
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereospecific access to α-haloalkyl esters via enol ester epoxides and synthesis of a C3–C21 fragment of bastimolide A†
Lucas W. Howell,
Jacob N. Hackbarth,
Jeffrey S. Abraham and
Gregory K. Friestad *
*
Department of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: gregory-friestad@uiowa.edu
First published on 11th February 2025
Abstract
We report a 14-step synthesis of a C3–C21 fragment of bastimolides A and B, antimalarial macrocyclic polyketides. A crucial ring-opening reaction of an enol ester epoxide showed previously unexplored reactivity, leading to an asymmetric synthesis of α-haloalkyl esters. The α-haloalkyl ester synthesis was shown to be stereospecific, and provided access to a key α-silyloxyaldehyde to initiate application of configuration-encoded 1,5-polyol synthesis. This strategy established the C11/C15 and C15/C19 remote stereochemical relationships of the bastimolides. The potential of this C3–C21 fragment for coupling to C22–C41 was established using a Mukaiyama aldol reaction with a simple enolsilane.
Introduction
Malaria continues to have a significant global impact causing an estimated 249 million malaria cases in 2022,1 and the threat of resistance emphasizes the continuing need for new treatment options. Marine cyanobacteria offer structurally interesting natural products that offer insights for drug discovery.2 Among these are bastimolide A (1),3 a 40-membered macrolide that has proven to exhibit potent antimalarial properties against four resistant strains of Plasmodium falciparum (IC50 = 80–270 nM), and its 24-membered macrolactone isomer, bastimolide B (2).4 Both 1,3- and 1,5-polyol motifs are combined in its structure (Fig. 1), which bears little apparent resemblance to any clinical antimalarial drugs or preclinical candidates.5 This suggests the possibility that biological evaluation of 1, 2, or analogs could uncover a new mode of action via a novel Plasmodium drug target.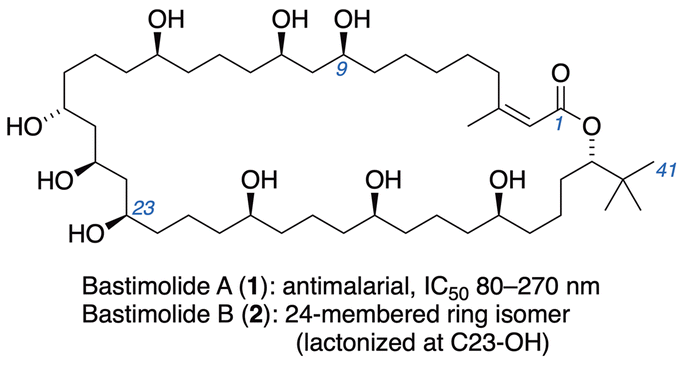 | ||
| Fig. 1 Structures of bastimolides A (1) and B (2). | ||
Synthesis of the bastimolides is therefore a high priority, and various strategies to access structural subunits6 and halogenated analogues7 have appeared. Smith8 and Aggarwal9 have reported the first total syntheses of 1 and 2, respectively, and Kirsch et al. reported a formal synthesis.10 We recently reported asymmetric synthesis of a 1,5-polyol comprising the C22–C41 fragment of the bastimolides.11
Polyols bearing 1,5-relationships between hydroxyl groups often cause complications for configurational assignments,12 stereocontrolled synthesis, and diastereomer separations.13 This prompted our development of a configuration-encoded synthetic strategy14 (Fig. 2) utilizing building blocks of defined hydroxyl configuration that are linked iteratively via Julia–Kocienski olefination.15 Subjecting α-silyloxyaldehydes to Julia–Kocienski olefination with γ-sulfononitrile building blocks (R)-3 or (S)-3 establishes syn- or anti-1,5-diol relationships, and subsequent reduction of the nitrile regenerates α-silyloxyaldehyde functionality at the chain terminus for another iteration. The programmed assembly allows synthesis of all possible diastereomers of 1,5-polyols with equal facility, and obviates analytical or preparative separations of diastereomers. Here we disclose the configuration-encoded synthesis of the C3–C21 subunit of the bastimolides (4), aided by the discovery of mild conditions for stereospecific transformation of enol ester epoxides into 1-haloalkyl esters.
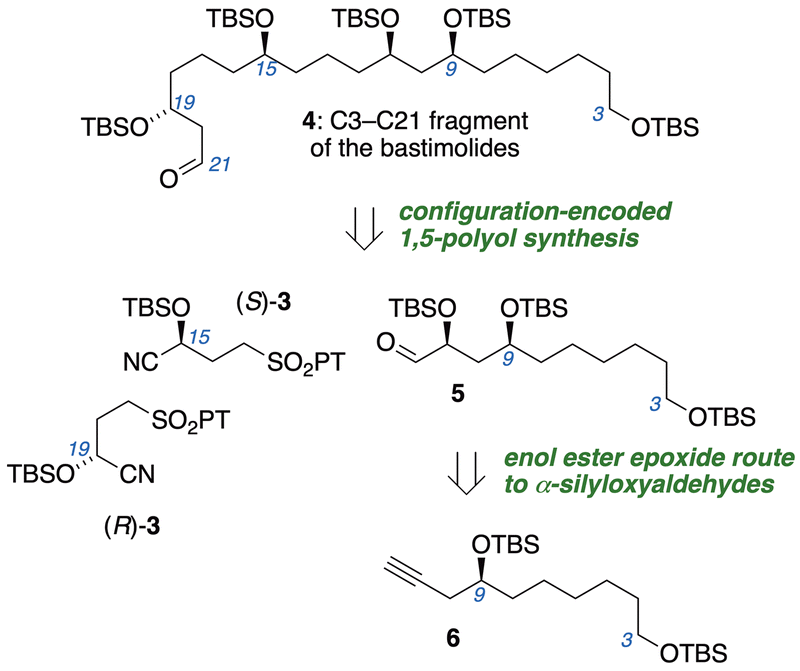 | ||
| Fig. 2 Two iterations of configuration-encoded 1,5-polyol synthesis to access an anti,syn-1,5,9 triol stereotriad. | ||
Results and discussion
Our retrosynthetic analysis (Fig. 2) involves two iterations of the configuration-encoded 1,5-polyol synthesis strategy. This suggesting α-silyloxyaldehyde 5 as a key precursor, which would be coupled successively with (S)-3 and (R)-3 to unambiguously establish C15 and C19 configurations. Our earlier successes in three-step synthesis of α-silyloxyaldehydes from alkynes via enol ester epoxides16 prompted a similar approach to 5 from alkyne 6.The synthetic sequence began with preparation of epoxide 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 (Scheme 1), obtained in 92% ee by enantioselective Katsuki epoxidation of alkene 7 with the Berkessel Ti catalyst A.18 Reaction of 8 with lithiated trimethylsilylacetylene, followed by alkyne desilylation and TBS protection of the 2° alcohol, furnished alkyne 6 in 72% yield over five steps, with only two column chromatography purifications.
17 (Scheme 1), obtained in 92% ee by enantioselective Katsuki epoxidation of alkene 7 with the Berkessel Ti catalyst A.18 Reaction of 8 with lithiated trimethylsilylacetylene, followed by alkyne desilylation and TBS protection of the 2° alcohol, furnished alkyne 6 in 72% yield over five steps, with only two column chromatography purifications.
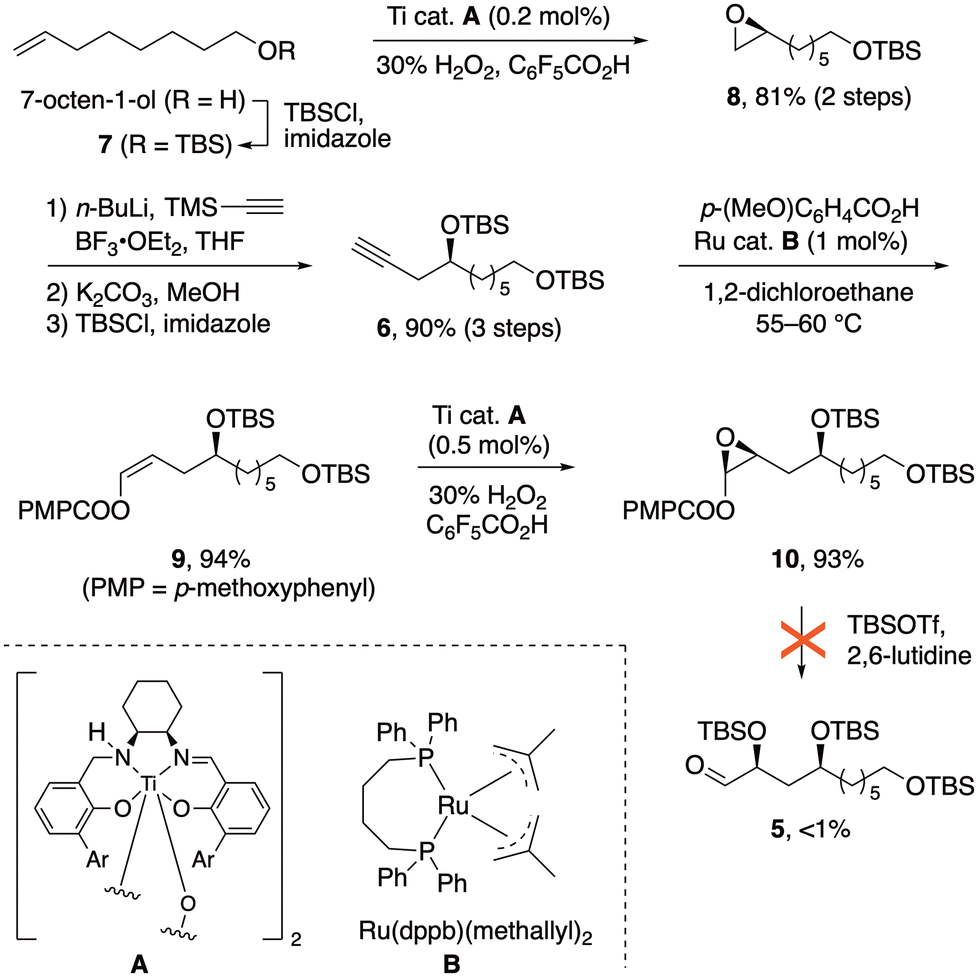 | ||
| Scheme 1 Synthesis of enol ester epoxide 10 and its anomalous reactivity. | ||
The next task was to implement our three-step route from alkynes to α-silyloxyaldehydes. Ruthenium-catalyzed addition19 of anisic acid to alkyne 6 (Scheme 1) gave (Z)-enol ester 9 in 90% yield, and another Berkessel–Katsuki epoxidation furnished enol ester epoxide 10 with excellent yield and selectivity (94%, 96:4 dr). The epoxide ring-opening, which had normally provided smooth access to α-silyloxyaldehydes upon treatment with silyl triflates and lutidine,11,14c,16 failed in this case; only traces of the aldehyde 5 were observed. In an effort to understand this anomaly, simplified enol ester substrate 11a (Fig. 3a) was subjected to the ring opening. Instead of α-silyloxyaldehyde 12a, a dimeric hydroxyfuran structure 13 was obtained. This could be rationalized by an oxocarbenium ion (or its equivalent) undergoing nucleophilic attack by the nearby silyloxy substituent as implied by structure C, followed by some combination of silyl transfer events and dimerization. The dimerization finds precedent in a similar structure formed from furanoses.20 Replacement of OTBS with less nucleophilic OBz would be expected to suppress the formation of dimer 13, and indeed with substrate 11b the expected ring-opening pathway to the α-silyloxyaldehyde 12b was restored (64% yield).11
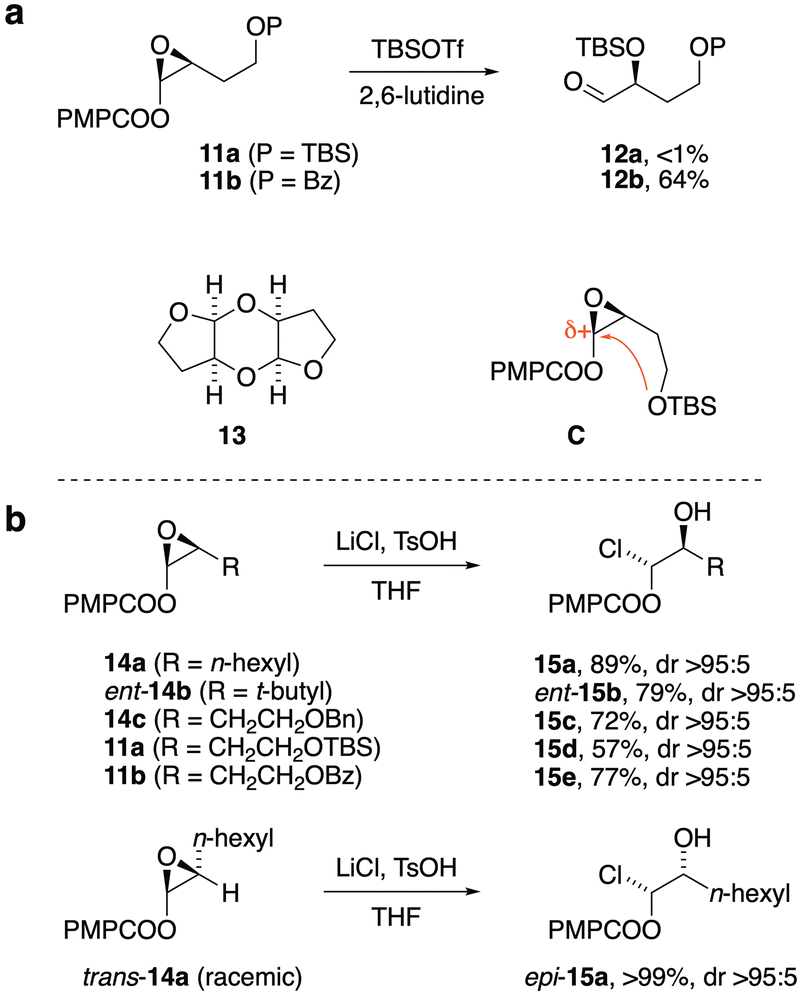 | ||
| Fig. 3 (a) Simplified analogs 11a and 11b reveal the reason for anomalous reactivity of 10. (b) Stereospecific enol ester epoxide ring-opening to 1-haloalkyl esters 15. | ||
Further comment about unexpected structure 13 is warranted. The cis configuration at both ring junctions of 13 was assigned by its apparent C2 symmetry (4 signals in its 13C NMR spectrum) and small coupling constants at the ring junction (J = 3.7 Hz observed at the anomeric C–H). A boatlike central ring would be accompanied by high torsional strain and lack of anomeric stabilization; we propose a chairlike conformation for the central ring of 13, with pseudo-C2 symmetry attributed to a rapid chair–chair conformational equilibrium.
Although changing to ester protection at the C9-OH could circumvent the anomalous reactivity of 10 and 11a, we sought to address the problem head-on, with the hypothesis that a large concentration of an external nucleophile could suppress the offending cyclization. Indeed, upon treatment of enol ester epoxide 14a with LiCl and TsOH, complete conversion to 1-haloalkyl ester 15a was observed within 5 min. Similar results were observed with enol ester epoxides 14b, 14c, 11a, and 11b, producing 1-haloalkyl esters 15b–15e respectively. All of these 1-haloalkyl esters were formed with complete stereospecificity despite the potential intermediacy of a planar oxocarbenium ion. Enol ester epoxide trans-14a, prepared via the corresponding E-enol ester, gave diastereomer epi-15a, providing further evidence of the stereospecific nature of the ring-opening. Crystallographic analysis of the 3,5-dinitrobenzoate derivative of 15b (see ESI†) confirmed the structural assignment suggesting inversion of configuration by the chloride nucleophile. To our knowledge there is only one related example leading from an enol ester epoxide to a 1-haloalkyl ester (SnCl4, 50% yield).21 Here we provide for the first time (a) evidence of stereospecificity, (b) preliminary evaluation of scope, and (c) subsequent reactivity studies in application to target-oriented synthesis. Racemic 1-haloalkyl esters22 have been used in various bond constructions,23 and this access to enantiopure samples presents new opportunities for reaction development and mechanistic study.
Turning our attention back to the synthetic route, we put the new method to the test in conversion of enol ester epoxide 10 to α-silyloxyaldehyde 5. Treatment of 10 with a chloride source (NBu4Cl was used for improved solubility) and camphorsulfonic acid, followed by O-silylation, cleanly furnished the α-haloalkyl ester 16 in 84.5% yield (Scheme 2). Transesterification with concomitant elimination of chloride then completed the alternative method for conversion of 10 to aldehyde 5. While the typical K2CO3/MeOH was effective for this transesterification on microscale, scale-up was problematic. A more reliable modification employed phase-transfer catalysis: in an immiscible mixture of benzene (or toluene) and ethylene glycol, exposure to K2CO3 along with phase-transfer catalyst NBu4HSO4 furnished aldehyde 5 in 74% yield, along with recovered 10 (88% yield based on conversion).24
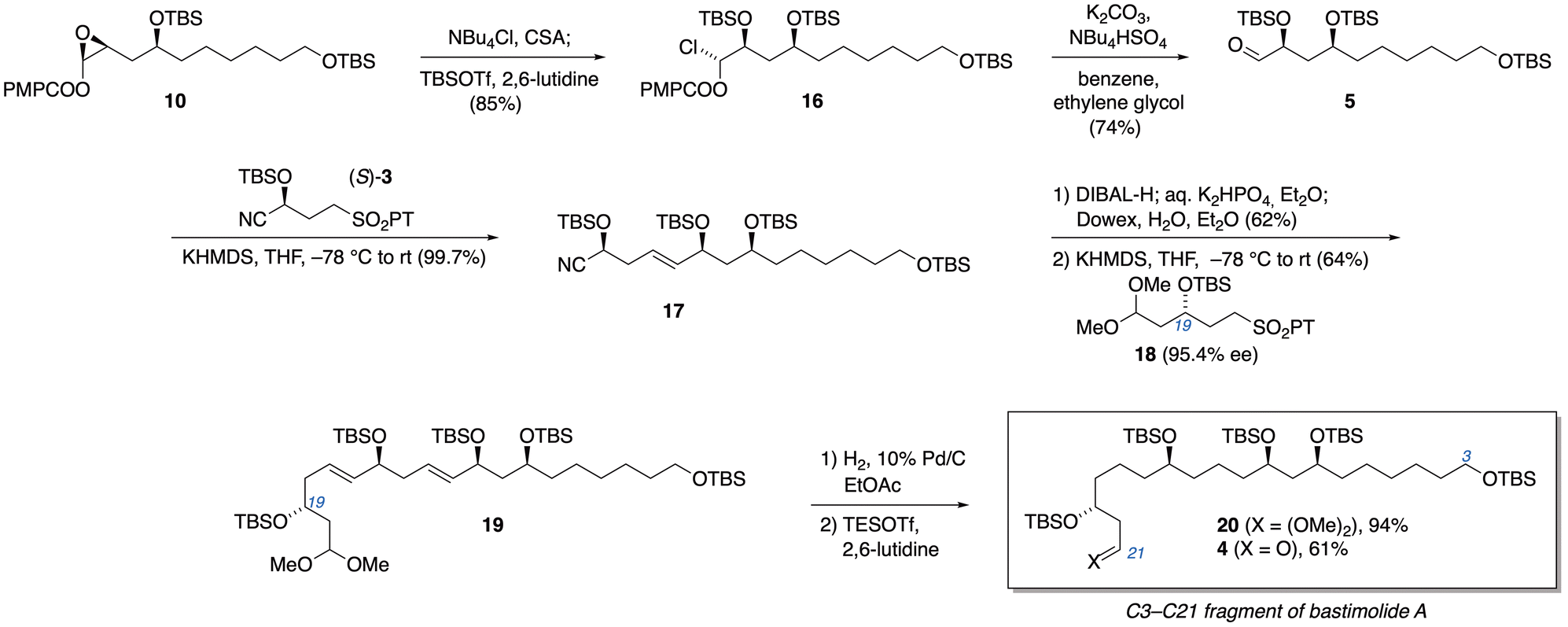 | ||
| Scheme 2 Access to α-silyloxyaldehyde 5 and configuration-encoded assembly of the C3–C21 subunit of bastimolides. | ||
With aldehyde 5 in hand, we applied our configuration-encoded 1,5-polyol synthesis. Julia–Kocienski olefination with (S)-3 smoothly established the syn-1,5-diol relationship between C11 and C15 with a quantitative yield of nitrile 17. Reduction of the nitrile with DIBAL-H and a second iteration of the Julia–Kocienski reaction with alternative building block 18 then established the desired anti,syn-1,5,9-triol stereotriad in 19, while placing masked aldehyde functionality at the terminus.25 Sulfone 18 was prepared in 6 steps from acrolein (see ESI†), using Keck allylation (95% ee) to encode the desired configuration at the carbon destined to become C19 of the bastimolides. Hydrogenation of the alkenes of 19 afforded acetal 20, which was converted to β-silyloxyaldehyde 4 through Fujioka–Kita acetal hydrolysis.26 This aldehyde is the C3–C21 fragment of bastimolides, ready for Mukaiyama aldol coupling to the C22–C41 subunit.
 | (1) |
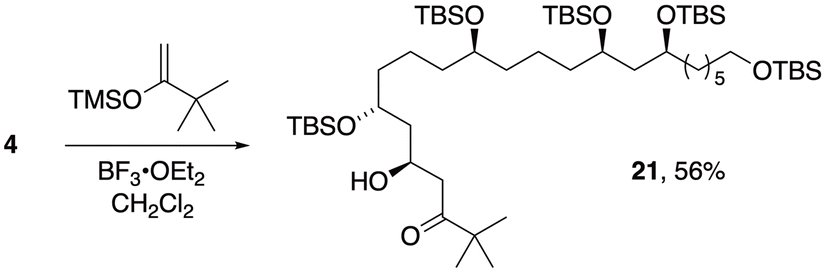
An initial assessment of Mukaiyama aldol conditions for coupling to the C3–C21 subunit (4) employed the trimethylsilyl enol ether derived from pinacolone (eqn (1)). In the presence of BF3·OEt2, this enolsilane added smoothly to aldehyde 4 to afford aldol 21 (dr 88:12) with close correspondence to the 1,3-diastereocontrol we previously observed in similar polyacetate-type aldol adducts.27 Diagnostic 1H NMR data for several closely related aldol products in that prior report enabled assignment of anti configuration to the major diastereomer 21 and demonstrated the potential of 4 as a viable intermediate en route to bastimolides and analogs.
Conclusions
We have developed a synthesis of the C3–C21 fragment of bastimolides using the configuration-encoded approach to 1,5-polyol assembly. The synthetic sequence encountered an unexpected structural incompatibility of our previously established three-step conversion of alkynes to α-silyloxyaldehydes. Solving this problem led to the discovery of a stereospecific synthesis of enantiopure 1-haloalkyl esters; these are richly functionalized synthetic building blocks with further synthetic potential.Data availability
Crystallographic data are available in CCDC 2403648. Preparative procedures and characterization data are provided in ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
With great appreciation, this article is dedicated to the memory of Professor Amos B. Smith, III. We thank the National Institutes of Health (AI-171379) for generously supporting this research. We gratefully acknowledge seed funding and fellowship support (LWH) from the University of Iowa Department of Chemistry and College of Liberal Arts and Sciences. We thank Prof. James Gloer and Dr Chris Knutson of the University of Iowa for assistance with HPLC instrumentation, and the University of Iowa NMR (G. Crull) and MATFab (D. Unruh) facilities for analytical support.References
- World Health Organization (WHO), World Malaria Report 2023. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2023, accessed 10/15/24.
- L. T. Tan, Bioactive natural products from marine cyanobacteria for drug discovery, Phytochemistry, 2007, 68, 954–979 CrossRef CAS PubMed.
- C.-L. Shao, R. G. Linington, M. J. Balunas, A. Centeno, P. Boudreau, C. Zhang, N. Engene, C. Spadafora, T. S. Mutka, D. E. Kyle, L. Gerwick, C.-Y. Wang and W. H. Gerwick, Bastimolide A, a Potent Antimalarial Polyhydroxy Macrolide from the Marine Cyanobacterium Okeania hirsuta, J. Org. Chem., 2015, 80, 7849–7855 CrossRef CAS PubMed.
- C.-L. Shao, X.-F. Mou, F. Cao, C. Spadafora, E. Glukhov, L. Gerwick, C.-Y. Wang and W. H. Gerwick, Bastimolide B, an Antimalarial 24-Membered Marine Macrolide Possessing a tert-Butyl Group, J. Nat. Prod., 2018, 81, 211–215 CrossRef CAS PubMed.
- (a) T. Umumararungu, J. B. Nkuranga, G. Habarurema, J. B. Nyandwi, M. J. Mukazayire, J. Mukiza, R. Muganga, I. Hahirwa, M. Mpenda, A. N. Katembezi, E. O. Olawode, E. Kayitare and P. C. Kayumba, Recent developments in antimalarial drug discovery, Bioorg. Med. Chem., 2023, 88–89, 117339 CrossRef CAS PubMed; (b) M. A. Phillips, J. N. Burrows, C. Manyando, R. H. van Huijsduijnen, W. C. Van Voorhis and T. N. C. Wells, Malaria, Nat. Rev. Dis. Primers, 2017, 3, 17050 CrossRef PubMed.
- N. S. Kumar, B. J. Ramulu and S. Ghosh, Stereoselective Synthesis of the C19–C39 Fragment of Bastimolide A, SynOpen, 2021, 5, 285–290 CrossRef CAS.
- A. Quintard, C. Sperandio and J. Rodriguez, Modular Enantioselective Synthesis of an Advanced Pentahydroxy Intermediate of Antimalarial Bastimolide A and of Fluorinated and Chlorinated Analogues, Org. Lett., 2018, 20, 5274–5277 CrossRef CAS PubMed.
- J. B. Cox, A. A. Kellum, Y. Zhang, B. Li and A. B. Smith, Total Synthesis of (−)-Bastimolide A: A Showcase for Type I Anion Relay Chemistry, Angew. Chem., Int. Ed., 2022, e202204884 CAS.
- D. Fiorito, S. Keskin, J. M. Bateman, M. George, A. Noble and V. K. Aggarwal, Stereocontrolled Total Synthesis of Bastimolide B Using Iterative Homologation of Boronic Esters, J. Am. Chem. Soc., 2022, 144, 7995–8001 CrossRef CAS PubMed.
- I.-E. Celik, F. Mittendorf, A. Gomez-Suarez and S. F. Kirsch, Formal synthesis of bastimolide A using a chiral Horner-Wittig reagent and a bifunctional aldehyde as key building blocks, Tetrahedron Chem, 2024, 9, 100065 CrossRef CAS.
- J. N. Hackbarth and G. K. Friestad, A Three-Step Catalytic Asymmetric Sequence from Alkynes to alpha-Silyloxyaldehydes and Its Application to a C22–C41 Fragment of Bastimolide A, Org. Lett., 2024, 25, 4492–4496 CrossRef PubMed.
- Assignment of the stereostructure of bastimolide A required X-ray diffraction analysis of the corresponding nona-p-nitrobenzoate derivative (see ref. 3).
- Review: R. M. Friedrich and G. K. Friestad, Inspirations from Tetrafibricin and Related Polyketides: New Methods and Strategies for 1,5-Polyol Synthesis, Nat. Prod. Rep., 2020, 37, 1229–1261 RSC.
- (a) G. K. Friestad and G. Sreenilayam, Versatile configuration-encoded strategy for rapid synthesis of 1,5-polyol stereoisomers, Org. Lett., 2010, 12, 5016–5019 CrossRef CAS PubMed; (b) R. M. Friedrich and G. K. Friestad, Access to an anti,syn-1,5,7-Triol by Configuration-Encoded 1,5-Polyol Synthesis: The C15-C25 Fragment of Tetrafibricin, Eur. J. Org. Chem., 2017, 1961–1964 CrossRef CAS; (c) R. M. Friedrich, G. Sreenilayam, J. Hackbarth and G. K. Friestad, Unified Strategy for 1,5,9- and 1,5,7-Triols via Configuration-Encoded 1,5-Polyol Synthesis: Enantioselective Preparation of γ-Sulfonyl-α-silyloxyaldehydes and Iterative Julia–Kocienski Coupling, J. Org. Chem., 2018, 83, 13636–13649 CrossRef CAS PubMed; (d) R. M. Friedrich, J. Q. Bell, A. Garcia, Z. Shen and G. K. Friestad, Unified Strategy for 1,5,9- and 1,5,7-Triols via Configuration-Encoded 1,5-Polyol Synthesis: Preparation and Coupling of C15–C25 and C26–C40 Fragments of Tetrafibricin, J. Org. Chem., 2018, 83, 13650–13669 CrossRef CAS PubMed.
- (a) Use of Julia–Kocienski reactions to establish 1,5-diol relationships has also been reported for late-stage fragment couplings (see ref. 13). Meanwhile, our iterative strategy is uniquely positioned for flexible access to various analogs including stereoisomers; (b) See also. L. C. Dias and E. C. de Lucca, Total Synthesis of (−)-Marinisporolide C, J. Org. Chem., 2017, 82, 3019–3045 CrossRef CAS PubMed.
- G. K. Friestad, G. Sreenilayam, J. C. Cannistra and L. M. Slominski, Preparation of enol ester epoxides and their ring-opening to α-silyloxyaldehydes, Tetrahedron Lett., 2012, 53, 5064–5067 CrossRef CAS . Also see ref. 11 and 14c.
- J. E. Robinson and M. A. Brimble, The first enantioselective total synthesis of the anti-Helicobacter pylori agent (+)-spirolaxine methyl ether, Chem. Commun., 2005, 1560–1562 RSC.
- (a) K. Matsumoto, C. Feng, S. Handa, T. Oguma and T. Katsuki, Asymmetric epoxidation of (Z)-enol esters catalyzed by titanium(salalen) complex with aqueous hydrogen peroxide, Tetrahedron, 2011, 67, 6474–6478 CrossRef CAS; (b) A. Berkessel, T. Gunther, Q. Wang and J.-M. Neudörfl, Titanium Salalen Catalysts Based on cis-1,2-Diaminocyclohexane: Enantioselective Epoxidation of Terminal Non-Conjugated Olefins with H2O2, Angew. Chem., Int. Ed., 2013, 52, 8467–8471 CrossRef CAS PubMed; (c) H. Engler, M. Lansing, C. P. Gordon, J.-M. Neudörfl, M. Schäfer, N. E. Schlörer, C. Copéret and A. Berkessel, Olefin Epoxidation Catalyzed by Titanium–Salalen Complexes: Synergistic H2O2 Activation by Dinuclear Ti Sites, Ligand H-Bonding, and π-Acidity, ACS Catal., 2021, 11, 3206–3217 CrossRef CAS.
- H. Doucet, B. Martin-Vaca, C. Bruneau and P. H. Dixneuf, General Synthesis of (Z)-Alk-1-en-1-yl Esters via Ruthenium-Catalyzed anti-Markovnikov trans-Addition of Carboxylic Acids to Terminal Alkynes, J. Org. Chem., 1995, 60, 7247–7255 CrossRef CAS.
- (a) S. Mukherjee, B. G. Roy, S. N. Das and S. B. Mandal, Deoxygenation/dimerization of sugar derivatives with BF3·Et2O–Et3SiH: synthesis of a β-isonucleoside, Tetrahedron Lett., 2012, 53, 4929–4932 CrossRef CAS; (b) L. Di, K.-J. Wang, C.-C. Zhu and N. Li, Chemical Constituents from Rhizomes of Curculigo capitulata, Bull. Korean Chem. Soc., 2010, 31, 2999–3002 CrossRef CAS.
- B. Peschke, J. Lussmann, M. Dyrbusch and D. Hoppe, Diastereo- and Enantioselective Synthesis of 1,2:5,6-Diepoxy-δ-hydroxyalkyl Carbamates. Regioselective Ring Opening and Their Transformation into Doubly C-Branched Deoxy Sugar Analogues, Chem. Ber., 1992, 125, 1421–1430 CrossRef CAS.
- For preparations of racemic 1-haloalkyl esters, see: F. Fache, I. de Azpiazu, B. Pelotier, O. Piva and C. Gozzi, Green Access to α-Haloalkyl and α-Halobenzyl Esters, Versatile Intermediates for the One-Pot Two-Step Synthesis of O,O’-Diacyl Acetals Using Zinc-Based Ionic Liquid Catalyst, Synthesis, 2019, 2430–2434 CAS , references therein.
- Selected examples: (a) K. Sakata, D. Urabe and M. Inoue, Preparation and palladium-mediated cross-coupling of α-benzoyloxyalkylzinc bromides, Tetrahedron Lett., 2013, 54, 4189–4192 CrossRef CAS; (b) L. Wang, J. M. Lear, S. M. Rafferty, S. C. Fosu and D. A. Nagib, Ketyl rasdical reactivity via atom transfer catalysis, Science, 2018, 362, 225–229 CrossRef CAS PubMed; (c) Z.-P. Yang and G. C. Fu, Convergent Catalytic Asymmetric Synthesis of Esters of Chiral Dialkyl Carbinols, J. Am. Chem. Soc., 2020, 142, 5870–5875 CrossRef CAS PubMed; (d) S. M. Rafferty, J. E. Rutherford, L. Zhang, L. Wang and D. A. Nagib, Cross-Selective Aza-Pinacol Coupling via Atom Transfer Catalysis, J. Am. Chem. Soc., 2021, 143, 5622–5628 CrossRef CAS PubMed; (e) H. Ji, D. Lin, L. Tai, X. Li, Y. Shi, Q. Han and L.-A. Chen, Nickel-Catalyzed Enantioselective Coupling of Acid Chlorides with α-Bromobenzoates: An Asymmetric Acyloin Synthesis, J. Am. Chem. Soc., 2022, 144, 23019–23029 CrossRef CAS PubMed; (f) C. Li, J. Cheng, X. Wan, J. Li, W. Zu, Y. Xu, Y. Huang and H. Huo, Ni/Photoredox-Catalyzed Enantioselective Acylation of α-Bromobenzoates with Aldehydes: A Formal Approach to Aldehyde-Aldehyde Cross-Coupling, J. Am. Chem. Soc., 2024, 146, 19909–19918 CrossRef CAS PubMed.
- Best results were obtained at incomplete conversion; longer reaction times led to partial decomposition of 5.
- Coupling with (R)-3 was also promising, but the modified building block 18 enabled more direct access to the C21 aldehyde terminus needed for fragment coupling.
- (a) H. Fujioka, T. Okitsu, Y. Sawama, N. Murata, R. Li and Y. Kita, Reaction of the Acetals with TESOTf−Base Combination; Speculation of the Intermediates and Efficient Mixed Acetal Formation, J. Am. Chem. Soc., 2006, 128, 5930–5938 CrossRef CAS PubMed.
- L. W. Howell and G. K. Friestad, On the Role of β-Silyloxy- and β-Alkoxyaldehyde Protecting Groups in Mukaiyama Aldol 1,3-Diastereocontrol, Tetrahedron Lett., 2022, 111, 154203 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2403648. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ob00070j |
| This journal is © The Royal Society of Chemistry 2025 |