
Progress in understanding triple ionic–electronic conduction in perovskite oxides for protonic ceramic fuel cell applications
Desheng
Feng
 *a,
Zhonghua
Zhu
b,
Dan
Li
ac and
Mengran
Li
*a
*a,
Zhonghua
Zhu
b,
Dan
Li
ac and
Mengran
Li
*a
aDepartment of Chemical Engineering, the University of Melbourne, Melbourne, 3010, Australia. E-mail: desheng.feng@unimelb.edu.au; Aaron.Li1@unimelb.edu.au
bSchool of Chemical Engineering, The University of Queensland, Brisbane, 4072, Australia
cDepartment of Chemical and Biological Engineering, The Hong Kong University of Science and Technology, Hong Kong, China
First published on 22nd April 2025
Abstract
Protonic ceramic fuel cells offer a promising route to effectively generate electricity from various fuels at reduced temperatures. However, the viability of this technology is impeded by the sluggish kinetics of the oxygen reduction reaction at the cathode. Recently, triple ionic–electronic conductors have shown their promise as cathode materials with improved catalytic activity because of their enhanced mixed electron and ionic conductivities that can maximise the active sites for the reaction. This review examines the transport mechanism of holes, oxygen ions, and protons within triple ionic–electronic conductors. This review highlights the equilibrium among these charge carriers and their requirement for specific cationic environments to facilitate rapid transport. As a result, triple ionic–electronic conductors need to balance the transport of these charges to realise optimum oxygen reduction reaction activity. The review further identifies the transport of oxygen ions or protons as the current limiting factor in triple ionic–electronic conductors. This review concludes by emphasizing the importance of understanding the role of ionic transport in the oxygen reduction reaction to enhance the performance of triple ionic–electronic conductors.
 Desheng Feng | Dr Desheng Feng is a postdoctoral research fellow in the Department of Chemical Engineering at the University of Melbourne, Australia. He completed his Bachelor of Engineering in the School of Chemical Engineering (2019) and Ph.D. (2024) from the University of Queensland. His current research focuses on the development of electrode materials and electrochemical processes for energy conversion. |
 Mengran Li | Dr Mengran Li is a lecturer and ARC DECRA fellow at the Department of Chemical Engineering, University of Melbourne (UoM). After obtaining his PhD from the University of Queensland (UQ) in 2016, he spent over five years as a postdoctoral research fellow at UQ and Delft University of Technology, working on developing CO2 electrolysis. In 2023, he joined UoM as a lecturer and led the Li Energy Lab, which focuses on advancing electrochemical technologies crucial for the global energy transition towards a greener future. His research includes CO2/water electrolysis, reactive carbon capture, fuel cells, and hydrogen separation and compression. |
1. Introduction
Protonic ceramic fuel cells (PCFCs) are electrochemical systems that generate electricity from fuels at reduced temperatures (e.g., <600 °C) with high efficiency.1–9 A PCFC contains a proton-conducting electrolyte, an anode for fuel oxidation, and a cathode for oxygen reduction reaction (ORR). Proton-conducting electrolytes, such as BaZr0.1Ce0.7Y0.2O3−δ and BaZr0.1Ce0.7Y0.1Yb0.1O3−δ show conductivity higher than 0.003 S cm−1 at temperatures as low as 400 °C, outperforming state-of-the-art oxygen ion-conducting electrolytes such as yttrium-stabilized zirconia and gadolinium-doped ceria.10 This advance in electrolyte material enables the potential operation of PCFCs at temperatures lower than 450 °C.11Oxygen reduction reaction at the cathode, as described in eqn (1), typically shows slow kinetics and is often the rate-limiting step for PCFC power density, particularly at reduced temperatures. Many studies report that most cell resistance in PCFCs originates from the ORR at reduced temperatures.2,4,5,7,9
| ½O2(g) + 2e− + 2H+ → H2O(g) | (1) |
As shown in eqn (1), ORR in PCFC is a multiple-step reaction.9,12,13 It begins with the adsorption of molecular oxygen onto the cathode surface.9,12,13 The adsorbed oxygen then dissociates and gains electrons to form oxygen ions.9,12,13 Simultaneously, protons react with these oxygen ions to produce water.9,12,13 Each step of the ORR can only take place in regions where the necessary charges and reactants are available. In cathode materials designed for solid oxide fuel cells (e.g., pure electronic or mixed electronic and oxygen-ion conductors, MIECs),14–17 the entire cathode surface is active for oxygen ion adsorption and reduction. However, the lack of proton conduction limits water formation to the contact line between the cathode, gas, and protonic-conducting electrolyte. As a result, most of the MIECs show limited ORR activities when serving as PCFC cathodes (as reflected by their high area-specific resistances, ASR), as shown in Fig. 1.2,4,5,9,13,18–35
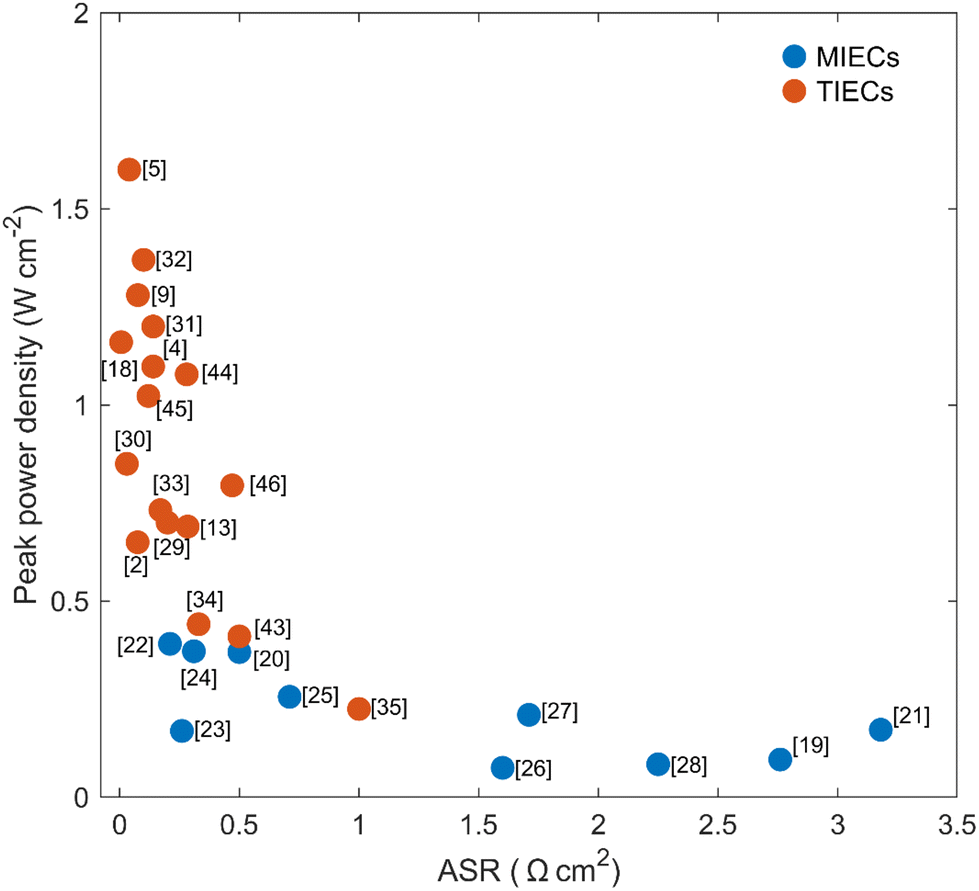 | ||
| Fig. 1 The ASR and peak power density of reported MIECs and TIECs on PCFCs at 600 °C. Data are collected from ref. 2, 4, 5, 9, 13, 18–35, 43–46. | ||
To address this critical issue, a new type of mixed conductors (or so-called triple ionic–electronic conductors, TIECs), such as BaCo0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY) and Ba0.875Fe0.875Zr0.125O3−δ, enabling simultaneous transport of electron, oxygen ions and protons, have emerged as alternative material candidates for the PCFC cathodes.2,9,13,36–38 Compared to traditional MIECs, TIECs can absorb water and form hydroxide groups within their structure under humid conditions, a process often evaluated using methods such as thermogravimetric analysis39 or H2O temperature-programmed desorption.40 Further, proton transport in TIECs can be further investigated using gas permeation methods41 or time-of-flight secondary ion mass spectrometry.42
The additional proton conductivity in TIEC materials allows protons to reach the cathode surface and participate in ORR. As such, using TIEC-based cathodes can extend the ORR active sites beyond the gas-electrolyte-cathode boundaries to the entire cathode surface. Such an increase in the density of ORR-active sites leads to a significant improvement of cathode activity as compared to the MIEC-based cathode for PCFCs: as presented in Fig. 1, most of the TIEC-based cathodes show an ASR value of <0.2 Ω cm2 at 600 °C, which is 5 to 10 times lower than those of MIECs. This advancement significantly increases the power output of PCFCs, from <500 mW cm−2 to higher than 1000 mW cm−2 at 600 °C.
However, even with TIECs-based cathodes, the cathode still accounts for more than 50% of the total cell resistance in current PCFCs. Especially at reduced temperatures, thermally activated charge transport in TIECs slows down,29 further increasing the overall cell resistance.5,7,9 For example, Bian et al. reported that the cathode contributes ∼50% of the resistance at 600 °C and this contribution increases to ∼90% at 400 °C.7 In another study, Liu et al. reveal that >90% of the PCFC cell resistance at 400 °C originates from electrodes, and a ∼60% resistance reduction can be achieved by just replacing the original cathode with an improved one, as shown in Fig. 2.5 These findings highlight the critical need for developing TIECs with improved ORR activity for PCFCs.1–9
 | ||
| Fig. 2 Electrochemical impedance spectroscopy of PCFCs with a PrBa0.5Sr0.5Co1.5Fe0.5O6−δ (PBSCF) and a Ba0.62Sr0.38CoO3−δ–Pr1.44Ba0.11Sr0.45Co1.32Fe0.68O6−δ (BSC + PBSCF) cathode at 400 °C. The polarization resistance in EIS is mainly attributable to the cathode. This figure has been adapted from ref. 5 with permission from Springer Nature, copyright 2025. | ||
Designing an active and stable TIEC-based cathode for PCFC is complicated due to the intricate interplay between structure, chemistry, and charge transport phenomena. This complexity is further exacerbated by reaction environments such as humidity, temperature, and gas atmosphere. Despite these challenges, the recent discovery of high-performance TIEC materials suggests that important physicochemical phenomena are only beginning to be uncovered. For readers seeking detailed insights into specific TIEC material design strategies, several recent review papers36,37,47–50 provide comprehensive summaries of advancements in TIEC materials tailored for PCFC cathodes. Instead, this review will focus on the current understanding of charge transport phenomena and their relationship to material design, complementing existing literature. Finally, we will discuss future directions for the rational design of TIEC materials in high-temperature electrochemical applications.
2. Structures of the TIECs
The most common TIEC cathodes for PCFC are metal oxides in a perovskite-type oxide structure.36,37,47–50 Perovskite oxides have a formula of ABO3, where A-site cations are typically large alkaline-earth cations (e.g., Sr2+ and Ba2+) or lanthanoid cations (e.g., La3+ and Pr3+).51 The B-site is occupied by smaller transition metal cations, such as cobalt and iron.51 While TIECs with alternative structures, such as spinel52,53 or Ruddlesden–Popper-type structures,54 have been explored, this review will primarily focus on perovskite oxide-based TIECs due to the extensive data available in the literature.Perovskite oxides are favoured as TIECs due to their exceptional versatility, offering extensive opportunities for A- and B-site substitution.51,55 This unique characteristic enables precise control over their electrical and ionic transport properties, allowing us to fine-tune their hole, oxygen ion, and proton conductivities.29,30,56,57 As a result, perovskite oxides are well-suited for tailoring materials to optimise ORR activity.
Additionally, perovskite-type oxides are notable for their high-symmetry crystal structures, such as cubic or layered forms.58 These symmetrical structures significantly enhance hole, oxygen ion, and proton conductivities by providing more isotropic sites for charge transport.55,59,60 Achieving such structures is often related to the Goldschmidt tolerance factor (τ) of perovskite oxides, which serves as an effective tool for guiding their design.61 Goldschmidt tolerance factor of perovskite oxides can be expressed as follows:
 | (2) |
3. Electronic and ionic transport
Charge transport is primarily governed by two key factors: the concentration of charge carriers and their mobility. This section will focus on the mechanisms responsible for charge carrier formation and transport within TIECs. A thorough understanding of these mechanisms is crucial for optimizing TIEC design and achieving well-balanced charge transport properties, which are essential for improving performance in practical applications.3.1 Electron transport
Electron transport can occur in materials containing free electrons or electron holes.63,64 Although some perovskite oxides could form free electrons under reducing atmospheres,65 these free electrons are quickly consumed by oxygen under atmospheric oxygen partial pressure. As a result, TIECs for ORR mainly rely on holes for electron transport.36Hole formation in oxides is primarily associated with the presence of redox-active cations.66,67 These cations show different oxidation states in perovskite oxides, such as Co2+/Co3+ for Co (ref. 68) and Ce3+/Ce4+ for Ce.69 The oxidation of these cations facilitates hole formation in perovskite oxides. Thus, redox-active cations in high oxidation states are crucial for hole formation, and their absence makes hole formation more difficult. The lack of holes could explain why perovskite oxides containing no redox-active cations, such as LaAlO3, show dielectric behaviour.70
Holes are transferred through the B–O bond network in perovskite oxides.71,72 This hole transport can be improved by increasing the overlap between B-site cations and oxygen ions.73 The larger overlap leads to stronger hybridisation between the d orbit of B-site cations and the p orbit of oxygen ions, which enhances the mobility of holes across the B–O lattice.73 Therefore, perovskite oxides with stronger B–O bond overlap, or have a more covalent B–O bond, typically show faster hole transport.
Further, hole transport also requires that redox-active cations form percolation pathways in perovskite oxides. The formation of percolation pathways requires the concentration of redox-active cations in the B-site to exceed a threshold, typically between 15–29%.74,75 Below this threshold, the redox-active cations are scattered and holes formed by these cations become less mobile. This could explain why the electrical conductivity of Ba(Zr, Fe)O3−δ drops by three orders of magnitude when Fe content reduces below 20% at 500 °C, as shown in Fig. 3a.75 Similarly, as presented in Fig. 3b, Raffaelle et al. report that the conductivity of La(Cr, Mn)O3 also increases by three orders of magnitude when Mn content increases from 10% to 40% at 700 K.76 It should be noted that although these materials are mixed conductors, the electronic conductivity of these materials is much higher than ionic conductivities.36,77 Therefore, the increase in conductivity of these materials should be primarily attributable to faster hole transport.
 | ||
| Fig. 3 (a) Electrical conductivity of Ba(Zr, Fe)O3−δ system under 1 atm oxygen partial pressure at 500 °C. This figure has been adapted from ref. 36 with permission from Springer Nature, copyright 2025. (b) Electrical conductivity of La(Cr, Mn)O3−δ system in air at 300–1300 K. This figure has been adapted from ref. 76 with permission from American Physical Society, copyright 2025. | ||
In addition to composition, external factors such as temperature, humidity, and oxygen partial pressure significantly influence hole transport in oxides. Elevated temperatures provide additional energy for hole transport but also reduce the oxidation state of cations, which decreases the number of holes. This dual effect explains why the electrical conductivity of (La, Sr)(Co, Fe)O3−δ initially increases and then decreases as the temperature rises from 200 to 1000 °C.78 Conversely, high oxygen partial pressures increase the cation oxidation state, thereby enhancing the number of holes. As a result, elevated oxygen partial pressures consistently promote faster hole transport.79 Furthermore, humidity also affects hole conductivity.80 Perovskite oxides can form hydroxyl groups at the expense of holes in the presence of humidity, which will be discussed later in Section 3.3.
In summary, hole transport dominates electron conductivity in perovskite oxides used for ORR. Holes form via redox-active cations transitioning between oxidation states. These redox-active cations must reach a threshold concentration to form percolation pathways for efficient transport. Below this threshold, conductivity drops sharply. External conditions, including temperature and oxygen partial pressure, are critical for optimising hole transport.
3.2 Oxygen ion transport
Oxygen transport in perovskite oxides is governed by two key steps: surface exchange and bulk diffusion.81,81 Surface exchange coefficient (k) describes the rate of oxygen surface exchange while oxygen ion diffusivity (D) determines the rate of oxygen ions transport through the bulk of perovskite oxides. Techniques such as isotope exchange82 or electrical conductivity relaxation83,84 are used to distinguish between these two steps. Notably, studies demonstrate that bulk transport and surface exchange are highly correlated: materials with fast oxygen ion bulk transport also show fast surface exchange.55 Therefore, this section mainly focuses on oxygen ion bulk transport.Oxygen ion transport typically involves two distinct types of charge carriers: oxygen vacancies and interstitial oxygen ions. Most state-of-the-art oxygen conductors rely on oxygen vacancy mechanisms.61,62 Even the state-of-the-art interstitial oxygen conductors that require less energy for migration (∼0.5 eV for interstitial oxygen cf. 1 eV for oxygen vacancies), such as La4Mn5Si4O22+δ as reported by Meng et al., they still appear to fall short of oxygen vacancy conductors, such as Ce0.9Gd0.1O1.95 or La0.8Sr0.2Ga0.83Mg0.17O3−δ.85–87
Oxygen vacancies are formed to compensate for charge imbalance in oxides.55 The concentration of oxygen vacancies can be quantified by oxygen non-stoichiometry, where a high oxygen non-stoichiometry indicates a high concentration of oxygen vacancies. When the positive charge in oxide is reduced, the lattice oxygen ions can react with holes to form oxygen gas while leaving an oxygen vacancy in the structure.88–90 Notably, the formation of oxygen vacancies reduces hole concentration, thereby impairing electronic conductivity. The formation of oxygen vacancies can be described by the following equation:
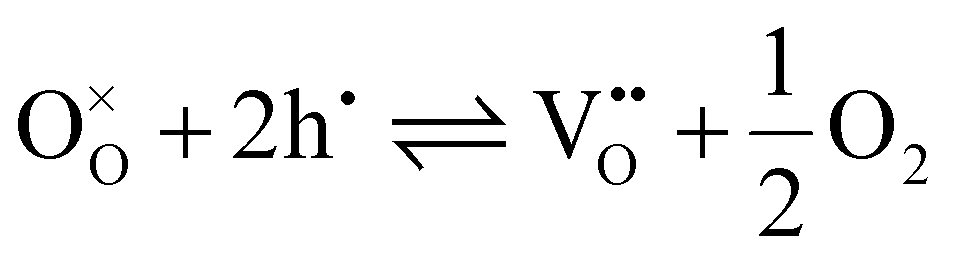 | (3) |
Both lattice cationic environment and external conditions can affect the formation of oxygen vacancies. Compositions, including radius, charge, stoichiometry and electronegativity of A- and B-site cations, are crucial for oxygen vacancy formation.15 In general, cations with large radius, low electronegativity and low valence can promote the formation of oxygen vacancies.
Aside from compositions, external factors play a crucial role in oxygen vacancy formation.15,55 Reducing atmospheres, such as those with elevated temperatures and lower oxygen partial pressures, tend to lower the oxidation states of cations and promote the formation of oxygen vacancies.14–16,91,92 Among these factors, temperature is the most widely studied in relation to oxygen vacancy concentrations. As temperature increases from ambient to elevated levels (e.g., 600 °C), oxygen non-stoichiometry can rise by more than 50%.15 Notably, humidity also reduces the concentration of oxygen vacancies, a topic that will be discussed in Section 3.3.
Experimental methods, including titration,93 electron paramagnetic resonance,94 X-ray photoelectron spectroscopy,95 and neutron powder diffraction,96 are often used to quantify the concentration of oxygen vacancies in perovskite oxides. Many of these methods need to first determine the oxidation state of ORR-active metal cations, such as Co and Fe, and then calculate the oxygen non-stoichiometry.
Recent efforts attempt to accelerate the discovery of perovskite materials with fast oxygen-ion transport through theory-derived descriptors or machine learning.15,97–101 Building on the study of numerous reported materials, our group recently developed a comprehensive approach to predict oxygen vacancies in perovskite oxides, as shown in Fig. 4.15 This method considers the key role of the cationic lattice environment in determining the states of the active metal centres such as Co and Fe at different temperatures.15 The states of the cationic lattice environment can be further mathematically described using fundamental properties such as electronegativity, polarization power, charge, and cation size.15
 | ||
| Fig. 4 (a) Workflow of the prediction of the oxygen vacancies in ABO3−δ perovskites. Comparison of the measured (δM) and predicted oxygen vacancy (δP) for (b) Co-based and (c) Fe-based perovskite oxides. This figure has been adapted from ref. 15 with permission from Springer Nature, copyright 2025. | ||
This work shows that these properties tend to inversely affect the properties of the active metal centres through the cation–cation inductive effects. For example, doping inert elements with high oxidation states tends to lower the oxidation state of Co and Fe, which originate from the delocalized electron distribution that is commonly present in perovskite-structured cathode materials.15 With the assistance of machine learning, this insight allows for accurate prediction of oxygen vacancy concentrations in perovskite oxides across different temperatures, even without detailed knowledge of the specific states of ORR-active metal centres.15 This finding, together with other recent reports,97–101 further highlights the important role of perovskite cations, structures, and temperatures in determining the concentrations of oxygen vacancies.
Notably, a high oxygen vacancy concentration also destabilizes the perovskite oxide structure and triggers phase transformation.102,103 These phase transformations profoundly impact oxygen ion transport as well. A typical example is that the loss of lattice oxygen in a reducing atmosphere converts the structure of Pr0.5Ba0.5MnO3−δ from cubic perovskite oxide to layered perovskite oxide.103 Consequently, it is not always possible to create oxygen vacancies in perovskite oxides to promote oxygen ion transport.
In addition to the concentration of oxygen vacancies, the mobility of oxygen ions through these vacancies also determines oxygen transport. However, our recent studies based on DFT calculations over multiple perovskite models reveal that a high concentration of oxygen vacancies tends to reduce the mobility of oxygen ions, as presented in Fig. 5.15 Such a phenomenon should be related to covalency of the bond between active metal cations and oxygen. Specifically, an increase in B–O covalency of the active cations, such as cobalt, promotes oxygen vacancy formation but may make the oxygen ion less ionic. Conversely, enhancing the ionic character of the B–O bond improves oxygen ion mobility but suppresses the formation of oxygen vacancies.
 | ||
| Fig. 5 (a) formation and (b) mobility of oxygen vacancies as a function of oxygen vacancy concentration. This figure has been adapted from ref. 15 with permission from Springer Nature, copyright 2025. | ||
Furthermore, the mobility of oxygen ions is also related to lattice-free volume, where a large lattice-free volume facilitates oxygen ion transport.104 In perovskite-type oxides, oxygen ions need additional energy to pass through the gap formed by the A-site and two B-site cations, which is referred to as a “saddle point”.105 Mogensen et al. report that the typical size of this saddle point is only ∼1.1 Å,104 far smaller than the size of oxygen ions (1.4 Å), emphasizing the importance of lattice relaxation in enabling oxygen ion migration.106 Oxygen ions therefore require additional energy to pass through this saddle point. A larger lattice can help lower the energy barrier for oxygen ions crossing this saddle point. Following this, Cook et al. report that the activation energy for oxygen ion transport reduces by 50% when the free volume in perovskite-type oxides increases from 20 to 30 Å3,107 as shown in Fig. 6a. Similar findings are reported in Ba(Fe, In)O3−δ system, as shown in Fig. 6b.108
 | ||
| Fig. 6 (a) high free volume significantly lowers the energy barrier for oxygen ion transport in various perovskite oxides. (b) Relationship between free volume and oxygen ion migration energy in Ba(Fe, In)O3−δ system. This figure has been adapted from ref. 55 with permission from Springer Nature, copyright 2025. | ||
To conclude this section, oxygen-ion transport normally requires oxygen vacancies as the charge carriers in most of the perovskite-based mixed conductors. The chemical composition, crystal structure, and operating conditions, all play crucial roles in determining the formation and mobility of these oxygen vacancies. The availability of the oxygen vacancies and the mobility of oxygen ions jointly determine the overall oxygen-ion transport in the lattice. However, trade-offs between the content of oxygen vacancies and oxygen mobility usually exist in these perovskite metal oxides, requiring careful fine-tuning of material compositions to maximise overall oxygen-ion transport.
3.3 Proton transport
Depending on the temperature, protons can be transported through TIECs in the bulk109 and on the surface.110,111 Proton surface transport primarily occurs in surface-adsorbed water at near-room temperatures (e.g., <50 °C).112–114 This surface transport mechanism is not limited to perovskite oxides115 but can also occur on the surface of other metal oxides, such as ZrO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 116 or TiO2.117 However, this surface transport mechanism tends to diminish at elevated temperatures due to the desorption of surface water.
116 or TiO2.117 However, this surface transport mechanism tends to diminish at elevated temperatures due to the desorption of surface water.
Instead, proton transport is governed by proton bulk diffusion at elevated temperatures (e.g., >400 °C).109,118 This proton bulk transport is mainly reported in perovskite oxides.115 At the same time, some studies also reported that other oxides, such as Ba7Nb3.9Mo1.1O20.05119 and La10Si6O27,120 also show high proton conductivity within this temperature regime. However, tuning these proton conductors into TIECs might be challenging.
A common approach to quantifying the concentration of hydroxyl groups in perovskite oxides is to measure their specific weight gain under humid conditions.128–130 This weight gain typically refers to water uptake by perovskite oxides and is related to the incorporation of both protons and oxygen ions in humid conditions.124 A higher weight gain generally indicates a greater concentration of hydroxyl groups in perovskite oxides. However, distinguishing the contributions of protons and oxygen ions to the water uptake remains challenging. This is due to the complexity of the reaction between water and perovskite oxides: perovskite oxides can incorporate the entire water molecule (hydration) or just protons from the water molecule (hydrogenation).80 Some studies also use in situ neutron powder diffraction (NPD) to directly probe the concentration of protons in the structure131,132 but NPD is less available than other techniques.
Incorporating protons into the lattice increases the positive charges in perovskite oxides. As a result, perovskite oxides must either incorporate negatively charged oxygen ions with protons or reduce the oxidation state of metal cations to compensate for the charge imbalance.80,122,133 When perovskite oxides form hydroxyl groups at the expense of oxygen vacancies, the hydration reaction occurs. Hydration reaction is an acid–base reaction, where perovskite oxides serve as a Brønsted–Lowry base. In the hydration reaction, the formation of two hydroxyl groups consumes one oxygen vacancy,122 which can be expressed by eqn (4) below:
 | (4) |
If oxygen vacancies are depleted, or holes are more available than oxygen vacancies, perovskite oxides tend to consume holes to maintain charge neutrality within their structure.134 This process is referred to as the hydrogenation reaction, which can be expressed by eqn (5).
 | (5) |
The hydrogenation reaction can be considered a combination of the oxygen vacancy formation reaction in eqn (3) and the hydration reaction in eqn (4). These two reactions both show that the formation of hydroxyl groups under humid conditions needs to consume electron holes for electron transport or oxygen vacancies for oxygen ion transport. Therefore, hydrated perovskite oxides tend to show compromised hole or oxygen vacancy concentrations.
These trade-offs between different charge carriers were further explored by Poetzsch et al.122 After considering other factors, such as charge neutrality and stoichiometry of perovskite oxides, they revealed the relationship between holes, oxygen vacancies and hydroxyl groups in perovskite oxides, as shown in Fig. 7.122 In their study, the authors found that the concentration of these charge carriers depends on the product of water partial pressure and the equilibrium constant of hydration reaction in eqn (4) (Kw·PH2O in Fig. 7) and the product of oxygen partial pressure and the equilibrium concentration of the reverse oxygen vacancy formation reaction in eqn (3) (KO·PO21/2 in Fig. 7).122 Their thermodynamic calculations show that materials tend to undergo the hydrogenation reaction when the concentration of holes exceeds that of oxygen vacancies and the hydration reaction when oxygen vacancies are more available.122 Further, since the total number of charge carriers is limited in perovskite oxides, achieving a high concentration of holes, oxygen vacancies and hydroxyl groups in TIECs is theoretically impossible. In other words, TIECs would need to achieve balanced transport properties to achieve optimum ORR activity.
 | ||
| Fig. 7 The concentration of holes, oxygen ions and hydroxyl groups in perovskite oxides as a function of water partial pressure (PH2O) oxygen partial pressure (PO) and equilibrium constants of hydration reaction (KW) and oxygenation reaction (KO). This figure has been adapted from ref. 122 with permission from John Wiley & Sons, copyright 2025. | ||
The hydration reaction is an acid–base reaction, with perovskite oxides acting as the base and water as the acid. As a result, increasing the alkalinity of the perovskite oxides tends to shift the hydration reaction towards the formation of hydroxyl groups.109,124,135 Consequently, materials with higher alkalinity tend to show increased water uptake. For example, SrFeO3 show negligible water uptake at 250 °C, whereas replacing Sr with Ba increases the water uptake to 3%.124 The increased alkalinity promotes hydroxyl group formation in Ba-based perovskite oxides, which may contribute to their higher proton conductivity compared to Sr- and La-based counterparts.136
Aside from the materials’ basicity, a high concentration of oxygen vacancies also favours the hydroxyl group formation. The most widely studied proton conducting systems are based on BaZrO3 and BaCeO3.137 These systems typically contain limited oxygen vacancies in their structure.132,138 A common approach to creating more oxygen vacancies in these proton conductors is doping low valence state rare earth metal cations (acceptors), such as Y3+ and Gd3+, into the perovskite oxide structure.139–142 One possible reason for choosing rare earth metal cations could be related to their relatively high alkalinity compared to other transition metals, such as cobalt. Therefore, doping rare earth metal cations can promote oxygen vacancy formation while retaining the alkalinity of materials. It should be noted that the high concentration of oxygen vacancies refers to the concentration of oxygen vacancies in dry conditions. Under humid conditions, water can consume the additional oxygen vacancies. As a result, materials with high oxygen vacancies under dry conditions do not necessarily show high oxygen vacancies under humid conditions.
To further elucidate the role of oxygen vacancies and alkalinity in hydroxyl group formation, Zohourian et al. compared the water uptake of 18 perovskite oxides at 250 °C, where high water uptake indicates a high concentration of hydroxyl groups. As shown in Fig. 8, they changed the materials’ alkalinity by substituting Sr2+ with Ba2+ in the A-site and increasing the concentration of oxygen vacancies by tuning B-site cations. They reported that replacing Sr2+ with more alkaline Ba2+ increases the water uptake of perovskite oxides by more than 15 folds while increasing oxygen non-stoichiometry from 0.4 to 0.6 also increases the water uptake by 5 to 10 folds at 250 °C and 16 mbar H2O. Similarly, Murphy et al. also reported that substituting Zr4+ with Hf4+ can increase the water uptake by the Ba(Ce, Zr, Hf, Y, Yb)O3−δ system.143 These findings emphasize the critical role of increasing material alkalinity and oxygen vacancies in boosting water uptake, which in turn benefits the formation of hydroxyl groups for proton transport.
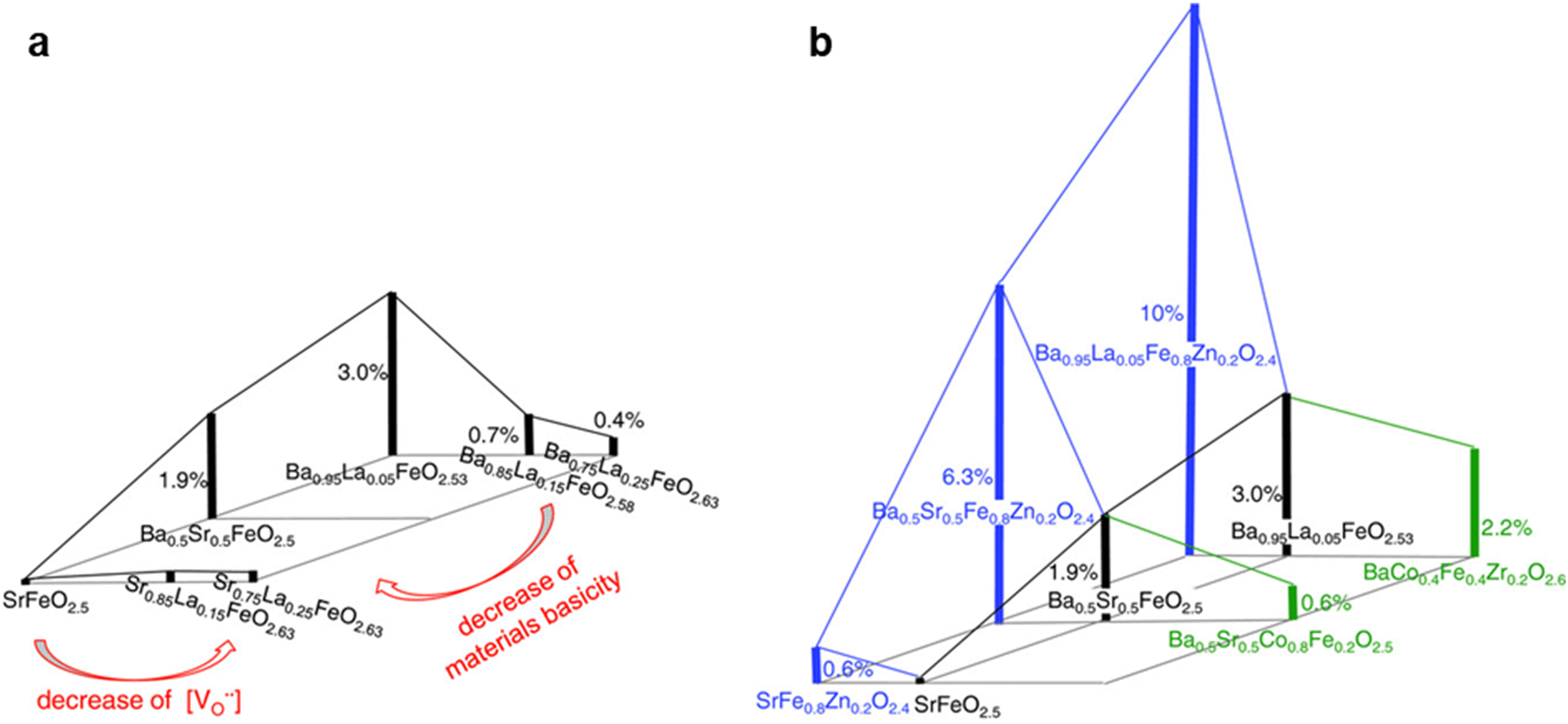 | ||
| Fig. 8 Proton concentration in perovskite oxides systems of (Ba, Sr, La)(Fe, Co, Zn)O3−δ system at 250 °C and 16 mbar H2O. (a) Effect of A-site cations (Ba, Sr and La) and (b) effect of B-site cations (Fe, Co and Zn). This figure has been adapted from ref. with permission from John Wiley & Sons, copyright 2025. | ||
It is generally accepted that hopping is the rate-limiting step in proton transfer,34,125,151 but it is challenging to determine the energy barrier for proton rotation and hopping experimentally. Therefore, modelling is the most common way to study hydroxyl transport in perovskite oxides. Modelling results typically show that the energy barrier is ∼0.2 eV for proton rotation and doubles to ∼0.4 eV for proton hopping.34,125,151 This energy barrier is substantially higher than the energy barrier for proton short-range transport determined by QENS, which is typically <0.1 eV.152 However, it should be pointed out that most QENS measurements are conducted at relatively low temperatures (e.g., <300 °C).152 In this temperature regime, it is unclear if the Grotthuss-like mechanism is still the dominant proton transport mechanism in perovskite oxide. Pulsed-field gradient nuclear magnetic resonance (PFG-NMR) can also probe proton diffusion in proton conductors such as BaCeO3 at elevated temperatures (e.g., >500 °C).153 However, most TIECs contain ferromagnetic elements, such as Co and Fe, which causes significant challenges in PFG-NMR measurements.
Similar to its influence on oxygen-ion transport, the local cationic environment also affects the proton hopping energy barrier. Since short-range proton transport mechanisms are challenging to measure experimentally, most studies rely on modelling to assess the impact of doping. For example, Draber et al. found that doping Y into BaZrO3 can reduce the proton hopping energy barrier from 0.41 eV to 0.22–0.35 eV.125 Similarly, substituting Sr with K also reduces the proton hopping energy barrier by ∼50% in Sr2Fe1.5Mo0.5O6−δ.138 These findings help explain the enhanced proton conductivity observed in doped materials.
While a lower energy barrier facilitates short-range proton transport between neighboring oxygen ions, it does not necessarily enhance long-range, macroscopic proton transport. This is because dopants that reduce the proton hopping energy also create local energy minima within the structure.121,125 As a result, protons transport rapidly within these dopant-rich regions but require additional energy to escape, leading to a phenomenon known as the trapping effect, commonly observed in doped proton-conducting systems.154–157 For example, Yamazaki et al. reported that proton hopping between different oxygen ions in the YO6 octahedra shows an activation energy of 0.13 to 0.17 eV, as shown in Fig. 9a.121 However, these protons require 0.44–0.47 eV to leave the YO6 octahedra.121 If protons lack sufficient energy to overcome this barrier, they remain trapped.121 This effect becomes more pronounced at lower temperatures (e.g., 400 °C), where protons have reduced thermal energy.121,158–160
 | ||
| Fig. 9 (a) Proton transport steps and the corresponding energies. This figure has been adapted from ref. 121 with permission from Springer Nature, copyright 2025. (b) Proton mobility in Ba(Zr, Y)O3−δ at 500, 700 and 1000 K. A low Y doping level reduces the proton mobility. This figure has been adapted from ref. 125 with permission from Springer Nature, copyright 2025. | ||
The traps are formed when dopants such as Y3+ are sparsely distributed within the host phase, particularly at low doping levels (e.g., <3%).125 At these low concentrations, increasing the doping concentration leads to more trap sites, which can hinder long-range proton mobility.125 For example, Draber et al. reported that when Y content in Ba(Zr, Y)O3−δ increases from 1% to 3%, proton mobility decreases from 1.2 × 10−6 cm2 V−1 s−1 to 0.9 × 10−6 cm2 V−1 s−1 at 500 K, as shown in Fig. 9b.125,161 This decrease in mobility is likely due to the greater number of traps associated with higher Y3+ concentrations. However, acceptor doping also promotes the formation of hydroxyl groups, increasing the overall charge carrier concentration. This effect can mitigate proton trapping, allowing the material to sustain or even slightly enhance proton conductivity despite the presence of traps.125
At higher doping levels, the trapping zones formed by dopants begin to connect, creating percolation pathways that facilitate fast proton transport.125 In Draber's study, proton mobility increases once the Y concentration exceeds 3%, as the interconnected trapping zones in the Ba(Zr,Y)O3 system form continuous percolation pathways.125 This effect becomes more pronounced when the Y concentration reaches ∼20%, as shown in Fig. 10.125 The formation of these fast percolation pathways is a key factor contributing to the highest proton conductivity observed in BaZr0.8Y0.2O3−δ, as shown in Fig. 11a.
 | ||
| Fig. 10 Number of percolation pathways (connected dots) in (a) BaZr0.97Y0.03O3−δ (b)BaZr0.95Y0.05O3−δ (c) BaZr0.91Y0.09O3−δ and (d) BaZr0.75Y0.25O3−δ in a 16 × 16 × 16 supercell. This figure has been adapted from ref. 125 with permission from Springer Nature, copyright 2025. | ||
 | ||
| Fig. 11 (a) Electrical conductivities of two proton conductors: Ba(Zr, Y)O3−δ and Ba(Zr, Sc)O3−δ at 400 °C. This figure has been adapted from ref. 165 with permission from Taylor & Francis Group, copyright 2024.158 (b) Electrical conductivity of Sc-based proton conductors, Ba(Sc, Mo)O3−δ (BSM) and Ba(Sc, W)O3−δ (BSW) outperform BaZr0.8Y0.2O3−δ. This figure has been adapted from ref. 168 with permission from Royal Society of Chemistry, copyright 2025. | ||
However, an excessively high dopant concentration could also cause proton trapping,162,163 possibly because, at this stage, the host cations themselves begin to act as trapping sites. As the doping level further increases, proton mobility decreases, ultimately hindering bulk proton transport.164,165 This type of trapping is reported in systems such as Y- or Gd-doped BaZrO3 and BaCeO3.121,155,166 This trapping effect could be the reason for the reduced proton conductivity of the materials with a high content of dopants, even though a high donor doping level benefits hydroxyl group formation.167 For example, the proton conductivity of BaZr0.7Y0.3O3−δ is only 60% of that of BaZr0.8Y0.2O3−δ at 400 °C, as shown in Fig. 11a.
However, an exception to this trend is the Sc dopant, which does not exhibit a trapping effect in BaZrO3.163 Unlike other dopants, increasing the Sc concentration consistently enhances the proton conductivity of the Ba(Zr, Sc)O3−δ system, as shown in Fig. 11a. Recent studies have even reported that BaScO3, when stabilized by Mo or W, can achieve proton conductivities exceeding 0.01 S cm−1 at temperatures as low as 250 °C, outperforming BaZrO3 or BaCeO3-based proton conductors, as presented in Fig. 11b.168,169 While these results are promising, the precise role of Sc in proton transport remains insufficiently understood and requires further investigation.
Finally, grain boundaries can play a role in assisting proton transport at near room temperatures, but they tend to impede proton transport at elevated temperatures.170 Some studies even report that grain boundaries contribute more than 90% of resistance for proton transport.171–173 Reducing the number of grain boundaries by increasing grain size can decrease the grain boundary resistance, which can be achieved by sintering materials at increased temperatures for prolonged periods,174 adding sinter aids,175,176 or using methods such as pulsed laser deposition.4 Recently, Liu et al. demonstrated that increasing grain size significantly reduces the ohmic resistance of the electrolyte layer.5 They further reported that compositing nanoscale BCFZY with a microscale proton conductor enhances cathode ORR activity fourfold, attributed to the microscale proton conductor reducing grain boundary resistance for proton transport in the cathode.177 Further, Bian et al. employed acid etching to increase electrolyte surface roughness, thereby enhancing the number of cathode–electrolyte–gas triple-phase boundaries, which doubled the cathode activity.7 Their findings suggest that these triple-phase boundaries show higher activity than the cathode–gas interface, possibly due to the high resistance of proton transport through grain boundaries limiting proton transport to the cathode surface.7 Collectively, these results indicate that grain boundary resistance may be a critical challenge for TIECs.
In summary, hydroxyl groups are the major charge carriers for proton transport in perovskite oxides at elevated temperatures. The formation of hydroxyl groups needs to consume electron holes or oxygen vacancies, which might negatively affect the transport of holes and oxygen ions. The mobility of the proton depends on several factors. The short-range proton transport can be described using the Grotthuss mechanism, which is significantly impacted by local cation environments. The formation of fast percolation pathways determines the long-range proton transport, especially in the doped proton conductors. The formation of percolation pathways is related to the distribution of cations in the lattice structure. Finally, grain boundaries generate high resistance to proton transport. The high resistance of grain boundaries might impair proton transport in porous TIECs for ORR. These results together highlight the importance of the cationic arrangement and material microstructures in enabling fast proton transport.
4. The importance of balancing charge transport in TIECs
It is generally accepted that transition metal cations, such as Co and Fe, are the active centres for catalysing ORR. Therefore, a high concentration of these cations in TIECs is critical for high ORR activity.9,178 Meanwhile, as discussed in the sections above, it seems to be challenging to design a TIEC with improved content of holes, oxygen vacancies and hydroxyl groups due to their competitive charge-carrier formation.122Different from many MIECs operated in the  region and proton conductors operated in the
region and proton conductors operated in the  region as indicated in Fig. 7, the TIEC materials need to operate at a narrow regime with moderate concentrations for all charge carriers.122 This stringent requirement of the TIECs leads to compromised transport properties compared to proton conductors or MIECs. For example, Wang et al. compared the hydration enthalpy and entropy of proton conductors (electrolyte) and TIECs (cathode) and find that the hydration enthalpy of cathodes is typically less negative than proton conductors, as shown in Fig. 12a and b.37 Therefore, the hydration reaction of cathode materials would have a less negative Gibbs free energy and a lower KW compared to electrolyte materials. In another example, the typical TIEC, BCFZY, shows electrical conductivity (∼1 S cm−1 at 500 °C) much lower than MIECs such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ (∼10 S cm−1 at 500 °C) and (La, Sr)(Co, Fe)O3−δ (∼300 S cm−1 at 500 °C), as shown in Fig. 12c.179 This might be partially attributable to the reduced hole concentration in BaCo0.4Fe0.4Zr0.1Y0.1O3−δ, given that Zr and Y are not redox active.
region as indicated in Fig. 7, the TIEC materials need to operate at a narrow regime with moderate concentrations for all charge carriers.122 This stringent requirement of the TIECs leads to compromised transport properties compared to proton conductors or MIECs. For example, Wang et al. compared the hydration enthalpy and entropy of proton conductors (electrolyte) and TIECs (cathode) and find that the hydration enthalpy of cathodes is typically less negative than proton conductors, as shown in Fig. 12a and b.37 Therefore, the hydration reaction of cathode materials would have a less negative Gibbs free energy and a lower KW compared to electrolyte materials. In another example, the typical TIEC, BCFZY, shows electrical conductivity (∼1 S cm−1 at 500 °C) much lower than MIECs such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ (∼10 S cm−1 at 500 °C) and (La, Sr)(Co, Fe)O3−δ (∼300 S cm−1 at 500 °C), as shown in Fig. 12c.179 This might be partially attributable to the reduced hole concentration in BaCo0.4Fe0.4Zr0.1Y0.1O3−δ, given that Zr and Y are not redox active.
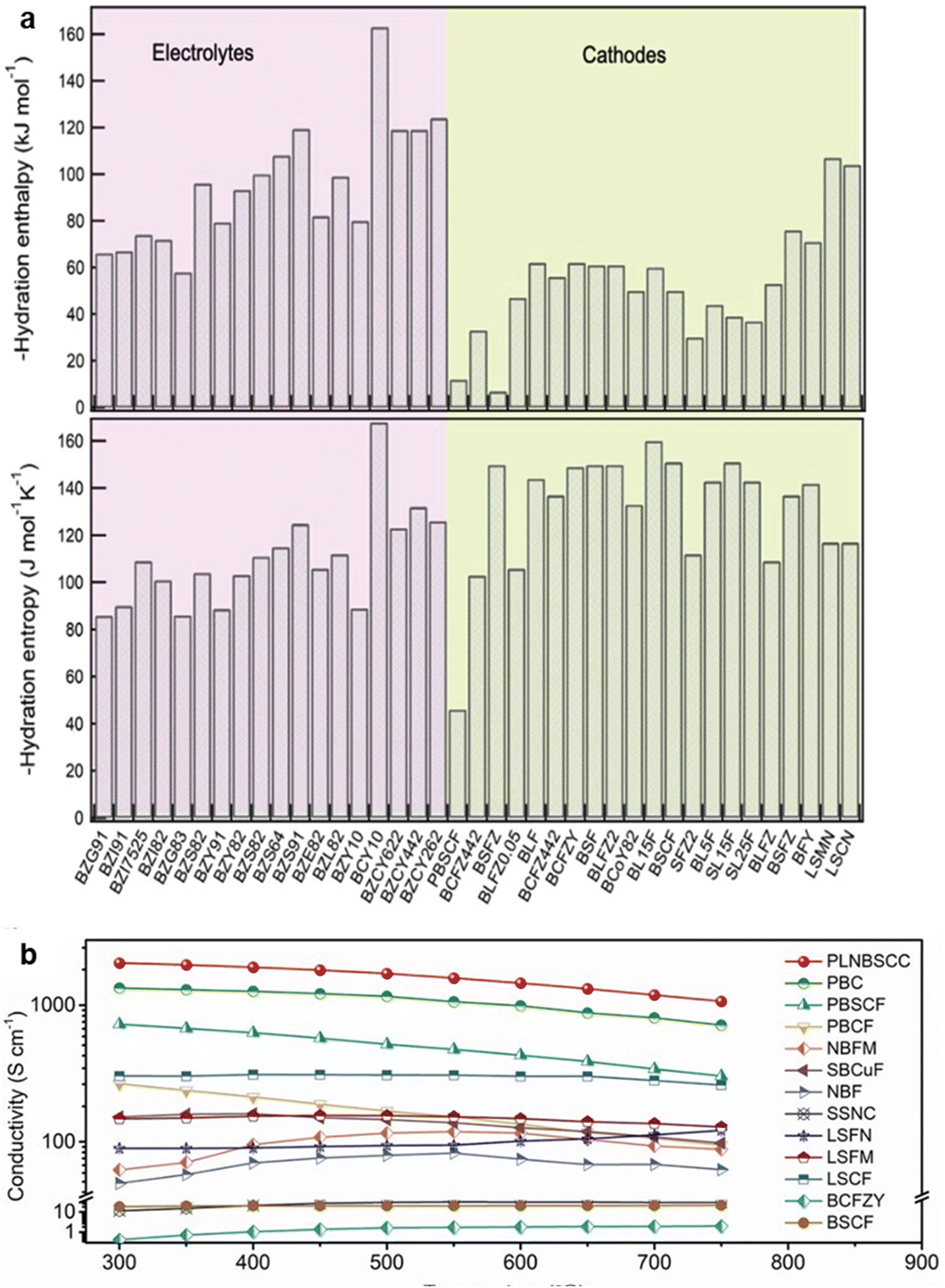 | ||
| Fig. 12 Comparison of hydration (a) enthalpy and (b) entropy of PCFC electrolytes and cathodes. Typical PCFC electrolytes are proton conductors while cathodes are TIECs. Typical cathodes show less negative hydration enthalpy than electrolytes. This figure has been adapted from ref. 37 with permission from John Wiley & Sons, copyright 2025. (c) The electrical conductivity of typical TIECs, MIECs, and BCFZY shows the lowest electrical conductivity among these materials. This figure has been adapted from ref. 125 with permission from Springer Nature, copyright 2025. | ||
Furthermore, the transport of these charge carriers also requires different cationic environments. In general, hole transport requires relatively high valence states, such as La3+ and Pr3+ at the A-site and redox-active cations, such as Mn or Ti at the B-site.180 These high valence state cations are essential for hole formation and suppress the oxygen vacancy formation reaction in eqn (3). The redox-active cations at the B-site also benefit hole percolation. However, oxygen ion transport might prefer cations with relatively low oxidation states, such as Sr2+ at the A-site and Co2+/3+ at the B-site.14,181 These cations can tune the cationic environment in perovskite oxides to realise the balance between high concentrations of oxygen vacancies and high mobility of oxygen ions.181 Further, most cations used in proton conductors are highly alkaline and redox-active inert, including Ba2+ at the A-site and Zr4+ and Y3+ at the B-site.125,182 These cations are critical for a cationic environment with high alkalinity for hydroxyl group formation and the formation of percolation pathways.125 Therefore, it is also challenging to design a composition that favours the transport of all three charge carriers.
4.1 Identifying dominant charge transport in accelerating the ORR kinetics
Since it is challenging to enhance all transport properties simultaneously, identifying the rate-limiting transport mechanisms in ORR becomes critical. Holes are the predominant charge carriers in most current TIECs. As a result, the electrical conductivity of these TIECs is very close to their electronic conductivities. Further, hole transport should be excessive in current TIECs. Liu et al. compared the electrical conductivity of several MIECs and TIECs. Their results reveal that BCFZY with the lowest electrical conductivity (∼1 S cm−1cf. ∼10 S cm−1 for Ba0.5Sr0.5Co0.8Fe0.2O3−δ and ∼300 S cm−1 for (La, Sr)(Co, Fe)O3−δ) shows the highest ORR activities, as shown in Fig. 12b.2,179 In another example, Liang et al. modified BCFZY via B-site nickel doping, which further reduces the electrical conductivity of BCFZY but enhances its ORR activity.29,183 Also, applying current collectors can enhance the electrical conductivity of cathodes, making slow hole transport of these materials less of a problem. In summary, even with electrical conductivity less than 1 S cm−1, the hole transport seems not to be the rate-limiting step in ORR.Unfortunately, the transport of oxygen and proton is often much slower than the holes in TIECs and should play a role in determining the ORR activity of the TIECs.184 However, it remains unclear by far which ionic transport (oxygen ion or proton) predominately limits the TIEC's ORR activity.184 Part of the reason is related to the challenges in directly quantifying the transport of these two charges in TIECs. Many studies rely on multimodal characterisation techniques to access the transport properties of oxygen ions and protons in TIECs.29,31,45,133,185–187 The reliance on multiple characterization techniques complicates the accurate assessment of their contributions to overall conductivity and transport behaviour. Thus, more precise and unified approaches are needed to better understand and quantify these transport mechanisms.
Nevertheless, as aforementioned in sections 3.2 and 3.3, increasing the concentration of oxygen vacancies in the perovskite oxide structure could benefit proton transport by promoting the hydration reaction, and oxygen ion transport, even though the hydration reaction would consume oxygen vacancies.29 Therefore, implying strategies that can promote oxygen vacancy formation can enhance the ORR activity of TIECs.29,178,188,189 However, increasing the oxygen vacancy concentration can reduce the oxygen ion mobility,181 and might also destabilise the perovskite oxide structure, converting perovskite oxides into brownmillerite structure102 or leading to the formation of a secondary phase.185 Therefore, the design of single-phase TIECs requires dedicated control over the protonic and oxygen-ion transport to achieve an optimal ORR activity for PCFCs.
4.2 Composite TIECs for mixed ionic–electronic conduction
The development of composite TIECs can be an alternative strategy to the single-phase TIEC material design.5,133,185,190,191 Composite TIECs contain more than one phase to facilitate the charge transfer. In early PCFC cathode studies, Fabbri et al. composited (La, Sr)(Co, Fe)O3−δ, an MIEC with proton conductors such as BaCe0.9Y0.1O3−δ192 and BaZr0.5Pr0.3Y0.2O3−δ.193 When different conductors are mixed, protons can transport in the cathode within these proton conductors while holes and oxygen ions transport through (La, Sr)(Co, Fe)O3−δ.192 The contact between these two phases becomes active for ORR.192 These increased ORR active sites improve the ORR activity of the cathode 2.5-fold.192 Similarly, Duan et al. also demonstrated that BCFZY, a TIEC cathode, also shows enhanced ORR activity after compositing with BaZr0.3Ce0.6Y0.1O3−δ.2 Since the contact between phases is active for ORR, composites with more phase boundaries often show increased ORR activity. Fig. 13 compares the performance of some reported single-phase and composite TIECs on PCFCs. In general, composite TIECs show similar ORR activity compared to single-phase TIECs.
 | ||
| Fig. 13 The ASR and peak power density of reported single phase and composite TIECs on PCFCs at 600 °C. Data are collected from ref. 2, 4, 5, 9, 13, 18, 22, 29–34, 133, 185, 190, 194–197. | ||
Traditional compositing methods, such as mechanical mixing,2,191,198 tend to generate composites with phases loosely in contact with each other. Recently, many studies have focused on self-assembly composites to improve the contact between phases.5,133,179,185,190,199,200 These self-assembled composites can form nanoscale particles (e.g., ∼100 nm) during synthesis.133,143,185,190 For example, Song et al. report a self-assembly composite, BaCo0.7(Ce0.8Y0.2)0.3O3−δ.190 During synthesis, this composite forms a Co-rich mixed oxygen ion-electronic conducting phase, a Ce-rich mixed proton-electronic conducting phase, and a BaCoO3−δ hole conducting phase.190 Protons can transport from the electrolyte to the Ce-rich phase, while oxygen ions can transport through the Co-rich phase.190 Protons and oxygen ions can combine to form water at the abundant boundaries of these two phases.190 As a result, their composite cathodes show ORR activity higher than single-phase TIECs such as BCFZY. Other studies also show that forming nanoparticles such as metallic silver133 or nickel oxide185 at BCFZY surface can facilitate oxygen ion surface transport. These surface nanoparticles also increase the ORR activity of BCFZY more than twofold.133,185,194 These studies together indicate that the superior ORR activity of self-assembled cathodes is closely related to the enhanced contact between these conducting phases.
5. Summary and outlook
In this review, we examined hole, oxygen ion, and proton transport in TIECs designed to catalyse the ORR in PCFC cathodes. Even the most advanced TIECs currently fall short of providing adequate ORR activity for practical PCFC applications, which could be related to imbalanced charge transport in TIECs. Charge transport depends on the concentration and mobility of charge carriers in TIECs, which are significantly impacted by the cationic environment in perovskite oxides. However, holes, oxygen ions and protons require distinct cationic environments for their fast transport, which creates trade-offs in the design of TIECs. Holes show the fastest transport among these charges, rendering it less likely to be the limiting transport for ORR, but whether oxygen ion or proton transport is the dominant contributor to the ORR activity in TIECs remains unclear. The transport of oxygen ions and protons is much slower than hole transport, and it is complicated to deconvolute their transports in TIECs under PCFC operating conditions. As a result, probing these ionic transports is challenging, but is necessary for understanding their role in ORR.Further research efforts are demanded to address challenges in the development of TIECs with enhanced ORR activity. First, it remains difficult to quantitively measure the formation and transport of hydroxyl groups in TIECs, especially under PCFC operating conditions. Protons are light in weight and do not contain any electrons. Consequently, probing the behaviour of protons with high accuracy is challenging but necessary to understand proton transport in TIECs. It is possible to take advantage of technology that relies on nuclei instead of electrons, such as pulsed-gradient spin-echo nuclear magnetic resonance, mass spectroscopy, time-of-flight secondary ion mass spectrometry, quasi-elastic neutron scattering, to study the formation and transport of hydroxyl groups in TIECs further. The new knowledge gained from these investigations can help us design the next generation of TIECs with balanced proton and oxygen ion transports for enhanced ORR activity.
Furthermore, oxygen ion transport in TIECs under humid conditions should be investigated. The transport of oxygen ions is often measured under dry conditions using techniques such as ECR. However, the presence of humidity can deplete oxygen vacancies and might impair oxygen ion transport in TIECs. As a result, fast oxygen ion transport under dry conditions does not necessarily lead to their fast transport in humid conditions. Therefore, it should take the impact of humidity into account when measuring the transport of oxygen ions and measuring oxygen ion transport under PCFC operating conditions is highly encouraged.
Machine learning should also play a role in the next generation of TIEC designs. Machine learning shows the possibility of predicting the structure and properties of materials, including phase evolution, oxygen vacancy formation, ionic conductivities and more. Therefore, ML can accelerate the development of single-phase and self-assembled TIECs with balanced ionic conductivity. By leveraging vast datasets of material properties and performance metrics, ML algorithms might identify patterns, predict material behaviour, and guide the design of compositions with optimal ionic conductivity and ORR activity.
Finally, the design of TIECs also needs to consider the stability issue. Current TIECs are mainly Ba- or Sr-based perovskite oxides. These alkaline-earth elements are critical for fast proton transport but also are susceptible to gases such as CO2 at reduced temperatures. The reaction between these alkaline-earth elements and trace amounts of CO2 in air can lead to the formation of the carbonate phase and the depletion of the TIEC phase. As a result, the practical application of TIECs on PCFCs requires stabilising these alkaline-earth elements, such as bulk doping or surface modification, or even replacing them with lanthanoid elements such as La and Pr.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no financial interests or personal conflict of interest.Acknowledgements
M. L. acknowledges the financial support from the Australian Research Council (DE230100637) and Australia Sunlight Group Pty Ltd (AUKRH000031). Z. Z. acknowledges the support from Australian Research Council Discovery Projects DP200101397 and DP250102334. D. F. and D. L. acknowledges the financial support from the Australian Research Council (FL180100029).References
- Y. Zhou, X. Guan, H. Zhou, K. Ramadoss, S. Adam, H. Liu, S. Lee, J. Shi, M. Tsuchiya, D. D. Fong and S. Ramanathan, Nature, 2016, 534, 231–234 CrossRef CAS PubMed.
- C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori and R. O'Hayre, Science, 2015, 349, 1321–1326 CrossRef CAS PubMed.
- C. Duan, R. J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E. J. Crumlin, D. Hook, R. Braun, N. P. Sullivan and R. O'Hayre, Nature, 2018, 557, 217–222 CrossRef CAS PubMed.
- S. Choi, C. J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji and S. M. Haile, Nat. Energy, 2018, 3, 202–210 CrossRef CAS.
- F. Liu, H. Deng, D. Diercks, P. Kumar, M. H. A. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, L. Fang, D. Chen, B. Liu and C. Duan, Nat. Energy, 2023, 8, 1145–1157 CrossRef CAS.
- H. An, H.-W. Lee, B.-K. Kim, J.-W. Son, K. J. Yoon, H. Kim, D. Shin, H.-I. Ji and J.-H. Lee, Nat. Energy, 2018, 3, 870–875 CrossRef CAS.
- W. Bian, W. Wu, B. Wang, W. Tang, M. Zhou, C. Jin, H. Ding, W. Fan, Y. Dong, J. Li and D. Ding, Nature, 2022, 604, 479–485 CrossRef CAS PubMed.
- C. Duan, R. Kee, H. Zhu, N. Sullivan, L. Zhu, L. Bian, D. Jennings and R. O'Hayre, Nat. Energy, 2019, 4, 230–240 CrossRef CAS.
- Z. Wang, Y. Wang, J. Wang, Y. Song, M. J. Robson, A. Seong, M. Yang, Z. Zhang, A. Belotti, J. Liu, G. Kim, J. Lim, Z. Shao and F. Ciucci, Nat. Catal., 2022, 5, 777–787 CrossRef CAS.
- L. Yang, S. Wang, K. Blinn, M. Liu, Z. Liu, Z. Cheng and M. Liu, Science, 2009, 326, 126–129 CrossRef CAS PubMed.
- R. J. Gorte, Science, 2015, 349, 1290–1290 CrossRef CAS PubMed.
- Z. Wang, W. Yang, Z. Zhu, R. Peng, X. Wu, C. Xia and Y. Lu, J. Mater. Chem. A, 2014, 2, 16707–16714 RSC.
- J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu and G. Kim, ChemSusChem, 2014, 7, 2811–2815 CrossRef CAS PubMed.
- M. Li, M. Zhao, F. Li, W. Zhou, V. K. Peterson, X. Xu, Z. Shao, I. Gentle and Z. Zhu, Nat. Commun., 2017, 8, 13990 CrossRef CAS PubMed.
- Z. Li, X. Mao, D. Feng, M. Li, X. Xu, Y. Luo, L. Zhuang, R. Lin, T. Zhu, F. Liang, Z. Huang, D. Liu, Z. Yan, A. Du, Z. Shao and Z. Zhu, Nat. Commun., 2024, 15, 9318 CrossRef CAS PubMed.
- Z. Shao and S. M. Haile, Nature, 2004, 431, 170–173 CrossRef CAS PubMed.
- W. Zhou, J. Sunarso, M. Zhao, F. Liang, T. Klande and A. Feldhoff, Angew. Chem., Int. Ed., 2013, 52, 14036–14040 CrossRef CAS PubMed.
- M. Saqib, I.-G. Choi, H. Bae, K. Park, J.-S. Shin, Y.-D. Kim, J.-I. Lee, M. Jo, Y.-C. Kim, K.-S. Lee, S.-J. Song, E. D. Wachsman and J.-Y. Park, Energy Environ. Sci., 2021, 14, 2472–2484 RSC.
- G. Taillades, J. Dailly, M. Taillades-Jacquin, F. Mauvy, A. Essouhmi, M. Marrony, C. Lalanne, S. Fourcade, D. J. Jones, J.-C. Grenier and J. Rozière, Fuel Cells, 2010, 10, 166–173 CrossRef CAS.
- Y. Lin, R. Ran, Y. Zheng, Z. Shao, W. Jin, N. Xu and J. Ahn, J. Power Sources, 2008, 180, 15–22 CrossRef CAS.
- L. Bi, E. Fabbri, Z. Sun and E. Traversa, Energy Environ. Sci., 2011, 4, 1352–1357 RSC.
- H. Shi, C. Su, X. Xu, Y. Pan, G. Yang, R. Ran and Z. Shao, Small, 2021, 17, 2101872 CrossRef CAS PubMed.
- L. Bi, E. Fabbri, Z. Sun and E. Traversa, Energy Environ. Sci., 2011, 4, 409–412 RSC.
- J. Dailly, G. Taillades, M. Ancelin, P. Pers and M. Marrony, J. Power Sources, 2017, 361, 221–226 CrossRef CAS.
- Q. Shao, W. Ge, X. Lu, Y. Chen, Y. Ding, B. Lin and Y. Ling, Ceram. Int., 2015, 41, 6687–6692 CrossRef CAS.
- K. Xie, R. Yan, X. Chen, D. Dong, S. Wang, X. Liu and G. Meng, J. Alloys Compd., 2009, 472, 551–555 CrossRef CAS.
- B. Lin, S. Zhang, L. Zhang, L. Bi, H. Ding, X. Liu, J. Gao and G. Meng, J. Power Sources, 2008, 177, 330–333 CrossRef CAS.
- L. Bi, S. Zhang, S. Fang, Z. Tao, R. Peng and W. Liu, Electrochem. Commun., 2008, 10, 1598–1601 CrossRef CAS.
- M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou and Z. Shao, Chem. Eng. J., 2021, 420, 127717 CrossRef CAS.
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS.
- K. Pei, Y. Zhou, K. Xu, H. Zhang, Y. Ding, B. Zhao, W. Yuan, K. Sasaki, Y. Choi, Y. Chen and M. Liu, Nat. Commun., 2022, 13, 2207 CrossRef CAS PubMed.
- W. Zhang, Y. Zhou, X. Hu, Y. Ding, J. Gao, Z. Luo, T. Li, N. Kane, X.-Y. Yu, T. Terlier and M. Liu, ACS Energy Lett., 2023, 8, 3999–4007 CrossRef CAS PubMed.
- C. Zhou, J. Sunarso, Y. Song, J. Dai, J. Zhang, B. Gu, W. Zhou and Z. Shao, J. Mater. Chem. A, 2019, 7, 13265–13274 RSC.
- X. Xu, H. Wang, M. Fronzi, X. Wang, L. Bi and E. Traversa, J. Mater. Chem. A, 2019, 7, 20624–20632 RSC.
- M. Shang, J. Tong and R. O'Hayre, RSC Adv., 2013, 3, 15769–15775 RSC.
- M. Papac, V. Stevanović, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed.
- N. Wang, C. Tang, L. Du, R. Zhu, L. Xing, Z. Song, B. Yuan, L. Zhao, Y. Aoki and S. Ye, Adv. Energy Mater., 2022, 12, 2201882 CrossRef CAS.
- T. P. Pandey, A. M. Maes, H. N. Sarode, B. D. Peters, S. Lavina, K. Vezzù, Y. Yang, S. D. Poynton, J. R. Varcoe, S. Seifert, M. W. Liberatore, V. D. Noto and A. M. Herring, Phys. Chem. Chem. Phys., 2015, 17, 4367–4378 RSC.
- D. Poetzsch, R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2014, 16, 16446–16453 RSC.
- Z. Luo, J. Tang, Z. Wang, G. Yang, T. Zhu, Z. Lin, S. Ping Jiang and Z. Shao, Energy Environ. Sci., 2024, 17, 4115–4125 RSC.
- Y. Huang, R. Qiu, W. Lian, L. Lei, T. Liu, J. Zhang, Y. Wang, J. Liu, J. Huang and F. Chen, J. Power Sources, 2022, 528, 231201 CrossRef CAS.
- A. Seong, J. Kim, D. Jeong, S. Sengodan, M. Liu, S. Choi and G. Kim, Adv. Sci., 2021, 8, 2004099 CrossRef CAS PubMed.
- B. Liu, Y. Zheng, X. Yang, Y. Liu, D. Yan, J. Li and L. Jia, Chem. Eng. J., 2025, 505, 159163 CrossRef CAS.
- Y. Feng, P. Su, H. Liu, X. Sun, B. Zhang, Q. Duan, J. Li, Y. Shen and F. Wang, Chem. Eng. J., 2024, 500, 156758 CrossRef CAS.
- P. Yao, J. Zhang, Q. Qiu, Y. Zhao, F. Yu and Y. Li, Adv. Energy Mater., 2024, 2403335 Search PubMed.
- P. Yao, J. Zhang, Q. Qiu, Y. Zhao, F. Yu and Y. Li, J. Power Sources, 2024, 606, 234591 CrossRef CAS.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- W. Zhang, X. Zhang, Y. Song and G. Wang, Sustainability, 2024, 3, 100028 Search PubMed.
- M. Wang, C. Su, Z. Zhu, H. Wang and L. Ge, Composites, Part B, 2022, 238, 109881 CrossRef CAS.
- J. Cao, Y. Ji and Z. Shao, Energy Environ. Sci., 2022, 15, 2200–2232 RSC.
- M. A. Peña and J. L. G. Fierro, Chem. Rev., 2001, 101, 1981–2018 CrossRef.
- J. Wang, Y. Lu, N. Mushtaq, M. A. K. Y. Shah, S. Rauf, P. D. Lund and M. I. Asghar, J. Rare Earths, 2023, 41, 413–421 CrossRef CAS.
- L. Huang and Y. Wu, Ceram. Int., 2024, 50, 5150–5159 CrossRef CAS.
- D. Huan, L. Zhang, X. Li, Y. Xie, N. Shi, S. Xue, C. Xia, R. Peng and Y. Lu, ChemSusChem, 2020, 13, 4994–5003 CrossRef CAS PubMed.
- Z. Li, M. Li and Z. Zhu, Electrochem. Energy Rev., 2022, 5, 263–311 CrossRef CAS.
- F. He, M. Liang, W. Wang, R. Ran, G. Yang, W. Zhou and Z. Shao, Energy Fuels, 2020, 34, 11464–11471 CrossRef CAS.
- Y. Shin, Y. Kim, M. Sanders, S. P. Harvey, M. Walker and R. O'Hayre, J. Mater. Chem. A, 2022, 10, 24839–24853 RSC.
- B. Wang and K. Ohgushi, Sci. Rep., 2016, 6, 37896 CrossRef CAS PubMed.
- E. Robens, R. Rauschen, J. Kaub, J. P. Parras, D. Kemp, C. L. Freeman and R. A. De Souza, J. Mater. Chem. A, 2022, 10, 2388–2397 RSC.
- K. D. Kreuer, Solid State Ionics, 1999, 125, 285–302 CrossRef CAS.
- V. M. Goldschmidt, Naturwissenschaften, 1926, 14, 477–485 CrossRef CAS.
- C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli and M. Scheffler, Sci. Adv., 2019, 5, eaav0693 CrossRef CAS PubMed.
- K. Yim, Y. Youn, M. Lee, D. Yoo, J. Lee, S. H. Cho and S. Han, Npj Comput. Mater., 2018, 4, 1–7 CrossRef CAS.
- H. He, Z. Yang, Y. Xu, A. T. Smith, G. Yang and L. Sun, Nano Converg., 2020, 7, 32 CrossRef CAS PubMed.
- R. Oja, M. Tyunina, L. Yao, T. Pinomaa, T. Kocourek, A. Dejneka, O. Stupakov, M. Jelinek, V. Trepakov, S. van Dijken and R. M. Nieminen, Phys. Rev. Lett., 2012, 109, 127207 CrossRef CAS PubMed.
- A. Gómez-Pérez, M. Yuste, J. C. Pérez-Flores, C. Ritter, M. T. Azcondo, J. Canales-Vázquez, M. Gálvez-Sánchez, K. Boulahya, F. García-Alvarado and U. Amador, J. Power Sources, 2013, 227, 309–317 CrossRef.
- A. Gómez-Pérez, M. T. Azcondo, M. Yuste, J. C. Pérez-Flores, N. Bonanos, F. Porcher, A. Muñoz-Noval, M. Hoelzel, F. García-Alvarado and U. Amador, J. Mater. Chem. A, 2016, 4, 3386–3397 RSC.
- A. Gómez-Pérez, M. Yuste, J. C. Pérez-Flores, C. Ritter, M. T. Azcondo, J. Canales-Vázquez, M. Gálvez-Sánchez, K. Boulahya, F. García-Alvarado and U. Amador, J. Power Sources, 2013, 227, 309–317 CrossRef.
- R. Ganguly, A. K. Singh, R. Kumar, A. Gupta, A. K. Pandey and A. K. Pandey, in Nanotechnology in Modern Animal Biotechnology, ed. P. K. Maurya and S. Singh, Elsevier, 2019, pp. 29–35 Search PubMed.
- M. Fabián, B. I. Arias-Serrano, A. A. Yaremchenko, H. Kolev, M. Kaňuchová and J. Briančin, J. Eur. Ceram. Soc., 2019, 39, 5298–5308 CrossRef.
- J. Richter, P. Holtappels, T. Graule, T. Nakamura and L. J. Gauckler, Monatsh. Chem., 2009, 140, 985–999 CrossRef CAS.
- D. Neagu and J. T. S. Irvine, Chem. Mater., 2011, 23, 1607–1617 CrossRef CAS.
- P. M. Raccah and J. B. Goodenough, Phys. Rev., 1967, 155, 932–943 CrossRef CAS.
- S. Kirkpatrick, Rev. Mod. Phys., 1973, 45, 574–588 CrossRef.
- D. Kim, S. Miyoshi, T. Tsuchiya and S. Yamaguchi, Solid State Ionics, 2014, 262, 875–878 CrossRef CAS.
- R. Raffaelle, H. U. Anderson, D. M. Sparlin and P. E. Parris, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 43, 7991–7999 CrossRef CAS PubMed.
- Z. Sherafat, M. H. Paydar, I. Antunes, N. Nasani, A. D. Brandão and D. P. Fagg, Electrochim. Acta, 2015, 165, 443–449 CrossRef CAS.
- L.-W. Tai, M. M. Nasrallah, H. U. Anderson, D. M. Sparlin and S. R. Sehlin, Solid State Ionics, 1995, 76, 259–271 CrossRef CAS.
- C. S. Kim, S. R. Bishop and H. L. Tuller, J. Electroceramics, 2018, 40, 57–64 CrossRef CAS.
- T. Hong, W. Lu, K. Ren and T. Liu, Ionics, 2020, 26, 5293–5297 CrossRef CAS.
- S. B. Adler, Chem. Rev., 2004, 104, 4791–4844 CrossRef CAS PubMed.
- E. N. Armstrong, K. L. Duncan and E. D. Wachsman, Phys. Chem. Chem. Phys., 2013, 15, 2298–2308 RSC.
- Z. Zhang, X. Xu, J. Zhang, D. Chen, D. Zeng, S. Liu, W. Zhou and Z. Shao, J. Membr. Sci., 2018, 563, 617–624 CrossRef CAS.
- A. Zomorrodian, H. Salamati, Z. Lu, X. Chen, N. Wu and A. Ignatiev, Int. J. Hydrogen Energy, 2010, 35, 12443–12448 CrossRef CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95–110 CrossRef CAS.
- K. Huang, R. S. Tichy and J. B. Goodenough, J. Am. Ceram. Soc., 1998, 81, 2565–2575 CrossRef CAS.
- J. Meng, M. S. Sheikh, R. Jacobs, J. Liu, W. O. Nachlas, X. Li and D. Morgan, Nat. Mater., 2024, 23, 1252–1258 CrossRef CAS PubMed.
- Q. Li, Y.-X. Deng, Y.-A. Zhu, Y. Li, Z.-J. Sui, D. Chen and W.-K. Yuan, Catal. Today, 2020, 347, 142–149 CrossRef CAS.
- T. Roohandeh and E. Saievar-Iranizad, Appl. Phys. A, 2019, 125, 552 CrossRef CAS.
- E. Olsson, J. Cottom, X. Aparicio-Anglès and N. H. de Leeuw, Phys. Chem. Chem. Phys., 2019, 21, 9407–9418 RSC.
- H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C. J. Orme, L. Wang, Y. Zhang and D. Ding, Nat. Commun., 2020, 11, 1907 CrossRef CAS PubMed.
- C. Duan, D. Hook, Y. Chen, J. Tong and R. O'Hayre, Energy Environ. Sci., 2017, 10, 176–182 RSC.
- M. Li, W. Zhou and Z. Zhu, ChemElectroChem, 2015, 2, 1331–1338 CrossRef CAS.
- C. Sun, Y. Kong, L. Shao, K. Sun and N. Zhang, J. Power Sources, 2020, 459, 228017 CrossRef CAS.
- T. J. Frankcombe and Y. Liu, Chem. Mater., 2023, 35, 5468–5474 CrossRef CAS.
- K. S. Knight, Solid State Ionics, 2001, 145, 275–294 CrossRef CAS.
- S. Zhai, H. Xie, P. Cui, D. Guan, J. Wang, S. Zhao, B. Chen, Y. Song, Z. Shao and M. Ni, Nat. Energy, 2022, 7, 866–875 CrossRef CAS.
- L. Giordano, K. Akkiraju, R. Jacobs, D. Vivona, D. Morgan and Y. Shao-Horn, Acc. Chem. Res., 2022, 55, 298–308 CrossRef CAS PubMed.
- R. Jacobs, J. Liu, H. Abernathy and D. Morgan, Adv. Energy Mater., 2024, 14, 2303684 CrossRef CAS.
- Q. Tao, P. Xu, M. Li and W. Lu, Npj Comput. Mater., 2021, 7, 1–18 CrossRef.
- Q. Tao, T. Lu, Y. Sheng, L. Li, W. Lu and M. Li, J. Energy Chem., 2021, 60, 351–359 CrossRef CAS.
- S. K. Jaiswal, V. K. Kashyap and J. Kumar, J. Asian Ceram. Soc., 2020, 8, 1018–1026 CrossRef.
- S. Sengodan, S. Choi, A. Jun, T. H. Shin, Y.-W. Ju, H. Y. Jeong, J. Shin, J. T. S. Irvine and G. Kim, Nat. Mater., 2015, 14, 205–209 CrossRef CAS PubMed.
- M. Mogensen, D. Lybye, N. Bonanos, P. V. Hendriksen and F. W. Poulsen, Solid State Ionics, 2004, 174, 279–286 CrossRef CAS.
- R. A. De Souza, Adv. Funct. Mater., 2015, 25, 6326–6342 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- R. L. Cook, J. J. Osborne, J. H. White, R. C. MacDuff and A. F. Sammells, J. Electrochem. Soc., 1992, 139, L19 CrossRef CAS.
- Y. Lu, H. Zhao, X. Cheng, Y. Jia, X. Du, M. Fang, Z. Du, K. Zheng and K. Świerczek, J. Mater. Chem. A, 2015, 3, 6202–6214 RSC.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- G. Gregori, M. Shirpour and J. Maier, Adv. Funct. Mater., 2013, 23, 5861–5867 CrossRef CAS.
- F. Maglia, I. G. Tredici, G. Spinolo and U. Anselmi-Tamburini, J. Mater. Res., 2012, 27, 1975–1981 CrossRef CAS.
- S. Miyoshi, Y. Akao, N. Kuwata, J. Kawamura, Y. Oyama, T. Yagi and S. Yamaguchi, Chem. Mater., 2014, 26, 5194–5200 CrossRef CAS.
- S. Kim, H. J. Avila-Paredes, S. Wang, C.-T. Chen, R. A. D. Souza, M. Martin and Z. A. Munir, Phys. Chem. Chem. Phys., 2009, 11, 3035–3038 RSC.
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- T. Norby, Prog. Energy, 2024, 6, 043002 CrossRef CAS.
- R. Sato, S. Ohkuma, Y. Shibuta, F. Shimojo and S. Yamaguchi, J. Phys. Chem. C, 2015, 119, 28925–28933 CrossRef CAS.
- Y. Zhao, T. Shi, J. Shang, L. Ding, X. Cao, C. Chen and J. Zhao, Appl. Catal., B, 2020, 277, 119234 CrossRef CAS.
- T. Norby, Solid State Ionics, 1999, 125, 1–11 CrossRef CAS.
- R. Morikawa, T. Murakami, K. Fujii, M. Avdeev, Y. Ikeda, Y. Nambu and M. Yashima, Commun. Mater., 2023, 4, 1–9 CrossRef.
- T. Nakao, A. Mineshige, M. Kobune, T. Yazawa and H. Yoshioka, Solid State Ionics, 2008, 179, 1567–1569 CrossRef CAS.
- Y. Yamazaki, F. Blanc, Y. Okuyama, L. Buannic, J. C. Lucio-Vega, C. P. Grey and S. M. Haile, Nat. Mater., 2013, 12, 647–651 CrossRef CAS.
- D. Poetzsch, R. Merkle and J. Maier, Adv. Funct. Mater., 2015, 25, 1542–1557 CrossRef CAS.
- T. Norby, in Perovskite Oxide for Solid Oxide Fuel Cells, ed. T. Ishihara, Springer US, Boston, MA, 2009, pp. 217–241 Search PubMed.
- F. M. Draber, C. Ader, J. P. Arnold, S. Eisele, S. Grieshammer, S. Yamaguchi and M. Martin, Nat. Mater., 2020, 19, 338–346 CrossRef CAS.
- R. Zohourian, R. Merkle, G. Raimondi and J. Maier, Adv. Funct. Mater., 2018, 28, 1801241 CrossRef.
- K. Arai, Y. Asai, Y. Kokubo, M. Saito, M. Inada, K. Hayashi and T. Motohashi, J. Solid State Chem., 2023, 323, 124026 CrossRef CAS.
- H. Maekawa, N. Kashii, J.-I. Kawamura, Y. Hinatsu and T. Yamamura, Solid State Ionics, 1999, 122, 231–236 CrossRef CAS.
- D. Poetzsch, R. Merkle and J. Maier, Phys. Chem. Chem. Phys., 2014, 16, 16446–16453 RSC.
- A. Løken, T. S. Bjørheim and R. Haugsrud, J. Mater. Chem. A, 2015, 3, 23289–23298 RSC.
- S. Fop, K. S. McCombie, E. J. Wildman, J. M. S. Skakle, J. T. S. Irvine, P. A. Connor, C. Savaniu, C. Ritter and A. C. Mclaughlin, Nat. Mater., 2020, 19, 752–757 CrossRef CAS PubMed.
- T. Nagasaki, S. Shiotani, N. Igawa, M. Yoshino, K. Iwasaki, H. Fukazawa and W. Utsumi, J. Solid State Chem., 2009, 182, 2632–2639 CrossRef CAS.
- G. C. Mather, G. Heras-Juaristi, C. Ritter, R. O. Fuentes, A. L. Chinelatto, D. Pérez-Coll and U. Amador, Chem. Mater., 2016, 28, 4292–4299 CrossRef CAS.
- J. H. Kim, J. Hong, D.-K. Lim, S. Ahn, J. Kim, J. K. Kim, D. Oh, S. Jeon, S.-J. Song and W. Jung, Energy Environ. Sci., 2022, 15, 1097–1105 RSC.
- H. Sakaguchi, K. Hatakeyama, S. Kobayashi and T. Esaka, Mater. Res. Bull., 2002, 37, 1547–1556 CrossRef CAS.
- K. D. Kreuer, Solid State Ionics, 1997, 97, 1–15 CrossRef CAS.
- N. Bonanos, in Encyclopedia of Applied Electrochemistry, ed. G. Kreysa, K. Ota and R. F. Savinell, Springer, New York, NY, 2014, pp. 1514–1520 Search PubMed.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- A. Perrichon, E. Jedvik Granhed, G. Romanelli, A. Piovano, A. Lindman, P. Hyldgaard, G. Wahnström and M. Karlsson, Chem. Mater., 2020, 32, 2824–2835 CrossRef CAS.
- D. Han, K. Shinoda, S. Sato, M. Majima and T. Uda, J. Mater. Chem. A, 2014, 3, 1243–1250 RSC.
- H. Iwahara, Solid State Ionics, 1996, 86–88, 9–15 CrossRef CAS.
- H. Iwahara, T. Yajima, T. Hibino, K. Ozaki and H. Suzuki, Solid State Ionics, 1993, 61, 65–69 CrossRef CAS.
- P. A. Stuart, T. Unno, J. A. Kilner and S. J. Skinner, Solid State Ionics, 2008, 179, 1120–1124 CrossRef CAS.
- R. Murphy, Y. Zhou, L. Zhang, L. Soule, W. Zhang, Y. Chen and M. Liu, Adv. Funct. Mater., 2020, 30, 2002265 CrossRef CAS.
- R. Hempehnann, Ch. Karmonik, Th. Matzke, M. Cappadonia, U. Stimming, T. Springer and M. A. Adams, Solid State Ionics, 1995, 77, 152–156 CrossRef.
- M. E. Björketun, P. G. Sundell, G. Wahnström and D. Engberg, Solid State Ionics, 2005, 176, 3035–3040 CrossRef.
- M. Karlsson, Phys. Chem. Chem. Phys., 2015, 17, 26–38 RSC.
- A. Perrichon, M. M. Koza, Z. Evenson, B. Frick, F. Demmel, P. Fouquet and M. Karlsson, Chem. Mater., 2023, 35, 6713–6725 CrossRef CAS.
- E. Naumovska, J. Orstadius, A. Perrichon, R. Lavén, M. M. Koza, Z. Evenson and M. Karlsson, J. Phys. Chem. C, 2023, 127, 24532–24541 CrossRef CAS.
- C. Xia, Y. Mi, B. Wang, B. Lin, G. Chen and B. Zhu, Nat. Commun., 2019, 10, 1707 CrossRef PubMed.
- Q. Li, Q. Yin, Y.-S. Zheng, Z.-J. Sui, X.-G. Zhou, D. Chen and Y.-A. Zhu, Langmuir, 2019, 35, 9962–9969 CrossRef CAS PubMed.
- W. Münch, G. Seifert, K. D. Kreuer and J. Maier, Solid State Ionics, 1996, 86–88, 647–652 CrossRef.
- D. Noferini, M. M. Koza and M. Karlsson, J. Phys. Chem. C, 2017, 121, 7088–7093 CrossRef CAS.
- K. D. Kreuer, Th. Dippel, Yu. M. Baikov and J. Maier, Solid State Ionics, 1996, 86–88, 613–620 CrossRef CAS.
- M. S. Islam, R. A. Davies and J. D. Gale, Chem. Commun., 2001, 661–662 RSC.
- D. Vignesh, B. K. Sonu and E. Rout, Energy Fuels, 2022, 36, 7219–7244 CrossRef CAS.
- C. Karmonik, R. Hempelmann, T. Matzke and T. Springer, Z. Naturforsch., A:Phys. Sci., 1995, 50, 539–548 CrossRef CAS.
- M. S. Islam, P. R. Slater, J. R. Tolchard and T. Dinges, Dalton Trans., 2004, 3061–3066 RSC.
- C. Y. Regalado Vera, H. Ding, J. Urban-Klaehn, M. Li, Z. Zhao, F. Stewart, H. Tian, X. Liu, Y. Dong, J. Li, M. Zhou, H. Luo and D. Ding, Chem. Mater., 2023, 35, 5341–5352 CrossRef CAS.
- K. Toyoura, W. Meng, D. Han and T. Uda, J. Mater. Chem. A, 2018, 6, 22721–22730 RSC.
- L. P. Putilov and V. I. Tsidilkovski, Phys. Chem. Chem. Phys., 2019, 21, 6391–6406 RSC.
- F. M. Draber, J. R. Denninger, P. C. Müller, I. K. Sommerfeld and M. Martin, Adv. Energy Sustainable Res., 2022, 3, 2200007 CrossRef CAS.
- C. Y. R. Vera, H. Ding, D. Peterson, W. T. Gibbons, M. Zhou and D. Ding, J. Phys. Energy, 2021, 3, 032019 CrossRef CAS.
- C. Y. Regalado Vera, H. Ding, J. Urban-Klaehn, M. Li, Z. Zhao, F. Stewart, H. Tian, X. Liu, Y. Dong, J. Li, M. Zhou, H. Luo and D. Ding, Chem. Mater., 2023, 35, 5341–5352 CrossRef CAS.
- S. Fujii, J. Hyodo, K. Shitara, A. Kuwabara, S. Kasamatsu and Y. Yamazaki, Sci. Technol. Adv. Mater., 2024, 25, 2416383 CrossRef PubMed.
- D. Han, N. Hatada and T. Uda, J. Am. Ceram. Soc., 2016, 99, 3745–3753 CrossRef CAS.
- E. Makagon, O. Kraynis, R. Merkle, J. Maier and I. Lubomirsky, Adv. Funct. Mater., 2021, 31, 2104188 CrossRef CAS.
- A. Fluri, A. Marcolongo, V. Roddatis, A. Wokaun, D. Pergolesi, N. Marzari and T. Lippert, Adv. Sci., 2017, 4, 1700467 CrossRef PubMed.
- K. Saito, K. Umeda, K. Fujii, K. Mori and M. Yashima, J. Mater. Chem. A, 2024, 12, 13310–13319 RSC.
- K. Saito and M. Yashima, Nat. Commun., 2023, 14, 7466 CrossRef PubMed.
- Y. Meng, J. Gao, Z. Zhao, J. Amoroso, J. Tong and K. S. Brinkman, J. Mater. Sci., 2019, 54, 9291–9312 CrossRef CAS.
- R. Pornprasertsuk, O. Kosasang, K. Somroop, M. Horprathum, P. Limnonthakul, P. Chindaudom and S. Jinawath, Solid State Sci., 2011, 13, 1429–1437 CrossRef CAS.
- S. M. Haile, G. Staneff and K. H. Ryu, J. Mater. Sci., 2001, 36, 1149–1160 CrossRef CAS.
- S. M. Haile, D. L. West and J. Campbell, J. Mater. Res., 1998, 13, 1576–1595 CrossRef CAS.
- Y. Yamazaki, R. Hernandez-Sanchez and S. M. Haile, Chem. Mater., 2009, 21, 2755–2762 CrossRef CAS.
- J. Li, C. Wang, X. Wang and L. Bi, Electrochem. Commun., 2020, 112, 106672 CrossRef CAS.
- I. A. Starostina, G. N. Starostin, M. T. Akopian, G. K. Vdovin, D. A. Osinkin, B. Py, A. Maradesa, F. Ciucci and D. A. Medvedev, Adv. Funct. Mater., 2024, 34, 2307316 CrossRef CAS.
- F. Liu, D. Diercks, P. Kumar, A. Seong, M. H. A. Jabbar, C. Gumeci, Y. Furuya, N. Dale, T. Oku, M. Usuda, P. Kazempoor, I. Ghamarian, L. Liu, L. Fang, D. Chen, Z. Wang, S. Skinner and C. Duan, Sci. Adv., 2025, 11, eadq2507 CrossRef CAS PubMed.
- Y. Shin, Y. Kim, M. Sanders, S. P. Harvey, M. Walker and R. O'Hayre, J. Mater. Chem. A, 2022, 10, 24839–24853 RSC.
- Z. Liu, Z. Tang, Y. Song, G. Yang, W. Qian, M. Yang, Y. Zhu, R. Ran, W. Wang, W. Zhou and Z. Shao, Nano-Micro Lett., 2022, 14, 217 CrossRef CAS PubMed.
- S. Tao and J. T. S. Irvine, Nat. Mater., 2003, 2, 320–323 CrossRef CAS PubMed.
- Z. Li, X. Mao, D. Feng, M. Li, X. Xu, Y. Luo, L. Zhuang, R. Lin, T. Zhu, F. Liang, Z. Huang, D. Liu, Z. Yan, A. Du, Z. Shao and Z. Zhu, Nat. Commun., 2024, 15, 9318 CrossRef CAS PubMed.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- D. Hu, J. Kim, H. Niu, L. M. Daniels, T. D. Manning, R. Chen, B. Liu, R. Feetham, J. B. Claridge and M. J. Rosseinsky, J. Mater. Chem. A, 2022, 10, 2559–2566 RSC.
- M. Papac, V. Stevanović, A. Zakutayev and R. O'Hayre, Nat. Mater., 2021, 20, 301–313 CrossRef CAS PubMed.
- M. Liang, Y. Zhu, Y. Song, D. Guan, Z. Luo, G. Yang, S. P. Jiang, W. Zhou, R. Ran and Z. Shao, Adv. Mater., 2022, 34, 2106379 CrossRef CAS PubMed.
- X. Zhang, C. Tang, Y. Yang, F. Zheng, Q. Su, H. Xiang, L. Meng, L. Du, Y. Aoki, D. Luo, N. Wang and S. Ye, Adv. Funct. Mater., 2025, 2421083 CrossRef.
- C. Tang, B. Yuan, X. Zhang, F. Zheng, Q. Su, L. Meng, L. Du, D. Luo, Y. Aoki, N. Wang and S. Ye, Adv. Energy Mater., 2025, 2402654 CrossRef.
- M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran and Z. Shao, Appl. Catal., B, 2022, 318, 121868 CrossRef CAS.
- R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D. W. Rooney and K. Sun, J. Mater. Chem. A, 2019, 7, 18365–18372 RSC.
- Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu and Z. Shao, Joule, 2019, 3, 2842–2853 CrossRef CAS.
- L. Yang, C. Zuo, S. Wang, Z. Cheng and M. Liu, Adv. Mater., 2008, 20, 3280–3283 CrossRef CAS.
- E. Fabbri, S. Licoccia, E. Traversa and E. D. Wachsman, Fuel Cells, 2009, 9, 128–138 CrossRef CAS.
- E. Fabbri, L. Bi, D. Pergolesi and E. Traversa, Energy Environ. Sci., 2011, 4, 4984–4993 RSC.
- K. Park, M. Saqib, H. Lee, D. Shin, M. Jo, K. M. Park, M. Hamayun, S. H. Kim, S. Kim, K.-S. Lee, R. O'Hayre, M. Choi, S.-J. Song and J.-Y. Park, Energy Environ. Sci., 2024, 17, 1175–1188 RSC.
- S. Lei, J. Ma, W. Li, M. B. Hanif, C.-X. Li, D. Wang and S. Wang, J. Power Sources, 2025, 631, 236257 CrossRef CAS.
- D. Zou, Y. Yi, Y. Song, D. Guan, M. Xu, R. Ran, W. Wang, W. Zhou and Z. Shao, J. Mater. Chem. A, 2022, 10, 5381–5390 RSC.
- S. H. Hwang, S. K. Kim, J.-T. Nam and J.-S. Park, Int. J. Hydrogen Energy, 2021, 46, 33551–33560 CrossRef CAS.
- L. Yang, Z. Liu, S. Wang, Y. Choi, C. Zuo and M. Liu, J. Power Sources, 2010, 195, 471–474 CrossRef CAS.
- W. Zhang, H. Muroyama, Y. Mikami, Q. Liu, X. Liu, T. Matsui and K. Eguchi, Chem. Eng. J., 2023, 461, 142056 CrossRef CAS.
- D. Feng, M. Li, V. K. Peterson, R. Lin, A. D’Angelo, O. Marenych, A. Yago, D. Appadoo, T. Zhu, M. Y. B. Zulkifli, S. Gao, Y. Wu, X. Tian, X. Du and Z. Zhu, Adv. Funct. Mater., 2025, 2417425 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |