
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Material characterization methods for investigating charge storage processes in 2D and layered materials-based batteries and supercapacitors
Albert
de Kogel
 a,
Ruocun (John)
Wang
*b,
Wan-Yu
Tsai
*cd,
Maciej
Tobis
ef,
Robert
Leiter
ef,
Ruipeng
Luo
g,
Evan Wenbo
Zhao
*g,
Simon
Fleischmann
*ef and
Xuehang
Wang
*a
a,
Ruocun (John)
Wang
*b,
Wan-Yu
Tsai
*cd,
Maciej
Tobis
ef,
Robert
Leiter
ef,
Ruipeng
Luo
g,
Evan Wenbo
Zhao
*g,
Simon
Fleischmann
*ef and
Xuehang
Wang
*a
aDepartment of Radiation Science and Technology, Faculty of Applied Sciences, Delft University of Technology, 2629JB, Delft, The Netherlands. E-mail: X.Wang-22@tudelft.nl
bUniversity of North Texas, 3940 N Elm St, Denton, TX 76207, USA. E-mail: Ruocun.Wang@unt.edu
cUniv. Lille, CNRS, Univ. Polytechnique Hauts-de-France, UMR 8520 – IEMN, F-59000 Lille, France. E-mail: wan-yu.tsai2@univ-lille.fr
dRéseau sur le Stockage Electrochimique de l'Energie (RS2E), CNRS FR 3459, 33 rue Saint Leu, Amiens, Cedex 80039, France
eHelmholtz Institute Ulm (HIU), 89081 Ulm, Germany. E-mail: simon.fleischmann@kit.edu
fKarlsruhe Institute of Technology (KIT), 76021 Karlsruhe, Germany
gMagnetic Resonance Research Center, Institute for Molecules and Materials, Radboud University, Nijmegen, AJ 6525, The Netherlands. E-mail: evanwenbo.zhao@ru.nl
First published on 9th May 2025
Abstract
Two-dimensional (2D) materials offer distinct advantages for electrochemical energy storage (EES) compared to bulk materials, including a high surface-to-volume ratio, tunable interlayer spacing, and excellent in-plane conductivity, making them highly attractive for applications in batteries and supercapacitors. Gaining a fundamental understanding of the energy storage processes in 2D material-based EES devices is essential for optimizing their chemical composition, surface chemistry, morphology, and interlayer structure to enhance ion transport, promote redox reactions, suppress side reactions, and ultimately improve overall performance. This review provides a comprehensive overview of the characterization techniques employed to probe charge storage mechanisms in 2D and thin-layered material-based EES systems, covering optical spectroscopy, imaging techniques, X-ray and neutron-based methods, mechanical probing, and nuclear magnetic resonance spectroscopy. We specifically highlight the application of these techniques in elucidating ion transport dynamics, tracking redox processes, identifying degradation pathways, and detecting interphase formation. Furthermore, we discuss the limitations, challenges, and potential pitfalls associated with each method, as well as future directions for advancing characterization techniques to better understand and optimize 2D material-based electrodes.
 Albert de Kogel | Albert de Kogel obtained his MSc in Chemical Engineering from Delft University of Technology in 2023, where he is currently pursuing a PhD degree at the Department of Radiation Science and Technology. His research focuses on the development of in situ characterization techniques for electrochemical energy storage systems. |
 Ruocun (John) Wang | Dr Ruocun (John) Wang is an Assistant Professor in the Department of Materials Science and Engineering at the University of North Texas (UNT). He received his B.S. from Purdue University and Ph.D. from North Carolina State University, both in Materials Science and Engineering. Before joining UNT, he was a Research Associate at the A. J. Drexel Nanomaterials Institute and the Department of Materials Science and Engineering at Drexel University. His research spans the discovery and synthesis of novel metastable transition metal oxides and MXenes, as well as the characterization of the interplay between structure and electrochemical properties in these materials. |
 Wan-Yu Tsai | Wan-Yu Tsai is a Junior Professor at IEMN in Lille University. She received her Ph.D. in Materials Science at Paul Sabatier University (Toulouse, France) in 2015. After graduation, she joined the Center for Nanophase Materials Sciences (CNMS) at Oak Ridge National Laboratory (ORNL, USA) as a postdoc. She then became a staff scientist in 2020 working with the Energy Storage Group at ORNL before she joined Lille University in 2023. Her research focuses on multimodal advanced characterizations, primarily using Atomic Force Microscopy and pairing up with other techniques, to investigate the interfacial properties in different energy storage applications. |
 Ruipeng Luo | Ruipeng Luo is currently a PhD candidate at Magnetic Resonance Research Center, Radboud University Nijmegen. He graduated from Harbin Institute of Technology (Harbin, China) with an ME degree. His research interests include the development of operando EC-NMR and its application in understanding the mechanism of electrocatalytic reactions such as ammonia synthesis. |
 Evan Wenbo Zhao | Evan Wenbo Zhao is a tenured assistant professor at Magnetic Resonance Research Center, Radboud University Nijmegen. He did his postdoctoral work at the Yusuf Hamied Department of Chemistry University of Cambridge, and obtained his PhD degree from the Department of Chemistry University of Florida. His research interest is in developing and applying operando NMR methods for studying redox flow battery and electrochemical synthesis. |
 Simon Fleischmann | Simon Fleischmann is a Principal Investigator and BMBF-Junior Research Group Leader at Helmholtz Institute Ulm (HIU) and Karlsruhe Institute of Technology (KIT), Germany. His group investigates the mechanistic origins and functional properties arising from nanoconfinement effects within solid-state host materials in the context of electrochemical energy storage and conversion. This involves developing syntheses for materials with well-defined nanoconfinement properties as well as innovative structural and electrochemical characterization on time & length scales relevant to processes in nanoconfinement. |
 Xuehang Wang | Xuehang Wang is an assistant professor in the Department of Radiation Science and Technology at Delft University of Technology. She earned her Ph.D. in chemical engineering from the Norwegian University of Science and Technology in 2016 and was a postdoctoral researcher at A.J. Drexel Nanomaterials Institute, Drexel University from 2017 to 2020. Her research focuses on gaining an understanding of the charge mechanisms of energy storage devices, particularly the electrolyte transport at the electrode–electrolyte interfaces of 2D MXenes and various carbon materials. |
1. Introduction
The growing demand for portable electronics, electric vehicles, and stationary energy grid storage is driving the development of electrochemical energy storage (EES) devices with higher energy and power densities and longer life. To meet these demands, new materials are continuously being developed to enhance performance, including improved redox activity for higher energy density, better kinetics for higher power output, and enhanced chemical and electrochemical stability for longer cycle life. Two-dimensional (2D) materials and thin layered materials, such as transition metal dichalcogenides (TMDs),1 graphene,2–4 borophene,5 phosphorene,6 layered double hydroxides,7 and 2D transition metal carbides/nitrides (MXenes),8 are a class of materials consisting of atomically thin layers with strong in-plane bonding and weak interactions between layers. Their unique structures offer many distinct advantages for EES applications compared to bulk materials, including a high surface-to-volume ratio which benefits the charge storage capacity, expandable interlayer spacing for enhanced ion transport,9 and high in-plane conductivity for fast electron transfer.10 Furthermore, some 2D materials exhibit abundant surface chemistry for redox activity, the ability to form heterostructures for synergistic performance enhancements, tunable morphology for improved transport properties, and excellent mechanical strength that enables flexible and free-standing structures.11 These advantages have led to the successful integration of 2D materials into various EES components, including electrodes, current collectors,12 electrolyte constituents,13 separators,14 and binders,15 where they have been reported to enhance device performance.2D materials have been widely explored as electrode materials in batteries, pseudocapacitors and supercapacitors, for example, the early use of graphene as an anode material in lithium-ion batteries16 and the application of MXene electrodes in aqueous supercapacitors.17 In batteries, charge is stored through diffusion-controlled faradaic reactions, which mostly involve phase transformation and at least one full electron transfer. This mechanism enables high energy density but limits power output due to slow ion diffusion. In contrast, supercapacitors rely on non-faradaic surface reactions, where ions rapidly adsorb onto the electrode surface to form an electrochemical double layer (EDL),18 resulting in exceptional power density but much lower energy density. Pseudocapacitors are expected to bridge this energy-to-power gap as they realize charge storage through fast surface redox reactions and pseudocapacitive intercalation.19 For 2D and layered materials, the charge storage mechanism cannot be solely classified based on their chemical composition, as it varies depending on many factors, such as the electrode–electrolyte combination, applied potentials and electrode thickness. For example, TMDs (TiS2,20 MoS2),21 layered transition metal oxides (TMOs) (V2O5,22 LiCoO2)23 and MXenes (Ti3C2Tx)24 have been widely studied as cation intercalation hosts for batteries. However, the charge storage behavior of these materials can change significantly when the electrode thickness is reduced or when the electrolyte composition is modified to favor fast, surface-based redox reactions. For example, pseudocapacitive behavior has been observed in TMDs (MoS2),25,26 MXenes,27–29 graphene with transition metal nanoparticles30 and N-doped graphene31 in various electrolytes. Moreover, the surface faradaic contribution in some 2D materials can be further suppressed by incorporating a high level of solvation, approaching double-layer behavior.32 To determine their charge storage mechanism, various electrochemical characterization techniques, including cyclic voltammetry, galvanostatic charge–discharge, and electrochemical impedance spectroscopy, are widely used. However, electrochemical methods alone do not provide detailed insights into electrode and electrolyte properties, offering little to no insight into ion intercalation, electron transfer, and other reaction processes.
Revealing the charge storage mechanisms in 2D materials and at 2D electrode–electrolyte interfaces is essential for establishing the correlation between electrode properties and EES performance, ultimately guiding the optimization and design of new 2D materials. For example, increasing the ion accessible redox-active electrode surface area, facilitating ion transport in the 2D interlayer space, and extending electrochemical stability can enhance the electrochemical performance of 2D materials. Since charge storage is always accompanied by changes in the chemical composition, structure, or electronic state of the electrode, a range of advanced characterization techniques is employed to measure these changes and elucidate the underlying charge storage mechanisms (Fig. 1). To investigate ion intercalation and solvent co-intercalation, techniques such as X-ray diffraction (XRD), electrochemical dilatometry (ECD), neutron diffraction (ND), transmission electron microscopy (TEM), and atomic force microscopy (AFM) are used to monitor variations in interlayer spacing.33–35 However, these methods do typically not directly identify the intercalated species at specific applied potentials. To obtain a microscopic picture of the intercalation process and the interlayer electrolyte composition, they must be combined with electrochemical quartz crystal microbalance (EQCM), inelastic neutron scattering (INS), and computational simulations.36,37 For understanding redox activities during ion intercalation, changes in the local chemical environment can be probed using X-ray photoelectron spectroscopy (XPS), nuclear magnetic resonance (NMR), TEM, scanning transmission X-ray microscopy (STXM), neutron total scattering (NTS), Raman spectroscopy, and Fourier transform infrared spectroscopy (FTIR).38,39 Meanwhile, the electron transfer process can be directly monitored using in situ UV-vis spectroscopy, X-ray absorption spectroscopy (XAS) and Raman spectroscopy. The diffusion kinetics of intercalated species, which is closely correlated with the rate performance of EES devices, can be analyzed using quasi-elastic neutron scattering (QENS) and NMR. Additionally, imaging techniques such as TEM, STXM, neutron tomography (NT), and neutron depth profiling (NDP) are valuable for chemical mapping and visualizing particle morphologies, enabling the monitoring of electrode degradation and interphase formation.40–43 It is important to note that several practical challenges complicate the characterization of 2D materials compared to bulk materials. Sample preparation demands extra care, as the physical properties of 2D materials can vary considerably with the number of layers and their uniformity. Additionally, 2D materials are often highly reactive with oxygen and moisture, increasing the risk of contamination. Furthermore, interpreting data from 2D materials requires special attention, as their properties can differ significantly from those of bulk materials, illustrating the necessity for comprehensive guidelines in the literature.
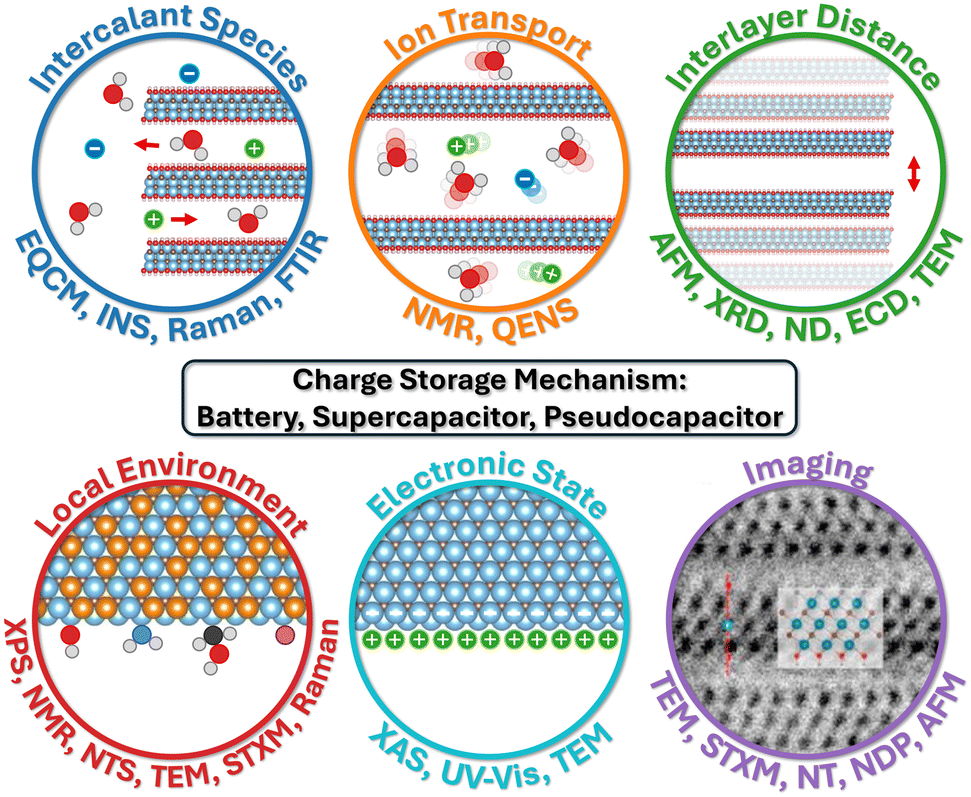 | ||
| Fig. 1 Characterization methods for 2D materials as EES electrodes, grouped by type of analysis. In these schematics, intercalant species indicate solvent molecules and ions that intercalate into the interlayers of 2D materials, the motion of which is defined by ion transport. Interlayer distance refers to the space between two layers of 2D material which varies when species intercalate. Local environment is the chemical composition and bonding structure at the nanoscale, whereas electronic state refers to the behavior and energy of electrons in the material. Also, imaging techniques produce a visual impression of the sample. Panel imaging is reproduced with permission from ref. 47, copyright 2021 Elsevier. | ||
In this review, we explore a wide range of in situ and ex situ characterization techniques used to investigate charge storage mechanisms in 2D and layered material-based EES devices. While previous review articles have covered characterization techniques for either 2D materials or bulk EES materials, they primarily focus on specific methods or broader applications without directly addressing the charge storage mechanisms in 2D electrodes. Additionally, other reviews focus on characterization methods for specific systems, such as lithium batteries,44,45 or on a single technique for a specific system.46 However, there is a lack of reviews that specifically cover characterization methods used to investigate the charge storage mechanism in various 2D-material-based electrodes. Our review fills this gap in the literature by uniquely integrating advanced characterization techniques with a focus on charge storage mechanisms in 2D materials, offering a comprehensive perspective on both their capabilities and limitations. For each method, we provide a general explanation of its working principle, followed by examples of studies demonstrating its application in uncovering key charge storage mechanisms, such as ion intercalation, redox activity, degradation mechanisms, and interphase formation in 2D material-based electrodes. Additionally, we highlight the challenges associated with each technique, offer practical guidelines to mitigate experimental pitfalls, and propose potential directions for future advancements in characterization methodologies for 2D material-based EES processes.
2. Optical spectroscopy
2.1 Raman spectroscopy
Raman spectroscopy is a fast, versatile, and non-destructive vibrational spectroscopy used to characterize lattice structure and, in some cases, the electronic, optical, and phonon properties of 2D and layered materials.48 A Raman spectrum offers rich information, where the band position identifies chemical structures, band intensity indicates the polarizability of chemical bonds, the orientation of phases, and the detectability of bonds, bandwidth informs the quality of the crystalline structure and the concentration of doping or defects, the band ratio gives relative concentrations of bonds, and band position shift advises changes to deformation, pressure, stress, and temperature. This abundant information, combined with the lab-scale Raman instrument's availability, made it one of the most popular techniques for characterizing 2D and layered materials. However, the simplicity of Raman measurements comes with the difficulty of spectral interpretation. It takes considerable experimental and theoretical effort to understand the structural roots of spectral features, which serve as the foundation for further investigations like understanding charge storage mechanisms. The groundwork was laid for graphene,48,49 MoS2,50 MXenes,51,52 and many others, and will be needed for new materials in the future. When it comes to energy storage, in situ Raman is helpful with identifying phase transformation,53–56 bond formation,52,57 and interfacial solvent orientations.58Here, we first review two examples of in situ Raman spectroscopy of graphene to showcase the depth of charge storage insights attainable from this powerful technique. Wang et al. characterized the potential-dependent transformation in the surface-confined interfacial water structure on graphene, which is a fundamental component of the electrical double layer.58 The authors overcame the weak interfacial water signal with shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS). They performed the experiments in setup as shown in Fig. 2a. Note that the signal enhancement here is achieved through special sample preparation. A similar setup can be used for any in situ Raman work, which requires the Raman laser to be focused on the electrode while performing the electrochemical experiments. With SHINERS, the authors identified three types of water near the interface: the most popular 2-coordinated hydrogen-bonded water (2-HB·H2O), 4-coordinated hydrogen-bonded water, and cation-hydrated water. With ab initio molecular dynamics (AIMD) simulations and surface selection rule, they found a good correlation between the potential-driven intensity change of 2-HB·H2O and water orientation at the interface (Fig. 2b). From the open-circuit potential (OCP) to more negative potentials, most interfacial water gradually transitions from being parallel to the surface into one H-down structure, with one OH bond facing the graphene and the other toward the solution and, eventually, the solution-facing OH bond also rotates toward the graphene surface. This finding deepens our understanding of surface-confined interfacial water, a crucible constituent of the electrical double layer, relevant to the development of future aqueous-based supercapacitors. In another example, Cohn et al. studied diglyme-solvated Na-ion intercalation in few-layered graphene (FLG) with in situ Raman, as shown in Fig. 2c. As the FLG intercalates diglyme-Na and reaches a voltage plateau (stage 2 to stage 1 transition happens, where the stage number represents the number of graphene layers in between intercalant layers), a new G band (in-plane optical phonon modes corresponding to sp2 carbon bonding) representing the charged graphene layers (Gc) increases drastically as the G band representing the uncharged graphene layers (Guc) red shifts and disappears. This drastic intensity change was attributed to the Pauli blocking of destructive interference Raman pathways, which happens when the Fermi level is at half the excitation laser energy. Using lasers of two different lasers (532 and 785 nm), they estimated the Fermi energy of ∼0.8 eV for the stage 2 compound and ∼1.2 eV for the stage 1 compound, which is close to the 1.5 eV Fermi energy of LiC6. They also found that the onset of the Gc band blue shift correlates well with the start of the staging reaction (Fig. 2d). The insight of staging behavior informs us of the phase transformation in the FLG, an important phenomenon related to the performance of energy storage materials. Phase transformation is typically related to nucleation overpotential, which lowers energy efficiency59 and mechanical deformation, which may lead to long-term mechanical failure of the material60 of the electrodes during charge storage. The change in the Fermi level indicates changes in the electronic conductivity of FLG, which in turn affects the power density of the electrode.
 | ||
| Fig. 2 (a) Schematic of an aqueous in situ electrochemical Raman cell, with Pt wire as the counter electrode and saturated calomel electrode (SCE) as the reference electrode. (b) intensity change of the OH stretching band and the vertical component of the OH bond of 2-coordinated hydrogen-bonded water confined at the surface of a single-layer graphene-Au(111) electrode in a 0.1 M NaClO4 solution collected by in situ shell-isolated nanoparticle-enhanced Raman spectroscopy (SHINERS).58In situ Raman spectroscopy of diglyme-solvated sodium ion intercalation in few-layered graphene: (c) potential profile of the intercalation with the corresponding microscope images, band diagrams, and pseudo heat map showing the Raman intensity using two lasers (1.58 eV = 785 nm and 2.33 eV = 532 nm). (d) The positions of graphene G peak components during Galvanostatic discharge at 0.2 A g−1, where Gc is the G mode of a charged graphene layer, Guc is the G mode of an uncharged graphene layer, triangles are data from the 1.58 eV laser, and circles are data from the 2.33 eV laser.54 Panel (a and b) reproduced with permission from ref. 58, copyright 2023 Elsevier. Panel (c and d) reproduced with permission from ref. 54, copyright 2016 American Chemical Society. | ||
In situ Raman can be applied to other 2D and layered materials to elucidate electrochemical ion intercalation mechanisms. Xiong et al. investigated Li-ion intercalation in a single MoS2 flake in a commercial lithium-ion battery electrolyte.56 As shown in Fig. 3a–i, Li-ion intercalation causes the MoS2 flake to be more translucent and broadens the E2g1 band (in-plane optical vibration of the Mo–S bond), which is a sign of structural defects caused by the lithiation of MoS2. When the flake is fully lithiated, the A1g band (out-of-plane optical vibration of S atoms) vanishes, indicating the dissociation of interlayer bonding due to Li-ion influx. Meanwhile, peaks at 150 and 200 cm−1 emerge, indicating a 2H to 1T phase transformation in MoS2. These effects appear to be reversible over multiple cycles, showing reversible structural changes in MoS2 caused by repeated Li intercalation and deintercalation. Similarly, the electrochemical proton surface redox reactions of Ti3C2Tx MXene in acidic electrolytes were investigated with in situ Raman. Shevchuk and Sarycheva et al. showed in Fig. 3j and k that during pseudocapacitive proton adsorption on the surface oxygen terminations of Ti3C2Tx MXene, the A1g(Ti, C, Tx) band (out-of-plane vibration modes of titanium atoms in the outer layer as well as carbon and surface groups) region diminishes and the A1g(C) band (out-of-plane vibration modes of the carbide core) red shifts with decreasing intensity.52 The reversible change of the latter was observed by Johnson et al. (Fig. 3i).57 The diminishing of the A1g(Ti, C, Tx) band is attributed to the change of symmetry due to the surface termination reaction from –O– to –OH, while the A1g(C) band is explained by the softening of the Ti–O bond. These Raman spectrum changes provide more insight into the structural evolution related to the redox-active intercalation of protons in Ti3C2Tx MXene.
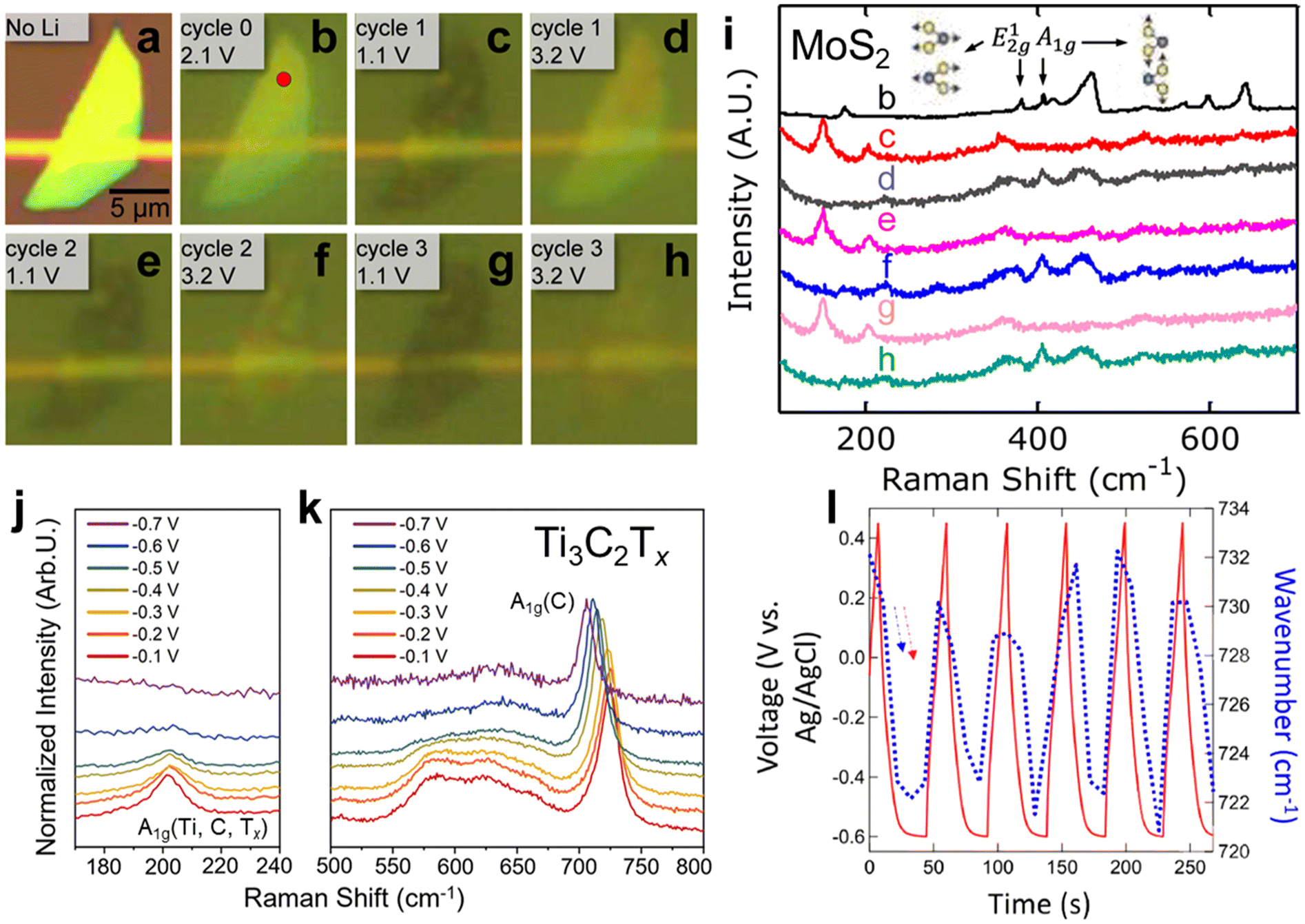 | ||
Fig. 3
In situ Raman spectroscopy of a MoS2 flake in a 1 M LiPF6 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 w/w ethylene carbonate/diethyl carbonate: (a–h) optical reflection images of the MoS2 flake at different cycles and potentials of Li-ion intercalations. (i) Raman spectra of the MoS2 flake at different cycles and potentials (potentials are indicated in the optical reflection image diagram).56In situ Raman spectroscopy of Ti3C2Tx MXene in acidic electrolytes: Raman spectra of (j) the A1g(Ti, C, Tx) vibration region and (k) the A1g(C) vibration region.52 (l) the shift in wavenumber of the A1g(C) peak and potential as a function of time.57 Panel (a–i) reproduced with permission from ref. 56, copyright 2015 American Chemical Society. Panel (j–k) reproduced with permission from ref. 52, copyright 2023 American Chemical Society. Panel (l) reproduced with permission from ref. 57, copyright 2022 Wiley. :1 w/w ethylene carbonate/diethyl carbonate: (a–h) optical reflection images of the MoS2 flake at different cycles and potentials of Li-ion intercalations. (i) Raman spectra of the MoS2 flake at different cycles and potentials (potentials are indicated in the optical reflection image diagram).56In situ Raman spectroscopy of Ti3C2Tx MXene in acidic electrolytes: Raman spectra of (j) the A1g(Ti, C, Tx) vibration region and (k) the A1g(C) vibration region.52 (l) the shift in wavenumber of the A1g(C) peak and potential as a function of time.57 Panel (a–i) reproduced with permission from ref. 56, copyright 2015 American Chemical Society. Panel (j–k) reproduced with permission from ref. 52, copyright 2023 American Chemical Society. Panel (l) reproduced with permission from ref. 57, copyright 2022 Wiley. | ||
With the advancement of instrumentations like faster detectors, coupling with atomic-force microscopes to enable tip-enhanced Raman spectroscopy (TERS),61 and more, Raman spectroscopy will bring more insights into charge storage mechanisms in 2D and layered materials. The challenge of spectral interpretation demands more development of theoretical frameworks to correlate spectral features to the structure of materials as well as coupling with additional characterization techniques to narrow down the factors that affect the spectral changes during charge storage.
2.2 Fourier transform infrared spectroscopy
Fourier Transform Infrared (FTIR) spectroscopy is a vibrational spectroscopy technique that characterizes the transmitted or reflected IR radiation from a sample. The spectrum carries information on molecular absorption, creating molecular fingerprints of materials. These fingerprints are used to identify chemical bonds that involve changes in dipole moments during molecular vibrations (stretching or bending), which is complementary to Raman spectroscopy. In 2D and layered materials, FTIR is useful for characterizing defects and confined water, as demonstrated in graphene oxides and the 12 most common MXenes.62–64 When operated in the attenuated total reflectance (ATR) mode, the infrared radiation penetrates the substrate and is reflected by the sample before it reaches the electrolyte, as shown in Fig. 4a. As a result, ATR-FTIR can get rid of much of the signal interference from the bulk electrolyte and allows for in situ or operando observation of molecular vibrational changes in materials during electrochemical cycling, giving insights into intercalation species, surface functional groups, defect evolution, and confined water dynamics.65–68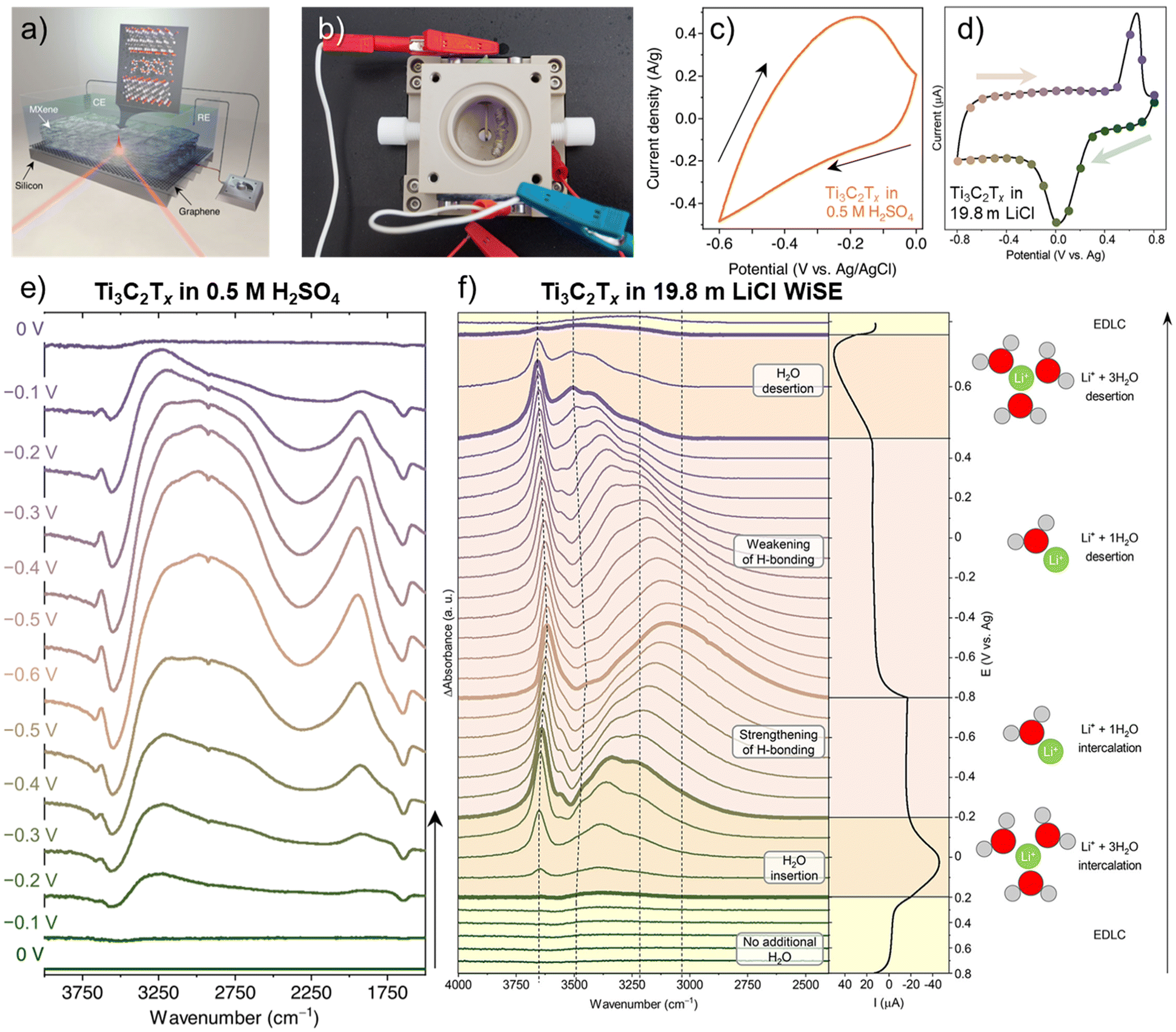 | ||
| Fig. 4 (a) Schematic of an operando attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy setup with Ti3C2Tx film drop cast on a monolayer-graphene-coated silicon wafer as the working electrode. (b) the physical instrument of the schematic in (a) showing the counter electrode of coiled Pt wire and a quasi-reference electrode of Ag wire.65 Cyclic voltammograms at 2 mV s−1 of Ti3C2Tx in (c) 0.5 M H2SO465 and (d) 19.8 m LiCl collected from this setup.66 Operando electrochemical FTIR difference spectra of Ti3C2Tx measured during electrochemical cycling in (e) 0.5 M H2SO465 and (f) 19.8 m LiCl.66 Panel (a–c and e) reproduced with permission from ref. 65, copyright 2023 Springer Nature. Panel (d and f) reproduced with permission from ref. 66, copyright 2023 American Chemical Society. | ||
Since many carbon bonds are active in IR spectra, FTIR is a powerful technique to identify surface functional groups of carbon materials. Yao et al. adopted operando ATR-FTIR in 2015 to study the structure and composition changes during electrochemical cycling in 0.1 M HClO4.68 This study used a Si ATR prism as a substrate and deposited mono- to few-layer graphene as the working electrode. With a temporal resolution of 1 s per spectrum and a spectral resolution of 4 cm−1, they could collect IR spectra at 20 mV s−1 with cyclic voltammetry. operando ATR-FTIR helped identify that the oxygen-containing functional groups such as C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and –COO− at the edge and defect sites of graphene are the active sites for water oxidation and graphene at the graphene at E >2 V vs. reversible hydrogen electrode (RHE). In addition, they found graphene is stable between 0.05 V and 1.5 V vs. RHE and provides high IR sensitivity of interfacial species, suggesting that graphene layers can be a stable support for amplifying the signal of other samples. Similarly, Bernicola et al. investigated the electrochemical activation of graphene oxide with operando ATR-FTIR.67 They revealed water uptake upon activation and the formation of organic intermediates like lactones and hydroquinones.
O, and –COO− at the edge and defect sites of graphene are the active sites for water oxidation and graphene at the graphene at E >2 V vs. reversible hydrogen electrode (RHE). In addition, they found graphene is stable between 0.05 V and 1.5 V vs. RHE and provides high IR sensitivity of interfacial species, suggesting that graphene layers can be a stable support for amplifying the signal of other samples. Similarly, Bernicola et al. investigated the electrochemical activation of graphene oxide with operando ATR-FTIR.67 They revealed water uptake upon activation and the formation of organic intermediates like lactones and hydroquinones.
Intercalation of hydrated ions is common among 2D and layered materials in aqueous media. The strong IR absorption of O–H bonds makes FTIR spectroscopy one of the most well-suited techniques for studying hydrogen bond networks and water solvation under the confinement of 2D materials. Recently, Lounasvuori et al. applied synchrotron operando ATR-FTIR to study the confined water in Ti3C2Tx MXene in acidic and water-in-salt electrolytes.65,66 Synchrotron IR offers a high flux in the far-IR and THz range and a high brightness in the mid-IR spectral range. The IRIS beamline at the Helmholtz-Zentrum Berlin could achieve an advantageous spectral resolution of 0.2 cm−1.69 A typical in situ IR cell used in the experiment is shown in Fig. 4b. When Ti3C2Tx MXene is cycled in 0.5 M H2SO4, the in situ cell showed a relatively resistive cyclic voltammogram at 2 mV s−1 (Fig. 4c), likely due to the higher resistance of dropcast film compared to vacuum-filtered ones. With this setup, they found fundamentally different H-bonding structures of hydrated protons confined in the interlayer of Ti3C2Tx MXene compared to the bulk solution. The evolutions of IR features in Fig. 4e highlighted two parallel processes: water reordering toward stronger H-bonding due to the applied potential in the electrical double layer and proton intercalation coupled with surface redox reactions. The intercalated protons/hydronium ions showed a reduction in the coordination number due to 2D confinement and possess an Eigen-like character. The water H-bonding was also strengthened at higher proton concentrations. However, challenges remain in identifying the surface hydroxyl groups in the IR spectra. In the 19.8 m LiCl water-in salt electrolyte (WiSE), the voltammogram of Ti3C2Tx MXene in Fig. 4d is almost identical to the standard Swagelok cell electrochemical characterization.32 Upon solvated lithium-ion intercalation, the difference FTIR spectra in Fig. 4f show the number of coordinated water molecules around Li-ions within MXene nanoconfinement as a function of electrochemical potentials, demonstrating a changing intermolecular hydrogen bonding network. The strongly H-bonded water has reduced activity and is consistent with the suppressed HER and a larger electrochemical window, typical in WiSE.
While numerous charge storage insights have been obtained from operando electrochemical ATR-FTIR, there are plenty of opportunities to investigate 2D and layered materials with this technique. One empty area is studying the structural change of MXenes, for example, through fingerprint regions, as previous studies focused on the confined water region. In addition, electrochemical conversion at the interface between the electrode and the electrolyte is a promising area.70 We expect to see continued development of this technique for studying 2D and layered materials.
2.3 UV-Vis spectroscopy
UV-Vis spectroscopy is a technique that measures the amount of discrete wavelengths of UV and visible light absorbed by or transmitted through a sample. It allows for concentration measurement of 2D and layered materials dispersed in solutions using Beer–Lambert's law, thickness measurement of thin films,71 degradation quantifications,72 and optical property investigations.73,74 Light absorption in the visible and near IR region can be related to electronic transitions and plasmonic effects in materials. Therefore, the electrochromic effect caused by electrochemical ion intercalation can be readily characterized by in situ UV-Vis spectroscopy, as demonstrated in multiple MXenes (Fig. 5a).75,76 Electrochromic effect and charge storage are two sides of the same coin. More recently, in situ UV-Vis spectroscopy was demonstrated as a powerful tool for distinguishing charge storage mechanisms and quantifying oxidation state changes.77 | ||
| Fig. 5 (a) in situ UV-Vis spectra of Ti3C2Tx, Ti3CNTx, Ti2CTx and Ti1.6Nb0.4CTx in a protic gel electrolyte comparing cathodic polarization (−1 V vs. Ag wire) with the open-circuit potential condition.76 (b) A general workflow to determine charge storage mechanism using in situ UV-Vis spectroscopy from experimental setup to data analysis. In situ UV-Vis spectroscopy Ti3C2Tx in 1 M H2SO4: (c) Electrochemical (red) and UV-Vis (blue) cyclic voltammograms (CVs), (d) electrochemical CV with the electrical double layer (EDL) contribution in red and the redox contribution in blue. (e) UV-Vis CV with the EDL contribution in red and the redox contribution in blue.77 Panel (a) reproduced from ref. 76 with permission from the Royal Society of Chemistry. Panel (b–e) reproduced with permission from ref. 77, copyright 2023 Springer Nature. | ||
A general workflow for using in situ UV-Vis spectroscopy to determine the charge storage mechanism and oxidation state is summarized in Fig. 5b. For a thin film sample, cost-efficient in situ cells can be constructed with glass slides, tape, the active material, an electrolyte, a counter electrode, and a reference electrode. The coupling between the electrochemical responses and UV-Vis signal can be determined by operando UV-Vis spectroscopy, where single-wavelength UV-Vis signal can be measured at a fast rate during a cyclic voltammetry experiment. In the case of Ti3C2Tx MXene in sulfuric acid, a clear correlation between the electrochemical cyclic voltammogram (CV) and the derivative of absorbance, which may be named UV-Vis CV, can be established, as shown in Fig. 5c. This correlation means that the evolution of UV-Vis spectra is related to redox activities, and allows for further investigations of charge storage mechanisms using UV-Vis. Then, in situ UV-Vis spectroscopy, where full UV-Vis spectra are collected at discrete potential steps, can be set up to distinguish between electrical double-layer (EDL), pseudocapacitive, and battery-type charge storage mechanisms in an electrochemical system. In a charge storage process, there are always regions dominated by electrical double-layer charging (red region in Fig. 5d). The comparison of absorbance change (or derivative) due to a redox-active region and an EDL region informs us of the extent of charge transfer (blue vs. red regions in Fig. 5e). In the study of Zhang et al.,77 between the potential region of interest and the EDL region, EDL, pseudocapacitive, and battery-type charge storage mechanisms typically have a ratio of ∼1, ∼10, and ∼50, respectively. This method can also quantify the extent of oxidation state changes (n) according to eqn (1):
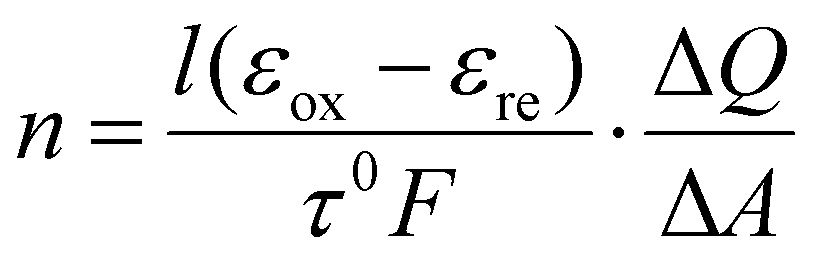 | (1) |
While in situ UV-Vis spectroscopy or spectroelectrochemistry is a mature technique especially in the field of electrochromic materials, determining charge storage mechanism and oxidation state changes is a new development that needs to be validated in more charge storage systems, including 2D and layered materials. Given the availability of UV-Vis spectrometers, this technique is expected to help make energy storage research more accessible.
2.4 Practical considerations for optical spectroscopy
There are several limitations to optical techniques, which require that the source light reaches and interacts with the materials of interest and then is received by the detector without significant interference throughout the process.First, the limited penetration depth of light in the UV-Vis-IR range in solids demands only optically transparent substances to be included in the optical path, making it challenging to probe the embedded interfaces in solids with Raman spectroscopy. Similarly, UV-Vis spectroscopy transmission mode needs optically transparent, thin samples. To circumvent this limitation, researchers may choose spectroscopy based on particles with extended penetration depth, such as neutrons, or light of different wavelengths, such as X-rays.
Second, the spectroscopic features of interest may be inactive in the materials of interest, outside the instrument's range, or overwhelmed by other sources. For example, metals are not Raman-active, charge storage-induced spectroscopic changes may be outside the UV-Vis range, fluorescence can interfere with Raman signal, and overlapping peaks of water can overwhelm material-specific vibrational information in FTIR. While studying metal requires fundamentally different techniques, range limitations may be overcome by installing a detector to cover the IR range, fluorescence interference in Raman spectroscopy can be overcome by switching the laser wavelengths, and overlapping water peaks in FTIR may be resolved by improved sample preparation or changing characterization techniques to those involving a vacuum environment, like vibrational electron energy loss spectroscopy.
Third, it is crucial to ensure that the observation is not caused by beam damage. For example, the focused laser beam in Raman spectroscopy may damage or alter the samples over time at high power. Control experiments can help rule out such effects.
Last, while optical spectroscopy provides valuable information on molecular vibrational signatures and oxidation state change quantifications, it does not characterise long-range order crystal structures, mechanical deformation, mass change, and elemental distribution. It is important to use the complementary techniques mentioned in this review to form a comprehensive understanding.
For example, XRD can be paired up with optics to get a full picture of the interlayer of 2D materials. While XRD requires long-range order in the structure, FTIR and Raman examine molecular vibrations, which represent local bonding environments. FTIR is suitable for studying interfacial molecules and functional groups, while Raman excels at probing the solid-state bonds in inorganic materials. Alternatively, for measuring the oxidation state change of electrodes, in situ UV-Vis, XAS, and Electron energy loss spectroscopy (EELS) can all be used; however, the sample requirements differ, and the availability of the instrumentation varies significantly.
3. Transmission electron microscopy
Transmission Electron Microscopy (TEM) is a powerful tool for highly localized investigation of 2D nanomaterials in EES applications down to atomic resolution. TEM involves a broad toolkit of analytical techniques, including imaging, diffraction and spectroscopy. Therefore, TEM is suitable for obtaining information on the structure/morphology, crystallography and chemical composition of materials. TEM measurements require thin samples that are transmissible for electrons (typically <100 nm), which generally brings both opportunities and challenges for the particular case of 2D materials. Because of their anisotropic shape, in-plane analysis is favoured with the electron beam normal to the (thin) 2D layers. The analysis of the interlayer region is complicated by the need for (often challenging) orientation of the 2D layers parallel to the electron beam. The latter case can require the preparation of thin lamellas, for example by focused-ion beam techniques, for TEM analysis.78,79TEM can provide insights into electrochemical mechanisms via in situ or ex situ/post-mortem analysis. While the former requires in situ TEM holders capable of biasing/electrochemically cycling samples, the latter often involves inert and/or cryogenic sample transfer workflows of cycled electrode materials in combination with low dose and/or cryogenic imaging. There are comprehensive reviews on both in situ41,80–82 and (cryogenic) ex situ/post-mortem TEM analysis.83–86 This chapter is intended to give a short overview of the particular uses for 2D nanomaterials with an analysis of the advantages and disadvantages of each technique.
The advantage of in situ TEM is that the dynamics of processes and the evolution of the 2D materials’ structure, crystallography and/or chemistry during an electrochemical process can be observed. The challenge is to replicate a “realistic” environment of an electrochemical cell in the microscope. For example, the ultra-high vacuum environment and the impact of a high-energy electron beam necessitate non-volatile and highly stable electrolytes (e.g., ionic liquids, polymer or solid electrolytes) or liquid cell TEM holders.
Prominent examples of such in situ TEM experiments are investigating the electrochemical lithiation of graphene sheets, using a custom silicon/silicon nitride chip with electrical contacts as a sample support.87,88 A device is constructed by placing a bilayer graphene sheet and solid electrolyte next to each other on the contacts (Fig. 6a). In combination with low-voltage aberration-corrected high-resolution TEM, this allows for a direct visualization of the intercalation of lithium upon electrical biasing on an atomic level. It was demonstrated that the lithium can assume a superdense close-packed phase in this configuration (Fig. 6b–d).87 However, while such in situ experiments allow us to investigate the dynamics of an electrochemical process on the atomic level, they may not necessarily represent exactly the conditions present in a bulk cell. For instance, the few-layer 2D material may extend the gap between the layers more easily than the bulk material. Also, the electron beam used for imaging may itself have a significant influence on the dynamics, as it provides energy and may even cause damage to the materials.
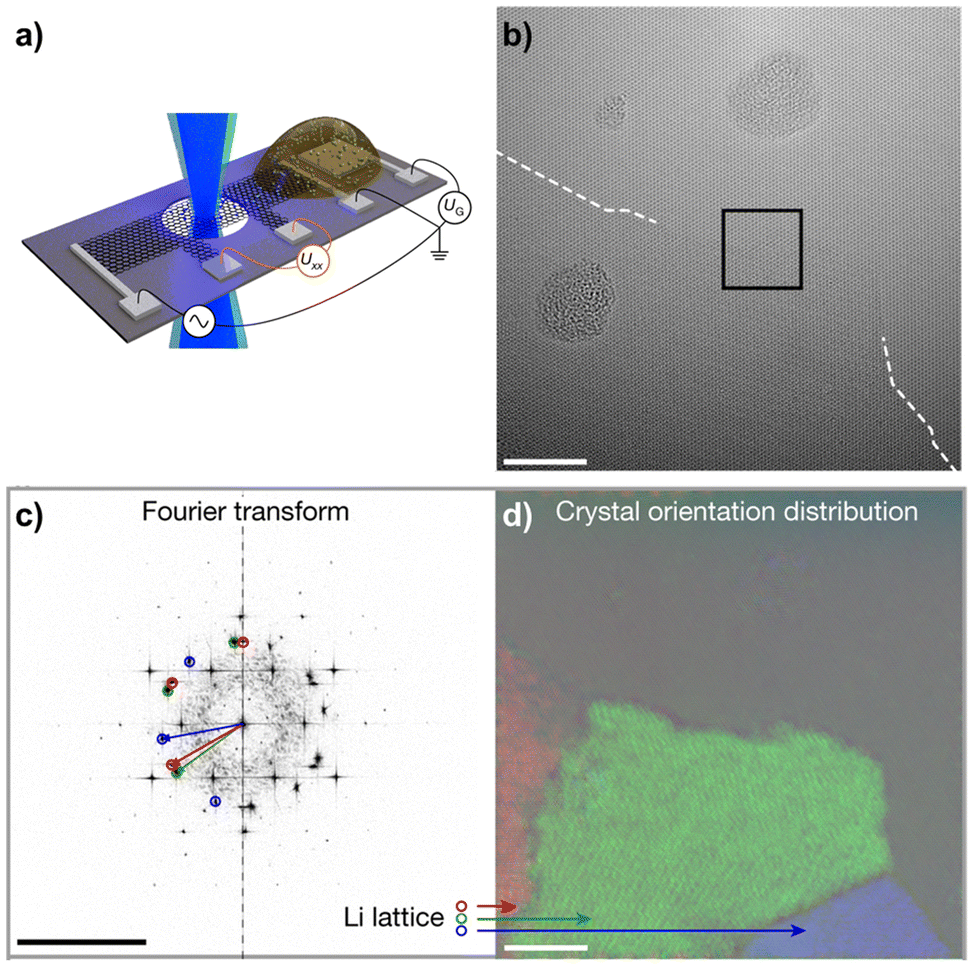 | ||
| Fig. 6 In situ TEM to study Li+ intercalation in bilayer graphene. (a) Shows the device layout on a Si/Si3N4 chip with a bilayer graphene bar placed across small holes in the substrate and on electrical contacts. The solid electrolyte is placed directly adjacent to the bilayer graphene on another set of contacts, allowing the lithium to intercalate upon electrical biasing. (b) Shows an aberration-corrected HRTEM image of a close-packed Li phase entering from the bottom during lithiation, scale bar is 5 nm. (c) shows a Fast Fourier Transform (FFT) of the image, revealing three distinct Li crystal orientations in addition to the graphene lattice, scale bar is 10 nm−1. Those Li crystal orientations are mapped in (d), with scale bar 5 nm. Figure reproduced with permission from ref. 87, copyright 2018 Springer Nature. | ||
An alternative, complementary strategy is to employ ex situ TEM characterization. This way, the 2D material is subjected to electrochemical operation in a standard electrochemical testing cell (e.g., coin cell or three-electrode cell) to reach a certain state of charge or aging. Then, the sample is removed from the electrochemical cell and loaded into the TEM holder, which subsequently has to be transferred to the microscope. Here, the main challenge is to avoid changes to the sample during loading and transfer, which requires specialized workflows with loading in an inert environment (e.g., glovebox) and transfer to the microscope with inert-/vacuum-/cryo-transfer holders.86 The main advantage of the strategy is that the sample undergoes electrochemical aging in realistic conditions and the setup involved is generally less complex compared to in situ analysis. However, dynamic information typical for in situ/operando measurements is not readily available. This shortcoming can be addressed, for example, by the use of identical-location TEM. Samples dispersed on identical-location TEM grids can be imaged in the pristine state, then be cycled in an electrochemical cell using the same TEM grid as a current collector, and then transferred back to the microscope to re-analyze the same particle.89,90
Yu et al. employed cryogenic ex situ TEM to study the conversion reaction of Li+ with MoS2 flakes. Chemical vapor deposition was employed to deposit a monolayer of MoS2 on the TEM grid. After lithiation in an electrochemical cell on the TEM grid, the sample was transferred in an argon-filled sample container and with liquid nitrogen cooling to the microscope, where they were loaded under liquid nitrogen to the TEM sample holder (Fig. 7a). Cryo-TEM analysis revealed that after lithiation, the MoS2 film wrinkled due to volume expansion and the formation of metallic Mo and Li2S domains was detected by both imaging and diffraction (Fig. 7b–e), confirming the electrochemical conversion reaction mechanism.91 Likewise, Zhao et al. used ex situ TEM to study the charge storage mechanism of Li+ in Co2(OH)2CO3 on graphene, finding that it phase separates into Co(OH)2 and CoCO3 after the 1st cycle, which increases the reversibility and stability of the electrode during lithiation.92
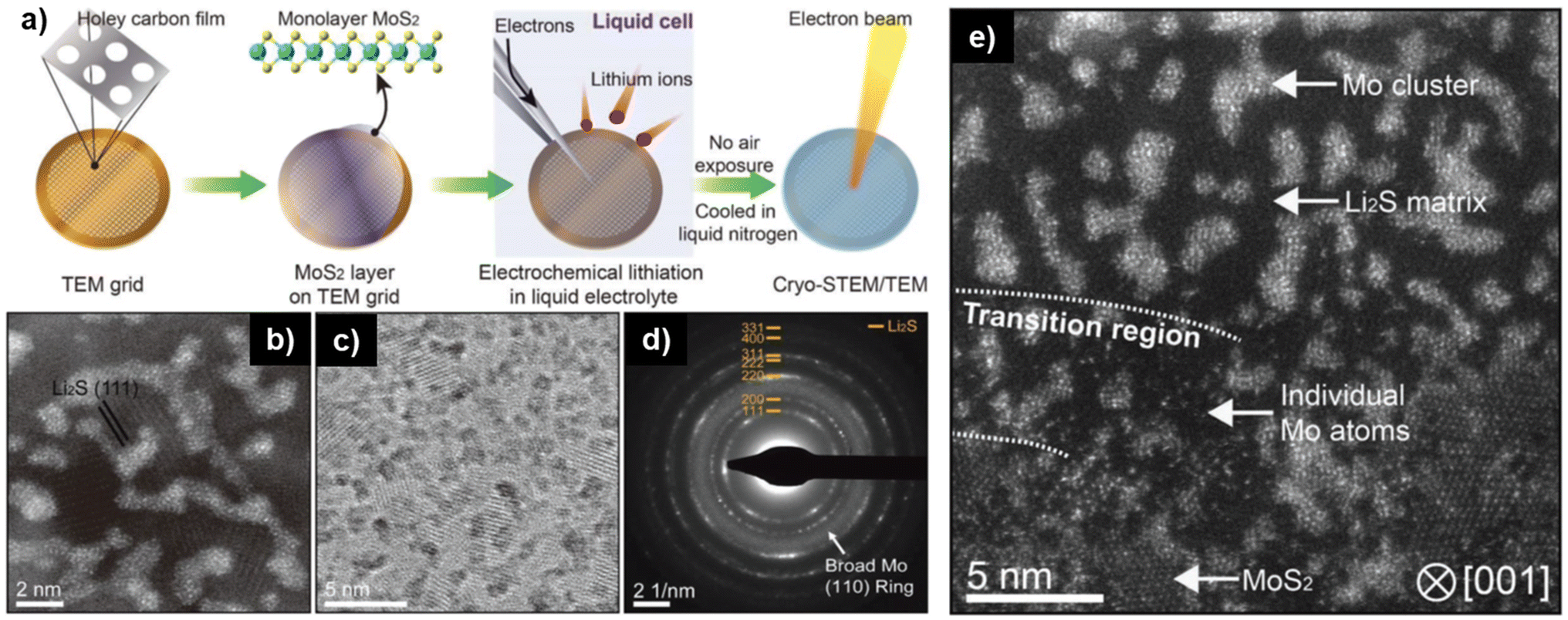 | ||
| Fig. 7 Ex situ cryo-TEM to study electrochemical conversion reaction of Li+ with monolayer MoS2. (a) Schematic illustration of the preparation and transfer of monolayer MoS2 from the TEM grid to the electrochemical cell to the microscope. (b) Annular dark-field cryo-scanning transmission electron microscopy (ADF cryo-STEM), (c) bright-field cryo-TEM, and (d) selected area electron diffraction (SAED) of lithiated MoS2 sample, detecting Li2S and Mo formation. (e) Cryo-STEM of partially lithiated MoS2 monolayer detecting both pristine MoS2 regions and converted Mo & Li2S regions. Figure reproduced with permission from ref. 91, copyright 2019 Wiley. | ||
The discussed cases illustrate how powerful in situ and ex situ TEM techniques can be for analyzing the charge storage mechanisms of 2D materials. When deciding whether to use in situ or ex situ methods, researchers need to evaluate whether dynamic information or electrochemical treatment like lithiation in realistic conditions is prioritized. The combination of both techniques can provide a full picture. The integration of Electron Energy Loss Spectroscopy (EELS) during TEM analysis can provide more detailed, localized information of the chemical environment and oxidation states of the sample. A big advantage of the technique is its ability to detect light elements like Li+ that are often relevant for electrochemical charge storage.
4. X-ray-based techniques
4.1 X-ray diffraction
X-ray diffraction (XRD) relies on X-ray scattering, where constructive interference produces diffraction patterns from crystalline or partially crystalline materials. Ex situ and in situ XRD are commonly used to monitor crystal structure changes in EES electrodes during charging and discharging. XRD is also valuable for studying phase transformations or crystal structure changes between 2D materials and their precursors, confirming the successful synthesis of 2D materials. XRD operates in two modes: reflection, where the X-ray exits on the same side it enters the sample, and transmission, where the exit is on the opposite side.93 In transmission mode, synchrotron XRD, with its higher photon brilliance and flux, is particularly effective at penetrating through cell components, enabling data collection with high spatial and temporal resolution while minimizing material damage. It is worth mentioning another X-ray technique commonly performed at synchrotron facilities and based on X-ray scattering: Grazing Incidence Wide-Angle X-ray Scattering (GIWAXS). Compared to XRD, this technique utilizes a shallow incidence angle, enabling precise analysis of the structural features of thin film 2D materials.94 Complementary to GIWAXS, X-ray Reflectivity (XRR) can be used on thin films to gather information on the interlayer space and electron density of 2D materials.95,96 Since this technique relies on low-angle reflection rather than diffraction, it is optimal for measuring the interlayer space change in very thin films for which the GIWAXS signal is too weak.For 2D material-based electrodes, irreversible crystal structure changes are primarily associated with variations in the interlayer spacing caused by the intercalation of ions or solvents.97 Different patterns of change reflect the intercalation of distinct species, providing insights into different charge storage mechanisms. For example, the d-spacing of 2D MXene, derived from the 002 peak in the XRD pattern, changes differently during the charging process in various electrolytes.27,98,99 In the H2SO4 electrolyte, when Ti3C2Tx is negatively charged, the d-spacing initially decreases by 0.1 Å from 0 V to −0.6 V vs. Ag wire before expanding rapidly by 0.5 Å from −0.6 V to −0.9 V (Fig. 8a).27 This change in interlayer spacing in the acidic aqueous electrolyte is primarily caused by the interactions between the intercalated ions and the surface groups. Specifically, H+ intercalation results in a contraction of the interlayer space in O-terminated MXenes, while it causes an expansion in OH-terminated MXenes. The co-intercalation of solvent molecules also significantly affects the charge storage mechanism, as reflected in the change in interlayer spacing. For example, cycling Ti3C2Tx in LiTFSI-based organic electrolyte with different solvents shows different interlayer space changes. Notably, the d-spacing of Ti3C2Tx MXenes at the most negative potential is 19.0, 13.0, and 10.7 Å in LiTFSI containing dimethyl sulfoxide (DMSO), acetonitrile (ACN), and propylene carbonate (PC), respectively. This indicates distinct arrangements of interfacial species in different solvents (Fig. 8b and c).100 Such variations in the ion and solvent arrangement within the interlayer space impact ion storage capacity and transport, leading to differences in energy storage and rate capabilities. Additionally, the amount of co-intercalated solvent can be observed via XRD. For example, during the ion intercalation process in Ti3C2Tx MXene with a WiSE, the interlayer space dramatically changes at a relatively positive voltage due to the co-insertion of three water molecules per Li-ion.32 At more negative potentials, however, the interlayer space remains almost unchanged when approximately one water molecule is intercalated per Li-ion. The co-insertion of more water per Li-ion weakens the Li-MXene interactions, leading to reduced surface redox activity at positive potentials. Furthermore, the intensity change in the in situ XRD pattern also provides information on irreversible phase changes during cycling. For example, the 200, 211, and 321 peaks of NiSe2 weaken and become obscured as the cell voltage increases from ∼0 to 3 V, indicating irreversible phase changes and structural deformation in NiSe2.101
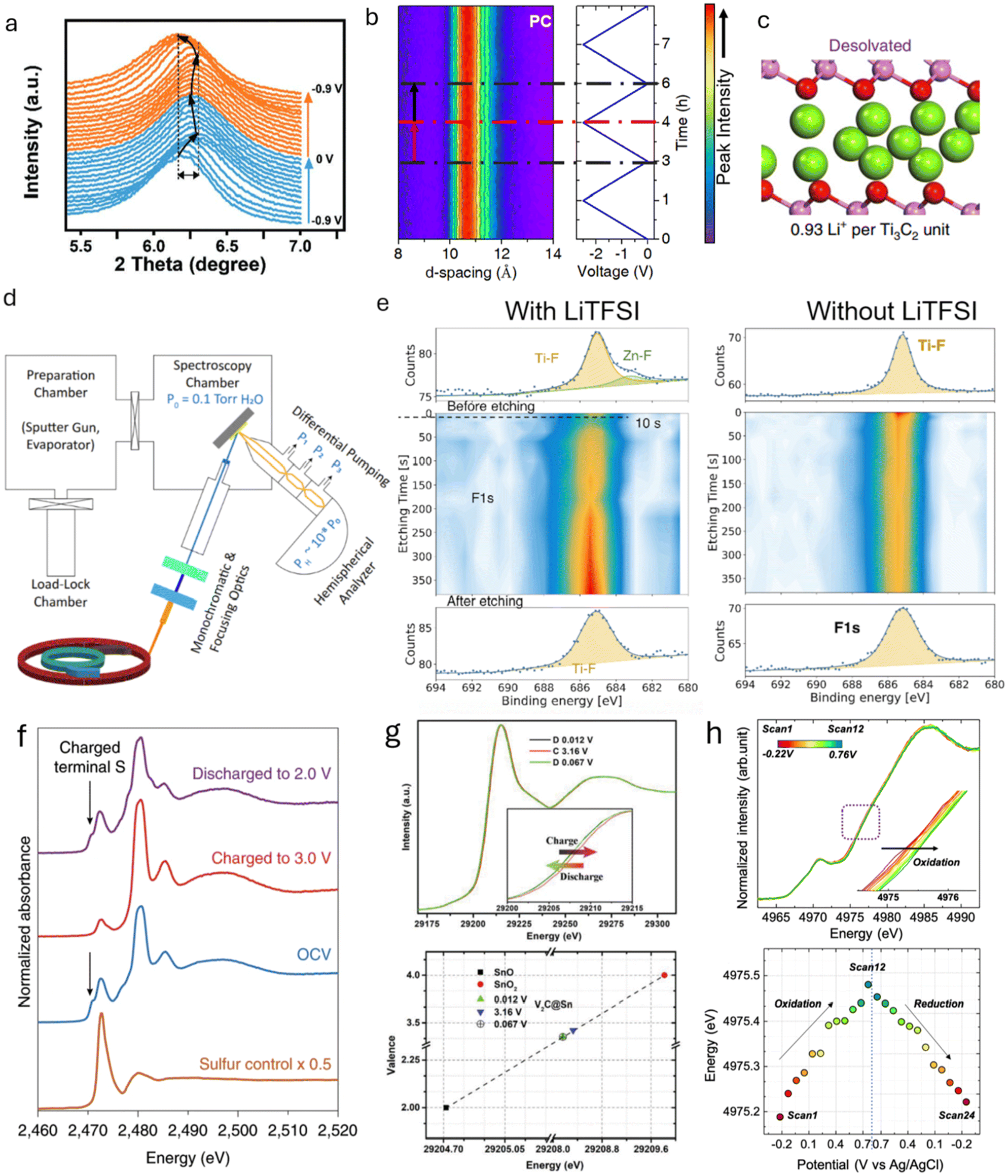 | ||
| Fig. 8 (a) In situ XRD patterns of MXene cycled in H2SO4 electrolyte.27 (b) In situ XRD patterns of MXene cycled in LiTFSI-PC organic electrolyte.100 (c) Illustration of the interlayer spacing of MXene cycled in LiTFSI-PC organic electrolyte, showing restricted access of PC molecules. (d) Schematic representation of synchrotron-based ambient-pressure X-ray photoelectron spectroscopy.116 (e) F 1s XPS depth profiling of MXene after Zn plating in LiTFSI-containing and LiTFSI-free electrolytes.119 (f) In situ S K-edge XANES spectra at various voltages during cycling.123 (g) XANES spectra of Sn K-edge at different voltages and the corresponding chemical valence changes of Sn atoms in various electrodes.124 (h) In situ Ti K-edge XANES spectra and the associated Ti valence changes at different voltages.122 Panel (a) reproduced with permission from ref. 27, copyright 2019 Wiley. Panel (b) reproduced with permission from ref. 100, copyright 2019 Springer Nature. Panel (c and d) reproduced with permission from ref. 116, copyright 2021 National Academy of Sciences. Panel (e) reproduced with permission from ref. 119, copyright 2025 Wiley. Panel (f) reproduced with permission from ref. 123, copyright 2020 Springer Nature. Panel (g) reproduced with permission from ref. 124, copyright 2019 Wiley. Panel (h) reproduced with permission from ref. 122, copyright 2022 American Chemical Society. | ||
The changes in interlayer space observed through XRD do not directly reveal the intercalated species when studying the mechanisms in 2D materials. To gain a deeper understanding of the intercalates’ structure and the molecular arrangement between layers, XRD must be combined with other techniques such as EQCM and simulations. Additionally, XRD has limitations for detecting small structures present only in trace amounts and is less effective for identifying amorphous materials. This restricts its application in identifying intermediate or final products of reversible/irreversible electrochemical reactions during cycling. For materials with low atomic numbers, the diffracted X-ray intensity may be low, making neutron diffraction a valuable alternative. Furthermore, the choice of method for analyzing XRD peaks—such as the Debye–Scherrer, Wilson, Williamson–Hall, and Halder–Wagner analyses—should be carefully considered, especially when high precision is required, as they can influence the accuracy of crystallite size and other microstructural parameters.102
4.2 X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS), a highly developed surface-sensitive technique based on the photoelectric effect, is widely employed to identify elements in a sample and provide information about their chemical bonding. In the context of 2D materials, XPS has been extensively used for elemental analysis and can also characterize the thickness of 2D films.103 Its quantitative nature and exceptional surface sensitivity (less than 10 nm) make it an indispensable tool for detecting surface composition changes induced by various processing methods, such as plasma treatment, thermal annealing, electrochemical processing, or chemical modification.104–106 For example, XPS has revealed that intercalating tetrabutylammonium into 2H-MoS2/MXene acts as an electron donor, increasing the local electron density around Mo and S in MoS2 and inducing a phase transition from 2H to 1T.106 XPS also revealed the degradation mechanism of MoS2 as a catalyst in Zn-air batteries, which deactivates over time as it gets oxidized to Mo6+.107 Additionally, the surface-sensitive feature allows XPS to determine the chemical formulae and bonding of surface groups. When combined with Ultraviolet Photoelectron Spectroscopy (UPS) and Resonant Photoemission Spectroscopy (PES), XPS provides detailed insights into the bonding environment and structural arrangement of termination species in Ti3C2Tx MXenes.108 This is achieved because the valence band intensity in UPS/XPS varies with photon energy, making valence band structures highly sensitive to the local environment of the probed species.When 2D materials are used in EES devices, XPS facilitates the understanding of charge storage mechanisms by analyzing changes in bonding structures and compositions during ion intercalation, redox reactions, and solid-electrolyte interphase (SEI) formation.109–111 By comparing XPS spectra at different charging states, researchers can gain valuable insights into surface reactions, interfacial phenomena, and degradation mechanisms, aiding the development of improved battery materials and optimized performance. As a non-destructive technique, XPS can be applied in situ under vacuum conditions. For example, in situ XPS has been used to study lithium deposition on graphene and N-doped graphene films prepared via CVD growth on Cu foil and transferred onto SiO2/Si substrates.112 Lithium was deposited within the XPS vacuum chamber, and analysis of the C 1s and Li 1s spectra showed that N-doped graphene exhibited a more pronounced shift in the C 1s peak compared to pristine graphene, indicating stronger lithium interaction. This was attributed to N-doping, which enhanced lithium adsorption. This enhanced interaction was attributed to the N-doping, which promoted stronger lithium adsorption. In situ XPS has also provided insights into SEI composition changes during cycling. For example, lithium electrochemical deposition on a garnet solid-state electrolyte was monitored to understand improved interfacial stability with the addition of 2D boron nitride.113 The presence of Li3N in the SEI layer between the electrolyte and lithium metal was observed, explaining increased ionic conductivity and enhanced stability against metallic lithium. While not yet applied to 2D materials, in situ XPS combined with imaging techniques holds great potential for investigating the SEI layer with high spatial resolution, enabling real-time tracking of its chemical composition, thickness, and evolution.114
For liquid electrolytes, in situ XPS is challenging because it requires open cells operated under ultra-high vacuum.114 However, in situ XPS has been successfully applied to study the stability of layered carbide-derived carbon/electrolyte interfaces during electrochemical polarization in ionic liquid electrolytes.115 Ionic liquid electrolytes can be used in open cells due to their non-volatile nature. By tracking changes in the C 1s and N 1s energy levels, Lust and co-workers identified the reaction mechanism responsible for electrolyte degradation at high potential (>3.6 V). This mechanism involves the oxidative dimerization of the imidazolium cation via N–N bond formation. For aqueous systems, ambient-pressure XPS enables researchers to study ion intercalation processes at water-solid MXene interfaces during ion exchange.116 Ambient-pressure XPS provides direct insights into how surface-terminating groups and solutes, such as inorganic cations, influence water uptake in layered materials, as illustrated in Fig. 8d.
In addition to the vacuum chamber challenge, XPS has other limitations, including high sensitivity to surface contamination and reduced sensitivity to insulating materials. To minimize contamination effects, samples are often cleaned by removing top layers with an ionized beam. For insulating samples, a charge neutralizer is used to prevent peak shifts and other complications caused by charging.117 In in situ XPS, the production of insulating materials during the observation process can also cause charging issues, affecting calibration and data acquisition.118 Finally, when bulk properties are of interest, XPS can be combined with ion-beam etching for line and depth profiling of elemental compositions. For example, in-depth XPS has been used to analyze SEI composition changes with depth113 in a Li-metal battery when an adhesive Li-BN composite is used in the anode. In another work, in-depth XPS revealed differences in SEI composition at various depths for different electrolytes during the Zn ion deposition process on a MXene-based anode. Comparison of the in-depth Zn 2p and F 1s XPS spectra showed that adding LiTFSI salt to the electrolyte led to the formation of ZnF2 in the SEI layer, which explains the suppressed Zn dendrite growth on MXene (Fig. 8e).119 However, the in-depth XPS technique is destructive and can only be applied ex situ.
4.3 X-ray absorption spectroscopy
X-ray absorption spectroscopy (XAS) measures the X-ray absorbance of a material as a function of X-ray energy. A sharp increase in absorption, known as the “absorption edge”, occurs when the X-ray energy matches the binding energy of the material. Analyzing the detailed shape of the absorption spectrum either around the edge (X-ray Absorption Near-Edge Structure, XANES) or beyond the edge (Extended X-ray Absorption Fine Structure, EXAFS) provides information about the chemical state and molecular structure.120 Unlike XPS, XAS does not require high vacuum conditions, making it more suitable for in situ analysis of EES devices with different electrolytes.45 However, XAS requires an intense, tunable, high-energy X-ray beam and is typically conducted at synchrotron radiation facilities, in contrast to UV-Vis spectroscopy mentioned earlier.In situ XAS provides insights into the chemical properties of electrode materials, particularly the oxidation state variations during charge and discharge cycles.121 Local structural analysis can be performed by examining oxidation states, bond lengths, or coordination numbers from the outer electron shell. Time-resolved XAS analysis enables continuous monitoring of oxidation state changes in electrodes during cycling, with the acquisition time of each XAS spectrum being crucial for uninterrupted observation without halting electrochemical cycling.122In situ XANES, combined with in situ optical microscopy, has been used to observe the formation of liquid sulfur species on a 2D MoS2 electrode in a Li–S battery. The sulfur K-edge XANES revealed the conversion of Sx2− to elemental sulfur, with key absorption features at 2471 eV and 2473 eV corresponding to negatively charged terminal sulfur atoms and internal atoms in the polysulfide chain, respectively. At 3.0 V, the absorption feature at 2471 eV diminished while the 2473 eV feature remained, confirming the formation of liquid sulfur droplets on the 2D material (Fig. 8f).123 XANES can also be used to observe the valence of intercalated ions in 2D materials, providing insights into the intercalation process. For example, during the Li+ intercalation/deintercalation in Sn-intercalated V2C MXene, the valence of Sn reversibly changed from +3.35 to +3.42 within a voltage window of 0.012 to 3.16 V vs. Li/Li+, contributing additional capacity to the Li-ion battery (Fig. 8g).124 Additionally, in situ XANES can probe the oxidation state changes of metal-containing 2D materials. A reversible metal valence change was observed when cycling MXenes in acidic, neutral, and water-in-salt aqueous electrolytes.122,125,126 This reversible valence change, along with less than one electron transfer per metal ion (Fig. 8h), indicates a pseudocapacitive charge storage mechanism for MXene electrodes in aqueous electrolytes.
XAS signals can be collected using the transmission mode of Scanning Transmission X-ray Microscopy (STXM), which allows simultaneous X-ray imaging with fluorescence mode acquisition. This technique has been used to investigate the degradation process of phosphorene during oxidation, as the absorption features of P(0), P(III), and P(V) differ.127 Furthermore, the absolute thickness of the phosphorene sample at each pixel can be determined by fitting the XANES spectra with reference spectra of fresh phosphorene. In contrast to AFM, which only provides morphological information, STXM offers detailed chemical imaging, providing a deeper understanding of the degradation process. For more real-time imaging, transmission X-ray microscopy (TXM), combined with a 2D camera, can be used, albeit with a trade-off in resolution. XANES combined with TXM has been employed to visualize and spectroscopically analyze the evolution of CuO during lithiation and delithiation, with voltage variation.128 Chemical mapping, along with line profiles of the sample, can provide insights into the morphological evolution and the chemical distribution of specific particles. These X-ray mapping techniques are still awaiting application in an appropriate system to effectively reveal the charge storage mechanisms in 2D material-based electrodes.
5. Neutron-based techniques
Neutron characterization techniques are useful for revealing the charge storage mechanism in 2D materials due to their ability to probe molecular interactions in tight interlayer spaces, their sensitivity to light elements often found in electrolytes, like H and Li, and their ability to penetrate bulk materials. Free neutrons interact with atomic nuclei through scattering or absorption. The selectivity of such interactions varies greatly between isotopes, giving a unique contrast to certain elements that other methods cannot match. For instance, X-rays are sensitive to elements with big electron clouds, making them mostly sensitive to heavy elements, while neutrons have particular sensitivity to light elements with small nuclei (H, Li, O, F). Furthermore, neutrons can distinguish between elements of similar atomic numbers,40 probe magnetic properties, and have better penetration properties than X-rays, allowing analysis of bulk processes.5.1 Neutron scattering
Neutron Scattering (NS) techniques can accurately reveal the structure and atomic interactions inside 2D materials and are particularly useful for measuring interactions between electrode materials and the electrolyte. Contrary to photon-based spectroscopy, neutrons can easily probe the confined interlayer spaces of 2D materials due to their strong penetrating power. The suitability of each scattering technique depends on the length scale of interest and the internal mobility of the sample. During Inelastic Neutron Scattering (INS), neutrons gain or lose energy when triggering vibrational transitions within the sample,37 revealing surface-solvent interactions of hydrogen species in confined spaces. Mashtalir et al. used INS to demonstrate the thermally reversible intercalation of N2H4 pillar molecules within Ti3C2Tx MXene layers by measuring the change in –OH/–NH vibrational modes (Fig. 9a).129 Furthermore, INS was used to monitor thermal NH3 intercalation in TiS2,130 prove water deintercalation in Nb2C MXene,131 and demonstrate that the water content and structural disorder in Ti3C2Tx interlayers decrease after metal ion intercalation.132 Quasi-Elastic Neutron Scattering (QENS) is similar to INS, but the energy transfer is much smaller, revealing the mobility and diffusivity of molecules in confined spaces. Osti et al. used QENS to study the effect of humidity exposure in [EMIm+][Tf2N−] ionic liquid with Ti3C2Tx (Fig. 9b and c), and found that humidity increases ionic diffusivity due to water-induced displacement of ions from the hydrophilic MXene surface.133 Furthermore, Sun et al. showed that Ti3CNTx has immobile intercalated water and almost no –OH surface groups when compared to Ti3C2Tx.134 QENS was also used to measure water mobility in porous Ti3C2Tx,135 and prove the stabilizing effect of K+ intercalation between Ti3C2Tx layers.136 For disordered or nanocrystalline samples, Neutron Total Scattering (NTS) is useful for determining the local atomic structure. Both inelastic and elastic scattering are collected to form a Pair Distribution Function (PDF). For instance, Brady et al. used NTS to elucidate the binding behavior of Na ions in Na2Ti3C2Tx MXene for Na-ion batteries (Fig. 9d and e), finding that the nature of Na-redox varies depending on the type of sodiation site.24 Moreover, NTS was used to assess the atomic structure of layered materials, such as the positional randomness of C and N in Ti3CNTx,134 and the difference in surface group distribution of Ti3C2Tx etched with various concentrations of HF.137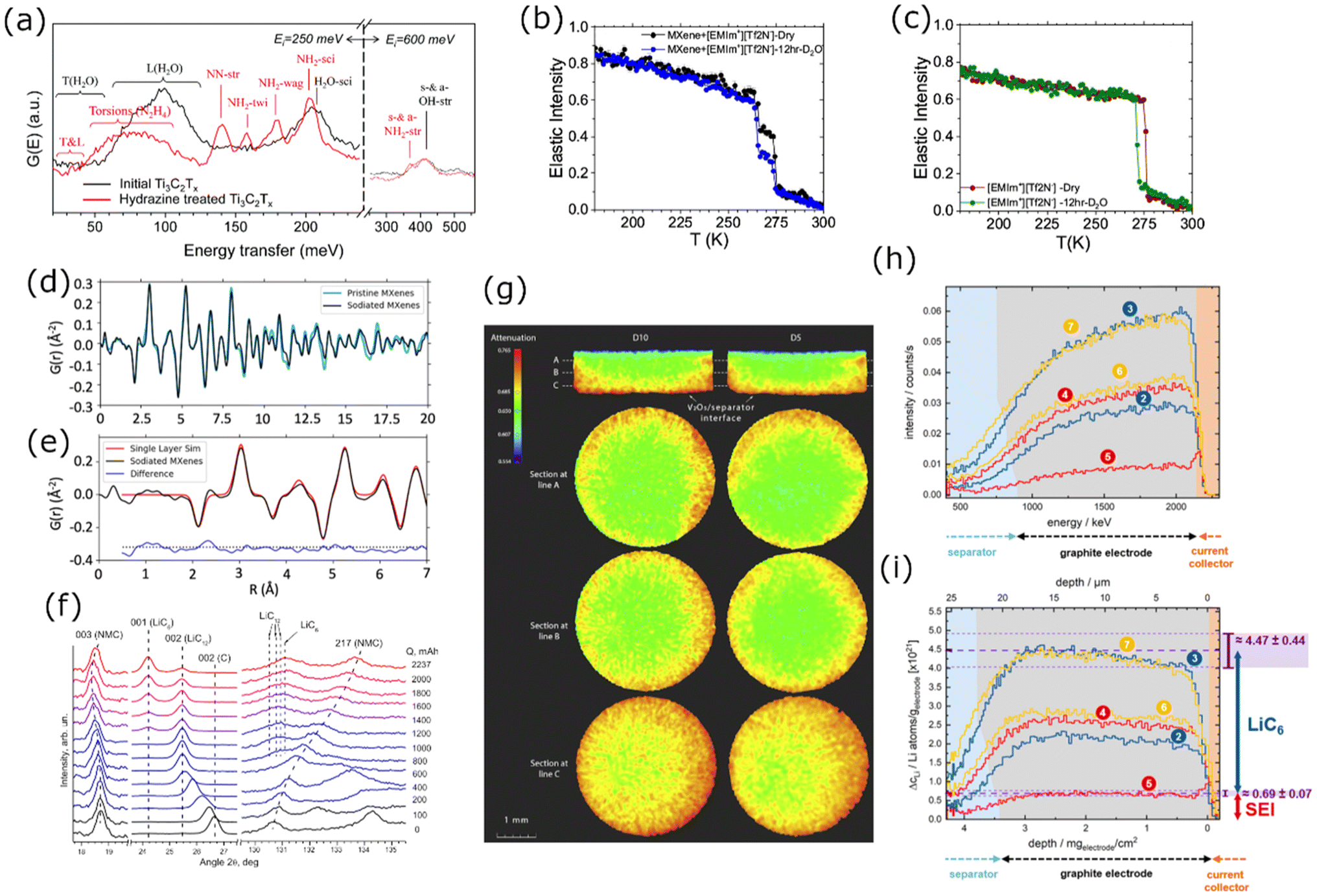 | ||
| Fig. 9 (a) INS spectra for Ti3C2Tx before (black curve) and after (red curve) hydrazine treatment.129 Normalized elastic intensities measured from dry and wet samples on heating at 2 K min−1 from 20 to 300 K for (b) MXene/[EMIm+][Tf2N−] and (c) bulk [EMIm+][Tf2N−] liquid.133 (d) Neutron PDFs of pristine and sodiated MXenes obtained by TNS. (e) A difference PDF analysis showing weak signals contributed from sodium-to-MXene pair correlations at the local scale.24 (f) Sections of ND patterns obtained from a commercial 18650-type NMC Li-ion cell upon different “equilibrated” charge states.138 (g) Neutron imaging data of sample D10 (left) and sample D5 (right), both cells were discharged (lithiation of V2O5) to 0% SOC.22 (h) Change of the NDP spectra for the pristine state of the cell. (i) Transformation of the NDP signal count rate into Li concentration in Li atoms per gram of electrode and transformation of the Triton energy into depth. The differently numbered spectra were collected at different points in the charge/discharge cycle.144 Panel (a) reproduced from ref. 129 with permission from the Royal Society of Chemistry. Panel (b and c) reproduced with permission from ref. 133, copyright 2018 American Chemical Society. Panel (d and e) reproduced with permission from ref. 24, copyright 2021 American Chemical Society. Panel (f) reproduced with permission from ref. 138, copyright 2014 Elsevier. Panel (g) reproduced with permission from ref. 22, copyright 2018 Elsevier. Panel (h and i) reproduced from ref. 144. with permission from IOP Publishing. | ||
5.2 Neutron diffraction
Neutron Diffraction (ND) is used to obtain structural information on crystalline solids through elastic neutron scattering. Similar to XRD, neutrons diffract on crystal planes and show Bragg peaks at well-defined angles. A shift in Bragg peak position relates to a changing crystal lattice length. Since neutrons are sensitive to lithium, ND is a popular tool for measuring lithium intercalation in crystalline 2D lithium host materials. Dolotko et al. used in situ ND to study structural changes in commercial battery cells containing LixNi0.5Mn0.4Co0.2O2 and graphite, and found that the NMC lattice parameters shift gradually during the charging cycle (Fig. 9f), corresponding to Li+ (de)intercalation. Since neutrons penetrate the whole cell, the signals for NMC and graphite are measured simultaneously, also revealing the lithiation of graphite. Besides, no Li/Ni cation mixing was observed at 80% SOC and beyond.138 Similar in situ studies were done on other layered TMOs, such as LixCoO223,139,140 and LixMoO2.141 Alternatively, Ex situ ND was done on LixTiS2 cathodes, demonstrating that Li preferentially occupies the octahedral sites in the 2D TiS2 van der Waals gaps while no 3D Li ordering was observed.42 Similar ex situ studies were done on LixMoS2142 and LixV2O5.143 The exceptional sensitivity of neutrons to lithium allows the detection of minuscule Li fractions in the sample, enabling in situ measurement of relatively small quantities of material.
5.3 Neutron imaging
Neutron Imaging (NI) is based on the differences in neutron contrast between elements. Since light elements (H, Li) strongly attenuate neutron beams, imaging techniques can reveal the position and concentration of such elements in the sample, which helps determine mesoscopic charge kinetics in 2D materials. Neutron Tomography (NT) renders a 3D image by rotating the sample in a neutron beam while measuring attenuation at each position. Zhang et al. used NT to track the spatial distribution of Li in V2O5 cathodes (Fig. 9g) and found that fast lithiation causes non-uniform Li distribution, while the uniformity of delithiation was independent of charge rate. The penetrating power of neutrons allowed them to use an unmodified commercial button cell.22 Similar work was done on LiCoO2 cells.139Neutron Depth Profiling (NDP) uses neutron absorption to measure spatial Li concentration with micrometer accuracy. When the isotope 6Li (7.5% natural abundance) absorbs a neutron, it decays into 4He2+ and 3H+ with well-defined kinetic energies, which slow down as they move through the sample. The remaining energy of these particles at the detector reveals the depth at which they formed. Linsenmann et al. used operando NDP to track the lithiation of graphite anodes (Fig. 9h and i), and found that the concentration of Li in the SEI is lower than predicted by models.144 NDP has been used to study Li plating behavior in batteries145,146 and Li motion in LiFePO4.147,148 While NDP is not commonly applied to 2D materials, it could be a valuable tool for probing spatial Li distribution with high precision. Its specific sensitivity to 6Li even enables measurement of aqueous systems, as hydrogen scattering cannot influence the detected signal.
NS techniques are regularly used with 2D and layered materials to elucidate the charge storage mechanism, while ND and NI techniques are more commonly used with layered materials only. Here lies an opportunity to apply these techniques to monitor the movement of Li in 2D electrode materials. Another possibility lies in Small Angle Neutron Scattering (SANS), a technique where elastic scattering at small angles is used to probe the average size, shape, and orientation of bulk nanostructures in the nm-μm size domain.149,150 This could be applied to 2D materials in which nanostructures form during cycling. Furthermore, Neutron Reflectometry (NR) which is not yet widely applied to 2D materials, could be used to study SEI formation in 2D material-based electrodes.151
Whereas neutrons are extremely sensitive to light isotopes, many other isotopes are hard to distinguish. Some elements will even become highly radioactive in a neutron beam. Besides, neutron facilities are uncommon and expensive, and most neutron beams have low fluxes requiring large amounts of sample material. These aspects limit the use of neutrons to cases where no alternatives are viable.
6. Mechanical techniques
6.1 Atomic force microscopy
Atomic Force Microscopy (AFM) is a force-based technique that exhibits high spatial resolution (sub-nanometer in out-of-plane direction and few nanometers in in-plane direction). Based on the interaction between the AFM probe and the sample, information about the morphology, mechanical properties (Young's modulus, plastic deformation, adhesion), and electrical properties (conductivity, surface potential) of the material can be extracted. For 2D or layered materials, AFM is particularly useful for verifying the morphology, dimension, thickness and other intrinsic information of the monolayer.152,153 More importantly, it is possible to conduct AFM measurements in a liquid environment, which allows for characterizing the materials under operando conditions and providing important insights into charge storage mechanisms.35,154During the operando AFM measurement, the electrode volume and morphological change are monitored. Their correlation with electrochemistry provides deeper insights into the charge storage mechanism and the ion intercalation process. For example, Aurbach et al. imaged the graphite electrode surface in the LiPF6 in EC-DMC electrolyte. It was observed that the electrode undergoes an irreversible morphological change during the first cycle, followed by an overall reversible surface change for the consecutive cycles, indicating a stable SEI was successfully formed during the first cycle.155 Apart from liquid imaging, Balke et al. developed another approach that uses the AFM probe as a nanometer-sized dilatometer.156,157 This approach obtains local (area under the tip) strain and global (entire electrode) current at the same time. Their correlation reveals details on electro-chemo-mechanical coupling behavior and the charge storage mechanism of the materials.156 This approach has been applied to a self-assembled 16-layer Ti3C2Tx electrode cycled in 0.5 M K2SO4 (Fig. 10), revealing that the MXene electrode volume remains constant from +0.25 V to −0.05 V vs. Ag/AgCl and starts to expand when the potential is below −0.05 V vs. Ag/AgCl.158 During the positive potential sweep, the electrode shrinks at a near-constant speed and back to its initial position. This result demonstrates that, during the negative sweep, there are two different charge storage mechanisms above and below −0.05 V vs. Ag/AgCl. The former does not involve volume change, while the latter does. Furthermore, the charging process is highly reversible, and the ions can access the interlayer space easily, as the deformation curves do not change much when cycling from 100 to 1000 mV s−1.
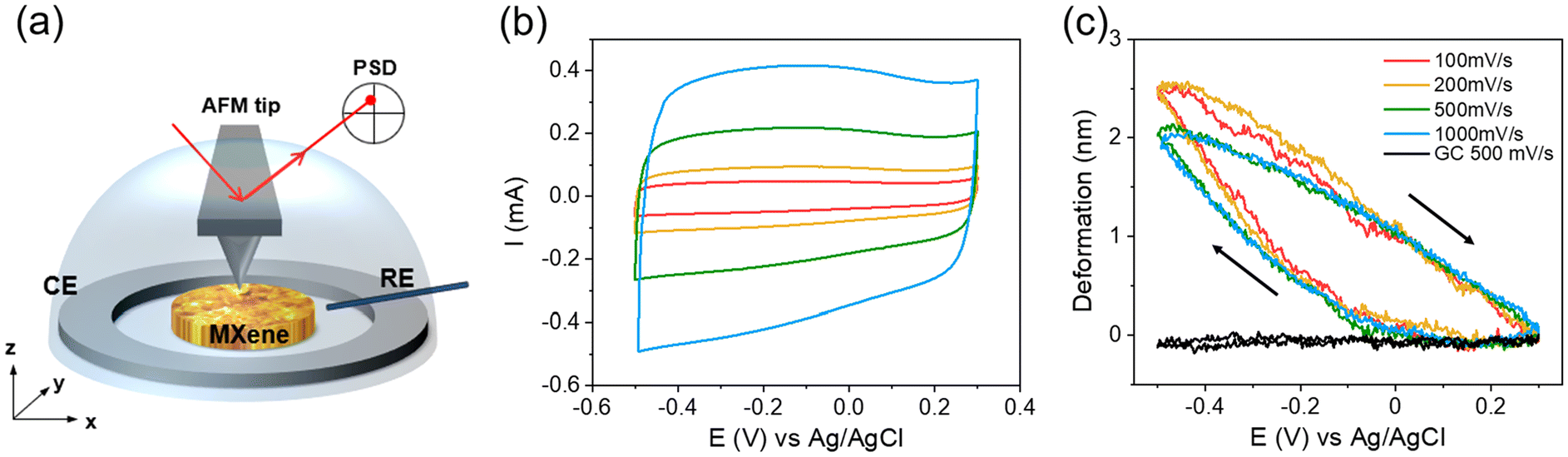 | ||
| Fig. 10 (a) Illustration of operando AFM set-up, (b) CV and (c) volume change of MXene electrode in 0.5 M K2SO4 at various scan rates. Reproduced with permission from ref. 158, copyright 2022 Wiley. | ||
The AFM can obtain the elastic modulus of materials through either force–distance (FD) measurement or contact-resonance AFM (CR-AFM). Traditionally, the FD measurement is performed with the sample lying on a hard, flat substrate (e.g. Si wafer). However, for a single or a few layer(s) of 2D material, the sample is too thin, and the result might be influenced by the substrate. Therefore, single-layer 2D materials are often specially prepared to be suspended on a grid or a patterned substrate to perform an AFM-nanoindentation (Fig. 11a–c).159 This allows accessing the 2D elastic modulus and fracture strength and strain of a single layer.160 The Young's modulus of the suspended MXene Ti3C2Tx was found to be 333 ± 30 GPa. A similar approach was also applied to graphene,161 MoS2162 and MoB2.153
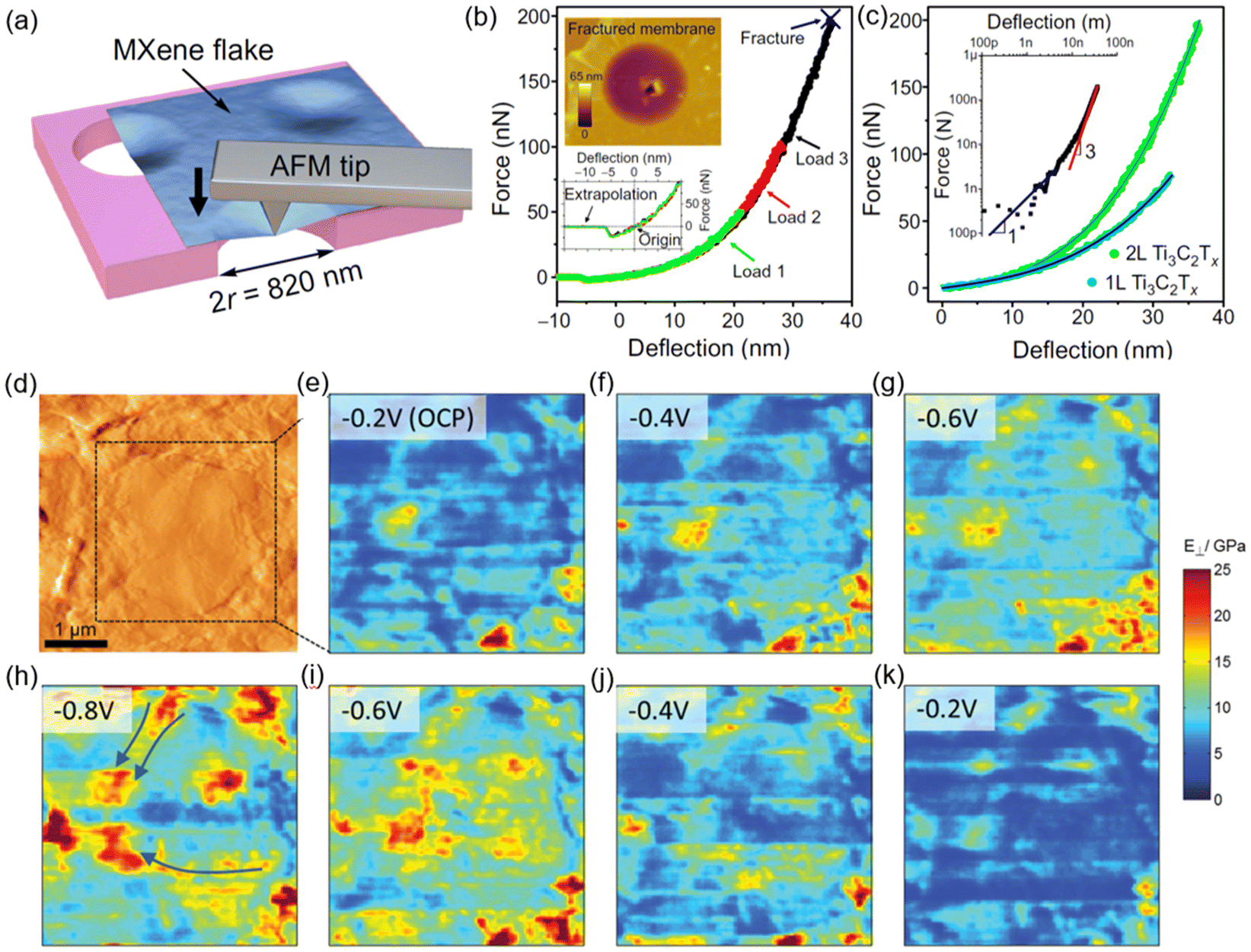 | ||
| Fig. 11 (a) Scheme of nanoindentation of a suspended Ti3C2Tx membrane with an AFM tip. (b) Force-deflection curves of a bilayer Ti3C2Tx flake at different loads. The bottom inset is a detailed view of the same curves showing the center of origin. The top inset shows an AFM image of the fractured membrane. (c) Comparison of loading curves for monolayer (1L) and bilayer (2L) Ti3C2Tx membranes.159 (d) Contact AFM topography image of a multilayer MXene flake at the surface of the electrode. (e–k) Elastic modulus images taken at potentials between −0.2 V and −0.8 V versus Pt during Li+ charge (e–h) and discharge (h–k).163 Panel (a–c) reproduced from ref. 159 with permission from the American Association for the Advancement of Science. Panel (d–k) reproduced with permission from ref. 163 copyright 2016 Wiley. | ||
However, at the device level, the electrode is composed of multilayer 2D material instead of a single layer. During ion intercalation, the stiffness of the electrode changes due to the additional ions in between the layers and the distance between the layer changes. Come et al. demonstrated that the elastic modulus of a Ti3C2Tx electrode, monitored by CR-AFM, exhibits an overall increase during Li-ion intercalation (Fig. 11d–h) and decreases when the ions leave the electrode (Fig. 11h–k).163 In addition, the modulus change of the electrode is inhomogeneous, suggesting that parts of the electrode do not participate in charge storage. A follow-up study monitoring the multivalent Mg2+ intercalation with CR-AFM shows that pre-intercalating K+ in the Ti3C2Tx electrode opens up the interlayer spacing and allows more electrode area to participate in the Mg2+ intercalation and charge storage.164 One should note that the elastic modulus measured by the AFM is dependent on the probe-sample contact area. It is thus important to carefully take into account the sample roughness, sample stiffness, probe size, and shape when extracting modulus values.
The recent development of AFM-based scanning electrochemical microscopy (AFM-SECM) makes it possible to simultaneously obtain topological information and spatially-resolved electrochemical activity, providing insights on local redox activities on Li2S2165 and hard carbon166 electrodes. However, it has not been applied to 2D electrode materials and would be a powerful tool to investigate the effect of heterogeneity on electrochemistry.
While AFM provides topological and mechanical information with high spatial resolution, it is not a chemically sensitive technique. To understand the chemical or molecular origin of the observed AFM results, it is crucial to pair AFM with other chemically sensitive techniques, such as XPS or Energy-Dispersive X-ray spectroscopy (EDX) to achieve a thorough understanding.
6.2 Electrochemical dilatometry
Electrochemical dilatometry (ECD) is an operando technique that can analyze the perpendicular, macroscopic dilatation/thickness change of an electrode in the z-direction (i.e., expansion or shrinkage) upon electrochemical stimulation. Electrochemical dilatometers can be custom-made or purchased commercially, and they are typically based on a setup (Fig. 12a) that decouples the volumetric response of the working electrode from the counter/reference electrode(s) by using a stiff glass frit as a separator. This allows for a detailed investigation of the electro-chemo-mechanical response of the individual electrode reaction, with a height resolution of commercial setups in the range of 5–10 nm. While Escher et al. provided an in-depth review of the ECD technique containing many technical details,167 this section will focus, in particular, on its use in the electrochemical investigation of 2D nanomaterials. | ||
| Fig. 12 (a) Cross-sectional view of a typical electrochemical dilatometry setup with 1: metal membrane, 2: working electrode (WE), 3: glass T-frit, 4: counter electrode (CE), 5: CE plunger, 6: sensor tip, 7: spacer disc, 8: reference electrode pin (RE).177 (b) Comparison of electrochemical operando XRD (left) and operando ECD (right) measurements of the Li+ intercalation reaction into hydrothermally synthesized nano-MoS2 in 1 M LiPF6 in ethylene carbonate/dimethyl carbonate (EC/DMC) electrolyte.168 (c) Cyclic voltammograms for both positive and negative polarization of Ti3C2Tx MXene electrode in 1-Ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide (EMIM-TFSI) ionic liquid electrolyte (left) and corresponding ECD response (right).172 Panel (a) reproduced with permission from ref. 177, copyright 2017 Elsevier. Panel (b) reproduced from ref. 168 with permission from Chemistry Europe. Panel (c) reproduced from ref. 172 with permission from Elsevier. | ||
To investigate the charge storage mechanism of 2D nanomaterials like MXenes or TMDs, it is necessary to analyze the change in interlayer spacing upon electrochemical ion intercalation and/or electrosorption reactions. The most common method to do this is operando X-ray diffraction (XRD), as discussed above, however, several challenges that are particular to 2D nanomaterials arise necessitating complementary methods.
First, 2D nanomaterials often exhibit small crystalline domain sizes and/or experience amorphization or exfoliation upon electrochemical cycling, leading to a loss in long-range crystalline order and, thus, in XRD signal. Tobis et al.168 investigated the Li-ion intercalation mechanism into hydrothermally-synthesized MoS2 with a nanocrystalline domain size, which only exhibits a weak diffraction signal. The authors found that operando XRD in transmission geometry was could not provide sufficient intensity to conclusively track the changes of interlayer spacing during cycling. As an alternative method, ECD was employed and demonstrated a reversible electrode expansion/contraction of ca. 2–3% for lithiation/delithiation (Fig. 12b). The results qualitatively demonstrate the electro-chemo-mechanical behavior of the host material that was not accessible through operando XRD, even though the quantitative expansion/contraction cannot be directly translated into changes of the d-spacing on a microscopic level. The example illustrates how ECD can be a superior method for investigating the charge storage mechanism of intercalation-based 2D electrode materials.
The second challenge for operando XRD measurements is that diffraction angles below ca. 8–10° 2θ are excluded due to the construction of operando cells for reflection measurements. This prevents the measurement of materials with large interlayer spacings or strong expansion upon cycling, where relevant diffraction angles are often below this threshold. Examples of such systems include MXenes, where diffraction signals related to the interlayer spacing are typically below 10° 2θ (based on Cu Kα radiation).169 ECD was used to demonstrate the volumetric response of Ti3C2Tx MXene electrodes upon ion intercalation or electrosorption.170–172 For example, Koudahi et al. demonstrated a reversible expansion of Ti3C2Tx MXene upon negative polarization in EMIM-TFSI electrolyte by ca. 2% as a consequence of EMIM+ electrosorption (Fig. 12c).172 No volumetric changes were observed upon positive polarization, which led to the conclusion that TFSI-anions are prohibited from entering the MXene interlayer spacing.172 Another phenomenon that may be prevalent in electrodes based on 2D nanomaterials is the intercalation of (partially) solvated ions instead of desolvated ions. Such ion-solvent cointercalation reactions33,173 can cause significant electrode expansion or even exfoliation with a loss of long-range order. In such cases, ECD was demonstrated as a suitable method to verify the cointercalation mechanism.174,175
While ECD enables the investigation of charge storage mechanisms in 2D nanomaterials where operando XRD is limited by structural disorder, there are several challenges associated with the interpretation of ECD results. The fact that the measurement is macroscopic means that inactive electrode components and electrode porosity influence the ECD signal.176 In the case of typical MXene-based electrode materials, this likely leads to different quantitative ECD results when comparing densely packed/restacked with few-layer/exfoliated electrodes. The preferential orientation of 2D (nano)materials parallel to the current collector and the possibility to prepare electrodes free-standing and additive-free generally favor them for ECD analysis. The height change information from ECD is comparable to the above-discussed microscopic operando AFM method. A combination of operando AFM and ECD, therefore, allows the obtaining of electro-chemo-mechanical information over several length scales.
6.3 Electrochemical quartz crystal microbalance
Electrochemical Quartz Crystal Microbalance (EQCM) is a powerful operando analytical tool that provides gravimetric information about an electrode during an electrochemical reaction. It has been widely used in the studies of electrodeposition, and polymer growth. In the last decade, it has gained increasing popularity in the energy storage field as it allows quantitative analysis of the electrolytic species that enter and leave the electrodes (via electrode mass change) and elucidates the charge storage mechanisms.The quartz used in EQCM is composed of a thin AT-cut quartz crystal with a thickness of a few hundred micrometers sandwiched by two thin metal electrodes. An alternating electric field is applied on the electrodes, causing a vibrational motion of the quartz due to its piezoelectric nature. The fundamental resonance frequency of the quartz under the alternating electric field is dependent on its thickness. If a deposited thin film is as rigid as the quartz itself and rigidly attached to the quartz surface, there is no dissipation of the oscillation energy across the deposited film (ΔD = 0). Under this condition, Sauerbrey's equation, which converts the resonance frequency (Δf) change into mass change (Δm), can be applied. However, for EQCM measurements in the energy storage field, the battery or supercapacitor electrode materials, usually made of irregularly shaped particles that are between a few nanometers to a few hundred micrometers in size, are mixed with polymer binders and deposited on the quartz. The electrode-deposited quartz is then used as a working electrode, immersed in the electrochemical cell for cycling. The thickness, morphology, porosity, and stiffness of the deposited electrode and the viscosity of the liquid electrolytes the quartz immersed in will affect the dissipation of the quartz oscillation.36 Verification of the dissipation (EQCM-D) and careful correction are needed to obtain quantitative gravimetric information on the electrode during cycling. For more details, the readers are referred to other reviews focusing on the fundamental aspects of the EQCM-D technique and the effect of a realistic battery electrode on the data analysis.178,179
The EQCM-D technique was used to investigate the amount of solvent that participated in the charge storage of an MXene electrode. Ti3C2Tx exhibits a rectangular CV with no redox peak when cycled in 2 M Li2SO4 (Fig. 13a),180 while with a clear set of redox peaks in saturated LiCl WiSE (Fig. 13c).32 The deposited MXene electrode was thin and rigid, and the dissipation change (ΔW/n) during cycling remains constant (Fig. 13b), suggesting the frequency change is of gravimetric origin. The Li-ions enter and leave the MXene with one water molecule in 2 M Li2SO4. The solvation number remains constant across the entire potential range. When cycled in saturated LiCl, the Li-ions first enter with almost no water molecules (S1: 0.45–0.17 V), followed by a sudden increase to 2.85 water molecules (S2: at the cathodic peak), and continue with 1.45 water molecules after passing the cathodic peak (S3) (Fig. 13d). The EQCM-D results quantify the solvent involvement during the electrochemical process and shed insights into the effect of solvation environment of the bulk electrolyte on charge storage mechanism.
 | ||
| Fig. 13 (a) Cyclic voltammogram recorded during EQCM measurement of a Ti3C2Tx electrode in 2 M Li2SO4 at different scan rates and (b) the corresponding EQCM-D (Δf/n and ΔW/n) results at 20 mV s−1.180 (c) Cyclic voltammogram recorded during EQCM measurement of a Ti3C2Tx electrode in 19.8 m LiCl electrolyte, and (d) the corresponding change of resonance frequency Δf/n during the insertion/extraction of Li-ion and water.32 Panel (a and b) reproduced from ref. 180, copyright 2017 American Chemical Society. Panel (c and d) reproduced from ref. 32, copyright 2017 American Chemical Society. | ||
It should be noted that EQCM measures the overall mass change of the electrode regardless of the species. When the individual electrolytic species (cations, anions and solvents) have similar molecular masses, EQCM cannot distinguish their contribution. Also, surface adsorption of solvents and/or parasitic side reactions can complicate precise determination of the species involved in charge storage. Thus, it is beneficial to pair up with another chemically sensitive technique, such as NMR, FTIR or Raman spectroscopy, to deconvolute the participation of each species in the charge storage mechanism.181
7. Nuclear magnetic resonance
7.1 Ex situ NMR
Nuclear Magnetic Resonance (NMR) is an element-specific, quantitative and non-destructive technique for studying materials. It is sensitive to both long-range and short-range ordered and disordered structures, as well as dynamics.182 Magic angle spinning solid-state NMR (MAS SSNMR) has been proven to be an informative tool for studying MAX phases and MXene, particularly excelling at revealing the structure and dynamics of surface functional groups. It has been applied to study V2AlC,183 V2CTx,183 Ti3C2Tx,134,184–186 Ti3AlC2,187,188 Nb2AlC,187 Nb4AlC3,187 Nb2CTx,187 Nb4C3Tx,187 Ti2AlC,188 Mo2TiAlC2,188 Mo2TiAlC3,188 Mo2Ga2C,188 Ti3C2(N-dda)78 and T3CNTz134 via probing nuclei 13C, 1H, 19F, 27Al, 93Nb, 47/49Ti and 15N.
In two seminal works applying MAS SSNMR to study MXene,183,184 the molecular details of the V2CTx and Ti3C2Tx surface terminations were analyzed. 1H NMR spectra were acquired and revealed an unusually large 1H chemical shift at 85 ppm and in the range of 10–20 ppm for the protons in close proximity to the V2CTx and Ti3C2Tx surfaces, respectively. The high values of chemical shifts were caused by the Knight shift effect through the Fermi contact interaction of nuclei with the conduction electrons from a metallic material. Therefore, the Knight shift is an indicator for a nucleus bound to a metallic surface within one or two-bond distance.190 The 1H spectra for the Ti3C2Tx prepared via the HF and LiF-HCl routes are shown in Fig. 14a.184 The 1H signals at 18.6 ppm and 12.5 ppm were assigned to Ti–OH groups. Besides 1H NMR, a recent work showed that 13C chemical shifts are sensitive to the surface terminations such as –F vs. –O, arising from the orbital contribution to the NMR shielding and the Knight shift.187
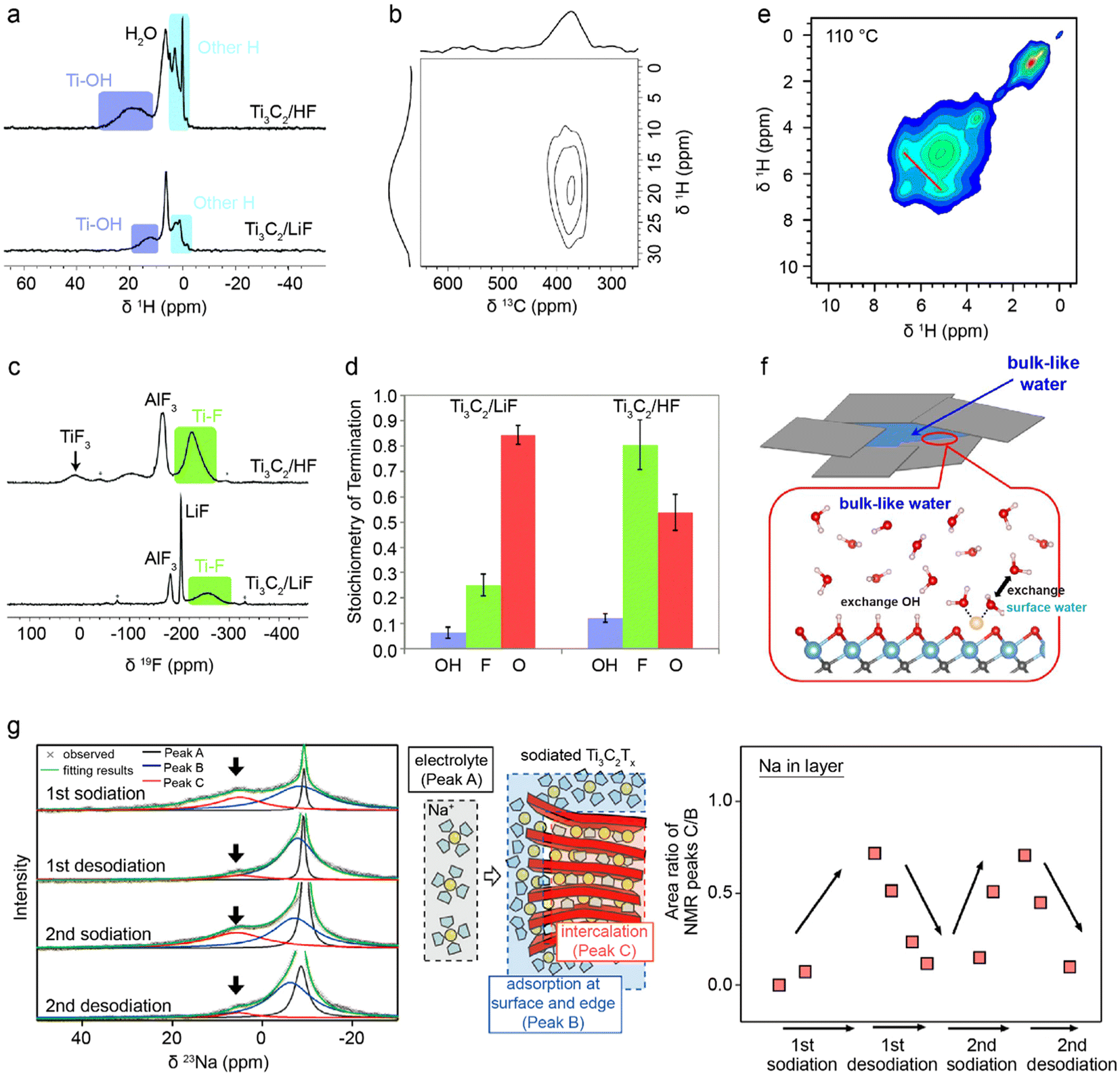 | ||
| Fig. 14 MAS SSNMR study of MXene. (a) 1H NMR spectra of HF and LiF-HCl synthesized T3C2Tx MXene recorded at 60 kHz MAS rate.184 (b) 1H–13C HETCOR NMR spectrum of HF synthesized Ti3C2Tx MXene recorded at 40 kHz MAS rate.184 (c) 19F NMR spectra of HF and LiF-HCl synthesized T3C2Tx MXene recorded at 60 kHz MAS rate.184 (d) NMR-derived composition of the Ti3C2Tx surface functional groups synthesized via the HF and LiF-HCl routes.184 (e) 2D 1H–1H homonuclear correlation NMR spectrum of Ti3C2Tx annealed at 110 °C.185 (f) Suggested model for Ti3C2(OH)x MXene with exchangeable water molecules between the interlayer bulk and surface. White: hydrogen, red: oxygen, orange: lithium, blue: titanium, grey: carbon.185 (g) 23Na NMR spectra of Ti3C2Tx after two cycles of sodiation and desodiation. The red component is assigned to the intercalated sodium, the blue component is assigned to the surface and edge sodium, and the black component is assigned to the electrolyte sodium ions. The plot on the right shows the signal integral ratio of the C to B component.189 Panel (a–d) reproduced from ref. 184 with permission from the Royal Society of Chemistry. Panel (e and f) reproduced with permission from ref. 185, copyright 2020 American Chemical Society. Panel (g) reproduced with permission from ref. 189, copyright 2016 American Chemical Society. | ||
To further confirm the connectivity of the surface hydroxyl groups to the Ti3C2Tx, a two-dimensional (2D) 1H–13C heteronuclear correlation (HETCOR) NMR was performed.184 HETCOR probes the connectivity of nuclei separated on the order of angstroms. The 1H–13C HETCOR spectrum for the Ti3C2Tx in Fig. 14b shows one signal centered at 20 ppm for 1H and 380 ppm for 13C, where the latter is from the bulk Ti3C2, confirming that the –OH group is in the proximity to the Ti3C2Tx. A similar 1D 19F and 2D 1H–19F correlation experiment, as shown in Fig. 14c, revealed the surface-bound Ti-F groups. A later work further measured the intermolecular distance between Ti-OH terminations to be within 5 Å via 2D 1H–1H correlation experiments.185
Since NMR is quantitative, the surface functional groups can be quantified by comparing them to the NMR signal of a known standard. With the quantitative information from the 1D 1H and 19F NMR spectra (Fig. 14d), the chemical formula for Ti3C2Tx via the LiF-HCl synthesis was determined to be Ti3C2(OH)0.06F0.25O0.84, and via the HF synthesis to be Ti3C2(OH)0.12F0.8O0.54, respectively.184
In addition to structural elucidation, NMR is sensitive to dynamic processes, such as chemical exchange. Exchangeable protons in bulk water and surface water in Ti3C2(OH)x MXene were unveiled by a 2D 1H–1H homonuclear correlation NMR experiment in which exchangeable protons give rise to off-diagonal signals.185 As shown in Fig. 14e, a 2D 1H–1H homonuclear correlation NMR spectrum showed an off-diagonal signal at 5.0 and 6.8 ppm, revealing the exchange between bulk-like water at 5.0 ppm and surface water at 6.8 ppm. Note that the bulk-like water refers to the water molecules trapped between MXene layers. The resonance frequency difference of these two signals puts a lower limit of exchange time to be above 1 ms.
Besides NMR studies of MXene structures and dynamics, intercalation of water into Ti3C2Tx,185 sodium ions into Ti3C2Tx189 and silica into Mo2TiC2191 were investigated via 23Na, 28Si and 1H NMR. Fig. 14g presents the 23Na NMR spectra of Ti3C2Tx that had undergone two cycles of sodiation and desodiation.189 The 23Na signal at 5 ppm increased after sodiation and decreased after desodiation, most likely arising from the intercalated sodium species. Although detailed quantification and structural elucidation were not further pursued, this work demonstrates the feasibility of applying MAS SSNMR for probing the intercalation of ionic species into MXene, and shows the prospects of applying advanced in situ NMR techniques to study intercalation as a function of electrochemical conditions.
7.2 In situ NMR
NMR is a low-frequency (in the radio-frequency regime) and low-energy technique, allowing deep penetration of the NMR excitation and non-destructive measurements, and thus is ideal for in situ/operando studies of an electrochemical device.192,193 Besides the high chemical specificity, temporal resolution for the commonly studied nuclei 1H, 19F, 7Li and 23Na is on the order of seconds to minutes, sufficient for following charge–discharge cycles of an EES device. Different probe designs based on solenoids,194 striplines,195 parallel plates,196 parallel lines detectors,197 and in situ cell designs, including the bag cell,194,195 plastic capsule cell,192 flow cell,198 and commercial coin cells199 have been demonstrated for studying different types of electrochemical devices. The solenoid probe combined with an electrochemical capsule cell is the most commonly used configuration, as illustrated in Fig. 15a.192 For example, it was applied to study Na-ion battery and captured the metastable metallic phase of sodium during (de)intercalation, as evidenced by the shifting signals towards 760 ppm in Fig. 15b, which, depending on the time for sample preparation, could escape ex situ characterizations due to self-discharge.200 The majority of in situ NMR experiments were performed in static mode, i.e. without MAS, and therefore the spectral resolution is sub-optimal. Nevertheless, well-separated resonances, such as the Knight-shifted metallic Na or Li resonances, can be readily observed.201–203 A recent work demonstrated the possibility of performing in situ MAS SSNMR on an intact battery via a jelly cell design.204 Although the battery was not (dis)charged during the NMR measurement, it represents a major step towards MAS SSNMR on an operating battery.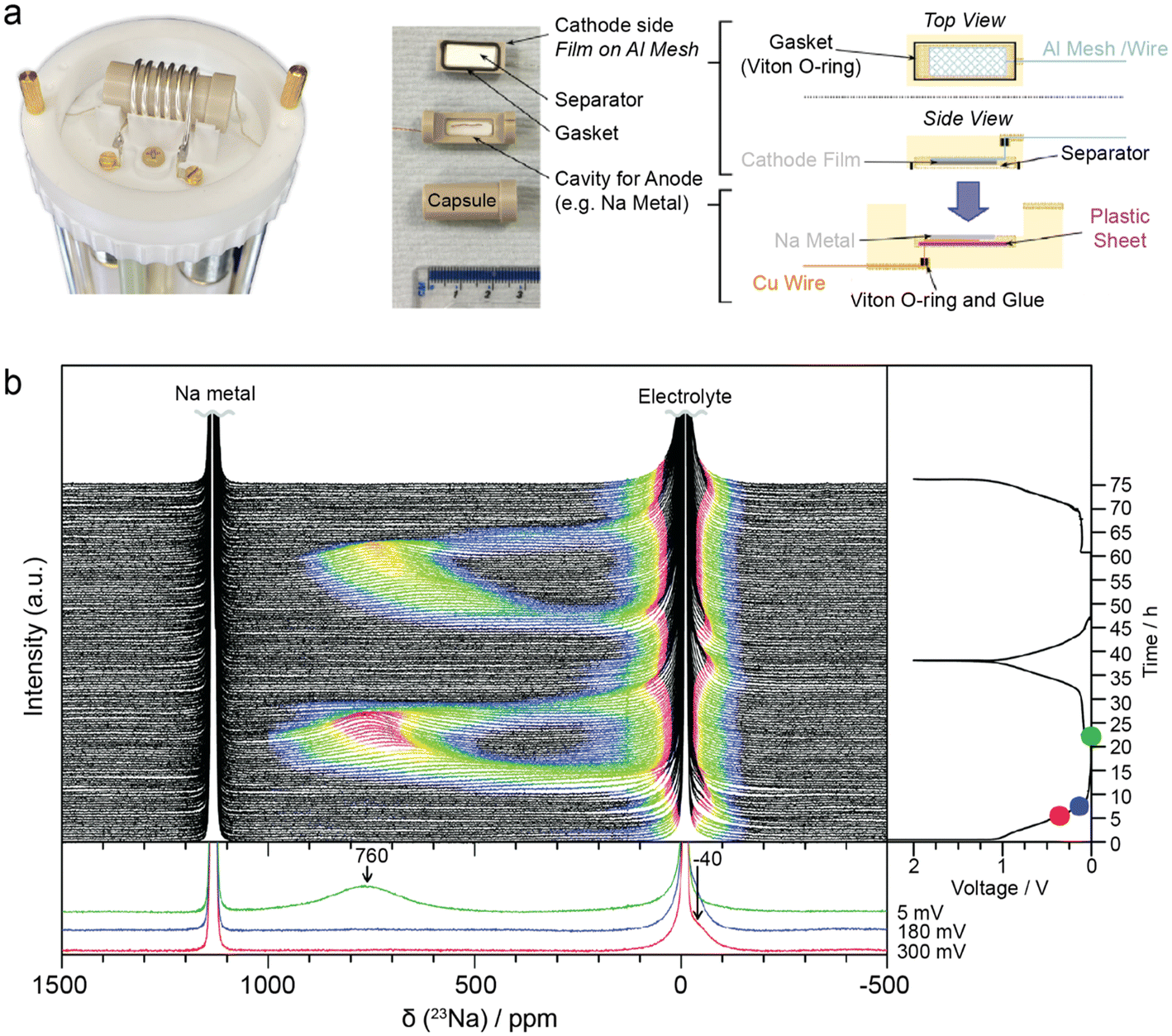 | ||
| Fig. 15 in situ NMR setup and an application for Na-ion battery. (a) A plastic electrochemical cell positioned in a solenoid-based in situ NMR probe.192,205 Components of the in situ cell are shown on the right.192 (b) In situ23Na NMR spectra for an electrochemical cell with sodium metal, hard carbon electrodes, and NaPF6 electrolyte. The corresponding voltage profile is shown on the right, and the selected spectra are shown below.200 Panel (a) reproduced from ref. 192 and 205 with permission from the American Chemical Society and ePROBE GmbH. Panel (b) reproduced from ref. 200 with permission from the Royal Society of Chemistry. | ||
While in situ NMR studies of 2D MXene-based electrode materials are yet to be demonstrated, the existing in situ NMR setups of various types could be readily transferrable to study Li-ion and Na-ion intercalation, for example, to probe the local environment of Li- and Na-ions, metallic phases and to investigate the effect of intercalation on surface functional groups and layered structures. Because of the rich structural and dynamic information obtainable via NMR, along with the existing and fast-developing in situ methods, in situ NMR for studying 2D MXene-based electrodes will likely emerge in the foreseeable future.
7.3 Practical considerations for NMR
NMR has a relatively low sensitivity compared to some other characterizations, such as optical or X-ray-based methods, and therefore, in some cases, requires isotopic labelling or hyperpolarization techniques.206 For studying surfaces, NMR requires a large surface area, therefore nanosized or 2D materials are the most suitable. Another challenge associated with NMR characterization is that the assignment of NMR signals often requires ancillary experiments on model compounds. If model compounds cannot be obtained easily, calculations of NMR shifts should be performed to aid the assignment.207 Furthermore, NMR is silent to paramagnetic species. Nevertheless, paramagnetic shift of the nearby nuclei can be used as an indirect probe.208 A technique that can directly detect unpaired electrons and characterize paramagnetic species is electron paramagnetic resonance (EPR) spectroscopy. NMR and EPR are complementary. Coupling these two methods will allow studies of paramagnetic and diamagnetic species, and is particularly useful when there is inter-conversion between paramagnetism and diamagnetism during battery cycling. The coupling has been demonstrated for in situ studies of redox flow batteries,209 but is yet to be applied for 2D material-based batteries.8. Summary
This review provides a comprehensive overview of advanced in situ and ex situ characterization techniques for investigating charge storage mechanisms in 2D and layered material-based EES devices. Understanding these mechanisms is crucial for optimizing EES performance, as it enables researchers to analyze how these materials interact with electrolytes, facilitate ion transport, undergo redox reactions, and form interphases. A summary of the characterization techniques described in this review, including their applications, is given in Table 1.| Detectable qualities | Technique | Main Application |
|---|---|---|
| Intercalant mass | EQCM | Measure mass change of electrodes |
| Imaging | TEM | Visualization of electrode structure and morphology |
| NI | Visualization of lithium transport | |
| Ion dynamics | NS | Diffusivity determination in confined spaces |
| NMR | Diffusion and exchange | |
| Local environment | Raman | Fingerprint chemical bonds in the electrode/electrolyte |
| FTIR | Fingerprint chemical bonds in the electrode/electrolyte | |
| TEM | Visualize differences between local structures | |
| NS | Fingerprint chemical bonds in the electrode/electrolyte | |
| XPS | Chemical composition of the electrode surface | |
| NMR | Chemical bonding of the surface and bulk of an electrode | |
| Structure & morphology | TEM | Visualization of electrode structure and morphology |
| AFM | Physically probe electrode morphology & expansion | |
| ECD | Thickness change of macroscopic electrode | |
| XRD | Measure interlayer space distance of 2D materials | |
| ND | Measure interlayer space distance of 2D materials | |
| Electronic state | XAS | Quantify oxidation states of active electrode elements |
| XPS | Quantify oxidation states of elements at the electrode surface | |
| UV-vis | Measure oxidation state shifts in the electrode |
We first discuss optical spectroscopy techniques, focusing on Raman, infrared, and UV-vis spectroscopy. These methods provide valuable insights into chemical bonding, phase transitions, and interfacial interactions by analyzing vibrational modes and light absorption characteristics. They are particularly useful for tracking the structural evolutions and oxidation states of 2D materials during electrochemical cycling, which is essential for understanding redox activities.
Next, we explore the imaging technique TEM, which allows for the direct visualization of morphological changes, ion diffusion pathways, and degradation processes in 2D materials, aiding in the correlation between structure and electrochemical performance.
We then discuss the role of X-ray and neutron-based methods in characterizing 2D material-based EES devices. X-ray and neutron scattering techniques are widely used to analyze crystallographic structures, interlayer spacing, and ion intercalation mechanisms. Additionally, X-ray absorption and photoelectron spectroscopy provide chemical state information. Compared to X-ray techniques, neutron-based methods offer unique sensitivity to light elements such as hydrogen and lithium, making them particularly useful for studying electrolyte interactions and ion transport.
For mechanical probing techniques, we examine AFM, ECD, and EQCM. These methods help reveal electrode expansion, mechanical properties, and mass changes, providing insights into ion intercalation dynamics and electro-chemo-mechanical coupling.
Finally, we provide an overview of NMR spectroscopy, an element-specific and non-destructive technique that enables the analysis of ion transport, interfacial interactions, and surface functionalization in 2D materials. NMR is particularly valuable for studying dynamic processes under both ex situ and in situ conditions.
As new 2D and layered materials continue to be developed and their applications in various EES devices expand, selecting appropriate characterization methods is essential for uncovering charge storage mechanisms and guiding material design. By systematically evaluating the strengths and limitations of these techniques in 2D material-based EES devices, we highlight existing gaps in understanding EES processes and identify opportunities for improving characterization methodologies. Future advancements in multi-modal and in situ/operando techniques, along with the integration of machine-learning-assisted data analysis, will play a crucial role in accelerating the discovery and optimization of 2D material electrodes.
Author contributions
Albert de Kogel: Main manuscript, Neutron methods. Ruocun (John) Wang: Raman, FTIR, UV-Vis. Wan-Yu Tsai: AFM, EQCM. Maciej Tobis: ECD. Robert Leiter: TEM. Ruipeng Luo: NMR. Evan Wenbo Zhao: NMR. Simon Fleischmann: ECD, TEM. Xuehang Wang: Main manuscript, X-ray methods.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
A. K. and X. W. acknowledge funding from the NWO open competition m (OCENW.M.22.303). R. J. W. acknowledges start-up funding from the University of North Texas. W.-Y. T. was supported by the French National Agency for Research (ANR) through Chaire de Professeur Junior funding and RS2E network (STORE-EX Labex Project ANR-10-LABX-76-01). R.-P. L. and E. W. Z. acknowledge funding from the Radboud University Start-up Fund and the uNMR-NL ROADMAP facilities grant (184.035.002). M. T., R. L. and S. F. acknowledge funding from the German Federal Ministry of Education and Research (BMBF) in the “NanoMatFutur” program (Grant No. 03XP0423) and basic funding from the Helmholtz Association. E. W. Z. is grateful to Dr Oliver Pecher (eProbe GmbH) for providing the picture of the in situ NMR probe. We also thank the Lorentz Center in Leiden (NL) for supporting our workshop, Electrochemical Energy Storage in 2D and Layered Materials, which offered us the opportunity to develop the ideas presented in this review.References
- E. A. Marseglia, Int. Rev. Phys. Chem., 1983, 3, 177–216 Search PubMed
.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed
- Y. Yan, P. Zhang, Z. Qu, M. Tong, S. Zhao, Z. Li, M. Liu and Z. Lin, Nano Lett., 2020, 20, 7662–7669 CrossRef CAS
- X. Bo, Q. Zhang, G. Li, J. Zhang, R. Wu, S. Wang, I. K. Tiwalade, S. Wang, Z. Lin and S. Zhao, Chem. Eng. J., 2024, 487, 150559 CrossRef CAS
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago, J. R. Guest, M. J. Yacaman, A. Ponce, A. R. Oganov, M. C. Hersam and N. P. Guisinger, Science, 2015, 350, 1513–1516 CrossRef CAS PubMed
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed
- S. Miyata and A. Okada, Clays Clay Miner., 1977, 25, 14–18 CrossRef
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS
- F. Liu, Y. Hu, Z. Qu, X. Ma, Z. Li, R. Zhu, Y. Yan, B. Wen, Q. Ma, M. Liu, S. Zhao, Z. Fan, J. Zeng, M. Liu, Z. Jin and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2303262120 CrossRef
- H. Cui, Y. Guo, W. Ma and Z. Zhou, ChemSusChem, 2020, 13, 1155–1171 CrossRef CAS PubMed
- F. Bonaccorso, L. Colombo, G. Yu, M. Stoller, V. Tozzini, A. C. Ferrari, R. S. Ruoff and V. Pellegrini, Science, 2015, 347, 1246501 CrossRef
- Z. Wang, C. Wei, H. Jiang, Y. Zhang, K. Tian, Y. Li, X. Zhang, S. Xiong, C. Zhang and J. Feng, Adv. Mater., 2024, 36, 2306015 CrossRef CAS
- Z. Chen, X. Li, D. Wang, Q. Yang, L. Ma, Z. Huang, G. Liang, A. Chen, Y. Guo, B. Dong, X. Huang, C. Yang and C. Zhi, Energy Environ. Sci., 2021, 14, 3492–3501 RSC
- S. Tian, Q. Zeng, G. Liu, J. Huang, X. Sun, D. Wang, H. Yang, Z. Liu, X. Mo, Z. Wang, K. Tao and S. Peng, Nano-Micro Lett., 2022, 14, 196 CrossRef CAS PubMed
- X. Zheng, Z. Wang, J. Li and L. Wei, Sci. China Mater., 2022, 65, 1463–1475 CrossRef CAS
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277–2282 CrossRef CAS PubMed
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS
- T. S. Mathis, N. Kurra, X. Wang, D. Pinto, P. Simon and Y. Gogotsi, Adv. Energy Mater., 2019, 9, 1902007 CrossRef CAS
- S. Fleischmann, J. B. Mitchell, R. Wang, C. Zhan, D. Jiang, V. Presser and V. Augustyn, Chem. Rev., 2020, 120, 6738–6782 CrossRef CAS
- Z. Hu, Z. Tai, Q. Liu, S.-W. Wang, H. Jin, S. Wang, W. Lai, M. Chen, L. Li, L. Chen, Z. Tao and S.-L. Chou, Adv. Energy Mater., 2019, 9, 1803210 CrossRef
- J. Xiao, D. Choi, L. Cosimbescu, P. Koech, J. Liu and J. P. Lemmon, Chem. Mater., 2010, 22, 4522–4524 CrossRef CAS
- Y. Zhang, K. S. R. Chandran and H. Z. Bilheux, J. Power Sources, 2018, 376, 125–130 CrossRef CAS
- M. A. Rodriguez, D. Ingersoll, S. C. Vogel and D. J. Williams, Electrochem. Solid-State Lett., 2003, 7, A8 CrossRef
- A. Brady, K. Liang, V. Q. Vuong, R. Sacci, K. Prenger, M. Thompson, R. Matsumoto, P. Cummings, S. Irle, H.-W. Wang and M. Naguib, ACS Nano, 2021, 15, 2994–3003 CrossRef CAS
- Q. Mahmood, S. K. Park, K. D. Kwon, S.-J. Chang, J.-Y. Hong, G. Shen, Y. M. Jung, T. J. Park, S. W. Khang, W. S. Kim, J. Kong and H. S. Park, Adv. Energy Mater., 2016, 6, 1501115 CrossRef
- J. Choi, H. Moon and S. Fleischmann, Electrochim. Acta, 2024, 476, 143774 CrossRef CAS
- X. Mu, D. Wang, F. Du, G. Chen, C. Wang, Y. Wei, Y. Gogotsi, Y. Gao and Y. Dall'Agnese, Adv. Funct. Mater., 2019, 29, 1902953 CrossRef
- S. Niu, Z. Wang, M. Yu, M. Yu, L. Xiu, S. Wang, X. Wu and J. Qiu, ACS Nano, 2018, 12, 3928–3937 CrossRef CAS PubMed
- C. Chen, A. de Kogel, M. Weijers, L. J. Bannenberg and X. Wang, 2D Mater., 2023, 11, 015001 CrossRef
- J. Fei, S. Zhao, X. Bo, F. Xie, G. Li, E.-A. M. A. Ahmed, Q. Zhang, H. Jin and Z. Lin, Carbon Energy, 2023, 5, e333 CrossRef CAS
- L. Li Zhang, X. Zhao, H. Ji, M. D. Stoller, L. Lai, S. Murali, S. Mcdonnell, B. Cleveger, R. M. Wallace and R. S. Ruoff, Energy Environ. Sci., 2012, 5, 9618–9625 RSC
- X. Wang, T. S. Mathis, Y. Sun, W.-Y. Tsai, N. Shpigel, H. Shao, D. Zhang, K. Hantanasirisakul, F. Malchik, N. Balke, D. Jiang, P. Simon and Y. Gogotsi, ACS Nano, 2021, 15, 15274–15284 CrossRef CAS
- H. Guo, M. Elmanzalawy, P. Sivakumar and S. Fleischmann, Energy Environ. Sci., 2024, 17, 2100–2116 RSC
- W. H. Bragg and W. L. Bragg, Proc. R. Soc. London A, 1913, 88, 428–438 CAS
- S. Wang, Q. Liu, C. Zhao, F. Lv, X. Qin, H. Du, F. Kang and B. Li, Energy Environ. Mater., 2018, 1, 28–40 CrossRef
- D. A. Buttry and M. D. Ward, Chem. Rev., 1992, 92, 1355–1379 CrossRef CAS
- B. S. Hudson, Vib. Spectrosc., 2006, 42, 25–32 CrossRef CAS
- P.-F. Guo, Y. Yang, B. Zhu, Q.-N. Yang, Y. Jia, W.-T. Wang, Z.-T. Liu, S.-Q. Zhao and X. Cui, Carbon Energy, 2024, 6, e532 CrossRef CAS
- X. Xu, H. Wu, Y. Yan, Y. Zheng, Y. Yan, S. Qiu, T. Liang, C. Deng, Y. Yao, J. Zou and M. Liu, Adv. Energy Mater., 2025, 2405236 CrossRef
- M. V. Avdeev, I. A. Bobrikov and V. I. Petrenko, Phys. Sci. Rev., 2018, 3(10), 20170157 Search PubMed
- B. L. Mehdi, M. Gu, L. R. Parent, W. Xu, E. N. Nasybulin, X. Chen, R. R. Unocic, P. Xu, D. A. Welch, P. Abellan, J. G. Zhang, J. Liu, C. M. Wang, I. Arslan, J. Evans and N. D. Browning, Microsc. Microanal., 2014, 20, 484–492 CrossRef PubMed
- J. R. Dahn, W. R. McKinnon, R. R. Haering, W. J. L. Buyers and B. M. Powell, Can. J. Phys., 1980, 58, 207–213 CrossRef CAS
- T. Köhler, J. Hanzig and V. Koroteev, Phys. Sci. Rev., 2019, 4(2), 20170154 Search PubMed
- D. Liu, Z. Shadike, R. Lin, K. Qian, H. Li, K. Li, S. Wang, Q. Yu, M. Liu, S. Ganapathy, X. Qin, Q.-H. Yang, M. Wagemaker, F. Kang, X.-Q. Yang and B. Li, Adv. Mater., 2019, 31, 1806620 CrossRef PubMed
- G. Bergman, E. Ballas, Q. Gao, A. Nimkar, B. Gavriel, M. D. Levi, D. Sharon, F. Malchik, X. Wang, N. Shpigel, D. Mandler and D. Aurbach, Adv. Energy Mater., 2023, 13, 2203154 CrossRef CAS
- S. Pattnaik, M. R. Kumar, S. K. Mishra and S. P. Gautam, Energy Storage, 2023, 5, e398 CrossRef CAS
- H. Alnoor, A. Elsukova, J. Palisaitis, I. Persson, E. N. Tseng, J. Lu, L. Hultman and P. O. Å. Persson, Mater. Today Adv., 2021, 9, 100123 CrossRef CAS
- J. B. Wu, M. L. Lin, X. Cong, H. N. Liu and P. H. Tan, Chem. Soc. Rev., 2018, 47, 1822–1873 RSC
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS
- J. L. Verble and T. J. Wieting, Phys. Rev. Lett., 1970, 25, 362–365 CrossRef CAS
- A. Sarycheva and Y. Gogotsi, Chem. Mater., 2020, 32, 3480–3488 CrossRef CAS
- K. Shevchuk, A. Sarycheva, C. E. Shuck and Y. Gogotsi, Chem. Mater., 2023, 35, 8239–8247 CrossRef CAS
- K. Share, A. P. Cohn, R. E. Carter and C. L. Pint, Nanoscale, 2016, 8, 16435–16439 RSC
- A. P. Cohn, K. Share, R. Carter, L. Oakes and C. L. Pint, Nano Lett., 2016, 16, 543–548 CrossRef CAS PubMed
- L. J. Hardwick, P. W. Ruch, M. Hahn, W. Scheifele, R. Kötz and P. Novák, J. Phys. Chem. Solids, 2008, 69, 1232–1237 CrossRef CAS
- F. Xiong, H. Wang, X. Liu, J. Sun, M. Brongersma, E. Pop and Y. Cui, Nano Lett., 2015, 15, 6777–6784 CrossRef CAS
- D. Johnson, K. Hansen, R. Yoo and A. Djire, ChemElectroChem, 2022, 9, e202200555 CrossRef CAS
- Y. H. Wang, X. Jin, M. Xue, M. F. Cao, F. Xu, G. X. Lin, J. B. Le, W. M. Yang, Z. L. Yang, Y. Cao, Y. Zhou, W. Cai, Z. Zhang, J. Cheng, W. Guo and J. F. Li, Joule, 2023, 7, 1652–1662 CrossRef CAS
- V. A. Nikitina, S. Y. Vassiliev and K. J. Stevenson, Adv. Energy Mater., 2020, 10, 1903933 CrossRef CAS
- V. Augustyn, R. Wang, N. Balke, M. Pharr and C. B. Arnold, ACS Energy Lett., 2020, 5, 3548–3559 CrossRef CAS
- A. Sarycheva, M. Shanmugasundaram, A. Krayev and Y. Gogotsi, ACS Nano, 2022, 16(4), 6858–6865 CrossRef CAS PubMed
- T. Parker, D. Zhang, D. Bugallo, K. Shevchuk, M. Downes, G. Valurouthu, A. Inman, B. Chacon, T. Zhang, C. E. Shuck and Y. Hu, Chem. Mater., 2024, 36(17), 8437–8446 CrossRef CAS PubMed
- M. Acik, G. Lee, C. Mattevi, A. Pirkle, R. M. Wallace, M. Chhowalla, K. Cho and Y. Chabal, J. Phys. Chem. C, 2011, 115, 19761–19781 CrossRef CAS
- K. G. Zhou, K. S. Vasu, C. T. Cherian, M. Neek-Amal, J. C. Zhang, H. Ghorbanfekr-Kalashami, K. Huang, O. P. Marshall, V. G. Kravets, J. Abraham, Y. Su, A. N. Grigorenko, A. Pratt, A. K. Geim, F. M. Peeters, K. S. Novoselov and R. R. Nair, Nature, 2018, 559, 236–240 CrossRef CAS PubMed
- M. Lounasvuori, Y. Sun, T. S. Mathis, L. Puskar, U. Schade, D.-E. Jiang, Y. Gogotsi and T. Petit, Nat. Commun., 2023, 14, 1322 CrossRef CAS PubMed
- M. Lounasvuori, T. S. Mathis, Y. Gogotsi and T. Petit, J. Phys. Chem. Lett., 2023, 14, 1578–1584 CrossRef CAS
- M. d. P. Bernicola, M. Lounasvuori, J. Padilla-Pantoja, J. Santiso, C. Debiemme-Chouvy, H. Perrot, T. Petit, J. A. Garrido and E. del Corro, Adv. Funct. Mater., 2024, 34(46), 2408441 CrossRef CAS
- Y. Yao, W. Chen, Y. Du, Z. Tao, Y. Zhu and Y. X. Chen, J. Phys. Chem. C, 2015, 119, 22452–22459 CrossRef CAS
- L. Puskar, E. Ritter, U. Schade and E. F. Aziz, Opt. InfoBase Conf. Pap., 2016, 99–101 Search PubMed
- Y. X. Chen, A. Miki, S. Ye, H. Sakai and M. Osawa, J. Am. Chem. Soc., 2003, 125, 3680–3681 CrossRef CAS PubMed
- P. Salles, E. Quain, N. Kurra, A. Sarycheva and Y. Gogotsi, Small, 2018, 14, 1–7 CrossRef
- T. S. Mathis, K. Maleski, A. Goad, A. Sarycheva, M. Anayee, A. C. Foucher, K. Hantanasirisakul, C. E. Shuck, E. A. Stach and Y. Gogotsi, ACS Nano, 2021, 15, 6420–6429 CrossRef CAS
- K. Maleski, C. E. Ren, M. Zhao, B. Anasori and Y. Gogotsi, ACS Appl. Mater. Interfaces, 2018, 10, 24491–24498 CrossRef CAS PubMed
- K. Maleski, C. E. Shuck, A. T. Fafarman and Y. Gogotsi, Adv. Opt. Mater., 2021, 2001563, 1–8 Search PubMed
- P. Salles, D. Pinto, K. Hantanasirisakul, K. Maleski, C. E. Shuck and Y. Gogotsi, Adv. Funct. Mater., 2019, 29(17), 1809223 CrossRef
- G. Valurouthu, K. Maleski, N. Kurra, M. Han, K. Hantanasirisakul, A. Sarycheva and Y. Gogotsi, Nanoscale, 2020, 12, 14204–14212 RSC
- D. Zhang, R. (John) Wang, X. Wang and Y. Gogotsi, Nat. Energy, 2023, 8, 567–576 CrossRef CAS
- C. Zhou, D. Wang, F. Lagunas, B. Atterberry, M. Lei, H. Hu, Z. Zhou, A. S. Filatov, D. Jiang, A. J. Rossini, R. F. Klie and D. V. Talapin, Nat. Chem., 2023, 15, 1722–1729 CrossRef CAS
- C. Liu, X. Zou, Y. Lv, X. Liu, C. Ma, K. Li, Y. Liu, Y. Chai, L. Liao and J. He, Nat. Nanotechnol., 2024, 19, 448–454 CrossRef CAS PubMed
- X. H. Liu, Y. Liu, A. Kushima, S. Zhang, T. Zhu, J. Li and J. Y. Huang, Adv. Energy Mater., 2012, 2, 722–741 Search PubMed
- C. Han, M. T. Islam and C. Ni, ACS Omega, 2021, 6, 6537–6546 CrossRef CAS PubMed
- Z. Sun, J. Pan, W. Chen, H. Chen, S. Zhou, X. Wu, Y. Wang, K. Kim, J. Li, H. Liu, Y. Yuan, J. Wang, D. Su, D. L. Peng and Q. Zhang, Adv. Energy Mater., 2024, 14, 2303165 Search PubMed
- X. Wang, Y. Li and Y. S. Meng, Joule, 2018, 2, 2225–2234 CrossRef CAS
- Z. Zhang, Y. Cui, R. Vila, Y. Li, W. Zhang, W. Zhou, W. Chiu and Y. Cui, Acc. Chem. Res., 2021, 54, 3505–3517 CrossRef CAS
- S. Weng, Y. Li and X. Wang, iScience, 2021, 24, 103402 CrossRef CAS
- Y. Shin, D. Stepien, M. Hepp, B. Butz, D. Bresser and S. Fleischmann, J. Power Sources, 2023, 556, 232515 CrossRef CAS
- M. Kühne, F. Börrnert, S. Fecher, M. Ghorbani-Asl, J. Biskupek, D. Samuelis, A. V. Krasheninnikov, U. Kaiser and J. H. Smet, Nature, 2018, 564, 234–239 CrossRef
- Y. Li, F. Börrnert, M. Ghorbani-Asl, J. Biskupek, X. Zhang, Y. Zhang, D. Bresser, A. V. Krasheninnikov and U. Kaiser, Adv. Funct. Mater., 2024, 34, 2406034 CrossRef CAS
- N. Hodnik and S. Cherevko, Curr. Opin. Electrochem., 2019, 15, 73–82 CrossRef CAS
- V. Shokhen, L. Strandberg, M. Skoglundh and B. Wickman, J. Mater. Chem. A, 2023, 11, 21029–21035 RSC
- S. Yu, M. J. Zachman, K. Kang, H. Gao, X. Huang, F. J. DiSalvo, J. Park, L. F. Kourkoutis and H. D. Abruña, Adv. Energy Mater., 2019, 9, 1902773 CrossRef CAS
- S. Zhao, Z. Wang, Y. He, H. Jiang, Y. W. Harn, X. Liu, C. Su, H. Jin, Y. Li, S. Wang, Q. Shen and Z. Lin, Adv. Energy Mater., 2019, 9, 1901093 CrossRef
- M. Xia, T. Liu, N. Peng, R. Zheng, X. Cheng, H. Zhu, H. Yu, M. Shui and J. Shu, Small Methods, 2019, 3, 1900119 CrossRef
- C. Chen, G. Quek, H. Liu, L. Bannenberg, R. Li, J. Choi, D. Ren, R. J. Vázquez, B. Boshuizen, B.-O. Fimland, S. Fleischmann, M. Wagemaker, D. Jiang, G. C. Bazan and X. Wang, Adv. Energy Mater., 2024, 14, 2402715 CrossRef CAS
- T. B. Sobyra, T. S. Mathis, Y. Gogotsi and P. Fenter, ACS Appl. Mater. Interfaces, 2021, 13, 43597–43605 CrossRef CAS PubMed
- T. B. Sobyra, K. Matthews, T. S. Mathis, Y. Gogotsi and P. Fenter, ACS Energy Lett., 2022, 7, 3612–3617 CrossRef CAS
- X. Wang and L. Bannenberg, MRS Bull., 2021, 46, 755–766 Search PubMed
- C. Yuan, C. Chen, Z. Yang, J. Cheng, J. Weng, S. Tan, R. Hou, T. Cao, Z. Tang, W. Chen, B. Xu, X. Wang and J. Tang, ACS Appl. Mater. Interfaces, 2024, 16, 55189–55197 CAS
- Z. Lin, P. Rozier, B. Duployer, P.-L. Taberna, B. Anasori, Y. Gogotsi and P. Simon, Electrochem. Commun., 2016, 72, 50–53 Search PubMed
- X. Wang, T. S. Mathis, K. Li, Z. Lin, L. Vlcek, T. Torita, N. C. Osti, C. Hatter, P. Urbankowski, A. Sarycheva, M. Tyagi, E. Mamontov, P. Simon and Y. Gogotsi, Nat. Energy, 2019, 4, 241–248 Search PubMed
- X. Ou, J. Li, F. Zheng, P. Wu, Q. Pan, X. Xiong, C. Yang and M. Liu, J. Power Sources, 2017, 343, 483–491 Search PubMed
- S. K. Sen, U. C. Barman, M. S. Manir, P. Mondal, S. Dutta, M. Paul, M. A. M. Chowdhury and M. A. Hakim, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2020, 11, 025004 CAS
- D. Y. Zemlyanov, M. Jespersen, D. N. Zakharov, J. Hu, R. Paul, A. Kumar, S. Pacley, N. Glavin, D. Saenz, K. C. Smith, T. S. Fisher and A. A. Voevodin, Nanotechnology, 2018, 29, 115705 Search PubMed
- I. Bertóti, M. Mohai and K. László, Carbon, 2015, 84, 185–196 CrossRef
- W. Eom, H. Shin and T. H. Han, Appl. Phys. Lett., 2023, 122, 211601 CrossRef CAS
- L. Wang, X. Zhang, Y. Xu, C. Li, W. Liu, S. Yi, K. Wang, X. Sun, Z.-S. Wu and Y. Ma, Adv. Funct. Mater., 2021, 31, 2104286 CrossRef CAS
- Y. Yan, S. Liang, X. Wang, M. Zhang, S.-M. Hao, X. Cui, Z. Li and Z. Lin, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2110036118 Search PubMed
- L.-Å. Näslund, M.-H. Mikkelä, E. Kokkonen and M. Magnuson, 2D Mater., 2021, 8, 045026 CrossRef
- C. Ye, D. Chao, J. Shan, H. Li, K. Davey and S.-Z. Qiao, Matter, 2020, 2, 323–344 CrossRef CAS
- C. Meng, P. Das, X. Shi, Q. Fu, K. Müllen and Z.-S. Wu, Small Sci., 2021, 1, 2000076 Search PubMed
- A. Benayad, J. E. Morales-Ugarte, C. C. Santini and R. Bouchet, J. Phys. Chem. A, 2021, 125, 1069–1081 CrossRef CAS PubMed
- L. G. Bulusheva, M. A. Kanygin, V. E. Arkhipov, K. M. Popov, Yu. V. Fedoseeva, D. A. Smirnov and A. V. Okotrub, J. Phys. Chem. C, 2017, 121, 5108–5114 Search PubMed
- J. Wen, Y. Huang, J. Duan, Y. Wu, W. Luo, L. Zhou, C. Hu, L. Huang, X. Zheng, W. Yang, Z. Wen and Y. Huang, ACS Nano, 2019, 13, 14549–14556 CrossRef CAS PubMed
- C.-Y. Tang, R. T. Haasch and S. J. Dillon, Chem. Commun., 2016, 52, 13257–13260 RSC
- J. Kruusma, A. Tõnisoo, R. Pärna, E. Nõmmiste, I. Kuusik, M. Vahtrus, I. Tallo, T. Romann and E. Lust, J. Electrochem. Soc., 2017, 164, A3393 Search PubMed
- W. Zaman, R. A. Matsumoto, M. W. Thompson, Y.-H. Liu, Y. Bootwala, M. B. Dixit, S. Nemsak, E. Crumlin, M. C. Hatzell, P. T. Cummings and K. B. Hatzell, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2108325118 CrossRef CAS PubMed
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS
- A. M. Tripathi, W.-N. Su and B.-J. Hwang, Chem. Soc. Rev., 2018, 47, 736–851 RSC
- C. Chen, R. Guo, S. Ganapathy, B. Terpstra, H. Wang, Z. Lei, F. Ooms, B. Boshuizen, M. Wagemaker, L. J. Bannenberg and X. Wang, Small, 2025, 2407226 CrossRef PubMed
- C. Wang, S. Chen and L. Song, Adv. Funct. Mater., 2020, 30, 2000869 CrossRef CAS
- M. Pin, J. Choi, J. H. Chang, A. S. Schenk, J. Han, S. Wacławek, Y. Kim and J. Y. Cheong, Energy Storage Mater., 2024, 73, 103798 CrossRef
- X. Wang, S.-M. Bak, M. Han, C. E. Shuck, C. McHugh, K. Li, J. Li, J. Tang and Y. Gogotsi, ACS Energy Lett., 2022, 7, 30–35 Search PubMed
- A. Yang, G. Zhou, X. Kong, R. A. Vilá, A. Pei, Y. Wu, X. Yu, X. Zheng, C.-L. Wu, B. Liu, H. Chen, Y. Xu, D. Chen, Y. Li, S. Fakra, H. Y. Hwang, J. Qin, S. Chu and Y. Cui, Nat. Nanotechnol., 2020, 15, 231–237 Search PubMed
- C. Wang, S. Chen, H. Xie, S. Wei, C. Wu and L. Song, Adv. Energy Mater., 2019, 9, 1802977 Search PubMed
- M. R. Lukatskaya, S.-M. Bak, X. Yu, X.-Q. Yang, M. W. Barsoum and Y. Gogotsi, Adv. Energy Mater., 2015, 5, 1500589 CrossRef
- A. Sugahara, Y. Ando, S. Kajiyama, K. Yazawa, K. Gotoh, M. Otani, M. Okubo and A. Yamada, Nat. Commun., 2019, 10, 850 CrossRef PubMed
- W. Li, Z. Wang, F. Zhao, M. Li, X. Gao, Y. Zhao, J. Wang, J. Zhou, Y. Hu, Q. Xiao, X. Cui, M. J. Eslamibidgoli, M. H. Eikerling, R. Li, F. Brandys, R. Divigalpitiya, T.-K. Sham and X. Sun, Chem. Mater., 2020, 32, 1272–1280 CrossRef CAS
- J. Wang, Y. K. Chen-Wiegart and J. Wang, Chem. Commun., 2013, 49, 6480–6482 RSC
- O. Mashtalir, M. R. Lukatskaya, A. I. Kolesnikov, E. Raymundo-Piñero, M. Naguib, M. W. Barsoum and Y. Gogotsi, Nanoscale, 2016, 8, 9128–9133 RSC
- W. S. Glaunsinger, M. J. McKelvy, E. M. Larson, R. B. Von Dreele, J. Eckert and N. L. Ross, Solid State Ionics, 1989, 34, 281–286 CrossRef CAS
- Y. Xie, M. Naguib, V. N. Mochalin, M. W. Barsoum, Y. Gogotsi, X. Yu, K.-W. Nam, X.-Q. Yang, A. I. Kolesnikov and P. R. C. Kent, J. Am. Chem. Soc., 2014, 136, 6385–6394 CrossRef CAS PubMed
- N. C. Osti, M. Naguib, K. Ganeshan, Y. K. Shin, A. Ostadhossein, A. C. T. van Duin, Y. Cheng, L. L. Daemen, Y. Gogotsi, E. Mamontov and A. I. Kolesnikov, Phys. Rev. Mater., 2017, 1, 065406 CrossRef
- N. C. Osti, M. W. Thompson, K. L. Van Aken, M. Alhabeb, M. Tyagi, J.-K. Keum, P. T. Cummings, Y. Gogotsi and E. Mamontov, J. Phys. Chem. C, 2018, 122, 27561–27566 CrossRef CAS
- W. Sun, H.-W. Wang, L. Vlcek, J. Peng, A. B. Brady, N. C. Osti, E. Mamontov, M. Tyagi, J. Nanda, S. G. Greenbaum, P. R. C. Kent and M. Naguib, Adv. Mater. Interfaces, 2020, 7, 1902207 CrossRef CAS
- M. L. Martins, T. S. Mathis, X. Wang, L. L. Daemen, Y. Gogotsi and E. Mamontov, Phys. Rev. Mater., 2022, 6, 034001 CrossRef CAS
- N. C. Osti, M. Naguib, A. Ostadhossein, Y. Xie, P. R. C. Kent, B. Dyatkin, G. Rother, W. T. Heller, A. C. T. van Duin, Y. Gogotsi and E. Mamontov, ACS Appl. Mater. Interfaces, 2016, 8, 8859–8863 CrossRef CAS PubMed
- H.-W. Wang, M. Naguib, K. Page, D. J. Wesolowski and Y. Gogotsi, Chem. Mater., 2016, 28, 349–359 CrossRef CAS
- O. Dolotko, A. Senyshyn, M. J. Mühlbauer, K. Nikolowski and H. Ehrenberg, J. Power Sources, 2014, 255, 197–203 CrossRef CAS
- A. Senyshyn, M. J. Mühlbauer, K. Nikolowski, T. Pirling and H. Ehrenberg, J. Power Sources, 2012, 203, 126–129 CrossRef CAS
- B. Vadlamani, K. An, M. Jagannathan and K. S. R. Chandran, J. Electrochem. Soc., 2014, 161, A1731 CrossRef CAS
- D. Mikhailova, N. N. Bramnik, K. G. Bramnik, P. Reichel, S. Oswald, A. Senyshyn, D. M. Trots and H. Ehrenberg, Chem. Mater., 2011, 23, 3429–3441 CrossRef CAS
- N. Sharma, G. Du, A. J. Studer, Z. Guo and V. K. Peterson, Solid State Ionics, 2011, 199–200, 37–43 CrossRef CAS
- H. Katzke and W. Depmeier, Adv. Mater. Res., 1994, 1–2, 129–136 CAS
- F. Linsenmann, M. Trunk, P. Rapp, L. Werner, R. Gernhäuser, R. Gilles, B. Märkisch, Z. Révay and H. A. Gasteiger, J. Electrochem. Soc., 2020, 167, 100554 CrossRef CAS
- M. Liu, Z. Cheng, K. Qian, T. Verhallen, C. Wang and M. Wagemaker, Chem. Mater., 2019, 31, 4564–4574 CrossRef CAS
- C. Wang, Y. Gong, J. Dai, L. Zhang, H. Xie, G. Pastel, B. Liu, E. Wachsman, H. Wang and L. Hu, J. Am. Chem. Soc., 2017, 139, 14257–14264 CrossRef CAS PubMed
- T. W. Verhallen, S. Lv and M. Wagemaker, Front. Energy Res., 2018, 6, 62 CrossRef
- S. C. Nagpure, R. G. Downing, B. Bhushan, S. S. Babu and L. Cao, Electrochim. Acta, 2011, 56, 4735–4743 CrossRef CAS
- A. C. Y. Yuen, T. B. Y. Chen, B. Lin, W. Yang, I. I. Kabir, I. M. De Cachinho Cordeiro, A. E. Whitten, J. Mata, B. Yu, H.-D. Lu and G. H. Yeoh, Compos., Part C: Open Access, 2021, 5, 100155 CAS
- S. Seidlmayer, J. Hattendorff, I. Buchberger, L. Karge, H. A. Gasteiger and R. Gilles, J. Electrochem. Soc., 2015, 162, A3116 CrossRef CAS
- J. E. Owejan, J. P. Owejan, S. C. DeCaluwe and J. A. Dura, Chem. Mater., 2012, 24, 2133–2140 CrossRef CAS
- K. L. Firestein, J. E. von Treifeldt, D. G. Kvashnin, J. F. S. Fernando, C. Zhang, A. G. Kvashnin, E. V. Podryabinkin, A. V. Shapeev, D. P. Siriwardena, P. B. Sorokin and D. Golberg, Nano Lett., 2020, 20, 5900–5908 CrossRef CAS PubMed
- J. Si, J. Yu, H. Lan, L. Niu, J. Luo, Y. Yu, L. Li, Y. Ding, M. Zeng and L. Fu, J. Am. Chem. Soc., 2023, 145, 3994–4002 CrossRef CAS PubMed
- Q. Gao, W.-Y. Tsai and N. Balke, Electrochem. Sci. Adv., 2022, 2, e2100038 CrossRef CAS
- M. Koltypin, Y. S. Cohen, B. Markovsky, Y. Cohen and D. Aurbach, Electrochem. Commun., 2002, 4, 17–23 CrossRef CAS
- W.-Y. Tsai, R. Wang, S. Boyd, V. Augustyn and N. Balke, Nano Energy, 2021, 81, 105592 CrossRef CAS
- J. M. Black, G. Feng, P. F. Fulvio, P. C. Hillesheim, S. Dai, Y. Gogotsi, P. T. Cummings, S. V. Kalinin and N. Balke, Adv. Energy Mater., 2014, 4, 1300683 CrossRef
- M. Mojtabavi, W.-Y. Tsai, A. VahidMohammadi, T. Zhang, Y. Gogotsi, N. Balke and M. Wanunu, Small, 2022, 18, 2105857 CrossRef CAS PubMed
- A. Lipatov, H. Lu, M. Alhabeb, B. Anasori, A. Gruverman, Y. Gogotsi and A. Sinitskii, Sci. Adv., 2018, 4, eaat0491 CrossRef PubMed
- P. Li, Z. Kang, Z. Zhang, Q. Liao, F. Rao, Y. Lu and Y. Zhang, Mater. Today, 2021, 51, 247–272 CrossRef CAS
- A. Zandiatashbar, G.-H. Lee, S. J. An, S. Lee, N. Mathew, M. Terrones, T. Hayashi, C. R. Picu, J. Hone and N. Koratkar, Nat. Commun., 2014, 5, 3186 CrossRef PubMed
- H. Pang, P. Huang, W. Zhuo, M. Li, C. Gao and D. Guo, Phys. Chem. Chem. Phys., 2019, 21, 7454–7461 RSC
- J. Come, Y. Xie, M. Naguib, S. Jesse, S. V. Kalinin, Y. Gogotsi, P. R. C. Kent and N. Balke, Adv. Energy Mater., 2016, 6, 1502290 CrossRef
- Q. Gao, W. Sun, P. Ilani-Kashkouli, A. Tselev, P. R. C. Kent, N. Kabengi, M. Naguib, M. Alhabeb, W.-Y. Tsai, A. P. Baddorf, J. Huang, S. Jesse, Y. Gogotsi and N. Balke, Energy Environ. Sci., 2020, 13, 2549–2558 RSC
- N. K. Thangavel, K. Mahankali and L. M. R. Arava, J. Electrochem. Soc., 2022, 169, 060501 CrossRef CAS
- S. Daboss, T. Philipp, K. Palanisamy, J. Flowers, H. S. Stein and C. Kranz, Electrochim. Acta, 2023, 453, 142345 CrossRef CAS
- I. Escher, M. Hahn, G. A. Ferrero and P. Adelhelm, Energy Technol., 2022, 10, 2101120 CrossRef PubMed
- M. Tobis, M. Elmanzalawy, J. Choi, E. Frąckowiak and S. Fleischmann, Batteries Supercaps, 2024, 7(11), e202400277 CrossRef CAS
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS
- N. Jäckel, S. P. Emge, B. Krüner, B. Roling and V. Presser, J. Phys. Chem. C, 2017, 121, 19120–19128 CrossRef
- N. Jäckel, B. Krüner, K. L. Van Aken, M. Alhabeb, B. Anasori, F. Kaasik, Y. Gogotsi and V. Presser, ACS Appl. Mater. Interfaces, 2016, 8, 32089–32093 CrossRef PubMed
- M. F. Koudahi, A. C. P. Tovar, F. Béguin and E. Frąckowiak, Energy Storage Mater., 2024, 72, 103771 CrossRef
- B. Jache and P. Adelhelm, Angew. Chem., Int. Ed., 2014, 53, 10169–10173 CrossRef CAS PubMed
- M. Goktas, C. Bolli, E. J. Berg, P. Novák, K. Pollok, F. Langenhorst, M. v. Roeder, O. Lenchuk, D. Mollenhauer and P. Adelhelm, Adv. Energy Mater., 2018, 8, 1702724 CrossRef
- M. Elmanzalawy, H. Song, M. Tobis, R. Leiter, J. Choi, H. Moon, W. Tsai, D. Jiang and S. Fleischmann, ChemRxiv, 2024. DOI:10.26434/chemrxiv-2024-bh4h4.
- I. Escher, M. Hahn, G. A. Ferrero and P. Adelhelm, Energy Technol., 2022, 10, 2101120 CrossRef PubMed
- J. Huesker, L. Froböse, A. Kwade, M. Winter and T. Placke, Electrochim. Acta, 2017, 257, 423–435 CrossRef CAS
- N. Shpigel, M. D. Levi and D. Aurbach, Energy Storage Mater., 2019, 21, 399–413 CrossRef
- N. Shpigel, M. D. Levi, S. Sigalov, L. Daikhin and D. Aurbach, Acc. Chem. Res., 2018, 51, 69–79 CrossRef CAS PubMed
- N. Shpigel, M. R. Lukatskaya, S. Sigalov, C. E. Ren, P. Nayak, M. D. Levi, L. Daikhin, D. Aurbach and Y. Gogotsi, ACS Energy Lett., 2017, 2, 1407–1415 CrossRef CAS
- J. M. Griffin, A. C. Forse, W.-Y. Tsai, P.-L. Taberna, P. Simon and C. P. Grey, Nat. Mater., 2015, 14, 812–819 CrossRef CAS PubMed
- B. Reif, S. E. Ashbrook, L. Emsley and M. Hong, Nat. Rev. Methods Primers, 2021, 1, 1–23 CrossRef
- K. J. Harris, M. Bugnet, M. Naguib, M. W. Barsoum and G. R. Goward, J. Phys. Chem. C, 2015, 119, 13713–13720 CrossRef CAS
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC
- T. Kobayashi, Y. Sun, K. Prenger, D. Jiang, M. Naguib and M. Pruski, J. Phys. Chem. C, 2020, 124, 13649–13655 CrossRef CAS
- M. Anayee, N. Kurra, M. Alhabeb, M. Seredych, M. N. Hedhili, A.-H. Emwas, H. N. Alshareef, B. Anasori and Y. Gogotsi, Chem. Commun., 2020, 56, 6090–6093 RSC
- K. J. Griffith, M. A. Hope, P. J. Reeves, M. Anayee, Y. Gogotsi and C. P. Grey, J. Am. Chem. Soc., 2020, 142, 18924–18935 CrossRef CAS PubMed
- F. Brette, D. Kourati, M. Paris, L. Loupias, S. Célérier, T. Cabioc'h, M. Deschamps, F. Boucher and V. Mauchamp, J. Am. Chem. Soc., 2023, 145, 4003–4014 CrossRef CAS PubMed
- S. Kajiyama, L. Szabova, K. Sodeyama, H. Iinuma, R. Morita, K. Gotoh, Y. Tateyama, M. Okubo and A. Yamada, ACS Nano, 2016, 10, 3334–3341 CrossRef CAS PubMed
- C. P. Slichter, Annu. Rev. Phys. Chem., 1986, 37, 25–51 CrossRef CAS
- P. A. Maughan, L. Bouscarrat, V. R. Seymour, S. Shao, S. J. Haigh, R. Dawson, N. Tapia-Ruiz and N. Bimbo, Nanoscale Adv., 2021, 3, 3145–3158 RSC
- O. Pecher, J. Carretero-González, K. J. Griffith and C. P. Grey, Chem. Mater., 2017, 29, 213–242 CrossRef CAS
- Z. Zhu, R. Luo and E. W. Zhao, Magn. Reson. Lett., 2024, 4, 100096 CrossRef CAS
- B. Key, R. Bhattacharyya, M. Morcrette, V. Seznéc, J.-M. Tarascon and C. P. Grey, J. Am. Chem. Soc., 2009, 131, 9239–9249 CrossRef CAS PubMed
- E. G. Sorte, N. A. Banek, M. J. Wagner, T. M. Alam and Y. J. Tong, ChemElectroChem, 2018, 5, 2336–2340 CrossRef CAS
- A. R. Aguilera, B. MacMillan, S. Krachkovskiy, K. J. Sanders, F. Alkhayri, C. A. Dyker, G. R. Goward and B. J. Balcom, J. Magn. Reson., 2021, 325, 106943 CrossRef CAS PubMed
- R. Luo, H. J. W. G. Janssen, A. P. M. Kentgens and E. W. Zhao, J. Magn. Reson., 2024, 361, 107666 CrossRef CAS PubMed
- E. W. Zhao, T. Liu, E. Jónsson, J. Lee, I. Temprano, R. B. Jethwa, A. Wang, H. Smith, J. Carretero-González, Q. Song and C. P. Grey, Nature, 2020, 579, 224–228 CrossRef CAS PubMed
- B. J. Walder, M. S. Conradi, J. J. Borchardt, L. C. Merrill, E. G. Sorte, E. J. Deichmann, T. M. Anderson, T. M. Alam and K. L. Harrison, Sci. Adv., 2021, 7, eabg8298 CrossRef CAS PubMed
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC
- S. Chandrashekar, N. M. Trease, H. J. Chang, L.-S. Du, C. P. Grey and A. Jerschow, Nat. Mater., 2012, 11, 311–315 CrossRef CAS PubMed
- J. M. Bray, C. L. Doswell, G. E. Pavlovskaya, L. Chen, B. Kishore, H. Au, H. Alptekin, E. Kendrick, M.-M. Titirici, T. Meersmann and M. M. Britton, Nat. Commun., 2020, 11, 2083 CrossRef CAS PubMed
- Y. Xiang, G. Zheng, Z. Liang, Y. Jin, X. Liu, S. Chen, K. Zhou, J. Zhu, M. Lin, H. He, J. Wan, S. Yu, G. Zhong, R. Fu, Y. Li and Y. Yang, Nat. Nanotechnol., 2020, 15, 883–890 CrossRef CAS PubMed
- A. I. Freytag, A. D. Pauric, S. A. Krachkovskiy and G. R. Goward, J. Am. Chem. Soc., 2019, 141, 13758–13761 CrossRef CAS PubMed
- ePROBE, https://eprobe.tech/, (accessed December 19, 2024).
- S. Haber and M. Leskes, Solid State Nucl. Magn. Reson., 2022, 117, 101763 CrossRef CAS PubMed
- G. A. de Wijs, R. Laskowski, P. Blaha, R. W. A. Havenith, G. Kresse and M. Marsman, J. Chem. Phys., 2017, 146, 064115 CrossRef CAS PubMed
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed
- E. W. Zhao, E. Jónsson, R. B. Jethwa, D. Hey, D. Lyu, A. Brookfield, P. A. A. Klusener, D. Collison and C. P. Grey, J. Am. Chem. Soc., 2021, 143, 1885–1895 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |