
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Removable dialkoxybenzyl linker for enhanced HPLC purification of peptide hydrazides†
Kohei
Sato
 *abcd,
Takaya
Yamamoto
a,
Tetsuo
Narumi
abcd and
Nobuyuki
Mase
abcd
*abcd,
Takaya
Yamamoto
a,
Tetsuo
Narumi
abcd and
Nobuyuki
Mase
abcd
aDepartment of Engineering, Graduate School of Integrated Science and Technology, Shizuoka University, 3-5-1 Johoku, Hamamatsu, Shizuoka 432-8561, Japan. E-mail: sato.kohei@shizuoka.ac.jp
bDepartment of Applied Chemistry and Biochemical Engineering, Faculty of Engineering, Shizuoka University, 3-5-1 Johoku, Hamamatsu, Shizuoka 432-8561, Japan
cGraduate School of Science and Technology, Shizuoka University, 3-5-1 Johoku, Hamamatsu, Shizuoka 432-8561, Japan
dResearch Institute of Green Science and Technology, Shizuoka University, 3-5-1 Johoku, Hamamatsu, Shizuoka 432-8561, Japan
First published on 6th January 2025
Abstract
High-performance liquid chromatography (HPLC) plays a crucial role in purifying peptides and proteins and monitoring their reactions. Peptide hydrazides are widely employed intermediates in modern peptide/protein chemistry. However, they often exhibit peak tailing during HPLC purification and analysis. Herein, we describe a dialkoxybenzyl-linker approach to improve HPLC performance in the purification and analysis of peptide hydrazides. A dialkoxybenzyl linker is designed for the solid-phase synthesis of peptide hydrazides, enabling selective cleavage under controlled global deprotection conditions to afford alkylated or free hydrazides. The peptides with this linker exhibit superior HPLC peak shapes, even on degraded columns, compared with those of free hydrazides. In addition, the dialkoxybenzyl linker enhances the overall yield in solid-phase peptide synthesis compared with that achieved using a conventional trityl-based linker. The utility of this linker is validated by the successful synthesis of ubiquitin from three peptide segments using native chemical ligation, achieving higher isolated yields of the peptide segments because of the improved purification efficiency. Thus, this approach is a powerful tool for the synthesis and purification of peptide hydrazides. In conclusion, the dialkoxybenzyl linker enhances HPLC peak symmetry, separation, and solid-phase peptide synthesis yields for peptide hydrazides, providing a practical solution to key purification challenges in peptide chemistry.
Introduction
The advent of high-performance liquid chromatography (HPLC) in the field of peptide chemistry has significantly impacted reaction monitoring, purity determination, and purification processes.1 Among several separation modes, reverse-phase chromatography is preferred for analysis because of its excellent resolution and reproducibility. Solid-phase peptide synthesis (SPPS) is commonly used for peptide production, necessitating the isolation of the desired peptide from the final crude product after cleavage since intermediate products cannot be separated during the process.2The crude material includes byproducts such as truncated, racemized, oxidized, or alkylated peptides, which often elute at retention times very similar to that of the desired product. Thus, the conditions that exhibit symmetrical and sharp peak shapes in HPLC must be explored to separate these byproducts. This requirement becomes especially significant in the production of high-purity products, such as peptide-based pharmaceuticals.3 However, peptide properties and column degradation can lead to tailing peaks, causing peaks to overlap and become inseparable, thereby hindering effective analysis and purification. Therefore, methods for maintaining good HPLC peak shapes for peptide materials are required.
Peptide Cα-hydrazides are valuable units due to their facile accessibility and unique reactivity, making them ideal intermediates for further transformations, such as native chemical ligation (NCL) to construct larger proteins,4,5 surface immobilisation,6 and solubilising tag attachment.7,8 In particular, the use of peptide hydrazides as a thioester precursor for NCL has played a significantly important role, replacing the traditional synthesis of peptide thioester through Boc SPPS.9
A peptide possessing a hydrazide moiety at the C-terminus was synthesised using various methods (Fig. 1A). Peptide hydrazides can be prepared using hydrazine-incorporated resins based on Wang, Cl-trityl (Trt), or Cl-Trt(2-Cl) resins, yielding the desired product during global deprotection.5,10 Other methods to obtain peptide hydrazides include post-chain-assembly photolysis11 and the on-resin hydrazinolysis of C-terminal ester-linked polypeptides.12 Hydrazinolysis methods for activated species have also been developed, including the hydrazinolysis of Gly–Cys or His–Cys motifs13 and Ser–Xaa–His–Zaa motifs in the presence of Ni(II).14 Importantly, both hydrazinolysis protocols are applicable to recombinant peptide/protein substrates, as their reactive motifs consist exclusively of proteinogenic residues. Recombinant synthesis of peptide/protein hydrazides has also been developed using intein-fusion5 or α-hydroxy acid-incorporating proteins.15 Peptide hydrazides synthesized by these methods are typically obtained as unsubstituted free hydrazides.
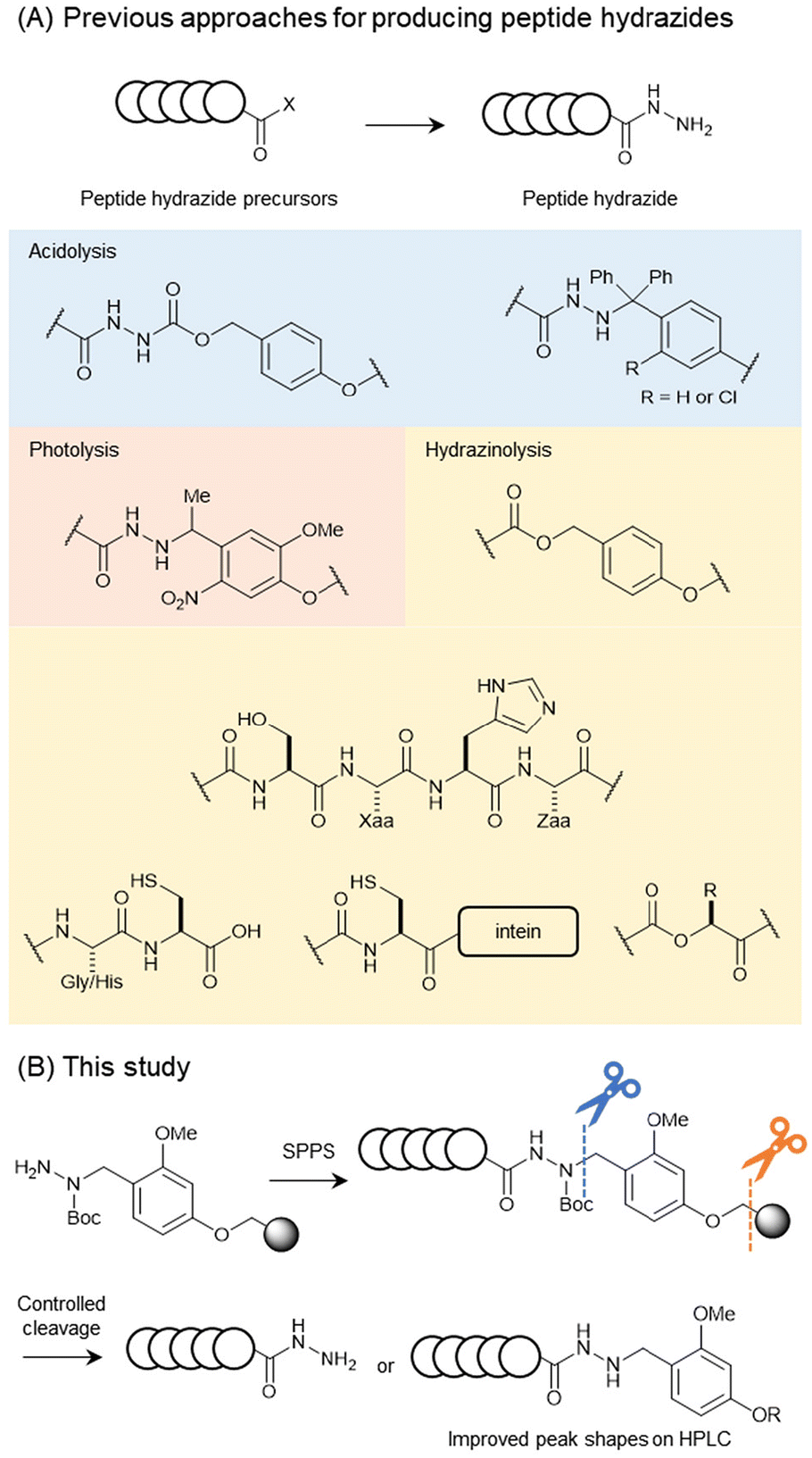 | ||
| Fig. 1 Various methods for preparing peptide hydrazides. | ||
The prepared peptide hydrazides are usually analysed and purified using reverse-phase HPLC. The peak shapes of peptide hydrazides in HPLC are sensitive to column conditions and often show tailing peaks. In our research on the reductive N-alkylation of peptide hydrazides with anisaldehyde, we discovered that alkylated products exhibit sharper HPLC peaks with higher symmetry.7 In this study, we investigate the alkylation impact on the peak shapes of peptide hydrazides. Encouraged by these results, we synthesised a linker for peptide hydrazides with a dialkoxybenzyl moiety at the terminal nitrogen atom, which exhibited controllable removal under different conditions (Fig. 1B). Then, we evaluated the peak shapes of the peptides with and without the linker moiety and discovered that the peptide hydrazides bearing the linker showed better peak shapes than those of the free hydrazides, even on a degraded C18 column. The applicability of the developed linker was validated through yield comparison with a conventional trityl (Trt) type linker and the synthesis of ubiquitin from the three peptide segments.
Results and discussion
Evaluation of HPLC peak shapes
To investigate the impact of alkylation on the peak shapes of peptide hydrazides, we calculated and compared the symmetry factors of free and alkyl hydrazides from previously reported data (Fig. 2).7 A symmetry factor of 1 indicated a perfectly symmetrical peak, values below 1 indicated leading peaks, and values above 1 indicated tailing peaks. Free hydrazides exhibited symmetry factors greater than 1, indicating tailing peaks, whereas the corresponding alkyl hydrazides showed smaller symmetry factors. This suggested that alkylation reduced HPLC peak tailing and improved the HPLC profile.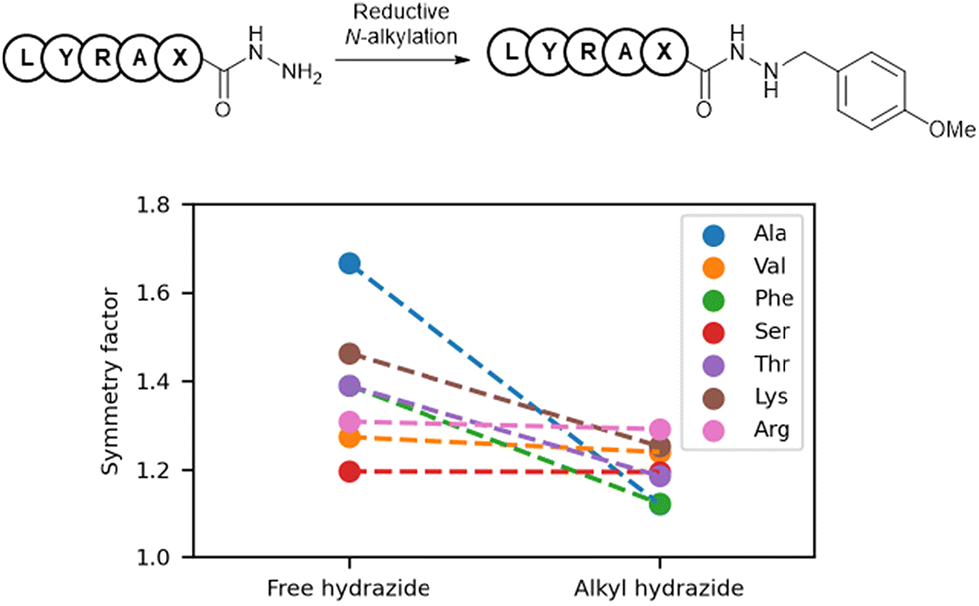 | ||
| Fig. 2 Comparison of the symmetry factors between free and alkyl hydrazides. | ||
In reverse-phase HPLC using silica gel columns attached to hydrophobic ligands, one cause of peak tailing was the gradual detachment of the alkyl groups from the stationary phase, resulting in the formation of silanol groups. Because hydrazides are weakly basic functional groups, their interactions with silanol groups may have caused peak tailing. It has been reported that the basicity of hydrazines is reduced by alkylation (pKa = 8.07 for NH2NH2·HCl; 7.87 for MeNHNH2·HCl),16 suggesting that the basicity of alkyl hydrazides may also be decreased. One potential reason for the improvement in peak tailing with alkyl hydrazides was the reduction in basicity, which led to decreased undesirable interactions with the silanol groups in the stationary phase.
Design and synthesis of the dialkoxybenzyl linker
Encouraged by the above results of the HPLC peak shape, we envisioned that the direct synthesis of alkylated hydrazides with a removable moiety at an appropriate synthetic stage would be a powerful tool, leading to easy peak separation during HPLC analysis and purification. A 2,4-dialkoxybenzyl (Dab) group on hydrazides can be removed to generate free hydrazides upon trifluoroacetic acid (TFA) treatment.8 In this case, elevated temperatures (37 °C) were necessary to complete the removal.Therefore, we reasoned that a Dab-type linker would have potential as a linker for the solid-phase synthesis of peptide alkyl hydrazides with tunable reactivities by temperature control.
The Dab-type linker was synthesised as shown in Scheme 1. The phenol derivative 1 was alkylated with ethyl 4-bromobutyrate, and the subsequent hydrolysis of the ester afforded carboxylic acid 3. Reductive amination with Fmoc carbazate afforded the linker compound 4. As an initial attempt, linker 4 was incorporated onto a Rink amide ChemMatrix resin using N,N′-diisopropylcarbodiimide and Oxyma, and a model peptide sequence (H-LYRAG) was elongated (Fig. 3A). However, HPLC analysis of the crude material after global deprotection revealed the doubly acylated peptide hydrazide 7 because of the nucleophilicity of both nitrogen atoms in the linker (Fig. 3B). Thus, to suppress undesired acylation, Boc protection was introduced, affording the Dab linker 5 in an overall yield of 62% from 1 without the need for column chromatography purification. When applied to SPPS, this linker yielded the desired peptide, hydrazide 6, without producing the doubly acylated peptide (Fig. 3C).
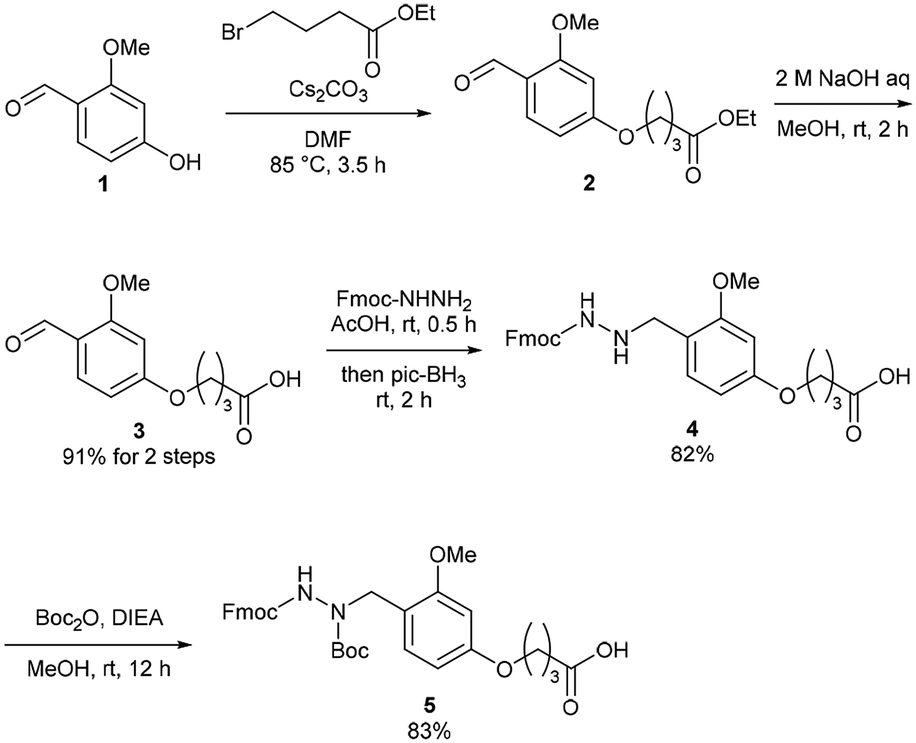 | ||
| Scheme 1 Synthesis of a 2,4-dialkoxybenzyl (Dab)-type linker. | ||
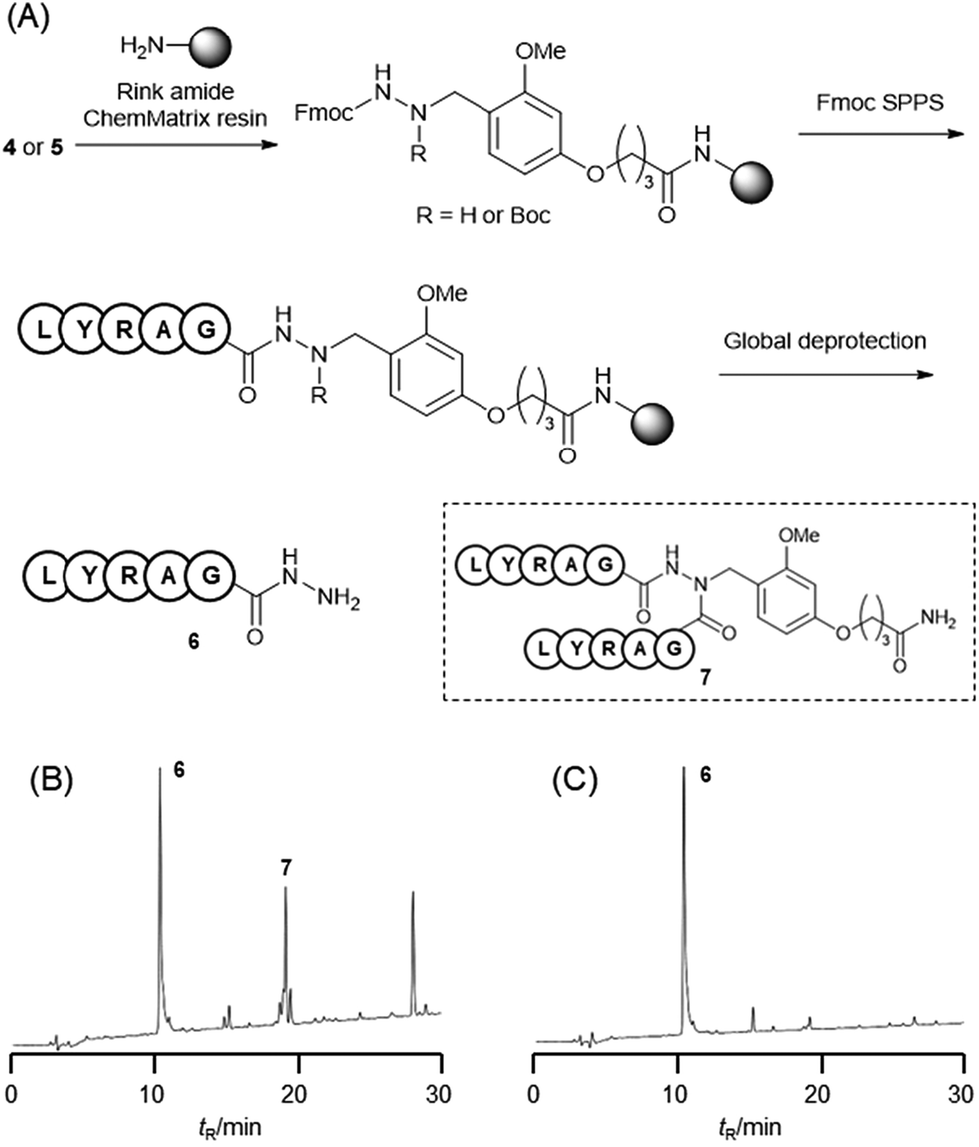 | ||
Fig. 3 (A) Solid-phase peptide synthesis using linker 4 or 5. UV trace of a crude peptide synthesized using linker 4 (B) and linker 5 (C). Global deprotection conditions: TFA–TIS–H2O = 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5 (v/v) at 37 °C for 2 h. :2.5:2.5 (v/v) at 37 °C for 2 h. | ||
Reactivity of the Dab linker
To evaluate the reactivity under global deprotection conditions, linker 5 was introduced onto the Rink Amide ChemMatrix resin using the usual carbodiimide chemistry, and a peptide sequence (H-LYRAG) was elongated using a conventional Fmoc SPPS protocol. The resulting resin was treated with TFA–triisopropylsilane (TIS)–H2O (95:2.5:2.5, (v/v)) at 25 °C for 2 h. HPLC analysis of the reaction mixture revealed the free hydrazide 6, whereas incomplete cleavage of the Dab linker was observed (Table 1, entry 1). Global deprotection at an increased temperature of 37 °C proceeded completely to yield free hydrazide 6 without a detectable amount of the Dab-linked hydrazide 8 (entry 2). Conversely, at a low temperature of 0 °C, the Dab moiety remained intact, whereas incomplete deprotection of the Pbf group on arginine was observed (entry 3). The thioanisole-containing deprotection cocktail accelerated Pbf removal,17 and 3 h of reaction yielded hydrazide 8 in 93% yield (entries 4 and 5).
|
|
||||||
|---|---|---|---|---|---|---|
| HPLC area ratioa (%) | ||||||
| Entry | Conditionb | Temp. (°C) | Time (h) | 6 | 8 | 9 |
|
a Detected at 220 nm.
b Condition A: TFA–TIS–H2O = 95:2.5:2.5 (v/v); condition B: TFA–thioanisole–TIS–H2O = 80:15:2.5:2.5 (v/v); condition C: 1 M TMSBr–thioanisole in TFA containing 5% (v/v) m-cresol and 5% (v/v) 1,2-ethanedithiol.
c n.d. = not detected.
|
||||||
| 1 | A | 25 | 2 | 45 | 54 | n.d.c |
| 2 | A | 37 | 2 | 92 | n.d. | n.d. |
| 3 | A | 0 | 2 | 1 | 39 | 45 |
| 4 | B | 0 | 2 | 3 | 86 | 9 |
| 5 | B | 0 | 3 | 3 | 93 | 2 |
| 6 | C | 0 | 1 | n.d. | >99 | n.d. |
| 7 | C | 25 | 1 | n.d. | 93 | n.d. |
Under harsh conditions, using 1 M TMSBr–thioanisole in TFA at 0 °C, which is a known deprotection cocktail for removing benzyl-type protections,18 we encountered an unexpected result: the Dab linker remained intact during deprotection, while all other side-chain protections were completely removed (entry 6). This deprotection at 25 °C also resulted in the Dab moiety remaining, indicating that the effect was due to the deprotection cocktail rather than the temperature (entry 7).
Investigation of the role of TMSBr
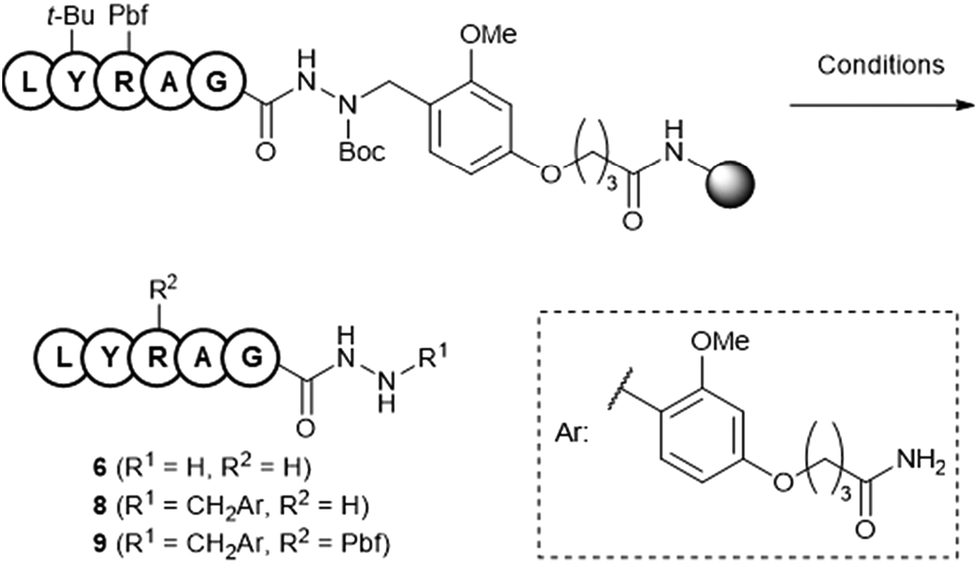
To investigate the role of the TMSBr-containing cocktail in maintaining the linker, NMR experiments using compound 4 were performed (Fig. 4A). In the 1H NMR spectra of compound 4 in TFA-d at 25 °C, two aromatic protons appeared at 6.51 and 6.59 ppm (Fig. 4B). However, these signals disappeared when 4 was dissolved in TFA-d containing 1 M TMSBr (Fig. 4C). This result indicated that DBr generated from TFA-d and TMSBr could protonate the aromatic ring (pKa = 0.23 for TFA; −9.0 for HBr), yielding the D-substituted compound. The D-substituted structure was confirmed using mass spectrometry (Fig. 4D). Cationic species derived from the protonation of the aromatic ring may have disturbed the deprotection of the benzyl group because of their lack of electron-donating properties (Fig. 4E).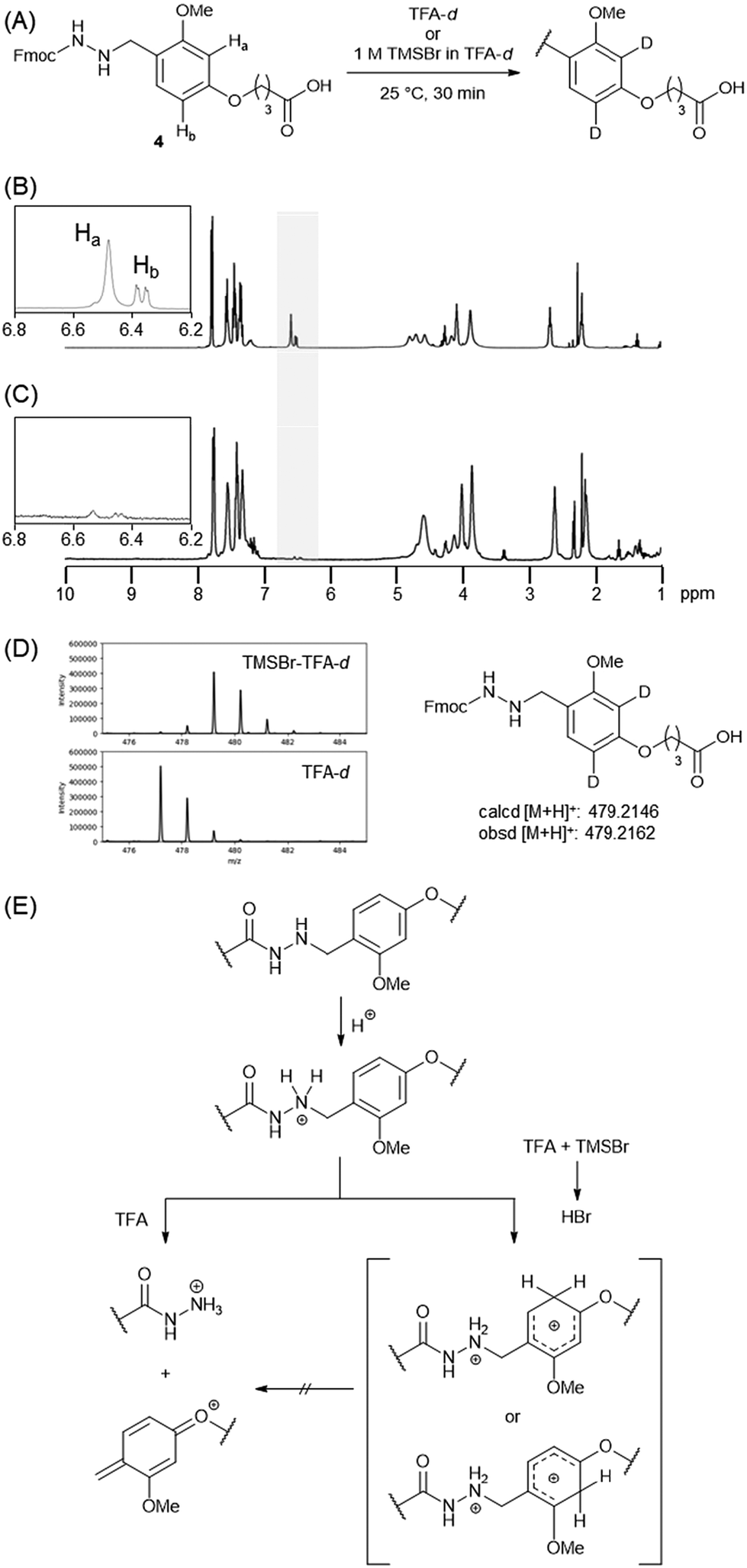 | ||
| Fig. 4 (A) NMR experiment with compound 4. (B) NMR spectra of 4 in TFA-d at 25 °C. (C) NMR spectra of 4 in 1 M TMSBr–TFA-d at 25 °C. (D) Mass spectra of 4. Top: treated with 1 M TMSBr–TFA-d; bottom: treated with TFA-d. (E) Possible role of TMSBr in maintaining the Dab linker during global deprotection. | ||
Evaluation of HPLC peak shapes using a degraded column
The tunable reactivity of the Dab linker during global deprotection was beneficial for HPLC purification. A peptide hydrazide without any substitution likely resulted in a broad peak in HPLC, and this behaviour was more apparent when a degraded column was used. This broadening led to reduced peak separation during HPLC purification, resulting in lower yields.We evaluated the HPLC peak shapes of hydrazides with and without the Dab linker. After coupling linker 5 to the Rink amide ChemMatrix resin, a myelin basic protein (MBP) 104–118 (H-GKGRGLSLSRFSWGA)19 was elongated. The peptidyl resin was treated with TFA–TIS–H2O at 37 °C to yield the free hydrazide or with 1 M TMSBr–thioanisole in TFA containing 5% m-cresol and 5% 1,2-ethanedithiol for the alkylated peptide (Fig. 5A). When using the new C18 column, we observed a good peak shape (symmetry factor S = 1.9 for free hydrazide and S = 1.6 for alkylated hydrazide; Fig. 5B). However, this behaviour was completely changed when using a degraded column treated with an aqueous solution of 0.1% TFA at 60 °C for several days. Alkylated hydrazide showed almost the same peak shape as S = 1.5, while free hydrazide was hardly detected (Fig. 5C and D). Peptide acids 13 and amide 14, which shared the same sequence, were eluted with the same peak shape as that of the alkylated hydrazide peptide (Fig. 5E and F). These results clearly showed that free peptide hydrazide was particularly sensitive to a degraded HPLC column, exhibiting a poor HPLC profile.
 | ||
| Fig. 5 (A) Preparation of the myelin basic protein 104–118 (MBP104–118) with and without the Dab moiety. (B) Symmetry factors of free or alkylated hydrazides using a degraded column. (C–F) UV trace of peptides using a degraded column treated with an aqueous solution of 0.1% TFA at 60 °C for 15 d. Spectra of (C) MBP104–118 free hydrazide 11, (D) MBP104–118 alkylated hydrazide 12, (E) MBP104–118 carboxylic acid 13, and (F) MBP104–118 amide 14. | ||
Comparison with a conventional Trt linker
We compared the synthetic efficiency and stability of the Dab linker with a widely used Trt-type linker.10 The MBP104–118 peptide was elongated on Fmoc–NHNH2-incorporated Cl-Trt(2-Cl) and Dab-incorporated Rink amide AM resins using an automated synthesiser at room temperature. The crude product yield was compared based on the weight of each peptidyl resin. The Dab linker afforded an almost quantitative yield, whereas the Trt linker exhibited only a 52% recovery, probably due to peptide loss during elongation, as the Trt group is more sensitive (Table 2, entries 1 and 2).20 Although the purity of the crude material using the Dab linker was lower than that using the Trt linker (59% for Dab; 71% for Trt), the higher recovery of the peptide material from the Dab linker resulted in an improved yield after the free hydrazide was isolated (34% for Dab; 21% for Trt).|
|
|||||
|---|---|---|---|---|---|
| Entry | Linker | Fmoc SPPS conditions | Crude yielda (%) | Crude purityb (%) | Isolated yieldc (%) |
| a (Weight of synthesised peptidyl resin)/(theoretical weight of resin with a fully protected peptide). b Determined by high-performance liquid chromatography at 220 nm after global deprotection. c Determined by the weight of the isolated peptide using preparative high-performance liquid chromatography. | |||||
| 1 | Trt | Rt | 52 | 71 | 21 |
| 2 | 5 | Rt | 97 | 59 | 34 |
| 3 | Trt | MW (75 °C) | 70 | 64 | 27 |
| 4 | 5 | MW (75 °C) | 97 | 65 | 39 |
A comparison under microwave-heating conditions (75 °C) was also performed, and the trend was identical to that observed under room-temperature conditions (entries 3 and 4). These results clearly indicated that the Dab linker could increase the overall yield of peptide hydrazides.
Synthesis of ubiquitin
To confirm the applicability of the new linker, ubiquitin was synthesised. Ubiquitin is a small protein that regulates a variety of cellular functions through modification with other proteins.21 The primary sequence consisting of 76 amino acid residues was divided into three peptide segments, namely Ub1–27, Ub28–45, and Ub46–76 (Fig. 6A). The alanine residues at the junction (Ala28 and Ala46) were substituted with cysteine to assemble the peptides using the NCL reaction. In our synthesis, we assembled three segments in the N-to-C direction using a Dab-attached hydrazide as the temporarily protected thioester equivalent. The isolated yield of the hydrazides was expected to improve by easy purification because of the better HPLC peak shapes compared with those of free hydrazides. The assembly of the three peptides, followed by desulfurization at the ligation junctions, yielded ubiquitin (Fig. 6C).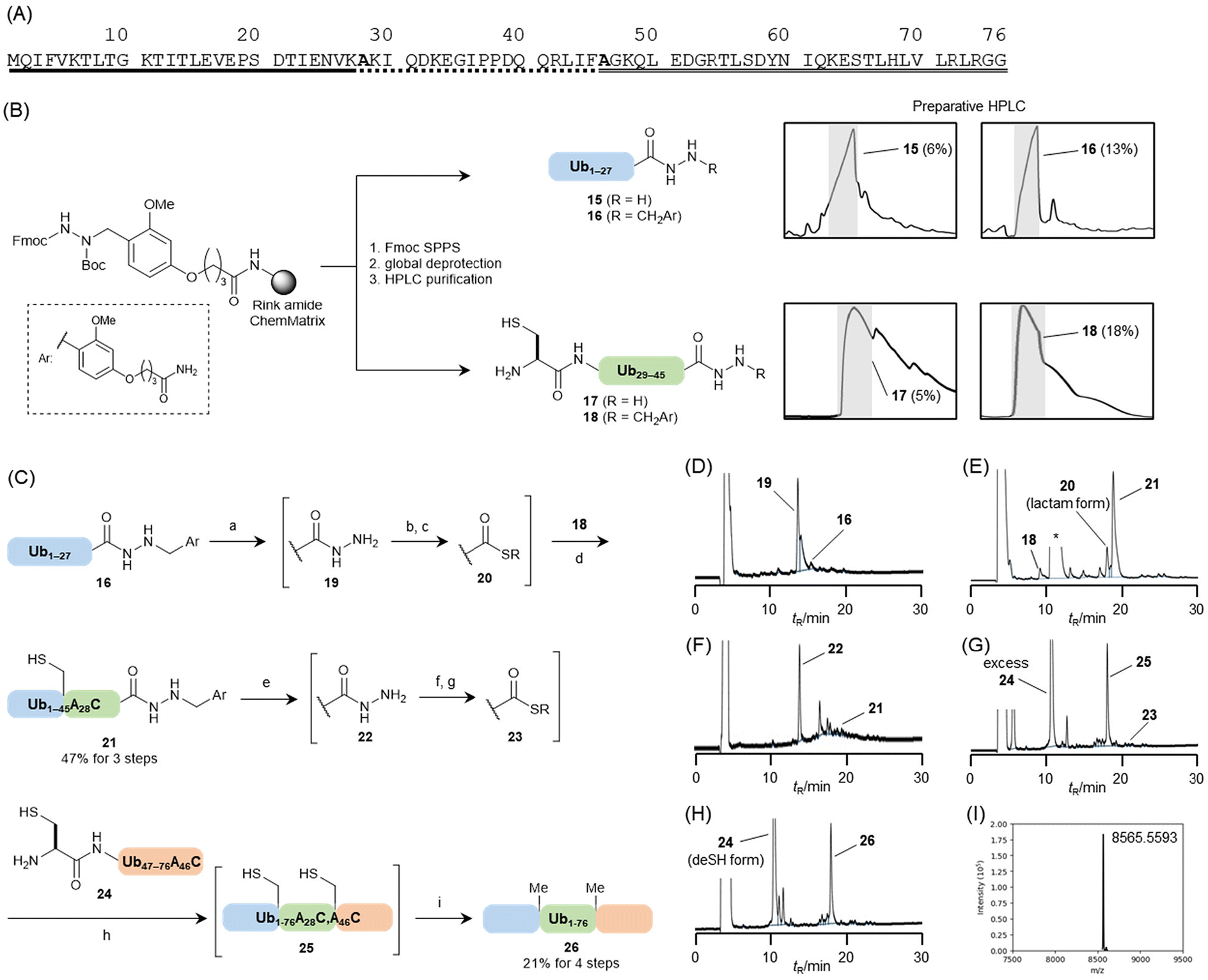 | ||
| Fig. 6 (A) Full sequence of ubiquitin, with underlined segments representing each peptide fragment. (B) Comparison of preparative HPLC for purifying Ub1–27 and Ub28–45A28C segments, with isolated yields shown in parentheses. (C) Synthetic scheme for ubiquitin assembly. Conditions: (a) TFA–TIS–H2O (94:3:3, (v/v)), 37 °C, 3 h; (b) addition of thioanisole and m-cresol (final ratio: TFA–thioanisole–m-cresol–TIS–H2O (80:10:5:2.5:2.5, (v/v))) followed by addition of NaNO2 aq, −10 °C, 30 min; (c) 6 M Gn·HCl, 0.1 M MPAA, 0.2 M Na phosphate buffer (pH 7.3); (d) addition of 18, 37 °C, 4 h; (e) TFA–TIS–H2O (94:3:3, (v/v)), 37 °C, 3 h; (f) addition of thioanisole and m-cresol (final ratio: TFA–thioanisole–m-cresol–TIS–H2O (80:10:5:2.5:2.5, (v/v))) followed by addition of NaNO2 aq, −10 °C, 30 min; (g) 6 M Gn·HCl, 0.1 M methyl thioglycolate, 10 mM TCEP·HCl, 0.2 M Na phosphate buffer (pH 7.3); (h) addition of 24, 37 °C, 2 h; (i) addition of 6 M Gn·HCl, 0.5 M TCEP·HCl, 0.2 M MESNa, 50 mM VA-044, 0.2 M Na phosphate buffer (pH 6.5, final concentration: 6 M Gn·HCl, 255 mM TCEP·HCl, 50 mM methyl thioglycolate, 0.1 M MESNa, 25 mM VA-044, 0.2 M Na phosphate), 37 °C, 10 h. (D) UV trace of Dab removal from 16 (3 h). (E) UV trace of NLC between 20 and 18 (4 h). Lactam formation of C-terminal Lys residue. *MPAA. (F) UV trace of Dab removal from 21 (3 h). (G) UV trace of NCL between 23 and 24 (2 h). (H) UV trace of desulfurization of 25 (10 h). (I) Deconvoluted mass spectrum of synthesized ubiquitin (26). Observed mass: 8565.5593; Calculated mass: 8565.8745 (average isotopes). | ||
The Dab linker 5 was introduced onto the Rink amide ChemMatrix resin, and each sequence corresponding to Ub1–27 or Ub28–45A28C was elongated on the resin. The resulting peptidyl resin was halved. Then, one was treated with TFA–TIS–H2O (95:2.5:2.5, (v/v)) at 37 °C for 2 h, and the other was treated with 1 M TMSBr–thioanisole in a TFA solution containing 5% (v/v) m-cresol and 5% (v/v) 1,2-ethanedithiol to yield free or Dab-attached hydrazides, respectively (Fig. 6B). For the Ub1–27 segments, each peptide with or without the Dab moiety was purified using preparative HPLC. In this case, both peptides were eluted as the main peak in the chromatogram. However, the free hydrazide exhibited a broader peak shape than that of the Dab-attached peptide (Fig. 6B). This resulted in an improved isolated yield of the Dab-attached hydrazide, which was twice that of the free hydrazide (13% for Dab; 6% for free).
For the Ub28–45A28C segments, the Dab-attached peptide showed sharper peaks on preparative HPLC, increasing the isolated yield by more than three times that of the free hydrazide segment (18% for Dab; 5% for free). These results clearly demonstrated that the alkylation of hydrazides could improve the HPLC peak shapes and purification efficacy, as anticipated.
The isolated Ub1–27–Dab segment (16) was subjected to Dab-group deprotection in TFA–TIS–H2O (94:3:3, (v/v)) at 37 °C. The reaction progress was monitored by HPLC analysis, which was completed within 3 h (Fig. 6D). The resulting free hydrazide (19) was directly converted to the corresponding thioester using hydrazide activation in TFA.22 In this protocol, thioanisole and m-cresol played critical roles by accelerating the reaction and suppressing nitration at a Tyr side chain, respectively. These reagents were added to the Dab-deprotection solution (final concentration: TFA–thioanisole–m-cresol–TIS–H2O (80:10:5:2.5:2.5, (v/v))). The mixture was treated with sodium nitrite to produce peptidyl azides, which were reacted with 4-mercaptophenylacetic acid (MPAA)23 in 6 M guanidine hydrochloride (Gn·HCl)–0.2 M sodium phosphate buffer (pH 7.3) containing the Ub28–45A28C–Dab segment (18) at 37 °C for 4 h, yielding the Ub1–45A28C–Dab segment (21) in an isolated yield of 47% (Fig. 6E). During this NCL, two equivalents of the Ub1–27 segment were used because this thioester segment formed a lactam derivative (Fig. 6E). Following Dab removal from Ub1–45A28C–Dab and conversion of the hydrazide to a peptidyl azide using the same procedure described earlier (Fig. 6F), the resulting peptidyl material was treated with 6 M Gn·HCl, 0.2 M Na2HPO4, 10 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl), and 100 mM methyl thioglycolate. This solution, containing Ub46–76A46C 24 prepared via a conventional Fmoc SPPS on an HMPB ChemMatrix resin, was added to the buffer solution at pH 7.3. Methyl thioglycolate was used as a thiol additive because of its compatibility with one-pot desulfurization conditions.24 The assembly reaction of the peptide segments was conducted at 37 °C for 2 h, yielding peptide 25 (Fig. 6G). Next, an equal-volume buffer solution (pH 6.1) containing 6 M Gn·HCl, 0.2 M Na2HPO4, 500 mM TCEP·HCl, and 200 mM sodium 2-mercaptoethanesulfonate (MESNa) was added to the reaction mixture. The radical initiator, 2,2′-azobis[2-(2-imidazolin-2-yl)propane]dihydrochloride (VA-044), was then introduced to a final concentration of 50 mM.25 After 10 h of reaction, all cysteine residues were converted to alanine, confirming the formation of ubiquitin (26), which was purified by HPLC and obtained in 21% yield (Fig. 6H). Characterisation of the synthesised ubiquitin was validated by MS analysis (Fig. 6I) and by comparing its circular dichroism spectrum with that of commercially available ubiquitin from bovine erythrocytes (ESI, Fig. S26†).
Conclusions
In this study, we developed a novel Dab linker to efficiently purify and synthesize peptide hydrazides. This linker exhibited controllable cleavage behaviour under selected global deprotection conditions. Through a systematic evaluation of the HPLC profiles of the peptide hydrazides, we confirmed the impact of N-alkylation on the HPLC analysis and purification of hydrazides. Peptide hydrazides with the Dab moiety showed improved HPLC peak symmetry and separation, even under degraded column conditions, and demonstrated superior yields in SPPS compared with those obtained using conventional linkers. The utility of this linker was demonstrated in the successful synthesis of ubiquitin from three peptide segments with enhanced purification efficiency. This approach provides a robust solution to overcome the analytical and preparative challenges associated with peptide hydrazides.Experimental
See the ESI† for details.Author contributions
KS: conceptualisation, funding acquisition, writing – original draft, writing – review, and editing. TY: investigation. TN and NM: writing – review and editing and supervision.Data availability
All data supporting the findings of this manuscript are available in the ESI,† which include detailed synthetic procedures, NMR spectra, MS spectra, HPLC chromatograms, and CD spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported in part by the JSPS KAKENHI (grant number 22K05349).References
- C. T. Mant, Y. Chen, Z. Yan, T. V. Popa, J. M. Kovacs, J. B. Mills, B. P. Tripet and R. S. Hodges, Peptide Characterization and Application Protocols, 2007, vol. 386, p. 3 Search PubMed.
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85, 2149 CrossRef CAS.
- C. De Luca, G. Lievore, D. Bozza, A. Buratti, A. Cavazzini, A. Ricci, M. Macis, W. Cabri, S. Felletti and M. Catani, Molecules, 2021, 26, 4688 CrossRef CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CrossRef CAS PubMed.
- G.-M. Fang, Y.-M. Li, F. Shen, Y.-C. Huang, J.-B. Li, Y. Lin, H.-K. Cui and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 7645 CrossRef CAS PubMed; S. Dong, J.-S. Zheng, Y. Li, H. Wang, G. Chen, Y. Chen, G. Fang, J. Guo, C. He, H. Hu, X. Li, Y. Li, Z. Li, M. Pan, S. Tang, C. Tian, P. Wang, B. Wu, C. Wu, J. Zhao and L. Liu, Sci. China: Chem., 2024, 67, 1060 CrossRef; Y.-K. Qi, J.-S. Zheng and L. Liu, Chem, 2024, 10, 2390 Search PubMed.
- R. Zitterbart and O. Seitz, Angew. Chem., Int. Ed., 2016, 55, 7252 CrossRef CAS PubMed.
- K. Sato, S. Tanaka, J. Wang, K. Ishikawa, S. Tsuda, T. Narumi, T. Yoshiya and N. Mase, Org. Lett., 2021, 23, 1653 CrossRef CAS.
- S. Tanaka, T. Narumi, N. Mase and K. Sato, Chem. Pharm. Bull., 2022, 70, 707 CrossRef CAS.
- V. Agouridas, O. El Mahdi, M. Cargoët and O. Melnyk, Bioorg. Med. Chem., 2017, 25, 4938 CrossRef CAS PubMed.
- G. Stavropoulos, D. Gatos, V. Magafa and K. Barlos, Lett. Pept. Sci., 1996, 2, 315 CrossRef CAS; J.-S. Zheng, S. Tang, Y. Guo, H.-N. Chang and L. Liu, ChemBioChem, 2012, 13, 542 CrossRef; Y.-C. Huang, C.-C. Chen, S.-J. Li, S. Gao, J. Shi and Y.-M. Li, Tetrahedron, 2014, 70, 2951 CrossRef; M. J. Bird and P. E. Dawson, Pept. Sci., 2022, 114, e24268 CrossRef.
- K. Qvortrup, V. V. Komnatnyy and T. E. Nielsen, Org. Lett., 2014, 16, 4782 CrossRef CAS.
- C. Bello, F. Kikul and C. F. W. Becker, J. Pept. Sci., 2015, 21, 201 CrossRef CAS.
- A. L. Adams, B. Cowper, R. E. Morgan, B. Premdjee, S. Caddick and D. Macmillan, Angew. Chem., Int. Ed., 2013, 52, 13062 CrossRef CAS PubMed.
- Y. Tsuda, A. Shigenaga, K. Tsuji, M. Denda, K. Sato, K. Kitakaze, T. Nakamura, T. Inokuma, K. Itoh and A. Otaka, ChemistryOpen, 2015, 4, 448 CrossRef CAS PubMed.
- Y.-M. Li, M.-Y. Yang, Y.-C. Huang, Y.-T. Li, P. R. Chen and L. Liu, ACS Chem. Biol., 2012, 7, 1015 CrossRef CAS PubMed.
- R. L. Hinman, J. Org. Chem., 1958, 23, 1587 CrossRef CAS.
- W. C. Chan and P. D. White, in Fmoc Solid Phase Peptide Synthesis, ed. W. C. Chan and P. D. White, Oxford, 1999, pp. 41–76 Search PubMed.
- N. Fujii, A. Otaka, N. Sugiyama, M. Hatano and H. Yajima, Chem. Pharm. Bull., 1987, 35, 3880 CrossRef CAS.
- E. H. Eylar, S. Brostoff, G. Hashim, J. Caccam and P. Burnett, J. Biol. Chem., 1971, 245, 5770 CrossRef.
- R. Subirós-Funosas, A. El-Faham and F. Albericio, Pept. Sci., 2012, 98, 89 CrossRef CAS; J. Spengler, A.-I. Fernandez-Llamazares and F. Albericio, ACS Comb. Sci., 2013, 15, 229 CrossRef PubMed.
- A. Hershko and A. Ciechanover, Annu. Rev. Biochem., 1998, 67, 425 CrossRef CAS PubMed; D. Komander and M. Rape, Annu. Rev. Biochem., 2012, 81, 203 CrossRef PubMed.
- K. Sato, S. Tanaka, K. Yamamoto, Y. Tashiro, T. Narumi and N. Mase, Chem. Commun., 2018, 54, 9127 RSC.
- E. C. B. Johnson and S. B. H. Kent, J. Am. Chem. Soc., 2006, 128, 6640 CrossRef CAS.
- Y.-C. Huang, C.-C. Chen, S. Gao, Y.-H. Wang, H. Xiao, F. Wang, C.-L. Tian and Y.-M. Li, Chem. – Eur. J., 2016, 22, 7623 CrossRef CAS PubMed.
- Q. Wan and S. J. Danishefsky, Angew. Chem., Int. Ed., 2007, 46, 9248 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of syntheses, including HPLC chromatograms, MS spectra, and NMR spectra. See DOI: https://doi.org/10.1039/d4ob01918k |
| This journal is © The Royal Society of Chemistry 2025 |