
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H amination of enolizable and nonenolizable ketones†
Oksana Holovko-Kamoshenkovaac,
Zdeněk Tošnera,
Ivana Císařováb and
Radim Hrdina *a
*a
aCharles University, Faculty of Science, Department of Organic Chemistry, Hlavova 8, 12840 Praha, Czech Republic. E-mail: hrdina@natur.cuni.cz
bCharles University, Faculty of Science, Department of Inorganic Chemistry, Hlavova 8, 12840 Praha, Czech Republic
cUzhhorod National University, Narodna ploshcha 3, 88000 Uzhhorod, Ukraine
First published on 19th February 2025
Abstract
We present a method for the amination of enolizable and non-enolizable ketones in the alpha (or beta) position to the carbonyl group. This approach is based on the conversion of the corresponding cyanohydrins to carbonazidates, precursors for thermal intramolecular nitrene insertion reactions into the adjacent C–H bond. Hydrolysis of the resulting carbamates under basic conditions with simultaneous regeneration of the carbonyl group yields amino ketones.
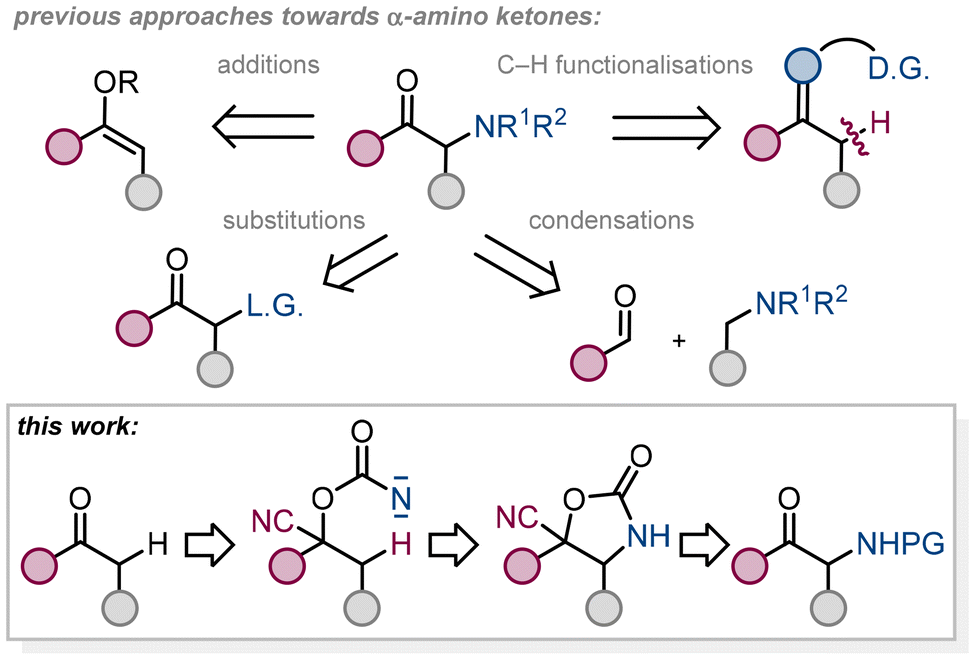
α-Amino ketones are common motifs in medicinal chemistry and drug design of antidepressants, appetite suppressants, and ACE-inhibitors and are versatile reagents in organic chemistry for the preparation of heterocycles and other synthetic building blocks.1 There are several methods for producing this type of structural pattern. α-Amino ketones, derived from enolizable precursors, are typically prepared by α-deprotonation of the starting material and a subsequent reaction with an electrophile.2–8 This electrophile is either a nitrogen-containing moiety or a group which is then subjected to a substitution reaction with nitrogen-based nucleophiles. Carbonyl α-aminations are performed also under reverse polarity mode using nitrogen nucleophiles.9 α-Amino ketones can be prepared by means other than functionalisation of the C–H bond adjacent to the carbonyl group, such as transformation of α-amino acids, aza-benzoin condensation reactions, transfer hydrogenation of alpha hydroxy imines, hydrolysis of substituted triazoles, ring opening of azetidine-3-ols, one pot oxidation of unsaturated carbon–carbon bonds, reductive coupling reactions between imines and nitriles, oxidation of enamines, functionalization of enones and other synthetic approaches.10 A further synthetic option to generate α-nitrogen substituted compounds is the conversion of a carbonyl to a transient directing group, which is then used for C–H functionalisation reactions.11 Preparation of β-amino ketones12 may rely on C–H activation strategies as well.13 To the best of our knowledge, the conversion of ketones to cyanohydrins and the use of such a moiety for intramolecular C–H amination reactions14 via nitrene insertions15–17 has not been explored. Intramolecular C–H amination reactions for the preparation of carbamates from monosubstituted alcohol derivatives as starting materials are well known and can be divided into two main categories; transition metal nitrenoid insertion reactions into C–H bonds18 and metal-free generation of reactive nitrene intermediates19 (under thermal or photochemical conditions)20 that insert into C–H bonds. There are various precursors for generation of alkoxycarbonylnitrene intermediates such as carbamates (ROCONH2)21–24 and their oxidation, carbonazidates (ROCON3)25–27 and their molecular nitrogen extrusion or N-oxidised carbamates (ROCONHOR′)28,29 and their R′OH elimination (Fig. 1).
 | ||
| Fig. 1 C–H insertion reaction of nitrenes derived from cyanohydrin carbonazidates. | ||
As a precursor for the generation of reactive nitrene intermediates, we decided to convert the hydroxy group of cyanohydrins to a versatile carbonazidate and use this moiety for molecular nitrogen extrusion reactions, which can theoretically be carried out under thermal or photochemical conditions or metal promotion, and a subsequent intramolecular insertion reaction of the generated nitrene species into the C–H bond, taking into account the influence of the incorporated cyano group on the reaction progress under different conditions. Eight different ketones 1 were converted to cyanohydrins 2 using various methods including the recent protocol of Zheng and Zhou.30 The prepared cyanohydrins do not require further purification and can be used directly in the next step, formation of carbonazidates (Fig. 2).
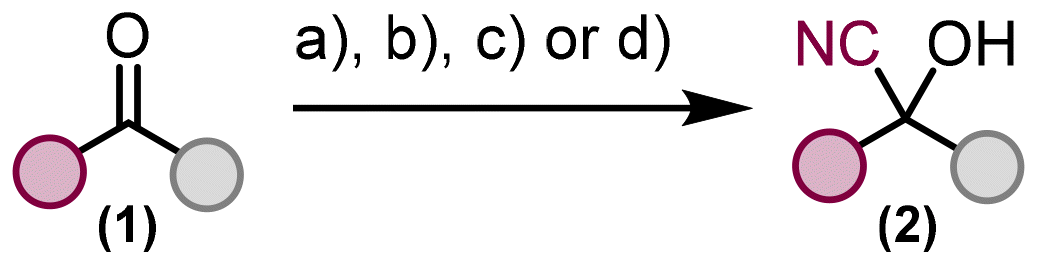 | ||
| Fig. 2 Methods for cyanohydrin formation from ketones: (a) I. NaHSO3, H2O, 45 °C, 10 min; II. KCN, 10 °C–24 °C, 10 min; III. 24 °C, 16 h; (b) I. CO2, EtOH, 24 °C, 10 min; II. KCN, 24 °C, 15 min; III. ketone addition, 24 °C, 18 h; (c) Me3SiCN/BF3·Et2O, DCM, 24 °C, 2 h; (d) Me3SiCN, phosphorus ylide as a catalyst (0.1 mol%), MeCN, 24 °C. | ||
To avoid multiple manipulations with hazardous chemicals such as triphosgene or sodium azide, we have used a procedure31 to prepare the corresponding carbonazidates in one pot (Fig. 3). Pyridine was chosen as the solvent, and all compounds, sodium azide, starting material 1 and triphosgene, were added subsequently at −20 °C and the reaction mixture was stirred first at 24 °C for 1 hour and at 50 °C for a further 5 hours. The inorganic compounds were extracted into water and the desired product 3 was extracted into ethyl acetate. All carbonazidates 3 prepared in this way are bench stable, can be concentrated under vacuum and were isolated in yields ranging from 67% to 84%.
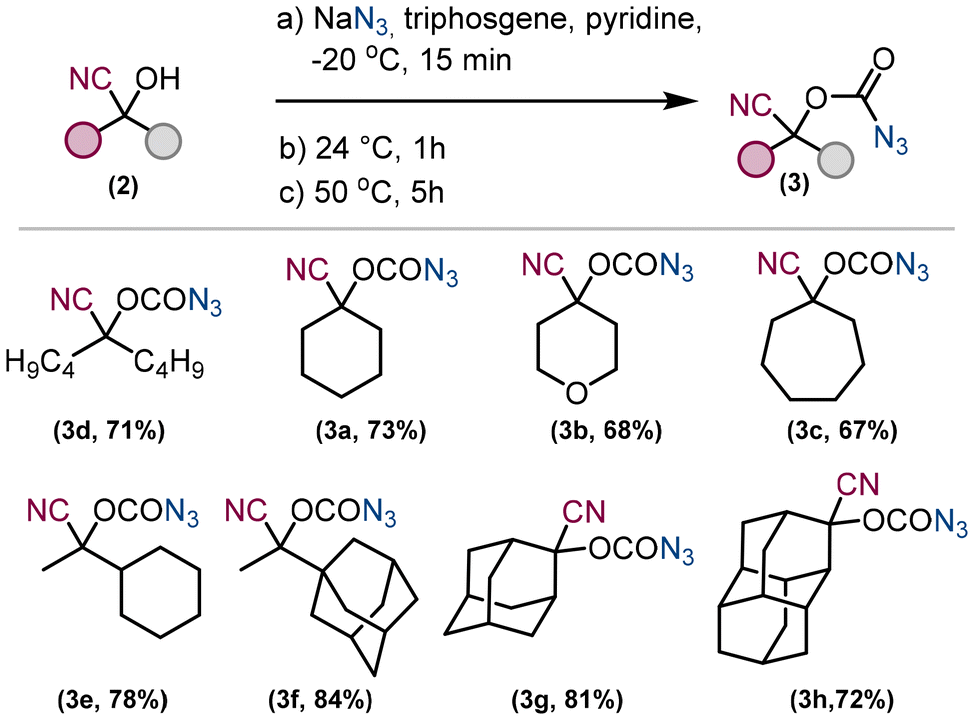 | ||
| Fig. 3 Preparation of carbonazidates from cyanohydrins. | ||
The diamantane32,33 derivative 3h was chosen as a model compound for the desired amination reaction (Fig. 4). All compounds 3 are well soluble in 1,2-dichloroethane, which is often the solvent of choice for such C–H amination reactions. The conditions for thermal reactions were first investigated. Two compounds of 4h were prepared and isolated in good yields. The structure of 4h2 was confirmed by X-ray crystallography. Dilution (0.7 mmol of the corresponding carbonazidate in 5 mL of dry DCE) and 130 °C were then used for all substrates 3.
 | ||
| Fig. 4 Amination reaction towards cyclic carbamates. All depicted compounds in the table were isolated and characterised. | ||
The reaction with 3h was then carried out under UV irradiation (254 nm) at 24 °C in CH2Cl2. The conversion was complete, and the ratio of isomers 4h1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4h2 was 1:1.3 (see ESI pages S17 and S18†). The cobalt catalysed azide decomposition of 3a using Sugbok Chang's catalyst and conditions26 did not give the desired product (see ESI page S23†). This is probably due to the coordination of the nitrile group to the Co atom in the complex.
:4h2 was 1:1.3 (see ESI pages S17 and S18†). The cobalt catalysed azide decomposition of 3a using Sugbok Chang's catalyst and conditions26 did not give the desired product (see ESI page S23†). This is probably due to the coordination of the nitrile group to the Co atom in the complex.
The properties of the catalyst and the reaction conditions were checked on standard published substrates to avoid any error in catalyst preparation or reaction performance. To test another catalytic approach to the preparation of cyclic carbamates, adamantan-2-one was converted to a cyanohydrin derivative and the resulting OH group was then converted to carbamate 13 (–OCONH2 group) (see ESI page S22†). Using compound 13 as a starting material, we tested typical conditions for dirhodium catalysed nitrenoid transfer (see ESI page S22†), generating the nitrene by oxidation of the OCONH2 group with PhI(OAc)2. Again, the reaction did not proceed to give the desired product for the same reason, coordination of the nitrile to the rhodium atom, which deactivates the catalyst.
The amination reactions were then all carried out thermally and the results are summarised in Fig. 4. The thermal reaction of 3a gave two diastereomers 4a1 and 4a2 in good yields. These isomers were separated by column chromatography on silica gel and their structures elucidated. The structure of 4a1 was confirmed by X-ray crystallography. The thermal decomposition of tetrahydropyran derivative 3b resulted in a complete conversion to a mixture of diastereomers 4b1 and 4b2 in a ratio of 1:0.8. These isomers were separated and characterised. The cycloheptane derivative 3c underwent the same transformation to give the isomers 4c1 and 4c2 in a ratio of 1:0.9. The acyclic derivative 3d gave a mixture of 4 compounds, two diastereomers of the 5-membered carbamate derivative 4d1a and 4d1b in the ratio 1:0.6 and two diastereomers of the 6-membered carbamate derivative 4d2a and 4d2b in the ratio 1:0.4. The structure of 4d2a was confirmed by X-ray crystallography. We then subjected the derivative 3e containing primary, secondary and tertiary C–H groups to thermal decomposition. The reaction gave a mixture of 5 isomers, five-membered carbamate derivative 4e1 and four isomers of the six-membered carbamate derivative 4e2. The isomer 4e2a was isolated and its structure confirmed by 2D-NMR. A similar substrate 3f with an adamantyl group instead of the cyclohexyl group (substrate 3e) provided a mixture of three compounds: the five-membered carbamate 4f1, as a result of nitrene insertion into the C–H bond of the methyl group (isolated in 12% yield) and two diastereomers of the six-membered carbamates 4f2a and 4f2b in a ratio of 1:0.8. These isomers were separated and characterised (their relative configuration was not determined). Finally, non-enolizable ketone derived substrates 3g and 3h were selected. Thermal decomposition of the adamantane derivative 3g afforded the insertion product 4g in high isolated yield. Thermal decomposition of the diamantane derivative 3f yielded two products (regioisomers) as a result of nitrene insertion to the C–H bond in the apical position, 4h1, and in the belt position, 4h2. The structure of product 4h2 was confirmed by X-ray crystallography. These results (regio- and diastereo-selectivity) reflect the high reactivity of the generated nitrene species (singlet and triplet) and low differences in dissociation energies of neighbouring C–H bonds. The synthesis of structurally diverse oxazolidinones bearing tetra-substituted carbon atoms is still considered to be a challenge.34
Model compound 4a1 was chosen to demonstrate the cleavage of the carbamate moiety (Fig. 5). Under acidic hydrolysis conditions, compounds 4 can be converted to α-hydroxy, β-amino carboxylic acid derivatives 5 and under basic hydrolysis conditions to α-amino ketones 6. To avoid side hydrolysis of the nitrile, 4a1 was first protected with Boc to allow carbamate cleavage under milder hydrolysis conditions. Boc protection of an amino group also prevents undesired self-condensation reactions of an α-amino ketone.
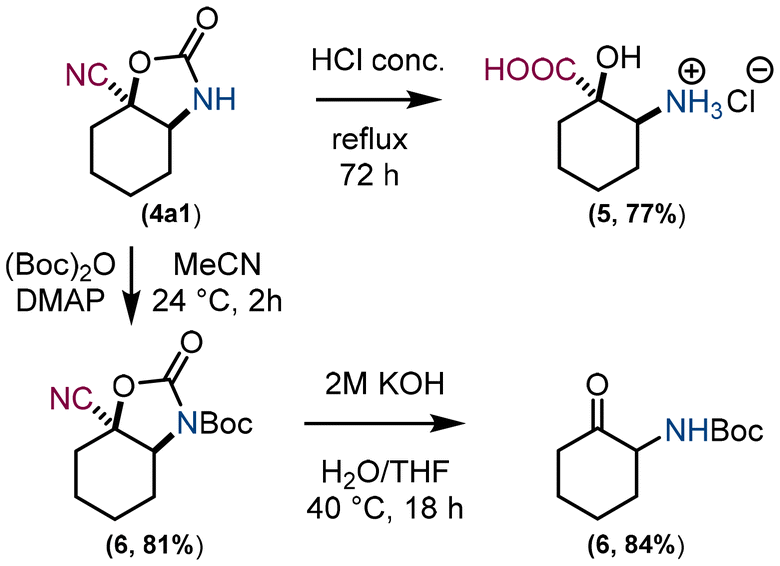 | ||
| Fig. 5 Cleavage of the carbamate group using acidic and basic hydrolysis. | ||
Our new approach to amino ketones was demonstrated using the natural compound estrone (Fig. 6). The commercially available O-methyl estrone derivative 7 was converted to the cyanohydrin derivative 8. In the next step, the hydroxy group of the cyanohydrin derivative 8 was converted to carbonazidate 9.
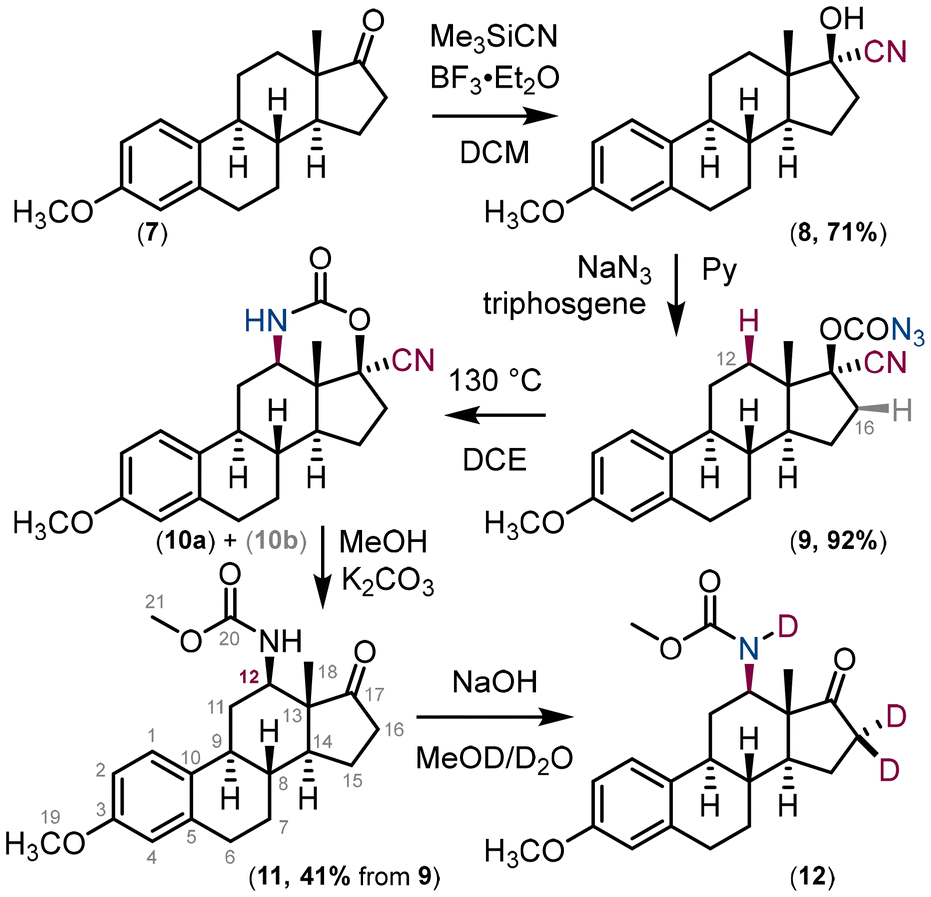 | ||
| Fig. 6 Synthesis of the C(12) N-substituted estrone derivative 11. Compound 10b is a result of nitrene insertion into the C(16)–H bond. | ||
Carbonazidate derivative 9 was used in the thermal nitrene insertion reaction to give a mixture of two insertion products – the six-membered carbamate 10a as a result of nitrene insertion into the C(12)–H bond and the five-membered carbamate 10b as a result of nitrene insertion into the C(16)–H bond in a 1:1 ratio. The formation of the corresponding diastereomers (for 10a and 10b) was not observed presumably due to the rigidity of the steroid structure. The mixture of inseparable (by column chromatography) isomers was used in the next step, the methanolysis of the carbamate under basic conditions. The major product 11 was isolated in 41% yield and its structure elucidated using 2D-NMR techniques (Fig. 7). Deuterium exchange of the acidic hydrogens in compound 12 confirmed the structure of the final compound 11. It is important to note that C(12) substitution reactions of estrone derivatives have so far been limited to copper-mediated hydroxylation reactions.35–39 Carbonazidates derived from C(17) alcohol, i.e. C(17)H–OCON3, yield ketone as the main product after hydrolysis, as the nitrene insertion reaction proceeds preferentially at the C(17)–H bond.27
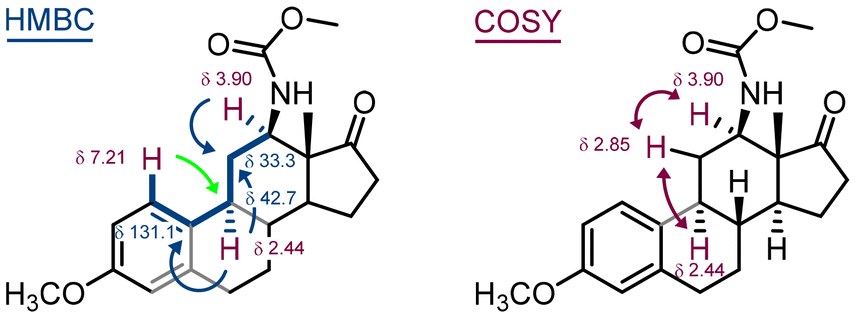 | ||
| Fig. 7 2D-NMR structure elucidation of the estrone derivative 11. HMBC 2-bond cross peaks are given in blue and the 3-bond cross peak is shown in green. Coupled hydrogens in COSY spectra are shown in red. | ||
In conclusion, we have developed a new protocol for the amination of enolizable and non-enolizable ketones. This method can also be used for the synthesis of various other compounds derived from cyclic carbamates attached to the α-position of nitriles.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.† The ESI includes experimental procedures, compound descriptions, X-ray data and copies of NMR spectra.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support of Charles University for the project 4EU+/UA/F4/16.References
- L. A. T. Allen, R.-C. Raclea, P. Natho and P. J. Parsons, Org. Biomol. Chem., 2021, 19, 498–513 RSC.
- A. M. R. Smith and K. K. (Mimi) Hii, Chem. Rev., 2011, 111, 1637–1656 CrossRef CAS PubMed.
- G. Guillena and D. J. Ramón, Tetrahedron: Asymmetry, 2006, 17, 1465–1492 CrossRef CAS.
- M. Marigo and K. A. Jørgensen, Chem. Commun., 2006, 2001–2011 RSC.
- T. Vilaivan and W. Bhanthumnavin, Molecules, 2010, 15, 917–958 CrossRef CAS PubMed.
- F. Zhou, F.-M. Liao, J.-S. Yu and J. Zhou, Synthesis, 2014, 2983–3003 CrossRef CAS.
- B. Maji and H. Yamamoto, Bull. Chem. Soc. Jpn., 2015, 88, 753–762 CrossRef CAS.
- P. Magnus, J. Lacour, I. Coldham, B. Mugrage and W. B. Bauta, Tetrahedron, 1995, 51, 11087–11110 CrossRef CAS.
- A. de la Torre, V. Tona and N. Maulide, Angew. Chem., Int. Ed., 2017, 56, 12416–12423 CrossRef CAS PubMed.
- A. Mukherjee, S. Mahato, D. S. Kopchuk, S. Santra, G. V. Zyryanov, A. Majee and O. N. Chupakhin, Russ. Chem. Rev., 2023, 92, RCR5046 CrossRef.
- Y. Li, R. Zhang, X. Bi and J. Fu, Org. Lett., 2018, 20, 1207–1211 CrossRef CAS PubMed.
- M. M. Hammouda and K. M. Elattar, RSC Adv., 2022, 12, 24681–24712 RSC.
- P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CAS.
- Y. Park, Y. Kim and S. Chang, Chem. Rev., 2017, 117, 9247–9301 CrossRef CAS PubMed.
- G. Dequirez, V. Pons and P. Dauban, Angew. Chem., Int. Ed., 2012, 51, 7384–7395 CrossRef CAS PubMed.
- C.-X. Ye and E. Meggers, Acc. Chem. Res., 2023, 56, 1128–1141 CrossRef CAS PubMed.
- D. Hernández-Guerra, A. Hlavačková, C. Pramthaisong, I. Vespoli, R. Pohl, T. Slanina and U. Jahn, Angew. Chem., Int. Ed., 2019, 58, 12440–12445 CrossRef PubMed.
- K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed.
- Y. Guo, C. Pei and R. M. Koenigs, Nat. Commun., 2022, 13, 86 CrossRef CAS PubMed.
- C. Wentrup, Angew. Chem., Int. Ed., 2018, 57, 11508–11521 CrossRef CAS PubMed.
- J.-P. Berndt, Y. Radchenko, J. Becker, C. Logemann, D. R. Bhandari, R. Hrdina and P. R. Schreiner, Chem. Sci., 2019, 10, 3324–3329 RSC.
- R. Hrdina, M. Larrosa and C. Logemann, J. Org. Chem., 2017, 82, 4891–4899 CrossRef CAS PubMed.
- R. Hrdina, O. M. Holovko-Kamoshenkova, I. Císařová, F. Koucký and O. Machalický, RSC Adv., 2022, 12, 31056–31060 RSC.
- J. L. Roizen, M. E. Harvey and J. Du Bois, Acc. Chem. Res., 2012, 45, 911–922 CrossRef CAS PubMed.
- R. Singh, J. N. Kolev, P. A. Sutera and R. Fasan, ACS Catal., 2015, 5, 1685–1691 CrossRef CAS PubMed.
- J. Lee, J. Lee, H. Jung, D. Kim, J. Park and S. Chang, J. Am. Chem. Soc., 2020, 142, 12324–12332 CrossRef CAS.
- P. C. Marais and O. Meth-Cohn, J. Chem. Soc., Perkin Trans. 1, 1987, 1553–1560 RSC.
- K. Huard and H. Lebel, Chem. – Eur. J., 2008, 14, 6222–6230 CrossRef CAS PubMed.
- Q. Guo, X. Ren and Z. Lu, Org. Lett., 2019, 21, 880–884 CrossRef CAS PubMed.
- W.-B. Wu, X.-P. Zeng and J. Zhou, J. Org. Chem., 2020, 85, 14342–14350 CrossRef CAS PubMed.
- B. Zonker, J. Becker and R. Hrdina, Org. Biomol. Chem., 2021, 19, 4027–4031 RSC.
- R. Hrdina, Synthesis, 2019, 629–642 CrossRef CAS.
- M. A. Gunawan, J.-C. Hierso, D. Poinsot, A. A. Fokin, N. A. Fokina, B. A. Tkachenko and P. R. Schreiner, New J. Chem., 2013, 38, 28–41 RSC.
- Z. Zhang, Z.-H. Zhang, Y. Sun, Y.-H. Tang, Y.-Z. Yang, F. Zhou and J. Zhou, Sci. China: Chem., 2024, 67 DOI:10.1007/s11426-024-2346-4.
- Y. Y. See, A. T. Herrmann, Y. Aihara and P. S. Baran, J. Am. Chem. Soc., 2015, 137, 13776–13779 CrossRef CAS PubMed.
- R. Trammell, Y. Y. See, A. T. Herrmann, N. Xie, D. E. Díaz, M. A. Siegler, P. S. Baran and I. Garcia-Bosch, J. Org. Chem., 2017, 82, 7887–7904 CrossRef CAS PubMed.
- B. Schönecker, T. Zheldakova, Y. Liu, M. Kötteritzsch, W. Günther and H. Görls, Angew. Chem., Int. Ed., 2003, 42, 3240–3244 CrossRef PubMed.
- B. Schönecker, T. Zheldakova, C. Lange, W. Günther, H. Görls and M. Bohl, Chem. – Eur. J., 2004, 10, 6029–6042 CrossRef PubMed.
- C. Zhao, Z. Ye, Z. Ma, S. A. Wildman, S. A. Blaszczyk, L. Hu, I. A. Guizei and W. Tang, Nat. Commun., 2019, 10, 4015 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2388739–2388741. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00009b |
| This journal is © The Royal Society of Chemistry 2025 |