
Strategies for industrial-grade seawater electrolysis: from electrocatalysts and device design to techno-economic analysis
Yuqing
Wang
 *ab,
Feng
Li
a,
Luoyin
Zhao
c,
Yuchen
Wang
a,
Guang
Yang
a,
Jinyan
Tian
d,
Shuaibing
Heng
a,
Xuewen
Sun
a,
Jianxu
Zhao
a,
Minghua
Chen
*a and
Qingguo
Chen
*a
*ab,
Feng
Li
a,
Luoyin
Zhao
c,
Yuchen
Wang
a,
Guang
Yang
a,
Jinyan
Tian
d,
Shuaibing
Heng
a,
Xuewen
Sun
a,
Jianxu
Zhao
a,
Minghua
Chen
*a and
Qingguo
Chen
*a
aKey Laboratory of Engineering Dielectric and Applications (Ministry of Education), School of Electrical and Electronic Engineering, Harbin University of Science and Technology, Harbin 150080, P. R. China. E-mail: wangyq@hrbust.edu.cn; mhchen@hrbust.edu.cn; qgchen@263.net
bDepartment of Chemical and Biomolecular Engineering, College of Design and Engineering, National University of Singapore, 117576, Singapore
cCollege of Intelligent Systems Science and Engineering, Harbin Engineering University, Harbin, 150001, P. R. China
dState Grid Jilin Electric Power Co., Ltd Baicheng Power Supply Company, Baicheng City, Jilin Province 137000, P. R. China
First published on 31st March 2025
Abstract
Seawater electrolysis offers a promising route for sustainable hydrogen production, utilizing abundant seawater to meet global energy demands while addressing environmental concerns. However, challenges such as inefficiencies, high costs, and reliance on noble metal catalysts hinder its practical implementation. This review examines the fundamental mechanisms of seawater electrolysis, focusing on the hydrogen and oxygen evolution reactions (HER and OER) at industrial-scale current densities. Key strategies for catalyst design, including interfacial engineering, structural optimization, and improved mass and electron transport, to enhance efficiency and stability are discussed. Additionally, device architecture and techno-economic considerations are explored to facilitate scalable, cost-effective deployment. By providing insights into advanced materials and system innovations, this review outlines pathways for integrating seawater electrolysis into large-scale sustainable energy solutions.
 Yuqing Wang | Yuqing Wang is an associate professor at the School of Electrical and Electronic Engineering, Harbin University of Science and Technology. She obtained her Ph.D. (2020) from the Harbin Institute of Technology. From 2023, she worked as a postdoctoral researcher at the National University of Singapore. Her main research interests are focused on the development of new catalytic materials for electrolytic water splitting and plastic upcycling and recycling, and the in-depth understanding of catalytic mechanisms. |
 Feng Li | Feng Li is a master's student at the School of Electrical and Electronic Engineering, Harbin University of Science and Technology. He graduated from East China Jiaotong University in 2019 and has been pursuing his master's degree at the Harbin University of Science and Technology since 2022. His main research interests focus on the development of new catalytic materials for the chlorine evolution reaction in water electrolysis and the in-depth understanding of catalytic mechanisms. |
 Luoyin Zhao | Luoyin Zhao is a Ph.D. candidate at the College of Intelligent Systems Science and Engineering, Harbin Engineering University. His main research interests are focused on the development of new energy materials related to the ocean, and research on autonomous underwater vehicles. |
 Minghua Chen | Minghua Chen has been a professor at Harbin University of Science and Technology since 2017. He received his Ph.D. from the Harbin University of Science and Technology, and was subsequently an exchange student and visiting scientist at Nanyang Technological University (NTU) from 2013 to 2016. His research interests include synthesis of 2D materials, carbon composites and heterostructure materials, and their applications for energy storage and conversion devices. |
1 Introduction
Pursuing sustainable energy solutions is increasingly critical in the context of rising global energy demands and environmental concerns. Hydrogen, recognized for its exceptional potential as a renewable energy source, stands out due to its plentiful availability and high energy density. Furthermore, it offers substantial environmental benefits, producing water vapor as its only byproduct during combustion, thus eliminating carbon emissions at the point of use. However, the production of hydrogen via conventional methods such as steam methane reforming is currently marred by high costs and significant CO2 emissions, exacerbating the greenhouse effect. This paradox highlights the need for a transition to cleaner production techniques. Electrolysis offers a highly viable solution, especially when driven by renewable energy sources like wind, solar, or tidal power.1 This method involves splitting water into hydrogen and oxygen using electricity and, if this electricity is derived from renewables, the entire process can be virtually emission-free. The scalability of electrolysis powered by renewable energy could radically transform hydrogen production into a truly sustainable practice. Looking ahead, integrating hydrogen production with these renewable technologies furthers the potential of hydrogen as a key component of a clean energy landscape and aligns with broader environmental goals. This strategic integration could play a pivotal role in building resilient, sustainable energy systems that cater to our growing energy needs while minimizing environmental impacts.Hydrogen production via seawater electrolysis poses notable challenges alongside considerable potential.2 Although this approach is currently less efficient and incurs higher costs compared with using purified water,3 its appeal lies in the vast availability of seawater, which constitutes approximately 97% of the Earth's water resources. Overcoming technical barriers could unlock this abundant resource for hydrogen generation.4 Traditional catalysts like RuO2 and IrO2, while effective, are both expensive and limited in supply, constraining the scalability of the process.5 Alternatively, transition metal-based electrocatalysts offer a promising solution due to their diverse structural and compositional properties, which may improve efficiency and reduce costs.6 These catalysts represent a promising shift from traditional noble metal catalysts, potentially catalyzing a more sustainable and economically feasible approach for hydrogen production from the vast reservoirs of seawater.
Developing noble metal-free catalysts for seawater electrolysis has become imperative to meet practical and industrial demands. Although existing catalysts perform adequately at low current densities, they fall short of the industrial benchmark, which requires exceeding 500 mA cm−2, revealing a considerable performance gap.7 Moreover, many of the reported electrocatalysts were tested at industrial-grade current densities with buffered electrolytes and alkaline environments, which only lasted a few tens of hours, while practical applications require direct electrolysis in seawater and stability for thousands of hours. While previous reviews have extensively discussed the fundamental electrochemical challenges of seawater electrolysis, including catalyst degradation, competition reactions, and scaling effects from dissolved cations such as Ca2+, Na+, and Mg2+, they have largely overlooked the critical aspect of techno-economic analysis, which ultimately determines the scalability and industrial adoption of this technology. Consequently, the review not only delves into the reaction mechanisms and key challenges of hydrogen generation from seawater electrolysis at industrial-grade current densities but also uniquely integrates a techno-economic perspective, evaluating cost factors, energy efficiency, and system feasibility for large-scale implementation. By providing a comprehensive analysis of catalyst design strategies alongside seawater electrolysis device designs, it offers insights into both scientific advancements and the economic realities necessary for the widespread adoption of seawater electrolysis in hydrogen production. Finally, it discusses the future development of catalysts optimized for large-scale seawater electrolysis. The review aims to provide valuable insights for researchers, advancing the design of high-performance catalyst materials and accelerating the transition of seawater electrolysis from research to practical application.
2 Mechanisms and evaluation
2.1 Mechanisms of seawater electrolysis
The seawater electrolysis reaction generally consists of the following reactions: the OER, the HER and the chlorine oxidation reactions (ClOR). The process of hydrogen generation through water electrolysis proceeds as follows:| 2H2O → 2H2 + O2 | (1) |
The occurrence of the HER at the cathode involves a two-electron reaction, comprising two steps: the Volmer, and Heyrovsky or Tafel steps, as shown in Fig. 1. Volmer step: adsorption of H2O molecules on the active site and hydration of protons (H3O+); Heyrovsky step: synthesis of hydrogen atoms on the electrocatalyst surface through the electrochemical pathway of hydrogen molecules; Tafel step: hydrogen atoms synthesize hydrogen molecules directly on the electrocatalyst surface via the chemical pathway. The process of hydrolysis, hydroxyl adsorption (OHads), and Hads exchange in alkaline seawater electrolytes take place through either the Volmer/Heyrovsky or Volmer/Tafel steps, with the reactions outlined below:
(I) Volmer step: the absence of protons leads to a scenario where the H2O molecule interacts with electrons rather than H+, resulting in the formation of an adsorbed hydrogen atom (Hads) species on the electrocatalyst,
| H2O → Hads + OH− − e− | (2) |
(II) Heyrovsky step: subsequently, Hads bonds with an H2O molecule and an electron, resulting in the formation of a hydrogen molecule,
| H2O + Hads → H2 + OH− − e− | (3) |
(III) Tafel step: alternatively, a pair of Hads atoms merge to create a hydrogen molecule,
| Hads + Hads → H2 | (4) |
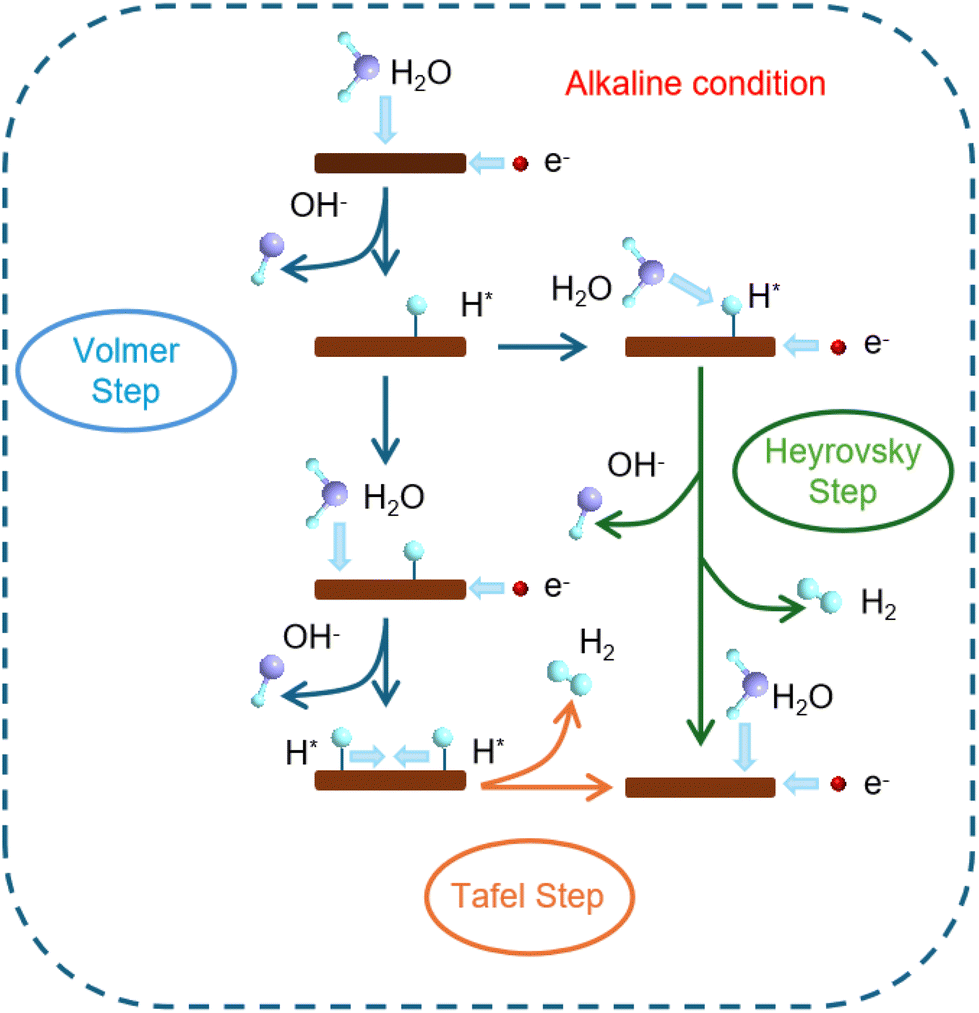 | ||
| Fig. 1 Schematic diagram of the HER principle of alkaline seawater electrolysis. | ||
| 2Cl− → Cl2 + 2e− | (5) |
While in alkaline conditions, they form hypochlorite:
| 2Cl− + 2OH− → ClO− + 2H2O + 2e− | (6) |
Although the OER is thermodynamically more favorable than the ClOR, the latter, as a two-electron reaction, benefits from faster kinetics compared with the four-electron OER pathway. Under alkaline conditions, a stable potential gap of 480 mV between the OER and ClOR allows for selective seawater electrolysis. To avoid triggering the ClOR, it is crucial to maintain the overpotential of OER electrocatalysts below this 480 mV threshold at high current densities. Therefore, developing OER electrocatalysts with high selectivity, corrosion resistance, and long-term stability is essential for achieving efficient seawater splitting under industrial conditions. Metal-based electrocatalysts in seawater electrolysis are particularly vulnerable to corrosion processes, including polarization, dissolution, and hydrolysis. During seawater electrolysis, chloride ions adsorb onto and penetrate the electrocatalyst surface, forming complexes with metal ions that lead to the dissolution of active metal sites. Additionally, hydroxide ions (OH−) in the electrolyte exacerbate this issue by promoting the formation of metal hydroxides from dissolved metal ions, further impairing the catalyst's performance and durability.
The OER represents an additional crucial half-reaction in the process of electrolyzing seawater. Occurring at the anode, this reaction differs from the HER's two-electron method by featuring a more intricate OER process, which includes a multi-stage four-electron transfer to generate O2 at a slower rate. The common consensus is that the OER can advance through two pathways: the lattice oxygen-mediated mechanism (LOM) and the traditional adsorbate evolution mechanism (AEM).
Adsorbate evolution mechanism. In an AEM system, the oxygen evolution reaction generally involves four coordinated proton and electron transfers, with the metal center (M) serving as the active catalytic site. This process facilitates the generation of oxygen molecules from water under both acidic and alkaline conditions. For the OER in alkaline seawater, the reaction sequence includes the following steps:
(I) The creation of an M–OH intermediate marks the initial phase in the OER procedure, focusing on OH adsorption on the electrocatalyst's surface,
| M + OH− − e− → M–OH | (7) |
(II) Continued interaction with OH− results in the formation of M–O intermediates, which then link with H2O and an electron.
| M–OH− + OH− − e− → H2O + M–O | (8) |
(III) Subsequently, two distinct routes can lead to the creation of the O2 molecule: the first involves the immediate attachment of two M–O species, resulting in the formation of O2 and M active sites.
| M–O + M–O → M + M + O2 | (9) |
(IV) Another method involves merging M–O and OH− to form M–OOH,
| M–O + OH− − e− → M–OOH | (10) |
(V) Subsequently undergoing electron oxidation, and then utilizing a proton-coupled electron transfer technique to synthesize O2 and active sites.
| M–OOH + OH− − e− → O2 + H2O + M | (11) |
As per the aforementioned reaction route, comprehending the critical rate-determining stages and verifying the kinetic characterization of the OER is typically challenging, owing to the multiple intermediates referenced earlier (Fig. 2).
 | ||
| Fig. 2 Schematic diagram of the AEM process for the OER during alkaline seawater electrolysis. | ||
Lattice oxygen-mediated mechanism. Within LOM, the catalyst's lattice oxygen plays a direct role in the reaction of oxygen evolution. Lattice oxygen's role has been recently shown in gas-phase catalytic oxidation processes involving alloy catalysts. Fascinatingly, this occurrence is viewed as a substitute reaction route in OER electrocatalysis, occasionally even the most advantageous (Fig. 3).
 | ||
| Fig. 3 Schematic diagram of the LOM process (oxygen vacancy site) for the OER during alkaline seawater electrolysis. | ||
2.2 Evaluation for industrial-grade seawater electrolysis performance
The efficiency of industrial-grade seawater electrocatalysts for HER and OER is assessed through critical parameters, including overpotential (η), Tafel slope, turnover frequency (TOF), Faraday efficiency (FE), stability (both mechanical and electrochemical), and the selectivity of anodic reactions.Eqn (12) outlines the operating cell voltage (Vop) necessary for breaking down water:
| Vop = IR + Veq + |ηc| + ηa | (12) |
Water electrolysis’ total overpotential comprises the overpotential values of the OER (ηa) and HER (ηc), along with the voltage reduction due to system resistance, with the OER's overpotential being the primary hindrance. In this context, Veq symbolizes the equilibrium potential (Veq = 1.23 V), ηa and ηc denote the necessary overpotential to surmount the kinetic obstacles of the OER on the anode and HER on the cathode, respectively, while IR indicates the voltage decrease to offset the system's inherent resistance.
The Tafel slope logarithmically connects overpotential with current density, expressible through eqn (13):
 | (13) |
In this context, m denotes the Tafel slope, j represents the current density, j0 is the exchange current density, R stands for the ideal gas constant, T is the absolute temperature, n indicates the number of electrons transferred during the redox reaction, F is the Faraday constant, and α refers to the charge transfer coefficient. Within the eqn (13), a minor Tafel gradient (m) suggests the necessity of a slight overpotential shift to accommodate the swift rise in current density, thereby demonstrating superior electrocatalytic kinetics. An optimal electrocatalyst ought to possess an industrial-grade current density and a minimal Tafel slope. Furthermore, utilizing the Tafel slope aids in pinpointing potential reaction processes.
| TOF = jNA/(nFΓ) | (14) |
In this context, NA represents Avogadro's constant, j denotes the current density, n signifies the electron count transferred during the redox reaction, F is the Faraday constant, and Γ indicates the overall concentration of surface or active sites or the total atoms involved. Practically speaking, acquiring a precise TOF value is challenging due to the unknown count of active sites. Up to this point, an appropriate technique for precisely determining the count of active sites in heterogeneous catalysis remains elusive. Certain research has straightforwardly determined TOF values using the total mass loading, presuming the electrocatalytic activity of all catalytic species. In certain instances, it's been hypothesized by scientists that solely atoms on the surface participate in the catalytic activity, with internal catalytic entities inactive, resulting in TOF measurements solely from surface catalytic locations, making precise TOF measurements a significant hurdle.
 | (15) |
In the domain of seawater electrolysis, ion polymers such as Nafion play a pivotal role at the electrode–electrolyte interface, primarily serving as adhesives that anchor catalysts onto the carrier. However, their application is not without significant drawbacks. The integrity of these polymers is compromised over time due to nucleophilic attacks by water and oxygen radicals, phenomena that catalyze the degradation of the membrane electrode. This degradation is a critical concern as it leads to the weakening of the structural and functional capacity of the electrolysis system. Moreover, the issues are compounded when there is excessive polymer loading. In such scenarios, the overly thick layer of polymer can prevent effective contact between the catalyst and its carrier, eventually leading to the detachment of the catalyst. This detachment undermines the efficiency of the electrolysis process, posing a substantial barrier to the sustainable use of ion polymers in energy conversion systems. Thus, while ion polymers like Nafion are indispensable for their initial functional contributions, their long-term viability and reliability in seawater electrolysis require careful consideration and management of their application to mitigate these degradative effects.
In addition, understanding the dynamics of bubble formation at the electrocatalyst interface is crucial for optimizing the stability and efficiency of electrochemical reactions. The research findings indicate that the process of bubble formation, which encompasses nucleation, growth, and eventual separation, is a disruptive force in electrocatalytic systems. As bubbles form, they obstruct ion pathways and deactivate crucial nucleation sites, leading to a diminished active surface area and compromised electrocatalyst performance. Particularly detrimental is the peeling of the electrocatalyst from the carrier at high current densities due to strong adhesion forces exerted by the separating bubbles. Additionally, at lower current densities, the loss of active sites predominantly results from dissolution rather than mechanical forces. However, the increased flow of electrolyte, intended to enhance performance, inadvertently accelerates catalyst peeling. Therefore, enhancing the bonding strength between the electrocatalyst and its carrier is essential, as it could significantly mitigate the mechanical stresses during bubble separation and improve the overall stability and efficacy of the electrocatalytic process. This emphasizes the need for improved design and management of electrocatalyst interfaces to counteract the adverse effects of bubble dynamics on electrode stability.9
3 Challenges faced
The majority of present-day seawater electrolysis studies take place in settings with buffered media or alkaline conditions, yet real-world use necessitates the direct electrolysis of seawater. The process of directly electrolyzing seawater encounters four additional significant obstacles.3.1 Pollutants
Initially, a range of dissolved cations exists, including Mg2+, Na+, and Ca2+, among others, as well as microbes, particulate substances, and various contaminants in natural seawater. During the electrolysis in seawater, these contaminants obstruct the electrodes, leading to poisoning and hastening the aging process of both electrodes and catalysts, thereby diminishing their longevity. For instance, commercial Pt-based electrocatalysts lose functionality in seawater within an hour due to the presence of toxic impurities in natural seawater.14 Due to the limited buffering capacity of seawater, the pH near the anode and cathode changes significantly during seawater electrolysis. At the anode, OH− ions are consumed, leading to localized acidity, while the cathode generates OH−, creating an alkaline environment. For instance, in seawater electrolysis with an initial pH ranging from 4–10, the local pH near the cathode can rise by approximately 5 pH units, while near the anode, it can drop by around 9 pH units at a current density below 10 mA cm−2.15 The alkaline environment reduces the potential gap between the surrounding cation reduction process and HER. At the same time, in the process of cathodic precipitation, the formation of MgO, Mg(OH)2, and Ca(OH)2 occurs as the cathodic region's pH rises during the electrolysis phase. Over time, the build-up of these insoluble precipitates will progressively influence the reaction mechanism, hinder the diffusion of water molecules, increase the charge transfer resistance, and reduce the reaction rate of seawater electrolysis. To tackle these challenges, incorporating supporting electrolytes (such as alkali or buffers) is suggested. However, alkali additives like KOH come with a substantial cost, reaching $800 per tonne.16 Consequently, the suggestion is for the seawater electrolysis catalyst to possess a selective permeation layer capable of eliminating contaminants from the seawater electrolysis process, selectively allowing seawater to pass through, and introducing extra electrolytes to maintain the cathode's pH balance.23.2 Competition reactions
In seawater electrolysis, the anode encounters competition between the OER and ClOR.17 Although the OER is thermodynamically more favorable, the ClOR, as a two-electron process, exhibits a kinetic advantage over the four-electron OER, making it more competitive under certain conditions.18 Specifically, Cl2 formation is favored at pH levels below 3.0, while HClO predominantly forms between pH 3.0 and 7.5, and ClO− becomes the dominant species at pH values above 7.5.19 Given that the OER potential diminishes as pH rises and the ClOR maintains a pH-neutral potential, chloride does not undergo substantial oxidation to ClO−, with only the OER taking place in alkaline environments when η is less than 480 mV, facilitating high OER selectivity.20 As previously stated, optimizing the thermodynamic potential difference between these reactions is essential for achieving selective OER, as it promotes OER current while preventing hypochlorite formation. Consequently, for the successful reaction of the OER in alkaline seawater electrolysis, creating an OER catalyst with high activity, capable of generating substantial current density even with minor overpotentials, is essential to circumvent the rival reaction of Cl−.23.3 Peel-off of electrocatalysts
Numerous advanced seawater electrolysis catalysts have been created so far, yet the majority function at low current densities (<100 mA cm−2), significantly lower than industrial standards. Creating catalysts for seawater electrolysis that possess industrial-grade current densities holds significant economic value and practical applications. Furthermore, the majority of these catalysts exist in a powdered state, and the active core might detach from the electrode upon the release of aggressive bubbles, necessitating regular substitution of catalytic substances during real-world industrial-grade current density industrial manufacturing. Consequently, employing a catalyst substrate that securely secures the catalyst and choosing electrode materials that can be easily substituted is an effective approach. These approaches focus on securely anchoring the catalyst to prevent detachment during vigorous bubble generation and enhancing durability under high-current-density operations. For example, Luo et al.21 reported that a hydroxide-mediated nickel-based catalyst loaded on nickel foam (h-NiMoFe) achieved high current density (1000 mA cm−2) at low overpotential and could be scaled up for industrial use. Similarly, Zhang et al.22 reported that a fluorine-doped cobalt–iron phosphide catalyst on iron foam (F-Co2P/Fe2P/IF) has demonstrated excellent performance and stability, highlighting the importance of integrating strong catalyst supports and replaceable electrode designs for economic and scalable hydrogen production.233.4 Bubbles
In the process of generating electrolytic hydrogen from seawater under industrial-grade current densities, numerous hydrogen gas bubbles quickly emerge. Accumulation of bubbles on the catalyst and electrolyte's contact areas significantly hinders the liquid's mass transfer, decelerates electron movement, and diminishes the count of active sites, leading to lower electrocatalytic efficiency and stability. Furthermore, the accumulation of H2 and O2 bubbles on the electrode creates a “bubble curtain”, temporarily obstructing electrolyte access to the catalyst surface and reducing energy efficiency by about 5–10%.24 Consequently, the primary difficulty lies in isolating the created hydrogen bubbles to preserve the electrode's catalytic efficiency during industrial-grade current density hydrogen generation in industry. It has been observed that surfaces with highly hydrophobic properties gradually emit bubbles, while electrodes with high hydrophilicity enhance the interaction between the catalyst and the liquid electrolyte. Consequently, a prevalent method for creating surfaces with both water-attracting and water-repellent qualities involves altering the texture of the catalyst electrode surface. This modification not only aids in releasing hydrogen but also expands the area exposed to the active site, thereby speeding up the electrolyte transfer mechanism.234 Design strategies for industrial-grade seawater electrolysis catalysts
In the development of industrial-grade seawater electrolysis catalysts, achieving high current densities and long-term stability is crucial. Membrane electrode assemblies (MEAs), commonly used in industrial applications, typically operate at current densities of several hundred mA cm−2 with catalyst loadings of 1–2 mg cm−2. For proton exchange membrane water electrolyzers (PEMWEs), current densities often exceed 1.6 A cm−2, while alkaline water electrolyzers (AWEs) generally reach around 0.5 A cm−2. Additionally, PEMWEs are expected to maintain operational stability for over 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours in industrial settings, serving as key performance benchmarks for seawater electrolysis catalysts.25 To meet these industrial requirements, catalyst design must focus on optimizing HER and OER kinetics through electronic structure modifications, alloying, and heterostructure engineering to achieve low overpotentials and high reaction rates. To meet these industrial requirements, interfacial engineering plays a crucial role in enhancing the catalyst–support interaction and stabilizing active sites. Optimizing mechanical structure through nanostructuring, porous architectures, and hierarchical morphologies ensures structural integrity, facilitates rapid gas bubble release, and enhances active site exposure, which is particularly important at high current densities. Efficient electron transfer paths can be achieved by tuning the electronic structure, introducing conductive supports, and leveraging alloying or defect engineering to accelerate charge transport and reduce overpotential. Meanwhile, mass transfer optimization is critical for minimizing diffusion limitations and ensuring efficient reactant delivery and product removal. By aligning catalyst design strategies with industrial benchmarks, researchers can develop seawater electrolysis catalysts that exhibit high efficiency in laboratory conditions while also demonstrating practical viability for large-scale hydrogen production.
000 hours in industrial settings, serving as key performance benchmarks for seawater electrolysis catalysts.25 To meet these industrial requirements, catalyst design must focus on optimizing HER and OER kinetics through electronic structure modifications, alloying, and heterostructure engineering to achieve low overpotentials and high reaction rates. To meet these industrial requirements, interfacial engineering plays a crucial role in enhancing the catalyst–support interaction and stabilizing active sites. Optimizing mechanical structure through nanostructuring, porous architectures, and hierarchical morphologies ensures structural integrity, facilitates rapid gas bubble release, and enhances active site exposure, which is particularly important at high current densities. Efficient electron transfer paths can be achieved by tuning the electronic structure, introducing conductive supports, and leveraging alloying or defect engineering to accelerate charge transport and reduce overpotential. Meanwhile, mass transfer optimization is critical for minimizing diffusion limitations and ensuring efficient reactant delivery and product removal. By aligning catalyst design strategies with industrial benchmarks, researchers can develop seawater electrolysis catalysts that exhibit high efficiency in laboratory conditions while also demonstrating practical viability for large-scale hydrogen production.
4.1 Interfacial engineering
Catalysts play a vital role in enhancing the efficiency of seawater electrolysis through interfacial engineering, as illustrated in Fig. 4a. Recent progress in electrocatalytic materials has been notably advanced by innovative energy research. Jiang et al.26 fabricated NiFe LDH/FeOOH heterostructure nanosheets through electrodeposition on foam nickel–iron substrates; they have not only achieved a robust structure but have also significantly enhanced seawater electrolysis performance. This novel electrocatalyst exhibits remarkable activity and stability, key attributes for the development of active NiOOH materials. The strong interactions within the heterostructure facilitate improved electrochemical processes essential for seawater electrolysis. In the research conducted by Xiu et al.,27 a novel approach for improving seawater electrolysis performance is detailed, centering on the use of multi-level hollow MXene as a support material for platinum catalysts. This method notably enhanced platinum utilization, which is critical in applications where cost and material efficiency are paramount. By optimizing the catalyst structure at the nano-scale, the study achieved not only increased conductivity but also improved hydrophilicity and anti-aggregation characteristics, ensuring the catalyst's stability and effectiveness across varying pH conditions. The unique synergistic interface between the ultrafine platinum particles and the MXene structure significantly boosted charge transfer rates, facilitated hydrogen and water adsorption activation, and enhanced both intermediate hydrogen binding and ion/mass exchange. These improvements collectively contributed to a superior seawater electrolysis process, particularly in low-platinum catalyst systems where maximizing active site availability and reaction dynamics is crucial for seawater electrolysis performance.28 | ||
| Fig. 4 (a) Interfacial engineering; (b) mechanical structure; (c) electron transfer; (d) mass transfer.31 | ||
4.2 Mechanical structure
In the realm of industrial-grade electrolyzed seawater, the majority of catalysts are commonly fashioned into thin films, necessitating interaction with the current collector. As a result, precise assembly is crucial for each compact catalyst unit. The mechanical structure of the catalyst, spanning nano, micro, and macro-scale features, significantly influences channel dimensions and the extent of active site immersion in the electrolyte (Fig. 4b). Augmenting the mechanical configuration of the catalyst can elevate its efficacy in water electrolysis.Enhancing the transport potential in industrial water electrolysis requires optimized channel designs that facilitate efficient electrolyte flow and bubble removal. Yu et al.29 recently proposed a Ni2P nanowire catalyst grown on a Ni foam substrate, showcasing remarkable electrocatalytic activity and stability in alkaline environments. This was attributed to efficient electron transfer and the robust integration of the nanowire array with the substrate. The Ni2P nanowire array's “super gas-permeable” surface ensured effective hydrogen release even at high current densities exceeding 1000 mA cm−2. By maximizing the exposure of catalytic sites to the electrolyte, this design enhances efficiency and provides abundant active sites for improved performance. Similarly, King et al.30 developed an economical cobalt phosphide catalyst, scaling it from a 1 cm2 laboratory setup to an 86 cm2 commercial PEM electrolyzer. Using a two-step synthesis on high-surface-area carbon supports, CoP integrates seamlessly into industrial PEM electrolysis systems. Under identical conditions (400 psi, 50 °C), CoP achieved superior performance with a current density of 1.86 A cm−2 and sustained hydrogen production for over 1700 hours, significantly lowering material costs compared with platinum-based alternatives.31
4.3 Electron transfer path
Reducing resistance by optimizing electron transfer pathways is a key strategy to improve catalyst efficiency in industrial seawater electrolysis. This can be achieved by enhancing electron transport rates within the catalyst and modifying the electron transfer mechanism at the interface between the conductive carrier and the catalyst–electrolyte (Fig. 4c). Employing highly conductive materials, such as carbon, metals, or metal compounds, as catalysts or support structures can significantly boost catalytic efficiency on an industrial scale. Techniques like phase engineering and defect engineering are commonly used to adjust material conductivity. Xu et al.32 reported a carbon-doped nanoporous cobalt phosphide (C–Co2P) electrocatalyst synthesized via electrochemical treatment for the hydrogen evolution reaction. This C–Co2P catalyst achieved an overpotential of 30 mV at 10 mA cm−2 in 1 M KOH, exhibiting excellent stability and industrial-grade current density in artificial alkaline seawater electrolytes. Experimental results and DFT calculations revealed that carbon doping tailored the electronic structure of Co2P, enabling the formation of C–Hads intermediates through the H-transfer pathway. This enhanced the HER by promoting water dissociation and hydrogen desorption. In artificial alkaline seawater containing NaCl, MgCl2, and CaCl2, the C–Co2P catalyst demonstrated exceptional activity and stability at industrial current densities, achieving an overpotential of 192 mV and a current density of 1000 mA cm−2. These results surpass those of commercial Pt/C catalysts, highlighting the potential of C–Co2P for industrial-scale applications.To enhance catalyst performance in industrial electrolysis, optimizing electron transfer at both catalyst–support and catalyst–catalyst interfaces is essential. The interaction between the catalyst and its support material significantly affects electron mobility, influencing interface resistance. Reducing this resistance requires careful consideration of catalyst thickness or particle size. Additionally, appropriate alignment of small structural units within the catalyst is critical to ensure efficient electron transfer over short distances. Yan et al.33 recently developed a method for electro-synthesizing metal hydroxides/oxides using reducing acidic anions on various substrates. On graphite, the introduction of nitrate ions into the graphene layer markedly improved the electrodeposition–support interface, resulting in enhanced loading capacity and improved hydrophilic/hydrophobic properties. In oxygen evolution electrocatalysis, co-depositing nanocrystalline cerium dioxide and amorphous nickel hydroxide on graphite achieved a low overpotential of 177 mV at 10 mA cm−2. Furthermore, this composite catalyst demonstrated exceptional durability, maintaining performance at an industrial-grade current density of 1000 mA cm−2 for over 300 hours, underscoring its potential for industrial-scale applications.31
4.4 Mass transfer path
Enhancing the mass transfer process in low-dimensional catalysts is an effective strategy to improve catalytic performance in industrial seawater electrolysis, as illustrated in Fig. 4d. These catalysts influence the concentration of reactants and products at the catalytic interface, modifying the local chemical potential and boosting efficiency. Liu et al.34 demonstrated this concept using a NixFe1−x alloy nanocone array with a thin NiO/NiFe(OH)2 layer (∼2 nm) to facilitate reactant transport. Finite element analysis revealed that the high-curvature tips of the nanocones significantly amplify the local electric field, increasing the concentration of hydroxide ions at active sites by an order of magnitude. This enhancement led to a 67% improvement in intrinsic oxygen evolution reaction activity at 1.5 V. Experimental data showed that the optimized nickel–iron alloy nanocone electrodes achieved overpotentials of 190 mV at 10 mA cm−2 and 255 mV at 500 mA cm−2. Electrochemical surface area calibration further confirmed the superior performance of these nanocone electrodes compared with leading OER electrocatalysts. Additionally, experiments comparing samples with varying tip curvature radii reinforced the role played by tip-enhanced local electric fields in improving mass transfer, highlighting their potential for industrial applications.315 Electrocatalysts for industrial-grade seawater electrolysis
Industrial-grade seawater electrolysis catalysts are currently available for OER, HER, and bifunctional catalysts, and each of these can be categorized into noble metal catalysts, transition metal compounds, and non-metallic catalysts (Fig. 5). | ||
| Fig. 5 Electrocatalysts for industrial-grade seawater electrolysis, including catalysts applied to the HER, OER and OWS (overall water splitting). | ||
5.1 OER electrocatalysts
 | ||
| Fig. 6 (a) SEM and TEM images of the as-prepared RuMoNi electrocatalyst.7 (b) Polarization curves of RuMoNi, RuO2, and Ni Foam in a 1.0 M KOH + seawater electrolyte at a scan rate of 5 mV s−1 with 85% iR correction.7 (c) Durability test for the OER for over 3000 h recorded at a current density of 500 mA cm−2 in a 1.0 M KOH + seawater electrolyte.7 (d) The electrochemical performance for the steam-fed anode and liquid-fed anode electrolysis cells.35 | ||
Transition metal oxides. Owing to their affordability, plentiful presence, and superior resistance to corrosion, transition metal oxides are extensively utilized as OER electrocatalysts in marine settings. In the course of the electrocatalytic procedure, they typically transform into hydroxyl oxides or hydroxides, thereby demonstrating superior OER efficiency. Significantly, recent studies have demonstrated that FeNi oxides excel in oxygen precipitation reactions during seawater electrolysis at elevated current densities. Li et al.36 suggested an uncomplicated dipping-and-heating technique for creating Fe2O3/NiO heterogeneous catalysts on nickel foam (FNE300) (Fig. 7a and b). Utilizing nickel foam as the catalyst's nickel source guaranteed substantial solid interaction between the substrate and the catalyst, leading to enhanced stability and rapid electron movement. Additionally, the electrode's porous and water-attracting nature aided in electrolyte diffusion and O2 release. The interaction between NiO and Fe2O3 aids in charge movement within the catalytic layer and enhances the OER kinetics at the active site.29,37,38 To attain current densities of 10 and 1000 A cm−2, the synthesized FNE300 necessitates overpotential of merely 182 and 267 mV in 1 M KOH, whereas 291 and 329 mV are needed in alkaline natural seawater and household wastewater, respectively (Fig. 7e).
 | ||
| Fig. 7 (a and b) SEM and further magnified images of FNE300.36 (c) SEM image of NiOx–FeOx@g-C3N4.39 (d) TEM image of NiOx–FeOx NCs.39 (e) Electrochemical performance of the FNE300.36 (f) Electrochemical performance of the NiOx–FeOx@g-C3N4.39 | ||
Due to their unique orbital confinement and compatible electronic configuration, nickel–iron oxides are considered cost-effective and selective anodes for seawater oxidation. However, their limited stability in corrosive seawater under high current densities remains a significant challenge, hindering efficient hydrogen production via seawater electrolysis. Addressing this issue, Haq et al.39 introduced a Ni–Fe oxide nanocluster (NiOx–FeOx@g-C3N4), supported by g-C3N4, as a durable and efficient anode for alkaline seawater oxidation (Fig. 7c and d). In pure seawater, the NiOx–FeOx@g-C3N4 anode achieved an industrial-grade current density of 1000 mA cm−2 at a low overpotential of 380 mV, maintaining this performance for over 100 hours without generating hypochlorite. The incorporation of nitrogen modifies the inherent properties of carbon, strengthening the catalyst–support interaction and enhancing electron transfer at the metal–carbon interface. This facilitates redox reactions at the electrode–electrolyte boundary. Additionally, g-C3N4 acts as a stabilizing agent by forming π and δ bonds, reducing interfacial resistance between the active sites and OER intermediates.40,41 It also protects the NiOx–FeOx anode from pitting and stress corrosion, significantly improving resistance to Cl− corrosion. These features make the NiOx–FeOx@g-C3N4 system a promising solution for stable and efficient seawater electrolysis under industrial conditions. NiOx–FeOx NCs, backed by g-C3N4, exhibit greater resistance to Cl− corrosion compared with unaltered NiOx–FeOx NCs. Their enhanced intrinsic activity and resistance to corrosion stem from the heterostructure's synergistic role in bolstering corrosion resistance, structural soundness, and rapid gas emission capabilities. This results in NiOx–FeOx@g-C3N4 having a larger surface area, reducing electrolyte infiltration, and controlling the chemisorption energy at the active site, thereby significantly boosting electrochemical efficiency.42 Additionally, its industrial-grade current density maintains robust stability without noticeable degradation or hypochlorite generation during continuous electrolysis over a period of 100 hours (Fig. 7f).
Transition metal (oxygen) hydroxide. Transition metal hydroxides and hydroxide oxides are highly efficient, corrosion-resistant, and cost-effective catalysts for oxygen evolution reactions in seawater electrolysis. Their catalytic performance can be optimized by tailoring crystal structure, chemical composition, and surface modifications, leading to enhanced activity and stability. Among these, Fe–Ni-based materials have garnered significant attention for their high intrinsic activity. To achieve industrial-grade current density, strategies such as nanostructure construction have been employed to accelerate bubble release and increase exposure of active sites. Zhang et al.43 developed a three-dimensional core–shell dendritic catalyst by vertically growing NiFe layered double hydroxide nanosheets (NiCo@NiFeLDH) on dendritic NiCo branches (Fig. 8a). This design offers a large surface area, efficient charge and mass transfer, and excellent corrosion resistance, making it highly active and stable in alkaline seawater. The NiCo@NiFeLDH catalyst achieved an industrial-grade current density of 500 mA cm−2 with a low overpotential of 266 mV. It also demonstrated nearly 100% OER faradaic efficiency, with no ClO− detected in the electrolyte after seawater electrolysis, confirming its high OER selectivity. Furthermore, the catalyst maintained stable performance with minimal overpotential fluctuations at 500 mA cm−2 over 100 hours of operation in seawater, highlighting its durability under industrial conditions (Fig. 8d).
 | ||
| Fig. 8 (a) SEM images of NiCo@NiFe LDH.43 (b) Schematic illustration of hydrogen production by AEM water electrolyzer in alkaline seawater.44 SEM images with corresponding EDX mappings of (c) after OER testing of CoFeOxHy.44 (d) Electrocatalytic performance of NiCo@NiFe LDH in 1 M KOH and 1 M KOH + seawater electrolytes.43 (e) Electrocatalytic performance of the Ni-doped FeOOH in different electrolytes.44 (f) Electrocatalytic performance of the CoFeOxHy/Ti.45 | ||
Catalyst performance can be enhanced by regulating the content of doped transition metals. Park et al.44 developed a nickel-doped FeOOH anode for efficient AEM alkaline seawater electrolysis (Fig. 8b and c). In half-cell tests, the Ni-doped FeOOH anode exhibited an overpotential of only 301 mV and achieved a current density of 100 mA cm−2 in a 1 M KOH + seawater electrolyte. The superhydrophobic nature of the Ni-doped FeOOH anode facilitates the rapid removal of generated oxygen, reducing mass transfer resistance and effectively utilizing the cell voltage. As a result, the AEM electrolyzer using this catalyst achieved a current density of 729 mA cm−2 with an energy conversion efficiency of 76.35% in 1 M KOH + seawater. This demonstrates the potential of Ni-doped FeOOH for high-efficiency seawater electrolysis under industrial conditions (Fig. 8e).
Iron–cobalt-based materials have recently garnered significant interest due to their high catalytic activity. Komiya et al.45 developed a CoFeOxHy/Ti electrode with a high double-layer capacitance, capable of stable oxygen evolution in a 0.5 mol kg−1 Cl− potassium borate solution at pH 9.2. This electrode features active sites composed of Co- or Fe-domains and benefits from a large reaction surface area, delivering strong OER selectivity even in Cl−-rich electrolytes. The CoFeOxHy/Ti electrode achieved a stable current density of 500 mA cm−2 at a pure potential of 1.67 V vs. RHE at 353 K. Extensive cycling tests and long-term performance evaluations confirmed consistent OER faradaic efficiency, highlighting the electrode's durability and suitability for applications requiring high activity and stability in challenging environments (Fig. 8f).
Transition metal sulfide. Transition metal sulfides are often combined with other compounds or doped with additional metal species to enhance OER activity, enabling their application in industrial-grade seawater electrolysis. Wang et al.46 developed a three-dimensional NiCoS NSAs catalyst anode for efficient and durable OER in alkaline seawater electrolysis.47 The NiCoS NSAs catalyst employed a conductive NiCoS NSAs backbone with a large surface area, promoting the formation and activity of NiCoOOH species (Fig. 9a and b). The in situ generation of an amorphous S-doped NiCoOOH coating enhanced OER selectivity and provided strong corrosion resistance to chloride ions during oxygen evolution. This catalyst achieved low overpotentials of 440 and 470 mV to produce current densities of 500 and 1000 mA cm−2, respectively, in alkaline natural seawater at 26 °C (Fig. 9f). Cyclic voltammetry analysis of NiCoS catalysts with varying Co/S ratios revealed that a Co/S ratio of 3
:10 delivered optimal OER performance (Fig. 9g). Compared with bare Ni foam and pure Ni3S2, the NiCoS catalysts demonstrated lower charge transfer resistance and higher electrochemically active surface area, attributed to their three-dimensional porous structure enriched with surface-active sites. The overpotential required to achieve industrial-grade current densities (up to 1000 mA cm−2) remained well below the 490 mV threshold for chloride oxidation, indicating thermodynamically favorable OER activity on the NiCoS anode while suppressing unwanted chloride evolution reactions. When paired with a NiMoS catalyst for HER, this system forms a monolithic seawater electrolyzer. Operating in alkaline natural seawater, the electrolyzer achieved an industrial-grade current density of 800 mA cm−2 at a low voltage of 2.08 V, maintaining good durability over 100 hours of operation (Fig. 9h). This demonstrates the potential of NiCoS NSAs for industrial seawater electrolysis applications.
 | ||
| Fig. 9 SEM images of (a and b) after growth of NiCoS NSAs.46 (c–e) S-(Ni,Fe)OOH.48 (f–h) Electrocatalytic performance of the NiCoS NSAs catalyst.46 (i and j) Electrocatalytic performance of the NiMoN||S-(Ni,Fe)OOH electrolyzer in different electrolytes.48 | ||
Sulfur doping into transition metal compounds via a one-step surface engineering approach significantly enhances their OER activity. Yu et al.48 reported high-performance S-doped Ni/Fe(oxy)hydroxide catalysts for seawater electrolysis. This method rapidly prepares commercial nickel foams at room temperature, transforming them into catalysts with multi-layered pores and large surface areas (Fig. 9c–e). Unlike conventional electrodeposition or hydrothermal methods, this approach directly reacts with nickel foam in solution, quickly etching a stable, hydrophilic S-doped Ni/Fe(oxy)hydroxide layer. This ensures robust contact, facilitating fast electron transfer and excellent stability. Simultaneously, sulfur is incorporated into both the surface and lattice of the Ni/Fe(oxy)hydroxide, tuning the valence states of Ni and Fe and optimizing the adsorption energy of OER intermediates, thereby boosting catalytic activity. The high density of active sites, combined with the catalyst's stability, results in outstanding OER performance in 1 M KOH + seawater electrolyte. When paired with the advanced HER catalyst NiMoN, the two-electrode electrolyzer achieves industrial-grade current densities at low voltages, demonstrating remarkable durability. Specifically, the system delivered current densities of 500 and 1000 mA cm−2 at low voltages of 1.837 V and 1.951 V, respectively (Fig. 9i and j). These results offer a practical and efficient solution for hydrogen production via seawater electrolysis under industrial conditions.
Transition metal phosphides. Transition metal phosphides, like transition metal sulfides, achieve efficient OER performance through phosphorus intercalation. Yu et al.49 introduced an anion-assisted strategy to enhance electrocatalyst performance, enabling rapid screening of electrolyte additives by analyzing the correlation between chloride rejection and anionic properties (Fig. 10a–c). This approach enabled highly stable alkaline seawater electrolysis at industrial-scale current densities of 0.5 A cm−2 for 500 hours by incorporating phosphate ions into transition metal hydroxides as anodic electrocatalysts. Using NiFe LDH as a prototype, the study explored the role played by anionic additives in OER catalysis within alkaline seawater electrolytes.50 Comparative electrochemical analysis revealed that the addition of PO43− ions significantly enhanced OER stability and faradaic efficiency compared with other anions with varying charges and radii. Increasing the concentration of PO43− effectively suppressed metal dissolution, and the activity–stability factor improved nearly 24-fold with the inclusion of 0.25 M Na3PO4, highlighting the anti-corrosion properties of phosphate ions (Fig. 10e). In a three- or two-electrode system using true alkaline seawater with 0.25 M Na3PO4, NiFe LDH@Ni sustained operation for 500 hours at a current density of 0.5 A cm−2 (Fig. 10f). When paired with Pt foil, the system exhibited negligible voltage attenuation in real seawater, showcasing exceptional corrosion resistance and performance comparable to the most advanced seawater splitting systems reported in recent years. This demonstrates the potential of phosphate-assisted electrocatalysts for industrial-scale seawater electrolysis.
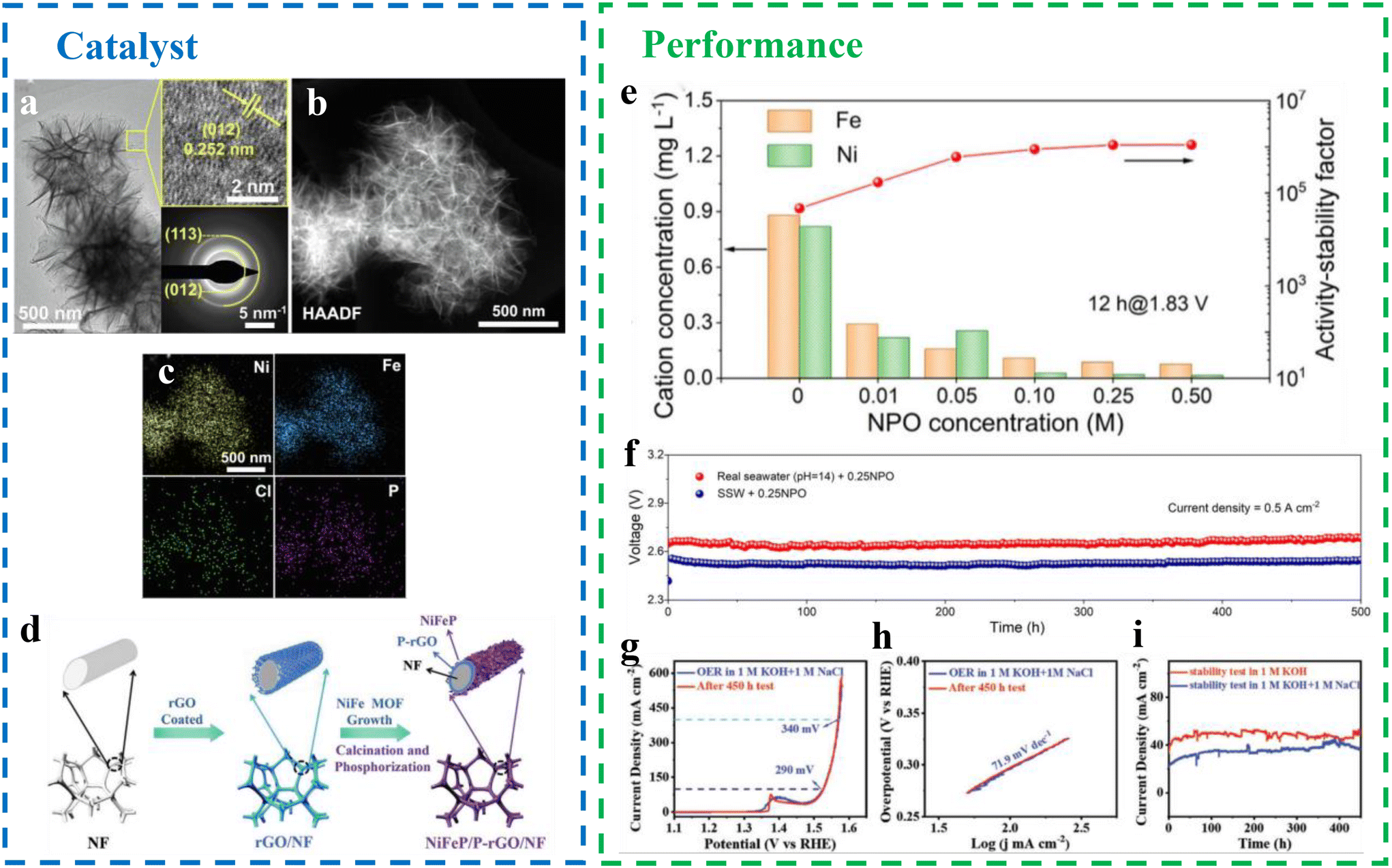 | ||
| Fig. 10 (a–c) The TEM, HAADF-STEM images and the corresponding elemental mappings of NiFe LDH after stability tests in SSW + 0.5NPO.49 (d) Schematic diagram of the fabrication (NiFeP/P-rGO/NF) process.51 (e) The concentration of Ni/Fe ions in electrolyte and the corresponding activity–stability factor after 12 h chronoamperometry at 1.83 V with different concentrations of NPO.49 (f) Long-term stability of NiFe LDH@Ni in SSW and real alkaline seawater with 0.25 NPO using two-electrode electrolysis cell.49 (g–i) Electrocatalytic performance of the NiFeP/P-rGO/NF electrocatalyst.51 | ||
Transition metal phosphides can also be synthesized by combining phosphorus with transition metal compounds to achieve efficient OER performance. Qi et al.51 fabricated nickel foam (NF)-enclosed, reduced graphene oxide (rGO)-supported subnanoscale nickel phosphide arrays (Fig. 10d). These arrays were prepared using metal–organic framework (MOF) precursors, which provide tunable active sites and a high density of exposed catalytic sites.52–54 The resulting NiFeP/P-doped rGO/NF (NiFeP/P-rGO/NF) catalyst demonstrated outstanding performance in seawater electrolysis. In a 1 M KOH + 1M NaCl electrolyte, the NiFeP/P-rGO/NF exhibited low overpotentials of 290 and 340 mV at current densities of 100 and 400 mA cm−2, respectively (Fig. 10g), significantly below the 490 mV threshold for initiating chloride oxidation. The catalyst also showed a low Tafel slope of 71.9 mV dec−1, indicating rapid reaction kinetics (Fig. 10h). Durability tests revealed no significant loss in current density after 450 hours of operation at 35 mA cm−2 (Fig. 10i). The excellent catalytic activity is attributed to the ultra-thin NiFeP arrays, which provide abundant active sites, and the conductive rGO network that facilitates efficient electron transfer. Additionally, the hydrophobicity of rGO ensures stability and durability in seawater, making NiFeP/P-rGO/NF a promising candidate for sustainable and efficient seawater electrolysis.
Noble metal catalysts demonstrate exceptional stability, high OER selectivity, and strong long-term performance, particularly in industrial applications. In contrast, transition metal compounds present cost-effective alternatives, offering enhanced corrosion resistance and significant efficiency gains through nanostructuring and doping strategies. Among them, NiFe-based materials exhibit notable intrinsic activity and stability, with modifications such as phosphate incorporation and sulfur doping effectively mitigating Cl− corrosion and optimizing performance at high current densities. The findings emphasize the critical role played by structural engineering, chemical composition tuning, and surface modifications in improving both activity and durability across all catalyst types. While transition metals provide economic advantages, noble metals ensure superior stability, making them complementary solutions adaptable to specific operational requirements. Table 1 summarizes the performance comparison of OER electrocatalysts for seawater electrolysis at industrial-grade current density.
| Catalyst type | Catalyst | Performance | Testing conditions | Ref. |
|---|---|---|---|---|
| Transition metal oxides | Fe2O3/NiO@NF | 291 mV@1000 mA cm−2/252 mV@1000 mA cm−2 | 1 M KOH + seawater/1 M KOH + 1M NaCl | 55 |
| Transition metal oxides | NiOx–FeOx @g-C3N4 | 380 mV@1000 mA cm−2 | 1 M KOH | 56 |
| Transition metal (oxygen) hydroxide | NiCo@NiFe LDH | 222 mV@100 mA cm−2 | 1 M KOH + seawater | 57 |
| 266 mV@500 mA cm−2 | ||||
| Transition metal (oxygen) hydroxide | Ni doped FeOOH | 1.7 Vcell@729 mA cm−2 | AEM + 1 M KOH + seawater | 58 |
| Transition metal (oxygen) hydroxide | CoFeOxHy/Ti | 1.67 V(vs. RHE)@500 mA cm−2 | 1.0 mol kg−1 K-borate + 0.5 mol kg−1 KCl, pH 9.2 + 353 K | 59 |
| Transition metal sulfide | NiCoS NSAs @NF | 360 mV@500 mA cm−2 | 1 M KOH + 0.5 M NaCl/1 M KOH + seawater | 60 |
| 430 mV@1000 mA cm−2/440 mV@500 mA cm−2 | ||||
| Transition metal sulfide | S doped (Ni,Fe)OOH | 339 mV@500 mA cm−2 | 1 M KOH + 0.5 M NaCl/1 M KOH + seawater | 48 |
| 378 mV@1000 mA cm−2/398 mV@500 mA cm−2 | ||||
| 462 mV@1000 mA cm−2 | ||||
| Transition metal phosphides | PO43− doped NiFe LDH@Ni | 2.674 V(vs. RHE)@500 mA cm−2 | Real alkaline seawater + 0.25 M Na3PO4 | 49 |
| Transition metal phosphides | NiFeP/P-rGO/NF | 340 mV@400 mA cm−2 | 1 m KOH + 1 m NaCl | 51 |
5.2 HER electrocatalysts
 | ||
| Fig. 11 (a) Schematic illustration of the formation process of Ru/P-NiMoO4 @NF.61 (b) FESEM images of NiRh-Cu NA/CF.62 (c) Schematic of the two-step synthesis process (Pt/Ni–Mo).63 (d) Electrochemical performance of the Ru/P–NiMoO4@NF catalysts.61 (e) LSV curves and (f) long-term stability test of NiRh-Cu NA/CF.62 (g) Electrochemical performance of the Pt/Ni–Mo electrocatalyst.63 | ||
The incorporation of platinum group metals (PGMs) significantly enhances the HER activity of transition metal alloy catalysts. Yang et al.63 synthesized a high-performance Pt/Ni–Mo electrocatalyst using a sequential reduction strategy. This catalyst consisted of PGM nanoparticles anchored on a high-surface-area, corrosion-resistant substrate designed for efficient electrolysis. The synthesis involved two key steps: first, a hydrothermal process was used to grow Pt/NiMoO4 micro-pillars on nickel foam by dissolving H2PtCl6, (NH4)2MoO4·7H2O, and Ni(NO3)2·6H2O in an aqueous solution. Next, the Pt/NiMoO4 was reduced using hydrogen gas (Fig. 11c). During this reduction, Pt nanoparticles and Ni4Mo nanocrystals were released and immobilized on the NiMoO4 substrate while maintaining the micropillar morphology. This design, with highly dispersed PGM nanoparticles and a tailored substrate, resulted in exceptional catalytic performance. The Pt/Ni–Mo catalyst achieved an overpotential of 42 mV for HER and 420 mV for OER at an industrial-grade current density of 2000 mA cm−2 in a 1 M KOH solution. In a brine electrolyte (1 M KOH and 0.5 M NaCl), the Pt/Ni–Mo electrocatalyst required only 113 mV to reach the same current density (Fig. 11g). The catalyst demonstrated high activity and durability across a range of challenging conditions, including strong alkaline and simulated seawater electrolytes. These characteristics highlight its potential for industrial-scale applications under harsh operating environments.
Transition metal phosphides. Transition metal phosphides have garnered significant attention as efficient HER catalysts for high-current-density seawater electrolysis. Nanostructured phosphides grown in situ on conductive substrates such as carbon cloth (CC), carbon fiber (CF), nickel foam (NF), and carbon paper (CP) have shown remarkable potential for these applications. These substrates provide large surface areas for active site growth, enhance charge transport across electrodes, and facilitate gas discharge and reactant–product exchange due to their porous structures. Chai et al.64 developed a bimetallic phosphide catalyst (Cu3P-FeP@CC) grown on carbon cloth for HER at ultra-industrial-grade current densities (Fig. 12a). The catalyst demonstrated excellent HER performance, achieving low overpotentials of 338 mV and 406 mV at a current density of 1000 mA cm−2 in alkaline and salt-alkaline electrolytes, respectively (Fig. 12g and h), and maintaining stability for up to 400 hours. Carbon cloth, with its high electrical conductivity, elasticity, and mechanical strength, provides numerous attachment points for the catalyst due to its woven structure. When tightly bonded with the catalyst, it enhances active site exposure and electrolyte diffusion, promoting efficient HER. In this system, FeP acts as the primary hydrogen adsorption site, while Cu3P adjusts the electronic structure and electron orbitals of FeP, shifting the d-band center closer to that of Pt.65 This synergy mimics Pt-like hydrogen evolution properties, enhancing catalytic kinetics and activity. Li et al.66 synthesized a P–Ni4Mo catalyst grown on copper foam (P–Ni4Mo/CF) (Fig. 12b–d), achieving appreciable HER performance in alkaline seawater with an overpotential of 260 mV at 100 mA cm−2 and 551 mV at 1 A cm−2, slightly underperforming compared with the Pt/C catalyst (453 mV) (Fig. 12i). The catalyst demonstrated strong durability, with negligible activity loss over 200 hours of testing (Fig. 12j). Yu et al.67 proposed incorporating small molecules like hydrazine (N2H4) into alkaline-saline electrolytes to shift anodic oxidation potentials negatively. This strategy increases the potential gap between hypochlorite formation and anodic small-molecule oxidation reactions, allowing for high cathodic HER current densities without generating corrosive hypochlorite species.68 Using bifunctional Co–Ni–P nanowires supported on carbon paper (Co–Ni–P/CP) as both cathode and anode (Fig. 12e), they achieved hydrogen production at 500 mA cm−2 in an alkaline brine electrolyte with a cell voltage of 0.533 V and excellent stability for 80 hours with minimal degradation (Fig. 12k). These studies underscore the potential of transition metal phosphides and advanced system designs to enhance HER performance and durability in industrial seawater electrolysis.
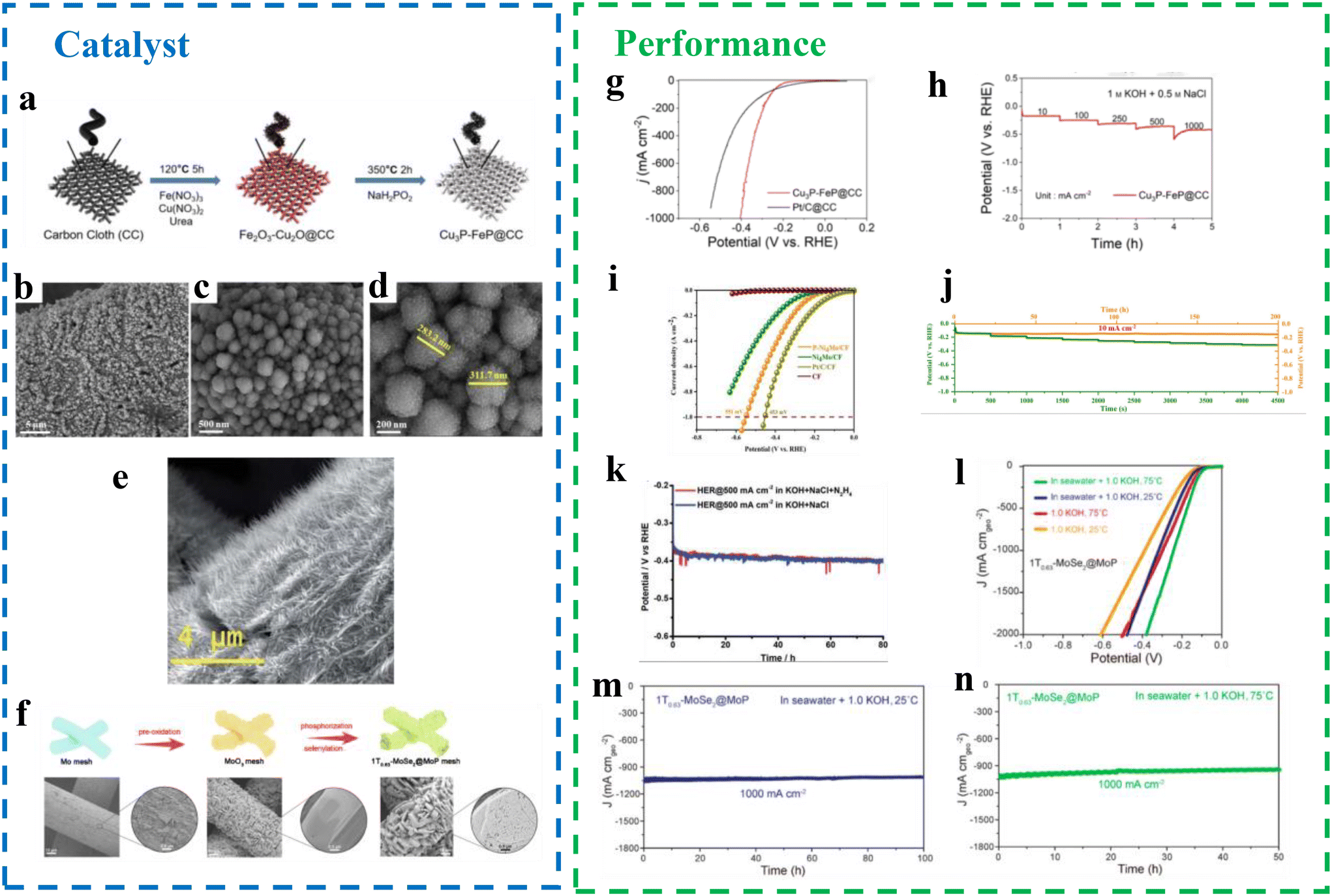 | ||
| Fig. 12 (a) Illustration of preparation procedures of the Cu3P-FeP@CC.64 (b–d) SEM of P-Ni4Mo/CF.66 (e) SEM of the Co–Ni–P/CP.67 (f) Schematic illustration of the fabrication process for self-standing 1T0.63-MoSe2@MoP MPIC and corresponding SEM images.69 (g) LSV curves, (h) Multicurrent chronopotentiometric curves of the Cu3P-FeP@CC.64 (i) LSV curves, (j) multipotential stability test of P–Ni4Mo/CF.66 (k) Chronopotentiometric curves of Co–Ni–P/CP at 500 mA cm−2.67 (l–n) Electrochemical performance of the 1T0.63-MoSe2@MoP MPIC.69 | ||
Recent advancements in tuning the intrinsic interfacial electronic structure of catalysts through multiphase synergistic interactions have drawn considerable attention for enhancing HER performance. Li et al.69 developed a multiphase interface catalyst (MPIC), 1T0.63-MoSe2@MoP, which exhibited exceptional HER activity in both alkaline freshwater and simulated seawater electrolytes. The catalyst demonstrated current densities of 1000 mA cm−2 with performance comparable to Pt-based materials (Fig. 12f). This MPIC leverages multiphase synergistic intrinsic interfacial electronic structure engineering to achieve efficient HER electrocatalysis. The unique modulation of interfacial electronic structures in the 1T0.63-MoSe2@MoP MPIC facilitates the formation of multiple heterostructures, including 2H-MoSe2, 1T-MoSe2, and MoP phases. These heterostructures enhance charge transfer and catalysis, enabling high activity. The catalyst is self-supporting and vertically grown on industrial-grade molybdenum meshes through a straightforward solid-state synthesis process under non-sensitive conditions. The method allows for scalable preparation of large MPICs, constrained only by the dimensions of the growth chamber and the molybdenum mesh. As a result, the 1T0.63-MoSe2@MoP MPIC achieved impressive current densities of 1000 mA cm−2 in both alkaline freshwater and artificial seawater electrolytes, requiring low overpotentials of 358 mV and 317 mV, respectively (Fig. 12l–n). This work highlights the potential of interfacial electronic structure tuning for developing high-performance, scalable HER catalysts.
Transition metal sulfide. Transition metal sulfides, known for their physical and chemical similarities to transition metal phosphides, have emerged as promising HER catalysts for high-current-density seawater electrolysis aimed at hydrogen production. Lv et al.70 developed self-supported ultrathin and metastable FeS nanolayers on a three-dimensional porous iron foam (IF) substrate using a one-step mild sulfation etching process, creating FeS@IF electrodes within just two hours (Fig. 13a). The FeS@IF electrodes demonstrated excellent and stable HER performance in various electrolytes, including alkaline simulated seawater (1 M KOH + 0.5 M NaCl), neutral electrolytes (1 M PBS), and other corrosive environments. The catalyst achieved low overpotentials of 63 mV and 78 mV to reach a current density of 10 mA cm−2 in 1 M KOH + 0.5 M NaCl and 1 M PBS, respectively (Fig. 13c). Furthermore, the FeS@IF electrode sustained operation at 0.2–0.4 A cm−2 for over 600 hours in neutral electrolyte (1 M PBS) with negligible performance degradation (Fig. 13d). The excellent durability and activity of the FeS@IF electrode can be attributed to the ultrathin, corrosion-resistant FeOxSy nanoscopic fan-shaped structures formed during etching. This uniform morphology enhances interfacial electron transport between active species and the substrate, exposes abundant active sites, and creates optimized channels for the rapid release of bubbles and efficient mass transfer. These properties make FeS@IF electrodes a highly effective and durable solution for HER in challenging seawater electrolysis environments.
 | ||
| Fig. 13 (a) The optimal synthesis route and condition of FeS@IF electrode.70 (b) Schematic illustration of the synthesis of VO2(p)–V2C host.71 (c) LSV curves of IF, Pt/C@IF and FeS@IF electrodes in neutral solution.70 (d) Chronopotentiometric measurement for FeS@IF electrodes in 1.0 M PBS.70 (e–g) Electrochemical performance of the VS2-based catalysts.71 | ||
The incorporation of heterogeneous structures between transition metal sulfides and other metallic materials can significantly enhance HER performance. Wang et al.71 utilized V2C, a material with high electrical conductivity, as the matrix for composites and developed a novel disulfide water electrolysis strategy based on a three-dimensional structure of vanadium-based two-dimensional carbides. Using a simple one-step hydrothermal method, they successfully synthesized VS2@V2C composites (Fig. 13b). Microstructural analysis revealed that vanadium disulfide (VS2) nanosheets were uniformly embedded vertically within multiple layers of V2C nanosheets. This architecture prevents aggregation and structural collapse of VS2 nanosheets, while the wide surface of V2C serves as a robust support for VS2. The tight bonding between the two phases facilitates efficient electron transfer, significantly enhancing the HER activity and stability of the composite compared with pure VS2 or V2C alone. In minimally treated seawater with varying pH levels, the VS2@V2C composites achieved industrial-grade current densities and low overpotentials, comparable to Pt-based catalysts. The biphasic interface enabled rapid charge transfer, while the high specific surface area and unique mosaic structure reduced the free energy of hydrogen ion adsorption. As a result, the composite exhibited excellent electrocatalytic HER performance across different pH environments. In an alkaline electrolyte (pH = 13.8), the composite achieved an overpotential of 164 mV and a Tafel slope of 37.6 mV dec−1 at 20 mA cm−2 (Fig. 13e). In highly acidic conditions (pH = 0), the overpotential was as low as 138 mV with a Tafel slope of 37.9 mV dec−1 (Fig. 13f). At industrial-grade current densities (>100 mA cm−2), VS2@V2C outperformed Pt-based catalysts, demonstrating superior efficiency and long-term stability. Additionally, the composite maintained excellent performance in neutral or mildly alkaline natural seawater, further establishing its potential for practical hydrogen evolution applications (Fig. 13g).
Transition metal oxides. Transition metal oxides have gained significant attention for their efficient HER activity. Du et al.72 developed a robust electrocatalyst composed of NiFe2O4 deposited on nickel foam for hydrogen production via HER-SMOR systems in alkaline water and seawater electrolytes (Fig. 14a). The study also introduced an in situ growth strategy for active catalysts on conductive substrates, such as NF, offering potential for large-scale applications. The in situ synthesis of NiFe2O4 on NF ensured high electrical conductivity while enhancing the electrocatalyst's durability under intense electrochemical operating conditions.73 The dual-electrode system, catalyzed by NiFe2O4/NF||NiFe2O4/NF, achieved industrial-grade current densities (>800 mA cm−2) and a high hydrogen production rate (>1.7 μmol s−1 cm−2) at a cell voltage of 2.0 V (Fig. 14c). The system also exhibited excellent faradaic efficiencies, exceeding 96% for HER at the cathode and 95% for formic acid oxidation at the anode (Fig. 14d). Additionally, the NiFe2O4/NF||NiFe2O4/NF HER-SMOR system demonstrated exceptional stability, maintaining performance after 48 hours of continuous operation (Fig. 14e). Importantly, the system performed well in seawater, exhibiting high activity and stability over 6 hours without significant side reactions, such as the ClOR or OER. Spectroscopic studies and DFT calculations revealed that the SMOR on NiFe2O4/NF follows a CO-free pathway, with Fe and Ni sites on the NiFe2O4 spinel acting as active centers for methanol and OH activation, respectively. This work highlights the potential of NiFe2O4/NF for efficient and durable hydrogen production in both freshwater and seawater electrolysis systems.
 | ||
| Fig. 14 (a) Schematic illustration of the synthesis of NiFe2O4/NF catalyst.72 (b) Illustration for preparing the Ti/TiO2@NiBx electrode toward overall water splitting.74 (c) Average rates of H2 generation at different potentials.72 (d) Faradaic efficiencies of formate generation at different potentials in KOH/MeOH.72 (e) I–t curves of NiFe2O4/NF|NiFe2O4/NF at 1.8 V in 1.0 M KOH for 48 h.72 (f) Chronopotentiometric measurement of HER at 500 mA cm−2.74 | ||
In addition, Fu et al.74 prepared TiO2 nanorods by a one-step alkali etching method and supported them on corrosion-resistant Ti plates (Ti/TiO2). Subsequently, they obtained Ti/TiO2@NiBx self-supported electrodes by modifying NiBx catalytic material on Ti/TiO2 by a simple electroless plating method (Fig. 14b). The modification of amorphous NiBx significantly enhances the catalytic properties of TiO2 semiconductors by increasing the number of reaction sites and improving both photoadsorption and electron–hole pair separation efficiency. The Ti/TiO2@NiBx electrode demonstrated improved catalytic performance, achieving increases of approximately 16.2% and 14.7% in HER and OER efficiency, respectively, at a current density of 10 mA cm−2 under xenon lamp irradiation and electrical energy input. Additionally, the electrode enabled solar-driven overall water splitting at lower potentials and higher current densities. The Ti/TiO2@NiBx electrode exhibited stable performance during 72 hours of continuous operation, highlighting its potential for industrial applications. Mechanistic analysis revealed that NiO-Had species were formed at the cathode, while NiOOH active species were detected at the anode, contributing to the enhanced catalytic performance and durability. Moreover, the electrode sustained efficient seawater electrolysis at industrial-grade current densities (500 mA cm−2) during the HER process for 72 hours, attributed to the high corrosion resistance of the TiO2 layer (Fig. 14f). The modification of amorphous NiBx increased the specific surface area of the electrode, facilitating the effective separation of electron–hole pairs within the TiO2 semiconductor structure, further improving its efficiency and stability for large-scale seawater electrolysis applications.
Precious metal-based catalysts consistently demonstrate high HER efficiency and stability, benefiting from outstanding catalytic activity and strong corrosion resistance. These findings align with the established role played by platinum group metals in enhancing transition metal alloy catalysts, enabling superior performance even under industrial conditions. Meanwhile, transition metal phosphides and sulfides offer complementary strategies by modulating electronic structures or forming heterostructures to emulate Pt-like activity and achieve high current densities. Additionally, advancements in multiphase interface engineering provide valuable insights into optimizing interfacial electronic properties, paving the way for scalable and durable catalysts. Similarly, studies on oxides and TiO2-based systems highlight alternative approaches, leveraging in situ growth techniques and light-assisted catalysis to enhance efficiency and long-term stability in seawater electrolysis. Table 2 summarizes the performance comparison of HER electrocatalysts for seawater electrolysis at industrial-grade current density.
| Catalyst type | Catalyst | Performance | Testing conditions | Ref. |
|---|---|---|---|---|
| Transition metal phosphides | Cu3P–FeP@CC | 291 mV@500 mA cm−2, 338 mV@1000 mA cm−2/406 mV@1000 mA cm−2 | 1 M KOH/1 M KOH + 0.5 M NaCl | 64 |
| Transition metal phosphides | P—Ni4Mo/CF | 551 mV@1000 mA cm−2, 422 mV@500 mA cm−2 | 1 M KOH + seawater | 66 |
| Transition metal phosphides | Co–Ni–P/CP | 0.533 V@500 mA cm−2 | 1 M KOH + 0.5 M NaCl + 0.1 M N2H4 | 67 |
| Transition metal phosphides | Ru/P–NiMoO4@NF | 0.173@1000 mA cm−2, 0.242 V@3000 mA cm−2/0.299 V@3000 mA cm−2 | 1 M KOH + 0.5 M NaCl/1 M KOH + seawater | 61 |
| Transition metal phosphides | C–Co2P | 192 mV@1000 mA cm−2 | 1 M KOH, 0.5 M NaCl, 41.2 × 10−3 M MgCl2, 12.5 × 10−3 M CaCl2 | 75 |
| Transition metal phosphides | 1T(0.63)-MoSe2@MoP | 358 mV@1000 mA cm−2/317 mV@1000 mA cm−2 | 1 M KOH/1 M KOH + seawater | 69 |
| Transition metal sulfide | FeS@IF | 490 mV@1000 mA cm−2 | 1 M KOH + 0.5 M NaCl | 70 |
| Transition metal sulfide | VS2@V2C | At higher current densities (>100 mA cm−2) | In natural seawater (pH = 8.6) | 71 |
| Transition metal alloy | NiRh-Cu NA/CF | 155 mV@300 mA cm−2 | Alkaline simulated seawater electrolyte | 62 |
| Transition metal alloy | Pt/Ni–Mo | 113 mV@2000 mA cm−2 | 1.0 m KOH + 0.5 m NaCl | 63 |
| Transition metal oxides | NiFe2O4/NF||NiFe2O4/NF | 2.0 V@700 mA cm−2 | Seawater | 72 |
| Transition metal oxides | Ti/TiO2@NiBx | 72 h@500 mA cm−2 | 1 M KOH + seawater | 74 |
5.3 Bifunctional electrocatalysts
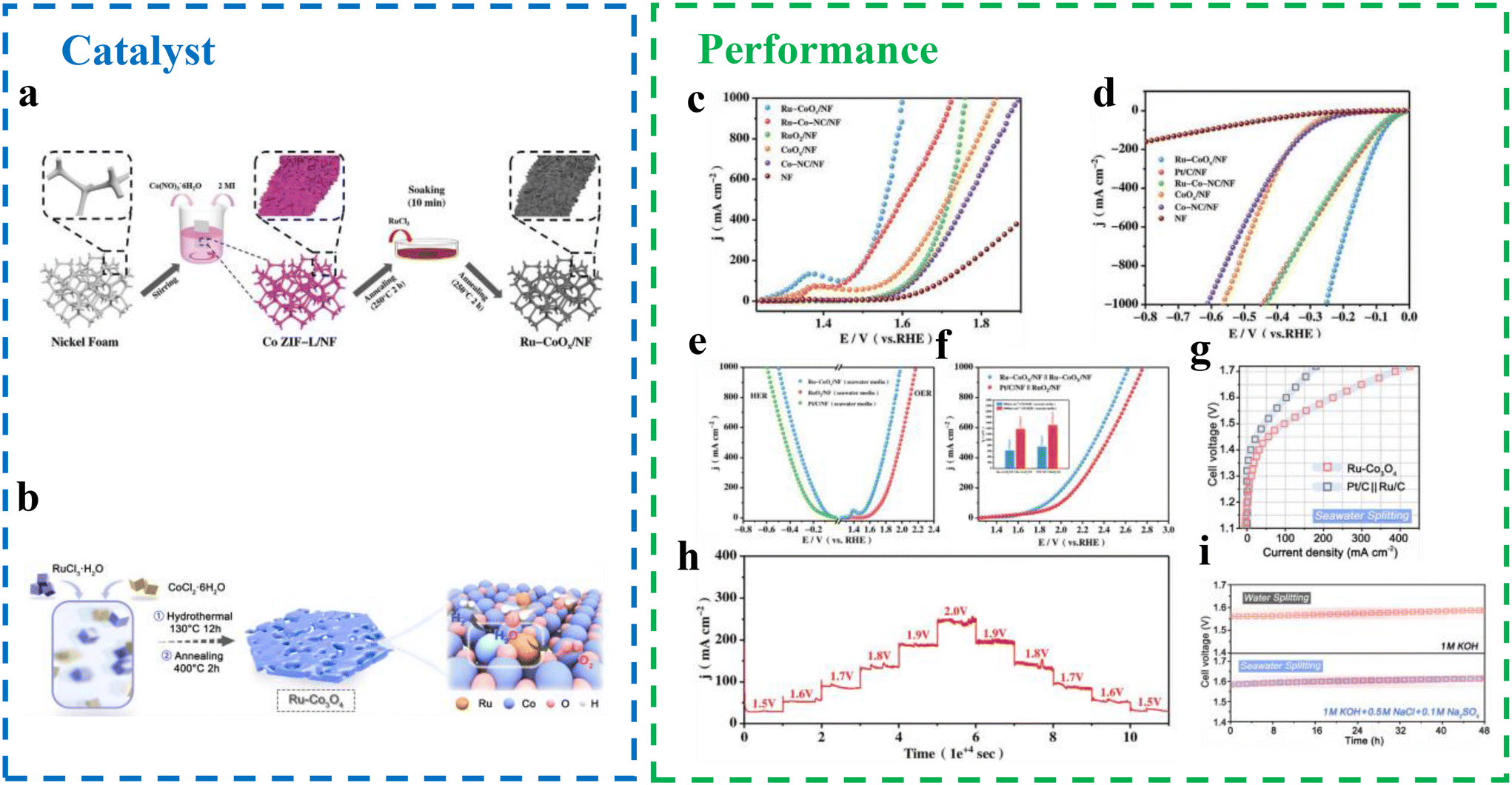 | ||
| Fig. 15 Schematic illustration of the fabrication of (a) Ru–CoOx/NF.76 (b) Ru–Co3O4 with Ru–O–Co moieties.77 (c–e) OER and HER polarization curves of Ru–CoOx/NF and other samples in 1 M KOH and seawater media.76 (f) Polarization curves of Ru–CoOx/NF||Ru–CoOx/NF and Pt/C/NF||RuO2/NF toward overall water splitting in seawater media.76 (g) Seawater splitting performance of Ru–Co3O4 and Pt/C||Ru/C.77 (h) Multipotential stability test chart of Ru–CoOx/NF in seawater media.77 (i) Chronopotentiometry stability tests of Ru–Co3O4 for water and seawater electrolysis.77 | ||
Transition metal oxide. Transition metal oxides exhibit high intrinsic electrocatalytic activity for the HER and OER due to their strong hydrolysis ability across various valence states. A hybrid strategy has been proposed to enhance the water electrolysis performance of transition metal oxide electrodes. Guo et al.78 introduced Lewis acid layers (e.g., Cr2O3) onto the surface of commonly used catalysts to regulate the local reaction microenvironment, enabling direct seawater electrolysis without the need for alkalization or acidification (Fig. 16a). This approach is versatile and can be applied to various catalysts without redesigning the catalyst or the electrolytic cell. Using Lewis acid-modified electrodes (Cr2O3–CoOx), the system achieved stable performance for over 100 hours in real seawater, splitting water molecules and capturing large amounts of hydroxyl anions (OH−) generated in situ around the catalyst. The performance was comparable to that of pure water PEM electrolytic cells, delivering a current density of 1.0 A cm−2 at 60 °C with a cell voltage of 1.87 V in a flowing seawater electrolyzer (Fig. 16b). The effectiveness of this strategy lies in the preferential enrichment of OH− ions around the catalyst, which suppresses chlorination reactions and minimizes carbon contamination on the catalyst surface. Additionally, the strong binding between OH− and the Lewis acid layer reduces the formation of precipitates, preventing the capture of OH− by Mg2+ and Ca2+ cations in seawater electrolytes. The properties of the anode and cathode in Lewis acid-modified flowing seawater electrolytic cells are comparable to those of pure water catalysts using RuO2 and Pt/C, respectively, demonstrating a promising pathway for efficient and sustainable seawater electrolysis.
 | ||
| Fig. 16 (a) Schematic diagram of local alkaline microenvironment generation on Lewis acid-modified cathode. LA = Lewis acid. EDL = electrical double layer.78 (b) Long-term durability test of 6 at% Cr2O3–CoOx anode at 1.80 V RHE in natural seawater.78 | ||
Transition metal phosphide. Transition metal phosphide catalysts often excel in either the OER or HER but rarely achieve high activity in both simultaneously. Chang et al.79 addressed this limitation by synthesizing Fe and phosphorus double-doped nickel selenide nanoporous films (Fe, P-NiSe2 NFs) as a bifunctional catalyst with excellent performance for seawater electrolysis (Fig. 17a). The enhanced performance of Fe, P-NiSe2 NFs is attributed to the weaker bond strength of Se–H compared with P–H, which allows selenides to trap reactants more effectively and accelerate the discharge step. Simultaneously, the slightly oxidized surface of Fe, P–NiSe2 prevents dissolution, ensuring stable and balanced HER and OER activities. Experimental results and DFT calculations revealed that Fe-dopants act as active HER sites, while Ni atoms adjacent to Fe-dopants serve as catalytic centers for the OER. The dual role played by P-dopants was identified as improving electrical conductivity and forming a surface passivation layer containing P–O species, which prevents selenium dissolution and enhances catalyst stability. Self-supported on carbon cloth, Fe, P–NiSe2 NFs can be directly utilized as bifunctional catalysts in seawater electrolyzers without the need for additional additives. The catalyst achieved an industrial-grade current density of 0.8 A cm−2 at 1.8 V, maintaining performance for over 200 hours with an oxygen faradaic efficiency exceeding 92% (Fig. 17c and d). In summary, Fe doping enhances OER selectivity and faradaic efficiency, while P doping improves electrical conductivity and stabilizes the catalyst by forming a protective P–O passivation layer. These features make Fe, P-NiSe2 NFs a promising candidate for efficient and durable bifunctional catalysis in seawater electrolysis.
 | ||
| Fig. 17 (a) Illustration for the HER and OER reaction pathways on different catalysts.79 (b) The synthesis of NiFe-P@NC.80 (c) Comparison of the HER and OER overpotentials required to achieve current densities of 10, 100, and 500 mA cm−2 for: (a1) Fe,P-NiSe2 NFs, (a2) Fe–NiSe2 NFs, and (a3) P–NiSe2 NFs.79 (d) Long-term operation test of asymmetric device I and symmetric device III seawater electrolyzers showing high selectivity and stability at 1.8 V.79 Plots of the (e) OER and (f) HER performance tests of the prepared electrocatalytic materials in KOH (1 M) + seawater.80 (g) Plots of NiFe-P@NC||NiFe-P@NC in alkaline seawater for overall water-splitting.80 | ||
Phosphorylation is a widely used technique to enhance the water electrolysis activity of transition metal catalysts. To further boost catalytic performance, Chen et al.80 developed a novel nitrogen-doped carbon-coated nickel phosphide catalyst (NiFe-P@NC) using corrosion engineering combined with a mild phosphorylation process (Fig. 17b). NiFe-P@NC is a bifunctional catalyst with exceptional electrocatalytic performance, enabling efficient HER and OER reactions. The unique hierarchical morphology of NiFe-P@NC, characterized by an interconnected three-dimensional porous structure, significantly increases the catalyst's surface area and the number of active sites, thereby enhancing its catalytic efficiency. Experimental results demonstrated that NiFe-P@NC achieved excellent HER activity, requiring an overpotential of only 40 mV to reach 10 mA cm−2. Additionally, its robust structure and chemical composition conferred remarkable stability in 1 M KOH, resisting corrosion and oxidation during prolonged operation. In a 1 M KOH + seawater electrolyte, NiFe-P@NC displayed outstanding bifunctional catalytic performance, with low overpotentials of 149 mV and 280 mV required for the HER and OER at 100 mA cm−2, respectively (Fig. 17e and f). When assembled into a NiFe-P@NC||NiFe-P@NC electrolyzer, the system achieved current densities of 100 mA cm−2 and 500 mA cm−2 at low voltages of 1.77 V and 1.93 V, respectively, in alkaline seawater electrolytes (Fig. 17g). This superior performance is attributed to NiFe-P@NC's high electrical conductivity and catalytic activity. Overall, NiFe-P@NC demonstrates exceptional potential as a bifunctional electrocatalyst for efficient HER and OER reactions, making it a promising candidate for industrial water electrolysis applications.
Transition metal sulfide. Transition metal sulfides are well known for their bifunctional HER and OER electrocatalytic activity. To optimize their performance for high-current-density seawater electrolysis, novel multifaceted electrocatalysts combining sulfur with other transition metals have been developed. Yao et al.81 synthesized a hybrid electrode with local photothermal effects (LPTE) via a one-pot hydrothermal reaction. This electrode features a ternary nickel–bismuth–sulfur nanosheet array supported on nickel foam (Ni3Bi2S2@NF) (Fig. 18a). The Ni3Bi2S2@NF electrode demonstrated significant efficiency improvements in HER and OER, with increases of 44% and 35%, respectively, under light irradiation. These enhancements are attributed to the intrinsic LPTE effect of bismuth, the strong phase stability of Ni3Bi2S2, and the synergistic properties of its layered structure. The LPTE effect facilitates localized heat generation, accelerating reaction kinetics and improving catalytic efficiency. The electrode also showed exceptional performance in a full water-splitting system. Using Ni3Bi2S2@NF as a bifunctional electrode, the system achieved an ultra-low voltage of 1.40 V under LPTE at a current density of 10 mA cm−2. It maintained stability for over 36 hours at industrial-grade current densities of 500–1000 mA cm−2 (Fig. 18c and d). These results highlight the superior electrocatalytic activity and durability of Ni3Bi2S2@NF, making it a promising candidate for seawater electrolysis and other water-splitting applications. The integration of LPTE with a robust ternary structure underscores its potential for energy-efficient and stable hydrogen production technologies.
 | ||
| Fig. 18 (a) Illustration for preparing Ni3Bi2S2@NF electrode and overall water splitting process with LPTE.81 (b) SEM image of NiFeSP-NT.82 (c) Long-time LPTE stability experiments of Ni3Bi2S2@NF under different lager current density each for 12 h in 1.0 M KOH and simulated seawater solutions.81 (d) Chronopotentiometric measurements of long-term stability of coupled-LPTE Ni3Bi2S2@NF under different constant voltages for 12 h in 1.0 M KOH and 1.0 M KOH + 0.5 M NaCl solution.81 Polarization curves of the (e) OER and (f) HER, recorded at 5 mV s−1.82 (g) Chronopotentiometric curves of the OSUWE and OSWE for the NiFeSP electrode pair.82 | ||
The sulfidation process is a powerful method to enhance the water electrolysis performance of transition metal phosphides. Yu et al.82 developed a self-supported nickel–iron phosphide (NiFeSP) nanotube array electrode using a two-step sulfidation/phosphorylation process. These NiFeSP nanotubes feature abundant NiFeS/NiFeP heterogeneous surfaces and low-coordination metal sites, which synergistically improve electrocatalytic activity (Fig. 18b). Experimental results showed that the NiFeSP electrode demonstrated outstanding HER and OER performance in simulated alkaline aqueous solutions (KOH + NaCl), achieving overpotentials of 380 mV for HER and 260 mV for OER at 500 mA cm−2 (Fig. 18e and f). The electrode exhibited excellent durability, maintaining performance for 1000 hours. To address the issue of competing chloride evolution reactions during the OER, the researchers replaced the OER with a urea oxidation reaction (UOR), significantly reducing the overall energy input required for alkaline water electrolysis. Theoretical calculations confirmed that the heterogeneous interfaces and low-coordination metal sites in the NiFeSP nanotubes lower the energy barrier for the rate-determining step, enhancing catalytic performance. Additionally, the electrode demonstrated excellent activity for the UOR, enabling efficient urea-mediated alkaline water electrolysis. By coupling the UOR with the HER, the bifunctional NiFeSP electrode effectively catalyzed alkaline water electrolysis at 500 mA cm−2 for 1000 hours at a voltage of 1.938 V without significant performance degradation (Fig. 18g). These findings highlight the potential of NiFeSP nanotube arrays for energy-efficient and durable water electrolysis systems.
Transition metal nitride. Transition metal nitrides are known for their strong electrocatalytic activity in the HER and OER, along with high corrosion resistance, electrical conductivity, and mechanical strength. However, their electrocatalytic performance in high-current-density seawater electrolysis still has room for improvement. One effective strategy to enhance bifunctional catalytic activity is designing heterogeneous structures with coupled functionalized species. Ning et al.83 successfully synthesized Fe-doped Ni&Ni0.2Mo0.8N on nickel foam as a highly active HER catalyst (Fig. 19a). The study revealed that varying Fe doping concentrations significantly influenced the rod size of the catalyst and its intrinsic HER activity. During rapid electrochemical reconstruction, Fex–Ni&Ni0.2Mo0.8N transformed in situ into Fex&Mo–NiO, exhibiting strong OER activity. Tafel analysis and DFT calculations indicated that Fe doping shifted the rate- and potential-determining steps of Mo–NiO in the OER mechanism, enhancing catalytic efficiency. Systematic investigations compared the performance of Fe0.01–Ni&Ni0.2Mo0.8N||Fe0.01&Mo–NiO electrolyzers in alkaline freshwater and seawater electrolysis. The electrolyzer achieved nearly 100% Faraday efficiency for H2 and O2 production in both 1 M KOH and seawater. It demonstrated record-high current densities in seawater electrolysis at 1.7 V (Fig. 19c). Under quasi-industrial conditions, its performance in highly alkaline seawater (60 °C) closely matched that in alkaline freshwater, with only a 1.49% voltage increase at 1000 mA cm−2. The electrolyzer maintained excellent stability in 6 M KOH and seawater at 60 °C, even under harsh conditions (Fig. 19d). These findings underscore the potential of Fe-doped Ni&Ni0.2Mo0.8N with Mo–NiO heterostructures as bifunctional catalysts for efficient and durable H2 and O2 production in seawater electrolysis. Moreover, the study highlighted the importance of addressing redox substance concentrations in alkaline seawater, which are higher than in alkaline freshwater and may lead to catalyst poisoning and corrosion. Careful electrolyte selection and treatment are crucial to ensuring the long-term stability and efficiency of catalysts in seawater electrolysis systems.
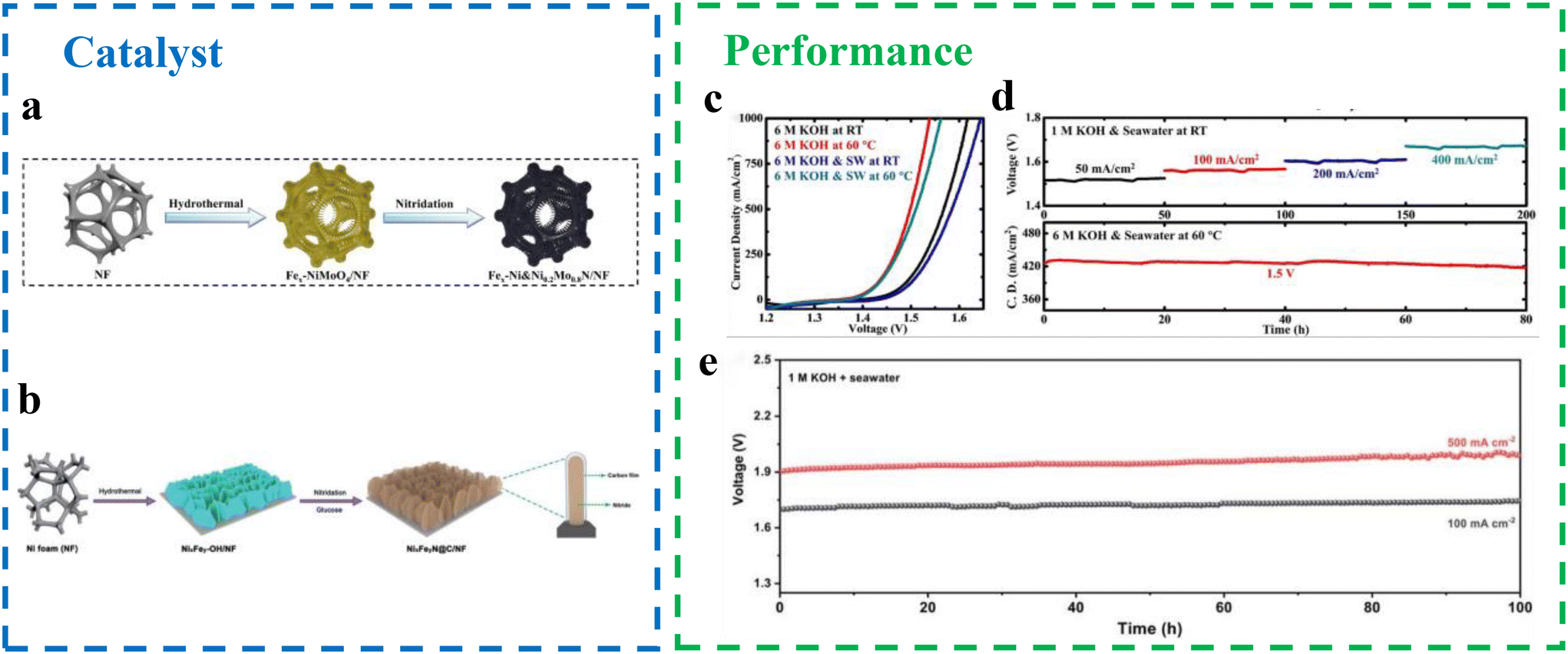 | ||
| Fig. 19 (a) Synthesis process for Fex–Ni&Ni0.2Mo0.8N microrods on NF.83 (b) The preparation procedure of NixFeyN@C/NF.84 (c) Alkaline water and seawater electrolysis performance of Fe0.01–Ni&Ni0.2Mo0.8N8Fe0.01&Mo–NiO under industrial conditions.83 (d) Stability measurements during alkaline seawater electrolysis.83 (e) Durability tests of this electrolyzer at 100 and 500 mA cm−2.84 | ||
Metal–N bonding in transition metal nitrides induces a contraction of the metal d-band, raising the energy levels near the Fermi energy. This redistribution of the density of states enhances the electrocatalytic activity of transition metal nitrides, making them comparable to noble metals. Wang et al.84 reported an advanced electrocatalyst, NiFeLDH@Ni3N/NF, which features a three-dimensional hierarchical heterogeneous structure composed of nano/micro sheets fabricated on nickel foam (Fig. 19b). This electrocatalyst demonstrates excellent electrical conductivity, abundant active sites, and a synergistic interaction between NiFeLDH and Ni3N, leading to superior electrocatalytic performance and stability. When used as a bifunctional electrocatalyst, NiFeLDH@Ni3N/NF enabled a two-electrode electrolyzer to achieve a current density of 500 mA cm−2 at a cell voltage of 1.80 V with remarkable stability over 100 hours (Fig. 19e). Additionally, Wang et al. developed a carbon-coated nickel nitride micro sheet array (NixFeyN@C/NF) as a highly efficient and durable electrocatalyst for seawater electrolysis. This electrocatalyst leverages a porous micro sheet array structure to provide abundant active sites, ideal surface superhydrophobicity and superhydrophilicity to enhance mass transfer, and efficient charge transport due to the synergistic coupling between NixFeyN and the carbon coating. The durability of NixFeyN@C/NF is attributed to the protective effect of the carbon coating and the intrinsic corrosion resistance of NixFeyN. These features make it a promising electrocatalyst for long-term and high-performance seawater electrolysis, offering a practical solution for sustainable hydrogen production in harsh environments.
 | ||
| Fig. 20 (a) Schematic illustration for the preparation of PGQDs and PGQDs/GH composites. (b) OER and (d) HER LSV curves of the prepared PGQDs/GH composites and commercial IrO2 in 1 M KOH. (c) LSV curves for electrolysis of fresh water and seawater using PGQDs/GH electrodes as both anode and cathode. (e) Chronopotentiometric stability of seawater electrolysis using PGQDs/GH electrodes for 1000 h.85 | ||
Noble metal-based catalysts demonstrate outstanding catalytic efficiency and stability, outperforming traditional Pt/C and RuO2 due to innovative structural enhancements and precise elemental doping strategies. In parallel, transition metal compounds leverage unique doping and surface engineering techniques to balance HER and OER activities, achieving excellent durability and industrial-level current densities. Contrastingly, approaches incorporating non-metallic catalysts provide a sustainable, cost-effective alternative with competitive performance metrics and long-term stability, emphasizing their potential for scalable hydrogen production. Furthermore, novel strategies, including photothermal effects and heterogeneous structures in transition metal sulfides and nitrides, showcase synergistic interactions and advanced reaction kinetics, pushing efficiency boundaries. While these studies collectively underline the versatility and potential of bifunctional catalysts, they also reveal differing optimization paths, highlighting the need for targeted designs tailored to specific operational conditions and sustainability goals. Table 3 summarizes the performance comparison of bifunctional electrocatalysts for seawater electrolysis at industrial-grade current density.
| Catalyst type | Catalyst | Performance | Testing conditions | Ref. |
|---|---|---|---|---|
| Transition metal oxides | Ru–CoOx/NF||Ru–CoOx/NF | 970 mV@1000 mA cm−2/1390 mV@1000 mA cm−2 | 1 M KOH prepared with deionized water and potassium hydroxide/1 M KOH prepared with seawater and potassium hydroxide | 76 |
| Transition metal oxides | Cr2O3–CoOx||Cr2O3–CoOx | 1.87 V@1000 mA cm−2 | In natural seawater, 60 °C | 78 |
| Transition metal phosphides | Fe, P-NiSe2 NFs||Fe, P-NiSe2 NFs | 1.8 V@0.8 A cm−2/(141 mV-HER, 317 mV-OER)@500 mA cm−2 | Natural seawater/1 M KOH | 79 |
| Transition metal phosphides | NiFe-P@NC||NiFe-P@NC | 217 mV-HER@1000 mA cm−2/340 mV-OER@500 mA cm−2, 1.93 V@500 mA cm−2 | 1 M KOH/Alkaline seawater electrolyte | 80 |
| Transition metal sulfide | Ni3Bi2S2@NF||Ni3Bi2S2@NF | 2.34 V@500 mA cm−2, 3.10 V@1000 mA cm−2 | 1.0 M KOH + 0.5 m NaCl | 81 |
| Transition metal sulfide | NiFeSP||NiFeSP | (380 mV-HER, 260 mV-OER)@500 mA cm−2, 2.225 V@500 mA cm−2 | 1.0 M KOH + 0.5 m NaCl | 82 |
| Transition metal nitride | Fe0.01-Ni&Ni0.2Mo0.8N||Fe0.01&Mo-NiO | 1.585V@500 mA cm−2, 1.644 V@1000 mA cm−2 | 6 M KOH + seawater | 83 |
| Transition metal nitride | Ni3FeN@C/NF(anode)||Ni3N@C/NF(cathode) | 1.91V@500 mA cm−2 | 1 M KOH + seawater | 84 |
| (NixFeyN@C/NF) |
6 Device designs and techno-economic analysis for direct seawater electrolysis
Designing direct seawater electrolysis (DSE) devices can pose greater challenges than catalyst design. Effective DSE operation necessitates innovative catalyst engineering and optimizing the catalyst layer characteristics. It also entails careful planning for components like porous transport layers, membranes, and operational parameters. Considering the unique requirements of different DSE setups for catalyst performance, this section will systematically examine the application and techno-economic analysis (TEA) of DSE devices to enhance their efficiency.866.1 Electrodes
Direct seawater electrolysis encompasses two distinct electrode preparation methods: the traditional method involves the use of ion membranes with catalyst-coated substrates and membranes, whereas the alternative method utilizes ion membrane-free self-supporting electrodes.
6.2 Membranes
Hydroxide ion-conducting membranes are critical components in alkaline direct seawater electrolysis, enabling hydroxide ion transport, establishing internal pathways, and preventing hydrogen gas infiltration to mitigate potential hazards. These membranes must possess key physical and chemical properties, including high ion conductivity, alkaline stability, mechanical strength, low gas permeability, an appropriate expansion ratio, and cost-effectiveness. In practical applications, hydroxide ion-conducting membranes for alkaline water electrolysis need to combine superior hydroxide ion conductivity, robust alkaline stability, mechanical durability, minimal hydrogen gas permeation, and affordability to support large-scale industrial use. Currently, commercial membranes for alkaline water electrolysis are classified into three categories: porous membranes (PMs), ion dissolution membranes (ISMs), and anion exchange membranes (AEMs) (Fig. 22). Porous membranes are typically composed of chemically inert polymer substrates and hydrophilic inorganic fillers, offering excellent resistance to alkalis. However, challenges such as high ohmic resistance and inadequate gas barrier properties in porous membranes have driven researchers to develop ion dissolution membranes tailored for alkaline liquid water electrolysis. Anion exchange membranes feature a polymer framework with nitrogen-containing cations that enable hydroxide ion transport through cationic sites within the membrane. This structure makes AEMs highly desirable due to their excellent hydroxide ion conductivity. A wide range of commercial anion exchange membranes is currently employed in water electrolysis, underscoring their significance in achieving efficient and reliable electrolysis processes.88 | ||
| Fig. 22 Three approaches for hydroxide conducting membranes, including PMs, ISMs, and AEMs.88 | ||
6.3 Direct seawater electrolysis system
AEMWE represents an innovative approach for water electrolysis, merging the strengths of AWE and PEMWE. It employs affordable non-precious metal catalysts to enhance energy efficiency. AEM plays a crucial role in halting cation diffusion from the anode to the cathode. Moreover, operating under high pH levels minimizes the competitive CER for OER, making AEMWE a more appealing option compared with PEMWE and generating significant enthusiasm for DSE.86 In practical applications, AEMWE continues to encounter issues like underutilization of catalysts, restricted mass transport, and elevated ohmic resistance. To mitigate polarization losses across kinetic, mass transport, and ohmic domains, the overall design of MEA needs to prioritize expanding three-phase boundaries and mass transport pathways within the CL, as well as enhancing the membrane/CL and CL/LGDL interfaces. Nanostructured MEAs with organized three-phase transport channels in the CL offer advantages over traditional MEAs, enhancing electrolysis performance and longevity. Strategies to boost hydroxide conductivity include increasing ion exchange capacity (IEC) and establishing ion conduits within the membrane. Transforming the CL/membrane interface structure from two-dimensional to three-dimensional can improve ion transport and stability at the interface. Additionally, optimizing MPL with suitable porosity, pore size, and thickness aids in creating advanced CL/LGDL interfaces, leading to improved catalyst utilization efficiency and reduced mass transport losses.886.4 Techno-economic analysis
TEA offers a method for evaluating the financial effects of industrial procedures, goods, or services. The integration of TEA and process modeling is vital for assessing the practicality of introducing new technologies and advocating for their possible enhancements and market uses. In particular, examining techno-economic elements is crucial for recognizing how new technologies are integrated into creative business frameworks and acts as a fundamental motivator for important research and development paths. The net present value (NPV) and levelized cost of hydrogen (LCOH) stand as two commonly utilized measures to demonstrate the outcomes of TEA analysis. Conventional NPV analysis employs the discount rate to gauge the true worth of upcoming investments, acting as an effective instrument for evaluating the profit prospects of investment endeavors, depending on their positive or negative correlation with NPV. Nonetheless, the considerable impact of selecting discount rates, capital expenditures, calculating cash flows, and investment magnitude on NPV makes its application in contrasting various hydrogen production systems difficult. Consequently, LCOH, serving as a benchmark for evaluating the cost-effectiveness of hydrogen production methods, has undergone extensive analysis and comparison.89DSE encounters difficulties in terms of both operational and capital expenditures. Table 4 displays a comparative analysis of the specific energy consumption (SEC) in seawater desalination versus traditional polymer electrolysis membrane water electrolysis. Only a mere 0.05–0.14% of the total SEC in H2 production is derived from the process of seawater desalination. Given the substantial role played by electricity in the economic and ecological facets of H2 generation, a comparable pattern in its influence on operational expenses and the LCOH is expected. Additionally, the latest studies show that the initial expenses for DSE (surpassing 6000 $ per kW) exceed double those of conventional electrolysis cell capital, ranging from 920–1725 USD per kW for alkaline water electrolysis and 1610–2668 USD per kW for PEMWE. The impact of seawater makeup, along with chlorine's cross-effects and corrosion, necessitates significant financial outlay for DSE, resulting in the regular substitution of membranes, catalysts, and system parts.
| Ref. | Water demand (kgH2O kgH2−1) | SEC of water demand via desalination (kW h kgH2−1) | SEC for H2 generation (kW h kgH2−1) | Percentage energy for water demand (%) |
|---|---|---|---|---|
| Beswick et al.90 | 9.0 | 0.03 | 63.97 | 0.05 |
| Lampert et al.91 | 25.7 | 0.09 | 63.97 | 0.14 |
| Hydrogenics92 | 11.126 | 0.04 | 74.54 | 0.05 |
| Siemens92 | 16.689 | 0.06 | 61.86 | 0.09 |
Utilizing pre-fading methods for the indirect breakdown of seawater allows us to avoid possible adverse effects and risks of corrosion. Yet, this method demands greater energy investment, diminishing its economic appeal. Moreover, the large size of an independent seawater desalination system restricts the flexibility of the seawater electrolysis system. Consequently, an analysis of processes and economics leads to the primary inference that DSE falls short in competing with advanced water purification and electrolysis technologies. Even with major technological progress, DSE, used as a preliminary treatment, manages to cut costs and energy use by merely 0.1%, presenting considerable difficulties with minimal advantages. Therefore, in producing H2, upcoming methods for treating seawater ought to integrate desalination processes with conventional electrolysis techniques.93 Xie et al.94 successfully utilized a water-repellent porous polytetrafluoroethylene (PTFE) membrane, which is both waterproof and breathable, as an interface for gas passage in seawater electrolysis. By selecting a dense potassium hydroxide solution as the self-dampening electrolyte (SDE), they integrated an in situ water purification mechanism. This system leverages a self-operating phase-change process within a seawater electrolysis apparatus, enabling efficient and innovative water electrolysis performance. This technique comprehensively tackles problems associated with adverse reactions and corrosion. Under real-world conditions, the test system functioned reliably for more than 3200 hours, maintaining a current density of 250 milliamps per square centimeter without any malfunctions. The approach effectively adopts a seawater direct electrolysis technique that is efficient, adaptable in size, and scalable, akin to freshwater electrolysis, while maintaining low operating expenses, thereby presenting vast opportunities for practical application.
Given these advancements, it is crucial to consider the broader context of seawater treatment technologies, particularly desalination. Over the past decades, the energy consumption of seawater reverse osmosis (SWRO) plants has declined considerably, from approximately 9–10 kW h m−3 to less than 3 kW h m−3, while the cost of desalinated water has decreased from $2.2 per m3 to below $0.6 per m3.95,96 Given these insights, it is essential to assess the role played by desalination within the broader framework of hydrogen production. While SWRO is often perceived as an energy-intensive process, its impact on the overall economics of hydrogen generation remains minimal. This becomes particularly evident when integrating SWRO with proton exchange membrane (PEM) water electrolysis, where the primary energy demand and cost contributions stem from the electrolysis process itself.
A cost analysis was performed for integrating seawater treatment with traditional PEM electrolysis. A typical PEM electrolysis system requires ∼10 kg of water to produce 1 kg of H2, equating to ∼500 m3 per day of SWRO water for a 50 ton per day H2 plant. The energy breakdown (Fig. 23a) shows SWRO energy demand (∼0.1% of the total) is negligible compared with electrolysis. Producing 10 kg of H2 requires ∼55.44 kW h, while desalinating the same amount of water consumes only 0.03 kW h.95 Capital expenditure for a 50 ton per day H2 plant is ∼$460 per kW installed, with 26% for balance of plant costs (Fig. 23b). SWRO capital costs depend on technology, location, and scale, but only contribute ∼3% of the total capital cost. Operational costs (Fig. 23c) show PEM's electricity costs dominate (∼95%), whereas SWRO accounts for ∼ 0.2%, assuming electricity at $0.05 kW per h. Including SWRO raises H2 production costs slightly, from $3.81 per kg to $3.83 per kg (Fig. 23d). These analyses indicate that SWRO's energy, capital, and operational costs are minimal compared with PEM electrolysis, making SWRO integration a low-cost solution for hydrogen production. Therefore, investing in SWRO–PEM systems offers a practical and deployable near-term pathway, compared with the uncertainties of direct seawater electrolysis.97
 | ||
| Fig. 23 Breakdown of the (a) daily energy requirement, (b) direct capital costs, (c) operating costs and (d) levelized cost of hydrogen for a SWRO-PEM electrolysis plant operating at 50 tons H2 per day capacity.97 | ||
7 Summary and prospect
Efficient hydrogen production from seawater faces significant challenges, primarily requiring the development of high-performance electrocatalysts made from abundant, cost-effective materials to replace precious metals. Such alternatives can reduce energy barriers, lower overpotentials, and enhance reaction rates, thereby improving seawater electrolysis efficiency. However, most reported catalysts operate at low current densities (1–100 mA cm−2), falling short of industrial demands. This review discusses HER and OER mechanisms, offering strategies for designing HER, OER, and bifunctional electrocatalysts. Key performance parameters—overpotential, Tafel slope, turnover frequency, Faraday efficiency, and stability—are also examined. Additionally, it highlights recent catalysts, including transition metal compounds and alloys, capable of sustaining industrial-grade current densities (500–1000 mA cm−2) with excellent durability. Design strategies for scalable, durable, and stable catalysts are emphasized to advance industrial seawater electrolysis.Significant progress has been made in developing electrocatalysts for industrial-grade seawater electrolysis, but challenges remain that require further systematic research. Impurities in natural seawater, including dissolved cations and microorganisms, necessitate electrocatalysts with selectively permeable layers and added electrolytes to stabilize pH at the cathode. Additionally, chloride ions interfere with the OER at the anode, requiring highly selective catalysts to suppress Cl− interference and prioritize oxygen evolution for industrial-scale hydrogen production. Excessive bubble generation compromises long-term stability, highlighting the need for binder-free, self-supporting electrocatalysts to enhance durability. Optimizing superhydrophilic or superhydrophobic properties is crucial for efficient proton transport and bubble detachment, improving gas evolution stability.
Seawater electrolysis catalysts have been extensively studied in laboratory-scale electrolyzers, but their application in seawater electrolysis devices remains limited, necessitating research on optimizing electrolyzer configurations. Proton exchange membrane and anion exchange membrane water electrolyzers offer advantages over conventional alkaline electrolyzers, such as compact designs, low ionic resistance, and reduced energy costs. PEMWE operates under acidic conditions with mature technology for commercial-scale use, but developing durable, Ir-free OER catalysts for industrial-grade current densities remains challenging. AEMWE, suited for alkaline environments, offers a broader range of OER electrocatalysts but requires improvements in membrane stability, support structure, and bubble management for long-term operation. Integrating electrolyzers with green energy systems is also recommended to lower hydrogen production costs.
Techno-economic analysis is vital for evaluating the feasibility and scalability of hydrogen production via water electrolysis at industrial-grade current densities. Advances in catalyst development and device design have reduced energy consumption and costs, but challenges persist, including high electrolyzer capital costs, energy losses from overpotentials, and the need for durable, efficient materials under high-current operations. Operational costs, such as energy input, water desalination, and maintenance, further add to expenses, emphasizing cost optimization as a priority. Future TEA should focus on integrating renewable energy to lower costs and enhance sustainability. Innovations in low-cost, Earth-abundant catalysts, along with improved membranes and electrode designs, are crucial for boosting efficiency and reducing degradation. Scaling from lab prototypes to industrial systems requires balancing efficiency, durability, and economic viability. Addressing these challenges can establish electrolysis as a key technology in the global energy transition.
Data availability
The data supporting the findings of this study are included within the article and its ESI. Any additional information or datasets used or analyzed during this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China [Grant No. 52122702] and Natural Science Foundation of Heilongjiang Province of China [No. JQ2021E005], China Postdoctoral Science Foundation Funded Project [No. 2021M690842], Fundamental Research Foundation for Universities of Heilongjiang Province [2020-KYYWF-0338].References
- H. Yang, M. Driess and P. W. Menezes, Adv. Energy Mater., 2021, 11, 2102074 CrossRef CAS
.
- L. Yang, L. Zhou, M. Wang, Y. Qi, D. Guo, H. Li, X. Chen and S. Wang, Adv. Mater. Interfaces, 2022, 9, 2201486 CrossRef CAS
- H. Hu, X. Wang, J. P. Attfield and M. Yang, Chem. Soc. Rev., 2024, 53, 163–203 RSC
- Z. Wang, W. Xu, X. Chen, Y. Peng, Y. Song, C. Lv, H. Liu, J. Sun, D. Yuan and X. Li, Adv. Funct. Mater., 2019, 29, 1902875 CrossRef
- F. Yang, Y. Luo, Q. Yu, Z. Zhang, S. Zhang, Z. Liu, W. Ren, H. M. Cheng, J. Li and B. Liu, Adv. Funct. Mater., 2021, 31, 2010367 CrossRef CAS
- C. Yang, K. Dong, L. Zhang, X. He, J. Chen, S. Sun, M. Yue, H. Zhang, M. Zhang and D. Zheng, Inorg. Chem., 2023, 62, 7976–7981 CrossRef CAS PubMed
- X. Kang, F. Yang, Z. Zhang, H. Liu, S. Ge, S. Hu, S. Li, Y. Luo, Q. Yu and Z. Liu, Nat. Commun., 2023, 14, 3607 CrossRef CAS PubMed
- H. Liu, R. Xie, Y. Luo, Z. Cui, Q. Yu, Z. Gao, Z. Zhang, F. Yang, X. Kang and S. Ge, Nat. Commun., 2022, 13, 6382 CrossRef CAS PubMed
- Q. Wang, Y. Cheng, H. B. Tao, Y. Liu, X. Ma, D. S. Li, H. B. Yang and B. Liu, Angew. Chem., 2023, 135, e202216645 CrossRef
- S. Li, E. Li, X. An, X. Hao, Z. Jiang and G. Guan, Nanoscale, 2021, 13, 12788–12817 RSC
- S. Wang, A. Lu and C. J. Zhong, Nano Convergence, 2021, 8, 4 CrossRef CAS PubMed
- Z. P. Wu, X. F. Lu, S. Q. Zang and X. W. Lou, Adv. Funct. Mater., 2020, 30, 1910274 CrossRef CAS
- X. Lyu and A. Serov, Ind. Chem. Mater., 2023, 1, 475–485 RSC
- H. Jin, X. Wang, C. Tang, A. Vasileff, L. Li, A. Slattery and S. Z. Qiao, Adv. Mater., 2021, 33, 2007508 CrossRef CAS PubMed
- M. Auinger, I. Katsounaros, J. C. Meier, S. O. Klemm, P. U. Biedermann, A. A. Topalov, M. Rohwerder and K. J. J. Mayrhofer, Phys. Chem. Chem. Phys., 2011, 13, 16384–16394 RSC
- L. Wu, F. Zhang, S. Song, M. Ning, Q. Zhu, J. Zhou, G. Gao, Z. Chen, Q. Zhou and X. Xing, Adv. Mater., 2022, 34, 2201774 CrossRef CAS PubMed
- X. Xiao, L. Yang, W. Sun, Y. Chen, H. Yu, K. Li, B. Jia, L. Zhang and T. Ma, Small, 2022, 18, 2105830 CrossRef CAS PubMed
- J. G. Vos, Z. Liu, F. D. Speck, N. Perini, W. Fu, S. Cherevko and M. T. M. Koper, ACS Catal., 2019, 9, 8561–8574 CrossRef CAS
- W. Tong, M. Forster, F. Dionigi, S. Dresp, R. Sadeghi Erami, P. Strasser, A. J. Cowan and P. Farràs, Nat. Energy, 2020, 5, 367–377 CrossRef CAS
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed
- Y. Luo, Z. Zhang, F. Yang, J. Li, Z. Liu, W. Ren, S. Zhang and B. Liu, Energy Environ. Sci., 2021, 14, 4610–4619 RSC
- X.-Y. Zhang, Y.-R. Zhu, Y. Chen, S.-Y. Dou, X.-Y. Chen, B. Dong, B.-Y. Guo, D.-P. Liu, C.-G. Liu and Y.-M. Chai, Chem. Eng. J., 2020, 399, 125831 CrossRef CAS
- L. Zhang, Z. Shi, Y. Lin, F. Chong and Y. Qi, Front. Chem., 2022, 10, 866415 CrossRef CAS PubMed
- D. Liu, Y. Cai, X. Wang, Y. Zhuo, X. Sui, H. Pan and Z. Wang, Energy Environ. Sci., 2024, 17, 6897–6942 RSC
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 CrossRef CAS PubMed
- K. Jiang, W. Liu, W. Lai, M. Wang, Q. Li, Z. Wang, J. Yuan, Y. Deng, J. Bao and H. Ji, Inorg. Chem., 2021, 60, 17371–17378 CrossRef CAS PubMed
- L. Xiu, W. Pei, S. Zhou, Z. Wang, P. Yang, J. Zhao and J. Qiu, Adv. Funct. Mater., 2020, 30, 1910028 CrossRef CAS
- H. Jin, J. Xu, H. Liu, H. Shen, H. Yu, M. Jaroniec, Y. Zheng and S.-Z. Qiao, Sci. Adv., 2023, 9, eadi7755 CrossRef CAS PubMed
- X. Yu, Z.-Y. Yu, X.-L. Zhang, Y.-R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M.-R. Gao and G. Wang, J. Am. Chem. Soc., 2019, 141, 7537–7543 CrossRef CAS PubMed
- L. A. King, M. A. Hubert, C. Capuano, J. Manco, N. Danilovic, E. Valle, T. R. Hellstern, K. Ayers and T. F. Jaramillo, Nat. Nanotechnol., 2019, 14, 1071–1074 CrossRef CAS PubMed
- Y. Luo, Z. Zhang, M. Chhowalla and B. Liu, Adv. Mater., 2022, 34, 2108133 CrossRef CAS PubMed
- W. Xu, G. Fan, S. Zhu, Y. Liang, Z. Cui, Z. Li, H. Jiang, S. Wu and F. Cheng, Adv. Funct. Mater., 2021, 31, 2107333 CrossRef CAS
- Z. Yan, H. Sun, X. Chen, H. Liu, Y. Zhao, H. Li, W. Xie, F. Cheng and J. Chen, Nat. Commun., 2018, 9, 2373 CrossRef PubMed
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef CAS PubMed
- R. Rossi, D. M. Hall, L. Shi, N. R. Cross, C. A. Gorski, M. A. Hickner and B. E. Logan, Energy Environ. Sci., 2021, 14, 6041–6049 RSC
- L. Li, G. Zhang, B. Wang, D. Zhu, D. Liu, Y. Liu and S. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 37152–37161 CrossRef CAS PubMed
- C. Liang, P. Zou, A. Nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC
- T. Haq and Y. Haik, ACS Sustainable Chem. Eng., 2022, 10, 6622–6632 CrossRef CAS
- H. Ge, G. Li, J. Shen, W. Ma, X. Meng and L. Xu, Appl. Catal., B, 2020, 275, 119104 CrossRef CAS
- L. Wang, Y. Li, X. Yin, Y. Wang, A. Song, Z. Ma, X. Qin and G. Shao, ACS Sustainable Chem. Eng., 2017, 5, 7993–8003 CrossRef CAS
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed
- F. Zhang, Y. Liu, L. Wu, M. Ning, S. Song, X. Xiao, V. G. Hadjiev, D. E. Fan, D. Wang and L. Yu, Mater. Today Phys., 2022, 27, 100841 CrossRef CAS
- Y. S. Park, J. Lee, M. J. Jang, J. Yang, J. Jeong, J. Park, Y. Kim, M. H. Seo, Z. Chen and S. M. Choi, J. Mater. Chem. A, 2021, 9, 9586–9592 RSC
- H. Komiya, T. Shinagawa and K. Takanabe, ChemSusChem, 2022, 15, e202201088 CrossRef CAS PubMed
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS
- C. Wang, X. Shao, J. Pan, J. Hu and X. Xu, Appl. Catal., B, 2020, 268, 118435 CrossRef CAS
- L. Yu, L. Wu, B. McElhenny, S. Song, D. Luo, F. Zhang, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2020, 13, 3439–3446 RSC
- M. Yu, J. Li, F. Liu, J. Liu, W. Xu, H. Hu, X. Chen, W. Wang and F. Cheng, J. Energy Chem., 2022, 72, 361–369 CrossRef CAS
- Y. Kuang, M. J. Kenney, Y. Meng, W.-H. Hung, Y. Liu, J. E. Huang, R. Prasanna, P. Li, Y. Li, L. Wang, M.-C. Lin, M. D. McGehee, X. Sun and H. Dai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 6624–6629 CrossRef CAS PubMed
- L. Qi, A. Li, M. Wang, Y. Zhang, K. Zhang and X. Li, Adv. Mater. Interfaces, 2022, 9, 2101720 CrossRef CAS
- J. Bian, Z. Song, X. Li, Y. Zhang and C. Cheng, Nanoscale, 2020, 12, 8443–8452 RSC
- C. Chen, Y. Tuo, Q. Lu, H. Lu, S. Zhang, Y. Zhou, J. Zhang, Z. Liu, Z. Kang, X. Feng and D. Chen, Appl. Catal., B, 2021, 287, 119953 CrossRef CAS
- Z. Lei, X. Jin, J. Li, Y. Liu, J. Liu, S. Jiao and R. Cao, J. Energy Chem., 2022, 65, 505–513 CrossRef CAS
- L. Li, G. Zhang, B. Wang, D. Zhu, D. Liu, Y. Liu and S. Yang, ACS Appl. Mater. Interfaces, 2021, 13, 37152–37161 CrossRef CAS PubMed
- T. U. Haq and Y. Haik, ACS Sustainable Chem. Eng., 2022, 10, 6622–6632 CrossRef CAS
- F. Zhang, Y. Liu, L. Wu, M. Ning, S. Song, X. Xiao, V. G. Hadjiev, D. E. Fan, D. Wang, L. Yu, S. Chen and Z. Ren, Mater. Today Phys., 2022, 27, 100841 CrossRef CAS
- Y. S. Park, J. Lee, M. J. Jang, J. Yang, J. Jeong, J. Park, Y. Kim, M. H. Seo, Z. Chen and S. M. Choi, J. Mater. Chem. A, 2021, 9, 9586–9592 RSC
- H. Komiya, T. Shinagawa and K. Takanabe, ChemSusChem, 2022, 15, e202201088 CrossRef CAS PubMed
- C. Wang, M. Zhu, Z. Cao, P. Zhu, Y. Cao, X. Xu, C. Xu and Z. Yin, Appl. Catal., B, 2021, 291, 120071 CrossRef CAS
- L. Guo, J. Chi, J. Zhu, T. Cui, J. Lai and L. Wang, Appl. Catal., B, 2023, 320, 121977 CrossRef CAS
- N. Q. Tran, B. T. N. Le, T. N.-M. Le, L. T. Duy, T. B. Phan, Y. Hong, T.-K. Truong, T. L. H. Doan, J. Yu and H. Lee, J. Phys. Chem. Lett., 2022, 13, 8192–8199 CrossRef CAS PubMed
- F. Yang, Y. Luo, Q. Yu, Z. Zhang, S. Zhang, Z. Liu, W. Ren, H.-M. Cheng, J. Li and B. Liu, Adv. Funct. Mater., 2021, 31, 2010367 CrossRef CAS
- C. Chai, J. Yang, C. Jiang, L. Liu and J. Xi, ACS Appl. Energy Mater., 2022, 5, 2909–2917 CrossRef CAS
- C. Wei, Y. Sun, G. G. Scherer, A. C. Fisher, M. Sherburne, J. W. Ager and Z. J. Xu, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS PubMed
- G. Li, S. Feng, J. Li, P. Deng, X. Tian, C. Wang and Y. Hua, Chin. J. Struct. Chem., 2022, 41, 2207068–2207073 CAS
- Z. Yu, J. Xu, L. Meng and L. Liu, J. Mater. Chem. A, 2021, 9, 22248–22253 RSC
- F. Dionigi, T. Reier, Z. Pawolek, M. Gliech and P. Strasser, ChemSusChem, 2016, 9, 962–972 CrossRef CAS PubMed
- C. Li, W. Hong, Q. Cai and C. Jian, ACS Appl. Mater. Interfaces, 2022, 14, 30683–30691 CrossRef CAS
- H. Lv, C. Fu, J. Fan, Y. Zhang and W. Hao, J. Colloid Interface Sci., 2022, 626, 384–394 CrossRef CAS PubMed
- Z. Wang, W. Xu, K. Yu, Y. Feng and Z. Zhu, Nanoscale, 2020, 12, 6176–6187 RSC
- X. Du, M. Tan, T. Wei, H. Kobayashi, J. Song, Z. Peng, H. Zhu, Z. Jin, R. Li and W. Liu, Chem. Eng. J., 2023, 452, 139404 CrossRef CAS
- J. Peng, Y. Li, Z. Chen, G. Liang, S. Hu, T. Zhou, F. Zheng, Q. Pan, H. Wang, Q. Li, J. Liu and Z. Guo, ACS Nano, 2021, 15, 11607–11618 CrossRef CAS PubMed
- C. Fu, S. Weng, J. Fan, Y. Zhang, Y. Guo and W. Hao, Chem. Eng. J., 2022, 430, 132881 CrossRef CAS
- W. Xu, G. Fan, S. Zhu, Y. Liang, Z. Cui, Z. Li, H. Jiang, S. Wu and F. Cheng, Adv. Funct. Mater., 2021, 31, 2107333 CrossRef CAS
- D. Wu, D. Chen, J. Zhu and S. Mu, Small, 2021, 17, 2102777 CrossRef CAS PubMed
- D. Kong, C. Meng, Y. Wang, X. Chen, J. Zhang, L. Zhao, J. Ji, L. Zhang and Y. Zhou, Appl. Catal., B, 2024, 343, 123578 CrossRef CAS
- J. Guo, Y. Zheng, Z. Hu, C. Zheng, J. Mao, K. Du, M. Jaroniec, S.-Z. Qiao and T. Ling, Nat. Energy, 2023, 8, 264–272 CAS
- J. Chang, G. Wang, Z. Yang, B. Li, Q. Wang, R. Kuliiev, N. Orlovskaya, M. Gu, Y. Du, G. Wang and Y. Yang, Adv. Mater., 2021, 33, 2101425 CrossRef CAS PubMed
- Z. Chen, Q. Li, H. Xiang, Y. Wang, P. Yang, C. Dai, H. Zhang, W. Xiao, Z. Wu and L. Wang, Inorg. Chem. Front., 2023, 10, 1493–1500 RSC
- D. Yao, W. Hao, S. Weng, M. Hou, W. Cen, G. Li, Z. Chen and Y. Li, Small, 2022, 18, 2106868 CrossRef CAS PubMed
- Z. Yu, Y. Li, V. Martin-Diaconescu, L. Simonelli, J. Ruiz Esquius, I. Amorim, A. Araujo, L. Meng, J. L. Faria and L. Liu, Adv. Funct. Mater., 2022, 32, 2206138 CrossRef CAS
- M. Ning, F. Zhang, L. Wu, X. Xing, D. Wang, S. Song, Q. Zhou, L. Yu, J. Bao, S. Chen and Z. Ren, Energy Environ. Sci., 2022, 15, 3945–3957 RSC
- B. Wang, M. Lu, D. Chen, Q. Zhang, W. Wang, Y. Kang, Z. Fang, G. Pang and S. Feng, J. Mater. Chem. A, 2021, 9, 13562–13569 RSC
- T. Van Tam, K. C. Bhamu, M. J. Kim, S. G. Kang, J. S. Chung, S. H. Hur and W. M. Choi, Chem. Eng. J., 2024, 480, 148190 CrossRef CAS
- H. Fei, R. Liu, T. Liu, M. Ju, J. Lei, Z. Wang, S. Wang, Y. Zhang, W. Chen, Z. Wu, M. Ni and J. Wang, Adv. Mater., 2024, 36, 2309211 CrossRef CAS PubMed
- J. E. Park, S. Y. Kang, S.-H. Oh, J. K. Kim, M. S. Lim, C.-Y. Ahn, Y.-H. Cho and Y.-E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS
- L. Wan, Z. Xu, Q. Xu, M. Pang, D. Lin, J. Liu and B. Wang, Energy Environ. Sci., 2023, 16, 1384–1430 RSC
- G. Zang, E. J. Graham and D. Mallapragada, Int. J. Hydrogen Energy, 2024, 49, 1288–1303 CrossRef CAS
- R. R. Beswick, A. M. Oliveira and Y. Yan, ACS Energy Lett., 2021, 6, 3167–3169 CrossRef CAS
- D. J. Lampert, H. Cai and A. Elgowainy, Energy Environ. Sci., 2016, 9, 787–802 RSC
- A. T. Mayyas, M. F. Ruth, B. S. Pivovar, G. Bender and K. B. Wipke, Manufacturing Cost Analysis for Proton Exchange Membrane Water Electrolyzers, National Renewable Energy Laboratory (NREL), Golden, CO (United States), United States, 2019.
- B. Lee, L. Wang, Z. Wang, N. J. Cooper and M. Elimelech, Energy Environ. Sci., 2023, 16, 714–722 RSC
- H. Xie, Z. Zhao, T. Liu, Y. Wu, C. Lan, W. Jiang, L. Zhu, Y. Wang, D. Yang and Z. Shao, Nature, 2022, 612, 673–678 CrossRef CAS PubMed
- N. Ghaffour, S. Lattemann, T. Missimer, K. C. Ng, S. Sinha and G. Amy, Appl. Energy, 2014, 136, 1155–1165 CrossRef CAS
- N. Ghaffour, T. M. Missimer and G. L. Amy, Desalination, 2013, 309, 197–207 CrossRef CAS
- M. Khan, T. Al-Attas, S. Roy, M. M. Rahman, N. Ghaffour, V. Thangadurai, S. Larter, J. Hu, P. M. Ajayan and M. G. Kibria, Energy Environ. Sci., 2021, 14, 4831–4839 RSC
| This journal is © The Royal Society of Chemistry 2025 |