
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Asymmetric thiol-modified hybrid polyoxometalates: building blocks for hierarchical nanostructured redox materials†
Elizabeth Hampsona,
Alexander J. Kiblerae,
Jamie M. Cameron a,
Julie A. Wattsb,
Abigail Bellamy-Carterc,
Alex Saywelld,
Lee R. Johnsona,
Darren A. Walsh*a and
Graham N. Newton*a
a,
Julie A. Wattsb,
Abigail Bellamy-Carterc,
Alex Saywelld,
Lee R. Johnsona,
Darren A. Walsh*a and
Graham N. Newton*a
aGSK Carbon Neutral Laboratory for Sustainable Chemistry, University of Nottingham, Nottingham, NG7 2GA, UK. E-mail: darren.walsh@nottingham.ac.uk; graham.newton@nottingham.ac.uk
bNanoscale and Microscale Research Centre, University of Nottingham, Nottingham, NG7 2RD, UK
cLiberal Arts and Natural Sciences, University of Birmingham, Birmingham, B15 2TT, UK
dSchool of Physics and Astronomy, University of Nottingham, Nottingham, NG7 2RD, UK
eAdvanced Propulsion Lab, Marshgate, University College London, London, E20 2AE, UK
First published on 22nd May 2025
Abstract
The development of hierarchical, redox-active nanostructures can drive new advances in fields ranging from energy storage to sensors and memristors. The self assembly of redox-active molecular building blocks into redox-active structures is especially promising, offering synthetic chemists the prospects of introducing unique chemical functionality into redox materials. In this contribution, we present an asymmetrically-functionalised organic–inorganic hybrid Wells–Dawson polyoxometalate bearing a chelating metal-binding group and a thiol-terminated aliphatic chain unit. The thiol-bearing group facilitates the solvent-dependent self-assembly of the cluster into soft nanostructures and grafting of the cluster onto Au surfaces and nanoparticles. We demonstrate how the redox properties of the molecular units are translated across the different classes of materials.
Introduction
Polyoxometalates (POMs) are discrete molecular metal oxides that can be isolated in a variety of morphologies and exhibit diverse physicochemical properties. Their properties make them promising building blocks for functional materials that could impact a wide range of fields, including catalysis, energy storage, sensors, memristors, and medicine.1–8 Key to their usefulness in many of these applications is their capability to undergo reversible multi-electron redox processes while retaining their structural integrity.9,10 The structural integrity of POMs can also allow the controlled preparation of covalently organofunctionalised analogues – so-called organic–inorganic hybrid POMs.11,12 Covalent hybridisation offers a robust route to enhanced functionality, endowing characteristics such as improved solubility,13 controllable self-assembly,14 or increased photosensitivity15 compared to their plenary (unmodified) parent POMs.Hybrid POMs typically contain one or more identical groups covalently bound to the POM core.16 However, a handful of examples of hybrid POMs bearing two different organic groups have been reported. The majority of these examples involve asymmetric hybridisation of the Anderson–Evans cluster [Hy(XO6)M6O18]n− and the hexamolybdate cluster [Mo6O19]2−.17,18 The most prominent examples featuring advanced functionality include that from She and co-workers comprising asymmetrically functionalised Anderson–Evans cluster, containing amine and carboxylic acid groups as an ‘inorganic amino acid’ in the automated synthesis of peptides.19 Another interesting example by Mialane and co-workers features a spiropyran-POM-BODIPY Anderson–Evans triad, with photo-switchable fluorescence and excellent photo-fatigue resistance.20 While the Anderson–Evans clusters are useful building blocks, their limited redox properties stymy their appeal as components in energy systems.
We recently reported the first example of a stable, asymmetrically bi-functionalised hybrid POM based on the Wells–Dawson cluster, a compound with multiple readily-accessible redox states and visible-light redox activity.21 The long chain alkyl and terpyridine (TPY) organic groups, in combination with the inorganic POM, allow the hybrid to function as a transition metal cation binder, a self-assembling surfactant, and a multi-redox active metal oxide cluster.22,23
The grafting of POMs to 2D and 3D surfaces permits the translation of their unique electrochemical and photophysical properties from the molecular to material level in a controlled and predictable manner.23,24 It is widely recognized that controlling the organization of electroactive molecules on surfaces and nanomaterials is a key requirement in designing nanoscale components for functional redox materials.25–27 Here, we report the synthesis of an asymmetric bi-functionalised hybrid polyoxometalate with non-equivalent metal binding sites (TPY and thiol groups – Fig. 1) and demonstrate that it can assemble into monolayers on nanoscale and bulk gold substrates. The retention of a free coordinative TPY anchor gives unprecedented opportunities for controlled fabrication of dual-component and multilayered POM-based materials with high degrees of structural control.
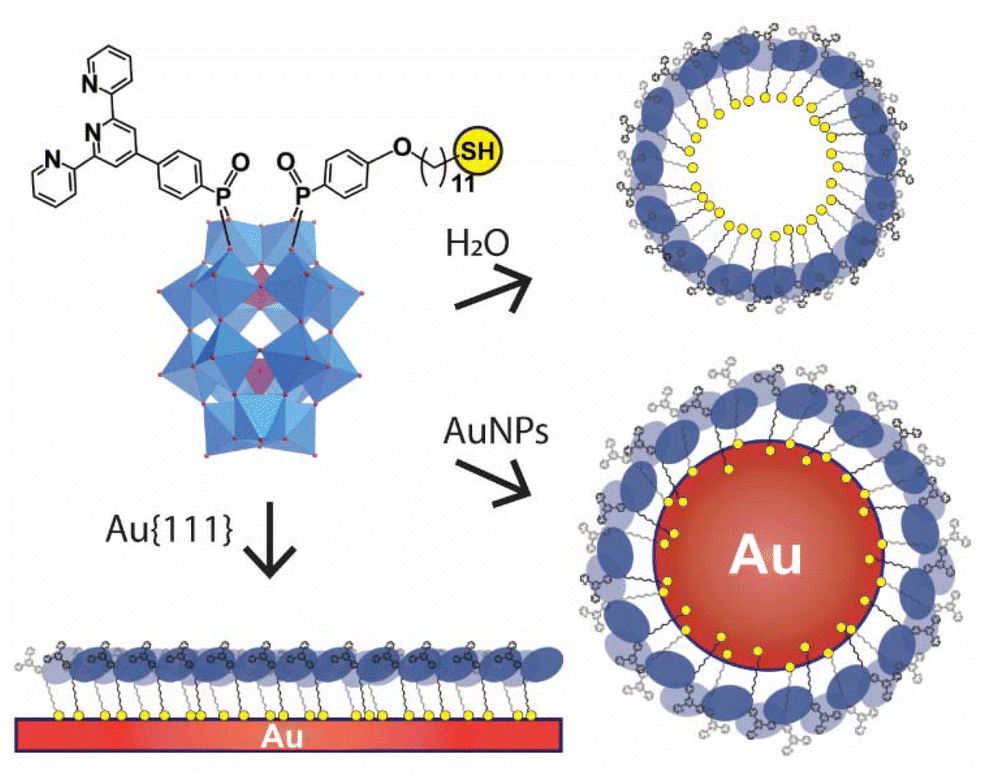 | ||
| Fig. 1 Schematic of the multi-functionality of the asymmetric hybrid POM (4). The long-chain alkanethiol group can be used to induce micelle formation, and assembly on Au surfaces, while the TPY group remains available as a metal cation binding site. | ||
Results and discussion
The synthesis of the desired asymmetric hybrid product was based on our previously reported methodology for the asymmetric TPY-C18 hybrid, K4(C2H8N)2[P2W17O57(PO3C21H14N3)(PO4C24H41)] (1).21 In a ‘one-pot’ acid-catalysed condensation reaction, 1 mol equivalent each of TPY and C11SH phosphonic acids were reacted with 1 mol equivalent of the potassium salt of the mono-lacunary Dawson-type anion, [P2W17O61]10−. 31P NMR spectroscopy of the crude product indicated the presence of three chemically-distinct species – a racemic mixture of the two enantiomers of the asymmetric hybrid, K5(C2H8N)[P2W17O57{(PO3C21H14N3)(PO4C17H26SH)}] (4) and the two symmetric hybrids, (C2H8N)6[P2W17O57(PO3C21H14N3)2] (2), and K3(C2H8N)3[P2W17O57(PO4C17H26SH)2] (3). In the 31P NMR spectrum of 4, the signals for the covalently-bound ligands, TPY and C11SH, appeared at 13.41 and 16.72 ppm, respectively. As observed for 1, the ligand signals were slightly shifted relative to the chemical shifts in the symmetric hybrids, 2 and 3, for which the corresponding peaks appeared at 14.20 and 15.99 ppm, respectively. 4 was isolated using a similar purification procedure as used for 1 – successive solvent extractions, taking advantage of the sparing solubility of 2 in acetonitrile and the superior solubility of 3 in diethyl ether (summarised in Fig. S1 and S2†). 4 was reliably isolated in 10–20% with excellent purity, as characterised by 1H NMR and 31P NMR spectroscopy, ESI-MS, elemental (CHN) analysis, FT-IR spectroscopy, and thermogravimetric analysis (TGA). Full experimental details and analyses are shown in Fig. S2–S7 in the ESI.† 1H NMR spectroscopy confirmed the presence of the thiol-terminated aliphatic chain group C11SH and the aromatic TPY group in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometric ratio.
:1 stoichiometric ratio.
We explored the functionality of the hybrid POM system, derived from its thiol-terminated aliphatic chain group and asymmetric structure. First, we probed whether the aliphatic chain C11SH imparts solvent-dependent self-assembly to the hybrid, as was observed for our previously-reported asymmetric system 1. After dissolving 4 in DMF, 9 eq. of water were added to facilitate its self-assembly in solution. Dynamic Light Scattering (DLS) experiments on a 1.4 mM solution confirmed the formation of nanoscale assemblies, with low dispersity and a hydrodynamic diameter (Dh) of approximately 7 nm (Fig. S8†). Cryo-TEM analysis of the solution of 4 deposited and frozen on a graphene-oxide and holey carbon-supported Cu grid showed spherical structures with diameters of approximately 6 nm (Fig. S9a†). Interestingly, long “worm-like” structures that appeared to be formed from many micelles joined end-to-end were also observed, resembling structures observed in work on similar hybrid POM systems (Fig. S9b†).22,28 This may be an effect of interactions between the large, highly charged POM polar headgroups and the counterions, a phenomenon that can promote cylindrical micelle formation.29
It has been shown that the redox chemistry of POMs changes when in their molecular (solvated) state compared to when in supramolecular assemblies or attached to surfaces.21,30,31 We first studied the electrochemistry of a solution of 4 in DMF containing 0.1 M NBu4PF6 as supporting electrolyte using cyclic voltammetry and the resulting voltammograms closely resembled that of 1, with four distinct quasi-reversible redox processes in the potential range from −0.5 to −2.0 V vs. Fc+|Fc (Fig. 3a, Table S2†). Cyclic voltammetry of 4 dissolved in an aqueous 0.1 M H3PO4:DMF (9:1, v/v) mixture was then carried out. These solvent conditions were shown separately to drive micelle formation by DLS (Fig. S10†). When compared to the solution voltammetry, the two redox processes in the range 0.5 to −0.5 V (vs. AgCl|Ag) appeared to partially coalesce (Fig. 3b). This is in line with previously observed behaviour for redox active Wells–Dawson hybrid micelle assemblies formed from shorter chain symmetric hybrids, and reflects the correlation between redox chemistry and inferred micelle stability.30
We also investigated the propensity of the terminal thiol of the ligand to tether 4 onto Au surfaces. This was first investigated by immersing a polished Au electrode in a stirred solution of 4 in DMF for 24 h. It was then thoroughly rinsed with DMF to remove any weakly-adsorbed material, before being placed in fresh electrolyte (0.1 M NBu4PF6 in DMF) and analyzed using cyclic voltammetry, analogous to the conditions in Fig. 3a. Three quasi-reversible redox processes were recorded in the range from +0.5 to −1.0 V (vs. Fc+|Fc), indicative of the presence of 4 (Fig. 3c). The peak shapes are more Gaussian and symmetric, consistent with surfaced confined redox, when compared with the peak shapes in Fig. 3a, which are consistent with diffusion-controlled redox events. However, the number of peaks is somewhat difficult to compare as the electrochemical stability window of the electrolyte appears to be significantly shortened when measurements are conducted at a gold working electrode.
The voltammogram did not change after multiple cycles, suggesting that the adsorbed compound was stably tethered and resistant to degradation during repeated redox cycling (Fig. S11†). The peak-to-peak separation for each redox event was significantly larger than the value of 0 mV expected for an ideally-behaved surface-confined redox species, which we attribute to sluggish electron transfer kinetics across the electrode/electrolyte interface.32 Surface confinement was corroborated through the linear relationship between the peak current and the scan rate (Fig. S12†). The charge (determined by integrating the first reduction peak), corresponded to a surface coverage of 6.64 × 10−11 mol cm−2, or 4.00 × 1013 molecules per cm2 (Fig. S13a†), which is close to that expected based on the crystallographic radius of the cluster.33 In contrast, cyclic voltammetry of an Au electrode prepared in the same way using our previously-reported system 1, which lacks a thiol functionality, did not reveal significant faradaic processes typical of a surface-bound redox-active species (Fig. S13b†).
AFM imaging of an Au/mica substrate that had been submerged in a DMF solution of 4 and rinsed thoroughly before air-drying revealed a dense array (∼1 monolayer coverage) of close-packed features. This is consistent with the expectation of a thiol–gold mediated self-assembled monolayer (SAM – see Fig. 2a).34 Individual features separated by 7.5 ± 1 nm could be resolved (Fig. S14a†). In contrast, substrates prepared using a solution of micellar assemblies of 4 (1.4 mM solution in water–DMF (9:1 v/v)) showed a sparsely populated surface, with 5 ± 1 nm diameter structures, as well as clustered groups and quasi-1D branched structures (Fig. 2b and S14b†). Etching/reconstruction of the Au step edges was visible by the prevalence of non-linear step-edges in the molecular sample, due to the stronger chemical interactions between the thiol groups and Au electrode, which were not present in the AFM images of the micellar sample (Fig. S15†). Cyclic voltammetry of a similarly prepared Au electrode (immersing a micellar solution of 4) in 9:1 H2O–DMF (v/v) (Fig. 3d) showed a very weak peak that was difficult to resolve above the charging current, as expected due to the lower coverage of the Au surface (Fig. 2b). In summary, the ability to tune the solvent of our system, driving formation of a SAM of 4 or assembly of micelles of 4 on Au, provides an interesting route to the stimulus-controlled nanostructuring of POMs onto surfaces via hierarchical assembly.
 | ||
| Fig. 2 AFM topographs of 4 on Au/mica, prepared by (a) immersion of the Au/mica in a 0.25 mM 4 in DMF, resulting in a uniform monolayer coverage, and (b) immersion of the Au/mica in 1.4 mM 4 in water–DMF (9:1 v/v), resulting in isolated and ‘worm-like’ micellar assemblies. | ||
 | ||
| Fig. 3 Cyclic voltammograms of (a) a solution of 4 in DMF containing 0.1 M TBAPF6 using a 3 mm diameter glassy carbon electrode, (b) assemblies of 4 in 9:1 0.1 M H3PO4/DMF (v/v) using a 3 mm diameter glassy carbon electrode, (c) 4 @ a 2 mm diameter Au electrode in DMF containing 0.1 M TBAPF6, and (d) assemblies of 4 @ at 2 mm diameter Au electrode in 0.1 M H3PO4. The scan rate was 100 mV s−1 in all cases. | ||
Considering different surface morphologies, POMs can act as effective capping agents for Au nanoparticles (AuNPs),35,36 enabling control in the reductive growth of NPs in solution37 and influencing the properties of the NPs.38,39 The terminal thiol functionalities of hybrid POM structures can anchor POMs to AuNPs, resulting in robust composite structure.40–42 The use of POMs as capping agents can tune the electronic and plasmonic properties of the hybrid nanoparticle, and is promising for the design of new electro- and photo-catalytic hybrid materials.42,43 We recently described composite AuNPs capped with a symmetric thiol-functionalised hybrid Wells–Dawson POM that exhibited stable redox chemistry and synergistic photoactivity.42 The unique asymmetric structure of 4 offers opportunities to introduce new functionality to the system while also providing a platform for post-functionalisation through the free TPY units. The preparation of 4-stabilised AuNPs was carried out by modifying our reported procedure.42 In brief, an aqueous solution of 10 nm citrate-stabilised AuNPs was added to a concentrated solution of 4 in acetonitrile and stirred at RT for two days. The NPs were then collected by precipitation with an excess of methanol and washed before characterization (see ESI† for full experimental details).
The UV-visible absorption spectrum of the solid material dissolved in DMF exhibited features characteristic of both AuNPs and 4 – an absorption band at 527 nm associated with surface plasmon resonance (SPR) and an intense band centered at ca. 275 nm attributed to the ligand-to-metal charge-transfer band of the POM anion (Fig. 4b). While the SPR peak was not shifted from that of the citrate-stabilized precursor AuNPs, unlike for the NPs modified with 1 (+4 nm shift),42 the intense absorptions in the low wavelength region in comparison to the AuNP precursor indicates that POMs were present. Moreover, Cryo-TEM imaging of the solution strongly indicates that 4 was attached to the AuNPs. Monodisperse NPs with an average diameter of 8.3 nm, each of which surrounded by well-resolved halos, were observed. The halos indicated that a layer of POMs was present on the Au surfaces (Fig. 4c). The thickness was approximately 2 nm, as expected for a monolayer of 4 (Fig. 4a). Commercial samples of citrate-stabilised Au nanoparticles lack this distinctive halo as we have shown previously.42 Energy-dispersive X-ray (EDX) mapping analysis of the sample showed regions of W, P and S concentrated at the AuNP locations, supporting the successful capping of AuNPs by molecules of 4 (Fig. S16†).
 | ||
| Fig. 4 (a) Estimation of the thickness of surface layer of 4 (4layer) from the diameter of 4-NPs (dtotal) and diameter of the AuNP core (dAuNP); (b) UV-Vis absorbance spectra of 4-NPs (black) and 4 (blue) and Citrate-NPs (red) in DMF, each as 0.5 mg in 5 mL; (c) Cryo-TEM image of 4-NPs in DMF with inset showing a single 4-modified AuNP. | ||
Conclusions
In summary, using a modular synthetic strategy we have designed and isolated an asymmetric organo-functionalised Wells–Dawson hybrid POM that can serve as a redox-active platform for the controlled preparation of a range of hierarchical nanoassemblies and functional materials. The thiol-terminated aliphatic chain enables both solvent-dependent self-assembly of the hybrid POM into redox-active soft nanostructures and the controlled formation of chemisorbed hybrid POM monolayers on gold surfaces. The existence of the second organic functional group on the asymmetric POM (in this case the terpyridine moiety) suggests that the POM can be further functionalized to act as a molecular semiconductor linker between gold surfaces and additional functional units. We will explore this in our future work. The modular design approach presents opportunities for asymmetric hybrid Dawson POMs as designer building blocks in new multifunctional molecular systems, nanomaterials and devices.Author contributions
EH and AJK contributed to data curation, methodology, visualization and manuscript preparation. JMC and LRJ contributed project supervision and data analysis. AB-C and AS performed the AFM analysis and data interpretation. JW performed the TEM data collection and analysis. The work was conceived by GNN and DAW and the manuscript was written by AJK, EH, DAW, and GNN.Data availability
The data supporting this article, including synthetic procedures, can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
EH thanks the Low Dimensional Materials and Interfaces Doctoral Training Programme at the University of Nottingham for support. JMC and GN thank the Leverhulme Trust (RPG-2016-442) and the University of Nottingham Propulsion Futures Beacon of Excellence. AS thanks the Royal Society for support via a University Research Fellowship. The authors thank the EPSRC [EP/L022494/1] and the University of Nottingham for funding access to the TEM instrumentation.References
- T. Ma, R. Yan, X. Wu, M. Wang, B. Yin, S. Li, C. Cheng and A. Thomas, Adv. Mater., 2024, 36, 2310283 CrossRef CAS PubMed.
- H. Hu, L. Lian, X. Ji, W.-L. Zhao, H. Li, W. Chen, H. N. Miras and Y.-F. Song, Coord. Chem. Rev., 2024, 503, 215640 CrossRef CAS.
- Z. Wei, J. Wang, H. Yu, S. Han and Y. Wei, Molecules, 2022, 27, 5212 CrossRef CAS PubMed.
- S. K. Petrovskii, E. V. Grachova and K. Y. Monakhov, Chem. Sci., 2024, 15, 4202–4221 RSC.
- L. Tang, F. Yu, B. Tang, Z. Yang, W. Fan, M. Zhang, Z. Wang, O. Jacobson, Z. Zhou, L. Li, Y. Liu, D. O. Kiesewetter, W. Tang, L. He, Y. Ma, G. Niu, X. Zhang and X. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 27558–27567 CrossRef CAS PubMed.
- C. Busche, L. Vilà-Nadal, J. Yan, H. N. Miras, D. L. Long, V. P. Georgiev, A. Asenov, R. H. Pedersen, N. Gadegaard, M. M. Mirza, D. J. Paul, J. M. Poblet and L. Cronin, Nature, 2014, 515, 545–549 Search PubMed.
- H. Lv, Y. V. Geletii, C. Zhao, J. W. Vickers, G. Zhu, Z. Luo, J. Song, T. Lian, D. G. Musaev and C. L. Hill, Chem. Soc. Rev., 2012, 41, 7572–7589 RSC.
- M. Zhang, H. Li, J. Zhang, H. Lv and G.-Y. Yang, Chin. J. Catal., 2021, 42(6), 855–871 CrossRef CAS; S. Sutter, B. Trepka, S. Siroky, K. Hagedorn, S. Theiß, P. Baum and S. Polarz, ACS Appl. Mater. Interfaces, 2019, 11, 15936–15944 CrossRef PubMed.
- J. M. Cameron, D. J. Wales and G. N. Newton, Dalton Trans., 2018, 47, 5120–5136 RSC.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- A. Proust, B. Matt, R. Villanneau, G. Guillemot, P. Gouzerh and G. Izzet, Chem. Soc. Rev., 2012, 41, 7605–7622 RSC.
- A. V. Anyushin, A. Kondinski and T. N. Parac-Vogt, Chem. Soc. Rev., 2020, 49, 382–432 RSC.
- C. L. Peake, A. J. Kibler, G. N. Newton and D. A. Walsh, ACS Appl. Energy Mater., 2021, 4, 8765–8773 CrossRef CAS.
- G. Izzet, B. Abécassis, D. Brouri, M. Piot, B. Matt, S. A. Serapian, C. Bo and A. Proust, J. Am. Chem. Soc., 2016, 138, 5093–5099 CrossRef CAS PubMed.
- W. Wang, L.-M. Chamoreau, G. Izzet, A. Proust, M. Orio and S. Blanchard, J. Am. Chem. Soc., 2023, 145, 12136–12147 CrossRef CAS.
- A. J. Kibler and G. N. Newton, Polyhedron, 2018, 154, 1–20 CrossRef CAS.
- Q. Zhuang, Z. Sun, C.-G. Lin, B. Qi and Y.-F. Song, Inorg. Chem. Front., 2023, 10, 1695–1711 RSC.
- C. Lv, R. N. N. Khan, J. Zhang, J. Hu, J. Hao and Y. Wei, Chem. – Eur. J., 2013, 19, 1174–1178 CrossRef CAS.
- S. She, N. L. Bell, D. Zheng, J. S. Mathieson, M. D. Castro, D.-L. Long, J. Koehnke and L. Cronin, Chem, 2022, 8, 2734–2748 CAS.
- A. Saad, O. Oms, A. Dolbecq, C. Menet, R. Dessapt, H. Serier-Brault, E. Allard, K. Baczko and P. Mialane, Chem. Commun., 2015, 51, 16088–16091 RSC.
- E. Hampson, J. M. Cameron, S. Amin, J. Kyo, J. A. Watts, H. Oshio and G. N. Newton, Angew. Chem., Int. Ed., 2019, 58, 18281–18285 CrossRef CAS PubMed.
- E. Hampson, J. M. Cameron, J. A. Watts and G. N. Newton, Chem. Commun., 2020, 56, 8237–8240 RSC.
- S. S. Amin, J. M. Cameron, R. B. Cousins, J. Wrigley, L. Liirò-Peluso, V. Sans, D. A. Walsh and G. N. Newton, Inorg. Chem. Front., 2022, 9, 1777–1784 RSC.
- M. Laurans, K. Trinh, K. Dalla Francesca, G. Izzet, S. Alves, E. Derat, V. Humblot, O. Pluchery, D. Vuillaume, S. Lenfant, F. Volatron and A. Proust, ACS Appl. Mater. Interfaces, 2020, 12, 48109–48123 CrossRef CAS.
- X. Lefèvre, F. Moggia, O. Segut, Y.-P. Lin, Y. Ksari, G. Delafosse, K. Smaali, D. Guérin, V. Derycke, D. Vuillaume, S. Lenfant, L. Patrone and B. Jousselme, J. Phys. Chem. C, 2015, 119, 5703–5713 CrossRef.
- S. Duhm, G. Heimel, I. Salzmann, H. Glowatzki, R. L. Johnson, A. Vollmer, J. P. Rabe and N. Koch, Nat. Mater., 2008, 7, 326–332 CrossRef CAS.
- P. Song, J. Shen, D. Ye, B. Dong, F. Wang, H. Pei, J. Wang, J. Shi, L. Wang, W. Xue, Y. Huang, G. Huang, X. Zuo and C. Fan, Nat. Commun., 2020, 11, 838 CrossRef CAS PubMed.
- M. Piot, B. Abécassis, D. Brouri, C. Troufflard, A. Proust and G. Izzet, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8895–8900 CrossRef CAS PubMed.
- K. Schäfer, H. B. Kolli, M. K. Christensen, S. Løland Bore, G. Diezemann, J. Gauss, G. Milano, R. Lund and M. Cascella, Angew. Chem., Int. Ed., 2020, 59, 18591–18598 CrossRef PubMed.
- K. Kastner, A. J. Kibler, E. Karjalainen, J. A. Fernandes, V. Sans and G. N. Newton, J. Mater. Chem. A, 2017, 5, 11577–11581 RSC.
- S. Amin, J. M. Cameron, J. A. Watts, D. A. Walsh, V. Sans and G. N. Newton, Mol. Syst. Des. Eng., 2019, 4, 995–999 RSC.
- C. E. D. Chidsey, C. R. Bertozzi, T. M. Putvinski and A. M. Mujsce, J. Am. Chem. Soc., 1990, 112, 4301–4306 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley Global Education, New York, 2nd unabridged edn., 2012 Search PubMed.
- P. Piotrowski, J. Pawłowska, J. Pawłowski, A. M. Czerwonka, R. Bilewicz and A. Kaim, RSC Adv., 2015, 5, 86771–86778 RSC.
- U. Jameel, M. Zhu, X. Chen and Z. Tong, J. Mater. Sci., 2016, 51, 2181–2198 CrossRef CAS.
- Y. Wang and I. A. Weinstock, Chem. Soc. Rev., 2012, 41, 7479–7496 RSC.
- S. Martín, Y. Takashima, C.-G. Lin, Y.-F. Song, H. N. Miras and L. Cronin, Inorg. Chem., 2019, 58, 4110–4116 CrossRef PubMed.
- S. G. Mitchell and J. M. de la Fuente, J. Mater. Chem., 2012, 22, 18091–18100 RSC.
- M. Zhang, J. Hao, A. Neyman, Y. Wang and I. A. Weinstock, Inorg. Chem., 2017, 56, 2400–2408 CrossRef CAS PubMed.
- C. R. Mayer, S. Neveu and V. Cabuil, Angew. Chem., Int. Ed., 2002, 41, 501–503 CrossRef CAS.
- S. Hegde, S. Joshi, T. Mukherjee and S. Kapoor, Mater. Sci. Eng., C, 2013, 33, 2332–2337 CrossRef CAS.
- C. Martin, K. Kastner, J. M. Cameron, E. Hampson, J. Alves Fernandes, E. K. Gibson, D. A. Walsh, V. Sans and G. N. Newton, Angew. Chem., Int. Ed., 2020, 59, 14331–14335 CrossRef CAS.
- S. Sutter, B. Trepka, S. Siroky, K. Hagedorn, S. Theiß, P. Baum and S. Polarz, ACS Appl. Mater. Interfaces, 2019, 11, 15936–15944 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00544b |
| This journal is © The Royal Society of Chemistry 2025 |