
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA molecular strategy for creating functional vesicles with balancing structural stability and stimuli-responsiveness†
Shoi Sasakia,
Hibiki Uenoa,
Noriyoshi Arai b,
Kouichi Asakuraa and
Taisuke Banno*a
b,
Kouichi Asakuraa and
Taisuke Banno*a
aDepartment of Applied Chemistry, Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan. E-mail: tbanno@keio.jp
bDepartment of Mechanical Engineering, Faculty of Science and Technology, Keio University, 3-4-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan
First published on 4th February 2025
Abstract
Vesicles, closed bilayer structures composed of amphiphiles, have attracted considerable attention as functional materials. Structural stability and stimulus responsiveness are required for next-generation functional vesicles. However, there is a dilemma between these properties because the desired membrane structure varies in terms of structural stability and stimulus sensitivity. Herein, we propose a new approach for the development of giant vesicles (GVs) through the molecular design and synthesis of amphiphiles with or without amide linkages, forming hydrogen bonding. From the 1H NMR analysis and fluorescence spectra of environment-responsive probes, intermolecular hydrogen bonding between the amide linkages in the membrane contributed to the enhanced structural stability of the GVs. Moreover, by adding amphiphiles containing a photoresponsive azobenzene moiety to GVs composed of amphiphiles with or without amide linkages, a distinct mechanism of photoresponsive deformation was observed: the former exhibited large and irreversible deformation, while the latter showed a modest and reversible manner due to the photoisomerisation of azobenzene under ultraviolet and subsequent visible light illumination. This difference was also attributed to the membrane structure affected by intermolecular hydrogen bonding. Based on these results, the finding provides a molecular methodology for developing highly functional vesicles.
Introduction
Vesicles are bag-like structures formed by the self-assembly of amphiphiles in water. Vesicles can contain both hydrophilic and hydrophobic substances because they have an internal water pool that can accommodate hydrophilic substances and a membrane that can hold hydrophobic substances. Because of these properties, vesicles attract significant attention as functional materials for environmental remediation and biomedical applications.1–4 For example, Sun et al. reported that vesicles composed of amphiphilic polymers with aromatic rings can be used to remove metal ions, polycyclic aromatic hydrocarbons, and cationic dyes from contaminated water.5 Furthermore, vesicles encapsulating silver or gold nanoparticles have been reported to reduce toxic nitro compounds to amine compounds.6,7 In biomedical applications, vesicles can also serve as drug carriers in drug delivery systems and can be engineered to display modified antigens on their surfaces for targeted interactions in cellular immunotherapy.8,9Balancing structural stability and stimuli-responsiveness is crucial for creating functional vesicles.10 Vesicles with exceptional structural stability or specificity to target substances or tissues have been developed through chemical modification of their surfaces or by incorporating various compounds, such as synthesised functional molecules, into the vesicle membrane.11–14 Additionally, since Kunitake et al. reported that double-chain synthetic amphiphiles with quaternary ammonium salts form vesicles in water,15 synthesised amphiphiles based on rational molecular design have been frequently reported. For example, Sawada et al. demonstrated that vesicles composed of double-chain amphiphiles with imine linkages and oleic acid were stable over a wide pH range but rapidly collapsed below a certain pH threshold.10 Furthermore, it was reported that giant vesicles (GVs) composed of hydrolysable cationic lipids with amide linkages exhibit temperature-dependent deformation.16 Vesicles made from stimuli-responsive polymers can control the uptake and release of substances by altering the membrane structure.17,18 For instance, Che et al. reported polymersomes that could regulate membrane permeability depending on the protonation of a tertiary amine,19 while Liu et al. reported polymersomes that release doxorubicin in response to temperature changes.20 Other examples include controlling membrane permeability using gas or light as stimuli.21,22 Vesicles composed of polymers exhibit excellent structural stability but relatively slow response to stimuli, whereas those composed of low-molecular-weight molecules respond quickly to stimuli but have poor structural stability.23,24 Vesicles with excellent structural stability and high substance retention capacity are preferred because they minimize the frequency and amount of dosage, reducing the burden on the natural and in vivo environments.25 Additionally, sensitivity to stimuli, such as deformation and regulation of substance release and uptake when the concentration of target components increases, is crucial.26–29
In this study, we designed and synthesised low-molecular-weight amphiphiles with amide linkages in the hydrophobic moiety (MonoAm and DiAm), as illustrated in Fig. 1. The amide and diamide structures are expected to form a hydrogen bonding network within the relatively hydrophobic membrane, resulting in vesicles with a rigid membrane structure, i.e., excellent structural stability.30 As a comparative substance, an amphiphile without an amide linkage (NoAm) was also synthesised, and its stability was evaluated. We further incorporated an amphiphile with an azobenzene moiety (Azo18) into a series of vesicles and assessed their photoresponsiveness. Azobenzene undergoes reversible isomerization from the trans-isomer to the cis-isomer under ultraviolet light (UV), and then back to the trans-isomer under visible light (Vis) illumination. Vesicles containing amphiphiles with azobenzene groups have been found to deform,31–35 fuse,36 and collapse37 under light illumination, all due to changes in the membrane structure caused by the photoisomerisation of azobenzene. Therefore, vesicles composed of amphiphiles with or without amide linkages and Azo18 are expected to exhibit distinct photoresponsive behaviours depending on the properties of their membranes. Since this study focuses on the real-time responsivity against adding electrolytes and light illumination, we carry out observations of GVs. This study presents a molecular design strategy for amphiphiles that can adjust the balance between structural stability and stimulus responsiveness of vesicles.
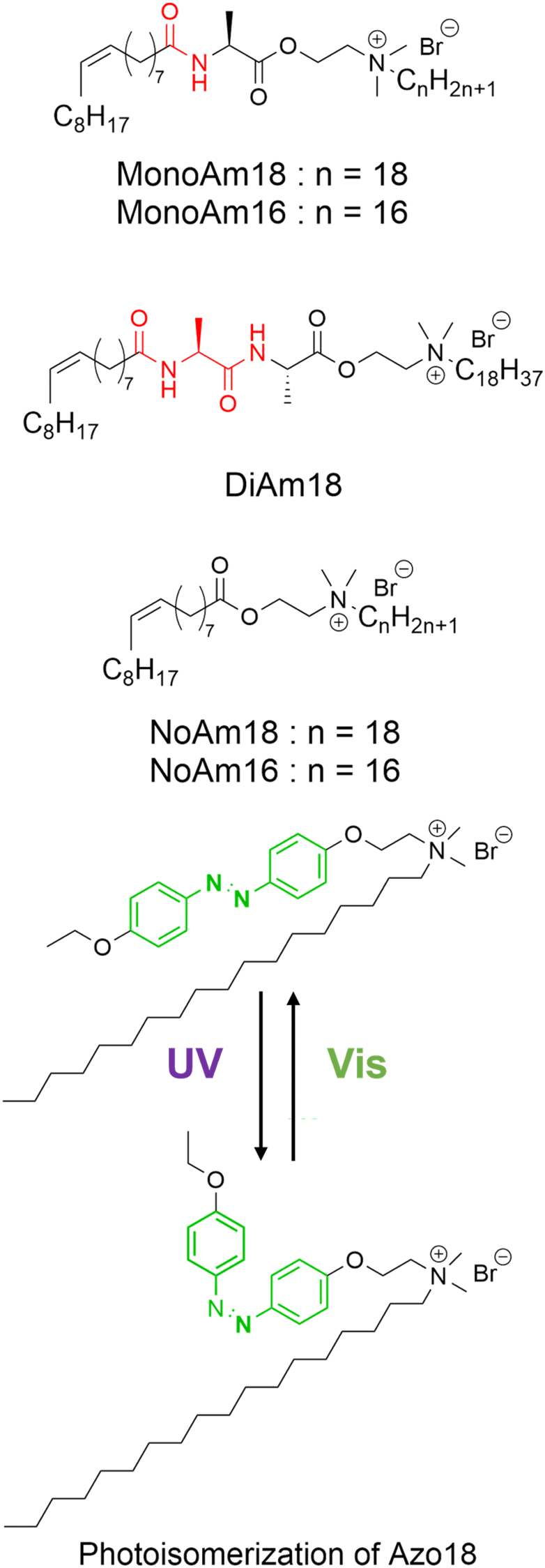 | ||
| Fig. 1 Molecular structure of amphiphiles with amide linkages (MonoAm18, MonoAm16, and DiAm18), without amide linkages (NoAm18 and NoAm16), and the azobenzene moiety (Azo18). | ||
Materials and methods
General
Commercial reagents and solvents were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan), Wako Chemical Industry Co., Ltd (Osaka, Japan), Kanto Chemical Co., Ltd (Tokyo, Japan), Thermo Fisher Scientific (Waltham, MA, USA), and Sigma-Aldrich (St Louis, MO, USA). The samples were used without further purification. The synthetic procedures for the amphiphiles used in this study are described in the ESI.† 1H NMR spectra were recorded using an ECA-500 spectrometer at 500 MHz or an ECA-400 Fourier transform spectrometer at 400 MHz (JEOL Ltd, Tokyo, Japan). The chemical shifts were calculated in parts per million (ppm) using tetramethylsilane as the standard (0 ppm). Mass spectrometry was performed by electrospray ionization using a TimsTOF instrument (Bruker, Massachusetts, USA).Preparation of microscopic observation specimens
The GV dispersions were prepared using a thin-film hydration method. Initially, an amphiphile chloroform solution and a 10 μM uranine aqueous solution were prepared. The amphiphile solution was added to a glass vial, and the amphiphile film was obtained by evaporating the solution under reduced pressure for more than 2 h. Then, a 10 μM uranine aqueous solution was added to the glass vial, and the vial was left for 2 h at room temperature (23–25 °C) to hydrate the film. The vial was agitated using a vortex mixer (TM-1; AS ONE, Osaka, Japan) to form the observation specimens. The final lipid and uranine concentrations were 2 mM and 1 μM, respectively.To investigate the photoresponsiveness of the GVs, specimens were prepared as follows: 10 mM solutions of MonoAm16 and Azo18 in chloroform, along with a 1 mM solution of Nile red, were prepared. MonoAm16, Azo18, and Nile red solutions were mixed in glass vials. The amphiphile film was obtained by evaporating the solutions under reduced pressure for more than 2 h. Then, 1 mL of deionised water was added to the glass vial, and the vial was left for 2 h at room temperature (23–25 °C) to hydrate the film. The vial was agitated using a vortex mixer to form GVs. The final concentrations were 2 mM MonoAm16/Azo18 = 1/1 (mM) and 1 μM Nile red. The specimens containing NoAm16 instead of MonoAm16 were prepared using a similar procedure.
Microscopic observation
The dispersions were sealed between two thin glass coverslips (24 × 60 mm; NEO CoverGlass, Matsunami Glass Industry, Osaka, Japan) using a frame-seal incubation chamber (15 × 15 × 0.28 mm; Bio-Rad, USA) as a spacer. The prepared sample was observed using an optical microscope (BX51, Olympus, Japan) or a confocal laser scanning microscope (FV10i-DOC, Olympus, Japan). When the specimens were illuminated with UV at 330–385 nm from the upper side for 5 min and subsequently visible light (Vis) at 460–495 nm from the upper side for 5 min, GVs were observed. All observations were conducted at room temperature (23–25 °C).Preparation of GVs with stability against osmotic shock
The GV dispersion was also prepared using a droplet transfer method. First, 10 mM amphiphile solutions were prepared using mineral oil. Next, 40 mL of 1 M sucrose, 1 M NaCl, and 100 mM uranine aqueous solutions were mixed with the amphiphilic oil solution to prepare a water-in-oil (w/o) emulsion. This emulsion was then gently loaded into 800 μL of a 1 M glucose solution in a microtube. The sample was incubated for 15 min at room temperature (23–25 °C) and then centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 rpm. The aqueous dispersion was collected from the bottom of the tube and observed under an optical microscope (BX51; Olympus, Tokyo, Japan) equipped with a CCD camera (DP22; Olympus, Tokyo, Japan). The specimens using KCl and MgCl2 instead of NaCl were prepared using a similar procedure. The concentrations of KCl and MgCl2 were 1 M and 0.66 M, respectively.
700 rpm. The aqueous dispersion was collected from the bottom of the tube and observed under an optical microscope (BX51; Olympus, Tokyo, Japan) equipped with a CCD camera (DP22; Olympus, Tokyo, Japan). The specimens using KCl and MgCl2 instead of NaCl were prepared using a similar procedure. The concentrations of KCl and MgCl2 were 1 M and 0.66 M, respectively.
Measurement of photoisomerisation of Azo18
A dispersion of 1/1 (mM/mM) MonoAm16/Azo18 was prepared similarly to the specimens for microscopic observations. The samples on Petri dishes were illuminated with UV and visible light for 5 min each. After the specimens were diluted 40 times with methanol, the absorption spectra were recorded using UV-Vis spectroscopy (UV-3600Plus, Shimadzu, Japan). The photoisomerisation ratio of Azo18 between the trans- and cis-isomers was calculated using the absorbance ratio, with the maximum absorption wavelength of trans-Azo18 being 354 nm. The photoisomerisation of Azo18 was also observed in the dispersion containing NoAm16 using a similar procedure.Estimating temperature increase due to light illumination
UV light was irradiated to thin glass coverslips (24 × 60 mm; NEO CoverGlass, Matsunami Glass Industry, Osaka, Japan) using a UV-LED lamp (LC-L1V3a, Hamamatsu Photonics) at 0.1 mW cm−2. The increase in surface temperature due to light illumination was monitored using an infrared thermography camera (FLIR ETS320; FLIR Systems, Inc., USA).Fluorescence spectroscopy using environment-responsive probes
Vesicle dispersions composed of synthesised cationic lipids and Laurdan were prepared using a thin-film hydration method. The final lipid and Laurdan concentrations were 2 and 1 mM, respectively. The dispersions were measured using a fluorometer (RF-6000, Shimadzu Corporation, Kyoto, Japan) at an excitation wavelength of 340 nm and a fluorescence wavelength range of 380 to 600 nm.The fluorescence intensity of vesicle dispersions containing Azo18 was also measured. The 2 mM amphiphile dispersion was sonicated for 2 min and extruded through a 0.8 μm pore-size polycarbonate membrane filter. The samples on a Petri dish were then illuminated with UV light and subsequently with Vis light for 5 min each. After illumination, a methanolic solution of Nile red was added. The final concentration of Nile red was 1 μM. The fluorescence intensity of these dispersions was measured using a fluorometer (RF-6000, Shimadzu Corporation, Kyoto, Japan) at an excitation wavelength of 554 nm and a fluorescence wavelength range of 580 to 800 nm.
Föster resonance energy transfer (FRET) analysis
A mixture of chloroform solutions comprising MonoAm16, Azo18, NBD-PE, and Texas-Red-DHPE was prepared and evaporated in a glass vial to form a thin film. The obtained thin films were hydrated using deionised water and maintained for more than 2 h. After gentle mixing using a vortex mixer, fluorescence spectra were recorded using a fluorometer (RF-6000, Shimadzu Corporation, Kyoto, Japan) before and after UV and Vis illumination for 5 min each. The final lipid concentrations were 2 mM MonoAm16/Azo18 = 1/1 (mM/mM), 5 μM NBD-PE, and 5 μM Texas-Red DHPE. The excitation and emission wavelengths were 460 and 480–780 nm, respectively. A similar analysis was carried out using NoAm16 instead of MonoAm16.Results and discussion
Microscopy observations and GVs’ stability against osmotic shock
Initially, a dispersion of synthesised cationic lipids was prepared using a thin-film hydration method at room temperature (23–25 °C). Immediately after specimen preparation, the spherical GVs were observed using confocal laser scanning microscopy (CLSM) (Fig. 2). Uranine was found to localize preferentially at the membrane, likely due to the strong coulombic interaction between the anionic uranine and the cationic amphiphiles. The GVs composed of MonoAm18 deformed from spherical to prolate shapes (Fig. 2a), while those composed of DiAm18 transitioned from spherical GVs to aggregates, resulting in the delocalization of uranine (Fig. 2b and Fig. S1†). In contrast, the GVs containing NoAm18 showed no significant changes even after 24 h (Fig. 2c). We quantitatively evaluated the deformation of GVs using the distortion index (D) suggested by Tomita et al.38 The distortion index is defined as D = l2/S − 4π, where l is the perimeter and S is the area of a GV on an image of the cross-section. As expected, the D values for MonoAm18-GVs were significantly higher than those for NoAm18-GVs, which were almost zero (Fig. S2†).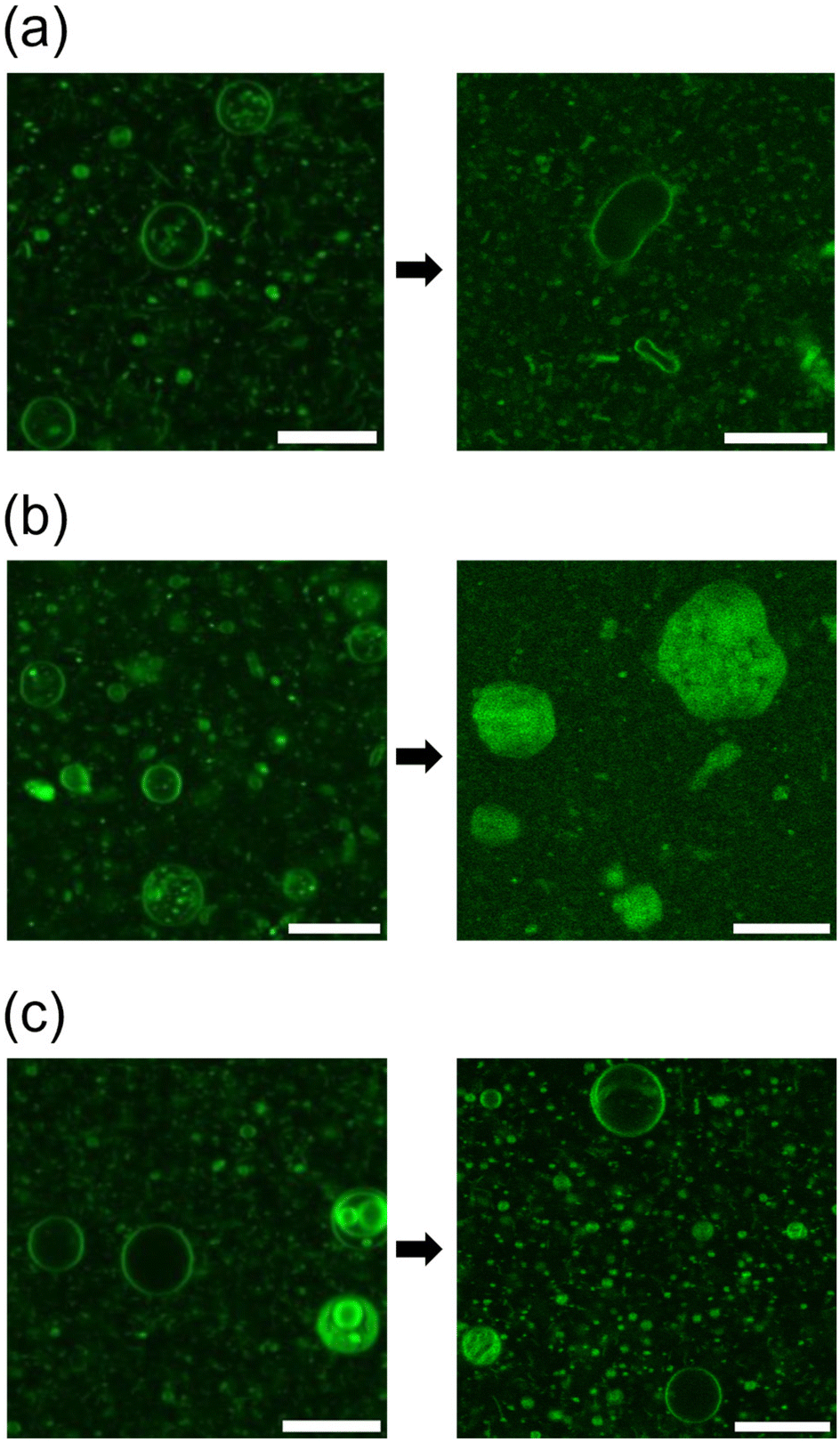 | ||
| Fig. 2 Microscopy images of GVs composed of (a) MonoAm18, (b) DiAm18, and (c) NoAm18, immediately after the sample preparation (left) and after 24 h (right). Scale bar: 20 μm. | ||
To investigate the structural stability of GVs composed of amphiphiles with amide linkages, osmotic stress was induced by adding NaCl as an osmolyte. The GVs were prepared using a droplet transfer method, a technique known to produce GVs with low lamellarity, which can influence vesicle stability.39,40 Unfortunately, GVs composed of MonoAm18 or DiAm18 were scarcely observed due to their low solubility in mineral oil. We thus carried out the time-course observation experiments under osmotic shock in three different trials for reproducibility. GVs composed of the amide-containing cationic amphiphiles MonoAm18 and DiAm18 survived in all the tested experiments, although shape changes occurred (Fig. 3a and b). Under osmotic shock, GVs composed of DiAm18 retained their spherical shape. In contrast, NoAm18-GVs completely collapsed within 60 min (Fig. 3c). GVs composed of MonoAm16 and NoAm16 showed similar tendencies to those of MonoAm18 and NoAm18, respectively (Fig. S3†). Additionally, similar experiments using KCl and MgCl2 were conducted. In the case of KCl and MgCl2, GVs composed of DiAm18 or MonoAm18 survived, and those composed of NoAm18 collapsed, which is consistent with the results using NaCl (Fig. S4 and S5†). These results strongly suggest that GVs composed of amide-containing MonoAm18 and DiAm18 have high stability to osmotic shock of various electrolytes.
 | ||
| Fig. 3 Typical sequential images of GVs containing (a) MonoAm18, (b) DiAm18, and (c) NoAm18 against osmotic shock using NaCl. (d) 1H NMR spectra depending on the DiAm18 concentration in CDCl3. Arrows indicate the shift of the peaks. Scale bar: 50 μm. | ||
Investigations on vesicular membrane structures
To quantitatively evaluate the membrane structure, fluorescence spectra were recorded using Laurdan, an environmentally responsive probe. It is empirically known that the emission intensity (I) at 440 nm and 490 nm correspond to the gel phase and the liquid crystalline phase, respectively.41 We calculated the GP values defined by (I440 nm − I490 nm)/(I440 nm + I490 nm) to estimate the ratio of the gel to liquid crystalline phases in the membrane. A higher GP value indicates a greater proportion of the gel phase, suggesting a more rigid membrane structure. The GP values for GVs composed of DiAm18, MonoAm18, and NoAm18 were −0.129, −0.143, and −0.250, respectively (Fig. S6†), indicating that vesicles composed of amide-containing amphiphiles have more rigid membrane structures.Next, we investigated the formation of hydrogen bonds between the amide linkages in the amphiphiles within the vesicular membrane. 1H NMR analysis performed in deuterium chloroform showed that, depending on the concentration of amphiphiles, the protons near the amide linkages in DiAm18 and MonoAm18 slightly but steadily shifted (Fig. 3d and Fig. S7†), suggesting intermolecular hydrogen bonding within a relatively hydrophobic environment.
Here, let us discuss the structural stability of the GVs against osmotic shock. Due to osmosis, water moves from the outer to the inner water phase, increasing the volume of the GVs and causing membrane expansion. When the membrane structure is rigid, this expansion is suppressed, preventing rupture.42 Synthesised cationic amphiphiles form hydrogen bonds within the hydrophobic membrane, contributing to a more rigid membrane structure and suppression of bursting. In other words, the hydrogen bonding of amide-containing amphiphiles contributes to the stabilisation of GVs against osmotic shock. The shape changes in GVs composed of amide-containing amphiphiles may be attributed to directional hydrogen bonding (Fig. 2a).43 It is assumed that the reorientation of amide-containing amphiphiles within the vesicular membrane leads to the formation of new hydrogen bonding, causing changes in spontaneous curvature.44 The mechanism behind the formation of aggregates from GVs composed of DiAm18 is thus thought to be a planar lamellar structure attributed to a diamide skeleton. It is considered that the planar membrane structure is formed by intermolecular hydrogen bonding.45 The planar structure is thought to be unfavorable in forming vesicles because the vesicular membrane has curvature.46 Therefore, vesicles composed of amphiphilic molecules with a diamide skeleton are considered to be kinetically unstable and transformed into thermodynamically stable aggregates. Given the high stability observed in vesicles composed of MonoAm-type amphiphiles, further investigations into their photoresponsiveness under light illumination were conducted.
Photoinduced GV deformation
To assess stimuli-responsiveness, Azo18 was mixed with GVs composed of MonoAm16 or NoAm16. Initially, spherical GVs were observed in both samples before illumination (Fig. 4a and b). Due to the low time resolution of confocal laser scanning microscopy, we used optical microscopy for further photoresponsiveness study. The photoresponsiveness was then tested under UV illumination, followed by Vis exposure. Both types of GVs deformed from spherical to non-spherical shapes under UV light. However, under subsequent Vis exposure, no significant deformation was observed in the GVs containing MonoAm16, whereas the GVs composed of NoAm16 and Azo18 almost reverted to spherical shapes (Fig. 4c and d). Additionally, GVs containing MonoAm16 deformed more slowly immediately after UV exposure, while those composed of NoAm16 showed abrupt deformation (Fig. 4e and f). Specifically, the degree of deformation of GVs containing MonoAm16 is more than one containing NoAm16, as confirmed by image analysis using the distortion index D (Fig. S8†). No deformations of both GVs were observed in the absence of Azo18 (Fig. S9†). Moreover, no photopolymerisation of the olefin bonds in MonoAm16 and NoAm16 occurred during UV illumination, as confirmed by 1H NMR (Fig. S10†). GVs containing DiAm-type amphiphiles collapsed in 30 seconds under UV illumination (Fig. S11†).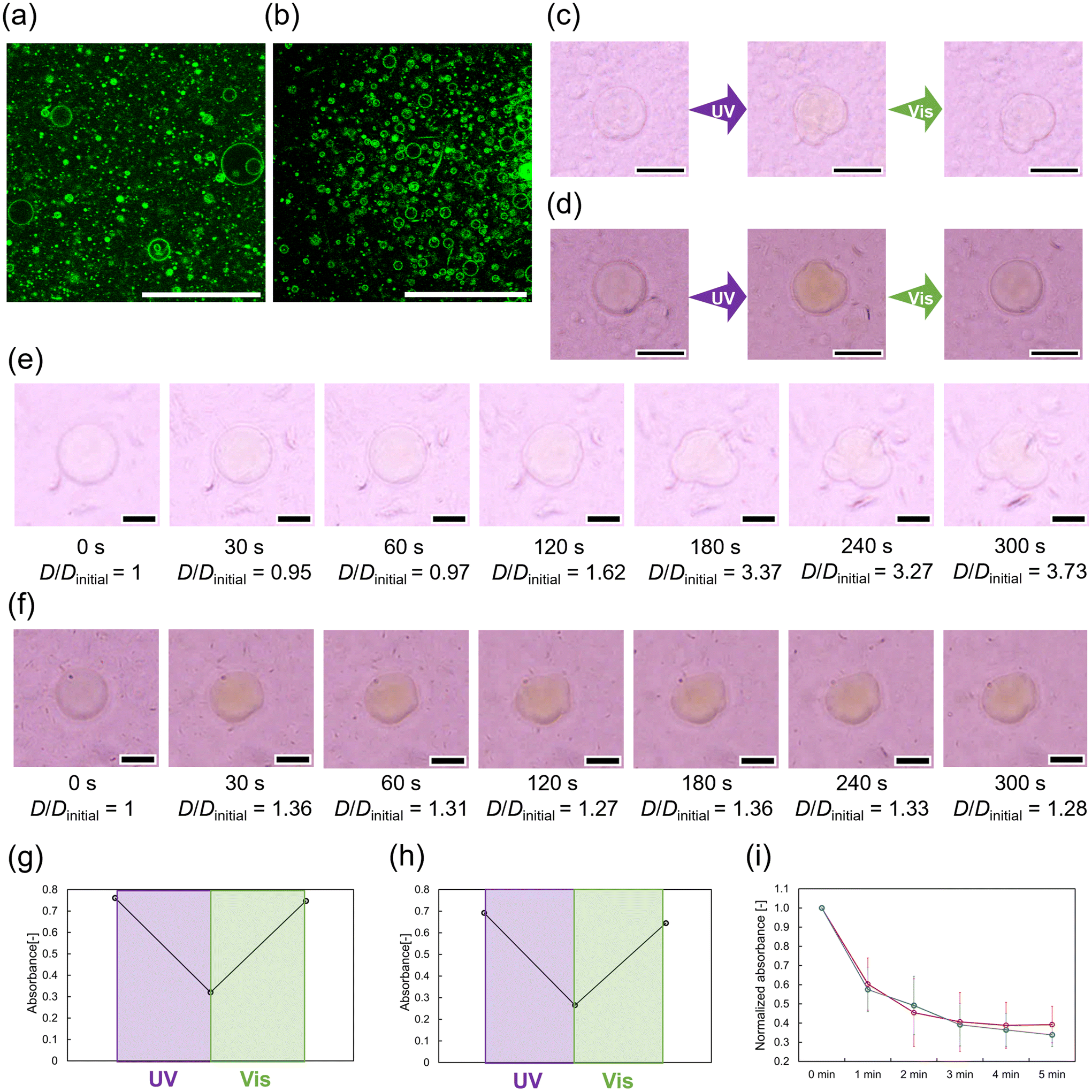 | ||
| Fig. 4 Photoinduced deformation of GVs. (a) Microscopic images of GVs composed of MonoAm16 and Azo18. Scale bar: 20 μm. (b) Microscopy images of GVs composed of NoAm16 and Azo18. Scale bar: 20 μm. (c) Typical sequential images of GVs containing MonoAm16 under UV and Vis illumination. Scale bar: 50 μm. (d) Typical sequential images of GVs containing NoAm16 under UV and Vis illumination. Scale bar: 50 μm. (e) Time-course of deformation of GVs containing MonoAm16. Scale bar: 10 μm. (f) Time-course of deformation of GVs containing NoAm16. Scale bar: 10 μm. (g) Maximum absorbance switching of Azo18 under light illumination in the GVs composed of MonoAm16. (h) Maximum absorbance switching of Azo18 under light illumination in the GVs composed of NoAm16. (i) Time-course of the normalized maximum absorbance of Azo18 in the vesicle dispersion of MonoAm16 (red) or NoAm16 (blue) under UV illumination. N = 3. | ||
Based on these results, the shape changes of the GVs could be attributed to the photoisomerisation of azobenzene-containing amphiphiles. Photoisomerisation of Azo18 in the vesicular membrane was estimated using UV-Vis absorption spectra. The vesicle dispersions were illuminated and then diluted with methanol to prepare the sample solution for absorption spectrum measurements. In samples containing MonoAm16 and NoAm16, reversible trans–cis photoisomerisation was confirmed (Fig. 4g and h). Furthermore, we measured the time-course of Azo18 absorbance in both samples. The rate of photoisomerisation of Azo18 was similar for GVs composed of both MonoAm16 and NoAm16 (Fig. 4i). The slower deformation of the GVs composed of MonoAm16 might be due to a more rigid membrane structure caused by intermolecular hydrogen bonding (Fig. S7†). Additionally, the turbidity of dispersion of Azo18 did not change during UV illumination, suggesting that cis-Azo18 is not water-soluble and is not released from the vesicular membrane during UV illumination (Fig. S12†).
To confirm the temperature increase caused by UV illumination, we monitored the temperature distribution on glass coverslips illuminated by UV light at 0.1 mW cm−2 using a thermal imaging camera. After 5 min of UV illumination, a 2.4 K temperature increase was observed in the glass coverslips (Fig. S13†). Given the high specific heat capacity of water relative to that of borosilicate glass, such a slight temperature increase due to light illumination is considered negligible.
Estimation of the membrane structure
Considering the differences in the vesicular membrane structures of MonoAm16 and NoAm16, the observed photoresponsiveness of the former could be attributed to intermolecular hydrogen bonding. To investigate these differences further, we analyzed the fluorescence spectra of Nile red and performed FRET using Texas red-DHPE and NBD-PE.Initially, the fluorescence spectra of Nile red in vesicle dispersions were recorded to investigate the effects of adding Azo18 to the MonoAm16 and NoAm16 vesicles on the vesicular structure. Nile red is an environmentally responsive probe used to characterize the properties of various molecular assemblies.47,48 The fluorescence intensity and maximum fluorescence wavelength of Nile red are sensitive to the surrounding environment; fluorescence intensity decreases, and maximum wavelength increases when the probe is in a polar medium.49–51 The addition of Azo18 to MonoAm16 vesicles resulted in a red shift of the maximum wavelength (from 629 nm to 633 nm) (Fig. 5a and b), indicating that the vesicular membrane becomes softer. This softening may be due to the interruption of hydrogen bonding between MonoAm16 molecules by Azo18. In contrast, no shift in the maximum wavelength was observed at 637 nm upon the addition of Azo18 to NoAm16 vesicles, suggesting no significant changes in intermolecular interactions between the amphiphilic molecules (Fig. 5a and c).
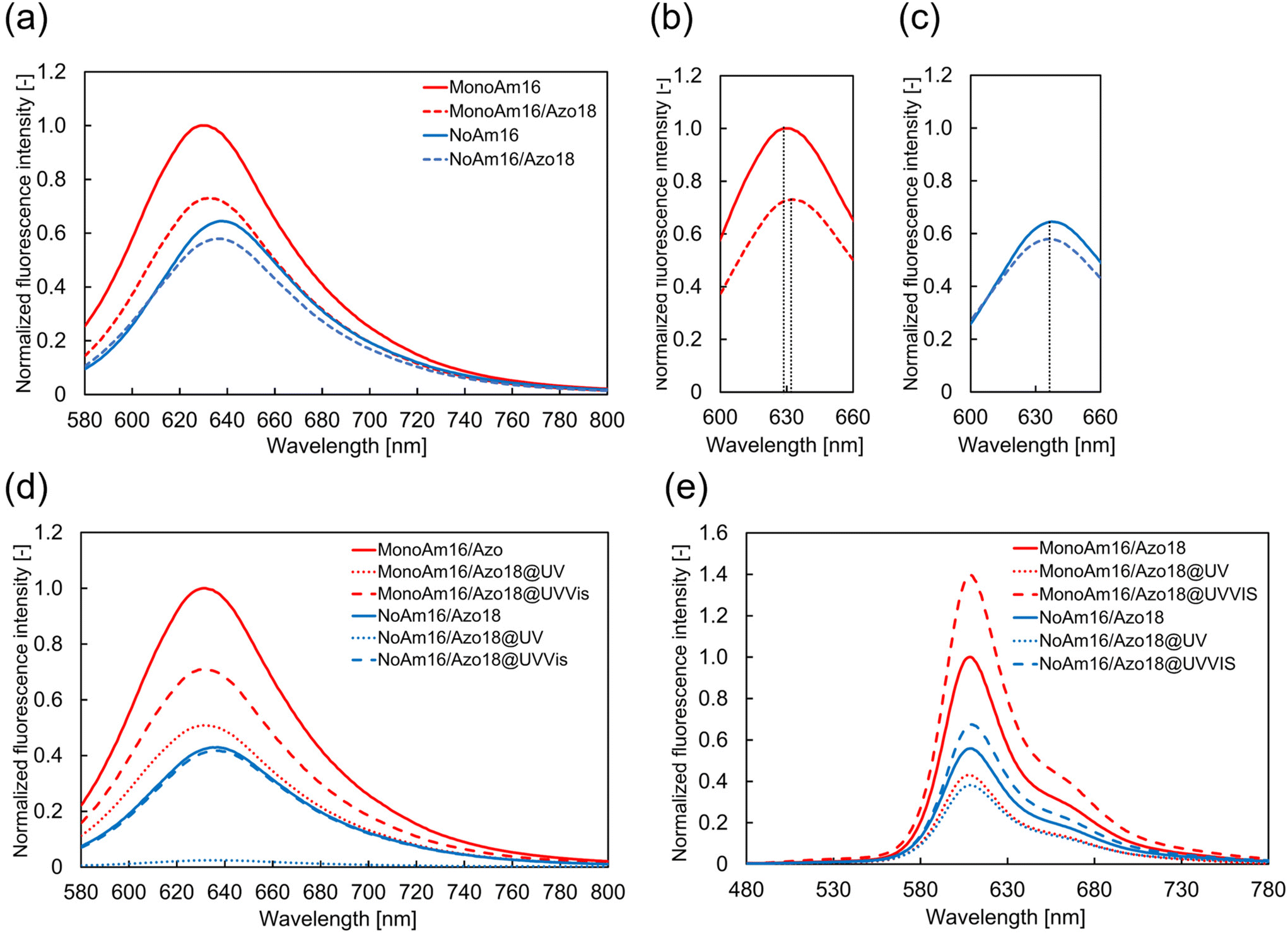 | ||
| Fig. 5 Estimation of the membrane structure. (a) Fluorescence spectrum of Nile red in vesicles. (b) Fluorescence spectra of Nile red in vesicles containing MonoAm16 and Azo18 near the peak of maximum fluorescence intensity. (c) Fluorescence spectra of Nile red in vesicles containing NoAm16 and Azo18 near the peak of maximum fluorescence intensity. (d) Fluorescence spectrum of Nile red in vesicles under light illumination. (e) Fluorescence spectrum of NBD-PE and Texas-red DHPE in vesicles before and after light illumination. | ||
The properties of the membrane before and after light illumination were estimated using Nile red. Before illumination, the fluorescence intensity of Nile red in vesicles composed of NoAm16 was lower than that in those composed of MonoAm16. Additionally, the maximum fluorescence wavelength for MonoAm16 was shorter than that for NoAm16 (Fig. 5d). These results suggest that water components were more likely incorporated into the vesicular membrane of NoAm16 than those of MonoAm16, indicating that the vesicular membrane containing MonoAm16 was more tightly packed due to intermolecular hydrogen bonding. After UV illumination, the fluorescence intensity decreased in both types of vesicles. This decrease is attributed to the change in the molecular structure of Azo18 into a bulky cis-isomer, which leads to looser packing of the membrane, allowing easier permeation of water and, thus, a decrease in fluorescence intensity. Following Vis illumination, the fluorescence intensity of the vesicles containing NoAm16 returned to its original value. However, in the case of MonoAm16, the intensity increased but did not ultimately return to the original level. This suggests that the membrane structure of the vesicles containing MonoAm16 shifted from its initial state to a different state during light illumination.
Therefore, to estimate how the membrane structure varied, FRET analysis was conducted using NBD-PE and Texas Red DHPE as the donor and acceptor, respectively. These fluorescent probes are not considered readily miscible due to the different unsaturated and saturated hydrophobic moieties.52 FRET efficiency increases with a shorter distance between the donor and acceptor within the vesicular membrane.53,54 Before light illumination, the fluorescence intensity of Texas Red-DHPE in the vesicles of MonoAm16 was higher than in those of NoAm16 (Fig. 5e), indicating tighter packing in the MonoAm16 membrane. After UV illumination, the fluorescence intensity of Texas red-DHPE decreased in both samples. Under subsequent Vis illumination, the fluorescence intensity was higher than in the initial state. To elucidate the fluorescence properties of NBD-PE and Texas-red DHPE without the effect of synthesized cationic amphiphiles, fluorescence spectra of vesicles composed of POPC and NBD-PE or Texas red-DHPE were measured. It was found that NBD-PE and Texas red-DHPE were photobleached during light illumination (Fig. S14 and S15†). Nevertheless, the higher intensity after Vis illumination suggests that the distance between the acceptor and donor was reduced because FRET is sensitive to the distance between the donor and the acceptor.53,54 This implies that the mixing degree of amphiphiles in the membrane changed due to the disturbance of the membrane structure during the photoisomerisation of Azo18.37
The results of Fig. 5d and e suggest that the membrane structure varies as follows. The mixing degree of MonoAm16 and Azo18 is low before light illumination, forming hydrogen bonding networks in the membrane. This indicates a rigid membrane structure. Under UV illumination, water components easily inflow to the vesicular membrane due to the photoisomerisation from the trans-isomer to the cis-isomer of Azo18, which partially breaks hydrogen bonding networks. Through UV illumination, the mixing degree of MonoAm16 and Azo18 is high since the membrane fluidity becomes high. Under subsequent Vis illumination, though the mixing degree does not vary, the hydrogen bonding between MonoAm16 molecules partially reforms, accompanied by the release of water components, due to the reverse photoisomerization of Azo18. The MonoAm16-GV membrane after UV and Vis illumination is thus softer than that before light illumination (Fig. 6).
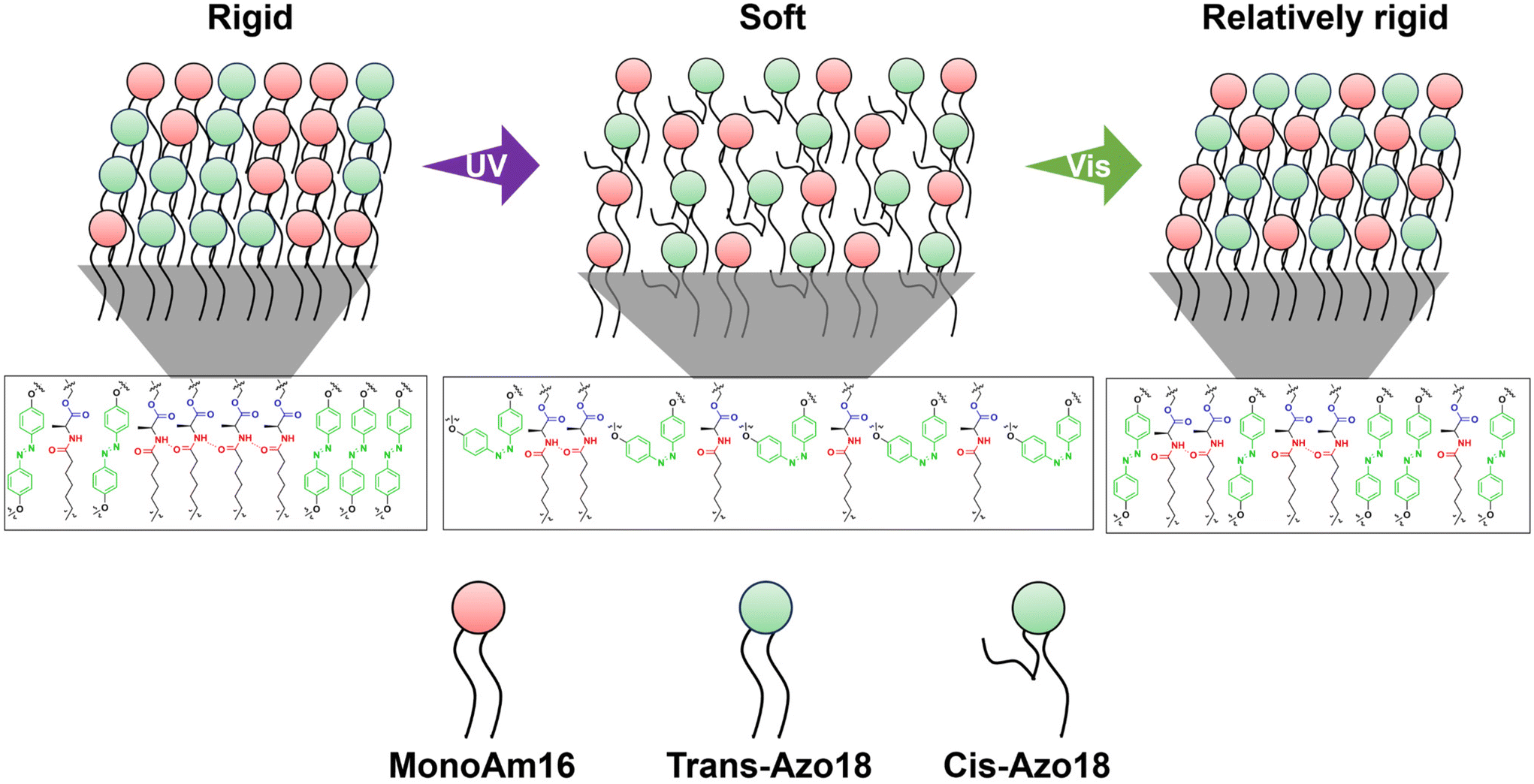 | ||
| Fig. 6 Proposed mechanism of MonoAm16-GVs deformation in response to UV and Vis illumination. | ||
Proposed mechanism for photoinduced deformation
We explore the mechanism of photoinduced deformation of GVs using the spontaneous curvature model.44 According to this model, the shape of the vesicles is determined by the minimum bending energy of the membrane. The bending energy, with surface area A and volume V of the vesicles, is expressed using the equation: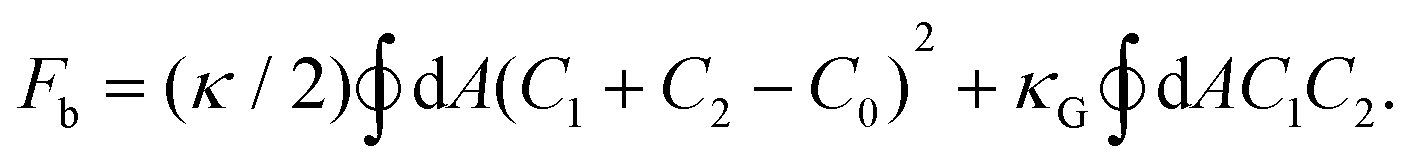
Here, C1 and C2 are the two principal curvatures and C0 is the spontaneous curvature, introduced to account for potential asymmetry in the bilayers. κ and κG denote the local and Gaussian bending constants, respectively. The second term of the equation can be neglected when no changes in vesicle topology occur.42 The phase diagram of the equilibrium shape, based on the spontaneous curvature model, is determined by two parameters: the reduced volume v and the reduced spontaneous curvature c0. The reduced volume v is calculated as follows
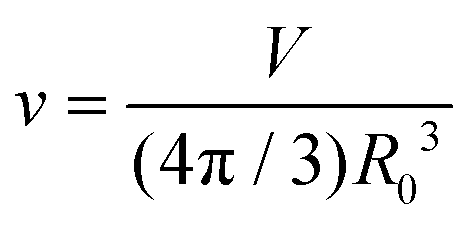
| R0 = (A/4π)1/2 |
| c0 = C0R0. |
It is estimated that introducing Azo18 into the membrane itself does not affect the membrane curvature because spherical GVs are observed before light illumination when Azo18 is added to the vesicles. Under UV illumination, the photoisomerisation of Azo18 from the trans-isomer to the bulky cis-isomer increases the membrane surface area A because the cis-isomer occupies a larger area in the membrane.35 This may decrease v. Moreover, photoisomerisation from the trans-isomer to the cis-isomer disrupts the orientation of amphiphiles, leading to membrane reorganisation37 and a change in spontaneous curvature C0.55 Therefore, the transformation occurs from spherical to non-spherical GVs. Intermolecular hydrogen bonding between MonoAm16 molecules causes further changes in the molecular orientation within the membrane due to its directionality, resulting in larger deformation. Membrane reorganisation also affords changes in the mixing degree within the membrane. Under subsequent Vis illumination, photoisomerisation of Azo18 from the cis-isomer to the trans-isomer occurs, leading to an instantaneous increase in v. As a result, the membrane structure of GVs containing NoAm16 returns to its original state after UV and subsequent Vis illumination, transforming from non-spherical back to spherical because the membrane of NoAm16-GVs is soft. On the other hand, a part of hydrogen bonding within the MonoAm16-GV membrane is quickly reformed after Vis illumination, resulting in a relatively rigid membrane (Fig. 6). Therefore, due to this reorganised membrane structure, their deformed non-spherical shape maintains.
Overall, the introduction of amide linkages provides both high structural stability and sensitive photoresponsiveness of vesicles. These findings may provide the molecular strategy of balancing contradictory properties, stability and sensitivity, and potentially contribute to the development of highly functional vesicles.
Conclusions
In this study, we investigated the structural stability and photoresponsiveness of GVs containing amphiphiles with amide linkages. GVs composed of amphiphiles with amide linkages exhibited higher durability against osmotic pressure compared to GVs composed of amphiphiles without amide linkages. From the 1H NMR analysis and fluorescence spectra of environmentally responsive probes, it was determined that the structural stability resulted from intermolecular hydrogen bonding among the amide-containing amphiphiles in the membrane. The addition of amphiphiles with an azobenzene moiety to GVs composed of amphiphiles with and without amide linkages led to different photoresponsive deformations: the former exhibited large and irreversible deformation, while the latter showed modest and reversible deformation. In both cases, the photoisomerisation of azobenzene under UV and subsequent Vis illumination and their ratio were similar. The different manner in photoresponsive deformation was attributed to the rigidity of the membrane due to intermolecular hydrogen bonding between amide-containing amphiphiles. These findings provide a new molecular strategy for resolving the longstanding dilemma of balancing structural stability with sensitivity to stimuli, potentially contributing to the development of highly functional vesicles.Author contributions
S. S., H. U. and T. B. designed the experiments. S. S. and H. U. performed the experiments. S. S., H. U., N. A., K. A. and T. B. discussed the results. S. S. and T. B. wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Exploratory Research Promotion Fund from the Keio Leading-edge Laboratory of Science and Technology. This was also partially supported by JSPS KAKENHI (Grant No. JP20H02712) and the JSPS Japan–Hungary Bilateral Joint Research Project (JPJSBP120213801).References
- R. S. Gupta, N. Padmavathy and S. Bose, Adv. Sustainable Syst., 2021, 5, 2100213 CrossRef.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS PubMed.
- I. A. Chacko, V. M. Ghate, L. Dsouza and S. A. Lewis, Colloids Surf., B, 2020, 195, 111262 CrossRef CAS PubMed.
- R. P. Brinkhuis, F. P. J. T. Rutjes and J. C. M. Van Hest, Polym. Chem., 2011, 2, 1449 Search PubMed.
- H. Sun, J. Jiang, Y. Xiao and J. Du, ACS Appl. Mater. Interfaces, 2018, 10, 713–722 CrossRef CAS PubMed.
- Y. Zhu, L. Fan, B. Yang and J. Du, ACS Nano, 2014, 8, 5022–5031 CrossRef CAS PubMed.
- J. Xiao and J. Du, Polym. Chem., 2016, 7, 4647–4653 RSC.
- Y. Zhou, R. Chen, H. Yang, C. Bao, J. Fan, C. Wang, Q. Lin and L. Zhu, J. Mater. Chem. B, 2020, 8, 727–735 RSC.
- A. C. Wauters, J. F. Scheerstra, I. G. Vermeijlen, R. Hammink, M. Schluck, L. Woythe, H. Wu, L. Albertazzi, C. G. Figdor, J. Tel, L. K. E. A. Abdelmohsen and J. C. M. Van Hest, ACS Nano, 2022, 16, 15072–15085 CrossRef CAS PubMed.
- D. Sawada, A. Hirono, K. Asakura and T. Banno, RSC Adv., 2020, 10, 34247–34253 RSC.
- U. Kauscher, M. N. Holme, M. Björnmalm and M. M. Stevens, Adv. Drug Delivery Rev., 2019, 138, 259–275 CrossRef CAS PubMed.
- V. De Leo, F. Milano, A. Agostiano and L. Catucci, Polymers, 2021, 13, 1027 CrossRef CAS PubMed.
- P. S. Zangabad, S. Mirkiani, S. Shahsavari, B. Masoudi, M. Masroor, H. Hamed, Z. Jafari, Y. D. Taghipour, H. Hashemi, M. Karimi and M. R. Hamblin, Nanotechnol. Rev., 2018, 7, 95–122 CAS.
- H. Moulahoum, F. Ghorbanizamani, F. Zihnioglu and S. Timur, Bioconjugate Chem., 2021, 32, 1491–1502 CAS.
- T. Kunitake and Y. Okahata, J. Am. Chem. Soc., 1977, 99, 3860–3861 Search PubMed.
- M. Tameyuki, H. Hiranaka, T. Toyota, K. Asakura and T. Banno, Langmuir, 2019, 35, 17075–17081 CAS.
- H. Che and J. C. M. van Hest, ChemNanoMat, 2019, 5, 1092–1109 CAS.
- Y. Zhu, S. Cao, M. Huo, J. C. M. Van Hest and H. Che, Chem. Sci., 2023, 14, 7411–7437 CAS.
- H. Che, S. Cao and J. C. M. Van Hest, J. Am. Chem. Soc., 2018, 140, 5356–5359 CAS.
- F. Liu, V. Kozlovskaya, S. Medipelli, B. Xue, F. Ahmad, M. Saeed, D. Cropek and E. Kharlampieva, Chem. Mater., 2015, 27, 7945–7956 CAS.
- Q. Yan and W. Sang, Chem. Sci., 2016, 7, 2100–2105 CAS.
- O. Rifaie-Graham, J. Yeow, A. Najer, R. Wang, R. Sun, K. Zhou, T. N. Dell, C. Adrianus, C. Thanapongpibul, M. Chami, S. Mann, J. R. De Alaniz and M. M. Stevens, Nat. Chem., 2023, 15, 110–118 CrossRef CAS PubMed.
- E. Rideau, R. Dimova, P. Schwille, F. R. Wurm and K. Landfester, Chem. Soc. Rev., 2018, 47, 8572–8610 RSC.
- P. Tanner, P. Baumann, R. Enea, O. Onaca, C. Palivan and W. Meier, Acc. Chem. Res., 2011, 44, 1039–1049 CrossRef CAS PubMed.
- C. Kurokawa, K. Fujiwara, M. Morita, I. Kawamata, Y. Kawagishi, A. Sakai, Y. Murayama, S. M. Nomura, S. Murata, M. Takinoue and M. Yanagisawa, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 7228–7233 CrossRef CAS PubMed.
- W. Li, S. Zhang, M. Sun, S. Kleuskens and D. A. Wilson, Acc. Mater. Res., 2024, 5, 453–466 CrossRef CAS PubMed.
- T. Nishimura and K. Akiyoshi, Adv. Sci., 2018, 5, 1800801 CrossRef PubMed.
- S. Cao, T. Ivanov, M. De Souza Melchiors, K. Landfester and L. Caire Da Silva, ChemBioChem, 2023, 24, e202200718 CrossRef CAS PubMed.
- Y. Wang, J. D. Byrne, M. E. Napier and J. M. DeSimone, Adv. Drug Delivery Rev., 2012, 64, 1021–1030 CrossRef CAS PubMed.
- H. Sakaino, J. Sawayama, S. Kabashima, I. Yoshikawa and K. Araki, J. Am. Chem. Soc., 2012, 134, 15684–15687 CAS.
- V. N. Georgiev, A. Grafmüller, D. Bléger, S. Hecht, S. Kunstmann, S. Barbirz, R. Lipowsky and R. Dimova, Adv. Sci., 2018, 5, 1800432 CrossRef PubMed.
- C. Pernpeintner, J. A. Frank, P. Urban, C. R. Roeske, S. D. Pritzl, D. Trauner and T. Lohmüller, Langmuir, 2017, 33, 4083–4089 CrossRef CAS PubMed.
- Y. Kageyama, N. Tanigake, Y. Kurokome, S. Iwaki, S. Takeda, K. Suzuki and T. Sugawara, Chem. Commun., 2013, 49, 9386 CAS.
- T. Hamada, R. Sugimoto, M. C. Vestergaard, T. Nagasaki and M. Takagi, J. Am. Chem. Soc., 2010, 132, 10528–10532 CrossRef CAS PubMed.
- M. Aleksanyan, A. Grafmüller, F. Crea, V. N. Georgiev, N. Yandrapalli, S. Block, J. Heberle and R. Dimova, Adv. Sci., 2023, 10, 2304336 CrossRef CAS PubMed.
- Y. Suzuki, K. H. Nagai, A. Zinchenko and T. Hamada, Langmuir, 2017, 33, 2671–2676 CrossRef CAS PubMed.
- A. Diguet, M. Yanagisawa, Y.-J. Liu, E. Brun, S. Abadie, S. Rudiuk and D. Baigl, J. Am. Chem. Soc., 2012, 134, 4898–4904 CrossRef CAS PubMed.
- T. Tomita, T. Sugawara and Y. Wakamoto, Langmuir, 2011, 27, 10106–10112 CrossRef CAS PubMed.
- Y. Zhang, H. Obuchi and T. Toyota, Membranes, 2023, 13, 440 CrossRef PubMed.
- L. Tayebi, D. Vashaee and A. N. Parikh, ChemPhysChem, 2012, 13, 314–322 CrossRef CAS PubMed.
- T. Parasassi, G. De Stasio, A. d'Ubaldo and E. Gratton, Biophys. J., 1990, 57, 1179–1186 CrossRef CAS PubMed.
- Y. Miele, G. Holló, I. Lagzi and F. Rossi, Life, 2022, 12, 841 CrossRef CAS PubMed.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- U. Seifert, K. Berndl and R. Lipowsky, Phys. Rev. A, 1991, 44, 1182–1202 CrossRef CAS PubMed.
- S. Ogi, K. Matsumoto and S. Yamaguchi, Angew. Chem., 2018, 130, 2363–2367 CrossRef.
- D. D. Lasic, Angew. Chem., Int. Ed. Engl., 1994, 33, 1685–1698 CrossRef.
- D. Sawada, K. Asakura and T. Banno, Chem. – Eur. J., 2021, 27, 13840–13845 CrossRef CAS PubMed.
- J. P. N. Silva, M. E. C. D. R. Oliverira and P. J. G. Coutinho, J. Photochem. Photobiol., A, 2009, 203, 32–39 CrossRef CAS.
- J. Sot, L. Gartzia-Rivero, J. Bañuelos, F. M. Goñi and A. Alonso, J. Mol. Liq., 2022, 363, 119874 CrossRef CAS.
- S. Mukherjee, H. Raghuraman and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 59–66 CrossRef CAS PubMed.
- A. Halder, B. Saha, P. Maity, G. S. Kumar, D. K. Sinha and S. Karmakar, Spectrochim. Acta, Part A, 2018, 191, 104–110 CrossRef CAS PubMed.
- S. L. Regen, Biochemistry, 2020, 59, 4617–4621 CrossRef CAS PubMed.
- L. M. S. Loura and M. Prieto, Front. Physiol., 2011, 2, 1–11 Search PubMed.
- C. Ghatak, V. G. Rao, R. Pramanik, S. Sarkar and N. Sarkar, Phys. Chem. Chem. Phys., 2011, 13, 3711–3720 RSC.
- R. Lipowsky, Adv. Biol., 2022, 6, 2101020 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00151j |
| This journal is © The Royal Society of Chemistry 2025 |