
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMorphology-tuned MnOx/TiO2 nanocatalysts for recycling PET plastic waste using biomass-derived ethylene glycol†
Bhattu
Swapna
a,
Madam Bobby
Barnabas
a,
Pragya
Moni Gogoi
b,
Pankaj
Bharali
b,
Giridhar
Madras
c and
Putla
Sudarsanam
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Kandi 502284, Telangana, India. E-mail: sudarsanam.putla@chy.iith.ac.in
bDepartment of Chemical Sciences, Tezpur University, Napaam 784028, Assam, India
cDepartment of Chemical Engineering, Indian Institute of Technology Hyderabad, Kandi 502284, Telangana, India
First published on 10th February 2025
Abstract
This study presents a decisive role of TiO2 morphology on the catalytic activity of MnOx/TiO2 nanomaterials for the chemical recycling of PET waste bottles using biomass-derived ethylene glycol to produce a valuable monomer, bis(2-hydroxyethyl) terephthalate (BHET). Three types of MnOx/TiO2 nanocatalysts were prepared by varying the TiO2 morphology (nanosheets: NS, nanotubes: NT, and nanorods: NR). The combination of MnOx nanoparticles and TiO2 nanorods (MnOx/TiO2-NR) showed significantly enhanced catalytic activity in PET glycolysis, with a 91% isolated yield of BHET at 190 °C in 3 h, whereas 74% and 82% yields of BHET were attained with MnOx/TiO2-NS and MnOx/TiO2-NT nanocatalysts, respectively. The morphology of TiO2 and the uniform dispersion of MnOx on TiO2-NR were confirmed by electron microscopic analysis. The MnOx/TiO2-NR catalyst contains optimum basic sites, which play a key role, along with surface hydroxyl species and Mn3+/Mn2+ species, in activating ethylene glycol and PET/its oligomers towards BHET formation. The excellent stability of the MnOx/TiO2-NR nanocatalyst, as confirmed by the hot-filtration test, good catalytic reusability up to four cycles, non-toxic nature, and the low cost of the MnOx/TiO2 materials indicate the practical feasibility of the developed catalytic protocol for the plastic recycling industry.
 Putla Sudarsanam | Dr Sudarsanam Putla is an Assistant Professor at the Department of Chemistry, Indian Institute of Technology Hyderabad, India. He completed his M.Sc. (Chemistry) at IIT Madras and Ph.D. at CSIR-IICT, Hyderabad. From 2014 to 2019, he worked as a postdoctoral fellow at RMIT University (Melbourne), LIKAT (Germany), and KU Leuven (Belgium). He has received several awards and fellowships, including the PECFAR award – 2023 (Indo-German Science & Technology Centre), the Associate Fellow of the Telangana Academy of Sciences – 2022, and the Australian Alumni Award – 2021. His research focuses on developing novel heterogeneous catalysts for plastic waste recycling and biomass valorization to fuels, monomers, and value-added chemicals. He has authored ∼80 journal articles (h-index: 41, ∼5800 citations), edited 4 books, and serves on editorial boards of ACS Sustainable Chemistry & Engineering, Molecular Catalysis, and Applied Catalysis O: Open journals. |
1. Introduction
Plastic is used widely because of its low cost, durability, and wide-range of applications.1–3 However, the use of plastic generates vast amounts of waste and causes environmental pollution. Current recycling processes only manage to recycle 9% of the plastic waste into valuable materials and chemicals, with the rest ending up in landfills or being incinerated.4–11 Among various plastics, polyethylene terephthalate (PET) is the most used polyester plastic due to its consistent properties and numerous applications.12–14 Recycling PET waste into materials, monomers, and chemicals is essential for mitigating plastic pollution and reducing the use of fossil feedstock to produce polyester-based materials.15–17 Recycling one ton of PET can save ∼5.6 barrels of oil that would otherwise be used to produce virgin PET polymers.18,19Mechanical and chemical recycling methods have been widely investigated for PET waste valorization.20,21 Mechanical recycling can transform PET waste bottles into new plastic products with minimal changes to the chemical structure. However, this time-consuming method needs multiple steps, resulting in low-quality products.21,22 In contrast, chemical recycling is an efficient process for transforming PET waste into high-quality products under mild conditions.22–24 PET glycolysis using biomass-derived ethylene glycol (EG) is both a cost-effective and environmentally sustainable process to produce bis(2-hydroxyethyl) terephthalate (BHET), a vital monomer for various plastic products.22,25 EG can be produced in greater quantities through the catalytic hydrogenolysis of renewable cellulosic biomass.26,27 Catalysts are essential for activating the ester bonds in PET and the hydroxyl groups in EG during the glycolysis reaction to achieve higher yields of the BHET monomer.22,28–32 In this regard, applying low-cost and efficiently recyclable heterogeneous catalysts can provide industrially compatible methods for PET recycling, thus promoting the circular plastic economy (Fig. 1).
 | ||
| Fig. 1 Catalytic glycolysis of post-consumer PET bottles to the valuable BHET monomer towards the circular plastic economy. | ||
Nanostructured metal oxide-based catalysts are particularly effective for heterogeneous catalysis, including plastic waste recycling, due to their size and shape-tunable surface chemistry, higher surface area, and abundant acid–base sites.33–36 The shape-tuning of metal oxide particles can optimize the coordination geometry of metal and oxygen sites, along with more surface-exposed sites like edges, corners, and kinks, which can provide a high concentration of selective active sites for achieving enhanced catalytic activity with desirable products. Titanium dioxide (TiO2) is a commonly used support material for anchoring catalytically active sites for various energy and environmental applications.37–40 The surface features/geometries, defects, and crystalline phases (anatase and rutile) of TiO2 nanomaterials can control the adsorption/activation of reactants/intermediates, influencing reaction pathways and catalytic performance. The morphology of TiO2, including nanosheets, nanotubes, and nanorods, offers distinct advantages in heterogeneous catalysis due to variations in surface area, exposed facets, and unsaturated surface-Tix+ sites. Controlling these morphologies and elucidating their role in the structure–activity properties of nanocatalysts can offer potential strategies to advance the heterogeneous catalysis field for plastic waste recycling applications.
Manganese oxides (MnOx) are widely utilized in heterogeneous catalysis due to their low cost, diverse crystal structures, multiple oxidation states, and facile redox Mn sites.21,34,41,42 These characteristics make MnOx a versatile catalyst for various acid–base and redox reactions.43 Our recent work emphasized the catalytic role of a MnOx nanomaterial for PET glycolysis.43 The combination of MnOx with redox metal oxides like TiO2 can lead to synergistic properties with enhanced catalytic activity and selectivity for PET glycolysis. This combination inspired us to develop advanced MnOx/TiO2 nanocatalysts by varying the morphology of TiO2 (nanosheets, nanotubes, and nanorods) for PET glycolysis using biomass-derived EG. The MnOx catalyst supported on TiO2 nanorods (NR) demonstrated superior catalytic performance, achieving a 91% isolated yield of BHET under mild conditions compared to the existing literature.22,44 The structure–activity relationship of MnOx/TiO2 nanocatalysts was elucidated by comparing the properties of the catalysts investigated using various techniques (powder XRD, BET, XPS, TEM, STEM-EDAX, and CO2-TPD) with catalytic activity data. The role of basic sites, surface hydroxyl groups, and Mn3+/Mn2+ species in activating PET and EG was elucidated.
2. Experimental section
2.1. Catalyst synthesis
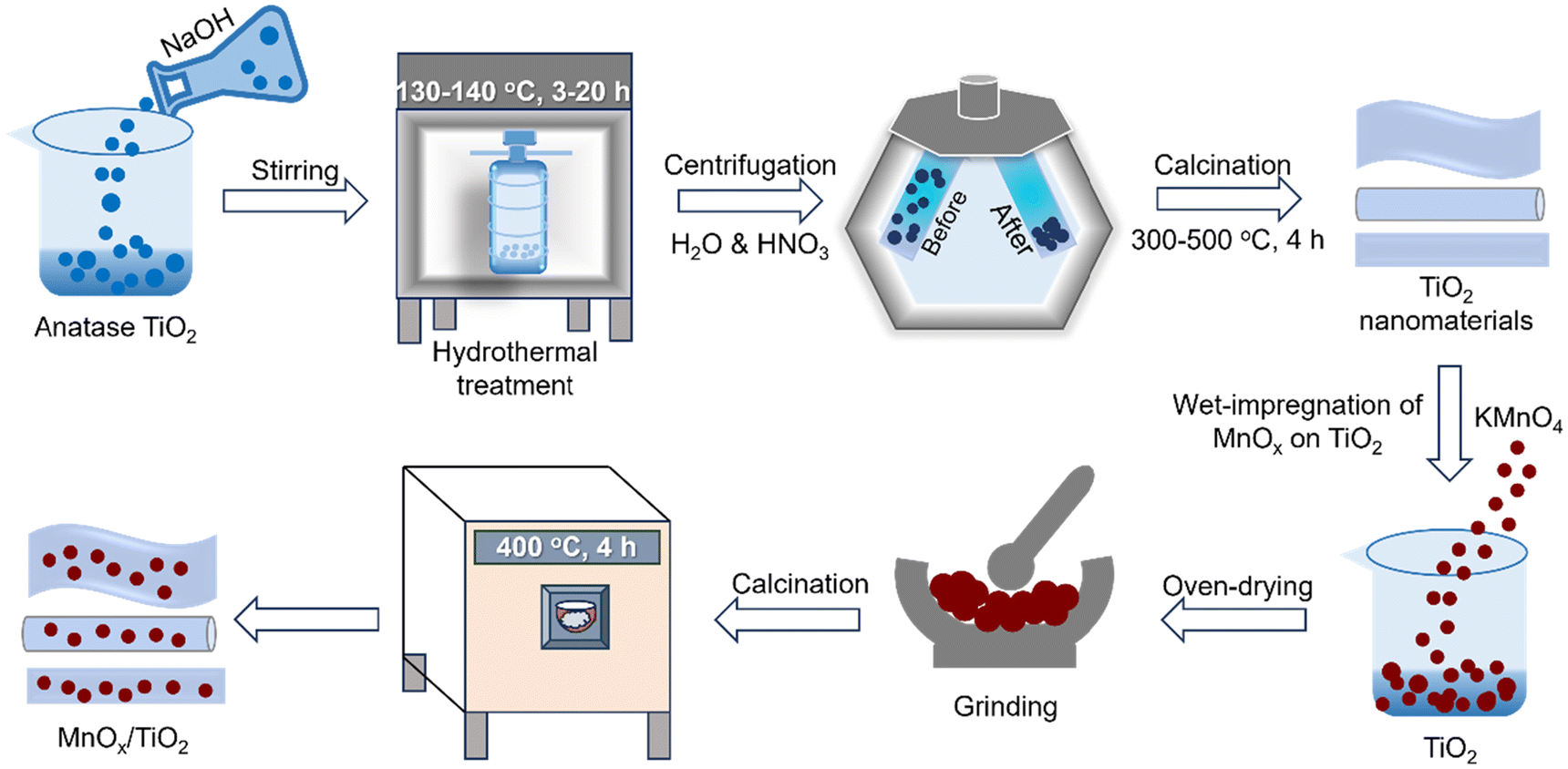 | ||
| Fig. 2 The synthesis procedure of TiO2 and MnOx/TiO2 nanocatalysts. | ||
A similar hydrothermal method with modifications was used for synthesizing TiO2 nanotubes (TiO2-NT) and nanorods (TiO2-NR).40,46 Anatase TiO2 powder (Sigma Aldrich, 99.9%) was dispersed in 5 mL of distilled water and ultrasonicated for 5 min, followed by the gradual addition of 70 mL of NaOH solution (10 mol L−1) over a period of 10 min (Fig. 2). After stirring for 30 min, the solution was treated in a hydrothermal autoclave at 140 °C for 20 h in an oven. The solid product was centrifuged several times using a dilute HNO3 solution (0.1 mol L−1) and deionized water until the pH reached 7. The solid sample was oven-dried at 100 °C for 12 h and then calcined at 400 °C and 500 °C for 4 h to obtain TiO2-NT and TiO2-NR materials, respectively.
2.2. Characterization studies of the TiO2 and MnOx/TiO2 nanocatalysts
The details are provided in the ESI.†2.3. Reaction procedure for PET glycolysis
Used PET plastic bottles were washed with soap water and acetone to remove the impurities. The cleaned bottles were then cut into small pieces of ∼3 mm for the reaction, which was conducted at 190 °C for 3 h using the required amounts of PET, EG, and catalyst. After the reaction, the mixture was filtered to remove unreacted PET and catalyst. The filtrate was heated to 70 °C and stirred for 1 h to achieve supersaturation of the solvent, and then placed in a refrigerator for 24 h to obtain BHET monomer crystals.The conversion of PET and BHET yield were estimated using the following equations, wherein Wi and Wf are the initial and final weights of PET, respectively.
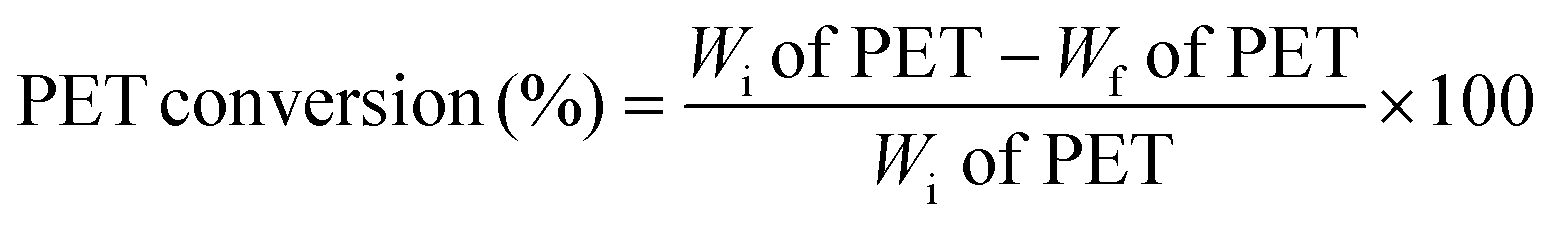

3. Results and discussion
3.1. TEM and powder XRD studies of TiO2 and MnOx/TiO2 nanocatalysts
The effects of hydrothermal treatment and calcination temperature on the morphology of TiO2 were confirmed by TEM analysis. The hydrothermal treatment at 130 °C for 3 h, followed by calcination at 300 °C for 4 h, gave the nanosheet morphology of TiO2 (TiO2-NS) with more than 1 micrometer size of side length (Fig. S1a, ESI†). In contrast, the hydrothermal treatment at 140 °C for 20 h, and then calcination at 400 °C and 500 °C for 4 h provided the nanotube (length = 55–235 nm and width = 9–12 nm, Fig. S1b, ESI†) and a major fraction of nanorod morphology particles (length = 35–174 nm and width = 7–9 nm, Fig. 3a), respectively. The TEM analysis of the most active MnOx/TiO2-NR catalyst (Fig. 3b and c) revealed the presence of TiO2 nanorods, along with a few nanotubes and MnOx nanoparticles (particle size of 6–11 nm). The STEM-EDAX elemental mapping analysis shows the homogeneous dispersion of Mn species in the MnOx/TiO2-NR catalyst (Fig. 3d). The homogeneous dispersion of Mn species can provide uniform surface-active sites, which can play a favorable role in heterogeneous catalysis to achieve higher reaction rates with enhanced product selectivity, including PET glycolysis reaction. | ||
| Fig. 3 Bright field TEM images of (a) TiO2-NR and (b) MnOx/TiO2-NR catalysts. (c) Dark-field image of the MnOx/TiO2-NR catalyst. (d) STEM-EDAX elemental mapping images of the MnOx/TiO2-NR catalyst. | ||
The powder XRD analysis showed various sharp diffraction peaks at 2θ = 25.3, 36.7, 37.7, 38.5, 48.1, 53.7, 55.1, 62.6, 68.7, 70.2, and 75.2°, corresponding to the anatase phase of TiO2 in all the materials40,45,46 (Fig. 4a). Pristine TiO2 nanomaterials exhibited a variation in the 2θ values of these peaks, attributed to the structural perturbations induced by the morphology of TiO2 (Fig. 4b). Similar diffraction peaks of anatase TiO2 were observed in MnOx/TiO2 nanocatalysts (Fig. 4a) with a noticeable peak shift compared to pristine TiO2 nanomaterials (Fig. 4b). This shift indicates the interaction of MnOx with TiO2, leading to structural perturbations in the lattice of anatase TiO2. Besides, the MnOx/TiO2 nanocatalysts have two diffraction peaks at 2θ = 12.3 and 28.8°, corresponding to the α-MnO2 phase (JCPDS file no. 44-0141).47
 | ||
| Fig. 4 (a) Powder XRD and (b) magnified powder XRD of TiO2 and MnOx/TiO2 nanocatalysts. | ||
3.2. XPS analysis of TiO2 and MnOx/TiO2 nanocatalysts
XPS analysis was conducted to determine the oxidation states of Mn, Ti, and O species, as well as their interactions in the catalysts. The Mn 2p XP spectra exhibited two spin–orbit doublets, namely Mn 2p3/2 and Mn 2p1/2, with the corresponding peaks at 636.1–644.8 and 647.5–655.1 eV, respectively (Fig. 5a).43,46,48–51 Deconvolution of the Mn 2p3/2 peak revealed two major peaks at 639.6–639.9 eV and 642.4–642.6 eV, indicating the presence of Mn2+ and Mn3+ oxidation states, respectively, with a major amount of Mn2+ in all MnOx/TiO2 catalysts.40,46 The MnOx/TiO2-NR catalyst has a higher percentage of Mn3+/2+ (71.8%) compared to the MnOx/TiO2-NS (15.8%) and MnOx/TiO2-NT (53.9%) catalysts. The Mn3+/2+ is a strong redox couple, and the more electron-deficient Mn3+ species can interact with the oxygen of the PET ester groups, inducing their susceptibility to the attack of EG. Though the powder XRD analysis showed the presence of MnO2 (Fig. 4a), using ultra-high vacuum conditions during XPS analysis can lead to the reduction of metal ions, which could be the reason for the absence of Mn4+ species in Mn 2p XPS spectra (Fig. 5a).52,53 The Ti 2p XPS spectra showed Ti4+ species with two strong peaks around 457.2 and 463.1 eV, corresponding to Ti 2p3/2 and Ti 2p1/2, respectively (Fig. 5b).54 Variation in BEs of the Ti4+ species was observed after adding MnOx, indicating the strong Mn–Ti interaction. The O 1s spectra showed different types of oxygen species (Fig. 5c). The first peak around 527.1 eV represents lattice oxygen (OL) bound to Ti4+ in pristine TiO2 or Mn3+/2+ in MnOx/TiO2 catalysts.43,46,48 The second peak around 529.1 eV corresponds to the adsorbed oxygenated species on oxygen vacancy sites (OV). The peak around 531.3 eV belongs to the surface hydroxyl (OOH) species in the catalysts. While the MnOx/TiO2 catalysts exhibited all three types of oxygen species; pristine TiO2 materials did not display the O 1s peak of the OOH species, which can function as Brønsted basic sites.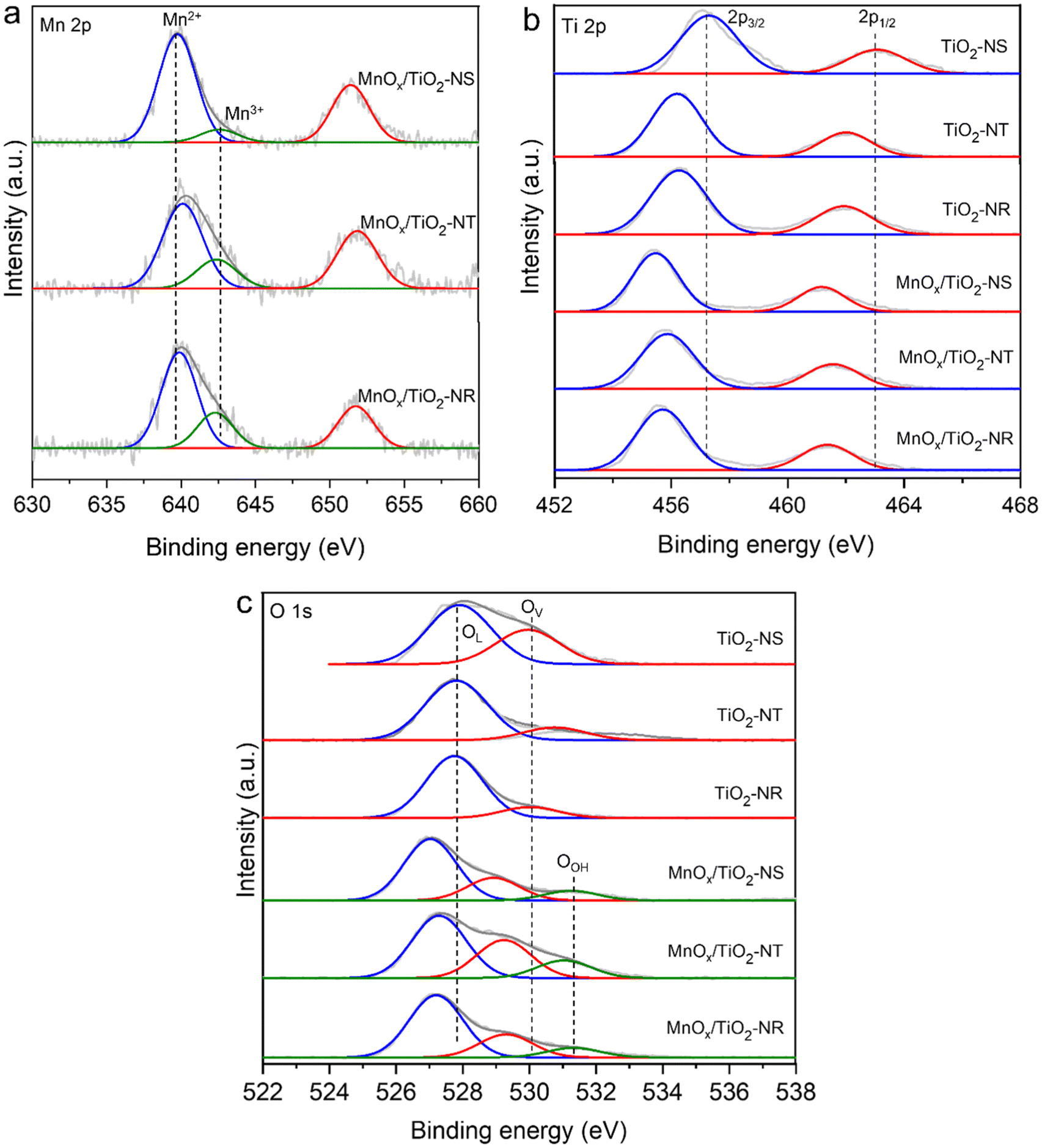 | ||
| Fig. 5 (a) Mn 2p, (b) Ti 2p, and (c) O 1s XPS spectra of TiO2 and MnOx/TiO2 catalysts. | ||
3.3. N2 adsorption–desorption and CO2-TPD studies of TiO2 and MnOx/TiO2 nanocatalysts
Both pristine TiO2 and MnOx/TiO2 nanocatalysts exhibited type IV isotherms with H3 hysteresis loops, indicative of slit-shaped pores/voids between the nanosized particles (Fig. S2, ESI†). Among the shape-controlled TiO2 materials (Table S1, ESI†), the TiO2-NS sample contains the highest BET surface area (198 m2 g−1), followed by TiO2-NT (185 m2 g−1) and TiO2-NR (129 m2 g−1). MnOx/TiO2 nanocatalysts showed lower BET surface areas compared to their corresponding TiO2 samples, which can be attributed to the aggregation of MnOx particles and/or blockage of the slit-shaped pores/voids by MnOx particles. The materials exhibited the average pore size and pore volume in the ranges of 25.06–26.18 nm and 0.288–0.604 cm3 g−1, respectively (Table S1, ESI†).Both the acid and base sites of the catalysts play a key role in the PET glycolysis reaction by activating the ester groups of PET and the alcohol group of EG, respectively.33,36,43,55,56 Electron-deficient metal species like Ti4+ and Mn3+/Mn2+ can activate the ester groups of the PET polymer. Although TiO2 can show a mild Lewis acidic nature, its role is minimal in PET glycolysis, as all the TiO2 materials showed very low activity compared with the MnOx/TiO2 catalysts (Table 1). The lattice oxygen (O2−) and/or surface hydroxyl species of metal oxides can act as Lewis and Brønsted basic sites, respectively, which can accelerate the PET glycolysis by activating EG. Thus, we investigated the basic properties of these catalysts using the CO2-TPD analysis (Fig. 6). All types of basic sites, namely weak (<200 °C), medium (200–400 °C), and strong (>400 °C) basic sites were found in the catalysts (Fig. 6a).17,43 The MnOx/TiO2 catalysts showed a higher concentration of basic sites than the pristine TiO2 materials (Fig. 6b). The surface hydroxyl species of MnOx/TiO2 catalysts (Fig. 5c), which are absent in TiO2, likely contribute to the higher concentration of basic sites in MnOx/TiO2 catalysts. Among all the catalysts, the MnOx/TiO2-NR catalyst exhibited the highest concentration of basic sites (Fig. 6b), which could be the reason for its higher catalytic activity in PET glycolysis as discussed in the following sections.
 | ||
| Fig. 6 (a) CO2-TPD profiles and (b) basic site concentration of TiO2 and MnOx/TiO2 catalysts. | ||
|
|
|||
|---|---|---|---|
| S. No. | Catalyst | PET conversion (%) | Isolated BHET yield (%) |
a Reaction conditions: 5 wt% of the catalyst concerning PET, 190 °C, 3 h, and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20 molar ratio of PET to EG.
b SRL, 99.9% purity. Error in isolated BHET yield: ±3%. :20 molar ratio of PET to EG.
b SRL, 99.9% purity. Error in isolated BHET yield: ±3%.
|
|||
| 1 | Blank | 12 | Trace |
| 2 | TiO2-NSa | 76 | 14 |
| 3 | TiO2-NT | 69 | 19 |
| 4 | TiO2-NR | 74 | 17 |
| 5 | MnOx/TiO2-NS | 99 | 74 |
| 6 | MnOx/TiO2-NT | 99 | 82 |
| 7 | MnOx/TiO2-NR | 99 | 91 |
| 8 | 3 wt% MnOx/TiO2-NR | 88 | 72 |
| 9 | 7 wt% MnOx/TiO2-NR | 99 | 81 |
| 10 | Commercial MnO2b |
99 | 67 |
| 11 | Commercial MnOb | 99 | 73 |
| 12 | Commercial Mn2O3b |
99 | 78 |

3.4. Catalytic glycolysis of PET waste
:12 molar ratio of PET:EG was taken, the PET chips were dissolved in EG over a period of 3 h at 190 °C, giving 89% PET conversion with 41% yield of BHET (Fig. 7b). A further increase of the EG amount (1:20 molar ratio of PET:EG) led to the rapid solubility of PET in EG, which can facilitate improved interaction of the catalyst's active sites with PET ester groups, giving 91% yield of BHET (Fig. 7b). Increasing the EG concentration beyond this ratio resulted in a decline in BHET yield. To determine the optimal reaction time, glycolysis of PET was carried out from 1 to 5 h in 30 min intervals (Fig. 7c). About 51% yield of BHET was attained with the 92% conversion of PET after 2 h. Further, an increase in the reaction time to 3 h led to the highest BHET yield (91%), but the backward reaction was initiated beyond 3 h.
 | ||
| Fig. 7 Optimization of the reaction conditions for PET glycolysis using the MnOx/TiO2-NR catalyst: (a) temperature, (b) PET/EG molar ratio, and (c) reaction time. Reaction conditions: 1:20 molar ratio of PET:EG, 5 wt% MnOx/TiO2-NR catalyst, 190 °C, and 3 h. Error in isolated BHET yield: ±3%. | ||
 | ||
| Fig. 8 (a) PET conversion with the reaction time, (b) the plot of −ln(1 − q) with the reaction time (q = 1 − CP/CP0), (c) conversion of PET at different temperatures and time intervals, and (d) Arrhenius plot of the PET glycolysis reaction over the MnOx/TiO2-NR catalyst. | ||
The kinetic analysis of PET glycolysis was carried out using a 5 wt% MnOx/TiO2-NR catalyst at various reaction time intervals in a temperature range of 160 to 190 °C. The effect of reaction temperature with time was scrutinized using the kinetic plots for PET glycolysis (Fig. 8c). The rate constants obtained at various temperatures were then represented in an Arrhenius plot (Fig. 8d). Using the rate constants, the activation energy (Ea) could be obtained using following the equation.
| lnk = lnA − Ea/RT |
The activation energy (Ea) was determined from the slope to be around 40 kJ mol−1 (Fig. 8d), which is much smaller than those reported in the literature. For instance, Schlüter et al. found the necessity of higher Ea (105 kJ mol−1) for PET glycolysis at 190 °C over a zinc acetate catalyst using γ-valerolactone as a co-solvent.57 In the case of Zn/Al mixed oxide, about 79.3 kJ mol−1 of Ea is needed to achieve an 82% yield of BHET under optimized reaction conditions.58 Though FAU-type zeolite is active for PET glycolysis reaction, it requires a higher Ea value (233.9 kJ mol−1) to obtain optimum yields of BHET.59 The mineral basic catalyst i.e., sodium carbonate showed an 80% yield of BHET at 196 °C for 1 h, and the Ea was estimated to be 185 kJ mol−1.60 The comparison of our results with the literature reports reveals that the MnOx/TiO2-NR catalyst is highly active for the PET glycolysis reaction with a much smaller Ea (40 kJ mol−1) to obtain a 91% yield of BHET.
:20 molar ratio of PET:EG) was conducted at 190 °C for 3 h over the 5 wt% MnOx/TiO2-NR catalyst. A 58% yield of BHET was attained with the 99% conversion of PET after 1.5 h. Afterwards, the catalyst was removed from the reaction mixture by filtration, and the reaction continued for another 1.5 h. The variation in the BHET yield (62%) was negligible after the catalyst removal at 1.5 h, confirming the high stability of the MnOx/TiO2-NR catalyst without any leaching of the active sites (Fig. 9a).
 | ||
| Fig. 9 (a) Hot filtration test of the MnOx/TiO2-NR catalyst for PET glycolysis (reaction conditions: 1:20 molar ratio of PET:EG, 5 wt% MnOx/TiO2-NR catalyst, 190 °C, and 3 h). (b) Reusability studies (reaction conditions: 1:20 molar ratio of PET:EG, 5 wt% MnOx/TiO2-NR catalyst, 190 °C, and 3 h). Error in isolated BHET yield: ±3%. | ||
The reusability of the MnOx/TiO2-NR catalyst for PET glycolysis was tested at 190 °C for 3 h with a 1:20 molar ratio of PET:EG using a 25 mg catalyst (5 wt% catalyst with respect to 0.5 g PET). The catalyst was reused for up to four cycles (Fig. 9b). About 20 mg of the catalyst was recovered after the reaction through filtration. To maintain the 25 mg catalyst for each cycle, we simultaneously carried out two batches of each reusability reaction, obtaining about 40 mg of the catalyst from two batches. The recovered catalyst was dried in an oven at 80 °C for 12 h, collected from the filter paper, washed with methanol to remove impurities, and then dried again at 80 °C for 12 h before calcination at 400 °C for 4 h. The calcined MnOx/TiO2-NR catalyst (25 mg) was used for the subsequent cycle. Only a small decrease in BHET yield was noticed after the 2nd (88%), 3rd (86%), and 4th (84%) cycles, demonstrating the catalyst's stability and consistent activity in PET glycolysis (Fig. 9b). The XPS analysis of the reused MnOx/TiO2-NR catalyst revealed that the peaks for both Mn2+ and Mn3+ were shifted to higher BEs compared to the fresh MnOx/TiO2-NR catalyst (Fig. S7a, ESI†). This shift is attributed to the participation of Mn3+/2+ species in the PET glycolysis. However, the BEs of Ti4+ species remained consistent in both fresh and reused MnOx/TiO2-NR catalysts (Fig. S7b, ESI†), indicating their role is negligible in PET glycolysis. Adequate amounts of surface hydroxyl species were present in the reused MnOx/TiO2-NR catalyst (Fig. S7c, ESI†). The reused MnOx/TiO2-NR catalyst exhibited 0.921 mmol g−1 basic sites, whose concentration is 1.002 mmol g−1 in the fresh catalyst (Fig. 6b). Thus, the presence of adequate basic sites and surface hydroxyl species in the MnOx/TiO2-NR catalyst is the reason for its good reusability in PET glycolysis (Fig. 9b).
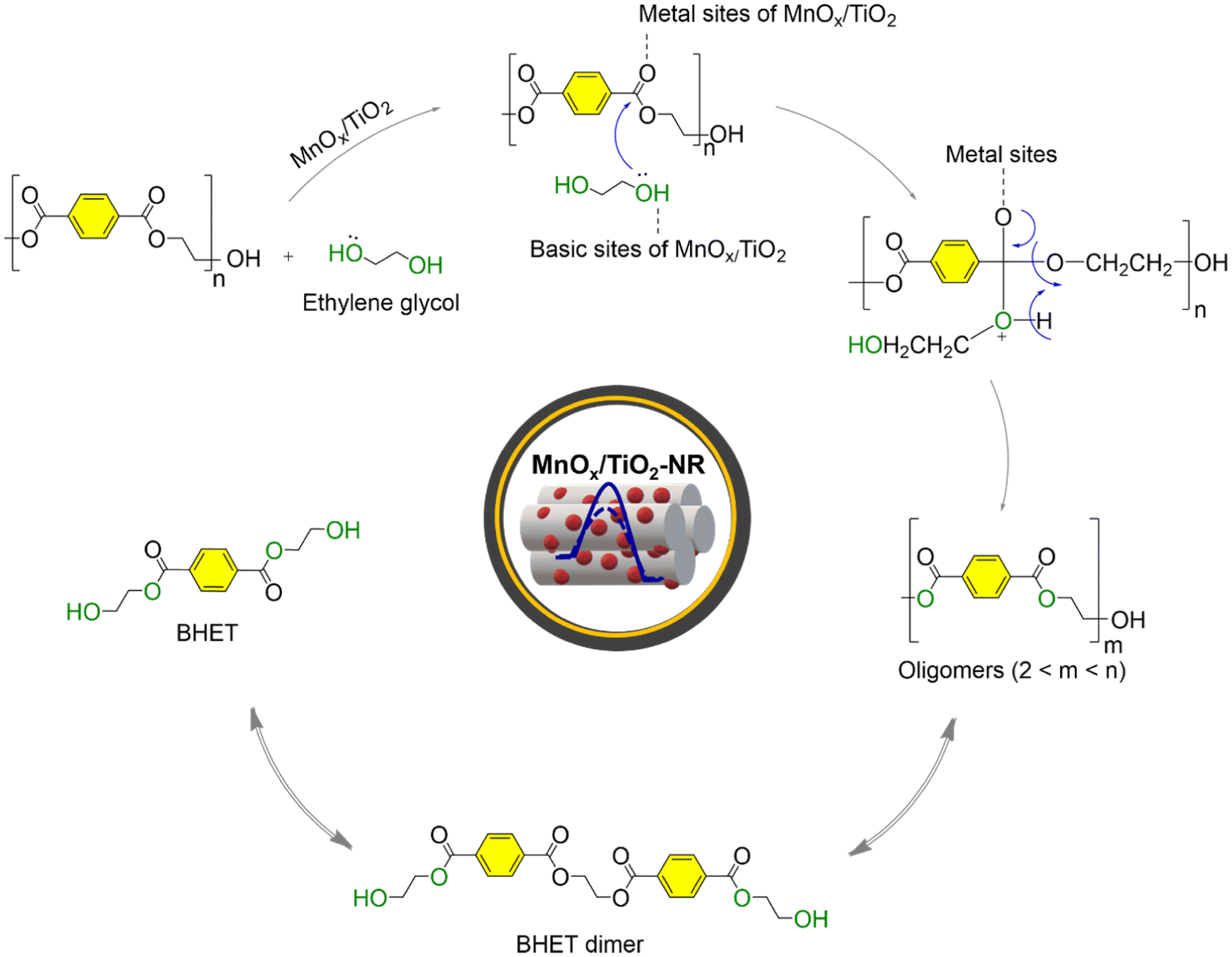 | ||
| Fig. 10 A possible mechanism for the PET glycolysis reaction over the MnOx/TiO2-NR catalyst. | ||
4. Conclusions
This study showed the effect of TiO2 morphology on the structure–activity correlation of MnOx/TiO2 nanocatalysts for PET glycolysis. The combination of MnOx with TiO2 nanorods showed the highest yield of BHET compared with the corresponding TiO2 nanosheet and nanotube-based MnOx catalysts. The role of basic sites, surface hydroxyl species, and Mn3+/Mn2+ in accelerating the PET glycolysis reaction is evident, elucidating the higher activity of the MnOx/TiO2-NR catalyst. The reaction rate and activation energy for PET glycolysis were estimated using kinetic studies at different reaction temperatures and time intervals. The MnOx/TiO2-NR catalyst was stable during the reaction, as investigated by the hot-filtration test, and the catalyst can be reused up to four times at least with complete conversion of PET while achieving good yields of BHET.Data availability
The data supporting this article has been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
B. S. is thankful to PMRF and MHRD for providing fellowship. P. S. acknowledges the funding support from MoE-STARS (Project ID: 2023-0038) and SERB (Project ID: CRG/2023/000197).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782, DOI:10.1126/sciadv.1700782.
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522, DOI:10.1002/cssc.202200522.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679, DOI:10.1021/acscatal.2c01286.
- J. Huang, A. Veksha, W. P. Chan, A. Giannis and G. Lisak, Renewable Sustainable Energy Rev., 2022, 154, 111866, DOI:10.1016/j.rser.2021.111866.
- G.-S. Ha, M. A. M. Rashid, D. H. Oh, J.-M. Ha, C.-J. Yoo, B.-H. Jeon, B. Koo, K. Jeong and K. H. Kim, Waste Manage., 2024, 174, 411–419, DOI:10.1016/j.wasman.2023.12.028.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, ACS Eng. Au, 2023, 3, 37–44, DOI:10.1021/acsengineeringau.2c00029.
- R. A. Clark and M. P. Shaver, Chem. Rev., 2024, 124, 2617–2650, DOI:10.1021/acs.chemrev.3c00739.
- H. Li, H. A. Aguirre-Villegas, R. D. Allen, X. Bai, C. H. Benson, G. T. Beckham, S. L. Bradshaw, J. L. Brown, R. C. Brown, V. S. Cecon, J. B. Curley, G. W. Curtzwiler, S. Dong, S. Gaddameedi, J. E. García, I. Hermans, M. S. Kim, J. Ma, L. O. Mark, M. Mavrikakis, O. O. Olafasakin, T. A. Osswald, K. G. Papanikolaou, H. Radhakrishnan, M. A. Sanchez Castillo, K. L. Sánchez-Rivera, K. N. Tumu, R. C. Van Lehn, K. L. Vorst, M. M. Wright, J. Wu, V. M. Zavala, P. Zhou and G. W. Huber, Green Chem., 2022, 24, 8899–9002, 10.1039/D2GC02588D.
- D. Sajwan, A. Sharma, M. Sharma and V. Krishnan, ACS Catal., 2024, 14, 4865–4926, DOI:10.1021/acscatal.2c02292.
- A. K. Manal, A. Shivhare, S. Lande and R. Srivastava, Chem. Commun., 2024, 60, 13143–13168, 10.1039/D4CC03261F.
- Q. Liu, S. Martinez-Villarreal, S. Wang, N. Ngoc Thanh Tien, M. Kammoun, Q. De Roover, C. Len and A. Richel, Chem. Eng. J., 2024, 498, 155227, DOI:10.1016/j.cej.2024.155227.
- T. Ren, H. Zhan, H. Xu, L. Chen, W. Shen, Y. Xu, D. Zhao, Y. Shao and Y. Wang, Environ. Res., 2024, 249, 118428, DOI:10.1016/j.envres.2024.118428.
- T. Muringayil Joseph, S. Azat, Z. Ahmadi, O. Moini Jazani, A. Esmaeili, E. Kianfar, J. Haponiuk and S. Thomas, Case Stud. Chem. Environ. Eng., 2024, 9, 100673, DOI:10.1016/j.cscee.2024.100673.
- C. Jehanno, M. M. Pérez-Madrigal, J. Demarteau, H. Sardon and A. P. Dove, Polym. Chem., 2019, 10, 172–186, 10.1039/C8PY01284A.
- Z. Gao, B. Ma, S. Chen, J. Tian and C. Zhao, Nat. Commun., 2022, 13, 3343, DOI:10.1038/s41467-022-31078-w.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322, 10.1039/D0GC02630A.
- B. Swapna, S. B. Putla, A. Ramesh, C. Subrahmanyam, G. Madras and P. Sudarsanam, Sustainable Energy Fuels, 2024, 8, 5170–5180, 10.1039/D4SE01136H.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070, DOI:10.1002/cssc.202100400.
- M. Rabnawaz, I. Wyman, R. Auras and S. Cheng, Green Chem., 2017, 19, 4737–4753, 10.1039/C7GC02521A.
- K. R. Delle Chiaie, F. R. McMahon, E. J. Williams, M. J. Price and A. P. Dove, Polym. Chem., 2020, 11, 1450–1453, 10.1039/C9PY01920K.
- N. George and T. Kurian, Ind. Eng. Chem. Res., 2014, 53, 14185–14198, DOI:10.1021/ie501995m.
- S. Lalhmangaihzuala, Z. Laldinpuii, C. Lalmuanpuia and K. Vanlaldinpuia, Polymers, 2020, 13, 37, DOI:10.3390/polym13010037.
- A. Bohre, P. R. Jadhao, K. Tripathi, K. K. Pant, B. Likozar and B. Saha, ChemSusChem, 2023, 16, e202300142, DOI:10.1002/cssc.202300142.
- K. Kumari, P. Choudhary and V. Krishnan, Catal. Sci. Technol., 2024, 14, 5352–5363, 10.1039/D4CY00468J.
- T. D. J. te Molder, S. R. A. Kersten, J. P. Lange and M. P. Ruiz, Ind. Eng. Chem. Res., 2021, 60, 7043–7049, DOI:10.1021/acs.iecr.1c01063.
- H. Xin, H. Wang, S. Li, X. Hu, C. Wang, L. Ma and Q. Liu, Sustainable Energy Fuels, 2022, 6, 2602–2612, 10.1039/D2SE00386D.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386, DOI:10.1021/ar3002156.
- Q. Wang, X. Yao, S. Tang, X. Lu, X. Zhang and S. Zhang, Green Chem., 2012, 14, 2559–2566, 10.1039/C2GC35696A.
- N. H. Le, T. T. Ngoc Van, B. Shong and J. Cho, ACS Sustainable Chem. Eng., 2022, 10, 17261–17273, DOI:10.1021/acssuschemeng.2c05570.
- B. Liu, W. Fu, X. Lu, Q. Zhou and S. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 3292–3300, DOI:10.1021/acssuschemeng.8b05324.
- Z. Wang, Y. Jin, Y. Wang, Z. Tang, S. Wang, G. Xiao and H. Su, ACS Sustainable Chem. Eng., 2022, 10, 7965–7973, DOI:10.1021/acssuschemeng.2c01235.
- D. Lei, X.-L. Sun, S. Hu, H. Cheng, Q. Chen, Q. Qian, Q. Xiao, C. Cao, L. Xiao and B. Huang, Ind. Eng. Chem. Res., 2022, 61, 4794–4802, DOI:10.1021/acs.iecr.1c05022.
- A. M. Al-Sabagh, F. Z. Yehia, D. R. K. Harding, Gh. Eshaq and A. E. ElMetwally, Green Chem., 2016, 18, 3997–4003, 10.1039/C6GC00534A.
- G. Park, L. Bartolome, K. G. Lee, S. J. Lee, D. H. Kim and T. J. Park, Nanoscale, 2012, 4, 3879–3885, 10.1039/C2NR30168G.
- M. R. Nabid, Y. Bide and M. Jafari, Polym. Degrad. Stab., 2019, 169, 108962, DOI:10.1016/j.polymdegradstab.2019.108962.
- Z. Fehér, J. Kiss, P. Kisszékelyi, J. Molnár, P. Huszthy, L. Kárpáti and J. Kupai, Green Chem., 2022, 24, 8447–8459, 10.1039/D2GC02860C.
- F. Tanos, A. Razzouk, G. Lesage, M. Cretin and M. Bechelany, ChemSusChem, 2024, 17, e202301139, DOI:10.1002/cssc.202301139.
- T. Kim, H. Nguyen-Phu, T. Kwon, K. H. Kang and I. Ro, Environ. Pollut., 2023, 331, 121876, DOI:10.1016/j.envpol.2023.121876.
- M. Ge, C. Cao, J. Huang, S. Li, Z. Chen, K.-Q. Zhang, S. S. Al-Deyab and Y. Lai, J. Mater. Chem. A, 2016, 4, 6772–6801, 10.1039/C5TA09323F.
- P. Sudarsanam, H. Li and T. V. Sagar, ACS Catal., 2020, 10, 9555–9584, DOI:10.1021/acscatal.0c01680.
- Y. Zou, M. Zhang, F. Cao, J. Li, S. Zhang and Y. Qu, J. Mater. Chem. A, 2021, 9, 19692–19697, 10.1039/D1TA04415J.
- C. Liu, Q. Hu, Q. Chen, J. Wang, L. Zhang and Y. Ni, J. Cluster Sci., 2018, 29, 185–194, DOI:10.1007/s10876-017-1320-z.
- B. Swapna, N. Singh, S. Patowary, P. Bharali, G. Madras and P. Sudarsanam, Catal. Sci. Technol., 2024, 14, 5574–5587, 10.1039/D4CY00823E.
- M. E. Viana, A. Riul, G. M. Carvalho, A. F. Rubira and E. C. Muniz, Chem. Eng. J., 2011, 173, 210–219, DOI:10.1016/j.cej.2011.07.031.
- Y. Liu, P. Fang, Y. Cheng, Y. Gao, F. Chen, Z. Liu and Y. Dai, Chem. Eng. J., 2013, 219, 478–485, DOI:10.1016/j.cej.2012.12.098.
- P. Sudarsanam, A. Köckritz, H. Atia, M. H. Amin and A. Brückner, ChemCatChem, 2021, 13, 1990–1997, DOI:10.1002/cctc.202001870.
- Y. Wang, Z. Lu, P. Wen, Y. Gong, C. Li, L. Niu and S. Xu, Nanoscale, 2023, 15, 17850–17860, 10.1039/D3NR04274J.
- N. Singh, S. B. Putla, C. Pratap Singh, P. N. Kalbande, P. Choudhary, S. Krishnamurty, V. Krishnan, K. Bhatte and P. Sudarsanam, ACS Appl. Nano Mater., 2023, 6, 23442–23453, DOI:10.1021/acsanm.3c04820.
- Y. X. Zhang, X. D. Hao, F. Li, Z. P. Diao, Z. Y. Guo and J. Li, Ind. Eng. Chem. Res., 2014, 53, 6966–6977, DOI:10.1021/ie5002229.
- J. Li, J. Chen, R. Ke, C. Luo and J. Hao, Catal. Commun., 2007, 8, 1896–1900, DOI:10.1016/j.catcom.2007.03.007.
- G. Qi and R. T. Yang, Appl. Catal., B, 2003, 44, 217–225, DOI:10.1016/S0926-3373(03)00100-0.
- D. Song, X. Shao, M. Yuan, L. Wang, W. Zhan, Y. Guo, Y. Guo and G. Lu, RSC Adv., 2016, 6, 88117–88125, 10.1039/C6RA20999H.
- A. Sathe, M. A. Peck, C. Balasanthiran, M. A. Langell, R. M. Rioux and J. D. Hoefelmeyer, Inorg. Chim. Acta, 2014, 422, 8–13, DOI:10.1016/j.ica.2014.08.011.
- J. Liu, B. Liu, Y. Ren, Y. Yuan, H. Zhao, H. Yang and S. (Frank) Liu, J. Mater. Chem. A, 2019, 7, 14761–14775, 10.1039/C9TA00736A.
- D. Thomas, R. Ranjan and B. K. George, RSC Sustainability, 2023, 1, 2277–2286, 10.1039/D3SU00304C.
- S. Najmi, B. C. Vance, E. Selvam, D. Huang and D. G. Vlachos, Chem. Eng. J., 2023, 471, 144712, DOI:10.1016/j.cej.2023.144712.
- M. Schlüter, S. Bhutani, K. Wohlgemuth and C. Held, Ind. Eng. Chem. Res., 2024, 63, 15458–15465, DOI:10.1021/acs.iecr.4c02382.
- F. Chen, Q. Zhou, R. Bu, F. Yang and W. Li, Fibers Polym., 2015, 16, 1213–1219, DOI:10.1007/s12221-015-1213-4.
- G. Yang, H. Wu, K. Huang, Y. Ma, Q. Chen, Y. Chen, S. Lin, H. Guo and Z. Li, J. Polym. Environ., 2024, 32, 5071–5085, DOI:10.1007/s10924-024-03298-2.
- R. López-Fonseca, I. Duque-Ingunza, B. de Rivas, L. Flores-Giraldo and J. I. Gutiérrez-Ortiz, Chem. Eng. J., 2011, 168, 312–320, DOI:10.1016/j.cej.2011.01.031.
- L. O. Mark, M. C. Cendejas and I. Hermans, ChemSusChem, 2020, 13, 5808–5836, DOI:10.1002/cssc.202001905.
- S. Javed and D. Vogt, ACS Sustainable Chem. Eng., 2023, 11, 11541–11547, DOI:10.1021/acssuschemeng.3c01872.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images of TiO2-NS and TiO2-NT, N2 adsorption-desorption isotherms, BET surface area, pore size, and pore volume of the catalysts, NMR spectra and HR-MS analysis of BHET, and XPS analysis of fresh and spent MnOx/TiO2-NR catalysts. See DOI: https://doi.org/10.1039/d4nr05373g |
| This journal is © The Royal Society of Chemistry 2025 |