRef CAS PubMed.
- C. L. Zhang, Y. Xu, M. Zhou, L. Y. Liang, H. S. Dong, M. H. Wu, Y. Yang and Y. Lei, Adv. Funct. Mater., 2017, 27, 1604307 CrossRef.
- H. Yu, J. Xu, C. Deng, M. Xia, X. Zhang, J. Shu and Z. Wang, ACS Appl. Energy Mater., 2021, 4, 9594 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05272b |
| This journal is © The Royal Society of Chemistry 2025 |