
Rational synthesis of carbon-rich hollow carbon nitride spheres for photocatalytic H2O2 production and Cr(VI) reduction†
Yong
Hu
a,
Zhenchun
Yang
b,
Dandan
Zheng
*a,
Wandong
Xing
 *b and
Guigang
Zhang
*b
*b and
Guigang
Zhang
*b
aCollege of Environment and Safety Engineering, Fuzhou University, Fuzhou, 350108, PR China
bState Key Laboratory of Photocatalysis on Energy and Environment, College of Chemistry, Fuzhou University, Fuzhou, 350108, PR China. E-mail: guigang.zhang@fzu.edu.cn
First published on 21st February 2025
Abstract
Hollow carbon nitride spheres with a well-designed architecture and excellent optical properties serve as promising polymers for solar fuel production. In this study, carbon-rich hollow carbon nitride nanospheres were rationally designed for photocatalytic hydrogen peroxide production. Experimental results revealed that the doping of carbon species in the heptazine unit enhanced light absorption and promoted charge separation and transport. Accordingly, the optimized carbon-rich hollow carbon nitride nanospheres exhibited significantly enhanced photocatalytic performance for solar-driven hydrogen peroxide production and Cr(VI) reduction in comparison with pristine polymeric carbon nitride and hollow carbon nitride nanospheres.
 Guigang Zhang | Guigang Zhang received his PhD from Fuzhou University in 2016 under the supervision of Prof. Xinchen Wang and Prof. Xianzhi Fu. Afterwards, he obtained the Humboldt fellowship and then moved to Germany for postdoctoral research at the Max Planck Institute of Colloids and Interfaces (2016–2019, with Prof. Markus Antonietti). Since 2020, he has been a Full Professor at Fuzhou University. His current research interest is mainly focused on the rational synthesis of novel visible light semiconductors and their applications in artificial photosynthesis. |
1. Introduction
Hydrogen peroxide (H2O2), a high-value chemical that is widely used in environmental remediation, chemical synthesis, and energy conversion, has been regarded as one of the most important chemicals.1–4 In recent years, the demand for H2O2 production has notably increased, especially during and after the COVID-19 pandemic.5 Conventional H2O2 production mainly relies on the anthraquinone method, but it faces many drawbacks, such as high energy consumption, high cost, generation of many organic by-products, and high environmental impact.6–8 To achieve energy and environmental sustainability, there is an urgent need for developing a clean and green H2O2 synthesis process. Photocatalysis, which is based on the clean and inexhaustible sunlight as the energy source, has attracted intense interest from a wide range of researchers,9 and it has been widely used for overall water splitting,10,11 CO2 reduction,12 pollutant degradation,13 conversion of organics,14 and production of H2O2via oxygen reduction.15,16 The efficiency of the photocatalytic process is highly related to the charge separation and transfer of the photocatalysts, and thus, it is highly desired to rationally control their local structure and charge carrier properties.Currently, a large number of photocatalysts have been developed for the photocatalytic production of H2O2via the 2e− dioxygen reduction reaction (ORR).17,18 The active sites of the carbon-based catalysts, such as covalent organic frameworks19 (COFs), phenolic resins (Pfs),20 and polymeric carbon nitride frameworks (PCNs),21 are particularly favorable for selective 2e− ORR. Owing to its unique physicochemical properties, polymerized carbon nitride (PCN) is considered one of the most promising photocatalysts since its application in the photocatalysis field.22 Compared with traditional inorganic semiconductor photocatalysts, PCN is considered one of the most promising materials for sustainable photocatalytic production of H2O2 owing to its advantages, including non-toxicity, earth-abundant elements, and simple synthesis process.23
Nevertheless, owing to its highly symmetric π-conjugated aromatic planar structure and abundance of structural defects, the undesired recombination of photogenerated electron–hole pairs severely limits the quantum efficiencies of PCN materials for photocatalytic reactions.24,25 Therefore, great efforts have been devoted to develop a series of methods, including nanostructure design,26 heterostructure building,27 noble metal deposition,28 elemental doping,29,30 and n-π* electron transfer engineering,31 for the rational control of the charge carrier behavior. Among these, nanostructure design, such as using oriented soft/hard templates to prepare materials with specific morphology, is proven to be an attractive and effective approach for improving photocatalytic performance. PCN nanohollow spheres (HCNS) with a mesoporous shell layer structure are polymer semiconductor photocatalysts prepared via a nanostructure design based on a SiO2 sphere template.32 Owing to its large specific surface area and multiple scattering effects of the surface as well as the ability to reduce the disordered stacking and agglomeration of the material, HCNS demonstrates a unique charm in photocatalytic reactions.33 However, due to the intrinsic high exciton binding energy of polymeric semiconductors, HCNS photocatalysts still suffer from poor exciton dissociation performance and severe recombination of photogenerated carriers.34
Elemental doping has been widely developed to modulate the local structure, optical properties, and charge separation of semiconductors. PCN is usually prepared by the polymerization of precursors at elevated temperatures, which enables the copolymerization of carbon-rich monomers during the polymerization process. The incorporation of carbon species into the PCN framework not only enhances optical absorption but also largely promotes charge separation and distribution.35 In particular, the substitution of C atoms with N atoms in the heptazine unit or the introduction of small C-rich units may lead to higher delocalization exhibited by the π-conjugated electrons in the PCN framework, which would narrow the bandgap and increase the optical absorption of the PCN.35–38 Additionally, the introduction of a small amount of carbon can change the distribution of electrons in the heptazine skeleton, increase the active sites of the molecular oxygen reduction reaction and further promote the photocatalytic reaction.39,40
Herein, based on the previous researach,32 mesoporous SiO2 spheres were prepared as oriented structural templates using the modified Stöber method. The synthesized mesoporous SiO2 spheres were fully mixed and preassembled with the precursor cyanamide and the small-molecule modifier glycosamide (GLD), and the C self-doped hollow spherical carbon nitride (HCNSGx) was synthesized under a nitrogen atmosphere. Also, one of the samples, HCNSG30, functions excellently for photocatalytic H2O2 production and reduction of hexavalent chromium, especially in comparison with previous studies, as listed in Tables S3 and S4.† Characterizations reveal that HCNSG30 prepared by the template-assisted pre-assembly of cyanamide with GLD successfully brings the C element into the heptazine structural unit while avoiding disordered stacking of the material. Therein, HCNSG30 is the most effective for photocatalytic H2O2 production, being 1.77 times and 5.24 times higher than HCNS and PCN, respectively. Moreover, HCNSG30 also exhibits excellent performance for photocatalytic reduction of Cr(VI) owing to the excellent separation and migration rate of the charge carriers.
2. Results and discussion
Scheme 1 shows the proposed synthesis process of the carbon-rich hollow carbon nitride spheres according to the modified Stöber method based on previous work. For simplicity, the obtained samples are denoted as HCNSGx. To elucidate the 3D morphology and significance of the C-doping effect, the GCN without the template and the HCNS without C-doping were also prepared using similar synthesis methods as references. | ||
| Scheme 1 (a) Schematic of the synthesis process of carbon-rich hollow carbon nitride spheres. (b) Schematic synthesis of HCNSGx from cyanamide and glycosamide. | ||
Firstly, the micro-morphology of the photocatalysts was characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). As shown in Fig. 1a, SiO2 prepared by the modified Stöber method exhibits a regular, dispersed spherical structure. With the assistance of the mesoporous SiO2 nanosphere templates, both HCNS and HCNSG30 exhibit uniformly sized and dispersed 3D nanosphere structures (Fig. 1b and c), whereas the sample prepared by the template-free thermal polymerization exhibits irregular lumpy aggregated stacks (Fig. 1d). In addition, HCNSG30 exhibits a hollow structure in high-resolution TEM (Fig. 1e), indicating that the doping of the C element did not impact the external morphology of HCNS. The energy dispersive spectroscopy (EDS) shows that the elements correspond well with the TEM dark field map (Fig. 1f), the material mainly comprises elements C and N (Fig. 1g–i), and the presence of the element O may be due to the adsorbed oxygen in the air. From the above Fig. 1b and c, it is clearly observed that HCNS materials prepared by the template-assisted methods are capable of spatially reducing the cluttered stacking, effectively increasing their exposed surface-active sites. The specific surface areas of HCNSG30, HCNS, GCN, and SiO2 were 77, 74, 17, and 23 m2 g−1 (Fig. S2†), respectively. These results indicate that the hard template design method significantly increased the specific surface area of the materials, which enables effective exposure of the active sites and facilitates the performance of the photocatalytic redox reactions.41
 | ||
| Fig. 1 (a–d) Scanning electron microscope images of SiO2 template, HCNS, HCNSG30, and GCN, respectively; (e) high-resolution transmission electron microscope image of HCNSG30 and (f) TEM dark field map and (g–i) EDS mapping of HCNSG30. | ||
The local chemical structures of the HCNSG30, HCNS, and GCN samples are then characterized by powder X-ray diffraction (XRD). As shown in Fig. 2a, HCNSG30 shows two distinct characteristic diffraction peaks at 13.4° and 27.5°, corresponding to the (100) and (002) planes of typical PCN, which are attributed to the periodic repeating heptazine units and the graphitic layer stacking of the PCN, respectively.42 Note that the diffraction peak of HCNSG30 at 27.5° is slightly shifted to a lower angle (about 0.31°) compared to HCNS, which is a result of the increased interlayer stacking distance of the sample due to the introduction of the carbon species into the HCNS framework. In addition, the reduction of the (100) plane is mainly due to the material presenting a regular spherical shape under the influence of the hard template, resulting in a plane with a certain radian.
 | ||
| Fig. 2 (a) Powder X-ray diffraction patterns, (b) Fourier transform infrared spectra, (c) survey XPS spectra, high-resolution XPS of C 1s (d) and (e) N 1s, and (f) 13C NMR spectra of GCN, HCNS and HCNSG30. | ||
The local chemical structures of the samples were further analyzed by Fourier transform infrared spectroscopy (FT-IR). As illustrated in Fig. 2b, the absorption peak at 802 cm−1 is associated with the out-of-plane bending vibration of the heptazine ring.43 The peaks at 2170 cm−1 correspond to the asymmetric vibrations of the cyano groups (–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).44 Note that, comparing GCN, HCNS, and HCNSGx, all present certain cyano vibrations (Fig. S3b†), and the cyano groups usually act as strong electron-withdrawing groups, which promote interfacial separation and migration of photo-induced charges.45 Moreover, the broad peaks at 3000–3700 cm−1 originate from the N–H and O–H bonds that are related to the surface adsorption of uncondensed amino groups and surface-adsorbed water molecules.46 In comparison with GCN, the main skeleton structure of HCNSGx prepared by the template-assisted pre-assembly of cyanamide and GLD remains unchanged, but its surface is enriched with cyano groups, consistent with the XRD analysis. Additionally, the Raman spectra (Fig. S3c†) show that the two samples have similar structures.
N).44 Note that, comparing GCN, HCNS, and HCNSGx, all present certain cyano vibrations (Fig. S3b†), and the cyano groups usually act as strong electron-withdrawing groups, which promote interfacial separation and migration of photo-induced charges.45 Moreover, the broad peaks at 3000–3700 cm−1 originate from the N–H and O–H bonds that are related to the surface adsorption of uncondensed amino groups and surface-adsorbed water molecules.46 In comparison with GCN, the main skeleton structure of HCNSGx prepared by the template-assisted pre-assembly of cyanamide and GLD remains unchanged, but its surface is enriched with cyano groups, consistent with the XRD analysis. Additionally, the Raman spectra (Fig. S3c†) show that the two samples have similar structures.
The local chemical environment of HCNSGx was further analyzed by X-ray photoelectron spectroscopy (XPS). Survey XPS scans confirmed the presence of the elements C, N, and O in all samples (Fig. 2c). As demonstrated in Fig. 2d, the C 1s spectrum of HCNSGx was deconvoluted to three peaks at 288.3 eV, 286.3 eV, and 284.8 eV, which were attributed to the sp2 hybridization of carbon in the heptazine ring (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), cyano carbons (C–NH/CN), and exotic carbons (C–C or CC), respectively.17 The N 1s spectra were deconvoluted into three peaks at 398.8 eV, 400.5 eV, and 401.4 eV, respectively, corresponding to the pyridine nitrogen (CN–C), the terminal amino groups (C–NHx), and the bridging nitrogen (N–(C)3) attached to the carbon atom in the heptazine unit47 (Fig. 2e). Notably, the peak area of 284.8 eV in the C 1s spectrum of HCNSG30 (35.25%) is significantly larger than that of HCNS (27.01%) (Fig. 2d), indicating that the C atoms were successfully doped into the heptazine structure of HCNSG30 and existed in the C–C and C–NH/CN forms (Table S1†). In addition, the contents of C and N elemental of HCNSG30 were measured by elemental analysis (Table S2†). Results reveal that the C/N ratio (0.596) of HCNSG30 was higher than that of HCNS (0.565), which further verified the above conclusion.
N), cyano carbons (C–NH/CN), and exotic carbons (C–C or CC), respectively.17 The N 1s spectra were deconvoluted into three peaks at 398.8 eV, 400.5 eV, and 401.4 eV, respectively, corresponding to the pyridine nitrogen (CN–C), the terminal amino groups (C–NHx), and the bridging nitrogen (N–(C)3) attached to the carbon atom in the heptazine unit47 (Fig. 2e). Notably, the peak area of 284.8 eV in the C 1s spectrum of HCNSG30 (35.25%) is significantly larger than that of HCNS (27.01%) (Fig. 2d), indicating that the C atoms were successfully doped into the heptazine structure of HCNSG30 and existed in the C–C and C–NH/CN forms (Table S1†). In addition, the contents of C and N elemental of HCNSG30 were measured by elemental analysis (Table S2†). Results reveal that the C/N ratio (0.596) of HCNSG30 was higher than that of HCNS (0.565), which further verified the above conclusion.
The chemical structure of HCNSG30 was further demonstrated by solid-state 13C NMR spectral analysis. As shown in Fig. 2f, HCNS bears two strong resonance signals at 156.2 ppm and 163.9 ppm, attributed to the C1 atom of the NC–N (2) and the C2 atom of the NC–N (NHx) coordination in the heptazine unit, respectively.48,49 For HCNSG30, the above two signals can still be found. It also can be observed that the chemical shift of the C1 and C2 species slightly moved to a higher chemical shift than that of HCNS, which is probably due to the substitution of the N atoms in the heptazine unit by the C atoms. In addition, an additional weak chemical shift was found at 148.5 ppm, which is usually derived from the NC–C(N)-coordinated C3 species into the heptazine unit.50,51
UV-Vis diffuse reflectance spectroscopy (UV-Vis DRS) was used to study the optical absorption properties of the samples. As shown in Fig. 3a, the light absorption intensity of HCNSG30 is significantly higher than that of HCNS and GCN, and the absorption band edge of HCNSG30 is red-shifted relative to HCNS. Based on the Tauc plot (Fig. S4a†), the bandgap energy of GCN, HCNS, and HCNSG30 were calculated to be 2.65 eV, 2.6 eV, and 2.52 eV, respectively. The valence bands of GCN, HCNS, and HCNSG30 determined by the XPS valence band spectroscopy (Fig. S4b†) were 2.02, 1.96, and 1.82 V (vs. NHE), with conduction bands of −0.63, 0.64, and 0.70 V (vs. NHE), respectively (Fig. 3b), which was in agreement with the redox potentials for the 2e− ORR. Notably, HCNSG30, which is enriched with the edge carbon species causing the change in electron distribution, exhibits enhanced reducibility relative to HCNS. The results indicate that the doping of C can slightly reduce its forbidden bandwidth, enhance the absorption of light, and improve the reduction ability of conduction band electrons, which is favorable for photocatalytic ORR.52
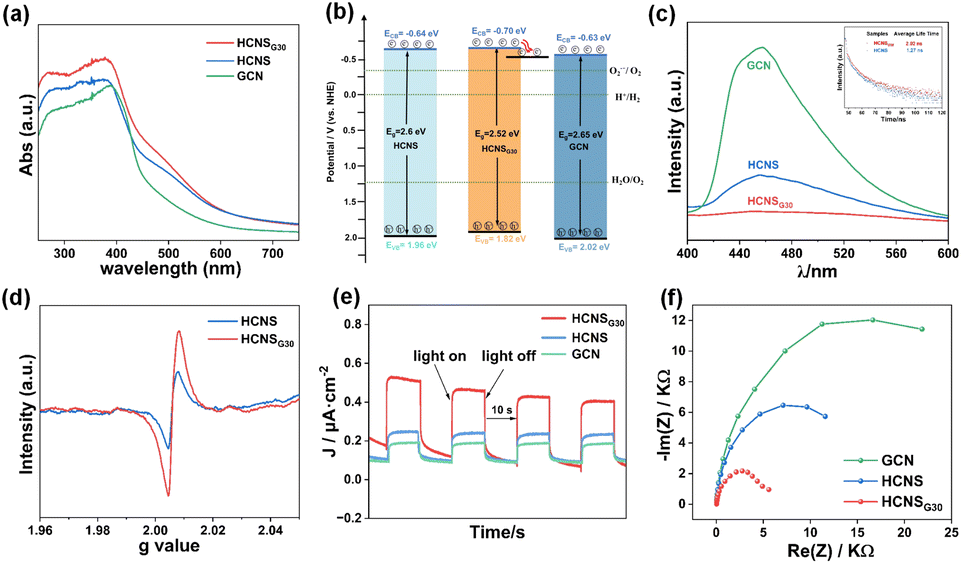 | ||
| Fig. 3 (a) UV-Vis diffuse reflectance spectra; (b) energy band structures of GCN, HCNS and HCNSG30 (ECB(NHE) = Φ + EVB − 4.5 (Φ is the electron work function of the analyzer), EVB(NHE) = Eg − ECB(NHE)); (c) room temperature steady-state photoluminescence spectra and time-resolved photoluminescence spectra of the samples; (d) solid-state electron paramagnetic resonance spectra. (e) Transient photocurrent spectra; and (f) electrochemical impedance spectra of GCN, HCNS and HCNSG30. | ||
The excited electrons at the conduction band either transit to separated polaritons and diffuse to the surface for processes competing with electron transfer kinetics or return to the ground state via photoluminescence (PL) as a radiative decay process and non-radiative processes.45 Therefore, steady-state photoluminescence (PL) and time-resolved photoluminescence (Tr-PL) spectroscopy were used to evaluate the relative extent of exciton dissociation. All three samples exhibited fluorescence emission peaks at about 450 nm but with different intensities. As shown in Fig. 3c, the PL intensity of HCNSG30 is significantly lower than those of GCN and HCNS, indicating an enhanced charge carrier separation efficiency. Furthermore, HCNSG30 exhibits longer photogenerated charge lifetimes of 2.02 ns than HCNS (1.27 ns) (Fig. 3c inset). The reduced PL intensity and prolonged lifetime of charges indicate that C doping effectively promotes the separation and migration of photogenerated electron–hole pairs,35 providing new evidence for improving photocatalytic performance.
The changes in chemical structure after the introduction of C atoms into the heptazine units of HCNS were verified by room temperature electron paramagnetic resonance (EPR) spectra (Fig. 3d). According to the results, the samples all show significantly similar Lorentz curves corresponding to a g-factor value of 2.006, which is mainly attributed to the unpaired electrons on the sp2 carbon atoms of the inner aryl ring in the heptazine unit.53 In particular, HCNSG30 exhibits a higher Lorentzian intensity compared to HCNS, which is probably derived from the more unpaired or delocalized electrons in the structure after the substitution of N with the C atoms in the heptazine units. Transient photocurrent and electrochemical impedance measurements were used to analyze the charge separation and transfer efficiency of the samples. As shown in Fig. 3e and f, compared with GCN and HCNS, the HCNSG30 electrode possessed a higher transient photocurrent as well as a smaller Nyquist circle radius, which demonstrated once again that the photogenerated carriers of the samples were efficiently separated and migrated.
In addition, the surface charge changes of the samples were examined using Kelvin probe microscopy (KPFM). Fig. 4 shows the images of the surface potential changes of the KPFM for HCNS and HCNSG30 in the dark and under light illumination, respectively. It can be seen that HCNSG30 exhibits higher surface potentials than HCNS both under darkness and illumination, namely, 22.26 mV and 13.71 mV, respectively. Meanwhile, the surface potential of HCNS is only 8.83 mV and 7.95 mV in darkness and light. In addition, after illumination, both samples show a decreasing trend, and the decreasing trend of the surface potential of HCNSG30 is more obvious, which is around 8.55 mV lower than that under darkness (Fig. S5†). Understandably, the photogenerated electrons transported to the surface are the main reason for the decrease in the surface potential of the sample under light. The C doping can change the charge distribution on the HCNS heptazine skeleton, promote the separation and migration of photogenerated charges, and improve the efficiency of photocatalytic reactions.54,55 All the above characterization results illustrate that C doping can change the physical and chemical properties of HCNS. For instance, the local charge distribution of the heterocycle structure slightly changes, which provides the basis for promoting carrier separation and migration.56
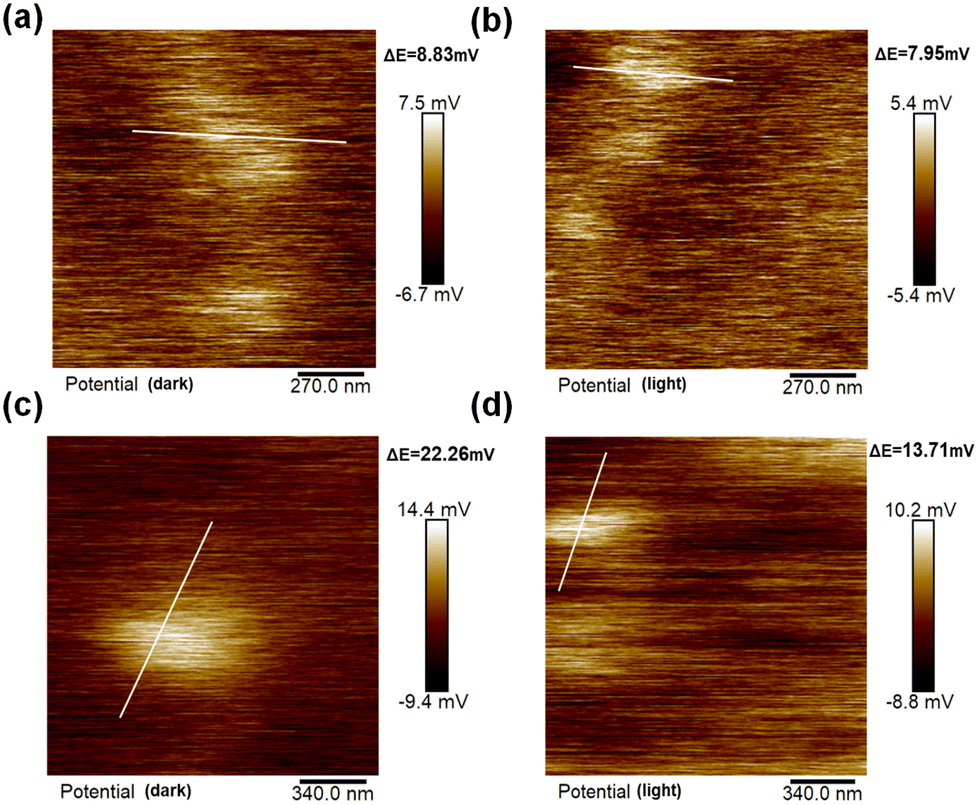 | ||
| Fig. 4 KPFM images of HCNS under (a) darkness and (b) 400 nm LED illumination and HCNSG30 under (c) darkness and (d) 400 nm LED illumination. | ||
DFT calculations were explored to gain molecular insight into the significantly altered electronic structure of HCNS and HCNSG30. Based on the above experimental results, the HCNS and HCNSG30 cells were modeled and optimized (Fig. 5a and d). With the introduction of C elements into the heptazine rings, a discernible decrease in the bandgap of the carbon nitride framework was perceptible, e.g., 2.52 eV of HCN, 2.03 eV of HCNSG30, conforming to the trend of the optical band gap changes (Fig. 5b and e). In comparison with HCNS, HCNSG30 exhibits an increased total density of states (TDOS), indicating the C elements were introduced into the heptazine rings, resulting in a redistribution of the charge density in the conjugated units (Fig. 5c and f). It is particularly noteworthy that the redistribution of charge density is an important factor in enhancing the separation of photogenerated electrons and holes.
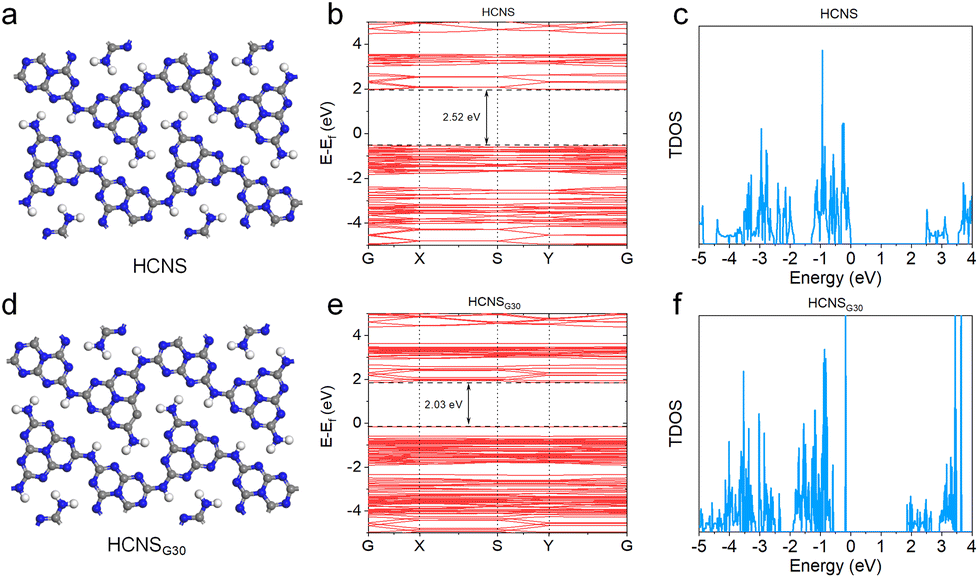 | ||
| Fig. 5 DFT calculations: optimized atomistic structures (a and d), theoretical calculation bandgap (b and e) and total density of states (c and f) of HCN and HCNSG30. | ||
The photocatalytic performance of the samples was evaluated by photocatalytic H2O2 production. As shown in Fig. 6a, under the light illumination, the yields of H2O2 of GCN, HCNS, and HCNSG30 (Fig. S6a†) were examined to be 444.8, 1250.1, and 2159.25 μM h−1, respectively, demonstrating a significant enhancement in photocatalytic H2O2 production performance for HCNSG30, being 4.85 times and 1.73 times higher than those of GCN and HCNS, respectively. HCNSG30 exhibited excellent apparent quantum yields (AQYs) of 12.8% at 400 nm and 4.1% at 420 nm (Fig. 6b). After five rounds (one round of 30 min) of cyclic reactions, HCNSG30 still retains good stability for H2O2 production (Fig. S6b†). Compared with other similar carbon-based materials, it exhibits superior performance (Table S3†). The XRD, FT-IR, and XPS patterns of the sample after the reaction basically did not change (the new peak of Cr was attributed to the adsorption of reduced Cr ions on the surface of the material, and the Cr ions were mainly trivalent), indicating robust stability of the HCNSG30 toward solution and light corrosion in photocatalytic reactions (Fig. S7 and S8†).
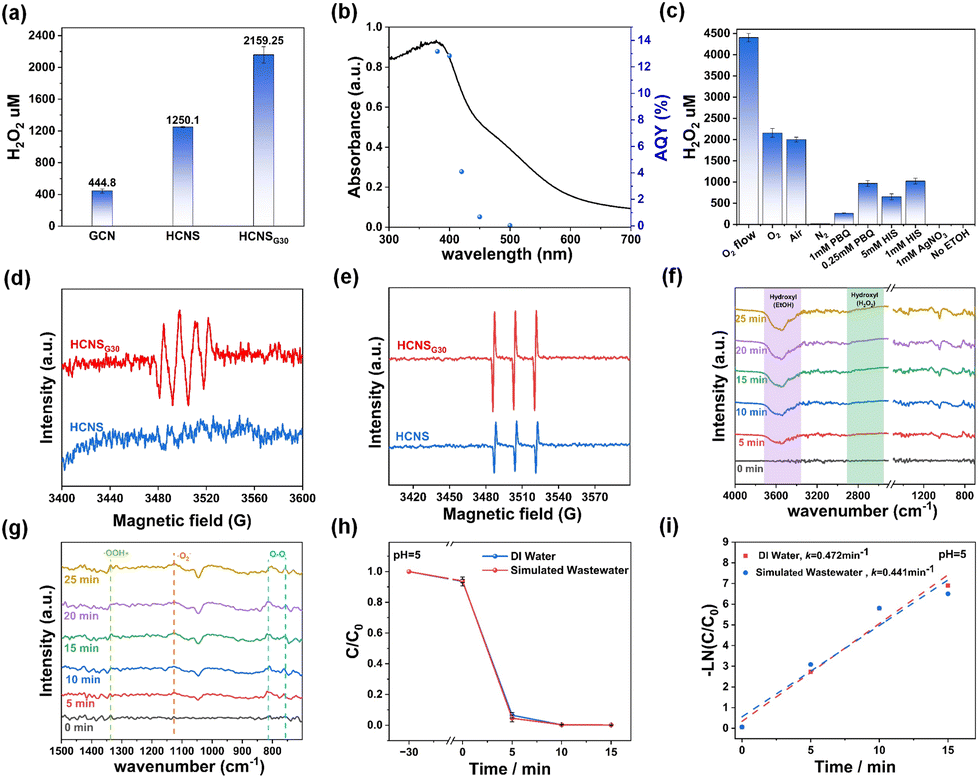 | ||
| Fig. 6 (a) Photocatalytic H2O2 yields of GCN, HCNS, and HCNSG30 samples. (b) Wavelength dependence of the AQY of HCNSG30. Mechanistic study of the photocatalytic production of H2O2 (c) quenching experiments. ESR measurements using a range of trapping agents, (d) superoxide radicals trapped by DMPO (6 min), (e) singlet oxygen trapped by TEMP (4 min). In situ DRIFT spectra of the HCNSG30 for (f) full spectrum (4000–700 cm−1), (g) spectrum from 1500–700 cm−1, (h) photocatalytic Cr(VI) reduction rate of HCNSG30 in different water environments, (i) first-order reaction kinetics of the photocatalytic reduction of Cr(VI) by HCNSG30 under different water environments. | ||
Subsequently, the photocatalytic H2O2 production performance of samples prepared by doping with monomers of different carbon contents was also evaluated. As shown in Fig. S9,† the prepared photocatalysts with different C contents present certain H2O2 production activities. However, the photocatalytic H2O2 production activity of the samples slightly decreased as the doping contents of carbon atoms were increased, mainly due to the excess amounts of structure defects.
To explore the mechanism of the photocatalytic reaction, a series of quenching reactions were carried out. As shown in Fig. 6c, the generation of H2O2 was strongly affected by the atmosphere of the reaction suspension. For instance, the amounts of H2O2 were determined to be 4405.6 μM, 2159.25 μM and 17.8 μM in the presence of oxygen flow, oxygen and nitrogen, respectively, indicating that the dissolved oxygen was necessary for photocatalytic H2O2 production. Besides, p-benzoquinone (PBQ) is usually reported to quench the superoxide radicals (˙O2−).35 Thus, it was found that the production of H2O2 was significantly reduced to 970 μM and 266 μM in the presence of 0.25 mM and 1 mM PBQ, respectively. Nitrotetrazolium Blue chloride (NBT) was further used as a superoxide radical detector to verify the widespread presence of superoxide radicals in the reaction. The result is shown in Fig. S11a–c,† and it was found that HCNSG30 shows a stronger ability to produce superoxide radicals under the same condition, which verifies our main conclusions. In addition, the H2O2 production rate was inhibited in the presence of L-histidine, a scavenger of singlet oxygen (1O2).15 For instance, after the addition of 1 mM and 5 mM L-histidine into the photocatalytic reaction system, the amount of H2O2 produced was measured to be 1023 and 652 μM, respectively. The above quenching experiments suggest that superoxide radicals and singlet oxygen radicals are both considered to be critical intermediates for photocatalytic H2O2 production (Fig. S10†).
The reactive oxygen species (ROS) in the reaction system were further investigated using the electron spin resonance (ESR) technique. As shown in Fig. 6d, using 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a superoxide radical spin trapping agent, characteristic peaks of trapped ˙O2− appeared in the HCNSG30 system under illumination, indicating the production of ˙O2− in the reaction system. The characteristic triplet state signal was observed in the system using 2,2,6,6-tetramethylpiperidine (TEMP) as a spin-trapping agent for 1O2 (Fig. 6e), confirming the presence of 1O2 in the photocatalytic reaction system. Compared with HCNS, the intensities of ˙O2− and 1O2 radicals of HCNSG30 are stronger, suggesting that the incorporation of carbon species into the HCNS framework favors the generation of reactive radicals. During the photocatalytic reaction, the ˙O2− radicals receive electrons from the electron donors and then combine with protons to form H2O2. Besides, 1O2 radicals are derived from the ˙O2− radicals, which are further oxidized by h+. Then, the 1O2 species extract protons from the proton donors (EtOH), which further combine with the excited electrons at the conduction band to generate H2O2.57
In situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements were carried out to further analyze the photocatalytic production of H2O2 in the presence of oxygen in HCNSG30. As shown in Fig. 6f, the spectra in the region of 2600–3000 and 3200–3700 cm−1 are attributed to the stretching vibrations of hydrogen peroxide and the O–H of ethanol,58 respectively. The enhancement of the absorbance signal at the 2600–3000 cm−1 region corresponded to hydrogen peroxide accumulation, and the diminution of the absorbance signal at the 3200–3700 cm−1 region corresponded to ethanol consumption, indicating ethanol was involved in the production of H2O2. As shown in Fig. 6g, the vibrations of O2, O2˙−, and ˙OOH species are located at 805, 1125, and 1334 cm−1, respectively, corresponding to the stepwise one-electron reduction reaction of oxygen and the production of hydrogen peroxide (O2 → O2˙− → OOH → H2O2).59,60 Ethanol acts as an electron donor, consuming holes and providing protons for hydrogen peroxide synthesis.
In addition, studies have shown that semiconductors such as MIL-101(Fe)/BiOCl,61 g-C3N4/BP/MoS2,62 and CCCN63 exhibit unique environmental friendliness, economy, and effectiveness in the photocatalytic treatment of Cr(VI) in industrial wastewater. When the CB position of the photocatalyst is more negative than the potential of Cr6+/Cr3+ (+1.05 eV vs. NHE), it will be converted to Cr3+ or Cr (OH)3 under different acid–base environments, which is favorable for further removal of Cr ions.64 Therefore, in order to investigate whether the on-site produced H2O2 could be further utilized for environmental purification, the performance of HCNSG30 and HCNS for photocatalytic reduction of Cr(VI) was also investigated (see experimental section for more details). The samples were evaluated under the irradiation of a 300 w Xenon lamp in the presence of 15 V% ethanol as hole scavengers (Fig. S13a and b†). As shown in Fig. S13c and d,† the Cr(VI) reduction activity of HCNSG30 was significantly improved in comparison with HCNS. This result indicates that the C-doping modification promotes charge separation, which also plays a significant role in improving the performance of the photocatalytic reduction of Cr(VI). In addition, the reaction rate of photocatalytic reduction of Cr(VI) by HCNSG30 in the air was larger than that in the nitrogen atmosphere, probably due to the generation of ˙O2− or H2O2 in the reaction system, which positively promotes the photocatalytic reduction of Cr(VI).65
The effect of pH on the reaction was further explored. It was found that the photocatalytic reduction efficiency of Cr(VI) was significantly improved under acidic conditions (Fig. S14a†). This may be due to the fact that Cr2O72−, HCrO4−, and CrO42− are the main states of Cr(VI) in the reaction solution, and Cr(VI) in the solution is easily surrounded by more protons under acidic conditions, resulting in a reduction reaction. In addition, the reduction effect of HCNSG30 on Cr(VI) under different light intensities was investigated. With the decrease in light intensity, the photocatalytic reduction efficiency gradually decreases, and the reduction efficiency of Cr(VI) within 15 min at 400 nm is 99% (Fig. S14b and c†). The photocatalytic reduction performance of Cr(VI) in simulated wastewater was tested. It is found that the samples in the simulated wastewater environment can still maintain a good Cr(VI) reduction efficiency and even have slightly enhanced performance, which may be due to the ions in the simulated wastewater environment promoting charge transfer, thus improving the photocatalytic Cr(VI) reduction performance (Fig. 6h and i). Moreover, in five cycles, HCNSG30 showed excellent cyclic stability for photocatalytic Cr(VI) reduction (Fig. S14d and Table S4†). Based on the above analysis, a proposed mechanism for photocatalytic H2O2 reduction and reduction of Cr(VI) is depicted in Fig. 7.
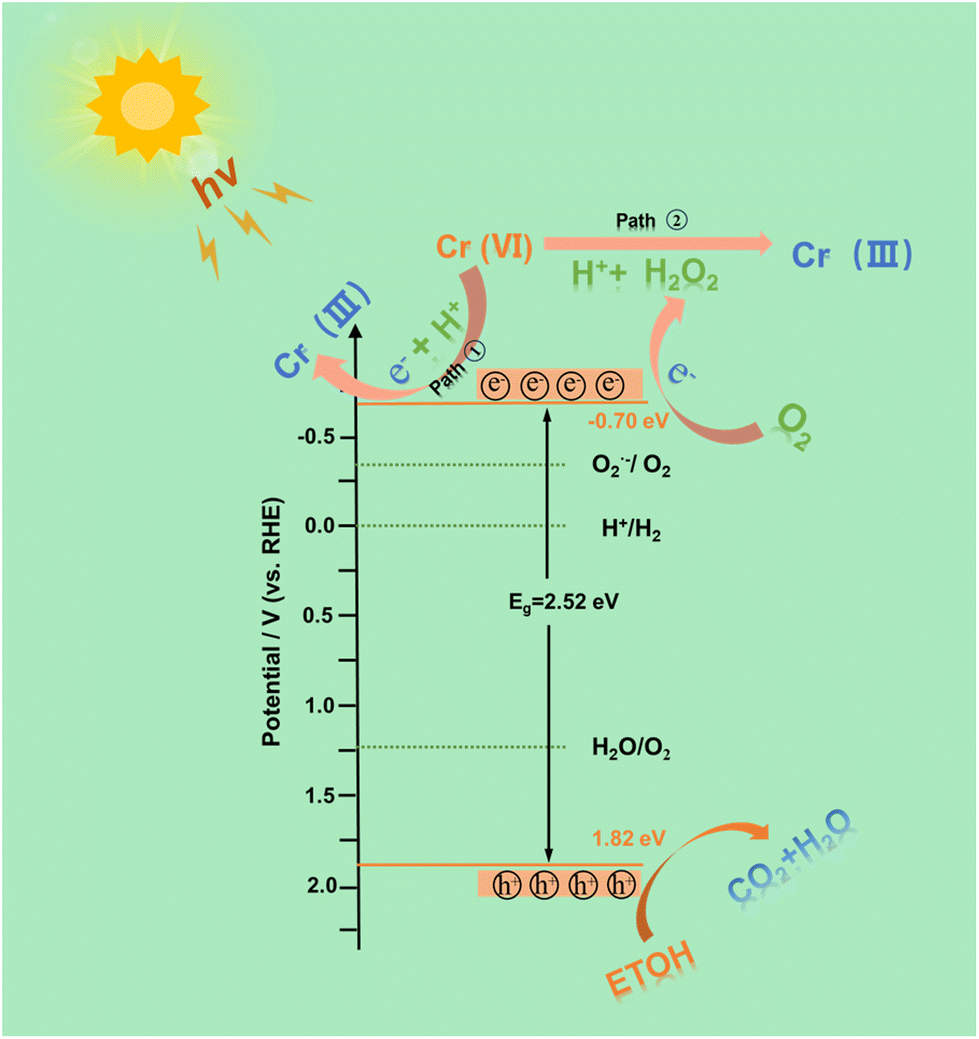 | ||
| Fig. 7 Proposed mechanism of HCNSG30 for photocatalytic hydrogen peroxide production and Cr(VI) reduction reaction. | ||
3. Experiment
3.1 Chemicals and materials
All chemicals and reagents were used as received without further purification. Diphenylcarbazide, glycosamide, ethanol sulfuric acid, phosphoric acid, 5,5-dimethyl-1-pyrroline N-oxide, 2,2,6,6-tetramethylpiperidine, p-benzoquinone, L-histidine, N,N-diethyl-p-phenylenediamine, Horseradish peroxidase, and sodium sulfate (Na2SO4) were acquired from China National Pharmaceutical Group Chemical Reagents Co., Ltd. Cyanamide was obtained from Shanghai Alfa Aesar Chemical Company Limited (China). Trimethoxy(octadecyl)silane, tetraethyl orthosilicate, and ammonium hydroxide solution were procured from Shanghai Sigma-Aldrich Company (China).3.2 Preparation of mesoporous SiO2@mSiO2 templates
In a conical flask, 3.1 g NH3·H2O, 10 g water and 58.5 g ethanol were mixed well, and 5.6 g ethyl orthosilicate was added, mixed well and left for 1 h. Subsequently, a certain amount of the tetraethyl orthosilicate and octadecyltrimethoxysilane mixture was slowly added to the above-mixed solution and left to stand for 3 h. Finally, the solution was dried by centrifugation, calcined in air at 550 °C for 6 h, removed and acidified in 1 M hydrochloric acid for 24 h, and collected by centrifugal drying.3.3 Preparation of HCNSGx
8 g of cyanamide, 1 g of SiO2 template and a certain amount of GLD were taken in a round-bottomed flask, and the “ultrasonic + evacuation” treatment was carried out at a temperature of 60 °C for 2 hours, and the mixture was stirred at 60 °C overnight. Then, deionized water was added and stirred, then centrifuged, dried and calcined at 550 °C under nitrogen atmosphere for 4 hours. The obtained powder was treated with 6 M NH4HF2 under magnetic stirring for 12 h to remove the silica template, then centrifuged and washed several times with distilled water and once with ethanol. The final yellow HCNSGx powder was obtained by drying in a vacuum oven at 70 °C overnight, where x (0, 10, 20, 30, 50, 80) refers to the mass of GLD.3.4 Preparation of GCN
A certain mass of cyanamide was weighed and calcined at 550 °C for 4 h under N2 to obtain a comparison sample and named as GCN.4. Conclusions
In this work, the carbon-rich hollow carbon nitride spheres (HCNSGx) were synthesized by a pre-assembly of GLD with cyanamide with the aid of a hard template. The incorporation of carbon species into the HCNS framework changes the charge distribution of the heptazine skeleton and promotes the effective separation and migration of photogenerated electrons and holes, reduces the band gap and enhances light absorption, thus improving the photocatalytic performance. During the photocatalytic oxygen reduction reaction, the HCNSG30 generated two different kinds of reactive species (1O2 and ˙O2−), which played an important role in the photocatalytic production of H2O2 and the photocatalytic reduction of Cr(VI). The H2O2 generation rate of HCNSG30 reached 4405.6 μM for 1 h under an O2 flow environment and the HCNSG30 almost reduced 100% Cr(VI) within 10 min in simulated wastewater. This study demonstrates the effect of non-metal doping and nanostructure engineering of PCN for simultaneous excellent photocatalytic H2O2 reduction and Cr(VI) reduction, which provides new ideas for the production of high-value chemical products and environmental purification.Author contributions
Yong Hu: conceptualization, data curation, methodology, writing – original draft, review & editing. Zhenchun Yang: formal analysis, investigation, resources. Dandan Zheng: formal analysis, methodology. Wandong Xing: software, formal analysis. Guigang Zhang: methodology, writing – review & editing.Data availability
The data that support the findings of this work are available from the corresponding author [G.-G. Zhang] on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The present work is financially supported by the National Natural Science Foundation of China (22172029, 22472029 and 22311540011), the Natural Science Foundation of the Fujian Province (2024J010014), and 111 Project (D16008). The authors acknowledge Beijing PARATERA Tech Co., Ltd. for providing access to high-performance computing resources.References
- S. P. Teong, X. Li and Y. Zhang, Green Chem., 2019, 21, 5753–5780 RSC.
- Y. Yi, L. Wang, G. Li and H. Guo, Catal. Sci. Technol., 2016, 6, 1593–1610 RSC.
- S. Yang, A. Verdaguer-Casadevall, L. Arnarson, L. Silvioli, V. Čolić, R. Frydendal, J. Rossmeisl, I. Chorkendorff and I. E. L. Stephens, ACS Catal., 2018, 8, 4064–4081 CrossRef CAS.
- Y. Xue, Y. Wang, Z. Pan and K. Sayama, Angew. Chem., Int. Ed., 2021, 60, 10469–10480 CrossRef CAS PubMed.
- M. Huang, Z. Cui, Z. Li and W. Sheng, ACS Catal., 2024, 14, 2095–2106 CrossRef CAS.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 5 Search PubMed.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192–9198 CrossRef CAS PubMed.
- G. Gao, Y. Tian, X. Gong, Z. Pan, K. Yang and B. Zong, Chin. J. Catal., 2020, 41, 1039–1047 CrossRef CAS.
- X. Yang, Z. X. Pan, J. Y. Yue, X. Li, G. Liu, Q. Xu and G. Zeng, Small, 2024, 47, 2405907 CrossRef PubMed.
- Q. Wang, G. Zhang, W. Xing, Z. Pan, D. Zheng, S. Wang, Y. Hou and X. Wang, Angew. Chem., Int. Ed., 2023, 62(37), e202307930 CrossRef CAS PubMed.
- G. Zou, Q. Wang, G. Ye, Z. Pan, S. Wang, M. Anpo and G. Zhang, Adv. Funct. Mater., 2025, 2420899 Search PubMed.
- B. Su, Y. Kong, S. Wang, S. Zuo, W. Lin, Y. Fang, Y. Hou, G. Zhang, H. Zhang and X. Wang, J. Am. Chem. Soc., 2023, 145, 27415–27423 CrossRef CAS PubMed.
- Y. Dong, D. Xu, J. Zhang, Q. Wang, S. Pang, G. Zhang, L. C. Campos, L. Lv, X. Liu, W. Gao, L. Sun, Z. Ren and P. Wang, J. Hazard. Mater., 2023, 445, 130364 CrossRef CAS PubMed.
- T. T. Mamo, M. Qorbani, A. G. Hailemariam, R. Putikam, C.-M. Chu, T.-R. Ko, A. Sabbah, C.-Y. Huang, S. Kholimatussadiah, T. Billo, M. K. Hussien, S.-Y. Chang, M.-C. Lin, W.-Y. Woon, H.-L. Wu, K.-T. Wong, L.-C. Chen and K.-H. Chen, Nano Energy, 2024, 128, 109863 CrossRef CAS.
- Z. Yang, B. Guo, Z. Hu, K. Wang, J. Cui, L. Li, C. Hu and Y. Zhao, Chin. Chem. Lett., 2024, 35, 109251 CrossRef CAS.
- Y. Jin, C. Zhang and G. Zhang, Chin. J. Struct. Chem., 2024, 43, 100414 CrossRef.
- J. Luo, C. Fan, L. Tang, Y. Liu, Z. Gong, T. Wu, X. Zhen, C. Feng, H. Feng, L. Wang, L. Xu and M. Yan, Appl. Catal., B, 2022, 301, 120757 CrossRef CAS.
- H. Zhang, L.-H. Guo, L. Zhao, B. Wan and Y. Yang, J. Phys. Chem. Lett., 2015, 6, 958–963 CrossRef CAS PubMed.
- W. Zhao, P. Yan, B. Li, M. Bahri, L. Liu, X. Zhou, R. Clowes, N. D. Browning, Y. Wu, J. W. Ward and A. I. Cooper, J. Am. Chem. Soc., 2022, 144, 9902–9909 CrossRef CAS PubMed.
- Q. Tian, X. K. Zeng, C. Zhao, L. Y. Jing, X. W. Zhang and J. Liu, Adv. Funct. Mater., 2023, 33, 2213173 CrossRef CAS.
- T. Gao, D. Zhao, S. Yuan, M. Zheng, X. Pu, L. Tang and Z. Lei, Carbon Energy, 2024, 6(11), e596 CrossRef CAS.
- M. Li, Q. Zheng, D. P. Durkin, H. Chen and D. Shuai, J. Hazard. Mater., 2022, 436, 129251 CrossRef CAS PubMed.
- A. Kumar, P. Raizada, A. Hosseini-Bandegharaei, V. K. Thakur, V.-H. Nguyen and P. Singh, J. Mater. Chem. A, 2021, 9, 111–153 RSC.
- D. Li, C. Wen, J. Huang, J. Zhong, P. Chen, H. Liu, Z. Wang, Y. Liu, W. Lv and G. Liu, Appl. Catal., B, 2022, 307, 121099 CrossRef CAS.
- J. Yuan, N. Tian, Z. Zhu, W. Yu, M. Li, Y. Zhang and H. Huang, Chem. Eng. J., 2023, 467, 143379 CrossRef CAS.
- D. Zheng, X.-N. Cao and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512–11516 CrossRef CAS PubMed.
- P. Basyach, P. K. Prajapati, S. S. Rohman, K. Sonowal, L. Kalita, A. Malik, A. K. Guha, S. L. Jain and L. Saikia, ACS Appl. Mater. Interfaces, 2022, 15, 914–931 CrossRef PubMed.
- M. Liu, G. Zhang, X. Liang, Z. Pan, D. Zheng, S. Wang, Z. Yu, Y. Hou and X. Wang, Angew. Chem., 2023, 135(37), e202304694 CrossRef.
- X. Dang, R. Yang, Z. Wang, S. Wu and H. Zhao, J. Mater. Chem. A, 2020, 8, 22720–22727 RSC.
- H. Wang, L. Yu, J. Jiang, Arramel and J. Zou, Acta Phys.-Chim. Sin., 2024, 40(5), 2305047 CrossRef.
- H. Liao, M. Li, Y. Wang, G. Wang, L. Yang, J. Shao, C. Zhang, H. Li and J. Lu, Sep. Purif. Technol., 2023, 325, 124603 CrossRef CAS.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3(1), 1139 CrossRef.
- Y. Yu and H. Huang, Chem. Eng. J., 2023, 453, 139755 CrossRef CAS.
- D. Zheng, C. Pang, Y. Liu and X. Wang, Chem. Commun., 2015, 51, 9706–9709 RSC.
- G. Yang, L. Qin, Y. Pan, J. Meng and Y. Guo, Sep. Purif. Technol., 2024, 329, 125230 CrossRef CAS.
- X. Zhang, G. Yang, J. Meng, L. Qin, M. Ren, Y. Pan, Y. Yang and Y. Guo, Small, 2023, 19(24), 2208012 CrossRef CAS PubMed.
- S. A. Hussain, J. Hu, H. Liu, F. Aslam, S. Khan, L. Khan and F. Jiao, Int. J. Hydrogen Energy, 2024, 87, 705–712 CrossRef CAS.
- H.-T. Liu, T.-J. Lin, D.-L. Tsai, C.-J. Wu and J.-J. Wu, Mater. Today Energy, 2024, 45, 101696 CrossRef CAS.
- Y. Kofuji, S. Ohkita, Y. Shiraishi, H. Sakamoto, S. Ichikawa, S. Tanaka and T. Hirai, ACS Sustainable Chem. Eng., 2017, 5, 6478–6485 CrossRef CAS.
- Z. Zhang, P. Luo, L. Gan, Y. Zhao, X. Wang, H. Peng and J. Peng, Appl. Surf. Sci., 2024, 649, 159118 CrossRef CAS.
- H. Chen, L. Nie, K. Xu, Y. Yang and C. Fang, Acta Phys.-Chim. Sin., 2024, 40, 2406019 CrossRef.
- J. Bai, P. Zhou, P. Xu, Y. Deng and Q. Zhou, Ceram. Int., 2021, 47, 4043–4048 CrossRef CAS.
- M. Yu, S. Chang, L. Ma, X. Wu, J. Yan, Y. Ding, X. Zhang, S. A. C. Carabineiro and K. Lv, Sep. Purif. Technol., 2025, 354, 128695 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29(16), 1605148 CrossRef PubMed.
- Y. Zhang, Z. Yang, D. Zheng, S. Wang, Y. Hou, M. Anpo and G. Zhang, Int. J. Hydrogen Energy, 2024, 69, 372–380 CrossRef CAS.
- H. Gao, S. Yan, J. Wang, Y. A. Huang, P. Wang, Z. Li and Z. Zou, Phys. Chem. Chem. Phys., 2013, 15, 18077–18084 RSC.
- W. Tang, Y. Tian, B. Chen, Y. Xu, B. Li, X. Jing, J. Zhang and S. Xu, ACS Appl. Mater. Interfaces, 2020, 12, 6396–6406 CrossRef CAS PubMed.
- J. Li, D. Wu, J. Iocozzia, H. Du, X. Liu, Y. Yuan, W. Zhou, Z. Li, Z. Xue and Z. Lin, Angew. Chem., 2019, 131, 2007–2011 CrossRef.
- W. Luo, Y. Li, J. Wang, J. Liu, N. Zhang, M. Zhao, J. Wu, W. Zhou and L. Wang, Nano Energy, 2021, 87, 106168 CrossRef CAS.
- H. Che, C. Li, P. Zhou, C. Liu, H. Dong and C. Li, Appl. Surf. Sci., 2020, 505, 144564 CrossRef CAS.
- Y. Huang, D. Li, Z. Fang, R. Chen, B. Luo and W. Shi, Appl. Catal., B, 2019, 254, 128–134 CrossRef CAS.
- C. T. Haile, K. H. Ng, C.-W. Chiu, N. Ahmad and C.-F. J. Kuo, Mater. Today Phys., 2024, 42, 101352 CrossRef CAS.
- D. Zheng, Q. Wang, Z. Pan, S. Wang, Y. Hou and G. Zhang, Sci. China Mater., 2024, 67, 1900–1906 CrossRef CAS.
- B. Xia, B. He, J. Zhang, L. Li, Y. Zhang, J. Yu, J. Ran and S.-Z. Qiao, Adv. Energy Mater., 2022, 12, 2201449 CrossRef CAS.
- P.-P. Ly, D.-V. Nguyen, T.-A. Luu, M.-C. Nguyen, P.-D.-M. Phan, H.-P. Toan, T.-V. Nguyen, M.-T. Pham, T.-D.-T. Ung, D.-D. Bich, H.-T. Pham, H.-T.-N. Nguyen, W.-J. Yu, S.-H. Hur, N.-Q. Hung and H.-T. Vuong, Chem. Eng. J., 2025, 504, 158657 CrossRef.
- Y. He, A. Wu, N. Wang, Y. Xie, C. Tian and H. Fu, Nano Res., 2024, 17, 6860–6869 CrossRef CAS.
- J. Luo, Y. Liu, C. Fan, L. Tang, S. Yang, M. Liu, M. Wang, C. Feng, X. Ouyang, L. Wang, L. Xu, J. Wang and M. Yan, ACS Catal., 2021, 11, 11440–11450 CrossRef CAS.
- K. Meng, J. Zhang, B. Cheng, X. Ren, Z. Xia, F. Xu, L. Zhang and J. Yu, Adv. Mater., 2024, 36, 2406460 CrossRef CAS PubMed.
- C. Shu, X. Yang, L. Liu, X. Hu, R. Sun, X. Yang, A. I. Cooper, B. Tan and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403926 CrossRef CAS PubMed.
- Y.-Y. Tang, X. Luo, R.-Q. Xia, J. Luo, S.-K. Peng, Z.-N. Liu, Q. Gao, M. Xie, R.-J. Wei, G.-H. Ning and D. Li, Angew. Chem., Int. Ed., 2024, 63, e202408186 CrossRef CAS PubMed.
- C. Wang, C. You, K. Rong, C. Shen, F. Yang and S. Li, Acta Phys.-Chim. Sin., 2024, 40(7), 2307045 CrossRef.
- A. Meng, W. Tian, H. Yang, X. Wang, X. Wang and Z. Li, J. Hazard. Mater., 2021, 413, 125400 CrossRef CAS PubMed.
- X. Chen, X. Li, L. Song, R. Chen, H. Li, J. Ding, H. Wan and G. Guan, Int. J. Hydrogen Energy, 2022, 47, 20803–20815 CrossRef CAS.
- G. He, Y. Xiao and H. Du, Int. J. Hydrogen Energy, 2024, 53, 633–646 CrossRef CAS.
- Y. Zhao, P. Zhang, Z. Yang, L. Li, J. Gao, S. Chen, T. Xie, C. Diao, S. Xi, B. Xiao, C. Hu and W. Choi, Nat. Commun., 2021, 12(1), 3701 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4nr05501b |
| This journal is © The Royal Society of Chemistry 2025 |