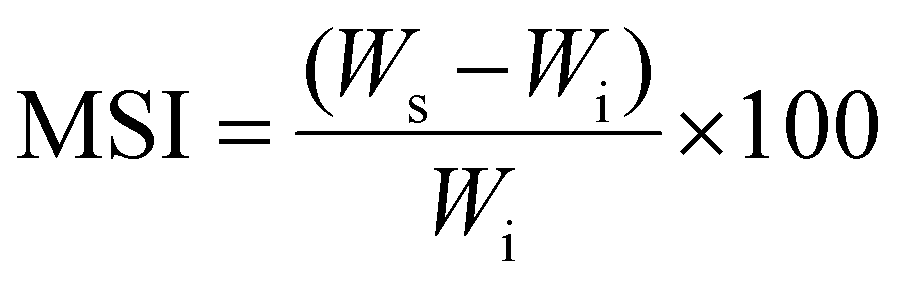DOI:
10.1039/D5NR00079C
(Paper)
Nanoscale, 2025,
17, 8544-8562
Poly[(N-acryloyl glycine)-co-(acrylamide)]-induced cell growth inhibition in heparanase-driven malignancies†
Received
8th January 2025
, Accepted 17th February 2025
First published on 21st February 2025
Abstract
In the present work, glycine, the monomer N-acryloylglycine (NAG), and polymeric units of poly[(N-acryloylglycine)-co-(acrylamide)] p(NAG-co-Ac) are examined using density functional theory (DFT), and experimental evidence is provided for their use in the therapy of cancer with a poor prognosis. Glycine plays a pivotal role in cell survival, and most anti-cancer agents alter glycine metabolomics and suppress cancer cell proliferation. Herein, we have utilized Frontier Molecular Orbital theory (FMO), and the results revealed that the introduction of acrylamide/divinyl benzene into the glycine-based polymer increased its biological activity by lowering the energy band gap. Heparanase and proteases are important in invasive tumor progression and worsening of prognosis. In this context, we have synthesized co-polymeric p(NAG-co-Ac) and revealed its protease inhibitory activities. It is revealed that the cross-linked homo-polymeric and cross-linked hetero-polymeric tetrameric arrangements inhibit heparanase activity via interacting at heparanase binding domain II (HBDII) with a docking score of ∼−11.08 kcal mol−1 (Ki) and at heparanase binding domain III (HBD III). The bathochromically shifted CD spectrum shows that the hydrogel interacts with heparanase and disturbs the secondary protein structure of the synthesized p(NAG-co-Ac) polymer. It is found that the synthesized p(NAG-co-Ac) hydrogel has anti-proliferative activity, acts as a migratory inhibitor of cancer cells, and favors programmed cell death. Further, the p(NAG-co-Ac) hydrogel exhibited anti-angiogenic behavior. In conclusion, p(NAG-co-Ac), with its anti-angiogenic and anti-tumorigenic capabilities, has a future as a potential anticancer polymer for the treatment of heparanase-driven invasive malignancies without using any additional anticancer drugs, and is promising for cancer treatment.
1. Introduction
Due to the aggressive phenotypes, vast heterogeneity, and unique pathophysiology of solid tumours, as well as the emergence of resistance and negative receptor expression, invasive and metastatic cancer are the second leading cause of death.1,2 Irradiation alters the invasion and migration of cancer stem cells, and thus local recurrence and metastases remain a therapeutic challenge.3 Chemo-resistance and radiation resistance contribute to treatment failure.4 Further, it was recently noted that heparanase is a signal protein that functions as an enzyme as well as in a non-enzymatic way; it is involved in chemo-resistance via its non-enzymatic function by accelerating angiogenesis.5–7 Proteases enzymatically cleave peptide bonds and generate six major moieties: cysteine, serine, threonine, glutamate, aspartate, proteases, and matrix metalloproteases. Matrix metalloproteases and urokinase plasminogen activators stimulate the tumour surface, rupture the basement membrane, and promote blood vessel sprouting.8 Therefore, protease-inhibitor-based anticancer therapy is considered to be an exciting option in cancer therapy. Another hallmark of aggressive cancers is the overexpression of heparanase, which is an endo-β-D-glucuronidase enzyme that cleaves the constituents of the fundamental extracellular matrix (ECM), i.e., heparan sulphate (HS) proteoglycan, releasing heparan sulphate and affecting the ECM integrity. Thus, in addition to its other activities, heparanase facilitates metastasis, neo-angiogenesis and cancer stem cell migration. Furthermore, heparanase has pleiotropic effects in tumour cells and is widely overexpressed in glioblastoma,9 TNBC,10 hepatocellular carcinoma, oral cancer11 and melanoma,12 which are aggressive types of cancer with poor prognoses and demonstrate poor outcomes with low survival rates. Heparanase-mediated cell invasion is enhanced in hypoxic conditions, along with induced angiogenesis and metastasis. It also regulates the expression CD24 (a surface biomarker of tumour stem cells), as the overexpression of heparanase consistently up-regulates CD24 and leads to significantly shortened survival, and heparanase-induced high-grade and low-grade glioma have been reported.13 Recently, the heparanase inhibitor CV122 was developed to manage sepsis and cytokine storm, but its use in cancer therapy is not known.14 Veider et al. developed a heparanase-responsive charge-converting nanocarrier for drug delivery and to overcome the intestinal barrier and achieve cellular uptake.15 Hence, the design of effective heparanase inhibitors and treatment using heparanase inhibitors are considered to be future potential targets for cancer therapy. To the best of our knowledge, to date, no heparanase inhibitors have been clinically approved; hence there is great scope to improve therapeutic efficiency using the amino-acid-based polymers reported in this work.
In cancer therapy, biopolymers, with their variable structures, provide advantages such as improved bioavailability, stability and target specificity, and co-delivery of multiple therapeutic agents.16 Conventional chemo-therapy drugs are inadequate due to limitations including low solubility in media, high toxicity, multidrug resistance, restricted bioavailability, inappropriate bio-distribution or pharmacokinetics, etc.17 Among the various synthetic polymeric systems, amino-acid-based polymeric systems are considered to be future green materials for therapeutic applications. In poly(amino acid) polymers, the β-sheet crystal structure formation results in a stabilized structure and the side chain cationic pendent structures facilitate the development of reactive oxygen species (ROS) to achieve significant in vitro/in vivo tumour inhibition and antimicrobial activity.18 The use of conventional ‘drug-free macromolecules’ for ‘anticancer activity’ is an interesting concept in cancer therapy. Polymer–peptide conjugates, polymer–oligonucleotides and glycopolymers have been used as drug-free macromolecules.17 Amino-acid-based polymers are used for various therapeutic purposes, and several have been reported as drug-free macromolecules having anticancer properties. For example, dual-acid-responsive polymeric-micelle-forming cinnamaldehyde prodrugs exerted a synergistic anticancer effect in a colon-tumour-bearing model.19 A sulphur- and methacrylate-based co-polymer synthesized via RAFT was reported as an anticancer cell membrane.20 However, to date, no polymers have been reported to have heparanase inhibitory properties and anticancer activity, and this area is still in the in silico stage.21
To design such a macromolecule, we adopted the in silico approach. The bioactivity prediction module in the software Molinspiration is a fragment-based method in which physicochemical properties, molecular features, and structural fragments are used to estimate potential biological activity against various target classes. Molinspiration uses the ZINC virtual screening database for property calculation (https://www.molinspiration.com/). Similarly, density functional theory (DFT) calculations are widely used to determine fundamental properties. Electronic band gap values are obtained from standard DFT calculations. Smaller band gaps represent higher electronic excitability, which ultimately indicates greater reactivity under physiological conditions and is related to ROS22,23 generation.24–26
Against this background, we first evaluated the biological activities of acrylate glycine and acrylate glycine polymers into which acrylamide units were introduced using the software Molinspiration. DFT calculations were then performed, and the electronic and solvation parameters for the monomeric glycine unit and different acrylate glycine polymers incorporating acrylamide and the cross-linker (divinylbenzene) were investigated by applying the B3LYP/6-311g* level of theory. In order to determine the extent of reactivity and anticancer properties of the polymeric system, the energy band gap (HOMO/LUMO), chemical hardness and electrophilicity were calculated. Furthermore, molecular docking, enzyme inhibitory assay, and CD spectroscopy were performed to investigate the heparanase inhibitory activity of the acrylate co-polymer. In vitro cell viability/migratory inhibitory assessment, flow cytometry analysis, and in ovo anti-angiogenic assessments were implemented to investigate the anticancer potential of this polymer for synergistic applications. Based on an extensive literature review, it was observed that only a few chemical moieties are known to exhibit heparanase inhibition, while no polymers or hydrogels are known to have heparanase inhibition activity. This is the first such work; herein, we have evaluated and compared the heparanase inhibition activity of glycine, glycine-acrylate monomers and their polymers. The dual effects of the polymers, i.e., protease inhibition and heparanase inhibition activity, result in a novel polymeric hydrogel for specific inhibition of heparanase-driven malignancies/aggressive cancer.
2. Experimental
2.1. Computational methodology
For the evaluation of physicochemical properties and bioactivity, random arrangements were designed using ChemDraw Ultra 8.0 software and categorized as: (1) linear homopolymers (polymers of N-acryloylglycine (G), denoted as G, G2…Gn), (2) linear heteropolymers (polymer of G and acrylamide (A), GA (GA1), G–A–G…(GAn)), (3) cross-linked homopolymers (polymers of G cross-linked by divinylbenzene (DVB), D1, D2…D6) and (4) arranged cross-linked heteropolymers (polymers of G and A cross-linked by DVB, also included in the series labelled as D1, D2…Dn); the cross-linked polymer that was finally synthesized is denoted as p(NAG-co-Ac) hydrogel. The bioactivities of all structures were first evaluated using the software Molinspration (https://www.molinspiration.com). The predicted structures were determined using the software Avogadro (version 1.2.0)27 and then optimized using the Merck molecular force field (MMFF 94) at the molecular mechanics level. DFT calculations were then carried out using the ORCA 5.3.2 computational package.28 The computing efficiency was accelerated by using the resolution of identity approximation with the basis set B3LYP/6-311g*/def2-SVP level. Further, the CPCM continuum solvation model was applied using the water, ethanol, hexane and chloroform level (ε = 80.4).
2.2. Molecular docking study
The crystal structure of the human heparanase complex with the inhibitor CB678 (PDB ID 7PRT) was retrieved from the RCSB protein data bank (https://www.rcsb.org/structure/1Q5K) having a resolution of 1.70 Å with the 100 steepest descent steps. The macromolecular structure preparation and energy minimization were conducted on UCSF Chimera by applying AMBER ff14SB and polar hydrogen atoms, and Kollman charges were added. Reference ligand molecules such as cyclophellitol, roneparstat and 2-acetamido-2-deoxy-beta-d-glucopyranose were retrieved from PubChem, and energy minimization was conducted using the PyrX interface by applying the MMff94 force field in Autodock v4.2.6 tool.29 Simulation was performed by generating the grid box, setting the genetic algorithms of 30 runs and the Lamarckian parameter. Furthermore, analysis of the data was performed using AutoDock and Discovery studio.30
2.3. Synthesis and biodegradation assessment of p(NAG-co-Ac) hydrogel
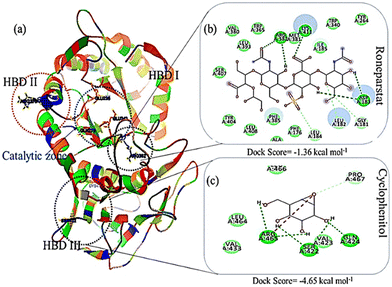
The p(NAG-co-Ac) hydrogel was synthesized; an Indian patent application has been filed for the method. The overall synthesis method involved a modified version of the free radical miniemulsion polymerization technique reported by the authors of this work and confirmed through FTIR and NMR, as previously described.31 After the synthesis and lyophilisation, the structural and surface morphology were studied using field-emission scanning electron microscopy (FESEM) (Model; Nova Nano SEM 450) with an accelerating voltage of 10 kV. The dynamic swelling behaviour and enzymatic degradation were then determined at pH 7.4 using phosphate saline buffer (PBS), 100 μg mL−1 lysozyme in PBS solution, 100 μg mL−1 proteinase K in PBS, and 100 μg mL−1 collagenase I by immersing nearly 10 mg of the p(NAG-co-Ac) hydrogel in 1 mL of the respective solutions. The swelling indexes were calculated using eqn (1).| | 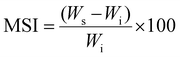 | (1) |
2.4.
In vitro cytotoxicity assessment in healthy and cancer cell lines
MTT colorimetric assay was used to investigate the effect of the synthesized p(NAG-co-Ac) material on cell viability. The MDA-MB-231, MCF7, LN229, L929, HEK 293, HepG2 and A549 cell lines were obtained from NCCS-Pune repository, India. All the cell lines were maintained in 10% fetal bovine serum (FBS) supplemented with Dulbecco's modified Eagle medium with 100 U of penicillin–streptomycin antibiotic in a humidified CO2 incubator at 37 °C. In brief, 1 × 104 cells were cultured for 24 h in a 96-well plate supplemented with complete medium followed by treatment with glycine, N-acryloyl glycine monomer or p(NAG-co-Ac) at different concentrations (from 2.5 mg mL−1 to 10 mL−1) for another 24 h. After incubation, the hydrogel was removed, and the cells were incubated with 5 μg mL−1 of MTT reagent in fresh medium for 4 h. Subsequently, the absorbance of the dissolved formazan crystals/DMSO was measured at λmax = 570 nm using a microplate reader (Bioteck).
2.5. Hemocompatibility of hydrogel
Fresh RBCs were isolated from whole blood samples obtained from Wister rats, and a homogenous suspension was obtained in 5% dextrose solution (pH 7.4). To evaluate the haemolytic impact of the p(NAG-co-Ac) hydrogel and N-acryloyl glycine monomer, the RBCs were exposed to different concentrations (1000, 500, 250, 125 and 62.5 μg mL−1) of p(NAG-co-Ac) hydrogel for 8 and 24 h at 37 °C (with 100 RPM). Treatment with 5% dextrose solution was used as a negative control, while treatment with 0.1–1% Triton-X was used as a positive control. Percent haemolysis was calculated using a spectrophotometer at the maximum absorption of λmax = 540 nm for each sample.
2.6. Wound scratch assay
The cell migration ability was analysed by performing wound scratch tests on the LN229, MDA-MB-231 and L929 cell lines. The cells were seeded in 12 well plates and cultured until an 80–90% confluent monolayer was obtained. Scratches were then made using 200 μL micropipette tips, and then cells were washed twice using PBS. Further, three different concentrations of hydrogel, monomer and glycine were added separately with untreated cells as the control, and the cells were cultured in complete medium for 48 h. Images were then acquired using an inverted microscope at hour 24 and 48 of incubation. The analytical tool ImageJ was used to estimate the percentage wound closure area at different time intervals.
2.7. Live and dead assay
MDA-MB-231 cells were incubated with glycine, NAG, or p(NAG-co-Ac) for 3 days to examine the comparative cellular death after the treatment. After 24 h and 72 h, the cells were stained with acridine orange and ethidium bromide (100 μg mL−1) for 30 min. Macroscopic fluorescence images were then acquired at 20× magnification.
2.8. Apoptosis and necrosis assay
After 24 h of treatment with glycine, NAG, or p(NAG-co-Ac) hydrogel, the percentages of viable apoptotic and necrotic cell death of LN229 cells (1 × 105 cells) were quantified using flow cytometry. After 24 h of treatment, the cells were detached using 1 mM EDTA solution and collected after washing with PBS followed by staining with Annexin V/PI as per the manufacturer's protocol (BD Biosciences FITC Annexin-V apoptosis detection kit).
2.9. Reactive oxygen species (ROS) estimation assay
Flow cytometry analysis was conducted to evaluate the intracellular ROS level. After treatment for 24 hours with glycine, NAG, or p(NAG-co-Ac) hydrogel, with H2O2 treatment as the positive control and untreated cells as the reference control, the adhered cells were collected (1200 RPM/5 min) and washed with PBS three times. The cells were then stained with a 50 μM solution of 2′,7′-dichlorofluorescin diacetate DCFDA and incubated at 37 °C for 30 min. The intracellular ROS production and peak shift were analysed using a flow cytometer (Beckman Coulter).
2.10. Chicken embryo angiogenesis (CEA) assay
The vascular sprouting inhibition potential of the N-acryloyl glycine monomer and p(NAG-co-Ac) polymer were evaluated in an in ovo system. Certified purchased (Ramana Hatchery, Varanasi, Uttar Pradesh, India) fertilized eggs were incubated in a humid environment at 37 °C for 4 days. A small circular examination window was generated, and 1.5 mL of albumen was removed for the detachment of the developing chick embryo chorioallantoic membrane. Three different concentrations (2.5 μg mL−1, 25 μg mL−1 or 100 μg mL−1) of the monomer and p(NAG-co-Ac) hydrogel were then applied via pipetting. The changes in the vasculature were examined at 0, 2, 4, and 8 h by obtaining images using a stereo-zoom-microscope-mounted Magnus camera (Magcam DC plus 10, Magnus Opto Systems India Pvt. Ltd, New Delhi, India). Images were analysed using the Image J angio tool software.
2.11. Fondaparinux heparanase inhibitory assay
To determine the heparanase inhibitory activity of glycine, N-acryloylglycine and p(NAG-b-Ac) hydrogel, a colorimetric fondaparinux heparanase inhibitory assay was performed using heparin as a positive control for heparanase inhibition. First, 100 μL of the reaction mixture was placed on a 4% BSA in TBST pre-treated plate. The 100 μL reaction mixtures consisting of 200 ng mL−1 of heparanase and 100 μM of fondaparinux sodium in 40 mM sodium acetate (pH 5) with/without inhibitor were incubated for 18 h at 37 °C. Subsequently, the reaction was quenched by incubation with 100 μL of 0.1 M NaOH with 30 μM of resazurin at 45 °C for one hour. The change in the fluorescence was recorded with λ = 560 nm excitation at λmax = 590 nm emission.
2.12. CD spectroscopy
Circular dichroism spectra for heparanase, N-acryloyl glycine, p(NAG-b-Ac) and heparin interaction with/without were obtained using a 1 mm quartz cuvette and CD polarimeter J-1500 (make JASCO). 1000 μl of reaction solution contained 750 ng mL−1 of heparanase (prepared in 20 mM sodium acetate of pH 5) and 200 mM NaCl containing buffer with or without the addition of 250 ng mL−1 of N-acryloyl glycine, p(NAG-b-Ac) and heparin. Samples were scanned at 25 °C over the range of 190–250 nm using a bandwidth of 1 nm with a scanning speed of 50 nm min−1. Spectra were recorded in triplicate, and the blank buffer spectrum was subtracted from the results.
2.13. Statistical analysis
Herein, all data are presented as the mean (±) standard deviation (SD). One-way ANOVA with Turkey's test was used to evaluate the statistical significance of the comparison groups by considering p < 0.05 to be a significant difference. All analysis was performed using the software Origin.
3. Results
3.1. Theoretical bioactivity assessment of p(NAG-co-Ac)/bioactivity assessment of p(NAG-co-Ac) through DFT calculation
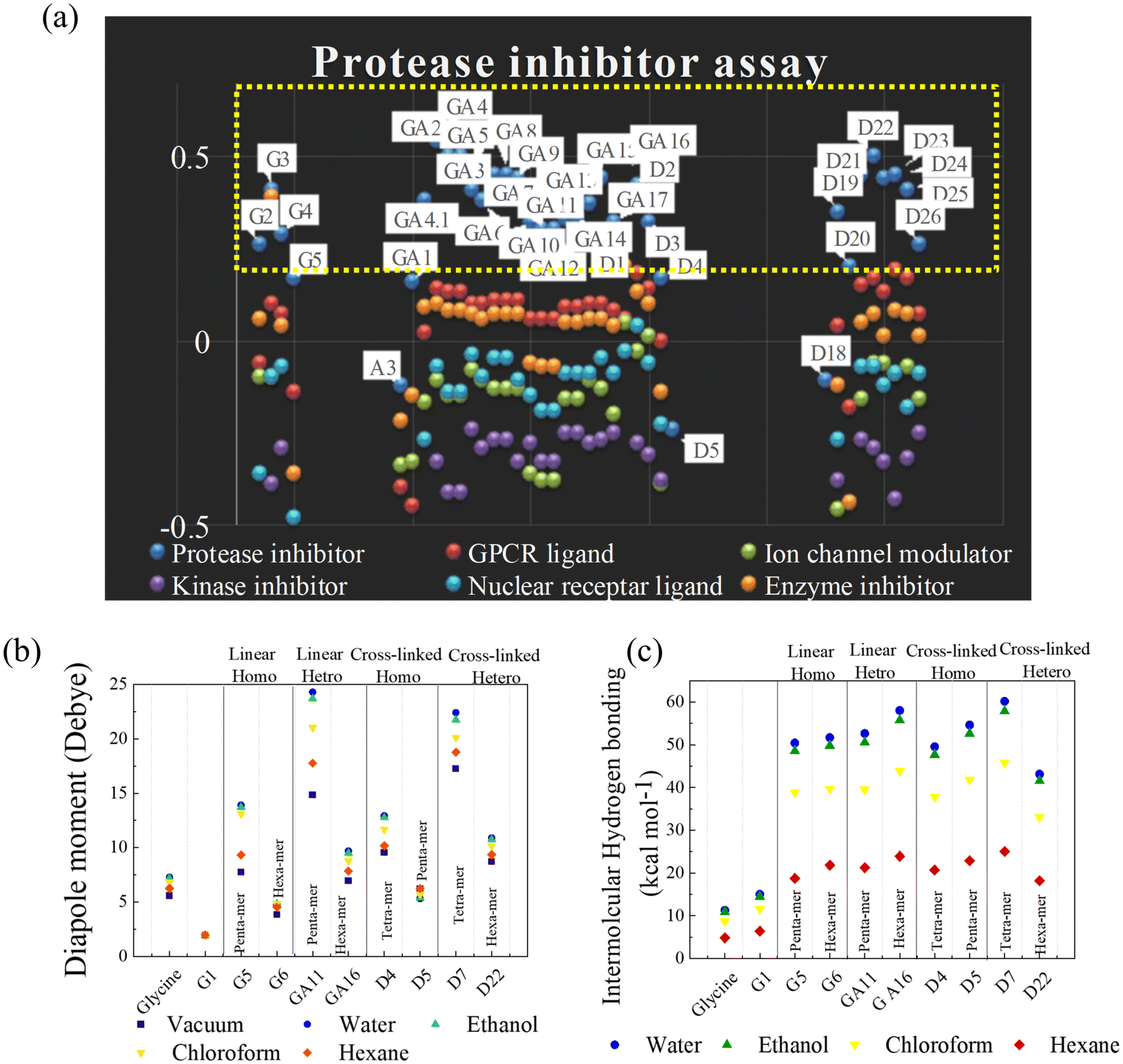
Synthesized polymers have wide applications in various medical domains. Any molecule that interacts with the native protein gives therapeutic effects. Therefore, to predict the biological activity of p(NAG-co-Ac), before synthesizing the polymer, a bioactivity score calculation was conducted using Molinspiration software, which employs a fragment-based method, and the obtained values are listed in Table S1.† Molecules with activity scores of more than 0.00 were considered to be bioactive. Those with scores from 0.50 to 0.00 were considered to be moderately active, and those with more than −0.50 were presumed to be inactive molecules. The 58 designed molecules were categorized into four groups: (1) linear homopolymers (LH) (G1 to G11), (2) linear heteropolymers (LE) (GA series), (3) cross-linked homopolymers (CH) (D1 to D6) and (4) cross-linked heteropolymers (CE) (D7 to D25). All these polymeric structures showed negative scores as kinase inhibitors, ion channel modulators and nuclear receptor ligands, and 55% of the polymeric structures showed positive scores for GPCR ligands (lower) and protease inhibitors (higher) (see Table S1†). Among the random structures, most of the compounds acted as potent protease inhibitors with the highest scores for the LH, CH, and CE groups; specifically, GA2, GA3, GA4 and D22 had the highest scores of 0.5 or more (see Fig. 1a).
 |
| | Fig. 1 Bioactivity prediction through DFT calculations. (a) Graphical representation of bioactivity scores of random arrangements of N-acryloyl glycine, acrylamide-containing and cross-linked structures, categorized as (1) linear homopolymer (LH) (G1 to G11), (2) linear heteropolymer (LE) (GA series), (3) cross-linked homopolymer (CH) (D1 to D6), and (4) cross-linked heteropolymer (CE) (D7 to D25). Bioactivity measured in terms of molecules with activity as protease inhibitors (blue), GPCR ligands (red), ion channel modulators (green), kinase inhibitors (violet), nuclear receptor ligands (sky blue), and enzyme inhibitors (orange). DFT calculations and graphical representations of dipole moment (b) and intermolecular hydrogen bonding (c) of glycine, N-acryloyl glycine, and LH, LE, CH, and CE random structures in polar solvents water, ethanol and the non-polar solvents chloroform and hexane. | |
3.2. DFT calculations for randomized structures of p(NAG-co-Ac) polymeric units
DFT offers a high level of accuracy with reduced time-consuming empirical processes for designing biologically active materials. It is a less computationally expensive approach than the ‘ab initio’ approach, which provides information about the geometric and electronic characteristics and subsequently gives an idea of the selected polymer as a drug delivery system.24 Herein, DFT was performed to calculate the solvation parameter and the energy band. The dipole moment and intermolecular/intermolecular hydrogen bonding in the presence of different solvents provide a correlation with the stability, polarity, reactivity and swelling behaviour of the polymers.32 We hypothesized that a reduction of the band gap might be observed with the acrylation of glycine and polymerization of acrylate glycine. A narrow band gap results in higher electronic excitability,25 promoting the generation of selected on the basis of torsion angle to allow flexibility for the simulation. Torsion angle directly impacts the dimensionality of the search space in the docking algorithm. A higher number of torsional degrees of freedom increases the computational complexity and reduces the accuracy. Autodock has a limit of 32 torsional angles for ligands, whereas DFT calculations are suitable for ligands having more than 30 torsional degrees of freedom.
DFT calculations were performed by relaxing the selected polymeric molecular structures in a vacuum and in polar/nonpolar solvents, e.g., water, ethanol, chloroform and hexane. The calculated intermolecular hydrogen bonding, dipole moment and free energy (in HF) are listed in Table S2.† High dipole moment values are obtained for glycine (5.58, 7.27, 7.2, 6.85 and 6.24 Debye in vacuum, water, ethanol, CHCl3 and hexane, respectively). The conversion of glycine to N-acryloyl glycine results in a significantly decreased dipole moment of 1.93 Debye (Fig. 1b). As the number of N-acryloyl glycine is increased up to the pentamer (i.e., homopolymer (G5)), an increase in the dipole moment is observed, while a sudden decrease in the dipole moment is observed for the hexamer unit (G6). A similar phenomenon is observed for all the LH, LE and CH. The introduction of acrylamide to form heteropolymers resulted in a significant increase in the dipole moment up to the pentamer unit (GA11), while the incorporation of cross-linkers decreased the dipole moment. The greatest dipole is observed for the CE hetero-hexamer (D7) (Fig. 1b). Based on these results, the variation in the dipole moment is well correlated with the polar (acrylamide) or nonpolar nature of the acrylamide/DVB in the polymeric unit. In all cases, the dipole moments followed the order water > ethanol > CHCl3 > hexane > vacuum.
A decrease in the number of hydrogen bonds corresponds to an increase in the swelling ratio of the polymers due to the reduction in intermolecular interactions. The conductor-like polarizable continuum model (CPCM) was used to calculate the intermolecular energy for hydrogen binding for all structures in both polar (water and ethanol) and nonpolar (chloroform and hexane) solvents. The intermolecular energy was found to follow the order CE > CH = LE > LH in water; as the polarity decreased from water to hexane, the extent of intermolecular hydrogen bonding also decreased (see Table S2† and Fig. 1c). The highest intermolecular hydrogen bonding energies were observed for D7 and GA16 with values of 60.18 kcal mol−1 and 58.02 kcal mol−1 in water, and 57.85 kcal mol−1 and 55.77 kcal mol−1 in ethanol, respectively.
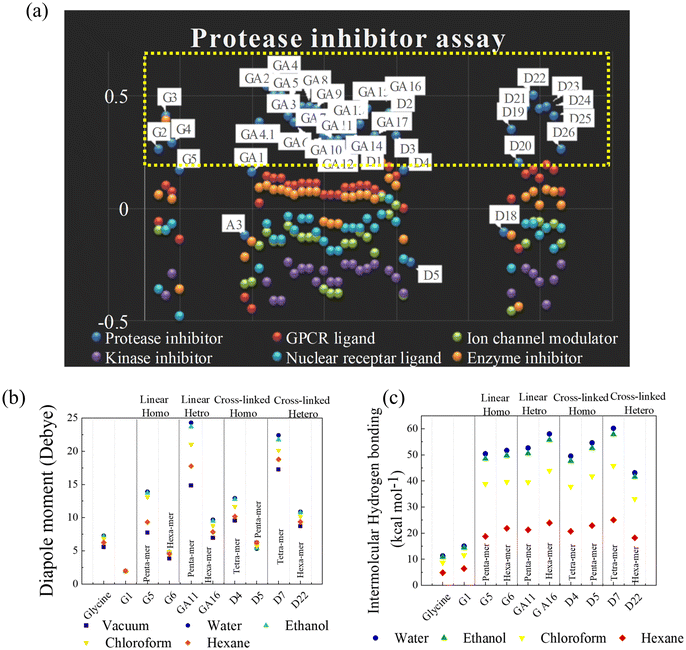
Furthermore, Koopmans’ theorem was useful in the identification of molecular reactivity in terms of ionization potential (I), chemical hardness (η), softness (σ), electronegativity (μ) and electrophilicity (ω ).25 Molecular kinetic stability is indicated in terms of the HOMO–LUMO energy gap (ΔE), as a higher value corresponds to greater chemical stability, while a lower value indicates more polarizability with higher biological activities.24,25 Glycine molecules contribute to chemical stability, while acrylate glycine exhibits chemical reactivity. The energy band gaps for acryloyl glycine, acrylamide, GA1 and D1 were calculated to be 6.006 eV, 5.99 eV, 6.95 eV and 5.018 eV, respectively (Table S3† and Fig. 2). In almost all the cross-linked structures, the energy band gap values are lower than those of the LH and LE polymer (see Table S3†). The well-suited HOMO–LUMO energy band gap values of the monomers and cross-linked polymers with molecular docking results are presented in Fig. 2. Higher electrophilicity corresponds with higher antimicrobial and anticancer properties.33 It was reported that ω > 1.5 eV represents good biological activity.25 However, in this work, the ω values for all structures were in the range of 1.6 eV–2.6 eV, except for GA17, for which a sudden increase in ω was observed obtained (9.4 eV).
 |
| | Fig. 2 Ground state HOMO, excited state LUMO and energy band gap of random polymer arrangements. The structural geometry and energy band gap of the tetramer and pentamer structures of the four groups LH, LE, and CH and CE were compared with those of glycine and N-acryloylglycine. The energy band gap decreased with increasing complexity of the structure. | |
3.3. Comparative heparanase inhibitory activity of p(NAG-co-Ac) polymeric units: molecular-docking-based investigation
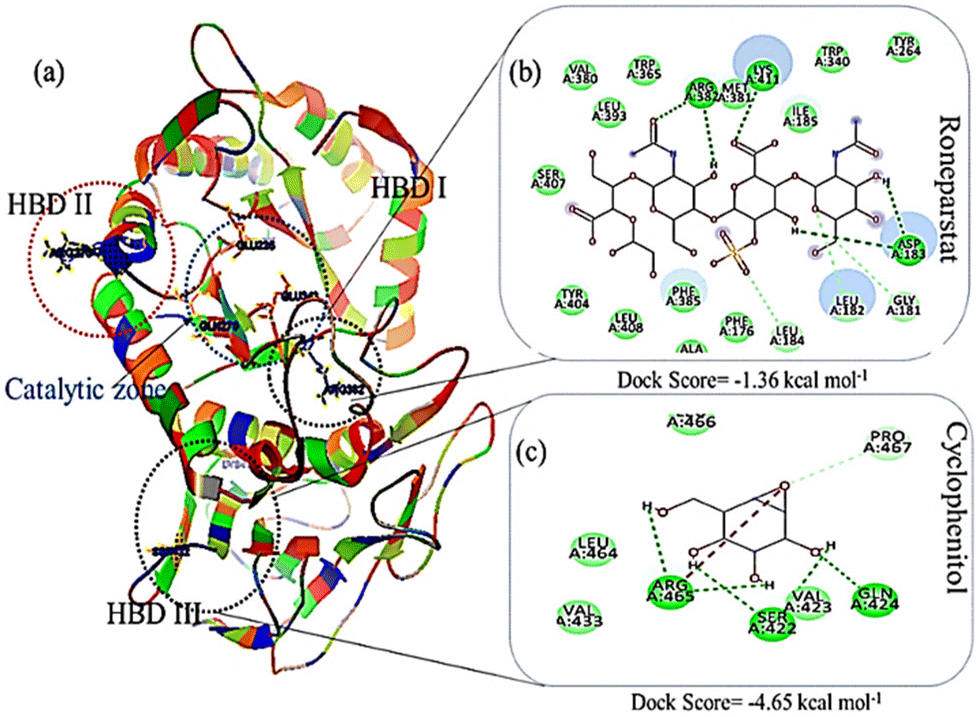
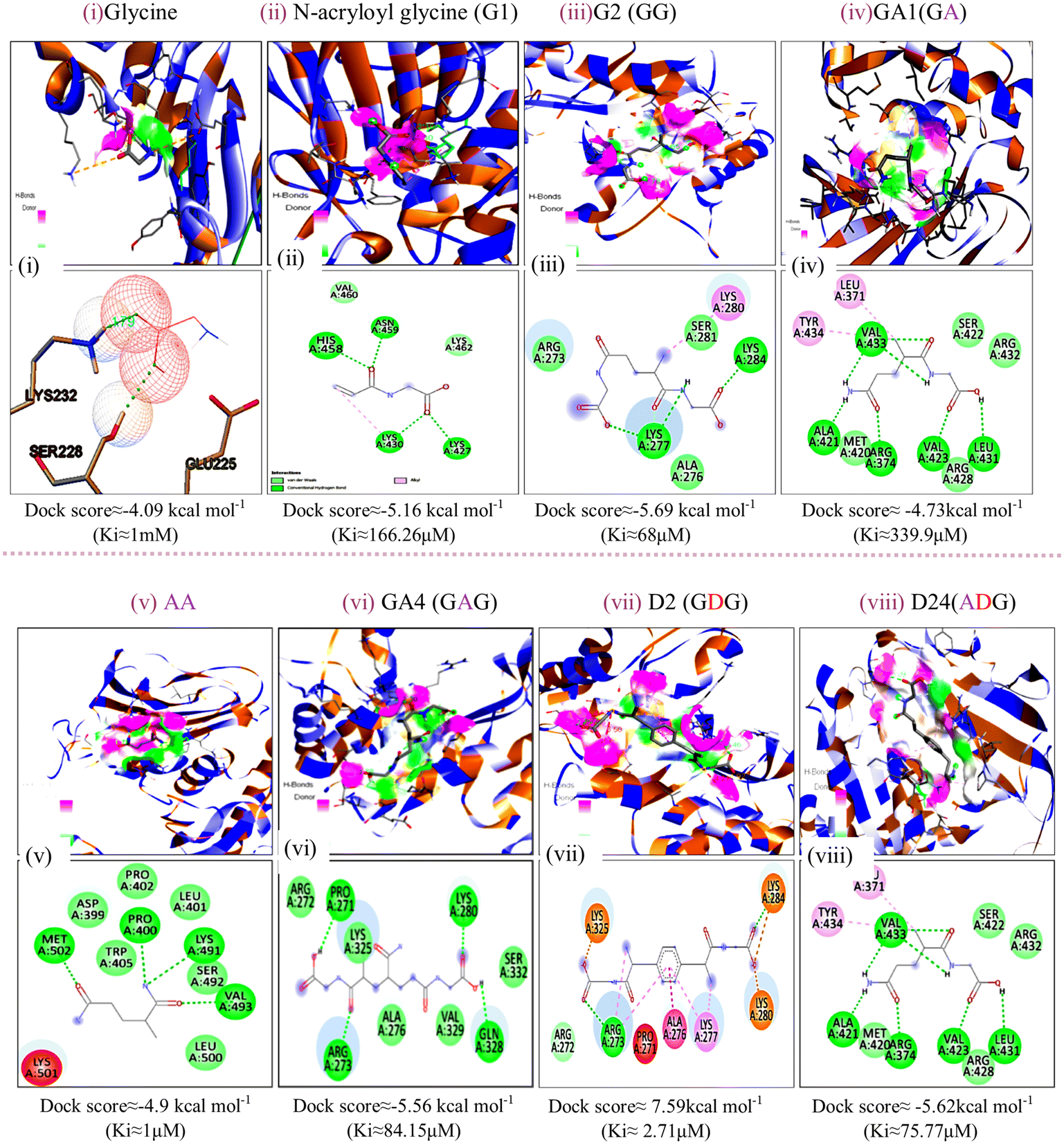
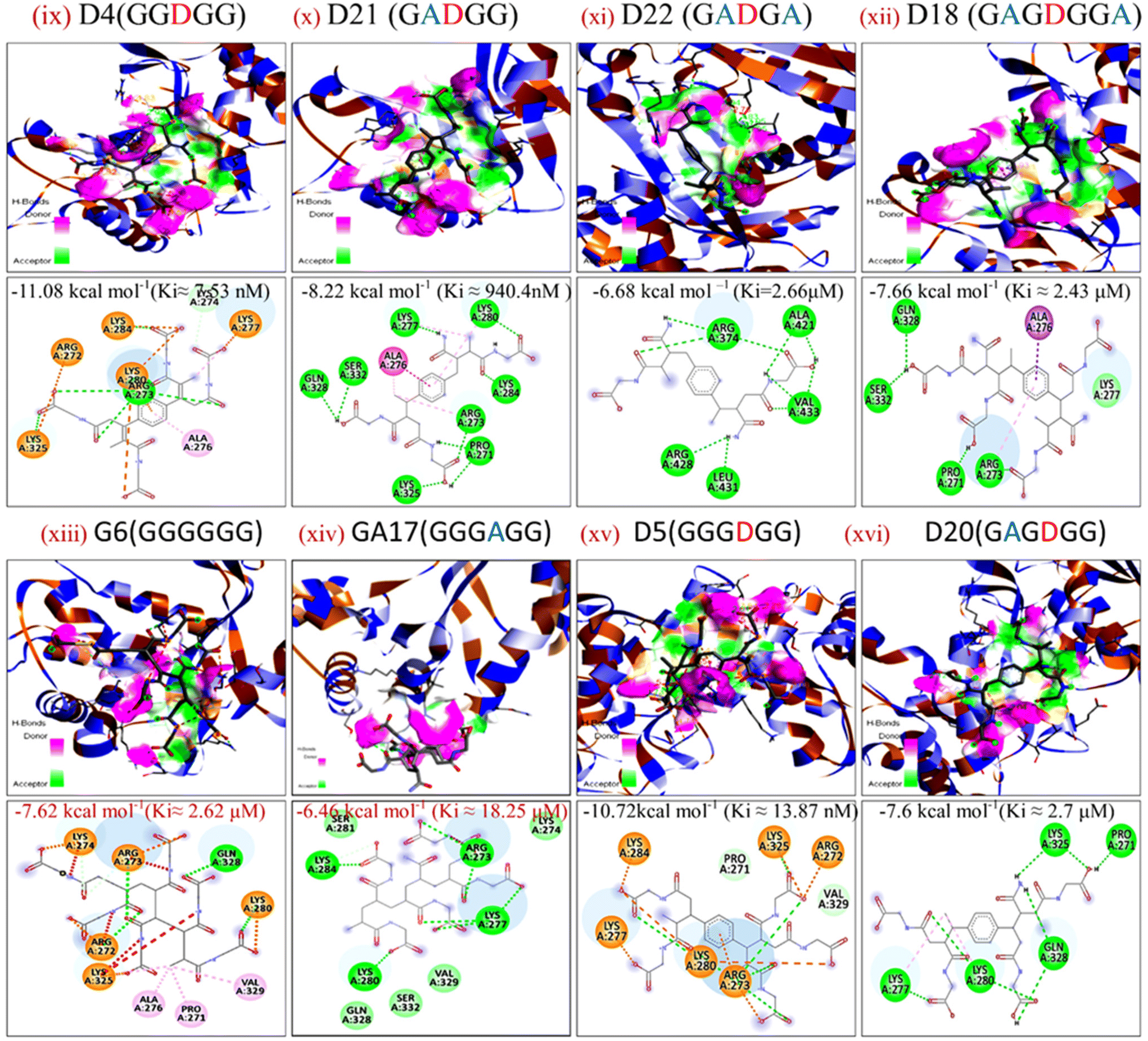
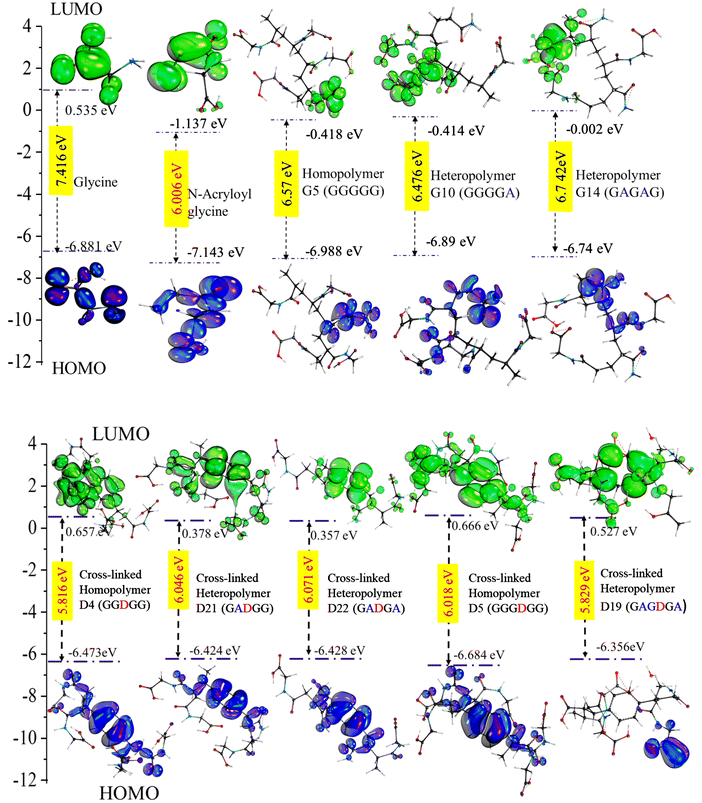
The energy band gap and electrophilicity of the cross-linked p(NAG-co-Ac) polymeric units provide an idea of the therapeutic activity.10,34 However, no heparanase inhibitors have been approved in clinical trials. Molecular interaction studies between 38 random arrangements and human heparanase (PDBID 7PRT) were performed using the geometrically optimized structures obtained after DFT calculation in a vacuum. The remaining structures were excluded from DFT calculation and for molecular docking due to their higher number of monomers (more than 6 units) showing the higher torsion angles such as θ = 32° (as the Autodock tool works best for ligands with 32° torsion angles). At the molecular level, negative binding energy and H-bonding correspond to strong protein–ligand interaction, while the inhibition constant measures the therapeutic activity of the drug. The active form of heparanase is composed of non-covalently linked 50 kDa and 8 kDa subunits, an enzymatic site near Glu343 and Arg272, and two heparin binding domains, heparin binding domain I (Lys159–Val170) and II (Gln270–Lys280), as shown in Fig. 3(a).35 Molecular docking was performed with the known potent inhibitor roneparstat,36 which is in Phase II trials, and with cyclophellitol,37 a natural glycosidase inhibitor, and its derivative with heparanase 7PRT using the Autodock tool (Fig. 3). Similarly, glycine, N-acryloylglycine and random LH, LE, CH and CE polymer structures of N-acryloylglycine and acrylamide were docked with heparanase 7PRT (Fig. 4 and 5). The interaction binding energies for the reference molecules and 38 interacting residues are listed in Table S4.† The inhibition constant (Ki) for cyclophellitol was calculated to be 823.53 μM, and five hydrogen bonds with Ser422, Gln424, Arg465 were formed with a binding energy of −4.2 kcal mol−1 (Fig. 3b and Table S4†). Roneparstat interacted with a binding energy value of −1.36 kcal mol−1 at a Ki of 100.76 mM by forming two hydrogen bonds with the Asp183 and Arg382 residues (Fig. 3c). Glycine interacted by forming only one hydrogen bond (Lys232 residue) with a binding energy of −4.09 kcal mol−1 at Ki = 1 mM (Fig. 4i) and N-acryloylglycine (Fig. 4ii) interacted at Ki = 166.36 μM, which is lower than the shown reference value (Table S4†), and by forming four hydrogen bonds, with a higher binding affinity value of −5.16 kcal mol−1 (Fig. 5). With the conversion of the N-acryloylglycine monomer to an LH of six units (G6), the binding affinity increases from −5.16 kcal mol−1 to −7.62 kcal mol−1 and for the CH of six units (D5), the binding affinity increases from −6.83 kcal mol−1 to −10.45 kcal mol−1 along with the increase in the number of hydrogen bonds. The LH and CH mostly formed hydrogen bonds at HBD II, with the most common interacting residue being Lys280 for LH and Arg272 for CH (Fig. 4, 5 and Table S4†). G6 formed 8 hydrogen bonds with a binding affinity of 6.76 kcal mol−1 (Ki = 11.16 μM) (Fig. 5xiii), and similarly, D5 formed 7 hydrogen bonds with a binding affinity of −10.72 kcal mol−1 (Ki = 13.87 nM) (Fig. 5(xv) ). The addition of acrylamide to the LH and CH yielded the LE and CE. The position and change in the number of acrylamide moieties affected the binding affinity and interaction with residues, as shown in Fig. 4 and Table S4.† The dimer of acryloylglycine (G2) interacted with HBDII by forming four hydrogen bonds, while upon the introduction of acrylamide (GA), it interacted at HBDIII by forming five hydrogen bonds. Similarly, as shown in Fig. S1,† with the introduction of acrylamide into p(NAG-b-Ac) structures, the interaction sites were found to be more random. Similarly, with the introduction of more than two units of acrylamide in the p(NAG-b-Ac) structures, the binding score decreased. For most of the copolymers, such as GA1 (Fig. 4(iv)), GA9, (Fig. S1a†), GA14 (Fig. S1c†) and GA15 (Fig. S1b†), with increasing the number of acrylamide units, they interacted at the residue Ser422, along with other residues. This alteration mainly occurred due to the acrylamide unit AA dimers that interacted at the HBDIII sites and random sites. As shown in Fig. 4(vi–viii), replacement of the central unit A in for GA4 with DVB (D) resulted in an increased docking score, whereas for the CE with ADG, the docking score decreased, but remained relatively higher than that of GA4 (D2 > D24 > GA4). All the CEs majorly interacted at heparin-binding domain III, interacting with the residues Lys280, Lys231, Arg272, Arg273, Gln328 and Lys227 and with the lowest inhibitory constant at nano-molar Ki. Recently, Zhang et al. reported the heparanase SAR for inhibitors, in which molecules interacted at residues Lys231, Glu225 and Ser228,38 similar to the CE interaction region. It was further observed that with increasing number of acrylamide units, the docking scores decreased, as shown in Fig. 5 (D4 > D21 > D22). We considered four units of glycine, one unit of acrylamide and one unit of cross linker to be the optimum ratio for designing the polymer, as in D20, which showed a docking score equivalent to that of G6 (Fig. 5xiii). LH above G3 do not show positive protease binding scores. The LE show maximum protease binding. Furthermore, in the CH series, a positive protease binding score was observed up to D4 (Fig. 5xi), while above this, the protease binding scores were negative (Fig. 1). The CE (D20) shows both protease inhibitory and heparanase inhibitory activities (Fig. 1 and 5xvi).
 |
| | Fig. 3 Heparanase interaction (a) 3D protein structure of heparanase (7PRT) depicting the catalytic domains and heparin binding domains HBDI, HBDII and HBDIII. Docking poses of complexes of (b) cyclophellitol and (c) roneparstat with 7PRT. | |
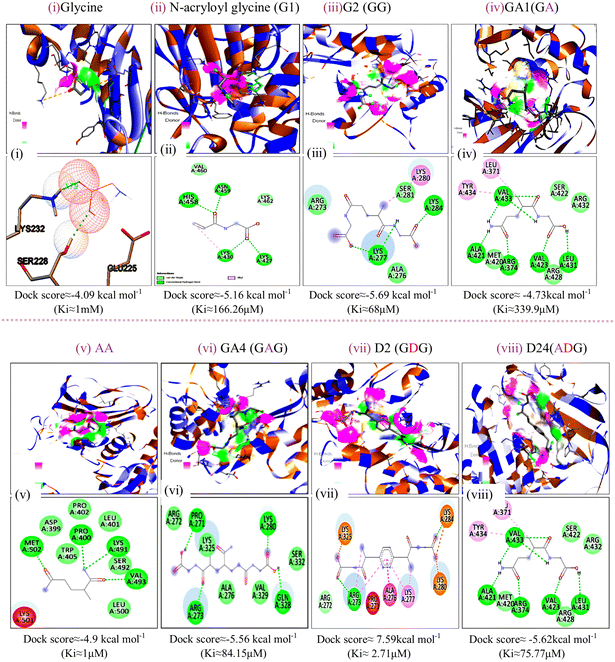 |
| | Fig. 4 Docking poses for complexes of (i) glycine, (ii) N-acryloyl glycine, (iii) G2, (iv) GA1, (v) AA, (vi) GA4, (vii) D2 and (viii) D24 with 7PRT. | |
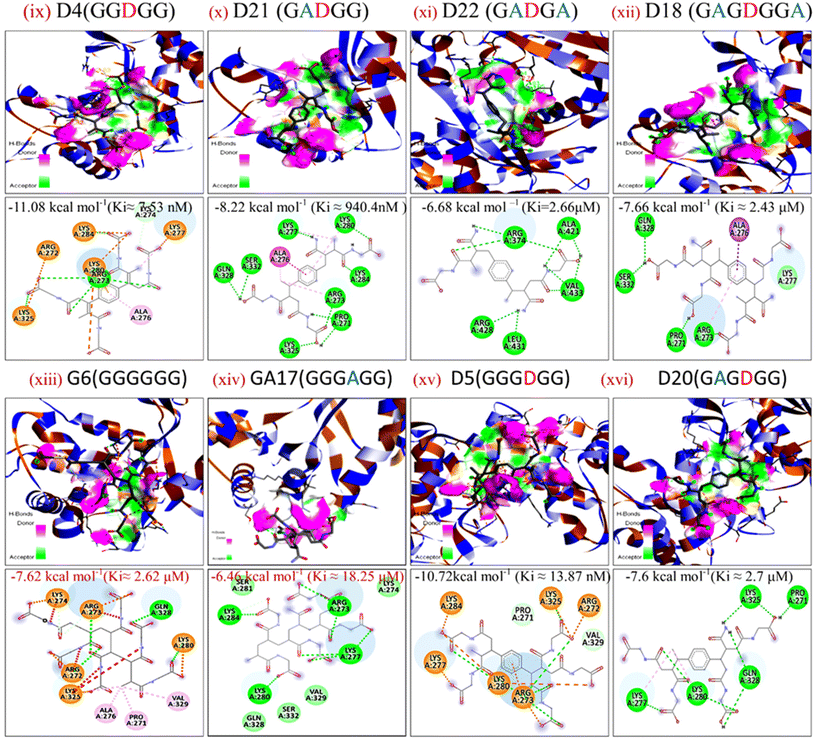 |
| | Fig. 5 Docking poses for complexes of (ix) D4, (x) D21, (xi) D22, (xii) D18, (xiii) G6, (xiv) GA17, (xv) D5 and (xvi) D20 with 7PRT. | |
3.4. Synthesis of p(NAG-co-Ac) hydrogel and Protease inhibition
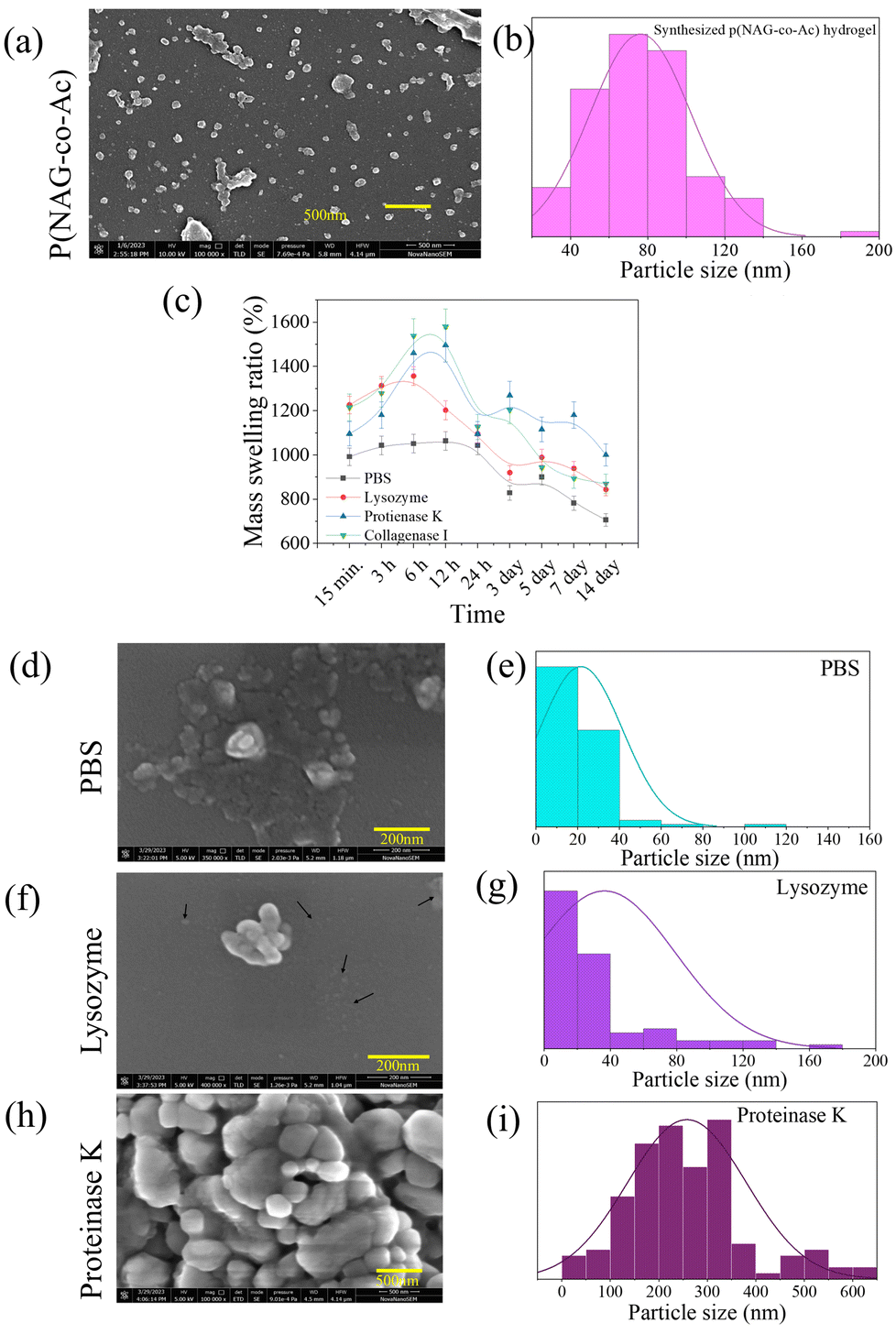
As reported in our previous studies, p(NAG-co-Ac) hydrogel was synthesized using a mini-emulsion polymerization approach through free-radical cross-linking polymerization.31Fig. 6a depicts the FESEM micrograph of p(NAG-co-Ac) hydrogel, showing spherical nano-hydrogels with a particle size of 20–140 nm (Fig. 6b) in diameter with an average particle size of 80 nm in diameter. It is difficult to obtain a narrow range of particle size distribution for polymeric nanoparticles; however, these particles are in an acceptable range for passive targeting. The incorporation of the DVB cross-linker and acrylamide results in the high swelling behaviour of the p(NAG-co-Ac) hydrogel particles (Fig. 6c). It is evident that the swelling behaviour of p(NAG-co-Ac) varied when different solution media were used, i.e., 1063.26 ± 46.53% in PBS, 1202.17 ± 42.53% in lysozyme, 1495.55 ± 74.77% in proteinase K, and 1579.59 ± 78.97% in collagenase I after 12 h of incubation. A decrease in the mass swelling ratio can be correlated with the degradation of p(NAG-co-Ac), as the mass swelling ratio was found to be 706.12 ± 28.24% in PBS, 843.49 ± 28.24% in lysozyme and 893.87 ± 43.46% in collagenase I on day 14th. The lowest degradation of ∼1280 ± 64.25% was observed in the solution of proteinase K at 5 min (see Fig. 6c ). On day 15th, after re-lyophilization of degraded p(NAG-co-Ac) hydrogel, FESEM micrographs were obtained and the results are shown in Fig. 6d, f and h, and the distributions of the particle sizes are shown in Fig. 6e, g and i. The decrease in the particle size of the degraded p(NAG-co-Ac) hydrogel in PBS and lysozyme is clearly shown in Fig. 6d and f, with a reduction in the particle size to 40 nm (Fig. 6e) and 80 nm (Fig. 6g) in diameter, respectively. However, in the presence of proteinase K, the p(NAG-co-Ac) hydrogel particles are agglomerated into larger particles with a size of 100–400 nm in diameter, which is also important evidence for the protease inhibition activity. The protease inhibition activity was further confirmed through in silico results (Table S1†).
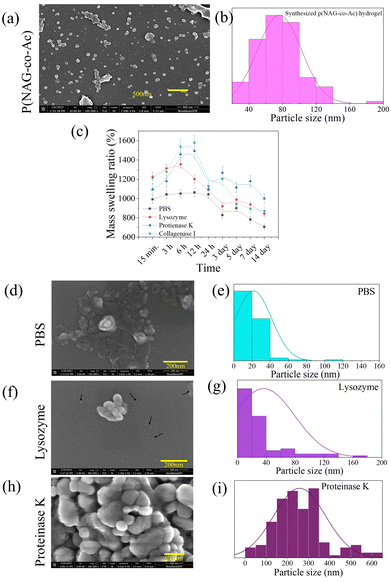 |
| | Fig. 6 Morphology, mass swelling ratio and degradation behavior of poly-[(N-acryloylglycine)-co-(acrylamide)] hydrogel in different enzymatic solutions. (a) FESEM micrograph of p(NAG-co-Ac); (b) particle size distribution obtained from Fig. 1(a). (c) Mass swelling ratios of p(NAG-co-Ac) in PBS and enzymatic solutions of lysozyme, proteinase K and collagenase I at a concentration of 100 μg mL−1 of the enzyme. (d–i) FESEM micrographs of degraded p(NAG-co-Ac) at the 15th day and particle size distribution from the respective micrographs: (d–e) p(NAG-co-Ac) in PBS, (f–g). p(NAG-co-Ac) in 100 μg mL−1 lysozyme, (h and i) p(NAG-co-Ac) in 100 μg mL−1 proteinase K. | |
3.5. p(NAG-co-Ac) hydrogel-induced enzyme inhibition and CD spectroscopy
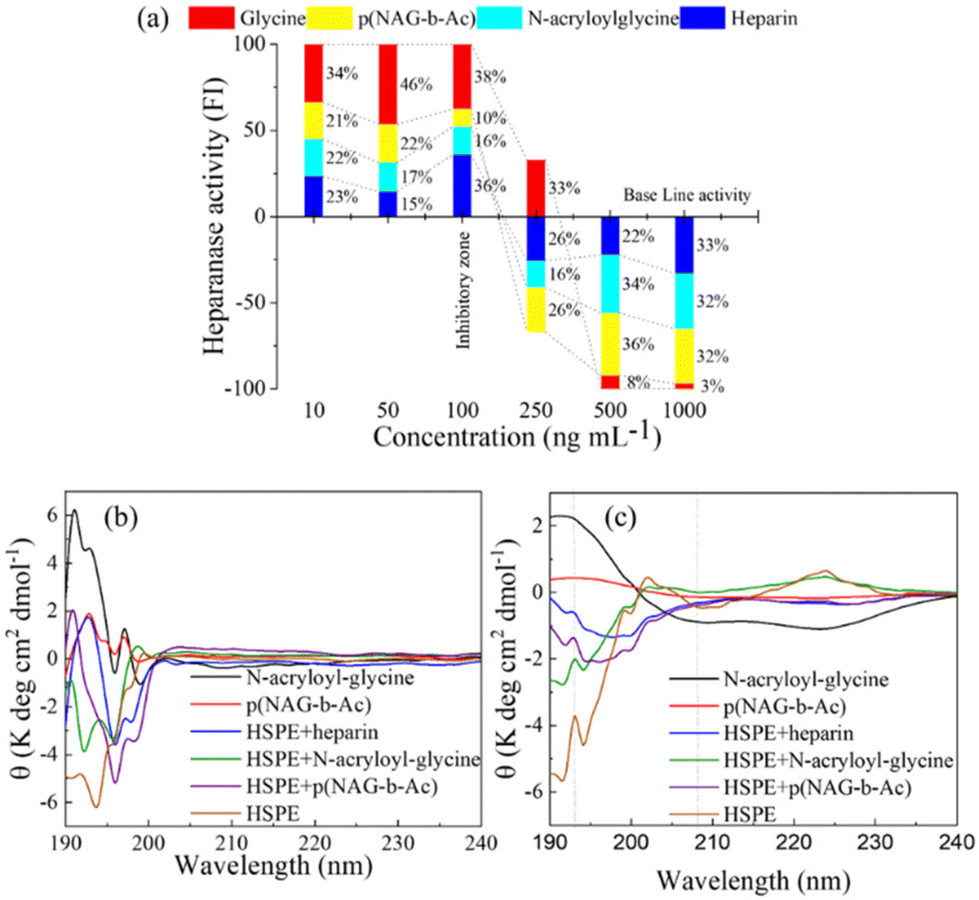
The synthetic pentasaccharide fondaparinux sodium with a relative molecular mass of 1728 Da is a commercially available substrate of heparanase. Heparanase hydrolyzes fondaparinux and generates the one-reducing terminal of glucuronic acid, which was detected using resazurin via the generation of fluorescent resofurin. Heparin and its derivatives, such as the N-acylated heparin derivative roneparstat, are well known heparanase inhibitors that show 70% inhibition of heparanase at a 1 μg mL−1 concentration of heparin. Therefore, in the present study, heparin was used as a positive control, and the inhibitory activity of glycine, N-acryloyl glycine and p(NAG-co-Ac) were tested via a fondaparinux-based inhibition assay. Glycine does not show heparanase inhibition, while the conversion of glycine to N-acryloylglycine and p(NAG-co-Ac) resulted in heparanase inhibition at a concentration of 250 ng mL−1. At particle concentrations above 250 ng mL−1, we observed an almost equivalent percentage of heparanase inhibition, as shown in Fig. 7a. These results corroborated that the N-acryloylglycine and p(NAG-co-Ac) hydrogels exhibited heparanase inhibitory activity.
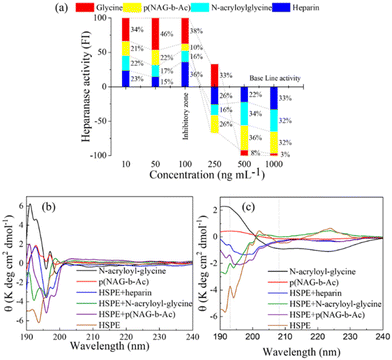 |
| | Fig. 7 Enzyme interaction and inhibition assay. (a) Heparanase inhibitory activity of glycine, N-acryloyl and p(NAG-co-Ac) compared using fondaparinux assay. (b) CD spectra of heparanase (HPSE) in presence of N-acryloyl glycine and p(NAG-co-Ac) alter the secondary structure of HPSE. (c) Comparison of the altered α-helix percentage of heparanase: N-acryloyl glycine and p(NAG-co-Ac) alone and with HSPE. | |
Furthermore, CD analysis shows a significant change in the protein secondary structure upon treatment with p(NAG-co-Ac) hydrogel. Polymerization of N-acryloyl glycine to p(NAG-co-Ac) hydrogel results in a decrease in the spectral intensity in the CD analysis. The CD spectrum of heparanase shows 29% α-helix (Fig. 7c) with a positive signal at 191 nm and negative signal at 193.5 nm (Fig. 7b), which were disturbed by the addition of the known heparanase inhibitor heparin, with a slightly hyposochromic shift for the positive signal to 192 nm and a bathochromic shift to 196 nm for the negative band intensity. The polymerization of N-acryloyl glycine to p(NAG-co-Ac) hydrogel results in the decrease in the intensity of the CD spectrum. Similar to heparin, upon the addition of p(NAG-co-Ac) to heparanase, there is a significant change in secondary structure of the protein, confirmed by the hyposochromic shift in comparison to p(NAG-co-Ac) alone to 190 nm and the bathochromic shift for the negative signal at 196 nm (Fig. 7b). This loss of α-helix (Fig. 7c) and large shift is the result of the disturbed secondary structure, and its occurrence may be due to the aggregation. A similar effect on the protein structure was also observed when it was treated with N-acryloyl-glycine. Furthermore, to evaluate the anticancer potential of p(NAG-co-Ac) hydrogel, its in vitro cytotoxicity and anti-migratory effects were investigated.
3.6. p(NAG-co-Ac) hydrogel enhances cytotoxicity in aggressive cancer
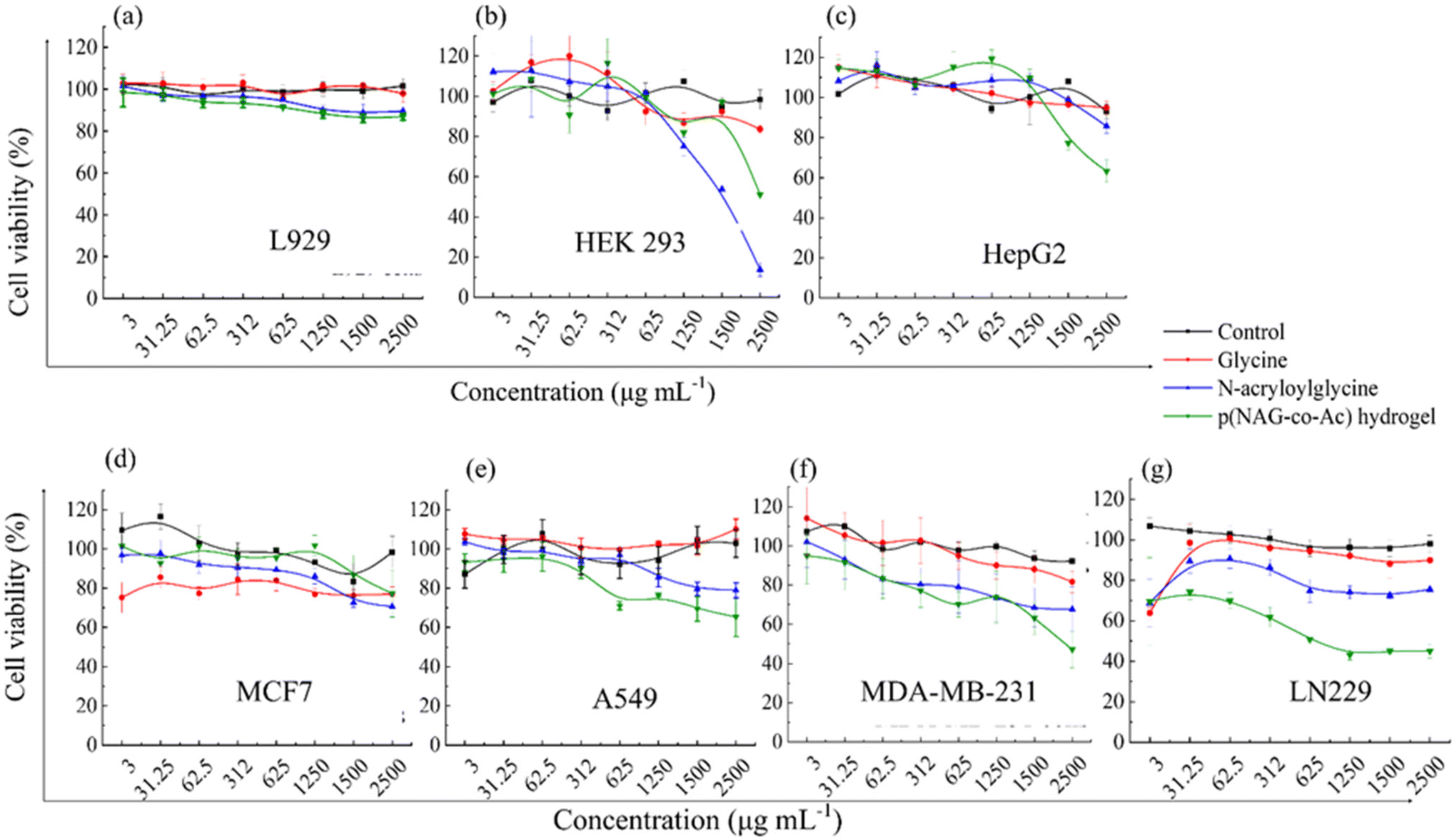
Glycine is an amino acid involved in cell proliferation and in tumor metabolomics, and therefore, upon treatment with glycine at concentrations from 3 μg mL−1 to 2.5 mg mL−1, all normal cells (L929, Hek293, HepG2) and cancer cells (A549, MDA-MB-231 and LN229 cell lines) showed 100% (P > 0.05) cell viability, except for MCF7 cells, which exhibited 80% cell viability at all the concentrations (P* < 0.05) (Fig. 8d). N-Acryloylglycine monomer had no effect on the cell viability of the L929, HepG2, and A549 cell lines (Fig. 8a, c and e), while significant toxicity was observed (P < 0.05) in Hek293 (Fig. 8b) at 1.25 mg mL−1 monomer concentration and the viability decreased to 80% in the MDA-MB-231 and LN229 cell lines (Fig. 8f and g). The viability of Hek293 was significantly improved for the polymerized form of N-acryloylglycine, i.e., p(NAG-co-Ac) (Fig. 8, green line). p(NAG-co-Ac) hydrogel possesses significant cellular toxicity toward the aggressive cancer cells in the order LN229 > MDA-MB-231 > A549. The observed IC50 for LN229 is 625 μg mL−1 (P* < 0.01) and for MDA-MB-231, it is 2.5 mg mL−1 (P* < 0.01) (Fig. 8g). However, the MCF7 cell line remains 80% viable, which could be due to the low expression heparanase (Fig. 8b and c).
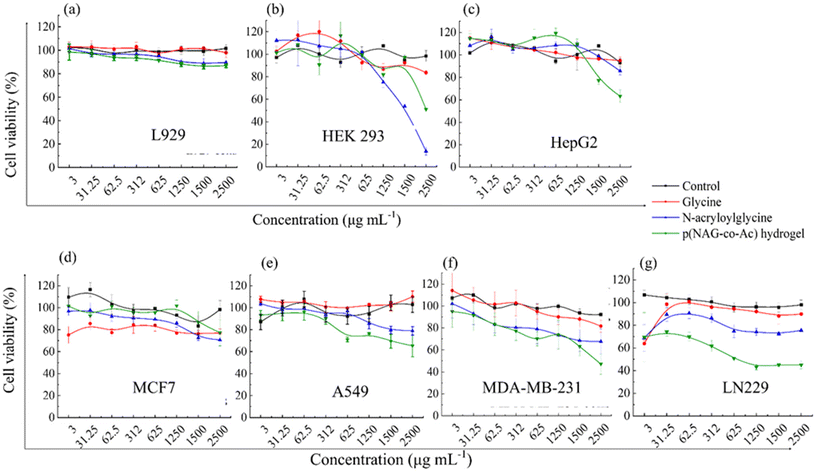 |
| | Fig. 8 MTT assay cell viability results for the (a) L929, (b) HEK 293, (c) HepG2, (d) MCF7, (e) A549, (f) MDA-MB-231, and (g) LN229 cell lines treated with different concentrations of glycine (red), N-acryloyl glycine (blue) and p(NAG-co-Ac) hydrogel (green). | |
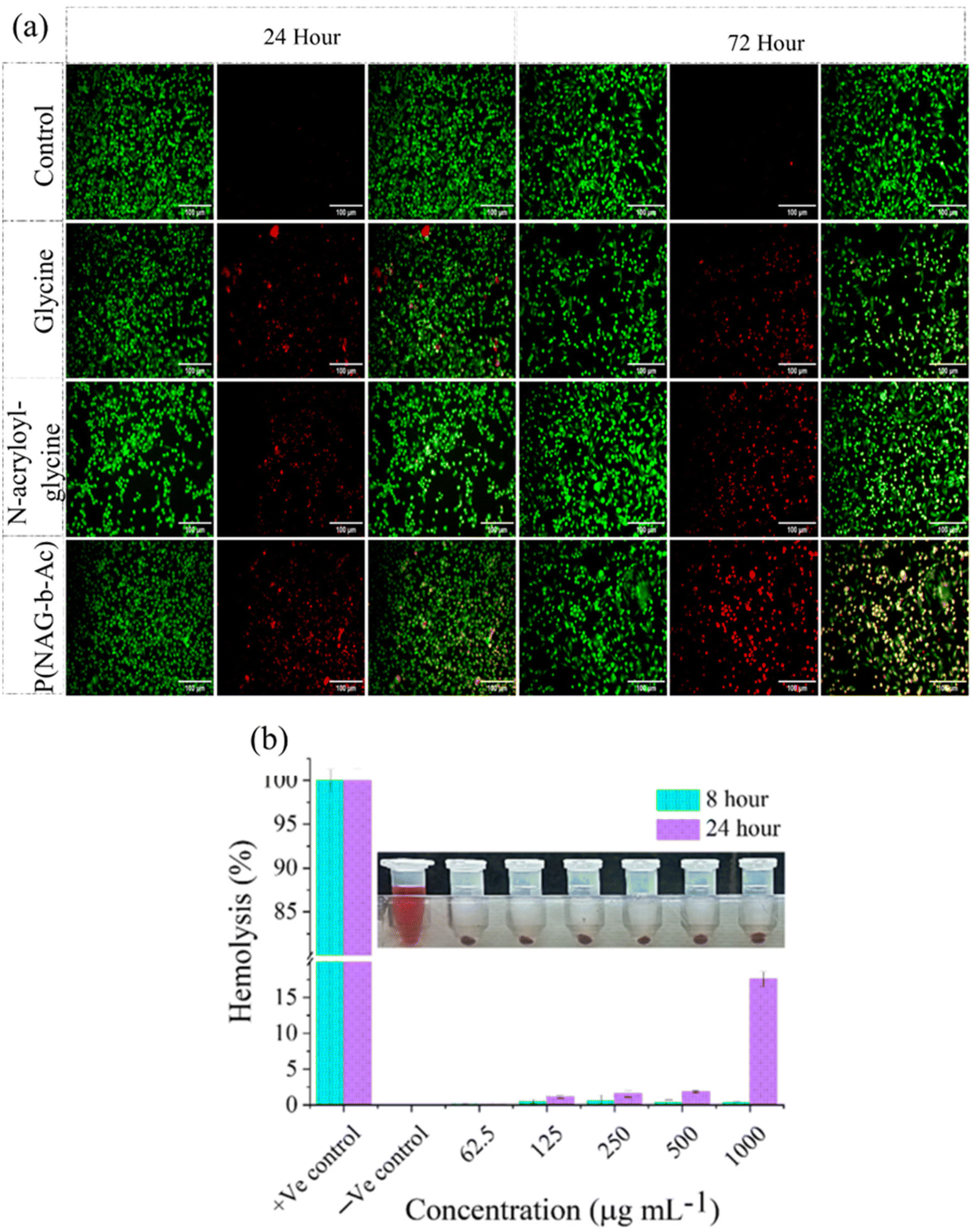
Next, live–dead assays were performed using acridine orange (AO) and propidium iodide (PI) staining (Fig. 9a). From the results, it is evident that the polymerized hydrogel exhibited significantly higher cytotoxicity towards cancer cells than the glycine and N-acryloyl glycine monomers. Further, the hemolytic activity of the N-acryloylglycine monomer at different concentrations was studied and found to be higher than that of p(NAG-co-Ac). The hemolytic percentage for the monomer at concentrations of 62.5, 125, 250, 500 and 1000 μg mL−1 was found to be −12.3580 ± 0.056, −30.0772 ± 0.0051, 2.8169 ± 0.014, 17.3130 ± 0.178, and 14.3116 ± 0.007, respectively (Fig. S2†). However, the hemolytic percentage of p(NAG-co-Ac) at concentrations of 62.5, 125, 250, 500 and 1000 μg mL−1 was found to be 0.14 ± 0.09, 1.13 ± 0.18, 1.58 ± 0.46, 1.88 ± 0.13 and 13.56 ± 1.04 at 24 h, respectively (Fig. 8b).
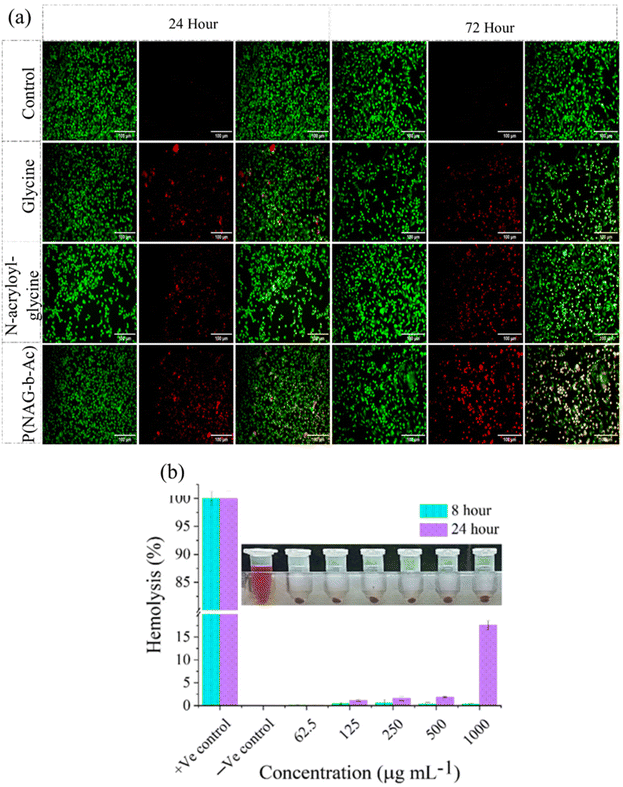 |
| | Fig. 9 (a) Live–dead assay performed via AO/PI staining of MDA-MB-231 cell line treated with glycine, N-acryloylglycine and p(NAG-co-Ac) hydrogel, and (b) hemolysis (%) in treatment with p(NAG-co-Ac) hydrogel. | |
3.7. p(NAG-co-Ac) hydrogel mitigates cancer cell migration
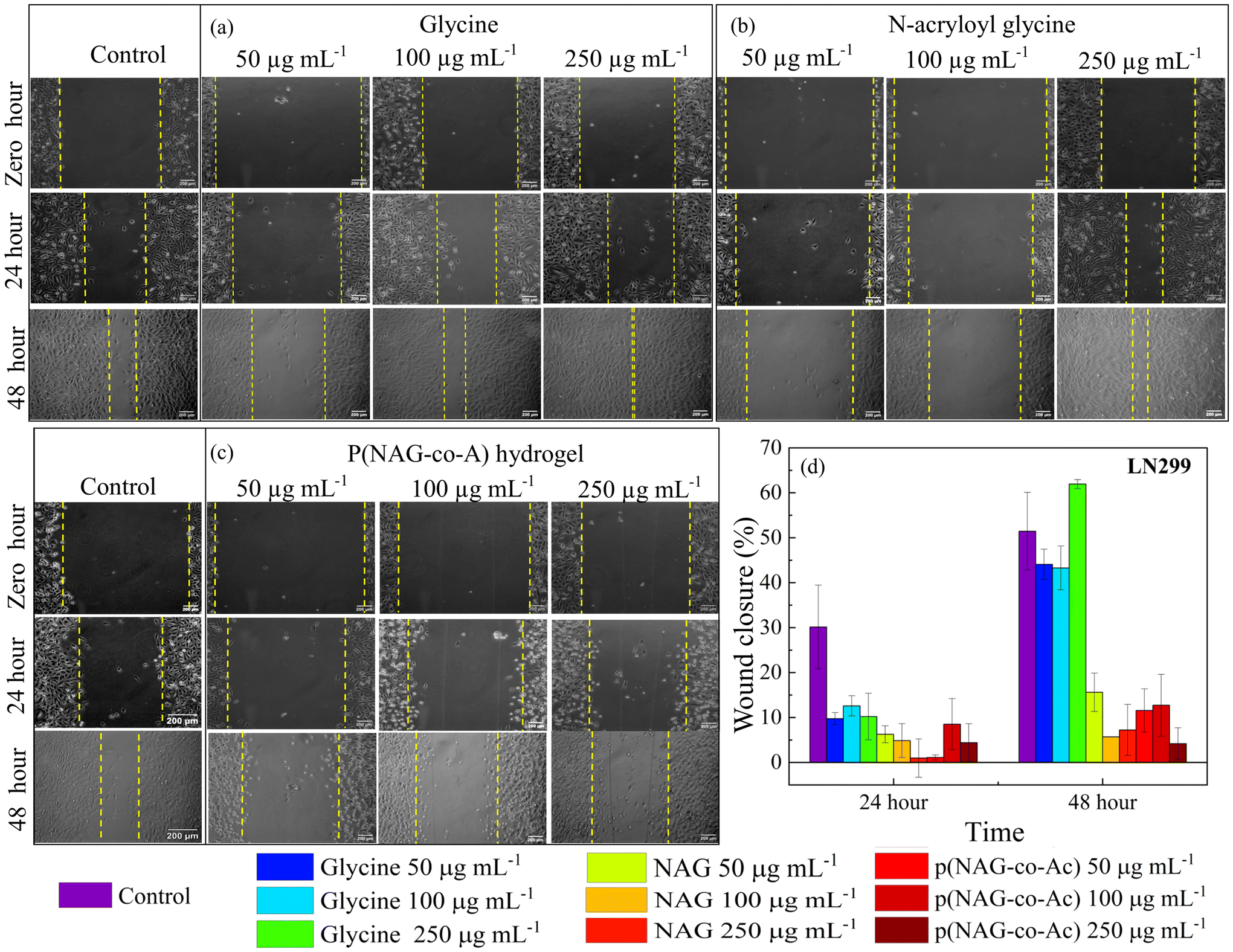
The prognosis of metastatic cancer worsens with the migration and invasion of cancer stem cells. Therefore, the migratory mitigation effects of p(NAG-co-Ac) were investigated by performing wound scratch assays on the MDA-MB-231 and LN229 cancer cell lines and L929 normal fibroblast cell lines. Glycine and N-acryloylglycine exhibited a non-significant effect on the migration of the MDA-MB-231 and LN229 cancer cell lines, while p(NAG-co-Ac) decreased the rate of migration for the MDA-MB-231 and LN229 cancer cell lines. The wound closure percentage for untreated LN229 was found to be 30.15 ± 9.3% and 51.44 ± 8.64% (Fig. 10), and that for MDA-MB-231 was 8.81 ± 4.53% and 48.30 ± 9.31% (Fig. 11), after 24 h and 48 h of treatment, respectively. Glycine enhances the LN229 cell migration in a dose-dependent manner: 50 μg mL−1: 44.08 ± 3.37%; 100 μg mL−1: 43.27 ± 4.87%; and 250 μg mL−1: 61.93 ± 0.98% at 48 h. In MDA-MB-231, glycine treatment inhibited wound closure with 15.13 ± 2.37% at 50 μg mL−1, 23.13 ± 3.53% at 100 μg mL−1 and 8.26 ± 3.28% 250 μg mL−1 at 48 h. N-Acryloylglycine and p(NAG-co-Ac) significantly inhibited the cellular migration of LN229 cells (P** < 0.001) (see Fig. 10) and MDA-MB-231 cells (P** < 0.001) (Fig. 11). For LN229, the extent of wound closure was observed to be 15.61 ± 4.26%, 6.79 ± 0.01%, and 7.22 ± 5.67% for N-acryloylglycine and 11.56 ± 4.82%, 12.72 ± 6.87%, and 4.18 ± 3.55% with p(NAG-co-Ac) at concentrations of 50, 100 and 250 μg mL−1, respectively, at 48 h. Similarly, in MDA-MB-231, the extent of wound closure was observed to be 4.22 ± 1.70%, 2.74 ± 0.55%, and 6.27 ± 4.76% for N-acryloylglycine and 8.05 ± 5.89%, 5.96 ± 3.11%, and 4.05 ± 0.20% for p(NAG-co-Ac) at concentrations of 50, 100 and 250 μg mL−1 at 48 h. The wound closure percentage in the untreated L929 control was calculated to be 16.56 ± 1.65% at 24 h, and p(NAG-co-Ac) showed a non-significant migration effect on L929 cells at 50 μg mL−1 (38.67 ± 3.86%), 100 μg mL−1 (19.88 ± 12.82%) and 250 μg mL−1 (20.15 ± 2.01%).
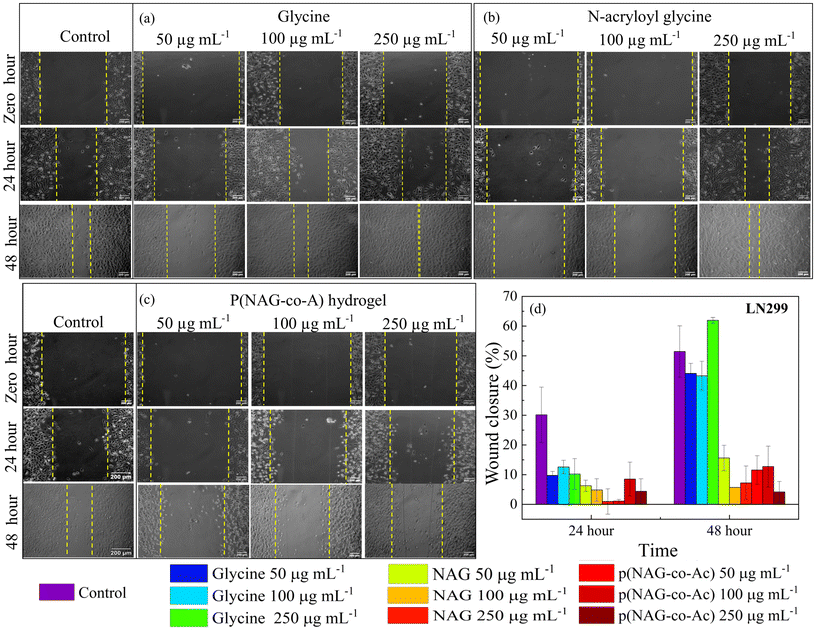 |
| | Fig. 10
In vitro wound scratch assay results obtained using LN229 glioblastoma cells and applying different concentrations (50 μg mL−1, 100 μg mL−1 and 250 μg mL−1) of samples. Results obtained for (a) glycine, (b) N-acryloyl glycine, and (c) p(NAG-co-Ac). (d) Effect on migration. Inhibition was measured in terms of wound closure percentage at 24 and 48 h. | |
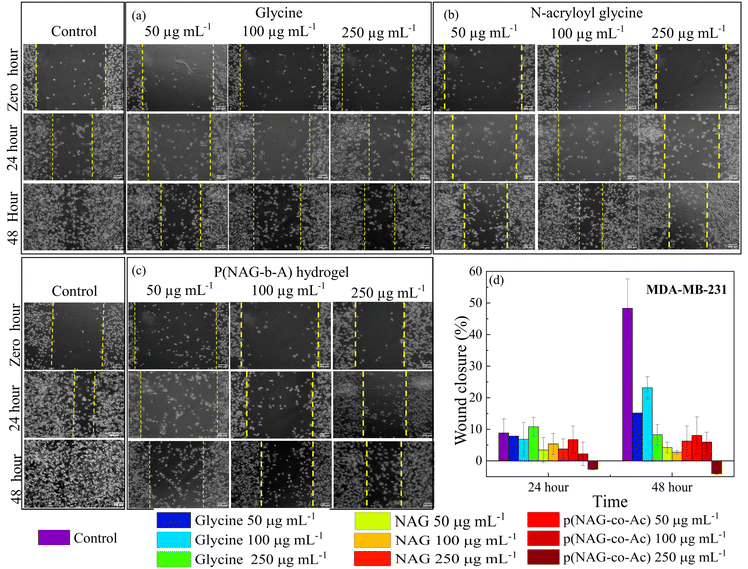 |
| | Fig. 11
In vitro wound scratch assay using MDA-MB-231 breast cancer cell line (TNBC) and applying different concentrations (50 μg mL−1, 100 μg mL−1 and 250 μg mL−1) of (a) glycine, (b) N-acryloylglycine, (c) p(NAG-co-Ac). (d) Effect on migration. Inhibition was measured in terms of wound closure percentage at 24 and 48 h. | |
3.8. p(NAG-co-Ac) hydrogel elevated reactive oxygen species (ROS) levels, leading to apoptotic cell death
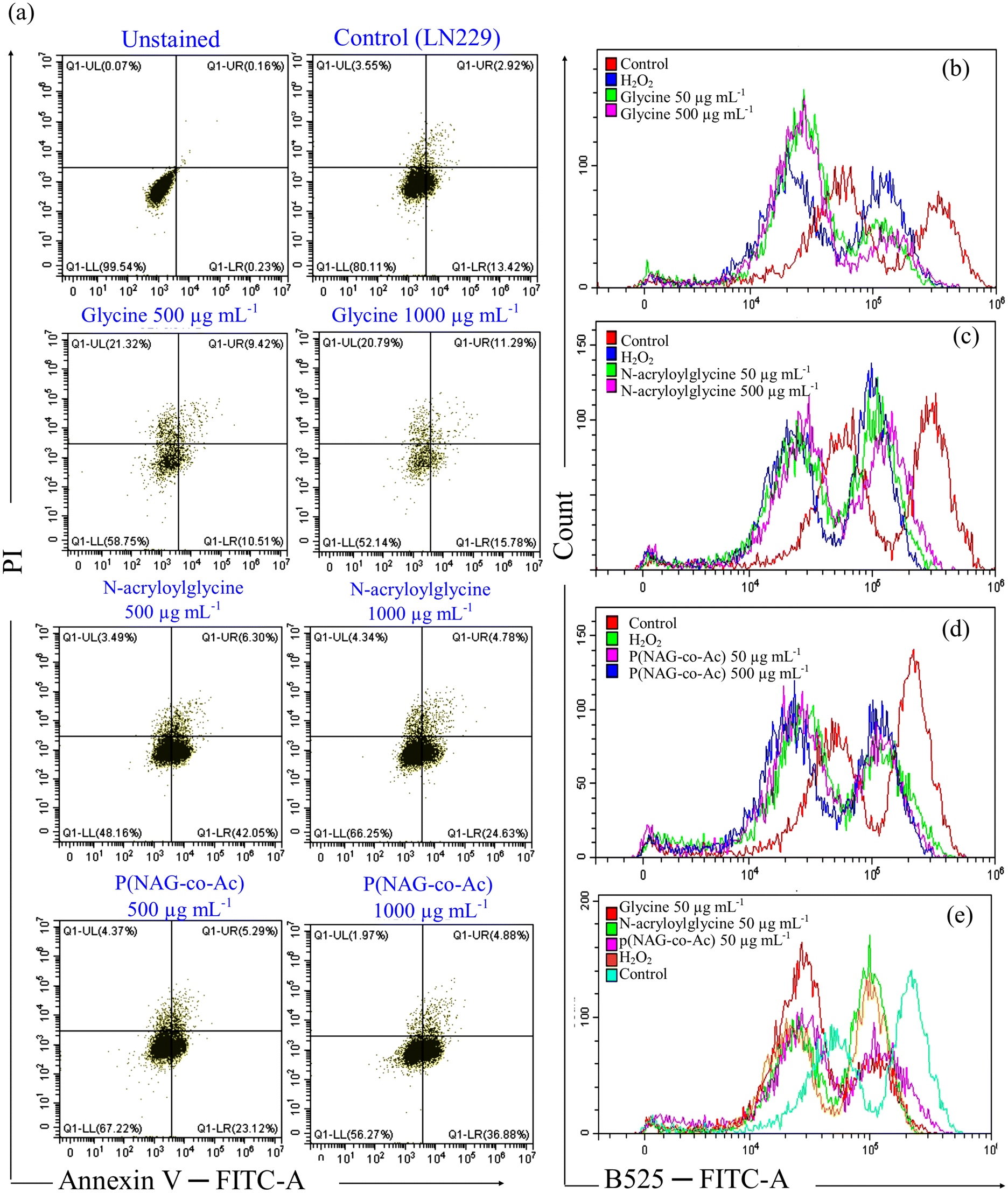
Furthermore, to evaluate the cell death mechanism, ROS generation was evaluated using dichlorofluorescein diacetate and annexin V/PI assay. The LN299 cell line was treated with 500 μg mL−1 and 1000 μg mL−1 of glycine, N-acryloyl glycine or p(NAG-co-Ac) hydrogel. In the control, it was observed that 80 ± 2.15% cells were healthy, 13.42 ± 1.83% were in early apoptosis, 2.92 ± 0.23% were in late apoptosis, and 3.45 ± 0.21% were in the necrotic phase. Glycine treatment induced necrosis, as the major cell populations shifted to the necrotic phase with 21.32% (500 μg mL−1) and 20.79% (1000 μg mL−1) at 24 h. With treatment with 500 μg mL−1N-acryloylglycine and p(NAG-co-Ac) hydrogel, the major cell population shifted to the early apoptotic zone (24.63% and 23.12%, respectively) and 4.78% and 5.29% were in the late apoptotic phase. Similarly, with 1000 μg mL−1 of N-acryloylglycine and p(NAG-co-Ac) hydrogel, 42.05% and 36.88% of the cell population were in the early apoptotic death phase at 24 h of treatment. As compared to treatment with 160 μM H2O2 (positive control), glycine treatment helped to generate more intracellular ROS (Fig. 12b). However, comparison indicated that N-acryloylglycine (Fig. 12c) and p(NAG-co-Ac) treatment induced similar levels of ROS generation to the positive control. Hence, this could be the reason for the necrotic cell death upon treatment with glycine. For N-acryloylglycine treatment, a relatively lower level of ROS production was observed compared to p(NAG-co-Ac) treatment, and hence apoptotic death was observed. However, this study was kept limited to flow cytometry and evaluation of molecular markers to reveal the clear inhibition mechanism.
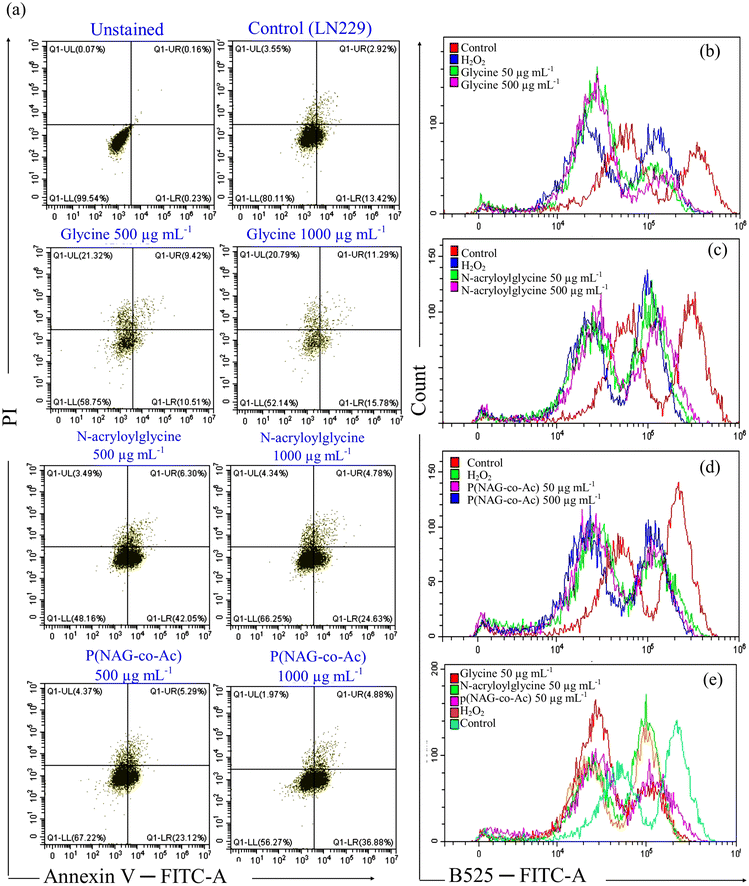 |
| | Fig. 12 (a) Flow-cytometry-based quantitative analysis (using annexin V/PI assay) of apoptotic and necrotic cell death induced by glycine, N-acryloylglycine and p(NAG-co-Ac) hydrogel, and comparison with the untreated reference control. (b–e) Intracellular ROS generation induced by (b) 50 and 500 μg mL−1 glycine, (c) 50 and 500 μg mL−1N-acryloylglycine, (d) 50 and 500 μg mL−1 p(NAG-co-Ac) hydrogel. (e) Comparison of intracellular ROS generation between b, c, and d. | |
3.9. Anti-angiogenic properties of p(NAG-co-Ac) hydrogel
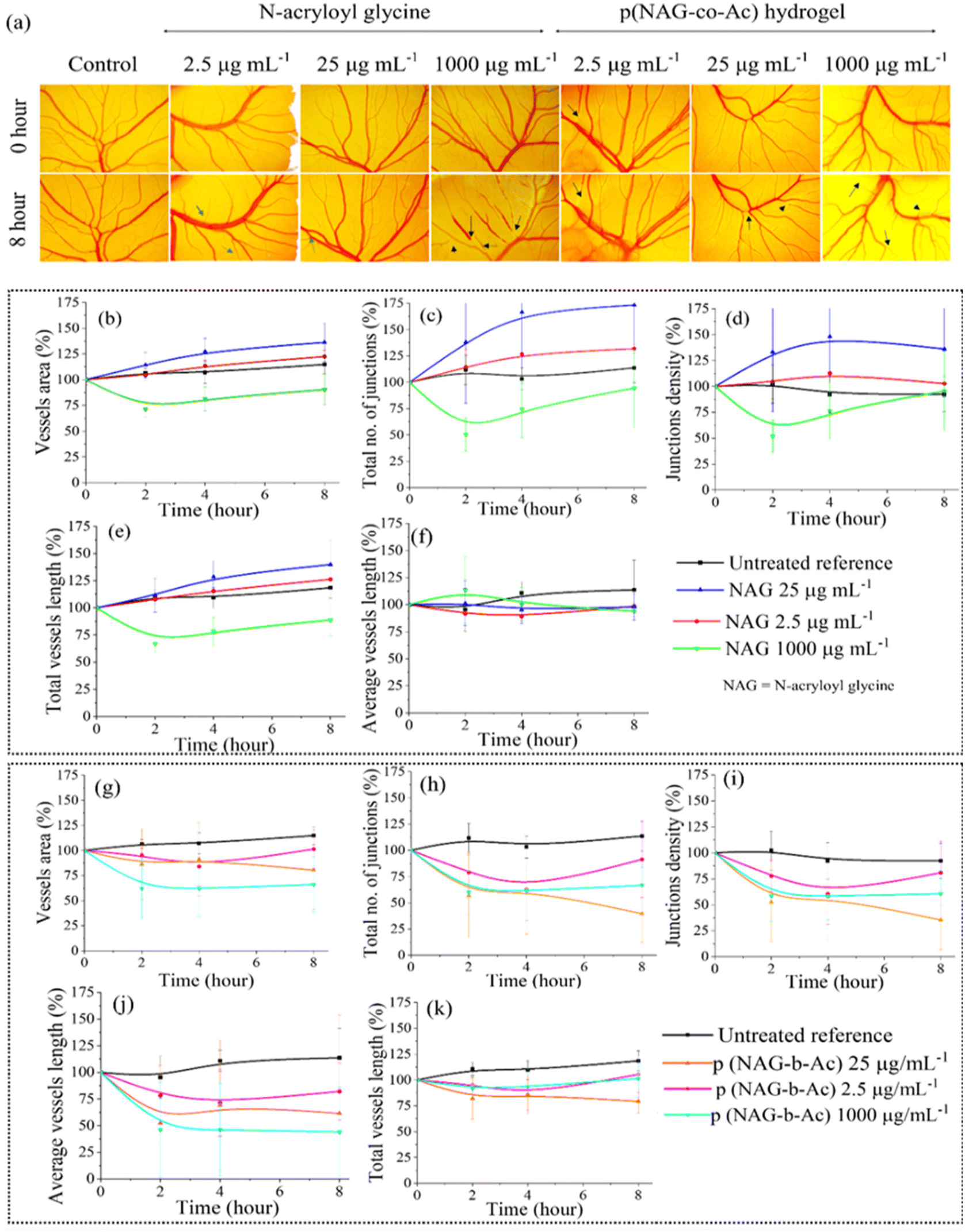
Cell surface expression of heparanase induces angiogenesis and metastasis of cancer. Therefore, a CAM assay was performed to estimate the anti-angiogenic potential of p(NAG-co-Ac) hydrogel, which was compared with that of the N-acryloyl glycine monomer. As shown in Fig. 13a, vessel sprouting was observed at lower doses of N-acryloylglycine, such as 2.5 and 25 μg mL−1; however, a higher dose of 1000 μg mL−1 led to irritation and blockage in the maturation of vasculature. In Fig. 13a, it is evident that there is a reduction in the number of vessels observed for p(NAG-co-Ac). Specifically, 8 h treatment with 2.5 μg mL−1 and 25 μg mL−1 concentrations of N-acryloylglycine (Fig. 13b–f) led to slight increases in vessel area (122.42 ± 3.32% at 2.5 μg mL−1, 136.23 ± 21.36% at 25 μg mL−1, 112.42 ± 95% for reference control), total vessel length (126.24 ± 0.57% at 2.5 μg mL−1, 139.84 ± 26% at 25 μg mL−1; 115.71 ± 10% for reference control), junction density (102.68 ± 2.12% at 2.5 μg mL−1, 136.14 ± 53.21% at 25 μg mL−1; 97.45 ± 8% for reference control) and total number of junctions (131.94 ± 5.58% at 2.5 μg mL−1, 176.23 ± 83% at 25 μg mL−1, 111.36 ± 19% for control). Treatment with 1 mg mL−1 of monomer did not result in any observed change in the mentioned parameters. However, the differences in these values are non-significant at the level of p < 0.05 compared to the control. Further, it was identified that treatment with different concentrations of p(NAG-co-Ac) hydrogel (Fig. 13g–k) led to statistically significant decreases in vessel area, total vessel length, junction density, total number of junctions and average vessel length at all concentrations. Specifically, for 2.5 μg mL−1, 25 μg mL−1 and 1 mg mL−1 concentrations of p(NAG-co-Ac), the vessel area decreased by 38.4 ± 51%, 54.03 ± 12.68% (p*** ≤ 0.001) and 57.74 ± 27.36% (p* ≤ 0.01); the total number of junctions decreased by 75.49 ± 19.48% (p** ≤ 0.005), 71.64 ± 19.09% (*p < 0.01), and 44.6 ± 19.92% (p* ≤ 0.01); junction density decreased by 63.12 ± 26.90 (p** ≤ 0.005), 78.46 ± 2.86% (p*** < 0.001), and 36.78 ± 25.48% (p = 0.05); total vessel length decreased by 26.24 ± 17.25% (p* < 0.05), 36.33 ± 12.87% (p** ≤ 0.005), and 14.71 ± 10.02% (p > 0.05) and average vessel length decreased by 81.86 ± 35.68% (p* < 0.05) 55.12 ± 34.72% (p > 0.05) and 72.64 ± 48.19% (P* < 0.05), respectively.
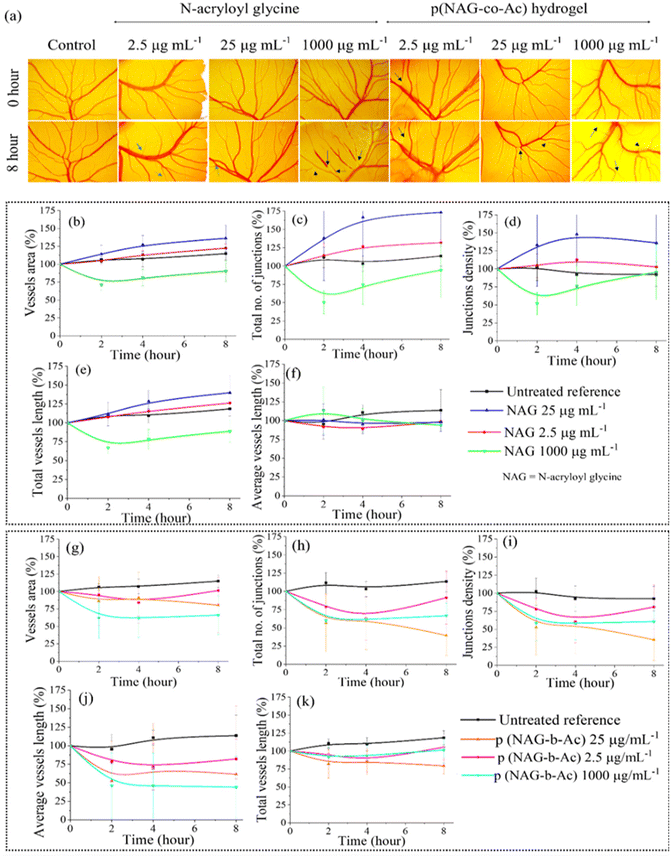 |
| | Fig. 13 Comparison of the anti-antigenic effect of N-acryloylglycine and p(NAG-co-Ac) hydrogel. (a) In ovo CEA assay in the presence of p(NAG-co-Ac) restricted vascular sprouting (marked with black arrows) in a dose-dependent manner compared to N-acryloyl glycine. Time-dependent changes in vessel area, total number of junctions, junction density, total vessel length and average vessel length at different dosages of (b–f) N-acryloylglycine and (g–k) p(NAG-co-Ac) hydrogels with a statistical significance level of p < 0.05. | |
4. Discussion
Designing an anti-tumorigenic polymer for heparanase-driven malignancies is a key research area because of the increase in chemoresistance and heterogeneity in cancer. Although chemotherapy is a primary successful treatment option, it has many limitations, as chemotherapy is a key factor in the induction of heparanase expression25,26 Heparanase promotes the aggressive malignant phenotype by creating the cancer stem cell niche within the bone marrow microenvironment25 and regulates inflammatory signalling molecules, leading to the reoccurrence of cancer with poor prognosis. Previously, Bentolia et al. developed a polymer possessing heparanase inhibitory activity. However, they restricted their study to only homopolymers. Among the various amino acids reported, glycine was selected for the present study due to the preventive effect of dietary glycine on cellular proliferation.27 Glycine shows a dose-dependent biphasic role; at lower concentrations, it shows pro-angiogenic activity, while at higher concentrations it possesses an anti-angiogenic effect28 and cardioprotective effect,39 and further plays an important role in one-carbon (1C) metabolism. However, the role of glycine in cancer is still contradictory because of the involvement of serine/glycine in the 1C metabolism pathway. We did not find any reports to date that have shown glycine activity against heparanase; however, benzoxazole is a heparanase inhibitor, and functionalization with glycine improved the heparanase inhibitory potency.40,41 Therefore, in the present study, a comparative assessment between glycine, N-acryloylglycine, homopolymers of N-acryloylglycine, heteropolymers of N-acryloyl glycine and N-acrylamide, and cross-linked homo- and heteropolymers was carried out.
Although computational approaches such as DFT for the understanding of the mechanism of chemical processes have various shortcomings, they can still provide many insights into the stability, chemical reactivity, and biological activity of newly designed polymers and give a brief idea of the underlying mechanism of the formation of the molecule.42 Here, the optimization of geometry and electronic properties for all the homo- and heteropolymeric structures were performed using a well-established basis set/force field combination (B3LYP/6-311g*). From the calculations, it is observed that introducing N-acrylamide into the homopolymer of N-acryloylglycine causes an increase in the polarity and subsequently increases the dipole moment. In contrast, the introduction of DVB to the homopolymer causes a decrease in the dipole moment value. However, the introduction of both acrylamide and DVB to the homopolymer causes a decrease in the dipole moment and accomplishes the formation of a stable structure. These dipole moment values are quite important, with the solvation parameters in selected solvents following a decreasing order of polarity. Simultaneously, the HOMO and LUMO results for the cross-linked heteropolymers showed a decrease in band gap energy as compared to the homopolymers (Fig. 2 and Table S3†). Further, the introduction of cross-linkers and acrylamide provides an increase in the biological activity of the polymers with a potential role in anticancer activity. Additionally, synthesized polymers in biological systems most commonly interact with G-coupled protein receptors, nuclear receptors, ion channels, kinase and protease, and express favourable pharmacological activity. Therefore, the prediction of bioactivity scores via DFT helps in the determination of potential applications and pharmacological categorizations. Among all the random arrangements considered, most fell under the protease inhibitory categories (Fig. 1). In malignant tumours, protease production, activation and inhibition are often disturbed, which further promotes tumour growth;26 hence, inhibitors are considered to be potent anticancer agents. In an attempt to establish heparanase inhibitory potential, blind molecular docking was performed in the present work. Glu225 and Glu343 are critical residues for functional catalytic activity. Gln270–Lys280 residues form the heparin-binding domain II, which is further responsible for the heparin binding, and Lys411–Lys417 and Lys427–Arg43 represent the third potential interactive sites (IIIrd interactive sites).24 Deletion of the Gln270–Lys280 residues results in an inactive enzyme; hence, it is one of the potential domains for heparanase inhibitor development. The homopolymers mainly interact at heparin binding domain II, whereas with the introduction of acrylamide, the hetero-linear polymer interacts at the third potential interactive site. The introduction of DVB to the homopolymer does not alter the binding sites; however, it induces increased binding affinity towards heparin binding domain II. Further, with the introduction of both DVB and acrylamide, the majority of the arrangements interact at heparin binding domain II with a higher affinity at lower inhibitory concentrations than the homo/hetero-linear polymers, and some cross-linked heteropolymers bind at the IIIrd interactive sites, which could be observed due to the formation of a stable structure. Additionally, the anticancer properties of p(NAG-co-Ac) were validated by in vitro studies. The synthesized p(NAG-co-Ac) is 40–120 nm in diameter and showed very high swelling behaviour (1579.59 ± 78.97%) in the presence of collagenase and 1063.26 ± 46.53% in PBS (Fig. 6). The swelling behaviour is well correlated with the computational findings, and we can infer that the highest swelling may have occurred due to the increase in the intermolecular hydrogen bonding energy (Table S2†). Similarly, in silico protease inhibitory activity was validated and is depicted in Fig. 1 and 6(h, i), where the p(NAG-co-Ac) hydrogel showed biodegradation in PBS and in lysozyme; however, in proteinase K solution, an increase in agglomeration was observed. A higher extent of degradation was observed in PBS. This is because the polymers mainly consist of NH2 and COOH groups and degrade primarily via hydrolysis, which is slow at neutral pH but can be accelerated by ionic interactions and temperature. In Molinspiration, the bioactivity scores are based on compound similarity with known bioactive molecules. Based on the positive bioactivity score, we can predict that certain functional groups of p(NAG-co-Ac) or structural features resemble those of known protease inhibitors. Proteinase K requires metal ions for bioactivity, which may interact with COOH groups of p(NAG-co-Ac). Furthermore, this group may chelate the metal ions or form covalent bonds with the active residues of Proteinase K, potentially could and a covalent bonds with the active residues of Proteinase K, potentially could disturb the enzyme structure. We consider that this could be the possible mechanism for the proteinase K inhibition, however, evaluation of the exact mechanism and kinetic study is a future extension of this study. We have also considered that the degradation products may yield similar arrangements of hetero/homo polymers and may give the heparanase activity.
The in vitro cytotoxicity of p(NAG-co-Ac) hydrogel was studied, and the results were compared with the cytotoxicity results for glycine and N-acryloyl glycine. The p(NAG-co-Ac) hydrogel showed higher cytotoxicity towards aggressive cancer cell lines than normal healthy cell lines. N-Acryloylglycine showed higher cell-killing efficiency in HEK293 (Fig. 8b); however, it was reduced by the formation of the p(NAG-co-Ac) hydrogel. We observed a lack of cell-killing effect of p(NAG-co-Ac) hydrogel in MCF7 cell lines (Fig. 8d), which may be due the lowered level of heparanase expression in MCF7 compared to that in MDA-MB-231 (Fig. 8f) and LN229 (Fig. 8g). The heparanase expression results in different cancer cell lines and in healthy cell lines have been presented in ESI Fig. S3 and S4,† which were analysed from supporting data obtained from the human protein atlas data bank. Further, p(NAG-co-Ac) hydrogel inhibits the migration of MDA-MB-231 (Fig. 10) and LN299 (Fig. 11) cells and induces apoptotic death in both cell lines. In LN299 cells, glycine induces necrotic death, which may be due to excessive ROS generation. In contrast, p(NAG-co-Ac) hydrogel induces programmed cell death by modulating ROS production at a therapeutic level, as shown in Fig. 12. We further observed reduced vessel area, total number of junctions, junction density and total vessel length, which demonstrate the anti-angiogenic behaviour of p(NAG-co-Ac) hydrogel (Fig. 13). This behaviour may be due to the heparanase inhibitory potency of p(NAG-co-Ac), as heparanase is tightly involved in angiogenesis. As the database shows, the highest expression of heparanase is observed in oesophageal cancers, bladder, cervical, and colorectal cancers, etc. Therefore, p(NAG-co-Ac) has huge potential for use in oesophageal, cervical and colorectal cancer therapy due to its heparanase binding capacity.
5. Conclusions
In this work, p(NAG-co-Ac) hydrogel has been successfully synthesized using N-acryloylglycine and acrylamide. It consists of a linear and cross-linked arrangement, and shows a positive score protease inhibitory in in silico investigation. With the increase in the dipole moment of the linear heteropolymer and cross-linked heteropolymers, a decrease in the energy band gap is observed. Cross-linking induces heparanase binding scores with lower Ki values in the nanomolar concentration range at heparin binding domain II. In vitro study revealed that the anti-migratory, anti-angiogenic and cancer-cell-killing potential of p(NAG-co-Ac) hydrogel in aggressive heparanase-over-expressing cancer cell lines is extraordinary. By increasing the ROS level, p(NAG-co-Ac) hydrogel promotes programmed cell death in cancer. Having revealed its anti-cancer mechanism at the molecular level, the high swelling and drug loading potential further enhance the future scope of this anticancer polymer in esophageal, cervical and colorectal cancer therapy.
Author contributions
P. P. is the main project investigator (PI). The manuscript was written and finalized by K. W. and P. P., and K. W. and P. P. contributed to ideation, experimental design, and experimental result analysis. P. S. G. assisted in characterization. Angiogenesis by G. S. In silico work by K. W., D. D. Y, A. O. and S. P. Flow cytometry by K. W. and S. K. Characterization helped by D. P. All the authors checked and approved the manuscript for publication.
Data availability
All the data in the original will be available with the communicating author of this article and they will be provided when required.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
Prof. Paik acknowledges the project funds awarded to him by I-DAPT Foundation (Ref. I-DAPT/IT(BHU)/2023-24/Project Sanction/47), STARS-IISc Bangalore (Ref. MoE-STARS/STARS-2/2023-0318), Indian Council of Medical Research (ICMR), India (Ref: EMDR/SG/12/2023-4724), Anusandhan National Research Foundation (ANRF), India (Ref: CRG/2023/005576) and Institute start-up grant/plan-OH, Instrument facilities and PARAM Shivay supercomputer facility of IIT-BHU and ISLS-CIF and Prof. Richa Arya for fluorescent microscopy facilities.
References
- E. K. Noch, R. Ramakrishna and R. Magge, World Neurosurg., 2018, 116, 505–517 CrossRef PubMed.
- R. Mahmoud, P. Ordóñez-Morán and C. Allegrucci, Cancers, 2022, 14, 4280 CrossRef CAS PubMed.
- M. Fujita, S. Yamada and T. Imai, Semin. Cancer Biol., 2015, 35, 45–52 CrossRef CAS PubMed.
- V. Jawalagatti, P. Kirthika and J. H. Lee, Mol. Ther. Oncolytics, 2022, 25, 350–363 CrossRef CAS PubMed.
- I. Vlodavsky, Y. Kayal, M. Hilwi, S. Soboh, R. D. Sanderson and N. Ilan, Proteoglycan Res., 2023, 1, e6 CrossRef PubMed.
- F. Yuan, H. Zhou, C. Liu, Y. Wang, J. Quan, J. Liu, H. Li, M. von Itzstein and X. Yu, Cancer Gene Ther., 2024, 31, 904–916 CrossRef CAS PubMed.
- Y. Song, D. Zhou, P. Zhang, N. Zhu, R. Guo, T. Wang, F. Zhuang and D. Sun, Cytotechnology, 2024, 76, 503–517 CrossRef CAS PubMed.
- A. Eatemadi, H. T. Aiyelabegan, B. Negahdari, M. A. Mazlomi, H. Daraee, N. Daraee, R. Eatemadi and E. Sadroddiny, Biomed. Pharmacother., 2017, 86, 221–231 CrossRef CAS PubMed.
- X. Hong, F. Jiang, S. N. Kalkanis, Z. G. Zhang, X. Zhang, X. Zheng, H. Jiang, T. Mikkelsen and M. Chopp, J. Exp. Clin. Cancer Res., 2008, 27, 23 CrossRef PubMed.
- T. Zahavi, M. Salmon-Divon, R. Salgado, M. Elkin, E. Hermano, A. M. Rubinstein, P. A. Francis, A. Di Leo, G. Viale, E. de Azambuja, L. Ameye, C. Sotiriou, A. Salmon, N. Kravchenko-Balasha and A. Sonnenblick, npj Breast Cancer, 2021, 7, 67 CrossRef CAS PubMed.
- A. A. N. Rodrigues, L. Lopes-Santos, P. A. Lacerda, M. F. Juste, B. A. Mariz, D. C. Cajazeiro, V. Giacobbe, R. Borges, A. Casarim, G. S. Callegari, F. A. M. Claret Arcadipane, I. Aprahamian, T. A. Salo, C. E. De Oliveira, R. D. Coletta, T. M. Augusto and N. K. Cervigne, Front. Cell Dev. Biol., 2022, 10, 742213 CrossRef PubMed.
- O. Vornicova, I. Boyango, S. Feld, I. Naroditsky, O. Kazarin, Y. Zohar, Y. Tiram, N. Ilan, O. Ben-Izhak, I. Vlodavsky and G. Bar-Sela, Oncotarget, 2016, 7, 74678–74685 CrossRef PubMed.
- U. Barash, A. Spyrou, P. Liu, E. Vlodavsky, C. Zhu, J. Luo, D. Su, N. Ilan, K. Forsberg-Nilsson, I. Vlodavsky and X. Yang, Int. J. Cancer, 2019, 145, 1596–1608 CrossRef CAS PubMed.
- D. Wang, K. Wang, Q. Liu, M. Liu, G. Zhang, K. Feng, K. Wang, X. Ding, H. Zhu, S. Yang, Y. Liu, T. Li, P. Gong, M. Wang, P. G. Wang, H. Jin, W. Zhao and F. Yu, Adv. Sci., 2024, 11, 2403337 CrossRef CAS PubMed.
- F. Veider, K. Zöller, A. Saleh and A. Bernkop-Schnürch, Int. J. Pharm., 2024, 651, 123817 CrossRef CAS PubMed.
- H. A. Yousefi Rizi, D. Hoon Shin and S. Yousefi Rizi, Iran. J. Public Health, 2022, 51, 226–239 Search PubMed.
- M. R. Tavares, M. Pechar, P. Chytil and T. Etrych, Macromol. Biosci., 2021, 21, 2100135 CrossRef CAS PubMed.
- I. Mukherjee, A. Ghosh, P. Bhadury and P. De, ACS Omega, 2017, 2, 1633–1644 CrossRef CAS PubMed.
- B. Kim, E. Lee, Y. Kim, S. Park, G. Khang and D. Lee, Adv. Funct. Mater., 2013, 23, 5091–5097 CrossRef CAS.
- H. Takahashi, K. Yumoto, K. Yasuhara, E. T. Nadres, Y. Kikuchi, L. Buttitta, R. S. Taichman and K. Kuroda, Sci. Rep., 2019, 9, 1096 CrossRef PubMed.
- S. Kocabay, M. A. Alagöz and B. Akkaya, J. Biomol. Struct. Dyn., 2024, 1–9, DOI:10.1080/07391102.2024.2317421.
- K. Sakthikumar, J. Dhaveethu Raja, R. Vijay Solomon and M. Sankarganesh, J. Biomol. Struct. Dyn., 2019, 37, 2498–2514 CrossRef CAS PubMed.
- B. Chen, Y. Ma, H. Li, X. Chen, C. Zhang, H. Wang and Z. Deng, J. Food Biochem., 2019, 43, e12968 Search PubMed.
- O. C. Adekoya, G. J. Adekoya, E. R. Sadiku, Y. Hamam and S. S. Ray, Pharmaceutics, 2022, 14, 1972 CrossRef CAS PubMed.
- N. T. El-Shamy, A. M. Alkaoud, R. K. Hussein, M. A. Ibrahim, A. G. Alhamzani and M. M. Abou-Krisha, Molecules, 2022, 27, 620 CrossRef CAS PubMed.
- E. O. Akintemi, K. K. Govender and T. Singh, Comput. Theor. Chem., 2022, 1210, 113658 CrossRef CAS.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 17 CAS.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2018, 8, e1327 CrossRef.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- D. S. Biovia, Discovery studio modeling environment, Release, 2017 Search PubMed.
- K. Wasnik, P. S. Gupta, S. Mukherjee, A. Oviya, R. Prakash, D. Pareek, S. Patra, S. Maity, V. Rai, M. Singh, G. Singh, D. D. Yadav, S. Das, P. Maiti and P. Paik, ACS Appl. Bio Mater., 2023, 6, 5644–5661 CrossRef CAS PubMed.
- M. Chen, B. Coasne, R. Guyer, D. Derome and J. Carmeliet, Nat. Commun., 2018, 9, 3507 CrossRef PubMed.
- R. Srivastava, ACS Omega, 2021, 6, 24891–24901 CrossRef CAS PubMed.
- A. J. Mayfosh, T. K. Nguyen and M. D. Hulett, Int. J. Mol. Sci., 2021, 22, 11096 CrossRef CAS PubMed.
- F. Levy-Adam, G. Abboud-Jarrous, M. Guerrini, D. Beccati, I. Vlodavsky and N. Ilan, J. Biol. Chem., 2005, 280, 20457–20466 CrossRef CAS PubMed.
-
A. Noseda and P. Barbieri, in Heparanase: From Basic Research to Clinical Applications, ed. I. Vlodavsky, R. D. Sanderson and N. Ilan, Springer International Publishing, Cham, 2020, pp. 523–538, DOI:10.1007/978-3-030-34521-1_21.
- E. B. de Melo, A. da Silveira Gomes and I. Carvalho, Tetrahedron, 2006, 62, 10277–10302 CrossRef.
- Y. Zhang, M. Xiong, Z. Chen, G. Seabra, J. Liu, C. Li and L. Cui, ACS Med. Chem. Lett., 2024, 15, 1032–1040 CrossRef CAS PubMed.
- J. Maneikyte, A. Bausys, B. Leber, N. Feldbacher, G. Hoefler, D. Kolb-Lenz, K. Strupas, P. Stiegler and P. Schemmer, Nutrients, 2020, 12, 2634 CrossRef CAS PubMed.
- S. Courtney, P. Hay and D. Scopes, PCT International Patent Applications, 2004 Search PubMed.
- A. Messore, V. N. Madia, L. Pescatori, F. Saccoliti, V. Tudino, A. De Leo, M. Bortolami, D. De Vita, L. Scipione, F. Pepi, R. Costi, S. Rivara, L. Scalvini, M. Mor, F. F. Ferrara, E. Pavoni, G. Roscilli, G. Cassinelli, F. M. Milazzo, G. Battistuzzi, R. Di Santo and G. Giannini, J. Med. Chem., 2018, 61, 10834–10859 CrossRef CAS PubMed.
-
Y. S. Mary, S. Mary, A. Bielenica, S. Armaković, S. Armaković, V. Chandramohan and M. Dammalli, Investigation of the reactivity properties of a thiourea derivative with anticancer activity by DFT, MD simulations, 2021 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1: docking poses for complexes of GA9, GA15 and GA14, Fig. S2: wound scratch assay on L929 cell line after treatment with p(Nag-co-Ac) hydrogel, Fig. S3: heparanase gene expression in cancer obtained from Human Protein Atlas, Fig. S4. Heparanase gene expression in brain cancer and in breast cancer obtained from Human Protein Atlas Table S1: bioactivity score of (a) homopolymer of n-acryloyl glycine (b) Linear hetero- polymer of n-acryloyl glycine and acrylamide and (c) cross-linked homopolymer and (d) cross-linked heteropolymer represent p(NAG-co-Ac) polymer. Table S2: DFT calculation for (a) homopolymer of n-acryloyl glycine (b) linear hetero-polymer of n-acryloyl glycine and acrylamide and (c) cross-linked homopolymer and (d) cross-linked heteropolymer represent p(NAG-co-Ac) polymer, changes in dipole moment and Intramolecular hydrogen bonding in different solvent. Table S3: HOMO LUMO energy difference with increase in size of monomer to polymer and in presence of cross. See DOI: https://doi.org/10.1039/d5nr00079c |
| ‡ Present Address: Datta Meghe Institute of Higher Education and Research, (Deemed to be University) Wardha (M.S.) India-442001. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Sukanya
Patra
a,
Prem S.
Gupta
a,
Alagu
Oviya
a,
Sandeep
Kumar
b,
Divya
Pareek
a and
Pradip
Paik
a,
Sukanya
Patra
a,
Prem S.
Gupta
a,
Alagu
Oviya
a,
Sandeep
Kumar
b,
Divya
Pareek
a and
Pradip
Paik