
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorinated metal–organic frameworks: hydrophobic nanospaces with high fluorine density and proton conductivity†
Ryota
Akai
 a,
Hitoshi
Kasai
a and
Kouki
Oka
*abc
a,
Hitoshi
Kasai
a and
Kouki
Oka
*abc
aInstitute of Multidisciplinary Research for Advanced Materials, Tohoku University, 2-1-1 Katahira, Aoba-ku, Sendai, Miyagi 980-8577, Japan. E-mail: oka@tohoku.ac.jp
bCarbon Recycling Energy Research Center, Ibaraki University, 4-12-1 Nakanarusawa, Hitachi, Ibaraki 316-8511, Japan
cDeuterium Science Research Unit, Center for the Promotion of Interdisciplinary Education and Research, Kyoto University, Yoshida, Sakyo-ku, Kyoto 606-8501, Japan
First published on 14th March 2025
Abstract
The current work revealed the relationship between the fluorine density of organic-based porous materials and water dynamics (proton conduction) in the hydrophobic nanospace. In detail, by focusing on UiO-66 with structural durability at high temperature/humidity based on strong Zr–O bonds, we prepared UiO-66-CF3 and UiO-66-(CF3)2 with different fluorine densities. The activation energies for proton conduction of UiO-66-CF3 (0.91 eV) and UiO-66-(CF3)2 (1.38 eV) were significantly larger than that in UiO-66 (0.47 eV) depending on their fluorine density. Introducing fluorine into organic-based porous materials allowed the hydrophobic nanospace to interact with protons, yielding a larger energy for proton conduction (activation energy). The fluorine density and activation energy were proportional. We clarified that the activation energy of proton conduction increased proportionally with the fluorine density in the nanospace. This indicated the possibility that the state of proton/water in the nanospace could be freely controlled by the fluorine density.
Introduction
Fluorinated organic-based materials exhibit high hydrophobicity and chemical durability owing to their specific interaction with water based on the high electronegativity of fluorine,1,2 and are suitable for various applications, such as hydrous proton conductors,3,4 water–oil separating materials,5,6 and electrocatalysts.5,7,8 Even among organic-based materials with low water resistance, those with fluorine have been extensively investigated for proton exchange membrane applications in fuel cells as hydrous proton conductors.3,4,9 However, the relationship between fluorine density and proton conductivity within the hydrophobic nanospace constructed by fluorine introduction has not been elucidated.Among organic-based porous materials with robust paths for proton conduction, metal–organic frameworks (MOFs) exhibit high crystallinity compared to covalent organic frameworks (COFs) and hydrogen-bonded organic frameworks (HOFs).10–14 Moreover, MOFs maintain high crystallinity even after introducing substituents to the linkers.10,11 Among them, UiO-66, which yields high chemical and thermal durability in water and organic solvents owing to strong Zr–O bonds,15–18 provides a platform for elucidating structure–property relationships.19–21 Focusing on proton conduction, Kitagawa et al. investigated the effect of the number of missing linkers in UiO-66 on the proton conductivity.22 Li et al. investigated the effect of the acidity of the functional groups introduced into UiO-66 on the proton conductivity.23 Li et al. investigated the relationship between the number of carboxy groups and proton conductivity in Ce-UiO-66 with Ce as the metal and carboxy groups as acidic functional groups.24 We have reported that highly fluorinated HOFs exhibited high water durability and proton conductivity.3 However, studies for improving proton conductivity by modulating the interaction with water and protons in the nanospace depending on the number of introduced fluorine substituents are limited.
In the current work, we prepared UiO-66-CF3 and UiO-66-(CF3)2 with different numbers of fluorine atoms in the linker to change the fluorine density in the nanospaces of UiO-66. We demonstrated that introducing fluorine into organic-based porous materials allowed the hydrophobic nanospace to interact with water and protons, yielding larger energy for proton conduction (activation energy) and a predominance of the vehicle mechanism. We revealed that the fluorine density and activation energies were proportional.
Experimental section
Preparation of UiO-66
We prepared UiO-66 with reference to a previous work.25 ZrCl4 (291 mg, 1.25 mmol) and terephthalic acid (208 mg, 1.25 mmol) were dissolved in a solution of N,N-dimethylformamide (10 mL), acetic acid (2 mL), and water (0.14 mL), and the mixture was sonicated at 20 °C for 5 min and stirred at 20 °C for 15 min. Finally, the mixture was placed in a microwave oven and irradiated at 120 °C for 15 min. The product was centrifuged, then washed with N,N-dimethylformamide and methanol, and, lastly, dried at 120 °C for 5 h, affording a white solid UiO-66 (68 mg).Results and discussion
Preparation and characterization of UiO-66-X
As shown in Fig. 1, based on previous works,25,26 we synthesized UiO-66-X (-X = -H, -CF3, and -(CF3)2) with terephthalic acid, 2-(trifluoromethyl)terephthalic acid, and 2,5-bis(trifluoromethyl)terephthalic acid as linker molecules, respectively, by a microwave method (the details are described in the Experimental section). Fig. S1† supports the introduction of trifluoromethyl groups in UiO-66-CF3 and UiO-66-(CF3)2. UiO-66-X were activated under vacuum at 120 °C for 5 h. As shown in Fig. S2,† all solvents used in the preparation of UiO-66-X were completely removed. As shown in the scanning electron microscopy (SEM) images in Fig. 1, the particle sizes of UiO-66, UiO-66-CF3, and UiO-66-(CF3)2 were about 200 nm, 200 nm, and 50–200 nm, respectively. | ||
| Fig. 1 Preparation and scanning electron microscopy (SEM) images (scale bar: 100 nm) of (a) UiO-66, (b) UiO-66-CF3, and (c) UiO-66-(CF3)2. | ||
In this section, we have characterized UiO-66-X. As shown in Fig. 2a and Table S1,† the Brunauer–Emmett–Teller (BET) specific surface area and pore volume of UiO-66-X were evaluated based on the adsorption/desorption isotherms of N2 at 77 K. As shown in Table S1,† the BET specific surface areas of UiO-66-X (-X = -H, -CF3, and -(CF3)2) were 2143 (×103 m2 mol−1), 1825 (×103 m2 mol−1), and 970 (×103 m2 mol−1), respectively. As shown in Fig. S3 and Table S1,† the pore volumes were 2220 (cm3 mol−1), 1836 (cm3 mol−1), and 395 (cm3 mol−1), respectively. As shown in the powder X-ray diffraction (PXRD) analysis in Fig. 2b, UiO-66-X (-X = -H, -CF3, and -(CF3)2) were isostructural, and therefore, as shown in Table S1,† their BET specific surface area and pore volume decreased according to the number of introduced trifluoromethyl groups. Fig. 2c and Table S2† depict Fourier transform infrared (FT-IR) spectra, in which four peaks at 1610 cm−1, 1410 cm−1, 1363 cm−1, and 1067 cm−1 were observed for UiO-66-X (-X = -H, -CF3, and -(CF3)2). These originate from COO− units.24 The peak at 1520 cm−1 was attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of benzene rings.27 Three peaks at 1300 cm−1, 1273 cm−1, and 1140 cm−1, which were observed only for UiO-66-CF3 and UiO-66-(CF3)2, were ascribed to the C–F bond.28,29 Two peaks at 909 cm−1 and 744 cm−1 were due to C–H bonds.30 Two peaks at 661 cm−1 and 604 cm−1 were attributed to Zr–Oμ3-O and Zr–(OC) bonds.27 The broad peak between 3000 cm−1 and 3500 cm−1 originated from absorbed water.27 Since UiO-66-X usually has missing linkers,20 as shown in Fig. S4,† the percentages of missing linkers were evaluated based on thermogravimetric analysis (TGA) (the details are described in section 2.3 of the ESI).† Defect-free UiO-66-X had six linkers per Zr cluster. UiO-66-X (X = -H, -CF3, and -(CF3)2) had 3.26 (-H), 4.58 (-CF3), and 4.21 (-(CF3)2) linkers, respectively. Introducing electron-withdrawing substituents into terephthalic acid enabled a smaller pKa of the carboxy group31 and easier coordination to Zr, yielding fewer structural defects in UiO-66-CF3 than in UiO-66 despite the similar synthesis conditions. Two isostructural UiO-66-X materials with different fluorine densities were successfully prepared.
C bonds of benzene rings.27 Three peaks at 1300 cm−1, 1273 cm−1, and 1140 cm−1, which were observed only for UiO-66-CF3 and UiO-66-(CF3)2, were ascribed to the C–F bond.28,29 Two peaks at 909 cm−1 and 744 cm−1 were due to C–H bonds.30 Two peaks at 661 cm−1 and 604 cm−1 were attributed to Zr–Oμ3-O and Zr–(OC) bonds.27 The broad peak between 3000 cm−1 and 3500 cm−1 originated from absorbed water.27 Since UiO-66-X usually has missing linkers,20 as shown in Fig. S4,† the percentages of missing linkers were evaluated based on thermogravimetric analysis (TGA) (the details are described in section 2.3 of the ESI).† Defect-free UiO-66-X had six linkers per Zr cluster. UiO-66-X (X = -H, -CF3, and -(CF3)2) had 3.26 (-H), 4.58 (-CF3), and 4.21 (-(CF3)2) linkers, respectively. Introducing electron-withdrawing substituents into terephthalic acid enabled a smaller pKa of the carboxy group31 and easier coordination to Zr, yielding fewer structural defects in UiO-66-CF3 than in UiO-66 despite the similar synthesis conditions. Two isostructural UiO-66-X materials with different fluorine densities were successfully prepared.
 | ||
| Fig. 2 (a) N2 adsorption/desorption isotherms of UiO-66 (black), UiO-66-CF3 (blue), and UiO-66-(CF3)2 (red) at 77 K. Their crystallinity was maintained after N2 adsorption (Fig. S5†). (b) PXRD patterns of simulated UiO-66 (upper black), UiO-66 (lower black), UiO-66-CF3 (blue), and UiO-66-(CF3)2 (red). (c) FT-IR spectra of UiO-66 (black), UiO-66-CF3 (blue), and UiO-66-(CF3)2 (red). | ||
Water vapor adsorption properties of UiO-66-X
To confirm whether water molecules can penetrate the hydrophobic nanospaces created by trifluoromethyl groups introduced into UiO-66, as shown in Fig. 3 and S6,† water vapor adsorption/desorption measurements were performed at 298 K. At high relative pressure (Pe/P0 > 0.80), UiO-66-X (-X = -H, -CF3, and -(CF3)2) readily adsorbed water vapor. The significant increases in the adsorption in the first step where 0.30 < Pe/P0 < 0.40 were attributable to water vapor clusters formed on the nanospace surfaces.32 The significant increases in adsorption in the second step where Pe/P0 > 0.97 were attributable to the liquefaction of the adsorbed water vapor.33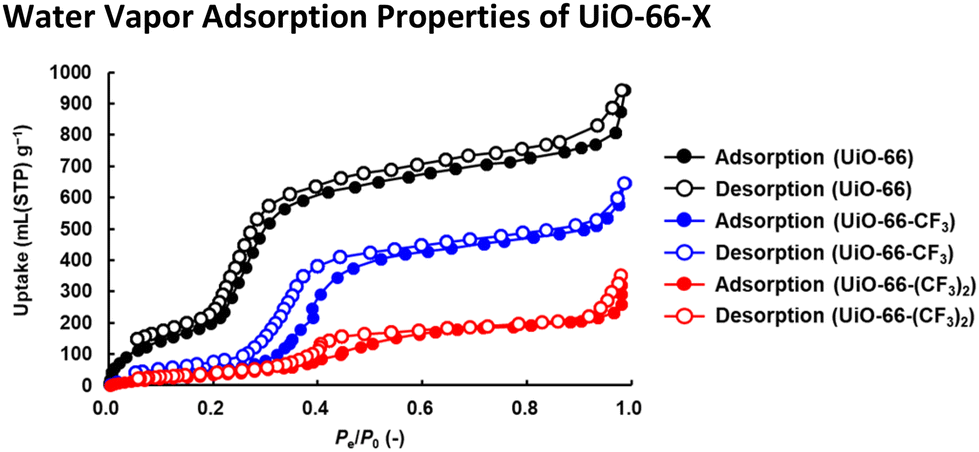 | ||
| Fig. 3 Water vapor adsorption/desorption isotherms of UiO-66 (black), UiO-66-CF3 (blue), and UiO-66-(CF3)2 (red) at 298 K. Their crystallinity was maintained after water vapor adsorption (Fig. S5†). | ||
It should be noted, as shown in Fig. S6,† that the adsorption in the first step by UiO-66-CF3 and UiO-66-(CF3)2 occured at higher relative pressure (Pe/P0 > 0.30) compared to UiO-66, while first-step adsorption for UiO-66-(CF3)2 with the highest fluorine density occured at Pe/P0 ≈ 0.40. Since the pore surface adsorbed water molecules sequentially from the more hydrophilic areas, the higher the hydrophobicity (fluorine density) of the pore surface, the higher the pressure required for water vapor adsorption.34 In other words, the larger the hydrophobic portion of the total pore surface, the less water vapor was adsorbed at low pressure, and the higher the pressure threshold for the first step of water vapor adsorption depending on the hydrophobicity of the pore surface. Since the trifluoromethyl groups introduced into UiO-66 were exposed on the pore surface based on the crystal structure of UiO-66,35 the hydrophobic portion of the total pore surface increased proportionally with fluorine density. Therefore, as shown in Fig. S7,† the pressure threshold for the first step of water vapor adsorption increased proportionally with fluorine density of UiO-66-X, indicating that the nanospaces have become more hydrophobic.32,36
Proton conductivity of UiO-66-X
As shown in Fig. 4a–c, the proton conductivities (σX) of pelletized UiO-66-X were measured by electrochemical impedance spectroscopy (EIS). The proton conductivities were calculated by fitting analysis assuming the equivalent circuit shown in Fig. S8.† As shown in Fig. 4a–c, dielectric relaxation in the high-frequency region indicated that the proton conductivities of UiO-66-X (-X = -H, -CF3, and -(CF3)2) under 95% relative humidity (RH) at 30 °C were σH = 3.34 × 10−4 S cm−1, σCF3 = 5.67 × 10−7 S cm−1, and σ(CF3)2 = 3.34 × 10−6 S cm−1, respectively. As shown in Fig. S9,† the direct current (DC) electrical conductivity values calculated from DC resistance of UiO-66-X (-X = -H, -CF3, and -(CF3)2) were 4.59 × 10−9 S cm−1, 2.14 × 10−9 S cm−1, and 1.44 × 10−9 S cm−1 under 95% RH at 30 °C, respectively. These values were much lower than those of σX, supporting σX as the proton conductivities of UiO-66-X. The coordinated water in the metal cluster site could be a proton source for proton conduction owing to partial dissociation caused by the Lewis acidity of the metal ions, yielding a lower pKa than that of pure water.24,37,38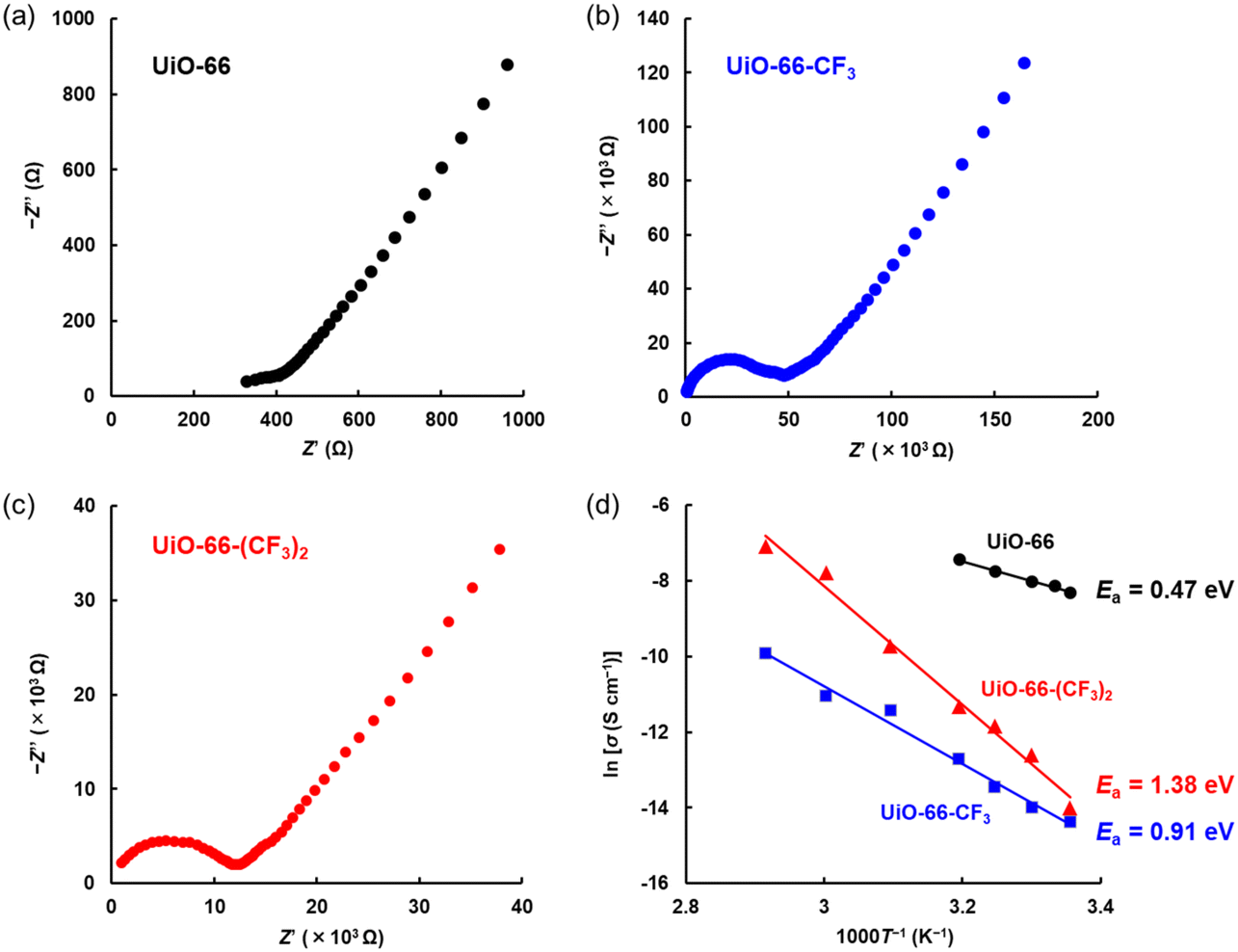 | ||
| Fig. 4 Cole–Cole plots of (a) UiO-66, (b) UiO-66-CF3, and (c) UiO-66-(CF3)2. Impedance spectrum of the disk-shaped pellet under 95% RH at 30 °C. (d) Arrhenius plots of proton conductivity under 95% RH. R2 values of UiO-66, UiO-66-CF3, and UiO-66-(CF3)2 were 0.994, 0.987, and 0.989, respectively. As shown in Fig. 1, small particles of UiO-66-(CF3)2 less than 200 nm were also produced, suggesting a slight decrease in proton conductivity.39 | ||
In UiO-66, a smaller σ value was reported for hydrophobic nanospaces where halogen substituents without proton-donating ability were introduced into the linker.23 A larger σ value was reported for a larger number of missing linkers in UiO-66.22 Therefore, σCF3 and σ(CF3)2 would have been smaller than σH. Introducing trifluoromethyl groups into the linker caused the pKa of the C–H of the benzene ring to follow the order pKa((CF3)2) < pKa(CF3).40 Thus, smaller pKa values yield larger σ values;41 therefore, the proton conductivity was σ(CF3)2> σCF3.
As shown in the Arrhenius plots in Fig. 4d, the activation energies (Ea) of UiO-66, UiO-66-CF3, and UiO-66-(CF3)2 were Ea(H) = 0.47 eV, Ea(CF3) = 0.91 eV, and Ea((CF3)2) = 1.38 eV, respectively. The proton conduction mechanisms differed depending on Ea and were classified as being either Grotthuss (Ea < 0.4 eV) or vehicle mechanisms (Ea > 0.5 eV).41,42 The Arrhenius plots shown in Fig. 4d indicated that UiO-66 followed the intermediate process between the Grotthuss and vehicle mechanisms (mainly vehicle mechanism),42 while UiO-66-CF3 and UiO-66-(CF3)2 followed the vehicle mechanism. UiO-66-CF3 and UiO-66-(CF3)2 could not form strong hydrogen bonding networks, like sulfo and carboxy groups,23 and the vehicle mechanism was predominant. Since the percentage of missing linkers in UiO-66 has been reported to have little effect on Ea,22 the fluorine density was considered to be mainly affected by Ea. The number of hydrogen bonds formed/broken when a proton moved a certain distance was proportional to the fluorine density. Therefore, as shown in Fig. S10,† the fluorine density and Ea exhibited a proportional relationship.
Conclusions
In the current work, we revealed the relationship between the fluorine density of organic-based porous materials and water dynamics (proton conduction) in the hydrophobic nanospace. In detail, focusing on UiO-66 with structural durability at high temperature/humidity based on strong Zr–O bonds, we prepared UiO-66-CF3 and UiO-66-(CF3)2 with different numbers of trifluoromethyl groups in the linker to change the fluorine density per unit volume, respectively. Compared to UiO-66, the hydrophobicity in nanospaces of UiO-66-CF3 and UiO-66-(CF3)2 were increased depending on their fluorine density owing to the higher relative pressure where steep adsorption in the first step occurred. The activation energies for proton conduction of UiO-66-CF3 (0.91 eV) and UiO-66-(CF3)2 (1.38 eV) were significantly larger than that in UiO-66 (0.47 eV) depending on their fluorine densities rather than the percentage of missing linkers. We demonstrated for the first time that introducing fluorine into organic-based porous materials allowed the hydrophobic nanospace to interact with water and protons, yielding a larger energy for proton conduction (Ea) and a predominance of the vehicle mechanism. The results revealed that, despite the small number of samples and the initial results, fluorine density and activation energy were proportional. We clarified that the activation energy of proton conduction increased proportionally with the fluorine density in the nanospace. This indicated the possibility that the state of proton/water in the nanospace could be freely controlled by the fluorine density. In our continuous work, we elucidated the detailed mechanism of proton conduction (e.g. vehicle or Grotthuss mechanism) by quasi-elastic neutron scattering and molecular dynamics simulation based on the crystal structures. The more detailed relationship between fluorine density and activation energy of proton conduction can be corroborated by using other UiO-66 derivatives such as UiO-66-F, UiO-66-F2 and UiO-66-F4.Author contributions
Ryota Akai: conceptualization, methodology, formal analysis, data curation, investigation, writing – original draft, funding acquisition, and resources. Hitoshi Kasai: writing – review & editing. Kouki Oka: conceptualization, methodology, formal analysis, data curation, project administration, validation, supervision, writing – original draft, writing – review & editing, funding acquisition, resources, and software.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Grants-in-Aid for Scientific Research (no. JP23K17945, JP23H03827, JP24K01552, and JP24KJ1576) from MEXT, Japan. This work was supported by JSPS Bilateral Program Number JPJSBP120258801. This work was partially supported by the Environment Research and Technology Development Fund (JPMEERF20241RA4) of the Environmental Restoration and Conservation Agency provided by the Ministry of the Environment of Japan. K. O. also acknowledges the support from the Shorai Foundation for Science and Technology, TEPCO Memorial Foundation, Amano Industry Technology Laboratory, Sugiyama Houkoukai, the Yamada Science Foundation, Kenjiro Takayanagi Foundation, the Kansai Research Foundation for Technology Promotion, the Yashima Environment Technology Foundation, JACI Prize for Encouraging Young Researcher, Iketani Science and Technology Foundation, Foundation for Interaction in Science & Technology, and Ozawa and Yoshikawa Memorial Electronics Research Foundation. R. A. also acknowledges the support from the Hosokawa Powder Technology Foundation and the Mishima Kaiun Memorial Foundation.References
- R. Berger, G. Resnati, P. Metrangolo, E. Weber and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- S. Kumar, B. Mohan, C. Fu, V. Gupta and P. Ren, Coord. Chem. Rev., 2023, 476, 214876 CrossRef CAS.
- T. Ami, K. Oka, S. Kitajima and N. Tohnai, Angew. Chem., Int. Ed., 2024, 63, e202407484 CrossRef CAS PubMed.
- P. G. M. Mileo, K. Adil, L. Davis, A. Cadiau, Y. Belmabkhout, H. Aggarwal, G. Maurin, M. Eddaoudi and S. Devautour-Vinot, J. Am. Chem. Soc., 2018, 140, 13156–13160 CrossRef CAS PubMed.
- W.-B. Li, Y.-Z. Cheng, D.-H. Yang, Y.-W. Liu and B.-H. Han, Macromol. Rapid Commun., 2023, 44, 2200778 Search PubMed.
- Y. Liu, Z. Lin, Y. Luo, R. Wu, R. Fang, A. Umar, Z. Zhang, Z. Zhao, J. Yao and S. Zhao, J. Cleaner Prod., 2023, 420, 138347 Search PubMed.
- M. Chatenet, S. Berthon-Fabry, Y. Ahmad, K. Guérin, M. Colin, H. Farhat, L. Frezet, G. Zhang and M. Dubois, Adv. Energy Mater., 2023, 13, 2204304 CrossRef CAS.
- J. Wang, R. Abazari, S. Sanati, A. Ejsmont, J. Goscianska, Y. Zhou and D. P. Dubal, Small, 2023, 19, 2300673 CrossRef CAS PubMed.
- S. Siracusano, C. Oldani, M. A. Navarra, S. Tonella, L. Mazzapioda, N. Briguglio and A. S. Aricò, J. Membr. Sci., 2019, 578, 136–148 CrossRef CAS.
- S. K. Bhardwaj, N. Bhardwaj, R. Kaur, J. Mehta, A. L. Sharma, K.-H. Kim and A. Deep, J. Mater. Chem. A, 2018, 6, 14992–15009 CAS.
- Z. Chen, X. Li, C. Yang, K. Cheng, T. Tan, Y. Lv and Y. Liu, Adv. Sci., 2021, 8, 2101883 CrossRef CAS PubMed.
- M. Åhlén, O. Cheung and C. Xu, Dalton Trans., 2023, 52, 1841–1856 Search PubMed.
- C.-Y. Xiao, T.-Y. Chen, R.-J. Chung, S. Yougbaré, L.-Y. Lin and Y.-F. Wu, J. Energy Storage, 2022, 55, 105622 Search PubMed.
- R. Medishetty, J. K. Zaręba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 CAS.
- E.-S. M. El-Sayed, Y. D. Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 Search PubMed.
- C. Xiao, J. Tian, Q. Chen and M. Hong, Chem. Sci., 2024, 15, 1570–1610 Search PubMed.
- X. Zhang, B. Wang, A. Alsalme, S. Xiang, Z. Zhang and B. Chen, Coord. Chem. Rev., 2020, 423, 213507 CAS.
- Y. J. Kim, S. Y. Ko, S. Kim, K. M. Choi and W.-H. Ryu, Small, 2023, 19, 2206561 CAS.
- F. Ahmadijokani, H. Molavi, M. Rezakazemi, S. Tajahmadi, A. Bahi, F. Ko, T. M. Aminabhavi, J.-R. Li and M. Arjmand, Prog. Mater. Sci., 2022, 125, 100904 CAS.
- J. Winarta, B. Shan, S. M. McIntyre, L. Ye, C. Wang, J. Liu and B. Mu, Cryst. Growth Des., 2020, 20, 1347–1362 CAS.
- D. Zou and D. Liu, Mater. Today Chem., 2019, 12, 139–165 CAS.
- J. M. Taylor, S. Dekura, R. Ikeda and H. Kitagawa, Chem. Mater., 2015, 27, 2286–2289 CAS.
- F. Yang, H. Huang, X. Wang, F. Li, Y. Gong, C. Zhong and J.-R. Li, Cryst. Growth Des., 2015, 15, 5827–5833 CAS.
- H.-M. Ren, B.-Y. Liu, B.-T. Zuo, Z.-F. Li and G. Li, Microporous Mesoporous Mater., 2023, 351, 112481 CAS.
- M. Taddei, P. V. Dau, S. M. Cohen, M. Ranocchiari, J. A. van Bokhoven, F. Costantino, S. Sabatini and R. Vivani, Dalton Trans., 2015, 44, 14019–14026 RSC.
- H. Reinsch, S. Waitschat, S. M. Chavan, K. P. Lillerud and N. Stock, Eur. J. Inorg. Chem., 2016, 2016, 4490–4498 CrossRef CAS.
- Y. L. Wang, S. Zhang, Y. F. Zhao, J. Bedia, J. J. Rodriguez and C. Belver, J. Environ. Chem. Eng., 2021, 9, 106087 CrossRef CAS.
- Z. Zhou, X. Cao, D. Lv and F. Cheng, Sep. Purif. Technol., 2024, 339, 126666 CAS.
- I. Das and G. De, Sci. Rep., 2015, 5, 18503 Search PubMed.
- R. A. Espinosa-Flores, M. D. Trejo-Valdez, M. E. Manríquez-Ramírez and F. J. Tzompantzi-Morales, Heliyon, 2023, 9, e17138 CAS.
- C. A. Hollingsworth, P. G. Seybold and C. M. Hadad, Int. J. Quantum Chem., 2002, 90, 1396–1403 CAS.
- B. Zhang, Z. Zhu, X. Wang, X. Liu and F. Kapteijn, Adv. Funct. Mater., 2024, 34, 2304788 CAS.
- Y. Byun, S. H. Je, S. N. Talapaneni and A. Coskun, Chem. – Eur. J., 2019, 25, 10262–10283 CAS.
- A. Metrane, A. Delhali, M. Ouikhalfan, A. H. Assen and Y. Belmabkhout, J. Chem. Eng. Data, 2022, 67, 1617–1653 CAS.
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gándara, H.-B. Bürgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CAS.
- E.-P. Ng and S. Mintova, Microporous Mesoporous Mater., 2008, 114, 1–26 CAS.
- N. C. Jeong, B. Samanta, C. Y. Lee, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 51–54 CAS.
- H. Ōkawa, A. Shigematsu, M. Sadakiyo, T. Miyagawa, K. Yoneda, M. Ohba and H. Kitagawa, J. Am. Chem. Soc., 2009, 131, 13516–13522 Search PubMed.
- D. A. Levenson, J. Zhang, B. S. Gelfand, S. P. Kammampata, V. Thangadurai and G. K. H. Shimizu, Dalton Trans., 2020, 49, 4022–4029 Search PubMed.
- K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Guo, Tetrahedron, 2007, 63, 1568–1576 CAS.
- A. Shigematsu, T. Yamada and H. Kitagawa, J. Am. Chem. Soc., 2011, 133, 2034–2036 CAS.
- X. Meng, H.-N. Wang, S.-Y. Song and H.-J. Zhang, Chem. Soc. Rev., 2017, 46, 464–480 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00342c |
| This journal is © The Royal Society of Chemistry 2025 |