
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of dihydrogambirtannine by alkyne [2 + 2 + 2]-cyclotrimerisation†
Pattarakiat Seankongsuk,
Patcharaporn Sae-Lao,
Shi He,
Veronica Pereira and
Roderick W. Bates*
and
Roderick W. Bates*
School of Chemistry, Chemical Engineering and Biotechnology, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: roderick@ntu.edu.sg
First published on 21st February 2025
Abstract
The first asymmetric synthesis of (−)-dihydrogambirtannine has been achieved using nucleophilic addition to a chiral sulfinyl imine to establish the stereogenic centre and [2 + 2 + 2] alkyne cyclotrimerisation to construct the aromatic E ring.
Introduction
Dihydrogambirtannine 1 is one of the yohimbine alkaloids,1 distinguished by the fact that the E ring is aromatic. It was isolated from Uncaria gambier,2 a plant that was formerly of economic importance in South East Asia as its extracts were used in dyeing and tanning. It has also been isolated from Ochrosia borbonica, a tree native to Mauritius and Réunion.3 The enantiomer of natural dihydrogambirtannine had earlier been prepared by degradation of deserpideine.4 (±)-Dihydrogambirtannine has been synthesized by Beisler,5 and by Wenkert et al.6 Each of these syntheses employed a late stage formation of the C-ring. (−)-Dihydrogambirtannine has been obtained as a minor product during transformations of secologanin.7 The analog lacking the methoxycarbonyl group has also been synthesized in racemic form.8,9 To the best of our knowledge, there has been no asymmetric synthesis of this alkaloid.Results and discussion
A challenge in the synthesis of dihydrogambirtannine 1 is the fact that the E-ring is a 1,2,3-trisubstituted benzene ring. One effective approach to the synthesis of this motif is by the [2 + 2 + 2]-cyclotrimerisation of alkynes, in which one partner is acetylene (Scheme 1).10,11 The other partner would be a suitably substituted diyne, derived, in turn, from a functionalised indole. We wish to report the successful application of this strategy to the asymmetric synthesis of this alkaloid. We note that the Knölker synthesis of the desmethoxycarbonyl analog9 does follow this strategy, but in a stepwise fashion and via an organoiron intermediate.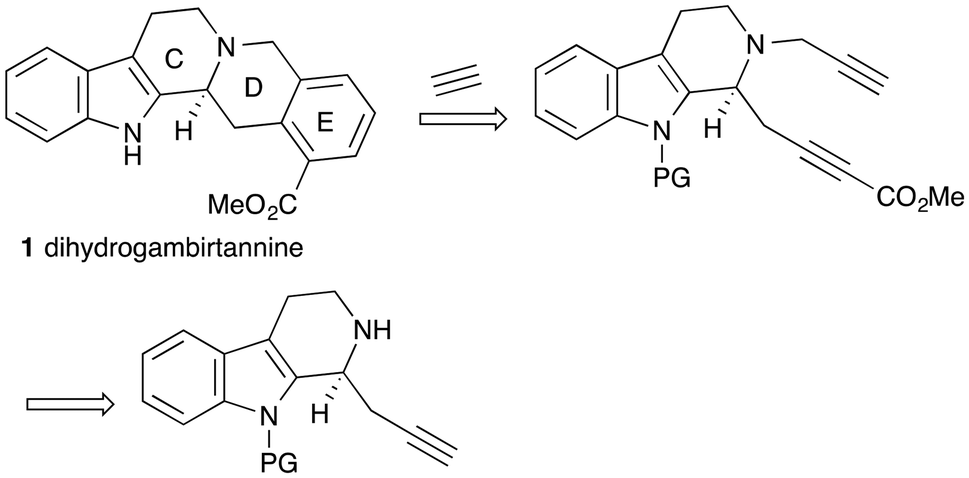 | ||
| Scheme 1 Dihydrogambirtannine retrosynthesis. | ||
One way to introduce the required stereogenic centre would be by asymmetric propargylation of the known imine 3 (Scheme 2). The imine was prepared from tryptamine 2 by the reported method,12 and subjected to asymmetric propargylation using the asymmetric catalyst reported by Snyder et al.13 In our hands, the results were capricious and poorly reproducible. In addition, we note that this reaction can also require a very high loading of a complex ligand.14
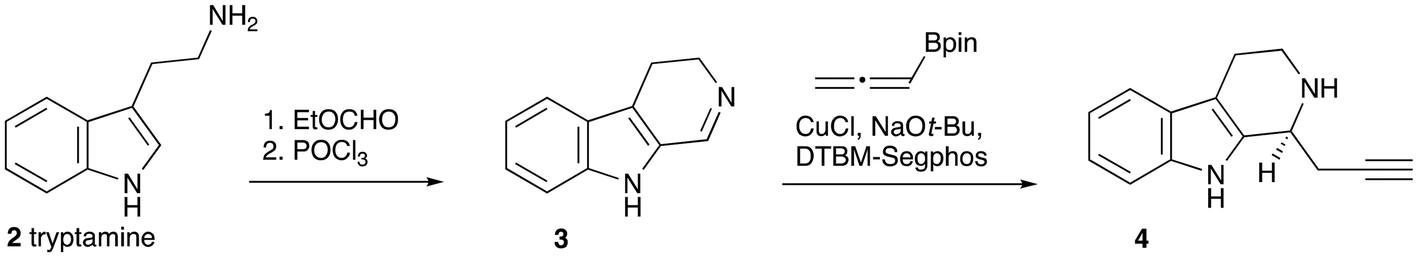 | ||
| Scheme 2 Asymmetric propargylation. | ||
We, therefore, turned to a more practical method (Scheme 3) using an imine bearing a sulfinyl chiral auxiliary,15 applying the propargylation reaction reported by Subba Reddy et al.16 Alcohol 6 was prepared by reduction of indole-3-acetic acid 5 and N-protected with a p-methoxybenzyl group using the conditions developed by Ley and Heaney.17 Alcohol 7 was then subjected to Vilsmeier–Haack formylation to give aldehyde 8 in modest yield. Aldehyde 8 was then converted to imine 9 under the reported conditions.16 Propargylation of imine 9 under carefully controlled conditions18 delivered a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereoisomers which could be separated by careful chromatography. Use of dichloromethane as the solvent for this reaction at −43 °C was essential to achieving this outcome. Use of either THF or toluene as the solvent resulted in the other diastereoisomer being the major product, but with ratios of just 1:2 and 1:4, respectively. The major diastereoisomer 10 was then treated with base to close the C-ring with formation of tricycle 11. Attempts to then introduce the methoxycarbonyl group using a combination of a strong base, n-butyllithium or LiHMDS, and methyl chloroformate or dimethyl carbonate were fruitless, yielding mixtures. However, palladium catalyzed methoxycarbonylation19 under carefully controlled conditions delivered the desired ester 12. The t-butylsulfinyl group was then removed under acidic conditions. The deprotected compound was isolated as its hydrochloride salt 13. Cleavage of the t-butylsulfinyl group under these conditions gives t-butylsulfinyl chloride as a by-product.20 It is essential that this material is fully removed otherwise sulfinamide 12 can subsequently re-form. The second alkyne moiety was introduced by N-alkylation with propargyl bromide to provide diyne 14 for the key [2 + 2 + 2]-cyclotrimerisation reaction, which would simultaneously create the D and E rings. While use of Wilkinson's catalyst in either ethanol or toluene resulted in little or no product formation, it was found that this reaction proceeded well using Cp*Ru(COD)Cl as the catalyst21,22 in 1,2-dichloroethane with slow addition of the substrate by syringe pump.11 Pentacycle 15 was formed cleanly and used directly in the next step. The synthesis was completed by removal of the methoxybenzyl group under acidic conditions in the presence of anisole as a carbocation scavenger to give (−)-dihyrogambirtannine 1.23 The spectroscopic data were in good agreement with that reported3 for the natural product.
:1 mixture of diastereoisomers which could be separated by careful chromatography. Use of dichloromethane as the solvent for this reaction at −43 °C was essential to achieving this outcome. Use of either THF or toluene as the solvent resulted in the other diastereoisomer being the major product, but with ratios of just 1:2 and 1:4, respectively. The major diastereoisomer 10 was then treated with base to close the C-ring with formation of tricycle 11. Attempts to then introduce the methoxycarbonyl group using a combination of a strong base, n-butyllithium or LiHMDS, and methyl chloroformate or dimethyl carbonate were fruitless, yielding mixtures. However, palladium catalyzed methoxycarbonylation19 under carefully controlled conditions delivered the desired ester 12. The t-butylsulfinyl group was then removed under acidic conditions. The deprotected compound was isolated as its hydrochloride salt 13. Cleavage of the t-butylsulfinyl group under these conditions gives t-butylsulfinyl chloride as a by-product.20 It is essential that this material is fully removed otherwise sulfinamide 12 can subsequently re-form. The second alkyne moiety was introduced by N-alkylation with propargyl bromide to provide diyne 14 for the key [2 + 2 + 2]-cyclotrimerisation reaction, which would simultaneously create the D and E rings. While use of Wilkinson's catalyst in either ethanol or toluene resulted in little or no product formation, it was found that this reaction proceeded well using Cp*Ru(COD)Cl as the catalyst21,22 in 1,2-dichloroethane with slow addition of the substrate by syringe pump.11 Pentacycle 15 was formed cleanly and used directly in the next step. The synthesis was completed by removal of the methoxybenzyl group under acidic conditions in the presence of anisole as a carbocation scavenger to give (−)-dihyrogambirtannine 1.23 The spectroscopic data were in good agreement with that reported3 for the natural product.
 | ||
| Scheme 3 Synthesis of (−)-dihydrogambirtannine. | ||
Conclusion
The first asymmetric synthesis of (−)-dihydrogambirtannine has been completed in 11 steps from acid 5 and in 11% overall yield, demonstrating again the power of the [2 + 2 + 2]-cyclotrimerisation of alkynes in the synthesis of densely substituted benzene rings.Experimental
General experimental
All reagents were obtained from commercial suppliers and used without further purification unless otherwise indicated. Reactions requiring anhydrous or air free conditions were performed under an atmosphere of nitrogen. Glassware was oven dried at 120 °C and cooled under vacuum. Anhydrous THF and ether were distilled from sodium metal and benzophenone under nitrogen. Anhydrous toluene was distilled from sodium and anhydrous dichloromethane was distilled from calcium hydride. Anhydrous methanol was distilled from magnesium. 1H and 13C NMR spectra were recorded on a Bruker Avance III 400 spectrometer, a 500 MHz ECZ Luminous (JNM-ECZL series) or a JEOL ECA400 UltraShield in CDCl3. Chemical shifts are given in parts per million (ppm) with residual protic solvent as the internal standard. Coupling constants are given in Hertz. High resolution mass spectra were recorded on a Waters Xevo G2-X2 MS in either positive or negative mode equipped with Waters Acquity UPLC. Analytical thin layer chromatography was performed on Merck DC pre coated TLC plates with 0.25 mm Kieselgel 60 F254. The plates were visualised with a 254 nm UV lamp, or by staining with ammonium molybdate or potassium permanganate. Flash chromatography was performed on silica gel 230–400 mesh.2-(1H-Indol-3-yl)ethan-1-ol (6)
Indole-3-acetic acid (1.75 g, 9.98 mmol) in anhydrous THF (15 mL) was added dropwise to a stirred suspension of lithium aluminium hydride (0.76 g, 20 mmol) in anhydrous THF (30 mL) at 0 °C. The mixture was stirred at 0 °C for 1 h, allowed to warm to room temperature and stirred for 18 h. The mixture was cooled to 0 °C and quenched with saturated Na2SO4 solution (20 mL). A white precipitate formed. The mixture was diluted with EtOAc (50 mL) and filtered through Celite, washing with EtOAc. The filtrate was dried (Na2SO4) and concentrated under reduced pressure to give alcohol (6) as a light brown solid (1.43 g, 89% yield) which was used directly without further purification. 1H NMR (400 MHz, CDCl3) δ 8.03 (brs, 1H), 7.63–7.61 (d, J = 7.6 Hz, 1H), 7.38–7.36 (d, J = 8.4 Hz, 1H), 7.25–7.08 (m, 2H), 7.04 (d, J = 2.4 Hz, 1H), 3.91 (t, J = 6.4 Hz, 2H), 3.05 (t, J = 6.4 Hz, 2H), 1.55 (brs, 1H). The spectroscopic data were in good agreement with literature values.242-(1-(4-Methoxybenzyl)-1H-indol-3-yl)ethan-1-ol (7)
1-(Bromomethyl)-4-methoxybenzene (5.2 mL, 37.5 mmol) was added to a stirred mixture of sodium hydroxide (3 g, 75 mmol) and 2-(1H-indol-3-yl)ethan-1-ol 6 (4 g, 25 mmol) in DMSO (60 mL) at 0 °C. The mixture was warmed to room temperature and stirred for 3 h. The mixture was diluted with EtOAc (100 mL), extracted with H2O (3 × 200 mL) and the organic layer was washed with brine (100 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 20% EtOAc/Hexanes) to give alcohol (7) as a yellow oil (5.75 g, 82% yield).1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 6.4 Hz, 1H), 7.31 (d, J = 6.4 Hz, 1H), 7.20–7.10 (m, 2H), 7.08 (d, J = 6.8 Hz, 2H), 6.99 (s, 1H), 6.85 (d, J = 6.8 Hz, 2H), 5.21 (s, 2H), 3.90 (t, J = 5.2 Hz, 2H), 3.78 (s, 3H), 3.03 (t, J = 5.2 Hz, 2H), 1.57 (brs, 1H). The spectroscopic data were in good agreement with literature values.24
3-(2-Chloroethyl)-1-(4-methoxybenzyl)-1H-indole-2-carbaldehyde (8)
Phosphoryl chloride (21 mL, 0.22 mol) was added dropwise to a stirred solution of 2-(1-(4-methoxybenzyl)-1H-indol-3-yl)ethan-1-ol (7) (4 g, 14.2 mmol) in DMF (28 mL) at 0 °C. The mixture was heated at 60 °C for 12 h. The mixture was poured into an ice water (400 mL) and the pH of the solution was slowly adjusted to pH 10 by addition of aq. NaOH solution (2 M, 150 mL). The solution was warmed to room temperature, and stirred for 1 h. The mixture was extracted with EtOAc (3 × 50 mL) and the combined organic layers were washed with brine (50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 15% EtOAc/Hexanes) to give aldehyde (8) as an off-white solid (2.69 g, 58% yield). 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1H), 7.74 (d, J = 8.4, 1H), 7.40–7.39 (m, 2H), 7.21–7.17 (m, 1H), 7.04 (d, J = 8.8 Hz, 2H), 6.78 (d, J = 8.4 Hz, 2H), 5.74 (s, 2H), 3.81 (t, J = 6.8 Hz, 2H), 3.74 (s, 3H), 3.57 (t, J = 7.2 Hz, 2H). The spectroscopic data were in good agreement with literature values.25(R,E)-N-((3-(2-Chloroethyl)-1-(4-methoxybenzyl)-1H-indol-2-yl)methylene)-2-methylpropane-2-sulfinamide (9)
Ti(OEt)4 (1.3 mL, 5.65 mmol) was added to a solution of aldehyde (8) (0.37 g, 1.13 mmol) and (R)-t-butanesulfinamide (164 mg, 1.36 mmol) in anhydrous THF (20 mL). The mixture was heated at reflux for 24 h. The mixture was cooled to room temperature and poured into water (50 mL). The resulting slurry was stirred for 30 min and filtered through Celite. The organic layer was washed with brine (30 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was triturated with hexane. The desired product was isolated by filtration, washed with 20% EtOAc/Hexanes (2 × 10 mL) to give sulfinyl imine (9) as an off-white solid (441 mg, 91% yield); Mp 104–105 °C.1H NMR (400 MHz, CDCl3) δ 8.79 (s, 1H), 7.72 (d, J = 6.8 Hz, 1H), 7.33–7.32 (m, 2H), 7.18–7.15 (m, 1H), 6.88–6.86 (m, 2H), 6.77–6.75 (m, 2H), 5.93 (d, J = 13.2 Hz, 1H), 5.70 (d, J = 13.2 Hz, 1H), 3.79–3.76 (m, 2H), 3.73 (s, 3H), 3.58–3.53 (m, 2H), 1.10 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 158.7, 151.7, 139.3, 130.1, 129.7, 127.0, 126.8, 126.2, 122.1, 120.7, 120.3, 114.0, 110.5, 57.5, 55.2, 47.3, 44.4, 27.9, 22.2.
[α]22D = −67.02 (c = 0.57, CHCl3).
FTIR (AT-IR) νmax 2962, 1581, 1510, 1243, 1077, 877, 751 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C23H27N2O2S35Cl + H]+ 431.1560, found 431.1562.
(R)-N-((S)-1-(3-(2-Chloroethyl)-1-(4-methoxybenzyl)-1H-indol-2-yl)but-3-yn-1-yl)-2-methylpropane-2-sulfinamide (10)
A 1 M solution of propargyl magnesium bromide was prepared using magnesium turnings (1.2 g, 49.4 mmol), 2 iodine balls and mercury chloride (120 mg, 0.89 mol%) in diethyl ether (25 mL). Propargyl bromide (80% w/w in toluene, 3.6 mL, 32.1 mmol) was added slowly. The mixture was stirred and heated to 35 °C for 2 h. A solution of propargyl magnesium bromide solution in Et2O (1 M, 8 mL, 8 mmol) was added dropwise to a solution of sulfinyl imine (9) (1.01 g, 2.35 mmol) in dichloromethane (25 mL) at −43 °C (ref. 26) over 15–20 min. The mixture was stirred at −43 °C for 16 h. The mixture was quenched with saturated aq. NH4Cl solution (10 mL) at −43 °C, warmed to room temperature and extracted with EtOAc (3 × 20 mL), and the combined organic layers were washed with brine (20 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 30% EtOAc/Hexanes) to give the sulfinamide (10) diastereomer A as an off-white amorphous solid (0.93 g, 85% yield) and its diastereomer as an off-white amorphous solid (0.16 g, 9% yield).Sulfinamide (10) (more polar, major diastereoisomer): 1H NMR (400 MHz, CDCl3) δ 7.64–7.62 (m, 1H), 7.19–7.11 (m, 3H), 6.88 (d, J = 8.8 Hz, 2H), 6.82 (d, J = 9.2 Hz, 2H), 5.70 (d, J = 17.2 Hz, 1H), 5.39 (d, J = 17.2 Hz, 1H), 5.01–4.99 (m, 1H), 4.17 (d, J = 2.4 Hz, 1H), 3.94–3.87 (m, 1H), 3.81–3.75 (m, 4H), 3.46–3.33 (m, 2H), 2.78 (ddd, J = 17.2, 10.0, 2.8 Hz, 1H), 2.60 (ddd, J = 16.8, 5.2, 2.8 Hz, 1H), 2.13 (t, J = 2.8 Hz, 1H), 1.21 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 159.6, 138.3, 132.9, 130.2, 128.0, 127.7, 123.6, 120.6, 119.6, 115.1, 114.8, 113.5, 111.0, 80.4, 73.2, 56.4, 56.0, 49.7, 47.8, 45.2, 29.2, 26.7, 23.4, 23.2.
[α]22D = −80.62 (c = 0.61, CHCl3).
FTIR (AT-IR) νmax 3292, 2959, 2302, 1613, 1512, 1466, 1245, 1059, 734 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C26H31N2O2S35Cl + H]+ 471.1873, found 471.1870.
Minor Diastereomer (less polar): 1H NMR (400 MHz, CDCl3) δ 7.62–7.60 (m, 1H), 7.20–7.12 (m, 3H), 6.86–6.77 (m, 4H), 5.54 (d, J = 17.2 Hz, 1H), 5.45 (d, J = 17.2 Hz, 1H), 5.04–5.00 (m, 1H), 3.83 (td, J = 8.4, 1.2 Hz, 2H), 3.74 (s, 3H), 3.62 (d, J = 2.0 Hz, 1H), 3.46–3.32 (m, 2H), 2.93 (ddd, J = 17.2, 10.0, 2.8 Hz, 1H), 2.78 (ddd, J = 16.8, 5.2, 2.8 Hz, 1H), 1.91 (t, J = 2.8 Hz, 1H), 1.11 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 159.3, 137.7, 134.5, 129.9, 127.6, 127.2, 123.3, 120.4, 119.3, 114.8, 111.9, 110.6, 80.2, 77.7, 71.9, 56.4, 55.7, 50.2, 47.5, 44.8, 29.0, 25.5, 22.8
[α]22D = −57.26 (c = 1.14, CHCl3).
FTIR (AT-IR) νmax 3290, 2958, 2355, 1612, 1512, 1464, 1245, 1175, 1059, 1034, 736 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C26H31N2O2S35Cl + H]+ 471.1873, found 471.1867.
(S)-2-((R)-tert-Butylsulfinyl)-9-(4-methoxybenzyl)-1-(prop-2-yn-1-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole (11)
Sulfinamide (10) (268 mg, 0.56 mmol) in anhydrous DMF (2 mL) was added dropwise to a stirred suspension of sodium hydride (36 mg of 57–63% w/w in mineral oil, 0.85 mmol) in anhydrous DMF (3 mL) at 0 °C. The mixture was stirred for 45 min and then quenched with saturated aq. NH4Cl solution (10 mL). The mixture was diluted with EtOAc (20 mL) and extracted with water (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 30% EtOAc/Hexanes) to give tricycle (11) as a colourless oil (252 mg, 97% yield).1H NMR (400 MHz, CDCl3) δ 7.52 (m, 1H), 7.17–7.08 (m, 3H), 6.92–6.76 (m, 4H), 5.26 (s, 2H), 4.67 − 4.64 (m, 1H), 3.74 (s, 3H), 3.71–3.69 (m, 1H), 3.50–3.42 (m, 1H), 3.09–2.99 (m, 1H), 2.88–2.78 (m, 3H), 2.65–2.59 (m, 2H), 1.12 (t, J = 2.8 Hz, 1H), 1.15 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 158.9, 137.1, 134.8, 129.2, 127.2, 126.9, 122.0, 119.5, 118.3, 114.2, 109.9, 108.6, 80.7, 72.2, 58.5, 55.2, 46.7, 24.6, 22.7, 21.0.
[α]22D = +90.40 (c = 1.00, CHCl3).
FTIR (AT-IR) νmax 3298, 2928, 2300, 2117, 1612, 1512, 1462, 1245, 1075, 1032, 734 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C26H30N2O2S + H]+ 435.2106, found 435.2105.
Methyl 4-((S)-2-((R)-tert-butylsulfinyl)-9-(4-methoxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)but-2-ynoate (12)
A solution of alkyne (11) (0.16 g, 0.36 mmol) in anhydrous MeOH (1.5 mL) was added to a stirred suspension of Pd(OAc)2 (25 mg, 0.11 mmol) and triphenylphosphine (58 mg, 0.22 mmol) in anhydrous DMF (3 mL) at room temperature. The mixture was then placed under a mixture of CO and O2 (∼1:1, 1 atm) and stirred at room temperature for 16 h. Water (10 mL) was added and the mixture was filtered through a plug of Celite. The filtrate was diluted with EtOAc (25 mL) and water (15 mL). The layers were separated and the organic layer was washed with water (2 × 10 mL). The organic layer was washed with brine (15 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 40% EtOAc/Hexanes) to give ester (12) as a brown gum (157 mg, 86% yield).
1H NMR (400 MHz, CDCl3) δ 7.52 (d, J = 6.8 Hz, 1H), 7.18–7.07 (m, 3H), 6.93–6.77 (m, 4H), 5.29 (d, J = 17.2 Hz, 1H), 5.23 (d, J = 17.2 Hz, 1H), 4.74 (dd, J = 8.0, 4.4 Hz, 1H), 3.74 (s, 6H), 3.67 (dd, J = 14.8, 6.4 Hz,1H), 3.51 (td, J = 11.6, 4.4 Hz, 1H), 3.11–3.03 (m, 1H), 2.95–2.72 (m, 3H), 1.17 (s, 9H).
13C NMR (100 MHz, CDCl3) δ 159.0, 153.7, 137.1, 134.2, 129.1, 127.2, 126.8, 122.2, 119.5, 118.4, 114.3, 109.9, 108.8, 85.6, 77.2, 75.7, 58.6, 55.2, 52.6, 46.7, 24.9, 22.5, 20.6.
[α]22D = +10.74 (c = 1.59, CHCl3).
FTIR (AT-IR) νmax 2952, 2237, 1711, 1512, 1246, 1074, 736 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C28H32N2O4S + H]+ 493.2161, found 493.2166.
Methyl (S)-4-(9-(4-methoxybenzyl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)but-2-ynoate hydrochloride salt (13)
HCl in dioxane (4 M, 0.2 mL, 0.8 mmol) was added dropwise to a solution of ester (12) (49.7 mg, 0.1 mmol) in anhydrous Et2O (3 mL) at 0 °C. The mixture was stirred for 45 min, concentrated to dryness, and triturated with a mixture of hexane and Et2O (1:1, 20 mL). The off-white hydrochloride salt was isolated by filtration, washing with Et2O (2 × 10 mL) to give hydrochloride salt (13) as an off-white powder (42 mg) which was used directly in the next step without further purification.
1H NMR (400 MHz, CDCl3) δ 10.32 (brs, 2H), 7.54 (d, J = 7.2 Hz, 1H), 7.26–7.10 (m, 3H), 6.87–6.77 (m, 4H), 5.24 (d, J = 17.2 Hz, 1H), 5.14 (d, J = 17.2 Hz, 1H), 5.03 (m, 1H), 3.81 − 3.75 (m, 1H), 3.73 (s, 3H), 3.66 (s, 3H), 3.57–3.54 (m, 1H), 3.30–3.22 (m, 2H), 3.04–3.00 (m, 1H), 2.83–2.77 (m, 1H).
13C NMR (100 MHz, CDCl3) δ 159.1, 153.2, 137.7, 128.3, 127.3, 126.9, 125.9, 123.3, 120.1, 118.9, 114.4, 110.1, 108.7, 82.6, 77.2, 55.2, 52.7, 48.2, 46.7, 38.6, 23.4, 18.3.
FTIR (AT-IR) νmax 2951, 2447, 2240, 1713, 1512, 1248, 1176, 741 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C24H24N2O3 + H]+ 389.1865, found 389.1868.
Methyl (S)-4-(9-(4-methoxybenzyl)-2-(prop-2-yn-1-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)but-2-ynoate (14)
Propargyl bromide (80% w/w in toluene, 9.6 µL, 0.1 mmol) was added dropwise to a solution of hydrochloride salt (13) (42 mg, 0.099 mmol) and potassium carbonate (35 mg, 0.25 mmol) in anhydrous CH3CN (3 mL) at 0 °C. The mixture was stirred at 0 °C for 2 h, allowed to warm to room temperature and stirred for 18 h. The mixture was diluted with EtOAc (20 mL) and water (10 mL). The layers were separated and the organic layer was washed with water (2 × 10 mL) and brine (15 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography (silica gel, 30% EtOAc/Hexanes) to give the diyne (14) as a pale-yellow gum (34.5 mg, 65% yield over 2 steps).1H NMR (400 MHz, CDCl3) δ 7.53 (d, J = 7.2 Hz, 1H), 7.21–7.08 (m, 3H), 6.89–6.76 (m, 4H), 5.26 (s, 2H), 4.25 (dd, J = 8.4, 4.4 Hz, 1H), 3.75 (s, 3H), 3.74 (s, 3H), 3.51 (dd, J = 16.4, 2.4 Hz, 1H), 3.40 (dd, J = 16.8, 2.0 Hz, 1H), 3.27–3.24 (m, 2H), 2.96–2.88 (m, 1H), 2.79–2.65 (m, 3H), 2.19 (t, J = 2.4 Hz, 1H).
13C NMR (100 MHz, CDCl3) δ 158.9, 154.0, 137.3, 133.3, 127.3, 127.1, 126.7, 122.0, 119.4, 118.4, 114.3, 114.2, 109.8, 108.4, 87.0, 80.1, 77.2, 74.3, 72.7, 55.2, 54.2, 52.5, 46.5, 43.5, 42.7, 25.1, 17.6.
FTIR (AT-IR) νmax 3282, 2924, 2302, 2239, 1713, 1513, 1275, 1260, 750 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C27H26N2O3 + H]+ 427.2022, found 427.2025.
Methyl (S)-13-(4-methoxybenzyl)-5,7,8,13,13b,14-hexahydroindolo[2′,3′:3,4]pyrido[1,2-b]isoquinoline-1-carboxylate (15)
A solution of Cp*RuCl(COD) (1.9 mg, 5.04 × 10−3 mmol, 5 mol%) in anhydrous 1,2-dichloroethane (2 mL) was purged with acetylene gas for 10 min. A solution of diyne (14) (43 mg, 0.1 mmol) in anhydrous 1,2-dichloroethane (2 mL) was similarly purged with acetylene gas for 10 min and then slowly added to the solution of Cp*RuCl(COD) at 70 °C over 4 h using a syringe pump. The mixture was cooled to room temperature and the mixture was filtered through a plug of Celite. The filtrate was concentrated under reduced pressure to give pentacycle (15) (48 mg) which was used directly in the next step without further purification.1H NMR (400 MHz, CDCl3) δ 7.75–7.72 (m, 1H), 7.56–7.54 (m, 1H), 7.24–7.11 (m, 5H), 6.97–6.94 (m, 2H), 6.81–3.79 (m, 2H), 5.39 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 17.2 Hz, 1H), 4.11–3.99 (m, 2H), 3.77–3.61 (m, 2H), 3.75 (s, 3H), 3.65 (S, 3H), 3.26–3.01 (m, 3H), 2.92–2.88 (m, 1H), 2.79–3.73 (m, 1H).
13C NMR (100 MHz, CDCl3) δ 167.8, 158.8, 137.9, 136.4, 135.5, 135.2, 130.4, 129.8, 129.6, 128.9, 127.2, 126.9, 125.6, 121.5, 119.3, 118.2, 114.1, 109.8, 109.2, 58.0, 55.3, 55.2, 51.7, 50.2, 47.3, 33.3, 22.1, 21.
FTIR (AT-IR) νmax 2952, 2929, 1717, 1513, 1249, 749 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C29H28N2O3 + Na]+ 475.1998, found 475.2000.
(−)-Dihydrogambirtannine (1)
Trifluoroacetic acid (2.5 mL) was added to a solution of the crude pentacycle (15) (48 mg) in anisole (0.5 mL) at 0 °C under N2 atmosphere. The mixture was warmed to room temperature and stirred for 3 days. The mixture was concentrated under reduced pressure and the residue was diluted with EtOAc (10 mL) and quenched with saturated aq. NaHCO3 to pH ∼10. The aqueous layer was extracted with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (basic alumina (200–300 mesh), 7:2:1 Hexanes/CH2Cl2/EtOAc) to give dihydrogambirtannine (1) as a pale yellow gum (21 mg, 63% yield over 2 steps).
1H NMR (400 MHz, CDCl3) δ 8.01 (brs, 1H), 7.85 (d, J = 7.6, Hz, 1H), 7.51 (d, J = 7.6, Hz, 1H), 7.35 (d, J = 8.0, Hz, 1H), 7.29–7.21 (m, 4H), 4.14 (d, J = 15.2 Hz, 1H), 3.90 (s, 3H), 3.89–3.65 (m, 3H), 3.31 (dd, J = 11.2, 4.8 Hz, 1H), 3.19 (dd, J = 17.2, 11.6 Hz, 1H), 3.08–3.03 (m, 1H), 2.86–2.81 (m, 1H), 2.73 (td, J = 11.6, 4.4 Hz, 1H).
13C NMR (100 MHz, CDCl3) δ 167.6, 136.3, 136.0, 135.6, 134.3, 130.9, 129.2, 128.9, 127.1, 125.7, 121.6, 119.4, 118.1, 110.9, 108.5, 58.1, 56.3, 52.4, 51.9, 33.5, 21.4.
[α]24D = –145 (c = 0.33, CHCl3).
FTIR (AT-IR) νmax 3391, 2948, 1716, 1457, 1289, 1268, 1141, 744 cm−1.
HRMS (ESI-Quadrupole) m/z calc'd for [C21H20N2O2 + H]+ 333.1603, found 333.1600.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Research Foundation, Singapore, NRF Competitive Research Programme (CRP), grant numbers NRF-CRP27-2021-0002 and NRF-CRP18-2017-01, for financial support. We thank NTU for support. V. P. thanks the URECA programme of NTU.References
- For a review, see: E. R. Miller and K. A. Scheidt, Synthesis, 2022, 54, 1217–1230 CrossRef CAS PubMed.
- L. Merlini, R. Mondelli and G. Nasini, Tetrahedron, 1967, 23, 3129–3145 CrossRef.
- Z. Yin, T. Liu and J.-X. Fu, J. Asian Nat. Prod. Res., 2019, 31, 257–261 CrossRef PubMed.
- E. Smith, R. S. Jaret, M. Shamma and R. J. Shine, J. Am. Chem. Soc., 1964, 86, 2083–2084 CrossRef CAS.
- J. A. Beisler, Tetrahedron, 1970, 26, 1961–1965 CrossRef CAS.
- E. Wenkert, K. G. Dave, C. T. Gnewuch and P. W. Sprague, J. Am. Chem. Soc., 1968, 90, 5251–5256 CrossRef CAS PubMed.
- R. T. Brown and S. B. Pratt, J. Chem. Soc., Chem. Commun., 1980, 165–167 RSC.
- A. Chatterjee, Pure Appl. Chem., 1986, 58, 685–692 CrossRef CAS.
- H.-J. Knölker and S. Cämmerer, Tetrahedron Lett., 2000, 41, 5035–5038 CrossRef.
- For a review, see: P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS.
- For a recent example from this laboratory, see: Rita, M. H. bin Abdul Rahman, S. S. M. Chong and R. W. Bates, Synlett, 2020, 31, 1479–1481 CrossRef CAS.
- U. Huan, B. Klin, F. H. Darras, J. Heilmann and M. Decker, Eur. J. Med. Chem., 2014, 81, 15–21 CrossRef PubMed.
- M. W. Smith, Z. Zhou, A. X. Gao, T. Shimbayashi and S. A. Snyder, Org. Lett., 2017, 19, 1004–1007 CrossRef CAS PubMed.
- D. R. Fandrick, C. A. Hart, I. S. Okafor, M. A. Mercadante, S. Sanyal, J. T. Masters, M. Sarvestani, K. R. Fandrick, J. L. Stockdill, N. Grinberg, N. Gonnella, H. Lee and C. H. Senanayake, Org. Lett., 2016, 18, 6192–6195 CrossRef CAS PubMed.
- (a) J. A. Ellman, T. D. Owens and T. P. Tang, Acc. Chem. Res., 2002, 34, 984–995 CrossRef PubMed. For the related p-tolyl sulfinylimines, see: (b) P. Zhou, B.-C. Chen and F. A. Davis, Syntheses and Reactions of Sulfinimines, in Advances in Sulfur Chemistry, ed. C. M. Raynor, JAI Press, Stamford, CT, 2000, vol. 2, pp. 249–282 Search PubMed.
- N. Siva Senkar Reddy, R. Anji Babu and B. V. Subba Reddy, Synthesis, 2016, 48, 1079–1086 CrossRef.
- H. Heaney and S. V. Ley, Org. Synth., 1974, 54, 58 CrossRef CAS.
- A. Liobat, J. Escorihuela, S. Fustero and M. Medio-Simón, Org. Lett., 2021, 23, 3691–3695 CrossRef PubMed.
- Y. Izawa, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2004, 77, 2033–2045 CrossRef CAS . See also ref. 13.
- V. K. Aggarwal, N. Barbero, E. M. McGarrihle, G. Mickle, R. Navas, J. R. Suárez, M. G. Unthank and M. Yar, Tetrahedron Lett., 2009, 50, 3482–3484 CrossRef CAS.
- Y. Yamamoto, T. Arakawa, R. Ogawa and K. Itoh, J. Am. Chem. Soc., 2003, 152, 12143–12160 CrossRef PubMed.
- R. S. Doerksen, T. Hodík, G. Hu, N. O. Huynh, W. G. Shuler and M. J. Krische, Chem. Rev., 2021, 121, 4045–4083 CrossRef CAS PubMed.
- Y. Ataka, M. Kitajima and H. Ishikawa, Org. Lett., 2023, 25, 7601–7605 CrossRef CAS PubMed.
- L. Han, C. Liu, W. Zhang, X.-X. Shi and S.-L. You, Chem. Commun., 2014, 50, 1231–1233 RSC.
- F. A. Davis, J. Y. Melamed and S. S. Sharik, J. Org. Chem., 2006, 71, 8761–8766 CrossRef CAS PubMed.
- The temperature was maintained using an Eyela PSL constant temperature bath.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra for compounds 6–8; 1H and 13C NMR spectra for compounds 1 and 9–15. See DOI: https://doi.org/10.1039/d5ob00162e |
| This journal is © The Royal Society of Chemistry 2025 |