
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescence polarization assays to study carbohydrate–protein interactions
José L. de Paz
 * and
Pedro M. Nieto
*
* and
Pedro M. Nieto
*
Glycosystems Laboratory, Instituto de Investigaciones Químicas (IIQ), cicCartuja, CSIC and Universidad de Sevilla, Americo Vespucio, 49, 41092 Sevilla, Spain. E-mail: pedro.nieto@iiq.csic.es; jlpaz@iiq.csic.es
First published on 23rd January 2025
Abstract
Fluorescence polarization (FP) is a useful technique to study the interactions between carbohydrates and proteins in solution, by using standard equipment and minimal sample consumption. Here, we will review the most recent FP-based approaches in this field, including the study of carbohydrate–lectin, carbohydrate–enzyme and glycosaminoglycan–protein interactions. Advantages and limitations of this methodology will be discussed. To develop a FP procedure for studying carbohydrate–protein interactions, the main requirement is the design and synthesis of a suitable fluorescent glycan probe showing high affinity for the protein of interest. Different synthetic strategies employed for this purpose will be described, including the conjugation of 2-aminoethyl glycosides with amine-reactive fluorescein derivatives, the cycloaddition reaction between azido-functionalized saccharides and alkynylated fluorescent derivatives, and the reaction of the reducing end aldehyde group of an oligosaccharide with a hydrazide-containing fluorescein molecule. Competition FP experiments are particularly interesting because they enable the rapid screening of hundreds/thousands of non-labelled compounds for the discovery of molecules that block carbohydrate–protein binding, potentially modulating the subsequent biological processes.
 José L. de Paz | José L. de Paz received his PhD degree in 2000 at the Instituto de Investigaciones Químicas (IIQ), Sevilla (Spain), under the supervision of Prof. Manuel Martín-Lomas and Prof. José M. Lassaletta Simon. In 2004, he joined the group of Prof. Peter H. Seeberger at ETH Zurich where he worked on the preparation and use of carbohydrate microarrays. In 2007, he returned to Spain, joining Dr Pedro Nieto's group. In 2008, he became tenured scientist at IIQ. His current research interests are focused on the chemical synthesis of glycosaminoglycan oligosaccharides and mimetics with potential biological activities. |
 Pedro M. Nieto | Pedro M. Nieto holds a PhD in supramolecular chemistry from UAM (1994), conducted post-doctoral studies in protein-NMR in London and returned to Spain in 1997 joining the Carbohydrate Group in Sevilla and assuming the leadership of the structural studies by NMR and MD. In 2002, he became Research Scientist and stablished the Glycosaminoglycan team at the Glycosystems lab. He was promoted to Senior Researcher in 2010. His research main stream has been the 3D structure and dynamics of glycans and their transient complexes with growth factors and lectins. He also has been involved in Spanish Royal Society of Chemistry directive roles. |
Introduction
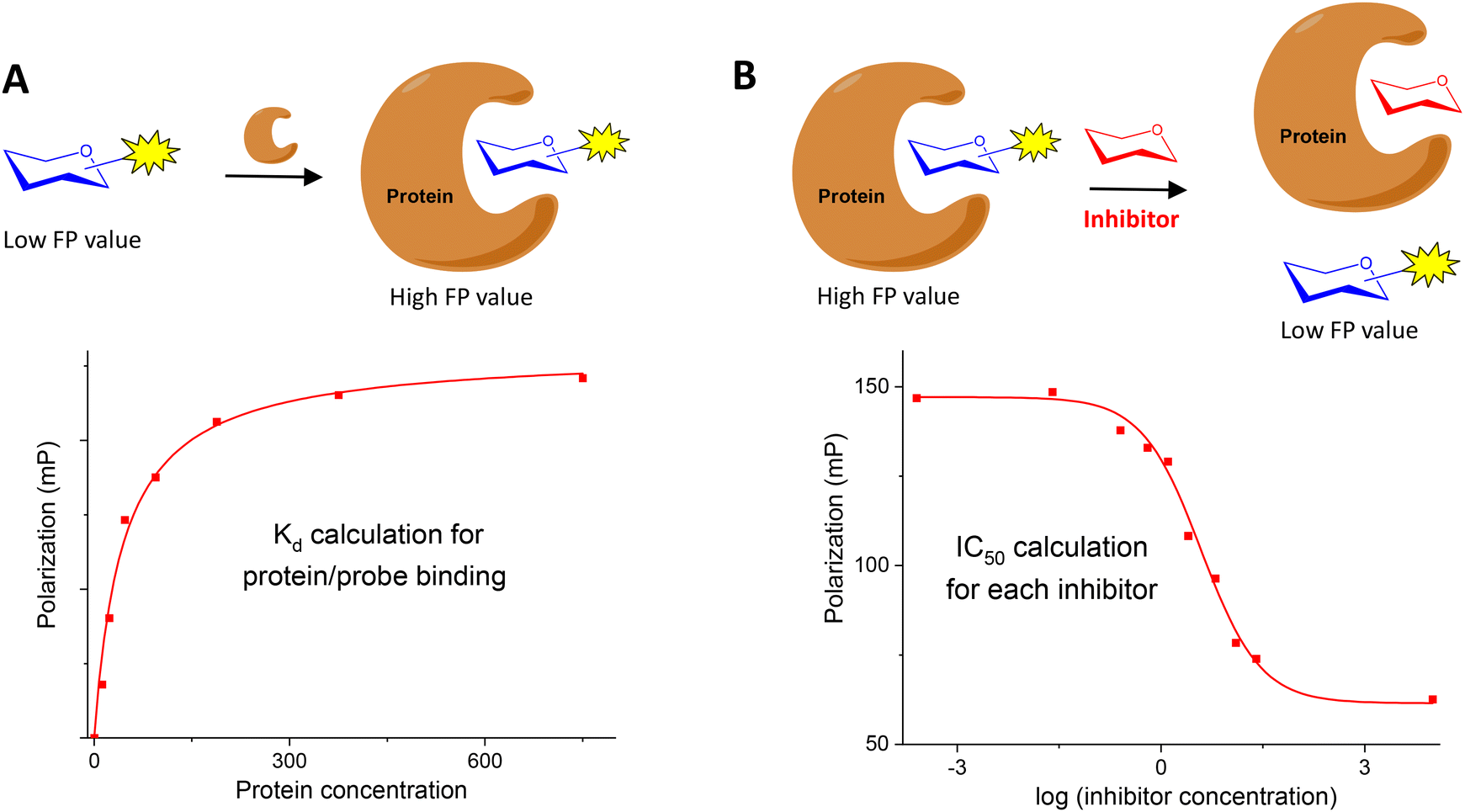
Fluorescence polarization (FP) is a powerful technique to study biomolecular interactions in solution. FP has been extensively used in drug discovery and the analysis of DNA–protein, DNA–DNA and protein–protein interactions.1–8 Its application in the field of carbohydrate–protein interactions9 has received less attention.10,11 The study of these interactions is fundamental to understand the functions of carbohydrates that are involved in biological processes such as inflammation, pathogen infection, cancer, etc. In this review, we will focus on the FP-based approaches to study glycan–protein interactions reported in the last 20 years.In a FP experiment, a fluorescent molecule is irradiated with polarized light (Fig. 1).1,10 The polarization of the emitted light depends on the apparent molecular weight of the fluorescent compound. In the case of a low molecular weight molecule (for example, a fluorescently labelled carbohydrate), which rapidly rotates in solution, the emitted light is highly depolarized due to the fast movement of the compound. However, when the small fluorescent glycan binds to a larger molecule (its protein receptor), the resulting complex rotates slower and, therefore, the emitted light largely remains polarized. This fact allows the use of FP measurements to analyse carbohydrate–protein binding events. Molecular size is directly proportional to FP: small carbohydrates show low FP values while large carbohydrate–protein complexes give high FP values. Relatively large changes in molecular size between the glycan probe and the complex are required for significant change in polarization values that allow the detection of binding.
 | ||
| Fig. 1 Basics of FP assays. | ||
FP measurements are recorded using commercially available microtiter plate readers equipped with appropriate excitation and emission filters. The FP value of the emitted light is calculated from the fluorescence intensity variations in its parallel and perpendicular components. Since the polarization value is a ratio of light intensities, it is a dimensionless number often expressed in millipolarization (mP) units (1 P unit = 1000 mP units). Anisotropy is sometimes employed instead of polarization.1,10 This analogous parameter, also derived from the variations between vertical and horizontal fluorescence intensities, is mathematically closely related with FP. In this review, these terms will be used interchangeably.
Two different types of FP experiments can be distinguished. In a direct binding assay (Fig. 2A), the interaction between a fluorescently labelled carbohydrate and the corresponding protein receptor is studied. Typically, FP of samples containing a fixed concentration of fluorescent ligand and increasing amounts of protein are measured. From these data, a binding isotherm can be constructed plotting the FP values against protein concentration. Nonlinear regression analysis can then be applied to obtain the dissociation constant (Kd) of the fluorescent glycan–protein binding. To calculate meaningful Kd measurements, it is important to use protein concentrations high enough to get a plateau in the saturation curve that indicates the maximum FP value approached, in which all the probe is bound to the protein.
 | ||
| Fig. 2 Types of FP experiments. (A) Direct binding assay; (B) competition experiment. | ||
Alternatively, a FP competition assay can be also established (Fig. 2B). In this case, the FP of samples containing fixed concentrations of fluorescent probe and protein are recorded in the presence of increasing concentrations of non-fluorescent, potential inhibitors. When added to the probe/protein mixture, an active inhibitor is able to displace the labelled saccharide from the protein binding pocket. In this way, a decrease of the FP signal is observed due to the displaced probe that can now move freely in solution. A competition curve can be generated by representing FP values against the logarithm of inhibitor concentration. From these data, IC50 values can be deduced by nonlinear regression analysis. The great benefit of these competition assays is the ability to determine relative binding affinities for compounds without the need to label them, using just one fluorescent labelled saccharide.
Typically, the experiments are performed at least in three replicate wells. In addition, several independent experiments are usually carried out and the reported IC50/Kd and error values represent, respectively, the average and the standard deviation from these independent assays.
Compared to other methods used in the analysis of glycan–protein interactions,9,11 such as surface plasmon resonance (SPR), ELISA-like assays, saturation transfer difference (STD) NMR and isothermal titration calorimetry (ITC), FP presents some advantages and also disadvantages. Like STD-NMR and ITC, FP is a homogeneous technique in which the binding partners are free in solution. FP does not require immobilization of protein or ligand to a solid support in contrast to SPR or ELISA-like assays. Therefore, washing steps are avoided and the experimental procedure is simple and fast. Once the equilibrium is reached, FP is usually recorded in a few minutes.
Another important advantages of FP include its suitability for high-throughput screening since 384-well or even 1536-well microtiter plates can be employed, and the low amount of samples (both protein and fluorescent probe) required (often, solutions into the nanomolar range) in comparison with other in-solution methods such as STD-NMR and ITC (typically, solutions in the micromolar/millimolar range). However, it is important to note that, in FP competition experiments, a protein concentration close to the Kd value of the probe/protein interaction is needed to get a high enough range of measurement, representing around 50% of the maximal shift. Therefore, the concentration of protein required depends on the affinity constant with the probe and this point can potentially limit FP studies when considering the low affinity of carbohydrate–protein interactions (see below).
Concerning the type of information obtained, FP experiments usually provide a quantitative value of the binding affinity, expressed as IC50 or Kd, in contrast to other techniques that give additional data on the glycan–protein interaction. Thus, SPR can afford association and dissociation kinetic constants, ITC provides all thermodynamic parameters and also the stoichiometry of the complex, and STD-NMR delivers information on the carbohydrate binding epitope in contact to the protein receptor by using an excess of carbohydrate ligand.
On the other hand, derivatization of the protein is not necessary in FP assays, although the fluorescent labelling of the carbohydrate probe is required. In competition experiments, the range of IC50 values that can be measured depends on the probe used: ligands displaying higher affinities than the probe cannot be analysed and, in these cases, STD or ITC studies are required (see below).
As mentioned before, a first requirement for the development of a FP assay is the preparation of a convenient fluorescently labelled carbohydrate probe. This compound should bind to the protein of interest with high affinity in order to minimize the amount of protein required for the experiments and expand the range of binding affinities which can be calculated.12 Fluorescein is the most commonly used fluorophore to label glycans in the FP field. Its properties (such as fluorescence lifetime) are adequate to observe significant FP variations between a small fluorescein labelled carbohydrate (for example, with a molecular weight of 1–2 kDa) and its complex with a 10–20 kDa protein. Alternative fluorophores have also been used. The relationship between molecular weight and FP depends on the fluorescence lifetime of the probe.1 Therefore, in cases where the change in molecular weight after binding is quite different from that mentioned before (for example, when considering larger fluorescent carbohydrates), the use of fluorophores with longer lifetimes can be useful.
The conjugation chemistry used for labelling, in particular the linker nature, can impact the assay performance. Long and flexible linkers are usually avoided because allow the local free rotation of the fluorophore upon protein binding, causing undesired depolarization (propeller effect).1 Different synthetic approaches used for the preparation of fluorescent saccharide probes are discussed below.
In FP assays, it is important to maintain a constant temperature since temperature affects the molecular movement in solution and therefore modifies the polarization of the emitted light. For the same reason, modifications in the buffer conditions resulting, for example, in variations of the solution viscosities, also change FP measures. A potential problem in the development of FP experiments is derived from the interference caused by auto-fluorescent compounds.1 For example, in competition experiments, the background fluorescence from the inhibitors can cause artifacts. This can be solved by pre-reading the fluorescence of samples before addition of the fluorescent probe. The background fluorescence can then be subtracted before FP calculation.
Hereafter, we discuss some of the most recent applications of FP for the analysis of carbohydrate–protein interactions. First, we review examples of FP experiments for the study of the binding between carbohydrates and lectins. Then, we present FP approaches towards the discovery of inhibitors for glycan-processing enzymes. Finally, we focused in the particular case of glycosaminoglycan (GAG)–protein interactions.
FP experiments to study carbohydrate–lectin interactions
Titz and coworkers reported a novel FP-based competitive binding assay for the discovery of inhibitors of LecB, a pathogenic carbohydrate–binding lectin produced by Pseudomonas aeruginosa.13,14 Although the interaction between carbohydrates and lectins is usually characterized by low to moderate binding affinities (in the micromolar/millimolar range), LecB binds L-fucosides with unusual high affinity and, therefore, fluorescein-labeled fucoside 4 was synthesized as probe for the FP measurements (Fig. 3).13 The synthesis started with the lanthanum triflate catalyzed glycosylation between L-fucose and 2-bromoethanol, followed by acetylation, nucleophilic substitution of the bromine atom by sodium azide, and deacetylation. Finally, after reduction of the azido group, the resulting 2-aminoethyl fucoside 3 was conjugated with fluorescein-5-isothiocyanate (FITC) to afford the fluorescent ligand 4 in excellent yield. Upon incubation of 4 with increasing concentrations of LecB, a dose-dependent increase of FP was observed due to the binding between the fucose probe and the lectin (see Fig. 2A). Compound 4 in the absence of LecB displayed FP values at the background level, as corresponds to low molecular weight compounds. Incubation of 4 with 16 μM LecB provided the maximum polarization value that corresponds to all fucose probe bound to the lectin. The binding was completely inhibited by the addition of excess fucose (1 mM). Fitting of the binding curve afforded a dissociation constant value of 697 nM in good agreement with the known dissociation constant for methyl α-L-fucoside (430 nM, obtained by ITC15). This binding affinity between 4 and LecB was ideal to develop a competition experiment for the analysis of inhibitors in the micromolar range.12 In the competition assay, serial dilutions of inhibitors were added to a stock solution containing a constant concentration of 4 (1.5 nM) and LecB (225 nM). A dose-dependent decrease of the FP value was observed when the tested compounds inhibited probe 4-LecB binding (see Fig. 2B). IC50 values for each inhibitor were calculated by nonlinear regression analysis of the competition curve. The assay was first validated using ligands with known binding affinities. Then, a series of mannoside derivatives containing different functions at position 6 were screened as potential LecB ligands. From this study, two different inhibitors were identified showing up to a 20-fold increase in affinity to LecB compared to the natural ligand methyl mannoside. The FP assay was run in a 384-well format, minimizing the sample consumption and avoiding the multiple handling steps associated with ELISA-like protocols.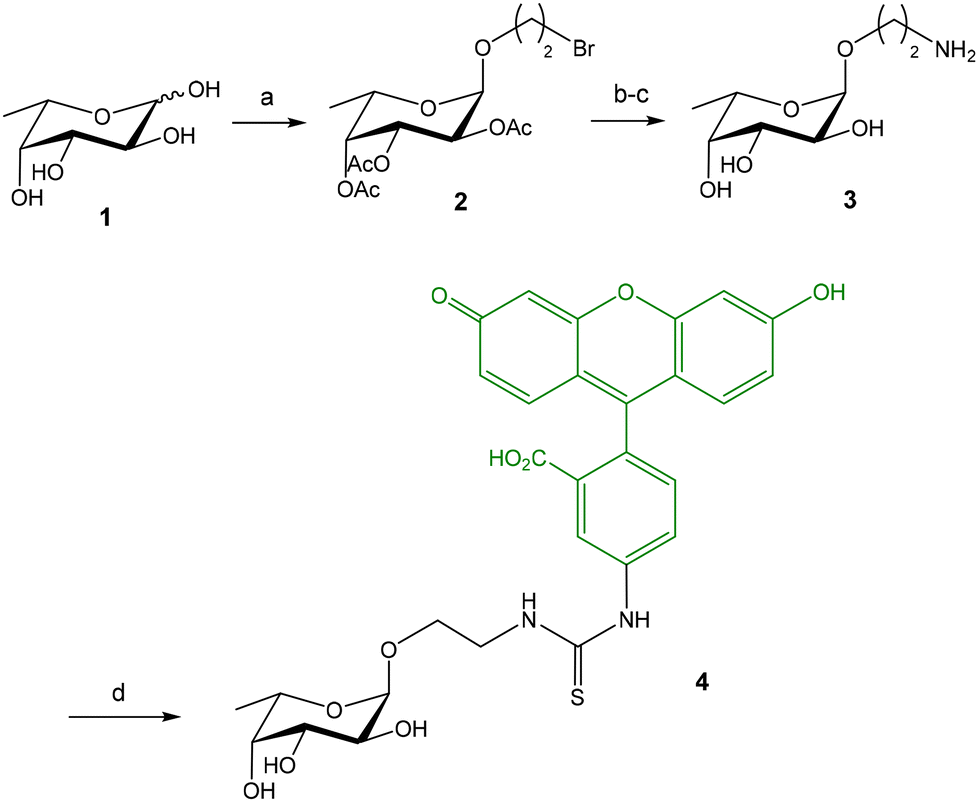 | ||
| Fig. 3 Synthesis of fluorescein-labeled fucoside probe 4. Reagents and conditions: (a) La(OTf)3, 2-bromoethanol, 70 °C; Ac2O, Py, 46%; (b) NaN3, DMF, 70 °C, 70%; (c) NaOMe, MeOH; H2, Pd/C, EtOH, 66%; (d) FITC, NaHCO3, DMF, 96%. | ||
The same research group also developed a competition assay for screening of potential LecA inhibitors.16–18 LecA is another C-type lectin produced by Pseudomonas aeruginosa which is important for biofilm formation causing antibiotic resistance. In contrast to LecB that shows a high affinity (nanomolar) for fucose residues, LecA binds specifically D-galactose units with only moderate affinity constants (Kd = 50 μM for methyl α-D-galactoside, obtained by ITC19). This fact complicates the design of a high affinity fluorescent probe for LecA. Several fluorescein-labelled galactoside probes were synthesized (Fig. 4) and their direct titration with increasing amounts of LecA afforded Kd values in the micromolar range (Kd = 7.4–27.4 μM).16 As discussed before, in competition FP experiments, the required amount of protein should be close to the Kd value for the probe/protein interaction. As a consequence, 10–15 μM protein was employed to screen a library of thiogalactosides and human blood group antigen epitopes as LecA inhibitors. In order to reduce protein consumption (up to 2 μM), the authors also prepared a fluorescein labelled divalent probe (9) showing a Kd of 1.54 μM.
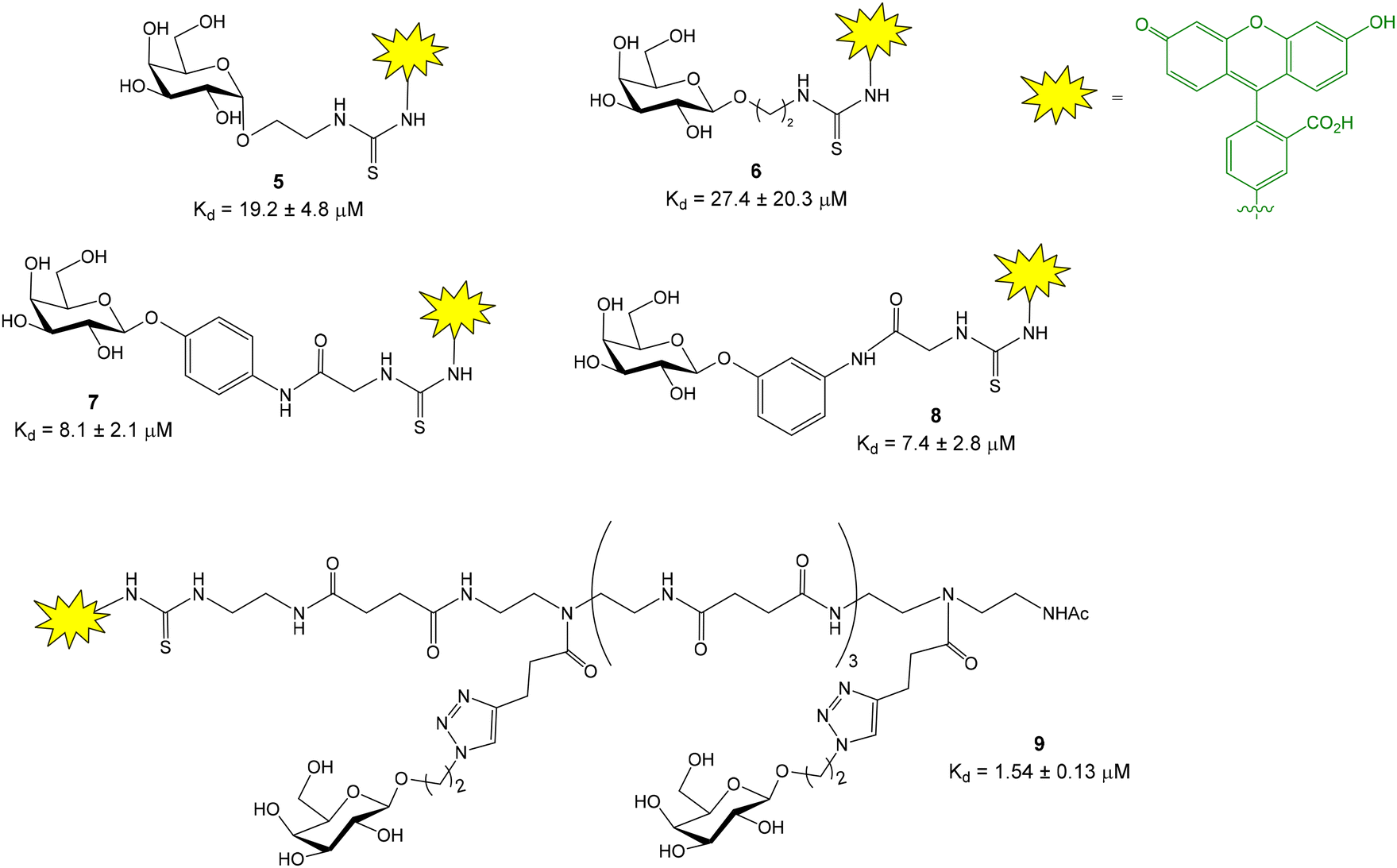 | ||
| Fig. 4 Fluorescein-labelled galactoside probes for LecA binding and their corresponding Kd values. Phenyl β galactoside tracers 7 and 8 and divalent compound 9, showing lower Kd's, were preferentially chosen as FP probes in competition experiments. | ||
This competitive LecA binding assay was also used to analyse a library of divalent galactoside derivatives.20,21 However, the inhibition curves exhibited very steep Hill slopes, indicating the high potency of divalent compounds and suggesting that the lower assay limit was reached. In fact, an alternative SPR study showed low nanomolar dissociation constants for the divalent inhibitors, confirming that ligands with much higher affinities than the fluorescent probe cannot be reliably analysed.12 In this context, the synthesis of high affinity (nanomolar) fluorescent probes is required to solve this limitation and measure high affinity nanomolar inhibitors.22,23
Additionally, a modified version of the competitive LecA binding assay was employed to screen a series of catechols derivatives as novel non-carbohydrate inhibitors.24 In this case, a Cy5 labelled galactoside was chosen as probe to avoid spectral overlap of some catechols with the fluorescein functionalized compounds.
Burkholderia cenocepacia is a Gram-negative bacterium that has several lectins with important roles in adhesion and biofilm formation. One of these lectins is mannose specific BC2L-A. In order to identify BC2L-A inhibitors with potential anti-infection activities, a FP competition experiment was developed.25 In this case, a mannose fluorescent tracer was prepared following a synthetic strategy analogous to the one depicted in Fig. 3 for the fucose probe 4.
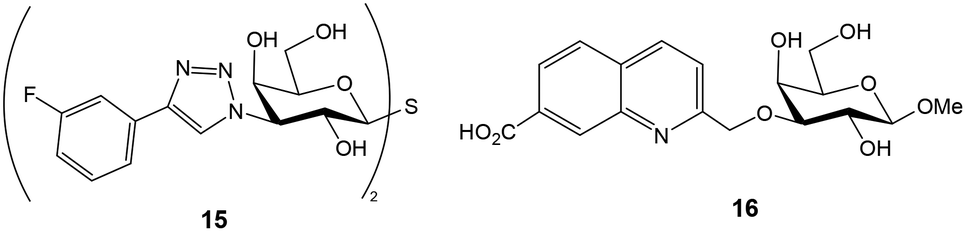
The groups of Leffler and Nilsson developed a FP approach to evaluate the interactions between galectins and natural or artificial saccharides and glycoconjugates.26 Galectins are a family of lectins that selectively recognize β-galactosides and play an important role in cancer biology and immune and inflammatory responses. A library of fluorescein-conjugated saccharides was synthesized and tested as fluorescent probes in direct binding assays with different galectins.26–28 In the design and synthesis of the probes, the structural features of galectin–carbohydrate interactions were considered and the 2-aminoethyl glycoside of N-acetyl lactosamine (or lactose) was selected as the starting material since it is known that the binding affinity of galactose alone is much lower than those of lactosamine (and lactose) derivatives. The 2-aminoethyl glycoside was treated with various amine-reactive fluorescein derivatives to generate conjugates containing thioureas (10), aminotriazines (11) or amide linkages (12, 13) (Fig. 5).26 Direct binding assays allowed the selection of the best probe, giving the lowest Kd value, for each galectin. For this, FP of wells containing 100 nM fluorescent probe and a series of different galectin concentrations was measured after incubating the plate in the dark for 5 min. Control wells containing only probe were included and provided the minimum FP value. An increasing polarization was observed with increasing protein concentrations until a plateau was reached, indicating the maximum FP value. Mathematical fitting of the binding curve gave the Kd value for the probe–galectin interaction. Introduction of additional saccharide units or functional groups at position 3 of galactose usually improved the binding affinity of the disaccharide. In general, amide linkers were preferred over thiourea and aminotriazine spacers due to stability issues. However, derivative 13 displaying a long amide linker gave poor results, probably because the fluorescein moiety can independently rotate even when the probe is bound to the protein. Alternatively, derivatives of thiodigalactosides, a class of small galectin inhibitors, such as 14, have also been employed as potent fluorescent ligands (Fig. 5).29 For some galectins, experiments were carried out at low temperature (8 °C) to increase the maximum anisotropy value reached due to slower tumbling of the probe/galectin complex. Background nonspecific binding was ruled out since a mannose nonbinding probe had the same anisotropy value in the presence of galectin as the free probe.
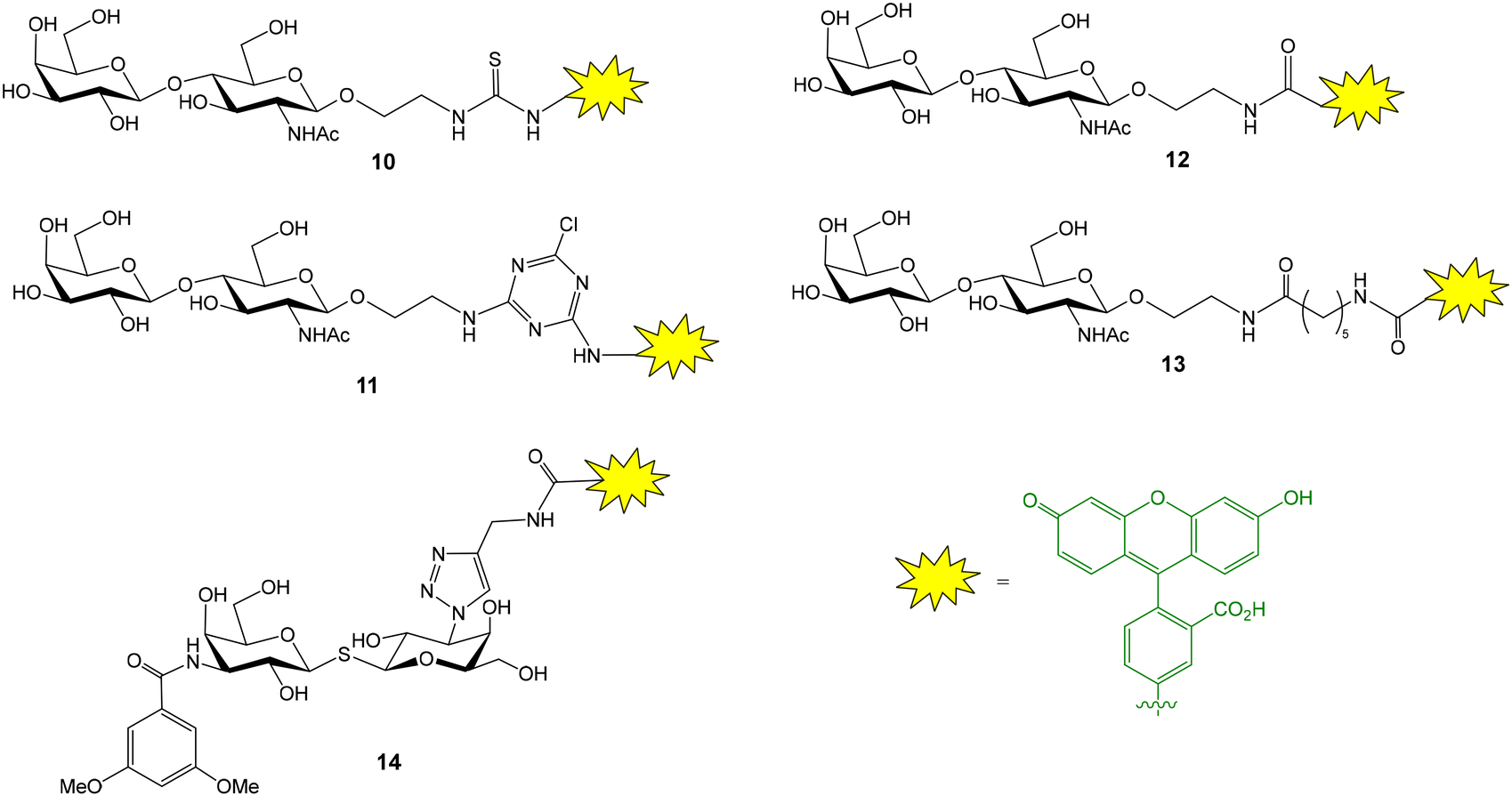 | ||
| Fig. 5 Structure of some fluorescent glycans employed for the evaluation of galectin interactions. | ||
Once the optimal fluorescent derivative was chosen, a competition assay was implemented for the analysis of a collection of non-labelled compounds. A first screening was performed by measuring the FP of wells containing fixed concentrations of galectin 3 (1 μM), probe (100 nM) and inhibitor (8 μM).26 This experiment allowed the quick identification of derivatives showing high inhibitory potencies. Selected inhibitors were then studied in more detail by titration with a fixed amount of galectin and probe. Anisotropy was plotted against inhibitor concentration and the resulting curves were mathematically fitted to the equation for a one-site competitive interactive to provide IC50 values. Using the same data, dissociation constants for galectin–inhibitor interactions were also calculated by solving a set of equations, since the concentrations of all interacting molecules are known.26 This competition experiment has been extensively used to study the interactions between different galectins and non-fluorescent molecules.22,23,30–37 For example, using this approach, some 3-3′-bis(aryltriazolyl)thiodigalactosides have been identified as high affinity ligands with selectivity for galectins 1 and 3 (Fig. 6).38 Compound 15 displaying low nanomolar Kd was able to attenuate lung fibrosis and block intracellular galectin 3 accumulation at damaged vesicles. On the other hand, carboxylic acid substituted quinoline–galactose derivatives, such as 16, have shown efficient and selective inhibition of galectin 8 with nearly 60-fold affinity enhancement over the reference compound methyl β-D-galactopyranoside (Fig. 6).39 These results provided a starting point for the development of galectin 8 inhibitors potentially interfering with tumor progression. Importantly, the preparation and use of second-generation, nanomolar affinity fluorescent probes allowed the minimization of sample consumption (galectin at 10 nM and probe at 4 nM) and the identification of inhibitors with single-digit nanomolar affinity.23
 | ||
| Fig. 6 Examples of high affinity galectin ligands identified in competition FP assays. | ||
A similar FP assay, using fluorescein labelled lactose as probe (compound 17, Fig. 7), was also applied to evaluate the binding between galectin 3 and several galactan oligosaccharides.40 This methodology, however, failed when larger pectic and galactan polysaccharides were tested as potential galectin 3 inhibitors. On the other hand, although it is outside the scope of this review, focused on FP studies employing fluorescent carbohydrate probes, it is worth mentioning that the presence of a tryptophane (Trp) residue in the carbohydrate recognition domain of all galectins also enabled the study of galectin–glycan interactions by intrinsic Trp fluorescence anisotropy spectroscopy.41
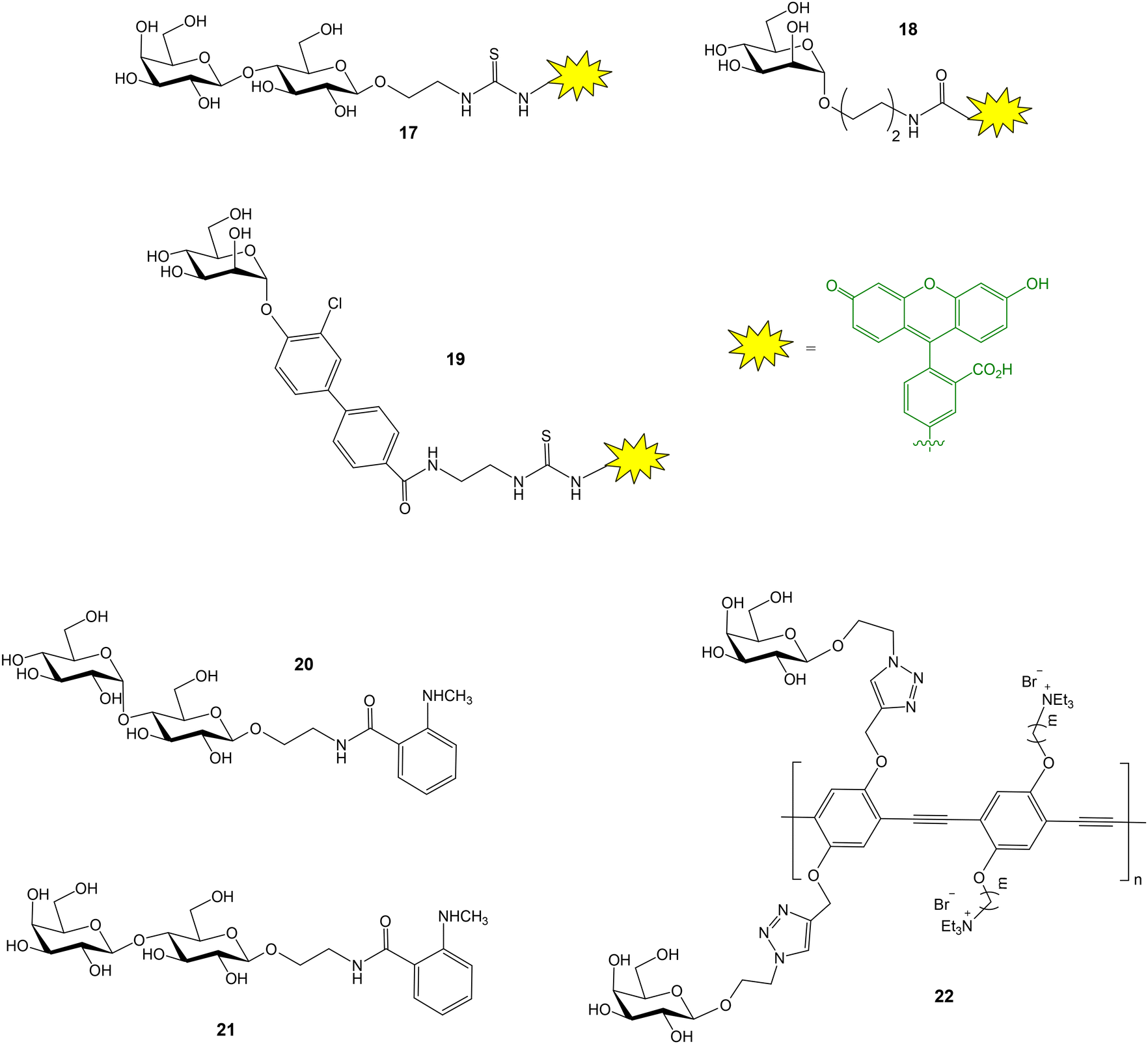 | ||
| Fig. 7 Fluorescent probes used for FP studies with galectin 3 (17), FimH (18, 19), maltose binding protein (20), galectin 1 (21) and PNA (22). | ||
FimH is a bacterial lectin that binds mannosylated proteins of host cells. This interaction is crucial for bacterial adhesion and subsequent Escherichia coli urinary tract infections. Blocking FimH binding with small molecules is a useful approach to prevent bacterial entry and infection. A FP competition experiment has been reported to study the interactions between FimH and mannoside derivatives.42 First, fluorescently tagged mannoside 18 was synthesized (Fig. 7). Then, 20 mannoside ligands with structural diversity were evaluated for their ability to inhibit binding between FimH and the fluorescent probe. Optimized biarylmannosides displayed low nanomolar IC50 values, representing excellent candidates for further development as novel therapeutics for the treatment of urinary tract infections. Ernst and coworkers also described a FP competition assay to characterize the binding affinities of novel biphenyl mannoside derivatives for FimH.43 Mannoside 19 was employed as probe (Fig. 7). Thus, a high affinity FimH ligand, showing improved pharmacodynamics and pharmacokinetics properties, was discovered.
A general strategy to label carbohydrates with N-methyl-anthranilic acid at the anomeric position has been reported.44 The generated glycoprobes were adequate for FP binding and competition experiments. As a proof of concept, using this approach, the binding of anthranilic acid-functionalized maltose and lactose (20 and 21, Fig. 7) to their corresponding protein receptors, maltose binding protein and galectin 1 respectively, were studied. A simple method to functionalize oligosaccharides at their reducing end, generating appropriate FP probes, has been also published.45 Thus, human milk oligosaccharides were treated with pyrenebutyric hydrazide and the resulting pyrene-labelled compounds were successfully applied in FP studies with Ricinus communis agglutinin 120. The interaction between galactose-functionalized fluorescent polymers (22, Fig. 7) and peanut agglutinin (PNA, a galactose–selective lectin) was also proved by fluorescence anisotropy experiments.46 Protein binding was detected by the enhancement of the anisotropy signal.
The interactions between Concanavalin A (ConA), an α-mannose specific plant lectin, and different glycans and glycomimetics have been also evaluated by FP. For instance, a glycosylasparagine derivative was fluorescently labelled (oligosaccharide 23, Fig. 8) and its interaction with ConA was analysed in a direct binding assay.47 In addition, the conjugation between a mannose monosaccharide equipped with an α-aminooxy group at the anomeric position and an aldehyde-functionalized fluorescein derivative furnished a suitable fluorescent probe (compound 24, Fig. 8) for the analysis of the binding between ConA and mannose multivalent systems based on a cyclic peptide scaffold.48 FP measurements also proved the interaction between ConA and collagen peptides displaying a mannose unit and a fluorescent dye.49 On the other hand, copper-catalyzed azide–alkyne cycloaddition reaction provided labelled mannose dendrimers (such as 25, Fig. 8) that enabled the study of ConA binding by fluorescence anisotropy.50
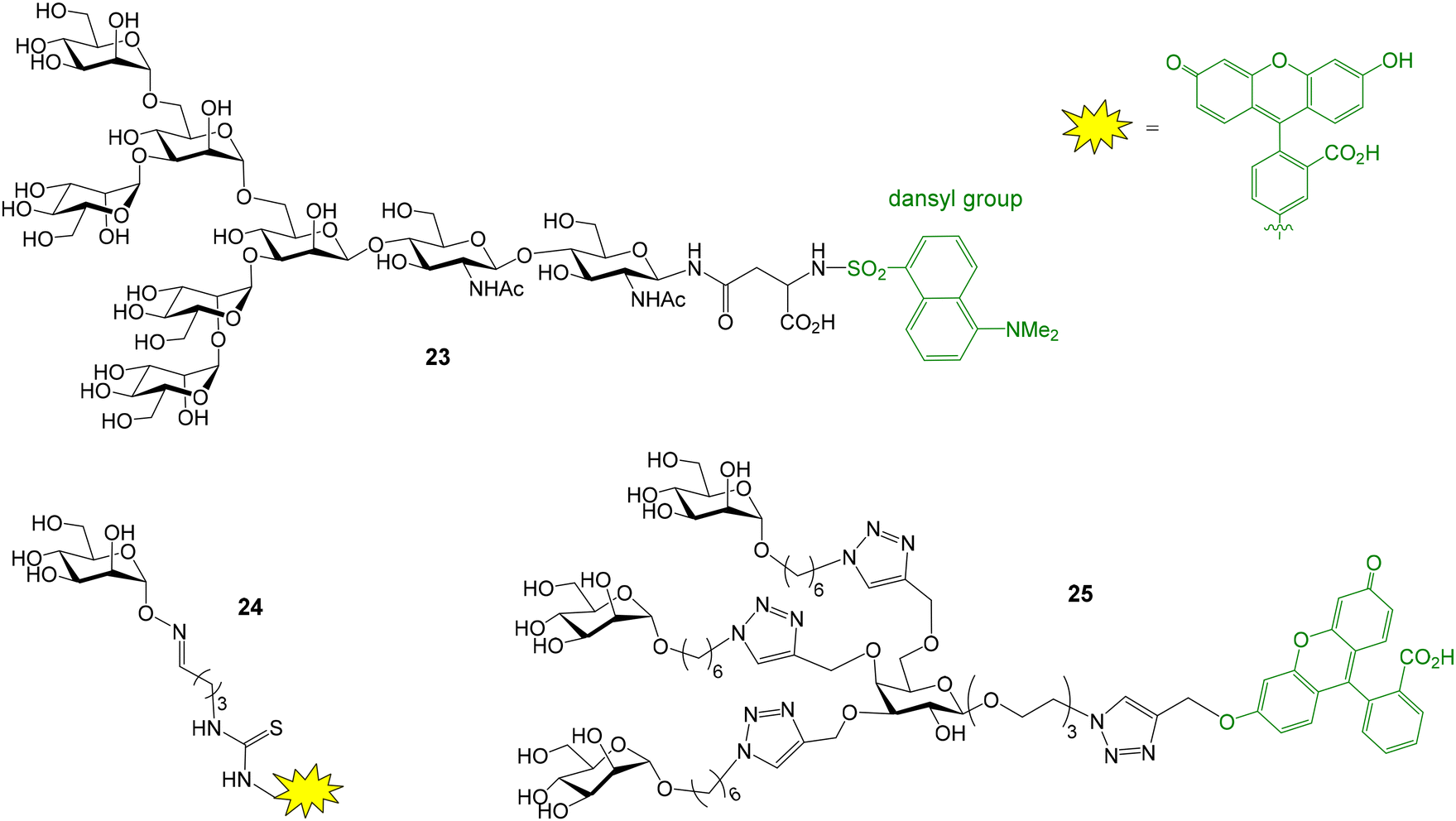 | ||
| Fig. 8 Structure of fluorescent probes employed to study ConA binding. | ||
The interaction between fluorescent ruthenium metalloglycoclusters and several lectins have also been demonstrated by FP. Thus, galactose metalloclusters showed specific binding to PNA while glucose systems preferentially bound to ConA.51
High-mannose Man9 oligosaccharide is present in several viral envelope glycoproteins. Its interaction with Dendritic Cell-Specific ICAM-3 Grabbing Non-integrin (DC-SIGN) receptor, found at the surface of dendritic cells, plays a crucial role in the attachment of viruses to cells. Rojo and coworkers reported a direct FP binding assay to calculate the dissociation constants for the binding between DC-SIGN and several fluorescent Man9 derivatives.52 Man9 oligosaccharides 26 and 27 displaying alpha and beta configurations at the reducing end, and their corresponding trivalent glycoclusters 28 and 29 were first synthesized (Fig. 9). Conjugation with commercially available alkynylated fluorescein derivative 30, using the Cu(I) azide–alkyne cycloaddition reaction, afforded fluorescent compounds 31–34 in excellent yield. The FP of microplate wells containing a 10 nM fluorescent ligand solution and different concentrations of DC-SIGN Extracellular Domain (ECD) were then recorded. In all cases, an increase in the polarization value was observed with increasing protein concentrations, due to the binding between DC-SIGN and the Man9 derivatives. Interestingly, a significant change in FP value was found upon binding, although the fluorescent ligands had a considerable size. From the binding curves, Kd's for each ligand were calculated by nonlinear regression analysis (Fig. 9). The results suggested that the anomeric configuration of the reducing end mannose does not significantly influence in the binding affinity for DC-SIGN, since similar values of Kd were obtained for α- and β-configurated compounds. Therefore, Man9 alpha epitope can be employed instead of more synthetically-challenging natural beta oligosaccharide as DC-SIGN ligand. On the other hand, the Kd's of trivalent systems 33 and 34 were lower than those for the monovalent oligosaccharides due to the multivalent effect.
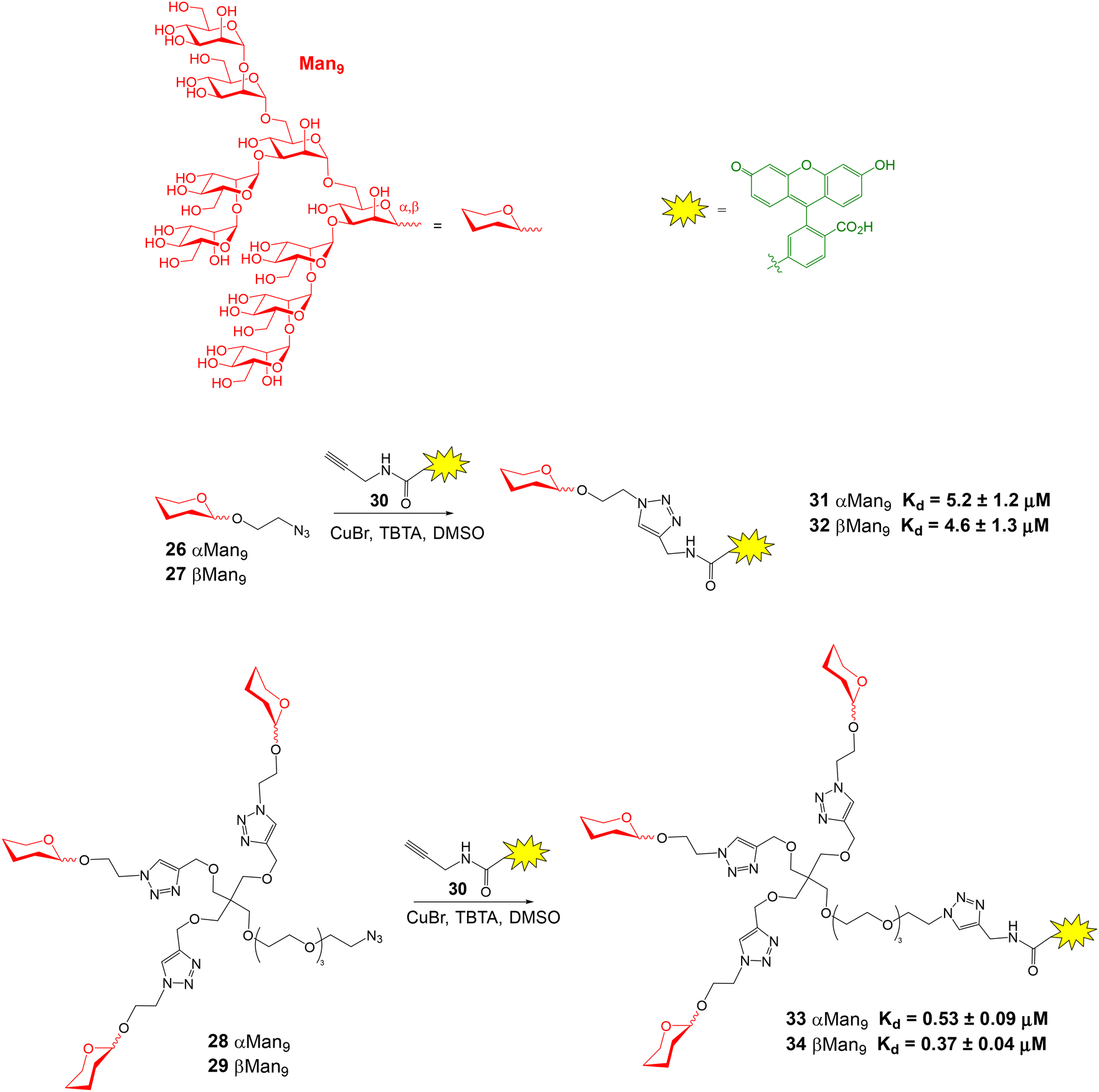 | ||
| Fig. 9 Synthesis of fluorescently labelled Man9 oligosaccharides and glycoclusters and Kd values obtained from FP direct binding assays with DC-SIGN ECD. TBTA = tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine. | ||
FP has also been employed to study carbohydrate–antibody interactions. Globo-series glycans are human cell-surface carbohydrates that include stage-specific embryonic antigen-4 and Globo H hexasaccharides. These oligomers were labelled with BODIPY and FP direct assays showed a low nanomolar interaction between the fluorescent conjugates and their corresponding monoclonal antibodies.53 Using fluorescein-labelled perosamine-containing oligosaccharides, a FP approach has also been developed to detect antibodies against Brucella infection as a new diagnostic tool.54
FP experiments for the discovery of glycan-processing enzyme inhibitors
Human nonlysosomal glucosylceramidase (GBA2) is a retaining glycosidase that controls levels of glycolipids and is involved in several human diseases. A fluorescence polarization activity-based protein profiling (FluoPol-ABPP) assay has been developed for the identification of GBA2 inhibitors with potential therapeutic applications.55 This assay is based on the covalent linkage of a reactive fluorescent probe to the active site of the enzyme that results in a high FP measurement (Fig. 10). The addition of an inhibitor that binds to the active site, blocking the reaction of the activity-based probe, provides lower FP values. Thus, tetramethylrhodamine (TAMRA) cyclophellitol aziridine 35 was synthesized and used as activity-based probe for GBA2. This fluorescent compound reacted with the active site nucleophile of the enzyme. In these experiments, extracts of cells overexpressing GBA2 were employed. The assay conditions (pH, protein and probe concentrations) were optimized in 96-well plates and the protocol was validated by calculating the IC50 values of several known GBA2 inhibitors that were in agreement with previously reported data. Then, an iminosugar library, containing more than 350 derivatives and including pyrrolidine and piperidine iminosugars displaying different configurations and N-alkylations, was screened in a 384-well plate format. Several potent and selective GBA2 inhibitors were identified, such as deoxynojirimicyn derivative 36 shown in Fig. 10.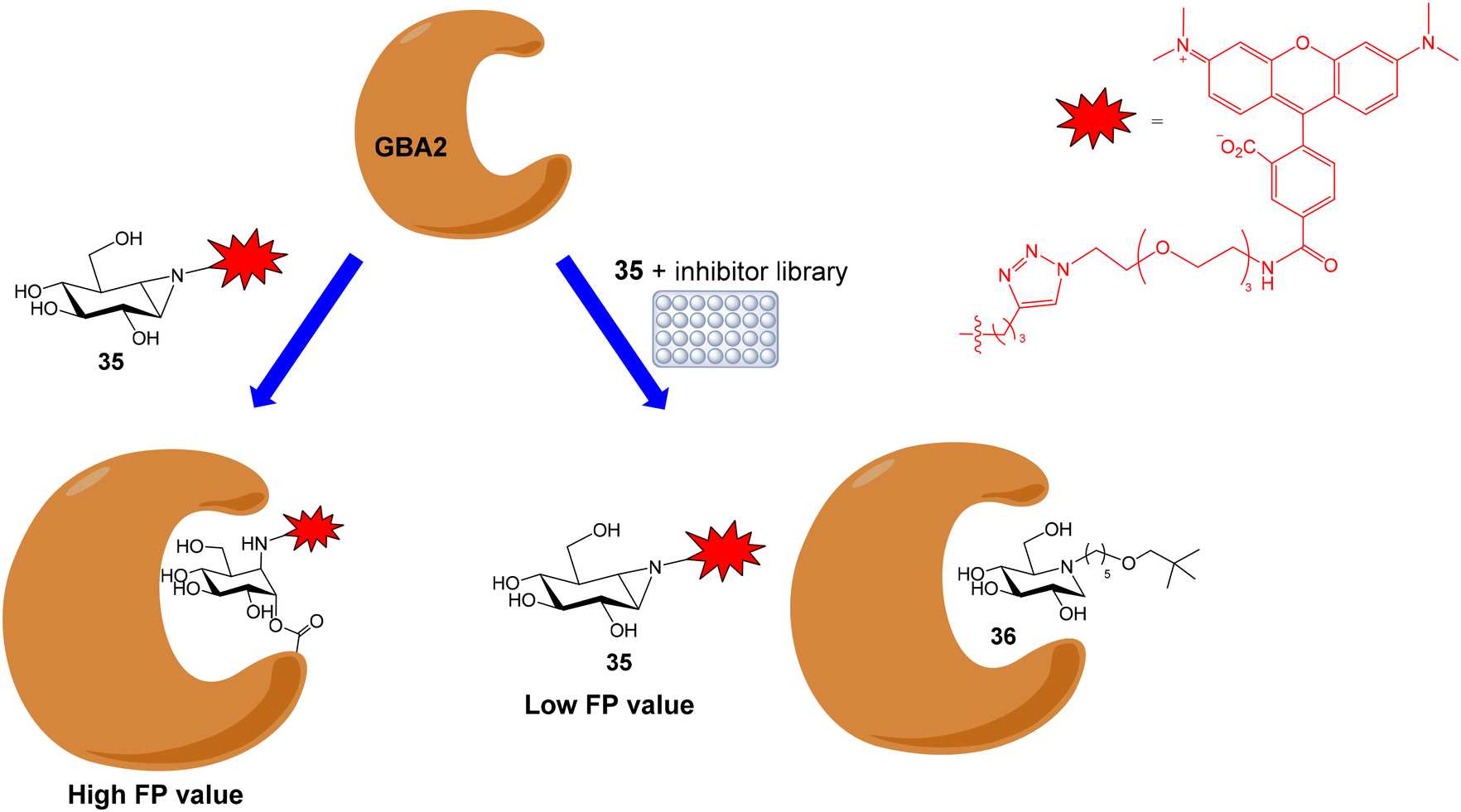 | ||
| Fig. 10 Schematic representation of the FluoPol-ABPP assay. | ||
Apart from GBA2, the FluoPol-ABPP strategy has also been applied to lysosomal glycosidases, such as GBA1 (a retaining β-glucosidase) and GAA (a retaining α-glucosidase).56 Fluorescent cyclophellitols derivatives (37 and 38, Fig. 11) were designed and synthesized for the selective covalent linkage to each enzyme and iminosugar library screening afforded selective inhibitors for GBA1 and GAA.
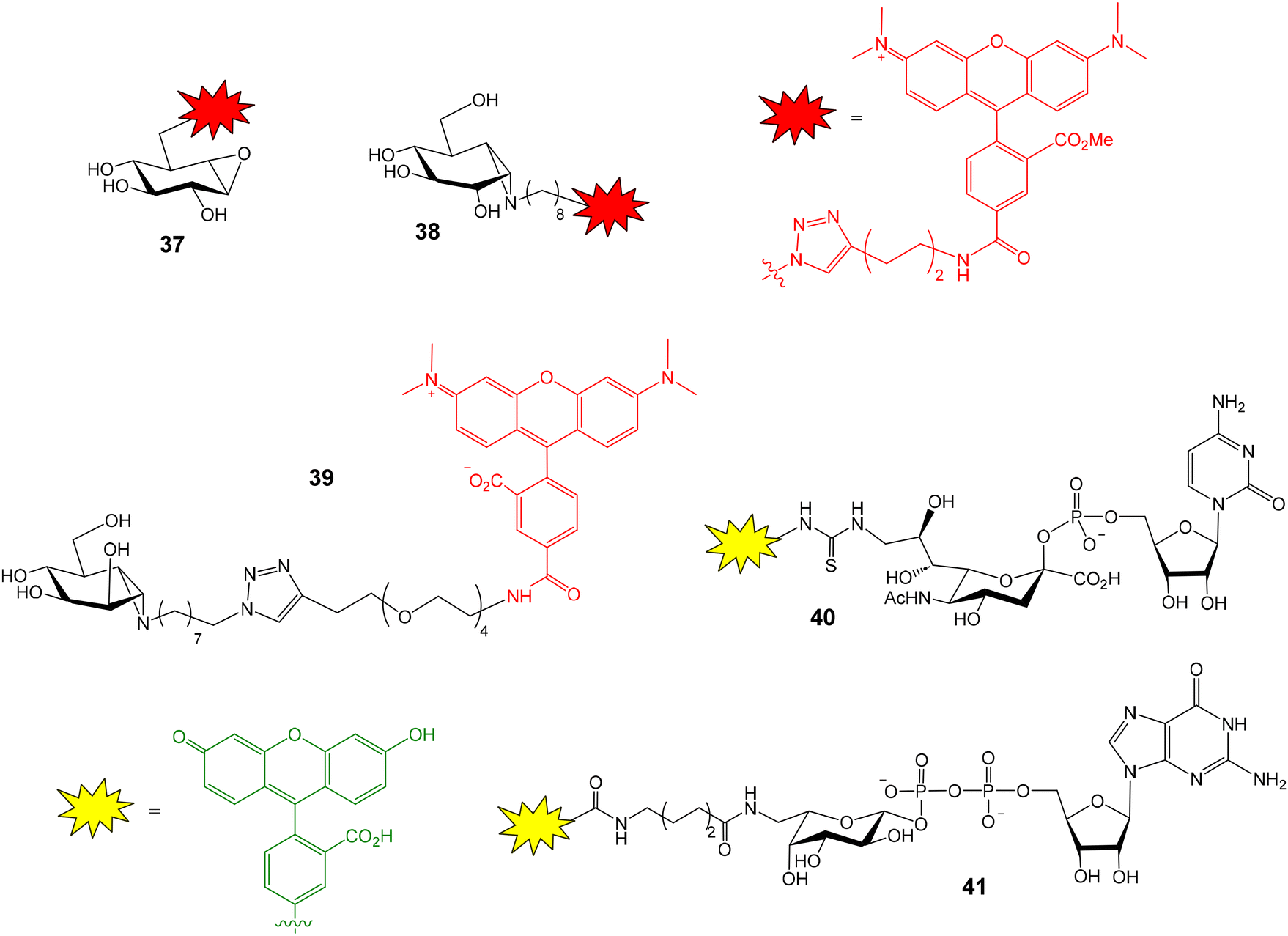 | ||
| Fig. 11 Fluorescent cyclophellitol derivatives for the covalent linkage to GBA1 (37), GAA (38) and GMII (39), and fluorescein-functionalized cytidine monophosphate sialic acid (40) and guanosine diphosphate fucose (41). | ||
This FluoPol-ABPP approach was also employed for the discovery of new α-mannosidases inhibitors.57 Golgi α-mannosidase (GMII) is a glycoside hydrolase that catalyses sequential hydrolysis of terminal α-1,3- and α-1,6-linked mannoses from N-glycans. GMII inhibitors are potential anticancer therapeutics. TAMRA-functionalized manno-epi-cyclophellitol aziridine 39 (Fig. 11) was prepared and used as activity-based probe for GMII. The experiment was again validated using previously reported inhibitors. As expected, these inhibitors reduced polarization of TAMRA-emitted light in a concentration-dependent manner. Next, the iminosugar library was screened and seven new inhibitors were identified. Remarkably, the experiment required very little material: 100 nM GMII and 25 nM probe solutions were needed. This aspect is important considering that mannosidases are usually expressed in small quantities. Therefore, the developed method could be applied to other different α-mannosidases.
Many human diseases are related to inappropriate glycosylation and, therefore, inhibition of glycosyltransferases is an attractive therapeutic approach. FP experiments have also been applied to the discovery of glycosyltransferase inhibitors.58,59 Paulson and coworkers reported a FP-based assay for the high-throughput screening of sialyl- and fucosyltransferase inhibitors (Fig. 12A).60 Fluorescein-containing analogues of the nucleotide carbohydrate donor substrates were first prepared (40 and 41, Fig. 11). The glycoprotein fetuin (50 kDa) was chosen as the acceptor substrate (Fig. 12A). Transfer of the fluorescent nucleotide donor to fetuin by the corresponding sialyl- or fucosyltransferase afforded a high FP signal.60,61 The addition of active inhibitors lowered the FP value, avoiding the formation of the fluorescent high molecular weight glycoprotein. Using this approach, a collection of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compounds was screened and several selective inhibitors were identified.60 This method allowed the identification of both donor and acceptor site inhibitors and required minimal amounts of enzymes. On the other hand, Rademann and coworkers also employed FP to develop nanomolar fluorescent inhibitors of sialyl transferases.62
000 compounds was screened and several selective inhibitors were identified.60 This method allowed the identification of both donor and acceptor site inhibitors and required minimal amounts of enzymes. On the other hand, Rademann and coworkers also employed FP to develop nanomolar fluorescent inhibitors of sialyl transferases.62
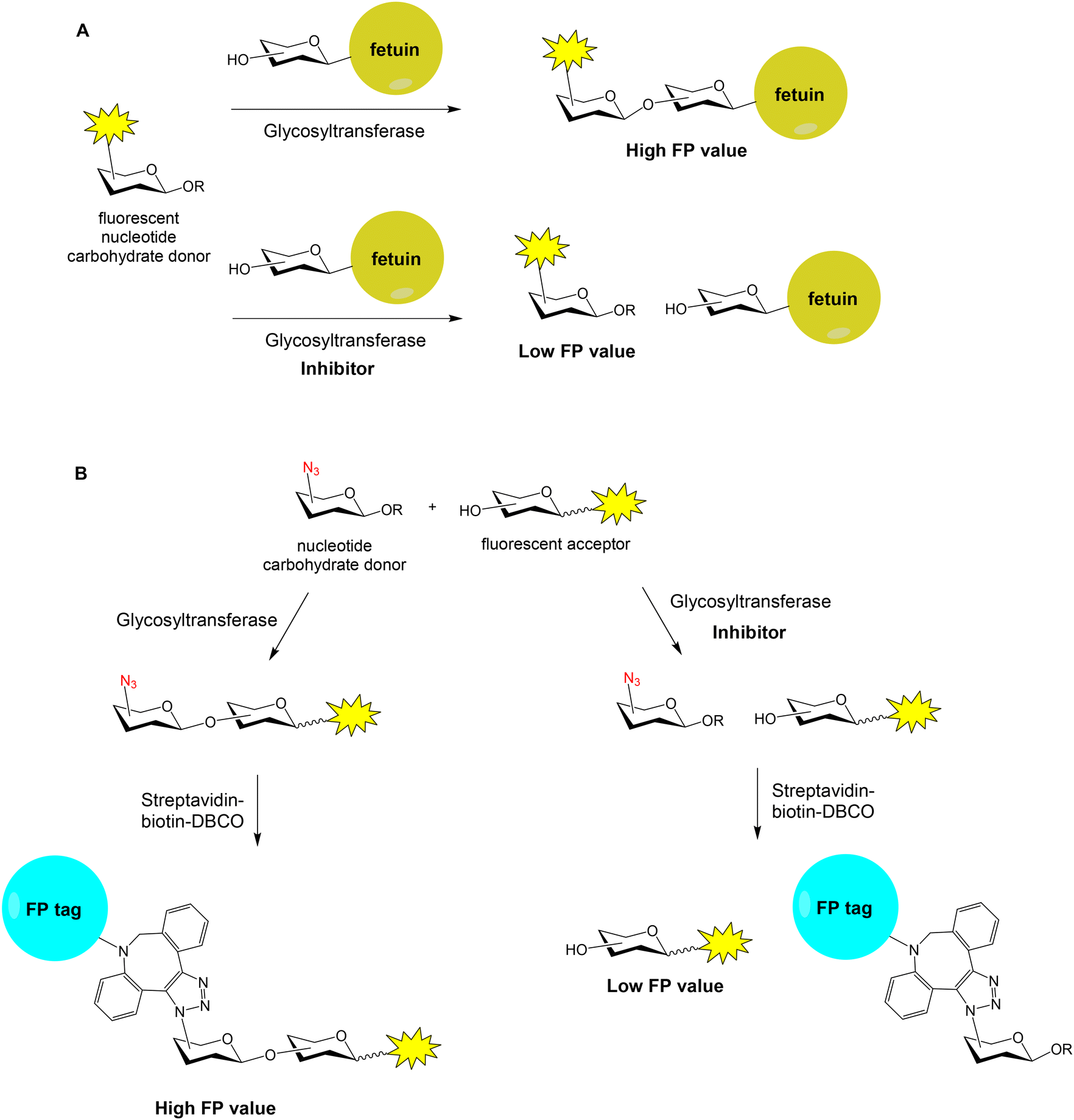 | ||
| Fig. 12 FP-based approaches for the discovery of glycosyltransferase inhibitors. | ||
A high-throughput “FP-tag” assay has been reported for the discovery of β-Kdo (3-deoxy-β-D-manno-oct-2-ulosonic acid) glycosyltransferase inhibitors.63 In this case, a fluorescent acceptor and an azido-functionalized donor were employed for the enzymatic reaction (Fig. 12B). Then, the reaction product was conjugated with streptavidin (52 kDa)–biotin–dibenzocyclooctyne (DBCO) by copper-free click chemistry, generating a high FP signal. If the enzymatic reaction was inhibited by a compound, the FP value lowered. Thus, a library of 1000 marine sponge derivatives was analysed in a 384-well format and several hits were identified. More recently, bovine serum albumin (66 kDa) was proposed as a low-cost and superior “FP-tag” for screening glycosyltransferase inhibitors, compared with streptavidin.64
The inhibition of enzymes involved in Mycobacterium tuberculosis cell wall biosynthesis, such as uridine diphosphate (UDP)–galactopyranose mutase (UGM), is an attractive strategy to block bacterial growth. Interestingly, FP measurements have been employed for the in situ screening of potential UGM inhibitors.65 A library of enamide structures was generated in a 384-well microplate and the binding affinities of the crude products were directly evaluated in the same wells by a FP competition assay using UDP-fluorescein as probe. FP measurements were also employed to detect the binding between fluorescently labelled chitooligosaccharides and lysozyme.66
FP approaches for the study of GAG–protein interactions
Glycosaminoglycans (GAG) are a family of negatively charged polysaccharides that includes heparin, chondroitin sulfate (CS) and hyaluronic acid (HA), among others.67 These polysaccharides usually present a high structural heterogeneity, for example, in terms of their sulfate groups distributions. GAG regulate a wide variety of biological processes due to their binding with numerous protein receptors. GAG–protein interactions are characterized by high binding affinities, often in the nanomolar range, in comparison with the low to moderate Kd of common carbohydrate–lectin interactions. In principle, this fact should facilitate the application of FP protocols for the study of the binding between GAG and proteins. Fluorescent GAG oligosaccharides can be prepared as high affinity FP probes, reducing the amount of protein required in competition experiments and increasing the range of inhibitor binding affinities that can be calculated.12 However, there are still few examples in the literature of FP studies for GAG–protein interactions, probably due to the complexity of GAG oligosaccharide synthesis.In our group, a competition assay for the analysis of GAG oligosaccharide–protein binding has been developed.68–70 First, we accomplished the synthesis of a suitable fluorescent probe (Fig. 13).68 For this purpose, commercially available heparin hexasaccharide 42, derived from the natural polysaccharide by enzymatic depolymerization, was conjugated with fluorescein hydrazide 43 to afford the corresponding glycosyl hydrazide 44 in excellent yield. We then tested the binding between 44 and basic fibroblast growth factor (FGF-2), a GAG–binding protein with a key role in angiogenesis and tumor cell growth. The FP of the fluorescent oligosaccharide was measured alone and in the presence of different concentrations of FGF-2. A binding curve was obtained by plotting the FP values against protein concentration. Nonlinear regression analysis afforded the dissociation constant of the interaction (Kd = 117 ± 10 nM) that was in good agreement with previously reported data on the binding affinity between a heparin hexasaccharide and FGF-2.
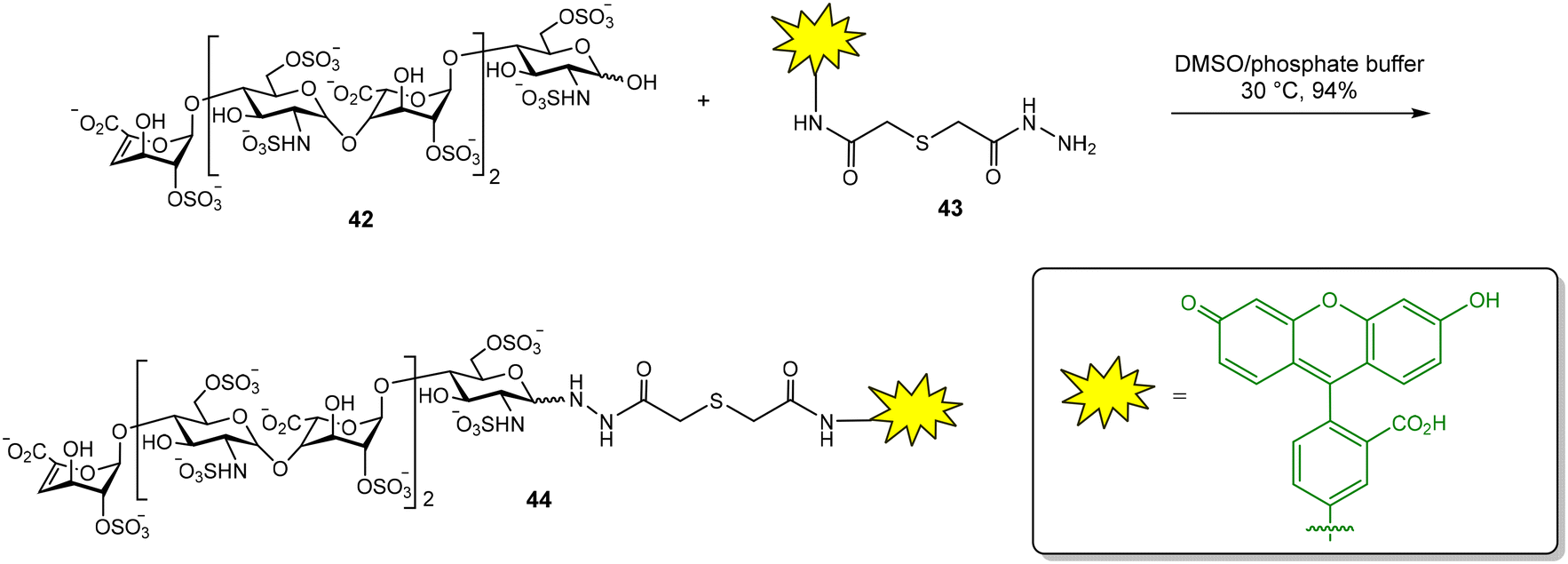 | ||
| Fig. 13 Synthesis of a fluorescent heparin hexasaccharide as FP probe. | ||
With a proper fluorescent probe in hand, the competition experiment was then implemented. The ability of a collection of synthetic GAG-like oligosaccharides to inhibit the interaction between FGF-2 and probe 44 was determined.68,69 An initial screening using 25 μM inhibitor concentration allowed to quickly identify those compounds displaying high inhibitory potencies. The displacement of fluorescent probe 44 by a nonfluorescent compound resulted in a decrease of the polarization value (Fig. 2B). Two control samples were included in this experiment. The first one contained only fluorescent probe 44 and indicated the expected FP value for 100% inhibition. The second one contained FGF-2 and 44 without any inhibitor and showed the FP value corresponding to 0% inhibition. Using these control values, the inhibition percentages achieved by each compound could be calculated. For compounds displaying a high inhibitory potency in the initial screening at 25 μM, the relative binding affinities, expressed as IC50 values, were calculated. For this, we measured the FP of samples containing 10 nM probe, 100 nM FGF-2 and increasing concentrations of the inhibitor. The analysis of the resulting competition curve afforded the IC50 value, defined as the compound concentration required for 50% inhibition. It is important to highlight the low protein and probe quantities required for these experiments: 384-well plates containing 40 μL per well with 10 nM probe and 100 nM protein solutions were employed. Bovine serum albumin (BSA) was included in the protein solutions to prevent nonspecific adsorption on the microplate wells.
This competition assay was also applied to midkine, another GAG–binding protein that plays an important role in the development of the central nervous system by promoting neuronal growth and is also involved in pathological processes such as cancer and inflammatory diseases. Midkine is a promising molecule for drug development and there is a great interest in the discovery of high affinity ligands for this protein. The direct binding assay between midkine and fluorescent heparin hexasaccharide 44 afforded a Kd of 44 ± 5 nM for the interaction.70 The competition protocol was then employed to screen the relative binding affinities of our library of synthetic compounds that included heparin and CS oligosaccharides and mimetics. Some of them are shown in Fig. 14. A midkine concentration close to the Kd for the probe/protein interaction was used in the experiments. Structure–activity relationships were deduced from these results.70–72 CS-E tetrasaccharide 46, presenting sulfate groups at positions 4 and 6 of the N-acetyl-galactosamine (GalNAc) unit, bound to midkine in the high micromolar range (IC50 = 254 μM). CS tetramer 48, displaying three more sulfates, showed higher binding affinity (IC50 = 10.6 μM), as expected for GAG–protein binding events, mainly driven by electrostatic interactions between positively charged amino acid residues of the protein and anionic sulfate and carboxylate moieties of the glycan. Interestingly, in both cases, the introduction of benzyl ethers at position 3 of the glucuronic acid (GlcA) gave lower IC50 values (compounds 45 and 47). The synthesis of natural CS sequences, such as 46, is very challenging. In order to facilitate the access to oligosaccharides showing affinity for midkine, we synthesized tetrasaccharide mimetics 49 and 50, where GlcA residues were replaced by glucose units.73 These compounds also bound to midkine in the high micromolar range, demonstrating that the carboxylate group is not essential for the interaction. The synthesis was further simplified replacing GalNAc by 2-amido-2-deoxy-glucose units and mimetics 51 and 52 were prepared and evaluated.74 Their IC50 values indicated much higher affinities for midkine than natural ligand 46. Our results suggested that increasing the hydrophobicity of sulfated oligosaccharides by introducing hydrophobic functionalities enhanced the binding affinity for midkine. In fact, sulfated, fully protected oligosaccharides showed IC50 values in the nanomolar range.75,76 These mimetics could potentially modulate the interactions between naturally occurring GAG and midkine, and the subsequent biological activities. Our FP competition experiment was also applied to pleiotrophin, another GAG-binding growth factor closely related to midkine.71,77 It has been also employed for the study of the binding affinities of multivalent systems displaying CS-E disaccharides.78
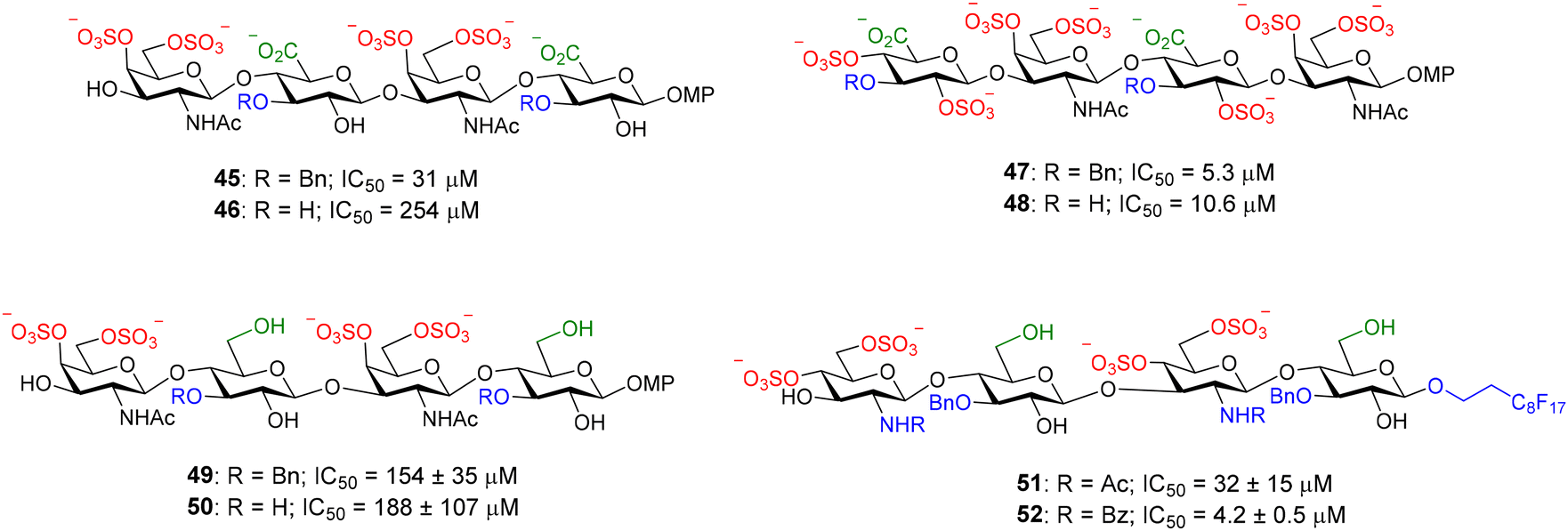 | ||
| Fig. 14 Examples of nonfluorescent CS tetrasaccharides and mimetics tested in our FP competition assay. Relative binding affinities for midkine are indicated by IC50 values. MP = 4-methoxyphenyl. | ||
The groups of Rademann and Pisabarro reported a FP protocol to study the interactions between sulfated HA oligosaccharides and ten representative regulatory proteins including cytokines, chemokines and growth factors.79 Compounds 58 and 59 were prepared and used as fluorescent probes (Fig. 15). HA tetra- and hexasaccharides 53 and 54 were derived from a chemoenzymatic approach followed by the introduction of the azido group with β configuration. Compounds 53 and 54 were conjugated with TAMRA-functionalized propargyl amide 55 using Cu-catalysed azide–alkyne cycloaddition reaction to afford fluorescent oligosaccharides 56 and 57 in good yield. Next, fully O-sulfation was achieved by treatment with SO3·pyridine complex in DMF to provide probes 58 and 59 displaying 9 and 13 sulfate groups, respectively. First, a direct FP binding assay gave the dissociation constants for the interactions between 58, 59 and the different proteins. The fluorescent, fully sulfated HA oligosaccharides bound to all tested proteins with high affinities (in most cases, Kd in the nanomolar range). Then, FP competition experiments allowed the determination of the binding affinities for a small library of unlabeled HA oligomers. FP results indicated that binding affinities depend on the oligosaccharide length, the position and number of sulfates, and the nature of the functional group at the reducing end anomeric position. Interestingly, the highest binding affinities were obtained with oligosaccharides containing a fluorophore moiety at the anomeric position (58, 59) and with a bivalent molecule showing two copies of the fully sulfated tetramer.
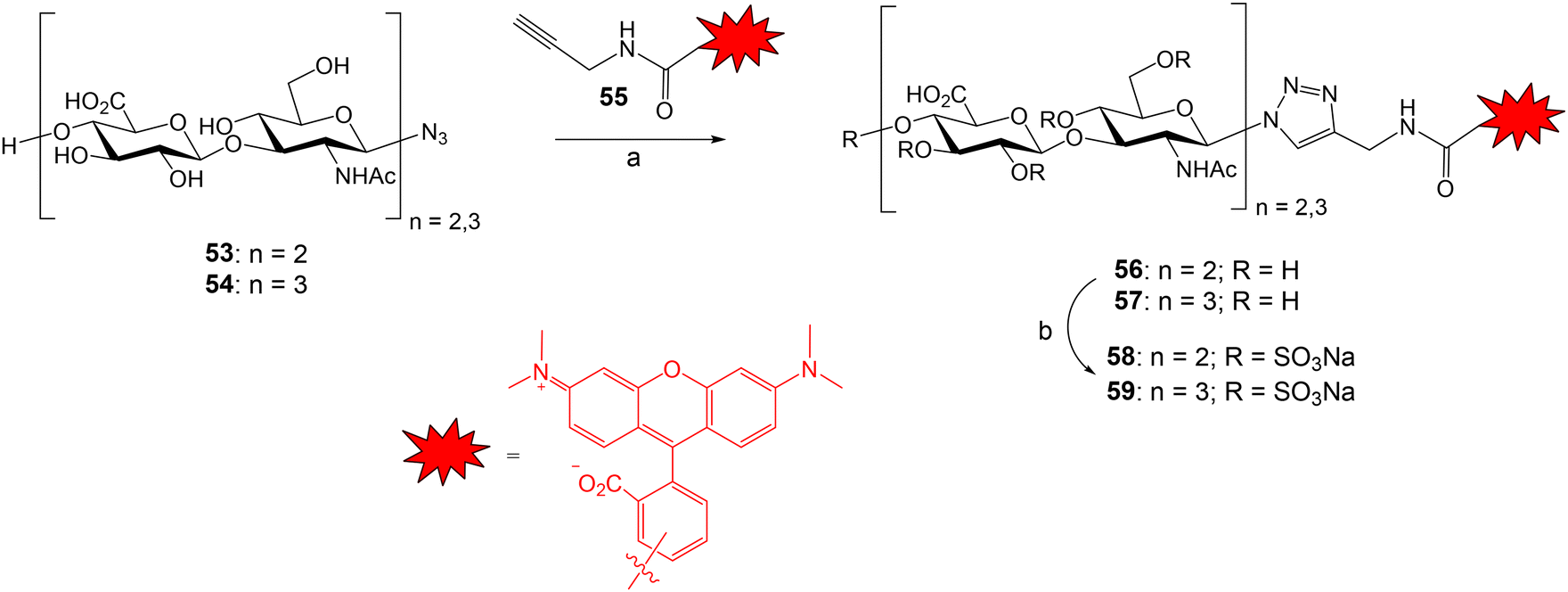 | ||
| Fig. 15 Synthesis of the fluorescent HA probes. Reagents and conditions: (a) TBTA, CuSO4·5H2O, sodium ascorbate, MeOH/H2O 3:1, 93% (56); 79% (57); (b) SO3·Py, DMF, 61–82% (58); 61–76% (59). | ||
The GAG binding properties of chemokine XCL1 has also been evaluated by FP.80 In this case, a fluorescein-tagged heparin tetrasaccharide (compound 60, Fig. 16), showing a dissociation constant value of 2.2 ± 0.2 μM for XCL1, was employed as a probe. Competition assays enabled the calculation of the binding affinities of several unlabeled heparan sulfate tetra- and octasaccharides. The results indicated that both length and sulfation pattern influence GAG–XCL1 interactions. Another example of FP experiments for GAG–protein interactions is the use of fluorescently labelled heparin to study the binding to bovine prion protein.81 The probe was prepared by conjugation of AlexaFluor 488 succinimidyl ester with the N-unsubstituted glucosamine residues of heparin. Fluorescent heparin showed enhanced FP values after binding to the protein and a Kd of 73 ± 4 nM was calculated for the interaction. This method also allowed to examine the effects of ionic strength, divalent metals, detergents and non-labelled GAG polysaccharides on the interaction. AlexaFluor 488-functionalized heparin (61, Fig. 16) was also employed in a FP competition experiment to study the interaction between bone morphogenetic protein 2 and a glycodendrimer displaying four copies of oversulfated maltose.82
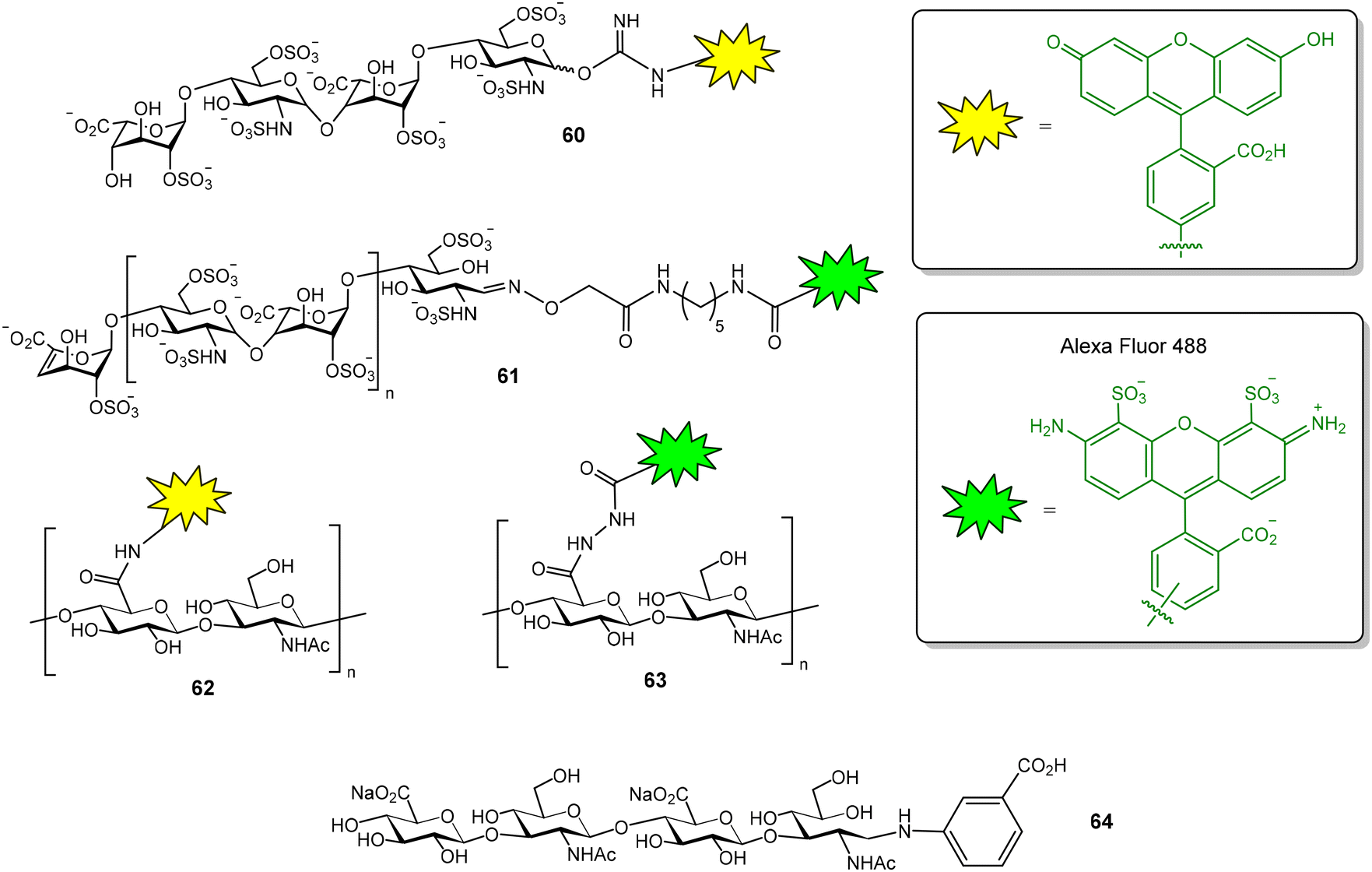 | ||
| Fig. 16 Heparin (60–61) and hyaluronic acid (62–64) fluorescent probes used in FP studies. | ||
FP experiments were recorded to evaluate the interactions between the transmembrane receptor CD44 and structurally modified HA samples.83 For this purpose, fluorescent HA was first prepared by activation of the carboxylate groups of the polysaccharide followed by treatment with fluoresceinamine. Using this probe (62, Fig. 16), the binding of N-deacetylated and 6-O-sulfated HA samples could be evaluated and the results suggested that both chemical modifications decrease CD44 interaction. A FP-based method has also been reported to measure the activity of hyaluronidase, the enzyme that hydrolyses HA to produce oligomers.84 In this case, fluorescent HA was prepared by reaction of the carboxylate groups of HA with Alexa Fluor 488 hydrazide (63, Fig. 16). The addition of hyaluronidase resulted in a decrease in the FP value due to the hydrolysis of the fluorescent HA polysaccharide and the subsequent production of low molecular weight oligomers. On the other hand, the interaction between hyaluronidase and fluorescent HA tetra- to decasaccharides (such as oligosaccharide 64, Fig. 16) was also studied by FP.85 Oligosaccharides were tagged at the anomeric position, using reductive amination reaction conditions, with 3-aminobenzoic acid and NaBH3CN. FP measurements were performed at 4 °C to reduce the enzyme hydrolysis of the oligomers.
Conclusions
FP experiments afford valuable data on the interactions between carbohydrates and proteins, in a very simple and fast way. In this method, a standard microplate reader is employed and minimal sample quantities are usually consumed. FP results complement the data obtained by other techniques. Unlike SPR and ELISA-like assays, FP does not require immobilization to a solid support, avoiding washing steps and complicated experimental procedures. Compared to other in-solution methods (ITC, STD-NMR), FP requires lower amounts of samples. It is important to note that some discrepancies may be found between the Kd/IC50 values obtained with FP or other bioanalytical techniques because the calculated parameters depend on the experimental setup. For example, binding affinities provided by FP assays in solution can be lower than those given by experiments involving carbohydrate-coated surfaces.76 The multivalent presentation of the carbohydrate on the surface increases glycan–protein affinities, in comparison with the solution-phase FP results, due to the cluster effect.The search for compounds that blocked carbohydrate–protein binding is highly demanded since these inhibitors could modulate these interactions and the subsequent biological processes. In this context, FP competition assays are particularly interesting because they allow the screening of hundreds or thousands of derivatives to quickly identify potent inhibitors.
The main requirement to develop FP-based assays is the design and preparation of a suitable, low molecular weight saccharide fluorescent probe that should interact with the protein of interest with high affinity (ideally in the nanomolar range). This fact has limited the use of FP in the study of glycan–protein interactions that are often characterized by low to moderate binding affinities. However, several research groups have solved this problem, reporting the design and synthesis of high affinity fluorescent probes that allowed the discovery of inhibitors with low nM affinities.23,43 We envisioned that the development of novel, high affinity saccharide fluorescent probes will increase the number of applications of FP in the carbohydrate–protein interactions field. For example, the discovery and synthesis of novel covalent activity-based probes will enable to consider different glycoprocessing enzymes, and advances in GAG oligosaccharide synthesis should facilitate the access to new GAG-like fluorescent probes for the analysis of the binding events involving this particular class of carbohydrate sequences.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank grant PID2021-125094NB-100 funded by MCIN/AEI/10.13039/501100011033 and by ERDF A way of making Europe, EU.References
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Methods Appl. Fluoresc., 2016, 4, 022001 CrossRef PubMed.
- W. A. Lea and A. Simeonov, Expert Opin. Drug Discovery, 2011, 6, 17–32 CrossRef CAS PubMed.
- L. Hua, D. Wang, K. Wang, Y. Wang, J. Gu, Q. Zhang, Q. You and L. Wang, J. Med. Chem., 2023, 66, 10934–10958 CrossRef CAS PubMed.
- X. Huang and A. Aulabaugh, in High Throughput Screening, ed. W. P. Janzen, Springer New York, New York, NY, 2016, vol. 1439, pp. 115–130 Search PubMed.
- H. Zhang, Q. Wu and M. Y. Berezin, Expert Opin. Drug Discovery, 2015, 10, 1145–1161 CrossRef CAS PubMed.
- J. A. Cisneros, M. J. Robertson, M. Valhondo and W. L. Jorgensen, J. Am. Chem. Soc., 2016, 138, 8630–8638 CrossRef CAS PubMed.
- J. P. Ludeman, M. Nazari-Robati, B. L. Wilkinson, C. Huang, R. J. Payne and M. J. Stone, Org. Biomol. Chem., 2015, 13, 2162–2169 RSC.
- Y.-C. He, B.-C. Yin, L. Jiang and B.-C. Ye, Chem. Commun., 2014, 50, 6236–6239 RSC.
- D. Solís, N. V. Bovin, A. P. Davis, J. Jiménez-Barbero, A. Romero, R. Roy, K. Smetana and H.-J. Gabius, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850, 186–235 CrossRef PubMed.
- K. Kakehi, Y. Oda and M. Kinoshita, Anal. Biochem., 2001, 297, 111–116 CrossRef CAS PubMed.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- X. Huang, J. Biomol. Screening, 2003, 8, 34–38 CrossRef CAS PubMed.
- D. Hauck, I. Joachim, B. Frommeyer, A. Varrot, B. Philipp, H. M. Moeller, A. Imberty, T. E. Exner and A. Titz, ACS Chem. Biol., 2013, 8, 1775–1784 CrossRef CAS PubMed.
- R. Sommer, S. Wagner, K. Rox, A. Varrot, D. Hauck, E.-C. Wamhoff, J. Schreiber, T. Ryckmans, T. Brunner, C. Rademacher, R. W. Hartmann, M. Brönstrup, A. Imberty and A. Titz, J. Am. Chem. Soc., 2018, 140, 2537–2545 CrossRef CAS PubMed.
- C. Sabin, E. P. Mitchell, M. Pokorná, C. Gautier, J. P. Utille, M. Wimmerová and A. Imberty, FEBS Lett., 2006, 580, 982–987 CrossRef CAS PubMed.
- I. Joachim, S. Rikker, D. Hauck, D. Ponader, S. Boden, R. Sommer, L. Hartmann and A. Titz, Org. Biomol. Chem., 2016, 14, 7933–7948 RSC.
- O. Metelkina, J. Konstantinović, A. Klein, R. Shafiei, M. Fares, A. Alhayek, S. Yahiaoui, W. A. M. Elgaher, J. Haupenthal, A. Titz and A. K. H. Hirsch, Chem. Sci., 2024, 15, 13333–13342 RSC.
- S. Wagner, D. Hauck, M. Hoffmann, R. Sommer, I. Joachim, R. Müller, A. Imberty, A. Varrot and A. Titz, Angew. Chem., Int. Ed., 2017, 56, 16559–16564 CrossRef CAS PubMed.
- J. Rodrigue, G. Ganne, B. Blanchard, C. Saucier, D. Giguère, T. C. Shiao, A. Varrot, A. Imberty and R. Roy, Org. Biomol. Chem., 2013, 11, 6906–6918 RSC.
- E. Zahorska, F. Rosato, K. Stober, S. Kuhaudomlarp, J. Meiers, D. Hauck, D. Reith, E. Gillon, K. Rox, A. Imberty, W. Römer and A. Titz, Angew. Chem., Int. Ed., 2023, 62, e202215535 CrossRef CAS PubMed.
- E. Zahorska, S. Kuhaudomlarp, S. Minervini, S. Yousaf, M. Lepsik, T. Kinsinger, A. K. H. Hirsch, A. Imberty and A. Titz, Chem. Commun., 2020, 56, 8822–8825 RSC.
- F. R. Zetterberg, K. Peterson, U. J. Nilsson, K. Andréasson Dahlgren, C. Diehl, I. Holyer, M. Håkansson, A. Khabut, B. Kahl-Knutson, H. Leffler, A. C. MacKinnon, J. A. Roper, R. J. Slack, R. Zarrizi and A. Pedersen, J. Med. Chem., 2024, 67, 9374–9388 CrossRef CAS PubMed.
- K. Peterson, R. Kumar, O. Stenström, P. Verma, P. R. Verma, M. Håkansson, B. Kahl-Knutsson, F. Zetterberg, H. Leffler, M. Akke, D. T. Logan and U. J. Nilsson, J. Med. Chem., 2018, 61, 1164–1175 CrossRef CAS PubMed.
- S. Kuhaudomlarp, E. Siebs, E. Shanina, J. Topin, I. Joachim, P. Da Silva Figueiredo Celestino Gomes, A. Varrot, D. Rognan, C. Rademacher, A. Imberty and A. Titz, Angew. Chem., Int. Ed., 2021, 60, 8104–8114 CrossRef CAS PubMed.
- G. Beshr, R. Sommer, D. Hauck, D. C. B. Siebert, A. Hofmann, A. Imberty and A. Titz, MedChemComm, 2016, 7, 519–530 RSC.
- P. Sorme, B. Kahl-Knutsson, M. Huflejt, U. J. Nilsson and H. Leffler, Anal. Biochem., 2004, 334, 36–47 CrossRef PubMed.
- S. Carlsson, C. T. Öberg, M. C. Carlsson, A. Sundin, U. J. Nilsson, D. Smith, R. D. Cummings, J. Almkvist, A. Karlsson and H. Leffler, Glycobiology, 2007, 17, 663–676 CrossRef CAS PubMed.
- C. T. Öberg, S. Carlsson, E. Fillion, H. Leffler and U. J. Nilsson, Bioconjugate Chem., 2003, 14, 1289–1297 CrossRef PubMed.
- E. Salomonsson, A. Larumbe, J. Tejler, E. Tullberg, H. Rydberg, A. Sundin, A. Khabut, T. Frejd, Y. D. Lobsanov, J. M. Rini, U. J. Nilsson and H. Leffler, Biochemistry, 2010, 49, 9518–9532 CrossRef CAS PubMed.
- R. Kumar, K. Peterson, M. Misini Ignjatović, H. Leffler, U. Ryde, U. J. Nilsson and D. T. Logan, Org. Biomol. Chem., 2019, 17, 1081–1089 RSC.
- F. Tobola, M. Lepšík, S. R. Zia, H. Leffler, U. J. Nilsson, O. Blixt, A. Imberty and B. Wiltschi, ChemBioChem, 2022, 23, e202100593 CrossRef CAS PubMed.
- K. Peterson, P. M. Collins, X. Huang, B. Kahl-Knutsson, S. Essén, F. R. Zetterberg, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, RSC Adv., 2018, 8, 24913–24922 RSC.
- I. Cumpstey, S. Carlsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2005, 3, 1922 RSC.
- S. Van Klaveren, M. Hassan, M. Håkansson, R. E. Johnsson, J. Larsson, Ž. Jakopin, M. Anderluh, H. Leffler, T. Tomašič and U. J. Nilsson, ACS Med. Chem. Lett., 2024, 15, 1319–1324 CrossRef CAS PubMed.
- J. Dion, F. Deshayes, N. Storozhylova, T. Advedissian, A. Lambert, M. Viguier, C. Tellier, C. Dussouy, F. Poirier and C. Grandjean, ChemBioChem, 2017, 18, 782–789 CrossRef CAS PubMed.
- J. Dion, T. Advedissian, N. Storozhylova, S. Dahbi, A. Lambert, F. Deshayes, M. Viguier, C. Tellier, F. Poirier, S. Téletchéa, C. Dussouy, H. Tateno, J. Hirabayashi and C. Grandjean, ChemBioChem, 2017, 18, 2428–2440 CrossRef CAS PubMed.
- S. G. Gouin, J. M. García Fernández, E. Vanquelef, F. Dupradeau, E. Salomonsson, H. Leffler, M. Ortega-Muñoz, U. J. Nilsson and J. Kovensky, ChemBioChem, 2010, 11, 1430–1442 CrossRef CAS PubMed.
- T. Delaine, P. Collins, A. MacKinnon, G. Sharma, J. Stegmayr, V. K. Rajput, S. Mandal, I. Cumpstey, A. Larumbe, B. A. Salameh, B. Kahl-Knutsson, H. van Hattum, M. van Scherpenzeel, R. J. Pieters, T. Sethi, H. Schambye, S. Oredsson, H. Leffler, H. Blanchard and U. J. Nilsson, ChemBioChem, 2016, 17, 1759–1770 CrossRef CAS PubMed.
- K. B. Pal, M. Mahanti, X. Huang, S. Persson, A. P. Sundin, F. R. Zetterberg, S. Oredsson, H. Leffler and U. J. Nilsson, Org. Biomol. Chem., 2018, 16, 6295–6305 RSC.
- T. Zhang, Y. Zheng, D. Zhao, J. Yan, C. Sun, Y. Zhou and G. Tai, Int. J. Biol. Macromol., 2016, 91, 994–1001 CrossRef CAS PubMed.
- T. Diercks, F. J. Medrano, F. G. FitzGerald, D. Beckwith, M. J. Pedersen, M. Reihill, A. Ludwig, A. Romero, S. Oscarson, M. Cudic and H. Gabius, Chem. – Eur. J., 2021, 27, 316–325 CrossRef CAS PubMed.
- Z. Han, J. S. Pinkner, B. Ford, R. Obermann, W. Nolan, S. A. Wildman, D. Hobbs, T. Ellenberger, C. K. Cusumano, S. J. Hultgren and J. W. Janetka, J. Med. Chem., 2010, 53, 4779–4792 CrossRef CAS PubMed.
- S. Kleeb, L. Pang, K. Mayer, D. Eris, A. Sigl, R. C. Preston, P. Zihlmann, T. Sharpe, R. P. Jakob, D. Abgottspon, A. S. Hutter, M. Scharenberg, X. Jiang, G. Navarra, S. Rabbani, M. Smiesko, N. Lüdin, J. Bezençon, O. Schwardt, T. Maier and B. Ernst, J. Med. Chem., 2015, 58, 2221–2239 CrossRef CAS PubMed.
- I. Bertin-Jung, A. Robert, N. Ramalanjaona, S. Gulberti, C. Bui, J.-B. Vincourt, M. Ouzzine, J.-C. Jacquinet, C. Lopin-Bon and S. Fournel-Gigleux, Chem. Commun., 2020, 56, 10746–10749 RSC.
- D. Sugahara, J. Amano and T. Irimura, Anal. Sci., 2003, 19, 167–169 CrossRef CAS PubMed.
- M. Zhang, P. Wu, W.-T. Dou, H.-H. Han, X.-P. He, C. Tan and Y. Jiang, Chem. Commun., 2017, 53, 5625–5628 RSC.
- M. Mizuno, M. Noguchi, M. Imai, T. Motoyoshi and T. Inazu, Bioorg. Med. Chem. Lett., 2004, 14, 485–490 CrossRef CAS PubMed.
- O. Renaudet and P. Dumy, Org. Lett., 2003, 5, 243–246 CrossRef CAS PubMed.
- T. Okada, C. Isobe, T. Wada, S. Ezaki and N. Minoura, Bioconjugate Chem., 2013, 24, 841–845 CrossRef CAS PubMed.
- C. Li, K. Hon, B. Ghosh, P. Li, H. Lin, P. Chan, C. Lin, Y. Chen and K. T. Mong, Chem. – Asian J., 2014, 9, 1786–1796 CrossRef CAS PubMed.
- T. Okada and N. Minoura, J. Biomed. Opt., 2011, 16, 037001 CrossRef PubMed.
- N. De La Cruz, J. Ramos-Soriano, J. J. Reina, J. L. De Paz, M. Thépaut, F. Fieschi, A. Sousa-Herves and J. Rojo, Org. Biomol. Chem., 2020, 18, 6086–6094 RSC.
- C. H. Eller, G. Yang, O. Ouerfelli and R. T. Raines, Carbohydr. Res., 2014, 397, 1–6 CrossRef CAS PubMed.
- L. I. Mukhametova, D. O. Zherdev, S. A. Eremin, A. N. Kuznetsov, V. I. Yudin, O. D. Sclyarov, O. V. Babicheva, A. V. Motorygin, Y. E. Tsvetkov, V. B. Krylov and N. E. Nifantiev, Biosensors, 2024, 14, 404 CrossRef CAS PubMed.
- D. Lahav, B. Liu, R. J. B. H. N. Van Den Berg, A. M. C. H. Van Den Nieuwendijk, T. Wennekes, A. T. Ghisaidoobe, I. Breen, M. J. Ferraz, C.-L. Kuo, L. Wu, P. P. Geurink, H. Ovaa, G. A. Van Der Marel, M. Van Der Stelt, R. G. Boot, G. J. Davies, J. M. F. G. Aerts and H. S. Overkleeft, J. Am. Chem. Soc., 2017, 139, 14192–14197 CrossRef CAS PubMed.
- D. Van Der Gracht, R. J. Rowland, V. Roig-Zamboni, M. J. Ferraz, M. Louwerse, P. P. Geurink, J. M. F. G. Aerts, G. Sulzenbacher, G. J. Davies, H. S. Overkleeft and M. Artola, Chem. Sci., 2023, 14, 9136–9144 RSC.
- Z. Armstrong, C.-L. Kuo, D. Lahav, B. Liu, R. Johnson, T. J. M. Beenakker, C. De Boer, C.-S. Wong, E. R. Van Rijssel, M. F. Debets, B. I. Florea, C. Hissink, R. G. Boot, P. P. Geurink, H. Ovaa, M. Van Der Stelt, G. M. Van Der Marel, J. D. C. Codée, J. M. F. G. Aerts, L. Wu, H. S. Overkleeft and G. J. Davies, J. Am. Chem. Soc., 2020, 142, 13021–13029 CrossRef CAS PubMed.
- J. S. Helm, Y. Hu, L. Chen, B. Gross and S. Walker, J. Am. Chem. Soc., 2003, 125, 11168–11169 CrossRef CAS PubMed.
- B. J. Gross, B. C. Kraybill and S. Walker, J. Am. Chem. Soc., 2005, 127, 14588–14589 CrossRef CAS PubMed.
- C. D. Rillahan, S. J. Brown, A. C. Register, H. Rosen and J. C. Paulson, Angew. Chem., Int. Ed., 2011, 50, 12534–12537 CrossRef CAS PubMed.
- K. Seelhorst, K. Pahnke, C. Meier and U. Hahn, ChemBioChem, 2015, 16, 1919–1924 CrossRef CAS PubMed.
- J. J. Preidl, V. S. Gnanapragassam, M. Lisurek, J. Saupe, R. Horstkorte and J. Rademann, Angew. Chem., Int. Ed., 2014, 53, 5700–5705 CrossRef CAS PubMed.
- Z. Gao, O. G. Ovchinnikova, B.-S. Huang, F. Liu, D. E. Williams, R. J. Andersen, T. L. Lowary, C. Whitfield and S. G. Withers, J. Am. Chem. Soc., 2019, 141, 2201–2204 CrossRef CAS PubMed.
- X. Yin, J. Li, S. Chen, Y. Wu, Z. She, L. Liu, Y. Wang and Z. Gao, ChemBioChem, 2021, 22, 1391–1395 CrossRef CAS PubMed.
- J. Fu, H. Fu, Y. Xia, I. N'Go, J. Cao, W. Pan and S. P. Vincent, Org. Biomol. Chem., 2021, 19, 1818–1826 RSC.
- L. I. Mukhametova, D. O. Zherdev, A. N. Kuznetsov, O. N. Yudina, Y. E. Tsvetkov, S. A. Eremin, V. B. Krylov and N. E. Nifantiev, Biomolecules, 2024, 14, 170 CrossRef CAS PubMed.
- S. Perez, O. Makshakova, J. Angulo, E. Bedini, A. Bisio, J. L. de Paz, E. Fadda, M. Guerrini, M. Hricovini, M. Hricovini, F. Lisacek, P. M. Nieto, K. Pagel, G. Pairardi, R. Richter, S. A. Samsonov, R. A. Vives, D. Nikitovic and S. R. Blum, JACS Au, 2023, 3, 628–656 CrossRef CAS PubMed.
- S. Maza, M. Mar Kayser, G. Macchione, J. Lopez-Prados, J. Angulo, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2013, 11, 3510–3525 RSC.
- G. Macchione, S. Maza, M. M. Kayser, J. L. de Paz and P. M. Nieto, Eur. J. Org. Chem., 2014, 3868–3884 CrossRef CAS.
- C. Solera, G. Macchione, S. Maza, M. M. Kayser, F. Corzana, J. L. de Paz and P. M. Nieto, Chem. – Eur. J., 2016, 22, 2356–2369 CrossRef CAS PubMed.
- J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2016, 14, 3506–3509 RSC.
- M. J. Garcia-Jimenez, S. Gil-Caballero, S. Maza, F. Corzana, F. Juarez-Vicente, J. R. Miles, K. Sakamoto, K. Kadomatsu, M. Garcia-Dominguez, J. L. de Paz and P. M. Nieto, Chem. – Eur. J., 2021, 27, 12395–12409 CrossRef CAS PubMed.
- M. Torres-Rico, S. Maza, J. L. de Paz and P. M. Nieto, Org. Biomol. Chem., 2021, 19, 5312–5326 RSC.
- J. L. De Paz, M. J. García-Jiménez, V. Jafari, M. García-Domínguez and P. M. Nieto, Bioorg. Chem., 2023, 141, 106929 CrossRef CAS PubMed.
- S. Maza, N. Gandia-Aguado, J. L. de Paz and P. M. Nieto, Bioorg. Med. Chem., 2018, 26, 1076–1085 CrossRef CAS PubMed.
- S. Maza, J. L. de Paz and P. M. Nieto, Molecules, 2019, 24, 1591 CrossRef CAS PubMed.
- M. J. García-Jiménez, M. Torres-Rico, J. L. De Paz and P. M. Nieto, Int. J. Mol. Sci., 2022, 23, 3026 CrossRef PubMed.
- P. Dominguez-Rodriguez, J. J. Reina, S. Gil-Caballero, P. M. Nieto, J. L. de Paz and J. Rojo, Chem. – Eur. J., 2017, 23, 11338–11345 CrossRef CAS PubMed.
- S. Kohling, J. Blaszkiewicz, G. Ruiz-Gomez, M. I. Fernandez-Bachiller, K. Lemmnitzer, N. Panitz, A. G. Beck-Sickinger, J. Schiller, M. T. Pisabarro and J. Rademann, Chem. Sci., 2019, 10, 866–878 RSC.
- J. C. Fox, R. C. Tyler, F. C. Peterson, D. P. Dyer, F. Zhang, R. J. Linhardt, T. M. Handel and B. F. Volkman, Biochemistry, 2016, 55, 1214–1225 CrossRef CAS PubMed.
- O. Andrievskaia, Z. Potetinova, A. Balachandran and K. Nielsen, Arch. Biochem. Biophys., 2007, 460, 10–16 CrossRef CAS PubMed.
- R. A. A. Smith, X. Luo, X. Lu, T. C. Tan, B. Q. Le, O. V. Zubkova, P. C. Tyler, V. Nurcombe and S. M. Cool, Biomater. Adv., 2023, 155, 213671 CrossRef CAS PubMed.
- D. S. Bhattacharya, D. Svechkarev, J. J. Souchek, T. K. Hill, M. A. Taylor, A. Natarajan and A. M. Mohs, J. Mater. Chem. B, 2017, 5, 8183–8192 RSC.
- T. Murai and H. Kawashima, Biochem. Biophys. Res. Commun., 2008, 376, 620–624 CrossRef CAS PubMed.
- M. Kinoshita, A. Okino, Y. Oda and K. Kakehi, Electrophoresis, 2001, 22, 3458–3465 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |