
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From Co to Co@Co2P and CoP nanorods: synthesis and performances in selective hydrogenation of phenylacetylene†
Brandon Azeredoa,
Jason Nguyen-Conga,
Mathieu Vidalb,
Seema Shafiqab,
Thi Thiet Vua,
Beatrice Muzzia,
Audrey Martya,
Yuanyuan Mina,
Fabio Ferrari a,
Fabien Delpecha,
Céline Nayrala,
Pier-Francesco Fazziniac,
Thomas Blona,
Philippe Serp*bd,
Guillaume Viau*a and
Katerina Soulantica*a
a,
Fabien Delpecha,
Céline Nayrala,
Pier-Francesco Fazziniac,
Thomas Blona,
Philippe Serp*bd,
Guillaume Viau*a and
Katerina Soulantica*a
aUniversité de Toulouse, Laboratoire de Physique et Chimie des Nano-Objets (LPCNO) UMR 5215, INSA, UPS, CNRS, 135 avenue de Rangueil, Toulouse F-31077, France. E-mail: ksoulant@insa-toulouse.fr; gviau@insa-toulouse.fr
bUniversité de Toulouse, Laboratoire de Chimie de Coordination (LCC) UPR 824, Toulouse INP, CNRS LCC, composante ENSIACET, 4 allée Emile Monso, Toulouse F-31030, France. E-mail: philippe.serp@ensiacet.fr
cUniversité de Toulouse, CEMES-CNRS, CNRS, 29 rue Jeanne Marvig, 31055 Toulouse, France
dInstitut Universitaire de France (IUF), France
First published on 9th May 2025
Abstract
Transition metal phosphides are emerging catalysts for reactions traditionally catalyzed by noble metals. Cobalt phosphide nanostructures are currently intensively studied as electrocatalysts, but their catalytic properties in thermocatalytic reactions are much less explored. Here we report the topochemical reaction of ferromagnetic metallic cobalt nanorods with tris(trimethylsilyl) phosphine (PTMS) at 165 °C, which leads to the conservation of the nanorod shape while transforming them to Co@Co2P core@shell nanorods. The Co2P shell thickness can be controlled through the PTMS/Co ratio. Modulation of the core–shell structure allows tuning the magnetic properties of the resulting nano-objects. Increasing both the temperature and PTMS/Co ratio results in the complete transformation of Co into well-crystallized CoP nanorods. The catalytic performances Co@Co2P and CoP nanorods in the selective hydrogenation of phenylacetylene have been evaluated. It is shown that increasing the P/Co ratio in the nanorods allows increasing both the activity and the selectivity to styrene. Comparison with cobalt phosphide nanoparticles reported in the literature for the selective hydrogenation of phenylacetylene shows the superior performance of the Co@Co2P and CoP nanorods prepared in this work.
1. Introduction
Phosphides of transition metals (TMPs) are active and stable heterogeneous catalysts for hydroprocessing processes such as hydrodesulfurization, hydrodenitrogenation and hydrodeoxygenation reactions.1–3 As the incorporation of phosphorus into transition metals induces electronic, geometric and synergetic effects, which are currently actively explored, TMPs are intensely studied as cost-efficient, Earth abundant electrocatalysts4–6 or alternative anode materials for lithium-ion batteries,7,8 and also in photo- and thermocatalysis.9–11As the transition from bulk to nanoscale offers the possibility to exploit new properties or performance improvements resulting from the nanoscale dimensions,12,13 different methods have been developed to prepare nanostructured TMPs with controllable size and morphology.9,14,15 In this context, solution phase synthesis methods are well-adapted for achieving a high degree of control over size, size monodispersity, morphology, crystal structure and composition of inorganic nanoparticles (NPs).16 Over the last two decades, CoxPy NPs of different sizes, shapes and crystal structures have been prepared by different methods.9,17–23 The most widely employed strategies for the synthesis of highly monodisperse shape controlled CoxPy NPs consist in (i) thermal decomposition in high boiling point solvents of molecular cobalt precursors in the presence of surfactants and a P source,24–26 or (ii) the use of preformed Co NPs as templates that are subsequently subjected to a reaction with a P source.27,28,34 In the latter case, hollow nanostructures usually result from the Kirkendall effect.27,28 Among the organophosphorus compounds which are largely employed for the synthesis of well-defined CoxPy nanostructures, tri-n-octylphosphine (TOP) is the most common one.22,25,26 Other phosphines such as triphenylphosphine,29 aminophosphines30 or P(SiMe3)331 have also been used. Tri-n-octyl phosphine oxide (TOPO)21,22,32,33 and triphenylphosphite34–36 can also act as organophosphorus sources. In most of the cases, reaction temperatures of about 300 °C are employed. Depending on the reaction conditions, the degree of phosphidization can be controlled toward Co2P or CoP nanostructures.18,22,28,30 Several syntheses afford Co2P nanorods (NRs),25,29,33–38 and less CoP NRs.39–41 However, no core shell Co@CoxPy has been reported up to now.
Among TMPs, cobalt phosphide (CoxPy) nanostructures have received increasing attention as electrocatalysts for the hydrogen evolution reaction (HER),27–29,32,42 the oxygen reduction reaction (ORR) over a wide pH range,33 and dual-function ORR and oxygen evolution reaction (OER) catalysts for zinc–air batteries.43 On the other hand, their performances in thermocatalytic reactions such as reductive molecular transformations are much less explored.24,34,35,44,45 The removal of phenylacetylene (PhA), an impurity present in the styrene (ST) feed monomer, by selective semi-hydrogenation is an important process for the polymer industry. Indeed, PhA constitutes a poison that deactivates polymerization catalysts in polystyrene production units.46 The major challenges in this selective hydrogenation reaction are avoiding the complete hydrogenation to ethylbenzene (EB) and maximizing the selectivity towards ST. Although Pd-based catalysts, in spite of their high cost, are regarded as the top-performing catalysts for this reaction,47 research is being carried out to find substitutes based on non-noble metals such as nickel48,49 or cobalt.50,51 In addition to the cost of the catalysts, these research studies are justified by the fact that, for many unmodified Pd-based catalysts, ST is easily hydrogenated to EB during the pre-desorption process, resulting in low ST selectivity at high PhA conversions.52,53 On the other hand, metallic (zero-valent) Co and Ni catalysts present serious air instability issues (pyrophoricity), which results in difficult catalyst handling, and chemical process safety issues.54 The use of their phosphides, which are stable in air, eliminates the need for strict anaerobic conditions or pre-reduction. Although non-noble metal phosphide catalysts have shown interesting performances for hydrogenation reactions,10,55,56 few results have been reported so far for PhA selective hydrogenation. The published studies mainly deal with nickel phosphide catalysts,54,57–61 and to a less extent with cobalt phosphide.62 In the case of nickel phosphide, it was shown that the electron transfer that occurs from Ni to P (resulting in a positively charged Ni) reduces the desorption energy of the alkene and thus improves the reaction selectivity.60 Indeed, the electron-withdrawing effect of phosphorus in metal phosphides is one of the most important electronic effects (ligand effects) that modulates the electronic environment surrounding the metal sites and distinguishes metal phosphides from other catalysts.10
We report here the synthesis of Co@Co2P and CoP nanorods by a topochemical reaction between tris(trimethylsilyl) phosphine (PTMS) and Co nanorods with a hexagonal close packed (hcp) structure, prepared by an organometallic route. This method allows tuning the thickness of the Co2P shell on Co up to the complete transformation of Co into Co2P and CoP. We also present the performances of the Co@Co2P core@shell and CoP nanorods in the selective hydrogenation of PhA.
2. Experimental section
All syntheses were carried out under an inert atmosphere, either using standard Schlenk techniques or in a glovebox. Oleylamine (Acros Organics 80–90%) and toluene (Fisher Scientific, >99%) were purged by Ar bubbling before introducing them into a glovebox. The tris(trimethylsilyl) phosphine (PTMS) was purchased from STREM (98%). They were introduced in the glove box and used without further purification. Note: tris(trimethylsilyl) phosphine (PTMS) catches fire spontaneously if exposed to air and causes eye, skin and respiratory irritation. It has to be handled under an inert atmosphere under a fume-hood, wearing protective gloves, protective clothing and eye protection.2.1. Bare Co nanorod synthesis
Bare Co nanorods (Co-NRs) were synthesized by a slightly modified published method.63 In a typical reaction, in a Fischer–Porter reactor in the interior of a glovebox, 1.642 g (6.8 mmol) of hexadecylamine (HDA) was dissolved in 60 mL of a toluene/anisole 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture. Once HDA was completely dissolved, 0.801 g (4 mmol) of lauric acid (LA) was dissolved in 8 mL of the same toluene/anisole mixture and then added to the HDA solution, forming a white gel-like suspension. A solution of 1.807 g (4 mmol) of the Co precursor [Co{N(SiMe3)2}2(thf)] in 12 mL of the toluene/anisole mixture was added to the HDA-LA mixture. The reactor was removed from the glovebox and transferred to a vacuum line where it was evacuated and then pressurized with H2 to 3 bar. The mixture was heated at 110 °C under stirring for 48 hours. At the end of the reaction, the H2 was removed, the reactor was transferred in the glovebox, and the nanorods were separated from the Co nanospheres formed during the synthesis, through several washing steps with toluene. Finally, the Co-NRs were dispersed in 20 mL of toluene and stored in a freezer inside the glovebox. Known volumes of the resulting dispersion were used for the subsequent reactions with PTMS. The Co content per mL of the Co-NR suspensions was calculated from magnetic measurements on a dried sample of known volume. The organometallic method gives rise to Co-NRs with diameters and lengths that can slightly vary from one batch to the other. The dimensions of the different batches of Co-NRs that were used as starting materials for each phosphidized product are shown in Table S1.†
:3 mixture. Once HDA was completely dissolved, 0.801 g (4 mmol) of lauric acid (LA) was dissolved in 8 mL of the same toluene/anisole mixture and then added to the HDA solution, forming a white gel-like suspension. A solution of 1.807 g (4 mmol) of the Co precursor [Co{N(SiMe3)2}2(thf)] in 12 mL of the toluene/anisole mixture was added to the HDA-LA mixture. The reactor was removed from the glovebox and transferred to a vacuum line where it was evacuated and then pressurized with H2 to 3 bar. The mixture was heated at 110 °C under stirring for 48 hours. At the end of the reaction, the H2 was removed, the reactor was transferred in the glovebox, and the nanorods were separated from the Co nanospheres formed during the synthesis, through several washing steps with toluene. Finally, the Co-NRs were dispersed in 20 mL of toluene and stored in a freezer inside the glovebox. Known volumes of the resulting dispersion were used for the subsequent reactions with PTMS. The Co content per mL of the Co-NR suspensions was calculated from magnetic measurements on a dried sample of known volume. The organometallic method gives rise to Co-NRs with diameters and lengths that can slightly vary from one batch to the other. The dimensions of the different batches of Co-NRs that were used as starting materials for each phosphidized product are shown in Table S1.†
2.2. Reaction of bare Co-NRs with PTMS
In a typical reaction, in a three-neck flask equipped with a refrigerant, thermocouple and mechanical stirrer, the bare Co-NRs (0.68 mmol Co) were dispersed in 12 mL of mesitylene. A solution of HDA 82.1 mg (0.34 mmol) in 3.3 mL of mesitylene was added to this mixture and the mixture was heated to 160 °C. Different PTMS amounts in 1.7 mL of mesitylene were injected into the Co-NR suspension when the temperature reached 160 °C. The temperature was increased to 165 °C and the mixture was then let to react at this temperature either for 90 min or for 3 h. The mixture was then allowed to cool down before transferring it to a glovebox where it was let to decant. The supernatant was removed and the precipitate was washed two times with 5 mL of toluene. Complete evaporation of the solvent gave rise to powders that were used for the characterization and for catalysis.In some cases, and in order to reach higher reaction temperatures, mesitylene was replaced by oleylamine. In these cases, PTMS in oleylamine was injected at 150 °C, and the mixture was kept at this temperature for 30 min, followed by an increase in the temperature to 250 °C, and continuation of the reaction at 250 °C for three hours. The rest of the procedure was identical to the procedure followed with mesitylene as a solvent.
No other nano-objects are formed during phosphidization and the reaction is quantitative with respect to the Co introduced in the reaction.
The samples are named as Co-Px/y where x corresponds to the P/Co ratio introduced and y to the reaction temperature (165 or 250 °C).
2.3. Characterization
The magnetic properties of the starting Co-NRs and the nanostructures obtained after the reactions were studied by vibrating sample magnetometry (VSM) using a Quantum Design PPMS 9T Evercool II magnetometer system. To avoid any oxidation, the samples were prepared in a glovebox, and transferred to a cryostat VSM in a Schlenk vessel. Standard VSM capsules were filled with a known amount (a few mg) of vacuum-dried sample, mixed with tetracosane as a solid matrix, and sealed. The VSM capsules containing the samples were rapidly introduced into the VSM in order to avoid air-exposure of the sample. A field-dependent (±6 T) magnetization cycle at 300 K was recorded in the first step. The saturation magnetization (Ms) of the Co nanorods was compared to the Ms value of Co (160 A m2 kg−1) at room temperature (RT), which allows the determination of the Co(0) wt% content in the sample. A second field-dependent magnetization cycle was recorded after cooling the sample down to 5 K (FC) while applying +6 T (FC) and allows the detection of Co-oxidation. The presence of CoO on the surface of Co provokes a shift of the 5 K magnetization loop because of an exchange bias between the antiferromagnetic CoO and the ferromagnetic Co.X-Ray diffraction (XRD) experiments have been performed on a Malvern Panalytical Empyrean diffractometer, equipped with a Co anticathode X-ray source (Co Kα = 1.790 Å), a Bragg–Brentano HD mirror and a PIXCel1D detector. To avoid any oxidation, the samples were encapsulated between two thin layers of Kapton® film. For the well-crystallized Co-P3/250 NRs, XRD analysis was carried out using the MAUD software64 based on the Rietveld method combined with Fourier analysis.
Routine transmission electron microscopy (TEM) analyses were performed in the Microcharacterization Center Raymond Castaing, on a JEOL 1011 microscope operating at 100 kV and a JEOL 1400 microscope operating at 120 kV equipped with a camera ORIUS 1000 GATAN. Both microscopes are equipped with a tungsten filament as the emission source. High-resolution TEM (HRTEM) was performed on two microscopes, an ARM200F Cold FEG spherical aberration corrected (Cs) at 200 kV (1.9 Å resolution) and a JEOL 2100F using a Schottky field emission gun at 200 kV (2.3 Å resolution). The images were collected using a GATAN ULTRASCAN 2k × 2k Model 994 (ARM) camera and a CMOS GATAN RIO16IS 4k × 4k (2100F) camera. Scanning TEM (STEM) images (0.78 Å resolution) and energy dispersive X-Ray spectroscopy (EDX) analyses were carried out on a JEOL-ARM200F Cold FEG mounted with a EDX SDD CENTURIO-X (129 eV resolution). The samples were prepared by dropping a diluted solution on a carbon-coated copper grid. The grids were systematically evaporated using a pump (P = 5.10−4 mbar) to remove excess volatile residues.
The X-ray photoelectron spectroscopy (XPS) analyses were performed on samples prepared by depositing a few drops of the suspended solids on a Si wafer followed by solvent evaporation. The spectra were recorded using a K-alpha plus system (Thermo Fisher Scientific, East-Grinstead, UK) fitted with a micro-focused and monochromatic Al Kα X-ray source (1486.6 eV, spot size of 400 μm). The spectrometer pass energy was set to 150 and 40 eV for the survey and the narrow high-resolution regions, respectively.
ICP analyses were performed by Microanalytical Laboratory Kolbe.
2.4. Catalysis
The catalyst performance was evaluated by measuring the site time yield (STY, in molPhA molCo−1 h−1) (eqn (1)) and styrene selectivity at isoconversion (SST-x%conversion in %) (eqn (2)).
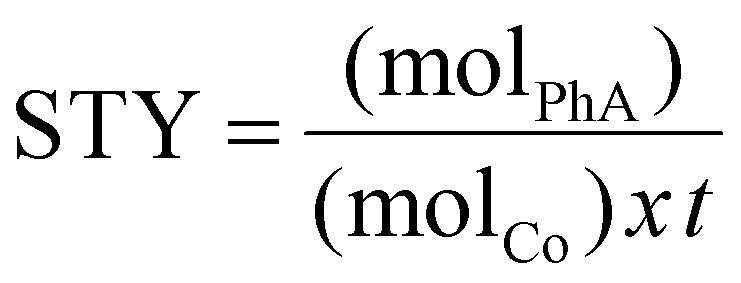 | (1) |
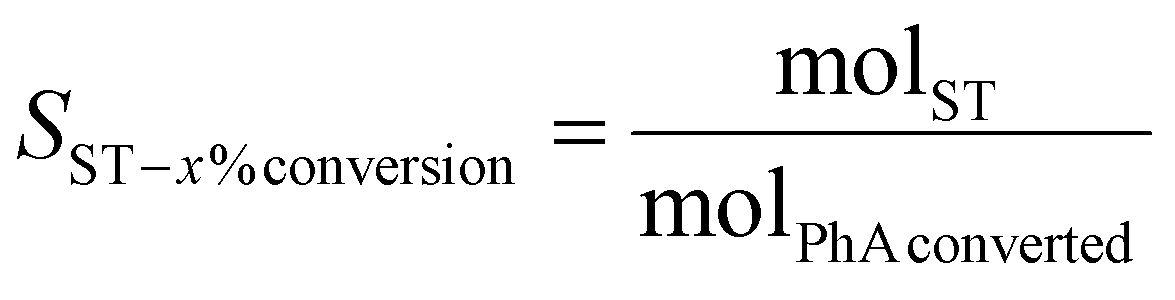 | (2) |
3. Results and discussion
The reaction of Co-NRs was explored with tris(trimethylsilyl) phosphine (PTMS), a reactive precursor that has been used efficiently for the synthesis of metal phosphides under relatively mild reaction conditions.31,66,67 Fig. S1a† shows a characteristic TEM image and the XRD pattern of the raw Co-NRs used as the starting material. Their mean diameter dm and mean length Lm were determined from TEM image analysis. The dimensions of the different batches of Co-NRs that were used are presented in Table S1.† The Co-NRs crystallize in the hcp structure of bulk Co. Their XRD pattern (Fig. S1b†) shows a strong anisotropy, the two broad peaks at 49° and 56° correspond to the (10![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and (101) reflections of hcp Co, while the narrow peak at 52° corresponds to the (0002) reflection, which shows a higher coherence length along the c axis indicating that the c-axis of the hcp structure lies along the nanorod long axis.
0) and (101) reflections of hcp Co, while the narrow peak at 52° corresponds to the (0002) reflection, which shows a higher coherence length along the c axis indicating that the c-axis of the hcp structure lies along the nanorod long axis.
The Co-NRs were first treated with PTMS at two Co:P ratios 1:0.2 (Co-P0.2/165) and 1:0.7 (Co-P0.7/165). PTMS was injected in the Co-NR dispersion in mesitylene at 160 °C, the temperature was increased to 165 °C and kept at 165 °C for 90 min. TEM observations showed that the resulting nano-objects retained their shape after the reaction for both ratios (Fig. 1a and b). While the Co-P0.2/165 NRs (Fig. 1a) exhibited the same contrast aspect as the starting Co-NRs; darker areas at the center of the Co-P0.7/165 NRs indicate the formation of a core@shell structure (Fig. 1b).
 | ||
| Fig. 1 TEM images of (a) Co-P0.2/165 NRs and (b) Co-P0.7/165 after 90 min reaction. (c) XRD diffractograms of the Co-P0.2/165 and Co-P0.7/165 NRs. Blue and olive-green lines correspond to the expected reflections of hcp-Co (COD file 9008492, P63/mmc, a = 2.507 Å, c = 4.068 Å) and orthorhombic Co2P (COD file 9009202, Pnam, a = 5.646 Å, b = 6.608 Å, c = 3.513 Å), respectively. | ||
The XRD data for both ratios are presented in Fig. 1c. For the Co-P0.2/165 NRs, the diffractogram mainly indicates the presence of hcp Co but some reflections of Co2P can be distinguished from the background, while these latter are more pronounced for the Co-P0.7/165 NRs. Only hcp Co could be observed in HRTEM for the Co-P0.2/165 NRs (Fig. S2a and b†), but scanning transmission electron microscopy-high angle annular dark field (STEM-HAADF) analysis (Fig. S2c†) clearly shows the presence of a denser core, which corresponds to hcp Co and a less dense shell attributed to poorly crystallized Co2P, in agreement with the XRD results. STEM-HAADF of Co-P0.7/165 NRs shows a very similar configuration (Fig. S2f†). EDX mapping of Co-P0.2/165 (Fig. S2d and e†) and Co-P0.7/165 (Fig. S2g and h†) lying flat on the TEM grid does not allow a clear distinction between the core and the shell. Indeed, the difference in the relative concentration of Co (higher in the core area but present also in the shell) is not high enough to clearly evidence the differences between the core and the shell. However, for the Co-P0.7/165 NRs, both the STEM-HAADF image (Fig. 2a) and the bright field image (Fig. 2b) on an NR array with the long NR axes oriented parallel to the electron beam clearly evidence the core–shell structure. EDX line analysis, confirmed the core–shell structure, with a Co core of roughly 2 nm and a 1.5 nm thick Co2P shell (Fig. 2a). As expected for a Co@Co2P configuration, the Co signal is maximized at the core area (brighter area highlighted by the white dotted line in Fig. 2a), and is reduced on the shell, while the opposite is observed for the P signal. In this configuration, the difference in Co and P concentrations between the core and the shell is revealed. As the beam crosses the whole length of the NRs, the Co core signal is much more intense, as compared to the minor contribution to the signal of P present on the shell of the extremities. These nanorods are composed of a crystalline hcp Co core and a partially crystalline Co2P shell. However, the Co core is sometimes fragmented as discerned in Fig. 1b, and as explicitly illustrated by HRTEM in Fig. 2c.
 | ||
| Fig. 2 Co-P0.7/165 NRs. (a) Overlay of an EDX line analysis (Co: cyan; P: yellow) and a dark field HAADF image of an assembly of nanorods oriented parallel to the electron beam (top view). (b) Top view bright field image of the same nanorods showing the core@shell Co@Co2P configuration. (c) HRTEM side view image of two NRs. The white rectangles represent domains where the Co hcp structure persists. | ||
The magnetic properties of the Co-P0.2/165 and Co-P0.7/165 NRs were compared to the initial Co-NRs (Fig. 3). As expected from the transformation of the ferromagnetic hcp Co into paramagnetic Co2P,68 the saturation magnetization Ms of the sample decreases as the amount of injected PTMS increases, that is, from 123 A m2 kg−1 for the starting Co-NRs to 59 and 24 A m2 kg−1 for Co-P0.2/165 and Co-P0.7/165, respectively (Fig. 3a and Table S2† entries 2 and 3 respectively). Considering that the magnetization of Co2P is negligible,68 it is obvious from these magnetic measurements that Co(0) is still present in the core, which indicates that the transformation reaction is not complete, in agreement with the core–shell structure observed by TEM. From the Ms values, we can evaluate the Co(0) amount that remains intact in each sample and from ICP analyses that give the total Co amount, we can deduce the percentage of Co transformed in each sample. Interestingly, while for Co-P0.2/165 (Table S2,† entry 2) the experimentally calculated composition is very close to the theoretical composition, for the Co-P0.7/165 NRs, the incorporation of P in the Co-NRs is incomplete (Table S2† entry 3). Indeed, the use of a slight excess of PTMS (Co/P = 1/0.7) as compared to the stoichiometric Co/P ratio of 1/0.5, is not sufficient to completely convert Co into Co2P under the conditions employed.
 | ||
| Fig. 3 Magnetization curves at (a) 300 K and (b) 5 K of the initial Co-NRs (blue), the Co-P0.2/165 NRs (black) and the Co-P0.7/165 NRs (red). | ||
The coercive fields follow the same trend as the magnetization. The values obtained at 300 K are 525, 347 and 126 mT for the starting Co-NRs, the Co-P0.2/165 NRs and the Co-P0.7/165 NRs, respectively. This change is attributed to a fragmentation of the hcp Co core, which is corroborated by the TEM observations for the sample Co-P0.7/165 (Fig. 1b and 2c). The Co core cannot be considered as a continuous cylinder anymore, thus, the contribution of shape magnetic anisotropy to coercivity is reduced. While the TEM images are less conclusive for the Co-P0.2/165 sample, the decrease in the Hc value leads to the same conclusion, albeit to a lesser extent. For both samples, the cycles at 5 K (Fig. 3b) do not present any exchange bias in any sample, which is in agreement with the absence of any significant amount of CoO in contact with metallic Co.
In order to understand whether the incomplete transformation can be attributed to a too low reaction temperature (165 °C) or to an insufficient PTMS amount, we performed two control reactions, allowing them to proceed for a longer period (3 h). The first reaction, leading to Co-P0.5/250 (Table S2† entry 4), was performed using a Co/P ratio of 1/0.5 but increasing the temperature to 250 °C. As the mesitylene boiling point did not allow for reaching this temperature, the experiment was performed in oleylamine. The second reaction, leading to Co-P3/165, (Table S2† entry 5) was performed at 165 °C with a Co/P ratio of 1/3. The TEM analyses of Co-P0.5/250 and Co-P3/165 NRs are shown in Fig. 4a and 5a respectively. While both samples retain their anisotropic shape, most of the Co0.5/250 NRs are bent and present irregular thickness or enlarged tips. EDX line analysis of two NRs shows that they both contain both Co and P (Fig. S3†). The HRTEM analyses of Co-P0.5/250 and Co-P3/165, are presented in Fig. 4b and 5b, respectively. The two areas analyzed in Co-P0.5/250 (Fig. 4b) correspond to two different NRs. We identified a different phase for each NR: orthorhombic Co2P in the area framed in yellow (Fig. 4b and c) and hcp Co for the area framed in cyan (Fig. 4b and d). On the other hand, Co-P3/165 presents a more homogeneous aspect (Fig. 5a) and the NRs examined showed only the presence of crystallized orthorhombic Co2P (Fig. 5b). Both Co-P0.5/250 and Co-P3/165, are polycrystalline, as evidenced by the HRTEM images presented in Fig. S4 and S5,† respectively.
 | ||
| Fig. 4 Co-P0.5/250: (a) (TEM), (b) HRTEM of two specimens; (c) and (d) FFT of the areas presented in (b), in yellow and cyan frames, respectively. The image zone-axis and the corresponding spots identification are indicated in the figure. The yellow and cyan-framed areas correspond to Co2P and hcp Co, respectively. | ||
 | ||
| Fig. 5 (a) TEM image and (b) HRTEM image of a Co-P3/165 sample. The inset in (b) shows the FFT obtained from the yellow-framed area. The spots in the FFT have been indexed and are in agreement with the Co2P bulk structure. | ||
The XRD patterns of the Co-P0.5/250 and Co-P3/165 NRs show the presence of both Co2P and hcp Co (Fig. 6a). For Co-P3/165, the peaks associated to the hcp Co are less intense. This is in agreement with the magnetic measurements (Fig. 6b and Table S2†) that reveal Ms values of 10.2 A m2 kg−1 and 3.3 A m2 kg−1 for the Co-P0.5/250 and Co-P3/165 NRs, respectively. These values correspond to 6.4 wt% and 2.1 Co wt%, respectively. For comparison, the starting Co-NRs present an Ms value of 134 A m2 kg−1. This indicates that when P is in large excess, even at low temperatures, homogeneous and quasi-complete transformation of the Co to Co2P can be achieved. On the other hand, under stoichiometric conditions, it is more difficult to transform completely Co into Co2P even at high temperature. The degree of phosphidization of the different experiments is summarized in Table S2.†
 | ||
| Fig. 6 (a) XRD diffractograms of Co-P0.5/250 and Co-P3/165; blue and olive green lines correspond to the expected reflections of hcp-Co (COD file 9008492, P63/mmc, a = 2.507 Å, c = 4.068 Å) and orthorhombic Co2P (COD file 9009202, Pnam, a = 5.646 Å, b = 6.608 Å, c = 3.513 Å) and (b) magnetization curves at 300 K and 5 K of starting Co nanorods (blue), Co-P0.5/250 (black) and Co-P3/165 (red). | ||
Interestingly, performing the reaction at 250 °C for 3 h and with a Co/P ratio of 1/3 (Co-P3/250) allows almost complete transformation of Co into CoP. TEM, XRD and magnetic measurements of the Co-P3/250 NRs are presented in Fig. 7. TEM images of Co-P3/250 NRs show conservation of the anisotropic shape without contrast differences (Fig. 7a). In Fig. 7b we present an NR array with the long NR axes oriented perpendicularly with respect to the TEM-grid plane. Similar self-organized superlattices have been regularly observed during the synthesis of the starting Co nanorods.63 The XRD diffractogram (Fig. 7c) shows only the presence of a well-crystallized CoP phase. The low magnetization and coercivity (Fig. 7d and Table S2† entry 6) can be attributed to metallic Co residues (less than 2 wt% of Co(0)). In Fig. S6† we present the EDX line analysis on a Co-P3/250 NR array with the long NR axes oriented perpendicularly with respect to the TEM-grid plane. As expected for an almost complete conversion to CoP, no core@shell structure was observed in contrast to the Co-P0.7/165 sample (Fig. 2). Due to the lower density of CoP and Co2P as compared to Co (ρCo = 8.90 g cm−3; ρCo2P = 7.54 g cm−3; ρCoP = 6.42 g cm−3), the chemical transformation to Co2P and CoP leads to an expansion of the nanorod volume, which is well-illustrated by an increase in the diameters of the phosphidized nanorods measured by TEM (Table S1†).
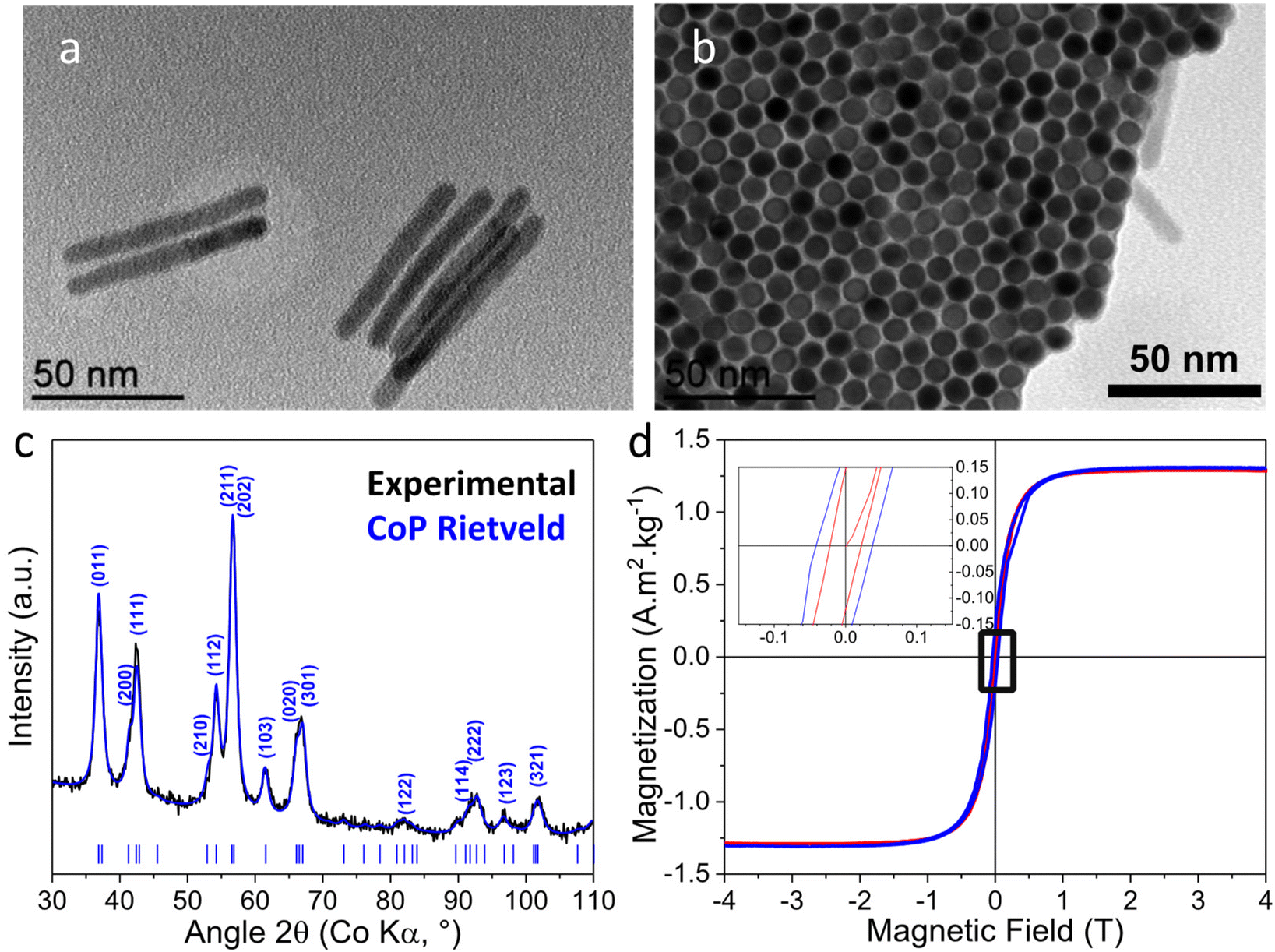 | ||
| Fig. 7 Co-P3/250 (a) TEM of NRs lying flat on the TEM grid (b) top view TEM of an NR array with the long NR axes oriented parallel to the electron beam; (c) XRD and Rietveld analysis (Rwp = 2.2%) of the CoP phase (COD file 9008928, Pnma, a = 5.076 Å, b = 3.281 Å, c = 5.599 Å) indexed with the most intense reflections for the given peak; (d) magnetization curves at 300 K (red) and 5 K (blue). The inset shows enlargement of the low-field area revealing a weak coercive field (5 K: 39 mT, 300 K: 22 mT). | ||
In order to investigate the crystallinity of the nanorods, HRTEM analyses have been performed on several specimens. In agreement with XRD, the HRTEM and FFT analyses (Fig. 8) revealed only the presence of crystallites of the orthorhombic CoP phase but in different zone axes. The NRs appear polycrystalline and no growth axis was identified. The HRTEM micrographs of several specimens of the nanorod array exhibit Moiré fringes which strongly supports the polycrystalline structure for the Co-P3/250 NRs. The absence of preferential growth direction and the polycrystallinity of the CoP NRs was confirmed by the Rietveld analysis of the XRD pattern. In Table S3† the coherence length/crystallite sizes for the main (hkl) reflections of CoP are reported. They are almost identical and close to the diameter of the rods.
 | ||
| Fig. 8 Co-P3/250 HRTEM (a–d) and the corresponding FFT (e–f) of different NRs: (a and e) an NR lying flat on the grid, and (b–d and f–h) different NRs of an array of NRs oriented perpendicularly to the TEM-grid and the electron beam (scale-bars = 2 nm). | ||
Our results indicate that phosphidization yields, expressed as % of Co converted either into Co2P or CoP, depend both on the temperature and the P/Co ratio. From Table S2,† we can make the following observations: at 165 °C, 0.2 eq. of P (entry 2) is enough to bring about the expected transformation into Co2P, however, an over-stoichiometric ratio (entry 3) does not achieve the complete transformation into Co2P. Transformation of Co into Co2P at this temperature progressed to 96.6% with an excess of P and by prolongation of the reaction (entry 5). This is most likely due to the inward diffusion of P being more favorable at the initial transformation steps. A Co/ratio of 3 at 165 °C (entry 5) seems to be more efficient in transforming Co into Co2P than a stoichiometric 0.5 ratio at 250 °C (entry 4). This indicates that a high P concentration favors the Co to Co2P transformation more than high temperature. Nevertheless, both P excess and high temperature are necessary to transform Co into CoP (entry 6). While, no co-existence of Co2P and CoP has been evidenced in any of the samples, we cannot exclude the presence of non-identified amorphous phases during the transformations. It has been postulated22 that the transformation of Co to Co2P initially proceeds by diffusion of P into the Co lattice, and when Co2P becomes majority, by diffusion of Co into the Co2P lattice, with a concomitant formation of voids due to the Kirkendall effect. Further transformation of Co2P into CoP, occurs by diffusion of Co in the lattice, the anion that theoretically should remain intact, but in practice accentuates the voids. In our case, no voids were detected in any of our phosphidized NRs.
The availability of cobalt phosphide-based NRs of different configurations/compositions incited us to explore their performances in the selective hydrogenation of PhA. In addition to the inorganic phosphorous ligand effect discussed in the introduction section, reported studies have shown that the catalytic performance of metal phosphide catalysts is affected by their structure and their composition, including the crystal facet, exposed termination, metal type and metal/P ratio. For Al2O3-supported Ni phosphide catalysts, it has been shown that the catalytic activity (100 °C, 3 bar H2) decreases gradually along with the decrease of the Ni/P ratio, the higher activity obtained for Ni/Al2O3 (8.25 molPhA molNi−1 h−1).60 This might be tentatively attributed to a slower H2 activation rate by heterolytic dissociation on the Ni phosphide catalysts as compared to a homolytic dissociation on the metallic particles.69,70 However, with the gradual incorporation of P, the selectivity toward ST increases dramatically from 0.7% for Ni/Al2O3 to a maximum of 88.2% on Ni2P/Al2O3, followed by a decrease to 65.9% for NiP2/Al2O3. The enhanced selectivity was attributed to: (i) the fact that the incorporation of P increases the Ni–Ni bond length, which modifies the alkene adsorption (from di-σ to π coordination), and (ii) the reduced electron density of Ni sites (Niδ+), which contributes to decreasing the desorption energy of the alkene. For unsupported Co1.2P catalysts, a modest activity of 0.03 molPhA molCo−1 h−1 was measured (100 °C, 7 bar H2) with a high ST selectivity of 95%.24 A higher activity (0.625 molPhA molCo−1 h−1) was reported for a Co2P catalyst modified by the addition of tri-n-butylphosphine.71
Our investigation focused first on the Co-P0.2/165 and Co-P0.7/165 NRs. Apart from the ICP and VSM analyses, XPS analyses were also performed on the NRs tested in catalysis (Table S4†). As expected, the Co/P ratio was higher for the Co-P0.2/165 NRs (Co/P = 2.2) than for the Co-P0.7/165 NRs (Co/P = 1.2). The electronic states of Co and P were probed by XPS, since electron transfer from Co to P could be expected. Indeed, XPS data on cobalt phosphide report Co 2p3/2 binding energy (BE) of 779–779.6 eV for CoP69,72 and 778.2 eV for Co2P.29,73 These BEs shift positively from that of metallic Co-NRs (777.9 eV).74 The Co 2p3/2 BE of CoP is higher than that of Co2P and metallic Co, indicating that a higher phosphorus content causes an electron density shift from Co to P due to the higher electronegativity of phosphorus. In the case of Co2P, the Co 2p3/2 binding energy is very close to that of metallic Co, suggesting a relatively low positive charge on cobalt for this phosphide (Coδ+, δ < 1), as predicted by DFT calculations (0.17 e−).75 Two pairs of peaks can be observed in the high-resolution Co 2p spectra of the cobalt phosphide comprising NRs (Fig. S7a and b†). The first pair of peaks at ∼778 eV and ∼793 eV corresponds to binding energies (BE) close to that of metallic cobalt, as expected for Co2P. The higher BE measured for the Co-P0.7/165 NRs (Co 2p3/2 778.2 eV) compared to the Co-P0.2/165 NRs (778.0 eV) should be related to a thicker layer of cobalt phosphide that seems to cover more efficiently the remaining Co0. The second pair of peaks at BE ∼780 eV and ∼796.5 eV can be assigned to surface cobalt oxidation, and could correspond to Co(OH)2.76 Interestingly, the ratio between non-oxidized and oxidized cobalt is higher for the Co-P0.7/165 NRs (Table S4†), suggesting that a thicker Co2P layer constitutes better protection towards air oxidation upon exposure to the air. For the deconvoluted P 2p spectra (Fig. S7d and e†), the two peaks centered at 129.3 eV and 130.2 eV corresponding to the P 2p3/2 and P 2p1/2 are assigned to P with a partial negative charge, characteristic of phosphides,77 while the peak located at 132.3 eV is ascribed to oxidized phosphorus species arising from surface oxidation of the air-exposed NRs.78
Supported, as well as free standing Co-P0.2/165 and Co-P0.7/165 NRs were tested for the selective hydrogenation of PhA and compared to bare Co-NRs. The selective hydrogenation of PhA was first performed at 90 °C under 5 bar of H2 in methanol. In the first series of experiments, the performances of bare Co, Co-P0.2/165 and Co-P0.7/165 NRs that were supported on oxidized few-layer graphene (FLG, 50 m2 g−1) were compared. This carbon support was used because: (i) its two-dimensional morphology should favor Co-NR deposition and stabilization, (ii) carbon supports are very commonly used in hydrogenation reactions, and (iii) oxidized carbon supports can favor hydrogen spillover (H-spillover), which can be involved in hydrogenation reactions.79 TEM micrographs of these three catalysts (5% Co w/w) are shown in Fig. S8.† All NRs were well dispersed on the FLG support. As shown in Fig. 9a, the space time yields (STY, molPhA molCo−1 h−1) of the three samples are quite similar at around 30 molPhA molCo−1 h−1 affording up to 90% conversion. This result does not support a reduced surface concentration of the H atom on the supported Co-P0.2/165 and Co-P0.7/165 NRs as compared to the hcp-Co,60 for which facile H2 dissociation has been reported.80 As far as selectivity is concerned, three products are formed: ST, EB and toluene. The production of toluene, a product of alkylation in the side chain during PhA hydrogenation, has already been reported for Ni catalysts modified with Keggin-type heteropoly compounds.81 In that work, the production of toluene was attributed to the acidity of the Keggin-type compound that favors the alkylation reaction. As the FLG support was oxidized with nitric acid, it contained significant amounts of carboxylic groups on its surface.82 We have independently evaluated the performance of unsupported Co-NRs and checked for the presence of toluene. In that case, no toluene was formed (Fig. S9†). Consequently, it is reasonable to propose that toluene (3–5% for all supported Co-NR catalysts) was formed because of the acidity of the FLG support. Fig. 9b shows the evolution of selectivity towards ST and toluene with conversion for the three catalysts. The selectivity towards ST remains very high (82.6%) at 99.6% conversion for the Co-P0.7/165/FLG, while it drops to 22% at 100% conversion for the Co/FLG catalyst. At a lower conversion (90%), the three catalysts behaved similarly, showing an SST-90% of 87%.
 | ||
| Fig. 9 (a) STY versus conversion curves for PhA hydrogenation on the FLG supported Co-NR catalysts. (b) Conversion versus selectivity curves for PhA hydrogenation on the FLG supported Co-NR catalysts. Black circles represent bare Co/FLG, red triangles represent Co-P0.2/165/FLG, and green squares represent Co-P0.7/165/FLG. (c) STY versus conversion curves for PhA hydrogenation on the Co-P0.7/165/FLG catalyst without and with water. Green squares represent Co-P0.7/165/FLG without water and blue diamonds represent Co-P0.7/165/FLG with water. (d) Conversion versus selectivity curves for PhA hydrogenation on the Co-P0.7/165/FLG catalyst without and with water. Green squares represent Co-P0.7/165/FLG without water and blue diamonds represent Co-P0.7/165/FLG with water. (e) STY versus conversion curves for PhA hydrogenation on the Co-P0.7/165/TiO2 catalyst without and with water. (f) Conversion versus selectivity curves for PhA hydrogenation on the Co-P0.7/165/TiO2 catalyst without and with water. Orange squares represent Co-P0.7/165/TiO2 without water, and cyan diamonds represent Co-P0.7/165/TiO2 with water. Conditions: PhA/Rh = 500, 90 °C, 5 bar H2, 6 h, methanol. | ||
As Co-P0.7/165/FLG, was the most interesting catalyst as far as the activity and selectivity to ST are concerned, the possibility to improve its performances was explored. The use of water as a promoter was first investigated. The positive role of water to enhance activity and selectivity in hydrogenation reactions has been attributed to water-involved hydrogen exchange83,84 and H-spillover.85 It has been shown that H-spillover can result in an enhanced activity in PhA hydrogenation.86–88 It is also known that gas phase H-spillover on carbon support can be enhanced by the presence of water, which acts as a shuttle for the spilt-over hydrogen species.89 Color change originating from the reduction of WO3 is considered as a simple verification method of H-spillover.89
The occurrence in the liquid phase of H-spillover was evaluated for the Co-P0.7/165/FLG catalyst in the presence or absence of water. To directly monitor the H-spillover in the liquid phase, the reduction behavior of WO3 in the presence or absence of the catalyst was followed under the same conditions where PhA hydrogenation was conducted (90 °C, 5 bar H2, methanol in batch reactor). Under these experimental conditions, no H-spillover was detected for the Co-P0.7/165/FLG sample after 30 min, as shown from the identical color of the samples before and after catalysis, either in the presence of absence of water (Fig. S10a†). A PhA hydrogenation test was performed on the Co-P0.7/165/FLG catalyst in the presence of 1 mL of water. Under these conditions, the catalyst is more active (Fig. 9c), exhibiting an STY of ∼45 molPhA molCo−1 h−1 (30 molPhA molCo−1 h−1 in the absence of water). Interestingly this catalyst also shows a selectivity towards ST of 76.7% at 100% conversion and a significantly lower selectivity towards toluene than in the case of the reaction performed without water addition (Fig. 9d). As H-spillover seems not to be operative with this catalyst, it is possible that water contributes to the hydrogenation of PhA by acting as a hydrogen donor.90
Another strategy to modulate the catalytic performance is to change the support. The use of TiO2, a reducible support allowing H-spillover, which has already shown interesting performance as a support for PhA hydrogenation91 was investigated. With such an oxide support, different metal–support interactions as compared to the carbon support can be expected, which can impact catalytic performances.92,93 The Co-P0.7/165 NRs were supported on a commercial TiO2-P25 support (BET surface area 57 m2 g−1), which consists of a mixture of 78% anatase and 22% rutile TiO2. Representative TEM micrographs of the Co-P0.7/165/TiO2 catalyst are shown in Fig. S11a.† The Co-P0.7/165/TiO2 catalyst shows a high STY (∼38 molPhA molCo−1 h−1, Fig. 9e) and a selectivity towards ST of 83.9% at 100% PhA conversion (Fig. 9f). The selectivity towards toluene was lower on TiO2 (∼2%) than on FLG (∼4.5%). Both Lewis and Brønsted acid sites, which could be responsible for the formation of toluene are present on TiO2-P25.94 H-spillover also operates on TiO2,95 and water-assisted proton diffusion on oxide surface is a known phenomenon.96,97 The surface H3O+ species can contribute to the reaction by decreasing the energy barrier of PhA hydrogenation.98 We have also evaluated the occurrence of H-spillover on the Co-P0.7/165/TiO2 catalyst in the presence or absence of water, and no H-spillover was detected (Fig. S10b†). The reactivity of the Co-P0.7/165/TiO2 catalyst was investigated in the presence of 1 mL of water (Fig. 9e and f). In the presence of water, the Co-P0.7/165/TiO2 catalyst is less active (Fig. 9e) and shows similar selectivity as when water is absent from the medium (Fig. 9f). As for the Co-P0.7/165/FLG catalyst, the presence of water has a negative impact on toluene formation, since no toluene was formed for the P0.7/165/TiO2 catalyst in the presence of water.
From this first series of experiments, we can conclude first that, compared to the Co-NRs and Co-P0.2/165 NRs, the Co-P0.7/165 NRs show the best compromise between activity and selectivity. Second, the tests carried out to optimize this system by playing with the nature of the support and/or the presence of water in the medium allowed us to identify the system “Co-P0.7/165/TiO2 without water in the medium” as the most interesting in terms of activity and selectivity (Fig. S12†).
In contrast to the Co-P0.2/165 and Co-P0.7/165 NRs that present Co2P on their surface, the Co-P3/250 NRs are composed almost exclusively of CoP (Table S2†). As the literature points to the influence of the modulation of the Co/P ratio on catalytic performances, the Co-P3/250 NRs supported on FLG were compared with FLG-supported Co-P0.2/165 and Co-P0.7/165 NRs.
As expected, the Co/P surface ratio is lower for the Co-P3/250 NRs than for the Co-P0.2/165 and Co-P0.7/165 NRs (Table S4†). The XPS Co 2p spectrum of Co-P3/250 NRs presents a single pair of peaks with BEs at 778.6 and 793.6 eV (Fig. S7c†). Contrary to the Co-P0.2/165 and Co-P0.7/165 NRs, the Co-P3/250 NRs do not show significant cobalt surface oxidation. This is expected, since only traces of oxidizable Co(0) is present in the Co-P3/250 NRs (Table S2†); and in addition, this Co(0) is most likely not on the surface, thus, protected by CoP. In Fig. 10a, the XPS-derived evolution of the Co(0)/CoOx ratio as a function of the Co/P atomic ratio is presented for the three types on NRs. For the P 2p spectrum (Fig. S8f†), the two peaks centered at 129.3 eV and 130.4 eV corresponding to P 2p3/2 and P 2p1/2 are well assigned to P with a partial negative charge of phosphides, while the peaks located at 132.8 and 134.9 eV correspond to the phosphate due to surface oxidation. These values are in good agreement with those reported for CoP nanorods.41 This suggests that the Co atoms in the Co-P3/250 NRs have a partial positive charge (Coδ+), while the P atoms have a partial negative charge, implying the transfer of electron density from Co to P.
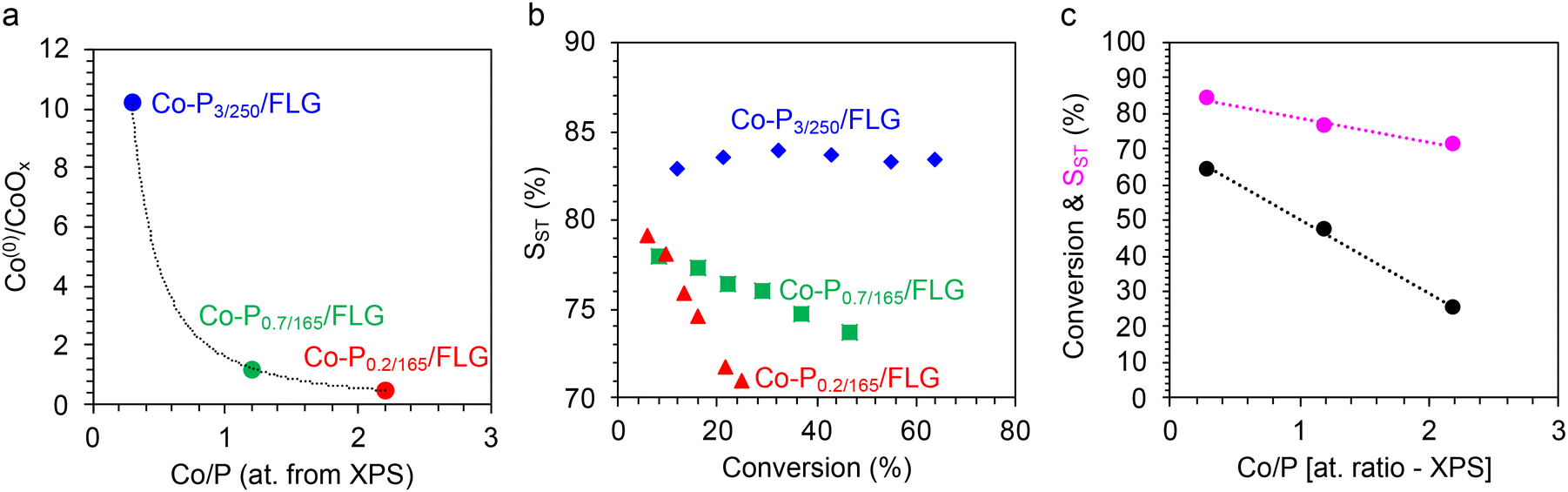 | ||
| Fig. 10 (a) Evolution of the Conon–oxidized/Cooxidized ratio determined by XPS with the atomic Co/P ratio determined by XPS. (b) STY versus conversion curves for PhA hydrogenation on Co-P0.2/165/FLG (red triangles), Co-P0.7/165/FLG (green squares), and Co-P3/250/FLG (blue diamonds) catalysts. (c) Correlation between the Co/P atomic ratio determined by XPS and the PhA conversion (for a 6 hours-test, black circles) and SST determined at 25% conversion (purple circles). Conditions: PhA/Rh = 500, 100 °C, 5 bar H2, 6 h, methanol. | ||
As the differences in activity and selectivity between the Co-P0.2/165/FLG and Co-P0.7/165/FLG catalysts were not marked at 90 °C (Fig. 9a and b), we performed a new series of tests including also FLG-supported Co-P3/250 NRs at 100 °C under 5 bar of H2 in methanol. The performances of the Co-P0.2/165/FLG, Co-P0.7/165/FLG, and Co-P3/250/FLG catalysts during these 6 hours-tests are presented in Fig. 10b. The Co-P3/250/FLG catalyst shows the best performance both in terms of conversion and selectivity. In fact, linear correlations were found between the Co/P ratio and the catalytic performances (Fig. 10c). The superior performance of the Co-P3/250/FLG catalyst in terms of activity could be rationalized by a favored H2 dissociation on CoP. Experimental results have shown that treatment of CoP with H2 introduced large amounts of reactive hydrogen onto CoP, of the order of one H per Co or the P surface atom.62 The improved performance of the Co-P3/250/FLG catalyst in terms of selectivity could be due to the reduced electron density of Co sites (Coδ+), which should contribute to decreasing the ST desorption energy. Considering the promising performances of the Co-P3/250 NRs, they were also supported on TiO2 (Fig. S11b† for a representative TEM micrograph). We have evaluated the occurrence of H-spillover on the Co-P3/250/TiO2 catalyst in the presence or absence of water (Fig. S10c†). Without water in the reaction medium, H-spillover was operative for this sample, as the color of the solution containing Co-P3/250/TiO2 and WO3 changed (WO3−x), indicating that the reduction of WO3 was promoted at 100 °C by the H-spillover in the liquid phase. This result also suggests a favored H2 dissociation on Co-P3/250 NRs as compared to Co-P0.7/165 NRs. In the presence of water, no H-spillover was observed, which could be related to the surface oxidation of the Co-P3/250 NRs at 100 °C in water or to the formation of a thin water layer of H2O molecules preferentially adsorbed on the surface of the catalyst.69 The Co-P3/250/TiO2 catalyst was evaluated for PhA hydrogenation in the presence or absence of 1 mL of water (Fig. S13†). Either in the presence of water or in its absence, this catalyst was less active but more selective than the Co-P3/250/FLG catalyst. In this case, the presence of water did not affect much the catalyst performances. The Co-P3/250/TiO2 catalyst shows an STY20% of ∼20 molPhA molCo−1 h−1 (STY20% of ∼41 molPhA molCo−1 h−1 for the Co-P3/250/FLG catalyst, Fig. S12a†) and an SST-20% of 93% (SST-20% of 83% for the Co-P3/250/FLG catalyst, Fig. S12b†).
Considering the superior performances of the Co-P3/250/FLG and Co-P3/250/TiO2 catalysts in terms of activity and selectivity, their stability was also evaluated through three recyclability tests. These tests were performed at low PhA conversions, typically less than 15%. As shown in Fig. S14,† the catalysts show quite good stability. TEM analyses were also performed on fresh and post-catalysis catalysts. The micrographs presented in Fig. S15† confirm the stability of these catalysts, nanorods retaining their integrity after catalysis.
While detailed mechanistic investigations and structure/property correlations deserve a dedicated exploration and are beyond the scope of this work, several important elements emerge from this preliminary study on the catalytic properties for semi-hydrogenation of PhA of the cobalt phosphide nanorods prepared. First, a number of parameters allowing to modulate the activity and selectivity of these catalysts have been identified, such as: (i) the Co/P ratio in the nanorods, which plays a key role on the catalytic performances and the resistance towards oxidation; (ii) the presence of a support whose acidic properties must be controlled to avoid the formation of toluene; and (iii) the presence of water in the medium, which can in some cases be beneficial both for the activity and the selectivity.
The prepared catalysts exhibit very promising catalytic performances, particularly in comparison with the results reported in the literature for the selective hydrogenation of PhA on Ni or Co phosphide catalysts (Table S5†).
4. Conclusions
The topochemical reaction of PTMS with metallic Co-NRs can be controlled to afford Co@Co2P core@shell NRs of tunable shell thickness, or even CoP nanorods, depending on the reaction conditions employed. The Co@Co2P nanorods present magnetic properties stemming from their ferromagnetic Co core. As the phosphidization reaction transforms a part of the ferromagnetic Co core to the non-magnetic Co2P and CoP phases, the magnetic properties can be modulated through the extent of phosphidization. No Kirkendall effect has been evidenced in any of the phosphidized samples, which is surprising considering the transformation mechanism proposed in the literature.22The supported Co@Co2P and CoP NRs are very promising catalysts for the selective hydrogenation of phenylacetylene to styrene, outperforming most Ni and Co phosphide catalysts reported in the literature, with supported CoP being the most active and selective catalyst. Finally, thanks to their magnetic core, the Co@Co2P NRs should present the advantage of an easy recovery, and last but not least, the possibility to be used in catalytic reactions induced by magnetic heating.99 In this context, we are confident that this strategy can be applied to shape-controlled nanoparticles of magnetic metals and alloys allowing tailoring their magnetic and catalytic properties.
Author contributions
Brandon Azeredo: investigation, data curation, formal analysis, and writing original draft. Jason Nguyen-Cong: investigation, data curation, and formal analysis. Mathieu Vidal: investigation, data curation, and writing original draft. Seema Shafiq, Thi Thiet Vu, Beatrice Muzzi, Audrey Marty, Yuanyuan Min, and Fabio Ferrari: investigation and data curation. Fabien Delpech and Céline Nayral: supervision and writing – review & editing. Pier-Francesco Fazzini and Thomas Blon: formal analysis and writing – review & editing. Philippe Serp, Guillaume Viau, and Katerina Soulantica: project administration, funding acquisition, supervision, methodology, formal analysis, and writing – review & editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank the European Union's Horizon 2020 research and innovation program for financial support of the SWIMMOT project under grant agreement no. 899612. This project has received funding from the French ANR national call (Project NIMRod ANR-21-CE09-0019). This work has received funding from the French Agence Nationale de la Recherche under grant agreements ANR-21-CE07-0021 (GRAAL) and ANR-22-CE07-0044 (LICORN).References
- S. T. Oyama, T. Gott, H. Zhao and Y.-K. Lee, Catal. Today, 2009, 143, 94–107 CrossRef CAS.
- S. T. Oyama, J. Catal., 2003, 216, 343–352 CrossRef CAS.
- R. Prins and M. E. Bussell, Catal. Lett., 2012, 142, 1413–1436 CrossRef CAS.
- J. F. Callejas, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Chem. Mater., 2016, 28, 6017–6044 CrossRef CAS.
- L. Chen, J.-T. Ren and Z.-Y. Yuan, Green Chem., 2022, 24, 713–747 RSC.
- H. Yin, F. Rong and Y. Xie, Int. J. Hydrogen Energy, 2024, 52, 350–375 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- B. Lan, Y. Wang, J. Lu, D. Liu, C. Wei, X. Zhang, X. Huang and G. Wen, Particuology, 2024, 88, 11–31 CrossRef CAS.
- Y. Shi, M. Li, Y. Yu and B. Zhang, Energy Environ. Sci., 2020, 13, 4564–4582 RSC.
- B. Liu, X. Lan, Q. Zhong and T. Wang, ACS Catal., 2024, 14, 757–775 CrossRef CAS.
- S.-H. Li, M.-Y. Qi, Z.-R. Tang and Y.-J. Xu, Chem. Soc. Rev., 2021, 50, 7539–7586 RSC.
- S. Carenco, D. Portehault, C. Boissière, N. Mézailles and C. Sanchez, Adv. Mater., 2014, 26, 371–390 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2013, 42, 2746–2762 RSC.
- Z. Pu, T. Liu, I. S. Amiinu, R. Cheng, P. Wang, C. Zhang, P. Ji, W. Hu, J. Liu and S. Mu, Adv. Funct. Mater., 2020, 30, 2004009 CrossRef CAS.
- S. L. Brock, S. C. Perera and K. L. Stamm, Chem. – Eur. J., 2004, 10, 3364–3371 CrossRef CAS PubMed.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- Y. Xie, H. L. Su, X. F. Qian, X. M. Liu and Y. T. Qian, J. Solid State Chem., 2000, 149, 88–91 CrossRef CAS.
- W. Maneeprakorn, M. A. Malik and P. O'Brien, J. Mater. Chem., 2010, 20, 2329 RSC.
- L. Yi, P. Shao and Z. Wen, New J. Chem., 2023, 47, 9545–9549 RSC.
- Y. Ni, J. Li, L. Zhang, S. Yang and X. Wei, Mater. Res. Bull., 2009, 44, 1166–1172 CrossRef CAS.
- H. Zhang, D.-H. Ha, R. Hovden, L. F. Kourkoutis and R. D. Robinson, Nano Lett., 2011, 11, 188–197 CrossRef CAS PubMed.
- D.-H. Ha, L. M. Moreau, C. R. Bealing, H. Zhang, R. G. Hennig and R. D. Robinson, J. Mater. Chem., 2011, 21, 11498 RSC.
- L. Karam, C. Farès, C. Weidenthaler and C. N. Neumann, Angew. Chem., Int. Ed., 2024, 63, e202404292 CrossRef CAS PubMed.
- A. Ropp, R. F. André and S. Carenco, ChemPlusChem, 2023, 88, e202300469 CrossRef CAS PubMed.
- J. Park, B. Koo, K. Y. Yoon, Y. Hwang, M. Kang, J.-G. Park and T. Hyeon, J. Am. Chem. Soc., 2005, 127, 8433–8440 CrossRef CAS PubMed.
- W. Yang, Y. Huang, J. Fan, Y. Yu, C. Yang and H. Li, Nanoscale, 2016, 8, 4898–4902 RSC.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 53, 5427–5430 CrossRef CAS PubMed.
- J. F. Callejas, C. G. Read, E. J. Popczun, J. M. McEnaney and R. E. Schaak, Chem. Mater., 2015, 27, 3769–3774 CrossRef CAS.
- Z. Huang, Z. Chen, Z. Chen, C. Lv, M. G. Humphrey and C. Zhang, Nano Energy, 2014, 9, 373–382 CrossRef CAS.
- M. E. Mundy, D. Ung, N. L. Lai, E. P. Jahrman, G. T. Seidler and B. M. Cossairt, Chem. Mater., 2018, 30, 5373–5379 CrossRef CAS.
- A. Sodreau, H. G. Zahedi, R. Dervişoğlu, L. Kang, J. Menten, J. Zenner, N. Terefenko, S. DeBeer, T. Wiegand, A. Bordet and W. Leitner, Adv. Mater., 2023, 35, 2306621 CrossRef CAS PubMed.
- E. J. Popczun, C. W. Roske, C. G. Read, J. C. Crompton, J. M. McEnaney, J. F. Callejas, N. S. Lewis and R. E. Schaak, J. Mater. Chem. A, 2015, 3, 5420–5425 RSC.
- V. V. T. Doan-Nguyen, S. Zhang, E. B. Trigg, R. Agarwal, J. Li, D. Su, K. I. Winey and C. B. Murray, ACS Nano, 2015, 9, 8108–8115 CrossRef CAS PubMed.
- T. Mitsudome, M. Sheng, A. Nakata, J. Yamasaki, T. Mizugaki and K. Jitsukawa, Chem. Sci., 2020, 11, 6682–6689 RSC.
- M. Sheng, S. Fujita, S. Yamaguchi, J. Yamasaki, K. Nakajima, S. Yamazoe, T. Mizugaki and T. Mitsudome, JACS Au, 2021, 1, 501–507 CrossRef CAS PubMed.
- J. Liu, M. Meyns, T. Zhang, J. Arbiol, A. Cabot and A. Shavel, Chem. Mater., 2018, 30, 1799–1807 CrossRef CAS.
- H. Hou, Q. Yang, C. Tan, G. Ji, B. Gu and Y. Xie, Chem. Lett., 2004, 33, 1272–1273 CrossRef CAS.
- Y. Ni, J. Li, L. Jin, J. Xia, J. Hong and K. Liao, New J. Chem., 2009, 33, 2055 RSC.
- P. J. Thomas, P. Christian, S. Daniels, Y. Li, Y. S. Wang and P. O'Brien, Pure Appl. Chem., 2006, 78, 1651–1665 CrossRef CAS.
- Y. Li, M. A. Malik and P. O'Brien, J. Am. Chem. Soc., 2005, 127, 16020–16021 CrossRef CAS PubMed.
- Q.-Q. Sun, M. Wang, S.-J. Bao, Y. C. Wang and S. Gu, Analyst, 2015, 141, 256–260 RSC.
- K. Bhunia, M. Chandra, S. K. Sharma, D. Pradhan and S.-J. Kim, Coord. Chem. Rev., 2023, 478, 214956 CrossRef CAS.
- Y. Liu, C. Li, M. Yuan, X. Zhang, H. Lan, Y. Chen, M. Tian, K. Liu and L. Wang, Catal. Sci. Technol., 2023, 13, 3084–3093 RSC.
- M. Sheng, S. Yamaguchi, A. Nakata, S. Yamazoe, K. Nakajima, J. Yamasaki, T. Mizugaki and T. Mitsudome, ACS Sustainable Chem. Eng., 2021, 9, 11238–11246 CrossRef CAS.
- H. Ishikawa, M. Sheng, A. Nakata, K. Nakajima, S. Yamazoe, J. Yamasaki, S. Yamaguchi, T. Mizugaki and T. Mitsudome, ACS Catal., 2021, 11, 750–757 CrossRef CAS.
- T. Vergunst, F. Kapteijn and J. A. Moulijn, Ind. Eng. Chem. Res., 2001, 40, 2801–2809 CrossRef CAS.
- S. Wang, T. Liu, Y. Zhu, X. Liu, Q. Luo, M. Zhu, T. Ding and T. Yao, J. Phys. Chem. C, 2023, 127, 5911–5919 CrossRef CAS.
- X. Chen, A. Zhao, Z. Shao, C. Li, C. T. Williams and C. Liang, J. Phys. Chem. C, 2010, 114, 16525–16533 CrossRef CAS.
- P. McNeice, M.-A. Müller, J. Medlock, W. Bonrath, N. Rockstroh, S. Bartling, H. Lund, K. Junge and M. Beller, ACS Sustainable Chem. Eng., 2022, 10, 9787–9797 CrossRef CAS.
- Q. Zhu, X. Lu, S. Ji, H. Li, J. Wang and Z. Li, J. Catal., 2022, 405, 499–507 CrossRef CAS.
- F. Chen, C. Kreyenschulte, J. Radnik, H. Lund, A.-E. Surkus, K. Junge and M. Beller, ACS Catal., 2017, 7, 1526–1532 CrossRef CAS.
- Q. Fan, S. He, L. Hao, X. Liu, Y. Zhu, S. Xu and F. Zhang, Sci. Rep., 2017, 7, 42172 CrossRef CAS PubMed.
- A. A. Shesterkina, O. A. Kirichenko, O. P. Tkachenko, A. L. Kustov and L. M. Kustov, Nanomaterials, 2023, 13, 2247 CrossRef CAS PubMed.
- D. Albani, K. Karajovic, B. Tata, Q. Li, S. Mitchell, N. López and J. Pérez-Ramírez, ChemCatChem, 2019, 11, 457–464 CrossRef CAS.
- D. Sharma, P. Choudhary, S. Kumar and V. Krishnan, Small, 2023, 19, 2207053 CrossRef CAS PubMed.
- T. Mitsudome, Catalysts, 2024, 14, 193 CrossRef CAS.
- X. Li, Y. Zhang, A. Wang, Y. Wang and Y. Hu, Catal. Commun., 2010, 11, 1129–1132 CrossRef CAS.
- W. Fu, L. Zhang, T. Tao and T. Tang, J. Ind. Eng. Chem., 2021, 95, 376–387 CrossRef CAS.
- S. Carenco, A. Leyva-Pérez, P. Concepción, C. Boissière, N. Mézailles, C. Sanchez and A. Corma, Nano Today, 2012, 7, 21–28 CrossRef CAS.
- Y. Chen, C. Li, J. Zhou, S. Zhang, D. Rao, S. He, M. Wei, D. G. Evans and X. Duan, ACS Catal., 2015, 5, 5756–5765 CrossRef CAS.
- Z. Guo, Z. Wang and M. Zhang, Chem. Eng. J., 2023, 463, 142505 CrossRef CAS.
- M. F. Delley, Z. Wu, M. E. Mundy, D. Ung, B. M. Cossairt, H. Wang and J. M. Mayer, J. Am. Chem. Soc., 2019, 141, 15390–15402 CrossRef CAS PubMed.
- B. Cormary, T. Li, N. Liakakos, L. Peres, P.-F. Fazzini, T. Blon, M. Respaud, A. J. Kropf, B. Chaudret, J. T. Miller, E. A. Mader and K. Soulantica, J. Am. Chem. Soc., 2016, 138, 8422–8431 CrossRef CAS PubMed.
- L. Lutterotti, D. Chateigner, S. Ferrari and J. Ricote, Thin Solid Films, 2004, 450, 34–41 CrossRef CAS.
- M. Vidal, J. Pandey, J. Navarro-Ruiz, J. Langlois, Y. Tison, T. Yoshii, K. Wakabayashi, H. Nishihara, A. I. Frenkel, E. Stavitski, M. Urrutigoïty, C. H. Campos, C. Godard, T. Placke, I. del Rosal, I. C. Gerber, V. Petkov and P. Serp, Chem. – Eur. J., 2024, 30, e202400669 CrossRef CAS PubMed.
- D. C. Gary, B. A. Glassy and B. M. Cossairt, Chem. Mater., 2014, 26, 1734–1744 CrossRef CAS.
- W.-S. Ojo, S. Xu, F. Delpech, C. Nayral and B. Chaudret, Angew. Chem., Int. Ed., 2012, 51, 738–741 CrossRef CAS PubMed.
- S. Fujii, S. Ishida and S. Asano, J. Phys. F, 1988, 18, 971–980 CrossRef CAS.
- X. Xu, Y. Lu, J. Shi, X. Hao, Z. Ma, K. Yang, T. Zhang, C. Li, D. Zhang, X. Huang and Y. He, Nat. Commun., 2023, 14, 7708 CrossRef CAS PubMed.
- D. R. Aireddy and K. Ding, ACS Catal., 2022, 12, 4707–4723 CrossRef CAS.
- A. Ropp and S. Carenco, Inorg. Chem., 2024, 63, 17077–17086 CrossRef CAS PubMed.
- M. Li, X. Liu, Y. Xiong, X. Bo, Y. Zhang, C. Han and L. Guo, J. Mater. Chem. A, 2015, 3, 4255–4265 RSC.
- D. Yin, J. Tang, R. Bai, S. Yin, M. Jiang, Z. Kan, H. Li, F. Wang and C. Li, Nanoscale Res. Lett., 2021, 16, 11 CrossRef CAS PubMed.
- K. Kaźmierczak, D. Yi, A. Jaud, P.-F. Fazzini, M. Estrader, G. Viau, P. Decorse, J.-Y. Piquemal, C. Michel, M. Besson, K. Soulantica and N. Perret, J. Phys. Chem. C, 2021, 125, 7711–7720 CrossRef.
- C. Waldt, H. Montalvo-Castro, A. Almithn, Á. Loaiza-Orduz, C. Plaisance and D. Hibbitts, J. Catal., 2023, 421, 403–418 CrossRef CAS.
- M. M. Alsabban, M. K. Eswaran, K. Peramaiah, W. Wahyudi, X. Yang, V. Ramalingam, M. N. Hedhili, X. Miao, U. Schwingenschlögl, L.-J. Li, V. Tung and K.-W. Huang, ACS Nano, 2022, 16, 3906–3916 CrossRef CAS PubMed.
- N. Jiang, B. You, M. Sheng and Y. Sun, Angew. Chem., 2015, 127, 6349–6352 CrossRef.
- M. Shimizu, Y. Tsushima and S. Arai, ACS Omega, 2017, 2, 4306–4315 CrossRef CAS PubMed.
- I. C. Gerber and P. Serp, Chem. Rev., 2020, 120, 1250–1349 CrossRef CAS PubMed.
- Q. Chen, I.-H. Svenum, L. Gavrilovic, D. Chen and E. A. Blekkan, Surf. Sci., 2019, 681, 24–31 CrossRef CAS.
- M. D. Navalikhina, N. E. Kavalerskaya, E. S. Lokteva, A. A. Peristyi, E. V. Golubina and V. V. Lunin, Russ. J. Phys. Chem. A, 2012, 86, 1800–1807 CrossRef CAS.
- J. Navarro-Ruiz, C. Rivera-Cárcamo, B. Machado, P. Serp, I. Del Rosal and I. C. Gerber, ACS Appl. Nano Mater., 2021, 4, 12235–12249 CrossRef CAS.
- Y. Dai, X. Gao, X. Chu, C. Jiang, Y. Yao, Z. Guo, C. Zhou, C. Wang, H. Wang and Y. Yang, J. Catal., 2018, 364, 192–203 CrossRef CAS.
- J. Tan, J. Cui, T. Deng, X. Cui, G. Ding, Y. Zhu and Y. Li, ChemCatChem, 2015, 7, 508–512 CrossRef CAS.
- H. Kim, S. Yang, Y. H. Lim, J. Lee, J.-M. Ha and D. H. Kim, J. Catal., 2022, 410, 93–102 CrossRef CAS.
- W. Chen, H. Xu, X. Ma, L. Qi and Z. Zhou, Chem. Eng. J., 2023, 455, 140565 CrossRef CAS.
- B. H. Arpini, J. L. Fiorio, J. V. F. da Costa, J.-O. Joswig and L. M. Rossi, Catal. Sci. Technol., 2024, 14, 1017–1025 RSC.
- L. Zhao, X. Qin, X. Zhang, X. Cai, F. Huang, Z. Jia, J. Diao, D. Xiao, Z. Jiang, R. Lu, N. Wang, H. Liu and D. Ma, Adv. Mater., 2022, 34, 2110455 CrossRef CAS PubMed.
- J. Navarro-Ruiz, J. Audevard, M. Vidal, C. H. Campos, I. Del Rosal, P. Serp and I. C. Gerber, ACS Catal., 2024, 14, 7111–7126 CrossRef CAS.
- A. W. Augustyniak and A. M. Trzeciak, Inorg. Chim. Acta, 2022, 538, 120977 CrossRef CAS.
- L. Xiang, H. Feng, M. Liu, X. Zhang, G. Fan and F. Li, ACS Appl. Nano Mater., 2021, 4, 4688–4698 CrossRef CAS.
- T. Kubota, H. Ogawa, Y. Okamoto, T. Misaki and T. Sugimura, Appl. Catal., A, 2012, 437–438, 18–23 CrossRef CAS.
- C. Rivera-Cárcamo, C. Scarfiello, A. B. García, Y. Tison, H. Martinez, W. Baaziz, O. Ersen, C. Le Berre and P. Serp, Adv. Mater. Interfaces, 2021, 8, 2001777 CrossRef.
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, Catal. Today, 2021, 373, 69–79 CrossRef CAS.
- C. Scarfiello, K. Soulantica, S. Cayez, A. Durupt, G. Viau, N. Le Breton, A. K. Boudalis, F. Meunier, G. Clet, M. Barreau, D. Salusso, S. Zafeiratos, D. P. Minh and P. Serp, J. Catal., 2023, 428, 115202 CrossRef CAS.
- L. R. Merte, G. Peng, R. Bechstein, F. Rieboldt, C. A. Farberow, L. C. Grabow, W. Kudernatsch, S. Wendt, E. Lægsgaard, M. Mavrikakis and F. Besenbacher, Science, 2012, 336, 889–893 CrossRef CAS PubMed.
- X. Zhao, J. Wang, L. Lian, G. Zhang, P. An, K. Zeng, H. He, T. Yuan, J. Huang, L. Wang and Y.-N. Liu, ACS Catal., 2023, 13, 2326–2334 CrossRef CAS.
- D. Gao, S. Wang, Y. Lv, C. Wang, J. Ren, P. Zheng, L. Song, A. Duan, X. Wang, G. Chen and C. Xu, ACS Catal., 2024, 14, 1939–1950 CrossRef CAS.
- J. Mazarío, S. Ghosh, V. Varela-Izquierdo, L. M. Martínez-Prieto and B. Chaudret, ChemCatChem, 2025, 17, e202400683 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5nr00618j |
| This journal is © The Royal Society of Chemistry 2025 |