
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAryl fucosides: synthesis and evaluation of their binding affinity towards the DC-SIGN receptor†
Rohit Chavan‡
a,
Maurice Besch‡ bcd,
Jonathan Lefèbrebcd,
Christoph Rademacher*bc and
Petra Ménová*a
bcd,
Jonathan Lefèbrebcd,
Christoph Rademacher*bc and
Petra Ménová*a
aUniversity of Chemistry and Technology, Prague, Technická 5, 16628 Prague 6, Czech Republic. E-mail: petra.menova@vscht.cz
bDepartment of Pharmaceutical Sciences, University of Vienna, Josef-Holaubek-Platz 2, 1090 Vienna, Austria. E-mail: christoph.rademacher@univie.ac.at
cDepartment of Microbiology, Immunobiology and Genetics, Max F. Perutz Laboratories, University of Vienna, Biocenter 5, 1030 Vienna, Austria
dVienna Doctoral School of Pharmaceutical, Nutritional and Sport Sciences, University of Vienna, Vienna, Austria
First published on 11th April 2025
Abstract
DC-SIGN, a C-type lectin receptor expressed on antigen presenting cells, is crucial for pathogen recognition and immune modulation. While most glycomimetic DC-SIGN ligands are mannose-based, fucose-based ligands offer enhanced selectivity, potentially reducing off-target effects. This study reports the stereoselective synthesis of aryl α-L-fucosides and their binding affinity for DC-SIGN. Using 1H–15N HSQC NMR and a competition assay, we identified 3-trifluoromethylphenyl α-L-fucoside as the best ligand, exhibiting both KD and IC50 values in a three-digit micromolar range and binding not only to the canonical site, but also to a secondary allosteric site.
DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin, CD209) is a C-type lectin receptor predominantly expressed on dendritic cells and some macrophages. It plays a crucial role in immune surveillance by recognizing and binding to a diverse range of glycoconjugates, facilitating pathogen capture, antigen presentation, and immune modulation.1 DC-SIGN is particularly known for its ability to interact with high-mannose and fucosylated glycans present on viral, bacterial, and parasitic pathogens. The interaction of mannose-containing glycans with DC-SIGN governs the binding of dendritic cells to HIV, Mycobacterium tuberculosis, Leishmania and many others,2,3 while fucose-containing DC-SIGN ligands, such as the Lewis X antigen, are highly expressed on Schistosoma mansoni and Helicobacter pylori.2
Beyond its role in pathogen recognition, DC-SIGN also mediates cell–cell interactions, contributing to immune homeostasis, inflammation, and immune evasion mechanisms exploited by certain pathogens.4 Due to its versatile binding capabilities and involvement in infectious diseases and immune regulation, DC-SIGN has emerged as a target for therapeutic intervention and drug design.5
The natural ligands of DC-SIGN primarily include pathogen-associated glycans4 and endogenous glycoproteins such as ICAM-2 and ICAM-3.6,7 In recent years, synthetic ligands have been extensively developed to modulate DC-SIGN activity, including small-molecule glycomimetics,8 multivalent carbohydrate-based constructs,9 and glycopolymers.10 These synthetic compounds offer valuable tools for studying DC-SIGN functions and hold potential for therapeutic applications, such as blocking pathogen entry, modulating immune responses, and developing targeted drug delivery systems.
To date, most glycomimetic DC-SIGN ligands have been derived from mannose.8 Reports on fucose-based glycomimetics, however, are rather scarce.11–15 Yet, they might bring in numerous advantages stemming from the higher selectivity of fucose towards DC-SIGN. Mannose interacts with a number of other lectin receptors, such as DC-SIGNR, langerin, dectin-2, the mannose receptor, and the mannose binding lectin, which may lead to unintended side effects. In contrast, fucose exhibits significantly greater selectivity, showing a strong preference for DC-SIGN over DC-SIGNR,16 langerin,13,17 and dectin-2.18 Nevertheless, it does interact with the mannose receptor19 and mannose-binding lectin.20 This potentially higher selectivity may lead to fewer side effects in therapeutic applications and a faster route to developing selective inhibitors.
L-Fucose occurs in nature mainly in the form of α-glycosides.21 Although a vast number of methods for glycosylations have been reported to date,22–25 achieving 1,2-cis α-glycosides remains a challenge,26 which is also the case for α-L-fucosides.27 Here, we report on the stereoselective synthesis of aryl α-L-fucosides using tetra-O-acetyl-L-fucopyranose as a glycosyl donor and various substituted phenols as acceptors and evaluation of their affinity for the DC-SIGN receptor.
We designed a small library of six aryl α-L-fucosides (1–6, Fig. 1), each featuring an aromatic moiety in the anomeric position. The compounds in the library differ in the nature of the aromatic substituent, its electron-donating or -withdrawing properties, and/or its position on the aromatic ring. Positioning of the aryl substituent at position 1 is supported by our recent study, which demonstrated that a substituent at the anomeric position of fucose does not interfere with the coordination of the Ca2+ ion.28 Additionally, insights from aryl mannosides suggest potential hydrophobic interactions with Phe313.29–31 All the compounds can be synthesized from a common fucosyl donor using a streamlined two-step methodology: glycosylation of the respective phenol followed by global deprotection (Scheme 1).
 | ||
| Fig. 1 Target fucosides 1–6. | ||
 | ||
| Scheme 1 Synthesis of target compounds 1–6. | ||
Initially, we screened three different fucosyl donors in a model reaction with 2-(trifluoromethyl)phenol: 2,3,4-tri-O-acetyl-α-L-fucopyranosyl trichloroacetimidate (7), 2,3,4-tri-O-acetyl-α-L-fucopyranosyl bromide (8), and tetra-O-acetyl-L-fucopyranose (9). Trichloroacetimidate 7 upon activation with TMSOTf at −15 °C yielded an inseparable mixture of anomers in a 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 ratio (α:β) (88% yield). Using fucosyl bromide 8, activated with TfOH and Ag2O, we obtained the desired α-fucoside exclusively, albeit in a low yield of 23%. Seeking further yield improvement, we investigated the use of the less reactive glycosyl acetate 9, activated by BF3·OEt2. This method yielded the desired α-anomer 10 exclusively in a 31% yield. Similarly, we synthesized compounds 11–13 by employing fucosyl acetate 9 and the respective substituted phenol acceptor and obtained exclusively the α-anomers in low to moderate yields (39% and 35% for the 3- and 4-CF3 derivatives, respectively, and 70% for the CH3 derivative).
:49 ratio (α:β) (88% yield). Using fucosyl bromide 8, activated with TfOH and Ag2O, we obtained the desired α-fucoside exclusively, albeit in a low yield of 23%. Seeking further yield improvement, we investigated the use of the less reactive glycosyl acetate 9, activated by BF3·OEt2. This method yielded the desired α-anomer 10 exclusively in a 31% yield. Similarly, we synthesized compounds 11–13 by employing fucosyl acetate 9 and the respective substituted phenol acceptor and obtained exclusively the α-anomers in low to moderate yields (39% and 35% for the 3- and 4-CF3 derivatives, respectively, and 70% for the CH3 derivative).
We next used halogen-substituted phenols, namely 2-chloro and 2-bromophenol, as aglycones and trichloroacetimidate donor 7 and observed exclusive α-anomer formation. However, the resulting products were contaminated with a hydrolysed donor by-product which we were not able to separate despite multiple column purifications. To avoid the purification issues, we explored the glycosylation reaction using the less reactive tetra-O-acetyl-L-fucopyranose (9) donor and successfully achieved pure α-anomers in low to moderate yields (40% for the bromo derivative 14 and 20% for the chloro derivative 15).
The observed stereoselectivity aligns with previous observations described in the literature. Studies have repeatedly shown an inverse correlation of selectivity with nucleophilicity.32–34 Highly nucleophilic alcohols react via diffusion-controlled trapping of the intermediate oxocarbenium ion, resulting in the lowest α/β ratios. In contrast, stereoelectronic control dominates reactions involving less nucleophilic alcohols. Consequently, phenols, particularly those bearing an electron-withdrawing substituent, tend to follow a more SN1-like mechanism. This leads to the preferential formation of the α-anomer, which is thermodynamically favoured due to the axial orientation of the anomeric oxygen, driven by the anomeric effect.22
Target compounds 1–6 were obtained by acetyl group cleavage employing Zemplén's conditions (0.2 M sodium methoxide in methanol) in yields of 38–78%. The general reaction scheme is depicted in Scheme 1. Detailed reaction procedures are given in the ESI.†
To characterize the DC-SIGN binding properties of the prepared fucosides 1–6, we employed protein-observed 1H–15N HSQC NMR to obtain binding site information and affinity data, alongside a plate-based competition assay to assess the impact of the compounds on receptor activity. The results of both assays are summarized in Table 1.
| No. | KD (μM) 1H–15N HSQC NMR | IC50 (μM) plate-based competition assay |
|---|---|---|
| 1 | 2240 ± 620 | 1130 |
| 2 | 600 ± 130 | 730 |
| 3 | 2600 ± 1240 | 1400 |
| 4 | 1860 ± 440 | N/A |
| 5 | 1480 ± 310 | 1140 |
| 6 | 1350 ± 510 | 650 |
Binding sites of the fucose-based compounds 1–6 were assessed using chemical shift perturbations (CSPs) on the 1H–15N backbone resonances of the 15N-labelled DC-SIGN CRD.35 The largest CSPs were detected in backbone resonances of residues located in the long loop in close proximity to the carbohydrate binding site (N349, N350, V351). The corresponding amino acids localize around Ca2+ II, directly involved in coordinating with the hydroxy groups of glycans interacting with the canonical binding site of the receptor. A comparison of the binding site of fucose and the aryl fucosides suggests a common binding mode.28
CSPs of residues located in β strands 3 and 4 (E358, S360, N365) aligned subjacent to the carbohydrate binding site are part of the conserved CSP pattern among all compounds. The comparison of resonance shift directions of carbohydrate binding site residues at increasing ligand concentrations supports a commonly shared binding mode among the different aryl fucosides, except for compound 2. Unlike all other compounds, compound 2 induces significant CSPs in resonances of M270 upon ligand titration. Since M270 was recently identified as part of a secondary allosteric binding site of DC-SIGN, we hypothesized the existence of a secondary binding site for 2.31,36 Shifting the CF3 position to the meta position enables the compound to bind to the secondary site, whereas the CF3 group in the ortho or para position, as in 1 and 3, respectively, does not permit efficient secondary site binding. 3 induces a minor CSP in M270 resonances and induces an elongated peak shape.
Affinities were estimated via residues displaying resonances in the fast exchange regime resulting from the addition of progressively increasing ligand concentrations. Selected CSP trajectories were used to fit the dissociation constants (KD) of compounds 1–6 (Table 1; Fig. 2a–d and Fig. S1–S5†).
 | ||
| Fig. 2 Interaction of 2 with the DC-SIGN CRD. (a) Superimposed 1H–15N HSQC NMR titration spectra of the 15N-labelled DC-SIGN CRD interacting with increasing concentrations of 2, which reveal several concentration-dependent CSPs. (b) Mapping of CSPs induced at 5 mM 2 to the structure of the DC-SIGN CRD (PDB code: 1SL4) suggesting that the ligand interacts with the carbohydrate binding site around Ca2+ site II in the long loop region and beta strands β3 and β4 as well as with the secondary site. (c) Examples of residues in the carbohydrate binding site showing fast exchanging resonances upon titration of increasing ligand concentrations. (d) Fitting of CSPs over the increasing concentration of 2 which enabled approximation of the affinity (KD = 600 ± 130 μM). | ||
Aryl fucoside 2 displayed the highest affinity (KD = 600 ± 130 μM), approximately 7-fold higher than that of fucose28 and 4-fold higher than that of 3 (KD = 2600 ± 1240 μM), the compound with the lowest measured affinity. As we have previously proposed allosteric activation via the secondary site around residue M270, we reasoned that the enhanced affinity of 2 might be attributed to its dual interaction with the allosteric binding site apart from the primary site binding.31 To rationalize our findings, we performed computational modelling of the binding pose of 2 in the secondary site. Using secondary site residue constraints, docking of the compound to the rigid receptor suggested orientation of the CF3 group towards a smaller cavity lined with residues of α helix 2 and β strands 1 and 5, including residue M270. While the aglycone orientation was similar across several binding poses, the fucoside moiety appeared flexible but was predicted to form interactions with polar residues adjacent to the secondary site (Fig. S6c†). Overall, binding poses suggested a binding mode similar to that of the previously identified biphenyl mannoside binding to this site.31
All compounds were evaluated in a plate-based competition assay in terms of their capability to inhibit the interaction between DC-SIGN and the glycans naturally present on horseradish peroxidase (HRP).37 The plate-based competition assay provides a more physiologically relevant model than HSQC NMR, as it enables the observation of compound-induced inhibition of multivalent interactions between DC-SIGN ECD and glycosylated HRP. Compounds 1, 2, 3, 5, and 6 were identified as inhibitors as they effectively impeded DC-SIGN binding to oligosaccharides (Table 1 and Fig. 3a, b). The determined half-maximal inhibitory concentrations (IC50) showed a strong correspondence with the dissociation constants (KD) obtained from 1H–15N HSQC NMR titrations (Fig. 2a–d and Fig. S1–S5†). Compound 2 has an IC50 of 730 μM and the lowest dissociation constant (KD = 600 ± 130 μM). Due to insufficient data quality, the binding parameters of compound 4 could not be fitted reliably.
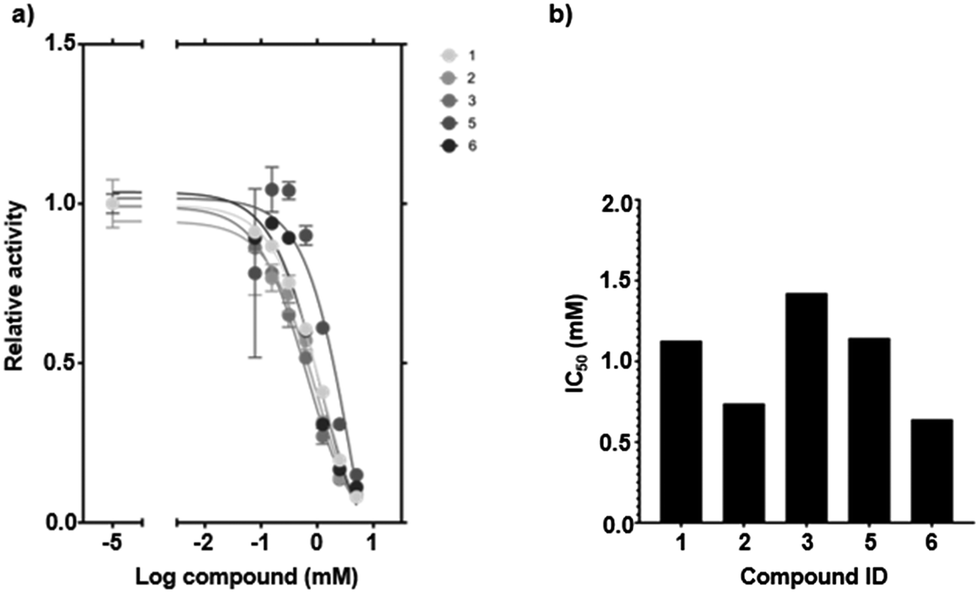 | ||
| Fig. 3 Activity evaluation of compounds 1–6 by means of a plate-based competition assay. (a) Normalized plot of the relative carbohydrate binding activity as a function of increasing ligand concentration. Curves were used to fit the respective IC50 values. (b) IC50 overview highlighting 2 and 6 as the most potent inhibitors of the measured compounds. | ||
In this study, we have synthesized and characterized a small library of aryl α-L-fucosides as potential selective ligands for DC-SIGN. Using a stereoselective glycosylation approach with tetra-O-acetyl-L-fucopyranose as the donor, we achieved exclusive α-anomer formation in moderate to good yields. The recognition of these compounds by DC-SIGN was assessed through 1H–15N HSQC NMR titrations, revealing a conserved Ca2+-dependent binding mode. Notably, compound 2 exhibited the highest affinity, likely due to its ability to interact with both the canonical carbohydrate binding site and an allosteric site. This is a remarkable finding, since selectivity for this secondary site was strongly dependent on the position of the CF3 group at the aromatic aglycon. It should also be noted that in the case of compound 2, two effects underlie the inhibitory mechanism: binding to the canonical carbohydrate binding site and binding to the activatory allosteric site. As 2 binds to both sites, its affinity for the primary site is enhanced, thereby facilitating increased overall ligand binding, which further inhibits interactions with glycosylated ligands such as HRP in the plate-based competition assay. This hypothesis aligns well with the higher affinity of 2 than that of other aryl fucosides and its notably lower IC50 value.
Overall, our findings support the potential of fucose-based glycomimetics as selective DC-SIGN ligands, potentially offering an advantage over mannose-based analogues by reducing off-target interactions with other lectins while displaying comparable binding affinities.8 Notably, aryl fucoside 2 emerges as a promising starting structure for the development of efficient ligands for targeted delivery, as our previous study has shown that engagement with the secondary site significantly enhances binding.31
Author contributions
R. C. synthesized the molecules. M. B. performed protein expression and purification, biophysical as well as biological assays and data processing. J. L. contributed to the biological assay and provided the Python script for HSQC NMR data processing. P. M. and C. R. supervised the work. R. C., M. B., J. L., P. M., and C. R. wrote the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
P. M. and R. C. express their gratitude to the Max Planck Society for support through their Partner Group programme. C. R. and J. L. are thankful for the funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska Curie grant agreement no. 956314 ALLODD. The present project is supported by the National Research Fund, Luxembourg (AFR PhD Grant 17929849).References
- U. Svajger, M. Anderluh, M. Jeras and N. Obermajer, Cell. Signalling, 2010, 22, 1397–1405 CrossRef CAS PubMed.
- B. J. Appelmelk, I. van Die, S. J. van Vliet, C. M. Vandenbroucke-Grauls, T. B. Geijtenbeek and Y. van Kooyk, J. Immunol., 2003, 170, 1635–1639 CrossRef CAS PubMed.
- Y. van Kooyk, B. Appelmelk and T. B. Geijtenbeek, Trends Mol. Med., 2003, 9, 153–159 CrossRef CAS PubMed.
- Y. van Kooyk and T. B. H. Geijtenbeek, Nat. Rev. Immunol., 2003, 3, 697–709 CrossRef CAS PubMed.
- J. Cramer, RSC Med. Chem., 2021, 12, 1985–2000 RSC.
- T. B. Geijtenbeek, D. J. Krooshoop, D. A. Bleijs, S. J. van Vliet, G. C. van Duijnhoven, V. Grabovsky, R. Alon, C. G. Figdor and Y. van Kooyk, Nat. Immunol., 2000, 1, 353–357 CrossRef CAS PubMed.
- M. a. Colmenares, A. Puig-Kröger, O. M. Pello, A. L. Corbí and L. Rivas, J. Biol. Chem., 2002, 277, 36766–36769 CrossRef CAS PubMed.
- S. Leusmann, P. Ménová, E. Shanin, A. Titz and C. Rademacher, Chem. Soc. Rev., 2023, 52, 3663–3740 RSC.
- J. J. Reina and J. Rojo, Braz. J. Pharm. Sci., 2013, 49, 109–124 CrossRef CAS.
- C. R. Becer, M. I. Gibson, J. Geng, R. Ilyas, R. Wallis, D. A. Mitchell and D. M. Haddleton, J. Am. Chem. Soc., 2010, 132, 15130–15132 CrossRef CAS PubMed.
- G. Timpano, G. Tabarani, M. Anderluh, D. Invernizzi, F. Vasile, D. Potenza, P. M. Nieto, J. Rojo, F. Fieschi and A. Bernardi, ChemBioChem, 2008, 9, 1921–1930 CrossRef CAS PubMed.
- M. Andreini, M. Anderluh, A. Audfray, A. Bernardi and A. Imberty, Carbohydr. Res., 2010, 345, 1400–1407 CrossRef CAS PubMed.
- M. Andreini, D. Doknic, I. Sutkeviciute, J. J. Reina, J. Duan, E. Chabrol, M. Thepaut, E. Moroni, F. Doro, L. Belvisi, J. Weiser, J. Rojo, F. Fieschi and A. Bernardi, Org. Biomol. Chem., 2011, 9, 5778–5786 RSC.
- B. Bertolotti, B. Oroszová, I. Sutkeviciute, L. Kniežo, F. Fieschi, K. Parkan, Z. Lovyová, M. Kašáková and J. Moravcová, Carbohydr. Res., 2016, 435, 7–18 CrossRef CAS PubMed.
- B. Bertolotti, I. Sutkeviciute, M. Ambrosini, R. Ribeiro-Viana, J. Rojo, F. Fieschi, H. Dvořáková, M. Kašáková, K. Parkan, M. Hlaváčková, K. Nováková and J. Moravcová, Org. Biomol. Chem., 2017, 15, 3995–4004 RSC.
- Y. Guo, H. Feinberg, E. Conroy, D. A. Mitchell, R. Alvarez, O. Blixt, M. E. Taylor, W. I. Weis and K. Drickamer, Nat. Struct. Mol. Biol., 2004, 11, 591–598 CrossRef CAS PubMed.
- H. Feinberg, A. S. Powlesland, M. E. Taylor and W. I. Weis, J. Biol. Chem., 2010, 285, 13285–13293 CrossRef CAS PubMed.
- H. Feinberg, S. A. F. Jégouzo, M. J. Rex, K. Drickamer, W. I. Weis and M. E. Taylor, J. Biol. Chem., 2017, 292, 13402–13414 CrossRef CAS PubMed.
- H. Feinberg, S. A. F. Jégouzo, Y. Lasanajak, D. F. Smith, K. Drickamer, W. I. Weis and M. E. Taylor, J. Biol. Chem., 2021, 296, 100368 CrossRef CAS PubMed.
- Functional Glycomics https://functionalglycomics.org/glycan-array/1001506, (accessed 25/02/2025, 2025).
- L. Wang, F. Fan, H. Wu, L. Gao, P. Zhang, T. Sun, C. Yang, G. Yu and C. Cai, Synlett, 2018, 29, 2701–2706 CrossRef CAS.
- X. Zhu and R. R. Schmidt, Angew. Chem., Int. Ed., 2009, 48, 1900–1934 CrossRef CAS PubMed.
- A. J. Fairbanks, S. L. Flitsch and M. C. Galan, Org. Biomol. Chem., 2020, 18, 6979–6982 RSC.
- R. Das and B. Mukhopadhyay, ChemistryOpen, 2016, 5, 401–433 CrossRef CAS PubMed.
- K. Toshima, Carbohydr. Res., 2006, 341, 1282–1297 CrossRef CAS PubMed.
- A. V. Demchenko, Curr. Org. Chem., 2003, 7, 35–79 CrossRef CAS.
- H. J. Vermeer, C. M. van Dijk, J. P. Kamerling and J. F. G. Vliegenthart, Eur. J. Org. Chem., 2001, 2001, 193–203 CrossRef.
- R. Chavan, J. Lefèbre, K. Jochová, H. Dvořáková, C. Rademacher and P. Ménová, Bioorg. Med. Chem., 2025, 123, 118164 CrossRef CAS PubMed.
- T. Tomašić, D. Hajšek, U. Švajger, J. Luzar, N. Obermajer, I. Petit-Haertlein, F. Fieschi and M. Anderluh, Eur. J. Med. Chem., 2014, 75, 308–326 CrossRef PubMed.
- A. Kotar, T. Tomašič, M. Lenarčič Živković, G. Jug, J. Plavec and M. Anderluh, Org. Biomol. Chem., 2016, 14, 862–875 RSC.
- R. Wawrzinek, E.-C. Wamhoff, J. Lefebre, M. Rentzsch, G. Bachem, G. Domeniconi, J. Schulze, F. F. Fuchsberger, H. Zhang, C. Modenutti, L. Schnirch, M. A. Marti, O. Schwardt, M. Bräutigam, M. Guberman, D. Hauck, P. H. Seeberger, O. Seitz, A. Titz, B. Ernst and C. Rademacher, J. Am. Chem. Soc., 2021, 143, 18977–18988 CrossRef CAS PubMed.
- P. O. Adero, H. Amarasekara, P. Wen, L. Bohé and D. Crich, Chem. Rev., 2018, 118, 8242–8284 CrossRef CAS PubMed.
- S. S. Nigudkar and A. V. Demchenko, Chem. Sci., 2015, 6, 2687–2704 RSC.
- M. Jacobsson, J. Malmberg and U. Ellervik, Carbohydr. Res., 2006, 341, 1266–1281 CrossRef CAS PubMed.
- M. P. Williamson, Prog. Nucl. Magn. Reson. Spectrosc., 2013, 73, 1–16 CrossRef CAS PubMed.
- J. Aretz, H. Baukmann, E. Shanina, J. Hanske, R. Wawrzinek, V. A. Zapol'skii, P. H. Seeberger, D. E. Kaufmann and C. Rademacher, Angew. Chem., Int. Ed., 2017, 56, 7292–7296 CrossRef CAS PubMed.
- S. Ng, N. J. Bennett, J. Schulze, N. Gao, C. Rademacher and R. Derda, Bioorg. Med. Chem., 2018, 26, 5368–5377 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00472a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |