
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Systematic studies toward the synthesis of D-galactosamine-containing coumarin glycosides†
Hannah S. Wootton and
Gavin J. Miller*
and
Gavin J. Miller*
Centre for Glycoscience and School of Chemical and Physical Sciences, Keele University, Keele, Staffordshire ST5 5BG, UK. E-mail: g.j.miller@keele.ac.uk
First published on 23rd May 2025
Abstract
An O-glycosylation method for accessing coumarin glycosides is presented. We report the reaction of 6,8-difluoro-7-hydroxy-4-methylcoumarin and 4-methylumbelliferone with a variety of glycosyl imidate donors using BF3·Et2O as activator to access a series of coumarin glycosides in 64%−76% isolated yields. Several reaction parameters are evaluated including promotors, temperature and reagent equivalents. Following initial methodology development using simple D-glucose donors, D-galactosamino mono- and disaccharides are explored as substrates, showcasing applicability towards late-stage transformation of biologically relevant chondroitin sulfate glycosides. Glycosylation diastereoselectivity trends were also considered, proposing that the identity of the D-galactosamino N-protecting group and the coumarin acceptor contribute to observed anomeric product ratios. This methodology provides a convenient access to D-galactosamino-coumarin glycoconjugates and provides a benchmark for the development of related systems for biological evaluation.
Introduction
Coumarins are naturally occurring heterocycles that have received attention due to their use as therapeutic agents. For example, warfarin and acenocoumarol are approved anticoagulants, derived from coumarin.1 Relatedly, coumarin glycosides, where coumarin is conjugated to a sugar, have been evaluated as anti-cancer,2 anti-diabetic,3 anti-inflammatory,4 anti-microbial,5 and anti-viral agents.6 Additionally, coumarin glycosides contain a fluorogenic reporter group, and have thus been deployed as probes/inactivators for enzymes.7,8 Notable examples include esculin, an anti-inflammatory therapeutic, which undergoes hydrolysis by esculin hydrolase as a rapid test to detect Gram-positive or negative bacteria.9 Furthermore, 4-methylumbelleriferone-α-D-galactopyranoside has been used to monitor α-galactosidase-A activity relating to Fabry disease.10 Access to coumarin-functionalised glycosides is thus of particular interest to advocate their use as biological tools in glycoscience. To achieve this, robust methods for their synthesis are required, particularly those that avoid harsh reaction conditions and are applicable beyond simple monosaccharides.Methods for coumarin attachment to sugars have been reviewed,11,12 and often involve heating glycosyl acetates (the Helferich method), exemplified for D-Gal in Fig. 1a, using an excess of Lewis acid to afford the glycoside. However, these reactions are often only moderately yielding and not applicable to more complex/precious saccharides.13 Methods for accessing coumarin conjugates of the aminosugar D-galactosamine (D-GalN) are surprisingly few. Glycosyl halide 3 has been employed as a D-GalN monosaccharide donor,14–16 undergoing appropriate phase-transfer activation to deliver coumarin glycoside 5 in 33% yield [Fig. 1b, method (a)].17 However, this method is incompatible with glycosyl halide substrates prone to elimination under basic conditions.18 Alternatively, coumarin glycoside 5 has been accessed by heating glycosyl acetate 4 with Cu(OTf)2 under microwave conditions,19 improving the yield to 53%. Finally, a D-GalN-containing α-linked 4-methylumbelliferone (4-MU) disaccharide (D-Gal-β(1,3)-D-GalN) 7 was prepared by Kiso and co-workers, who optimised a Mitsunobu reaction using glycosyl donor 6 containing an α-directing 4,6-O-di-tert-butylsilylene group. α-Selectivity was achieved, but as an inseparable mixture (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 α:β) in 80% yield, and required high temperature using an excess of 4-MU (Fig. 1c).20
:1 α:β) in 80% yield, and required high temperature using an excess of 4-MU (Fig. 1c).20
 | ||
| Fig. 1 Methods of O-glycosylation toward accessing D-galactosamine coumarin glycosides and the reaction conditions developed here for a range of mono- and disaccharides. LG = Cl (3) or OAc (4); X = H = 4-MU; X = F = DiFMU; P = Troc or Phth. | ||
As part of a wider programme concerning the chemical synthesis of glycosaminoglycan mimetics,21–27 we required a general synthetic approach to access structurally defined chondroitin sulfate fragments containing a D-GalN-coumarin reducing end conjugate, particularly toward materials with orthogonal protecting group patterns. Considering the relative sparsity of methods available, our approach sought glycosyl imidate donors, which are reactive, accessible under mild conditions and have been employed previously for saccharide-coumarin attachment. For example, Ferro and co-workers synthesised a fluorogenic heparan sulfate disaccharide (D-GlcN-α(1,4)-D-GlcA) using a glycosyl D-glucuronate imidate in 76% yield. Notably this required prior TMS protection of 4-MU, successive addition of BF3·Et2O and acetylation of the product to facilitate purification,28 highlighting further the challenges associated with this type of transformation. Reported herein is our development of a methodology to generate coumarin-functionalised chondroitin sulfate disaccharides (D-GlcA-β(1,3)-D-GlcN).
Results and discussion
D-Glucopyranosyl donors
To initiate a general method for coumarin attachment, a series of simple D-glucopyranosyl monosaccharide donors were synthesised (see ESI† for details) before exploring D-GalN derivatives. Briefly, commercially available peraacetylated β-D-glucose 8 was diversified at the anomeric position to generate six different donors, which were then subjected to glycosylation using 6,8-difluoro-7-hydroxy-4-methylcoumarin (DiFMU) as the acceptor (Table 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Conditions | Promotor | Equiv. | α:β |
Yield (%) |
| Conditions:a 1–2 h, −20 °C–0 °C.b 48 h, RT.c 2 h, RT. General conditions: glycosyl donor (1.0 equiv.); DiFMu (1.2 equiv.); DCM. | ||||||
| 1 | 11 | b | Ag2O | 20 | 0:1 |
30 |
| 2 | 11 | c | Ag2O/TMSOTf | 3.0 | 0:1 |
60 |
| 3 | 12 | a | BF3·Et2O | 0.2 | 0:1 |
69 |
| 4 | 12 | a | BF3·Et2O | 1.0 | 0:1 |
64 |
| 5 | 13 | a | BF3·Et2O | 1.0 | 0:1 |
63 |

Reactions using acetate 8, thioglycoside 9 and phosphate 10 donors were unsuccessful due to no reaction, decomposition or hemiacetal formation, respectively. Glycosyl bromide 11 and Ag2O activation was more promising, generating the required coumarin derivative 14 in 30% yield (Table 1, entry 1), and this could be improved to 60% using a shorter reaction time (2 h versus 48 h) and the addition of TMSOTf (Table 1, entry 2), as adopted from work by Demchenko and co-workers.29 Optimal results were achieved using D-glucopyranosyl imidate donors 12 and 13. Whilst initially no reaction or donor hydrolysis occurred when using N-trichloroacetimidate (TCAI) donor 12 with catalytic TMSOTf or tris(pentafluorophenylborane) (BCF), the adoption of catalytic BF3·Et2O gave the desired β-coumarin 14 in 69% yield (Table 1, entry 3). Increasing the amount of BF3·Et2O from 0.2 to 1.0 equivalent had little effect on reaction yield using TCAI donor 12 (Table 1, entry 4); these conditions could be similarly applied using donor 13 (Table 1, entry 5). Overall, D-glucopyranosyl donors 11–13 generated the derived difluorinated coumarin glucoside 14 in 60–69% yields with expected β-stereoselectivity and minimal by-product formation.
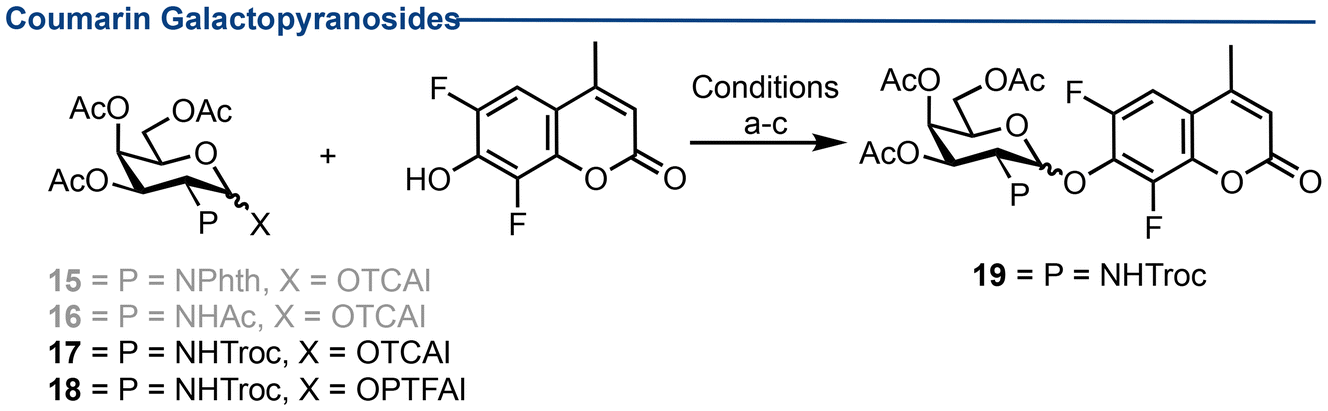
Towards D-galactosamine-coumarin conjugates
With a glycosylation method established, a panel of monosaccharide D-GalN imidate donors (15–18) were synthesised, including N-Phth, N-Ac and N-Troc amine protecting groups (see ESI† for details).Glycosylation using N-Phth donor 15 and N-Ac donor 16 were unsuccessful, either generating inseparable mixtures or no product. Using N-Troc imidate 17, glycosylation was successful, generating disaccharide 19 in yields of up to 71% (Table 2, entries 1–3). β-Diastereoselectivity was improved using a DCM/MeCN solvent system, from a 1:2 α:β mixture (Table 2, entry 1) to 1:4 (Table 2, entry 2). Such mixtures were separable via column chromatography and displayed clear differences in derived 3JH1–H2 1H NMR coupling constants [J = 3.2 Hz (α) versus 8.3 Hz (β)] and coupled HSQC [1JC1–H = 183.5 Hz (α) versus 168.8 Hz (β)]. Similar patterns were observed for PTFAI donor 18 (Table 2, entries 4–7), albeit in reduced yield and a stochiometric amount of BF3·Et2O proved optimal to deliver coumarin glycoside 19 in 65% yield (Table 2, entry 7). Finally, scalability was demonstrated (1.3 mmol, Table 2, entry 8). Despite a less-than-optimal diastereoselectivity, these results using TCAI or PTFAI D-GalN donors encouraged us to explore disaccharide substrates.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Promotor | Equiv. | α:β |
Yield |
| Solvent conditions:a DCM.b DCM/MeCN (10/1).c DCM/MeCN (5/1). General conditions: glycosyl donor (1.0 equiv.); DiFMu (1.2 equiv.); 1–2 h, −20–0 °C.d 1.3 mmol scale. | ||||||
| 1 | 17 | a | BF3·Et2O | 0.2 | 1:2 |
60% |
| 2 | 17 | b | BF3·Et2O | 0.2 | 1:4 |
71% |
| 3 | 17 | b | BF3·Et2O | 1.0 | 1:4 |
66% |
| 4 | 18 | a | BF3·Et2O | 0.2 | 1:2 |
50% |
| 5 | 18 | b | BF3·Et2O | 0.2 | 1:5 |
54% |
| 6 | 18 | c | BF3·Et2O | 0.2 | 1:3 |
52% |
| 7 | 18 | b | BF3·Et2O | 1.0 | 1:4 |
65% |
| 8d | 18 | b | BF3·Et2O | 1.0 | 1:4 |
66% |
Coumarin glycosylation with disaccharide D-GalN donors
The compatibility of late-stage disaccharide glycosylation with both DiFMu and 4-MU was explored next, utilising a selection of available chondroitin sulfate disaccharide precursors containing a variety of protecting groups (Table 3, see ESI† for details of donor preparation).21 Glycosyl donor 20 containing a N-Phth group generated β-24 in 70% yield (Table 3, entries 1 & 2) with stochiometric activator (using 0.2 equivalents of Lewis acid reduced the yield to 50%). Full characterisation of β-24 was obtained, with notable coupling constants deduced from the relevant 1D and 2D NMR spectra. The β-configuration at the reducing end was determined by a 3JH1–H2 coupling of 8.4 Hz and a large 1JC–H coupling of 169.4 Hz. Furthermore, characteristic 19F coupling was observed, with 1JC–F, 3JH–F and 4JF–F values of 251.5, 10.1 and 3.6 Hz respectively (Fig. 2). | ||
| Fig. 2 Chemical structure of coumarin glycoside 24, highlighting the key coupling constants observed in NMR spectroscopic data. | ||
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | Conditions | Promotor | Equiv. | Product | α:β |
Yield |
| Reaction conditions:a DCM, RT.b MeCN, RT.c DCM/MeCN (10/1, v/v), −20 °C to 0 °C.d DCM/MeCN (10/1, v/v), −50 °C to 0 °C. General conditions: glycosyl donor (1.0 equiv.); coumarin (1.2 equiv.); 1–2 h. | |||||||
| 1 | 20 | c | BF3·Et2O | 0.2 | 24 | 0:1 |
50% |
| 2 | 20 | c | BF3·Et2O | 1.0 | 24 | 0:1 |
70% |
| 3 | 21 | c | BF3·Et2O | 0.2 | 25 | 1:2 |
20% |
| 4 | 21 | c | BF3·Et2O | 1.0 | 25 | 1:2 |
28% |
| 5 | 22 | c | BF3·Et2O | 0.2 | 25 | 1:1 |
39% |
| 6 | 22 | d | BF3·Et2O | 0.2 | 25 | 1:1 |
42% |
| 7 | 22 | c | BF3·Et2O | 1.0 | 25 | 2:1 |
73% |
| 8 | 22 | c | BF3·Et2O | 1.0 | 26 | 1:2 |
76% |
| 9 | 23 | c | BF3·Et2O | 1.0 | 27 | 1:1 |
64% |
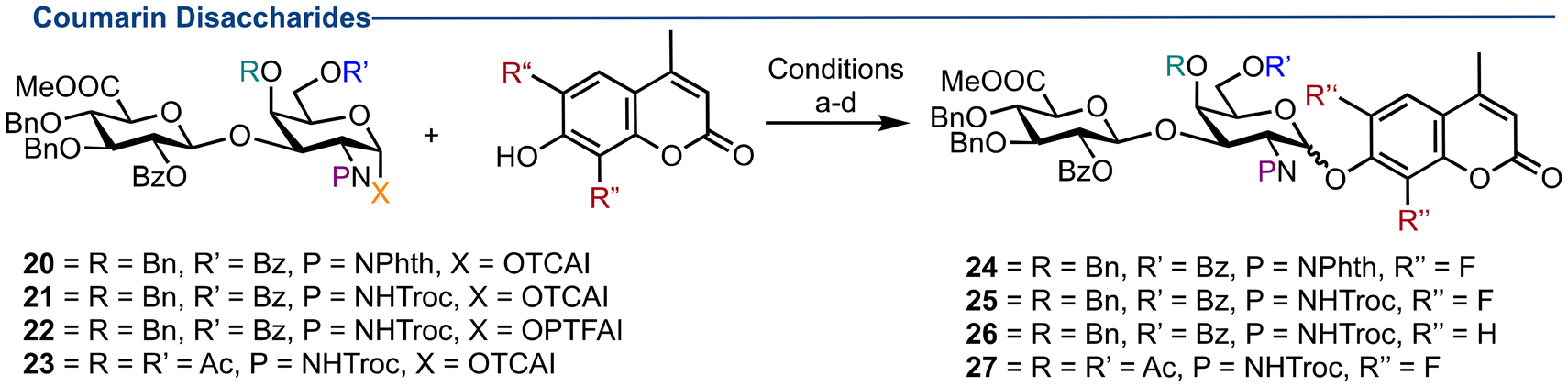
Reaction with N-Troc TCAI donor 21 generated glycoside 25 (1:2 α:β) firstly in 20% yield (Table 3, entry 3) then 28% yield (Table 3, entry 4) using stoichiometric activator. The major product isolated from this reaction (in 50% yield) was the N-glycoside, formed through return nucleophilic addition of the acetamide leaving group at C1. Switching to N-Troc PTFAI donor 22 and DiFMu generated disaccharide 25 in a moderately improved 39% yield (Table 3, entry 5). Reducing the temperature to −50 °C had a minimal impact on yield or diastereoselectively (Table 3, entry 6). Stoichiometric activator afforded 25 in an improved 73% yield but was poorly diastereoselective, affording a separable 2:1 α:β mixture (Table 3, entry 7). Furthermore, when donor 22 was reacted with 4-MU under identical conditions, the reaction again proceeded smoothly to generate 26 in 76% yield, but now as a 1:2 α:β mixture (Table 3, entry 8). Finally, 4,6-O-acetylated imidate 23 was reacted with DiFMu (Table 3, entry 9) which afforded disaccharide 27 in 64% yield in a 1.5:1 α:β ratio.
Several factors can influence the rate and stereochemical outcome of a glycosylation reaction, including the choice of anomeric leaving group, promotor system and acceptor nucleophilicity. For aminosugars, donor reactivity and glycosylation stereoselectivity can also be influenced by participation of a C2-amine protecting group. Indeed, this has been noted previously for D-GlcN derivatives, but less so for D-GalN.30 Herein we observed that the outcome (20%–76% yields; 2:1–0:1 α:β ratio) of glycosylation reactions to install a coumarin aglycon was dependent upon both the identity of the galactosamino N-protecting group and the phenol nucleophilicity. An N-Phth protecting group effected complete β-stereoselectivity and an N-Troc group also promoted β-stereoselectivity using a monosaccharide donor. However, at disaccharide level a decrease in β-selectivity was observed; reaction of N-Troc disaccharide donor 22 with DiFMU generated compound 25 as a 2:1 α:β mixture. Notably reaction with 4-MU saw an improvement in glycosylation diastereoselectivity (to 1:2, α:β for 26). Related studies have noted the effects of acceptor reactivity within glycosylation reactions, observing weaker nucleophiles to generate more α-product.31–34 Using a DiFMU acceptor, containing electron withdrawing fluorine appears to follow this trend, promoting α-selectivity. Shifting to 4-MU acceptor improved β-selectivity (2:1, α:β in 25 to 1:2, α:β in 26). Additionally, it has been proposed that an axial C4-O-acetate in D-Gal substrates promotes α-selectivity, due to participation.35 Our results partially support this, evidenced by β-selectivity being reduced when D-GalN monosaccharide donors are used versus D-GlcN (Tables 1 and 2). However, at disaccharide level, reaction of DiFMU with 4,6-O-acetyl donor 23 showed an increase in β-selectivity compared to C4-benzylated donor 22 (2:1, α:β for 22 to 1:1, α:β for 23). Whilst this methodology did not always afford diastereomerically pure glycosylation outcomes, disaccharide anomers were separable using conventional chromatography (see ESI† for details), providing access to stereopure materials for wider deprotection and biological evaluations.
Compared to previously reported methods (Table 4), this methodology requires low equivalents of both coumarin (1.2 equiv.) and glycosyl donor (1.0 equiv.) which is helpful for purification. Also, a low reaction temperature is employed compared to methods (a), (b) or (d) in Table 4 (−25 °C versus 60 °C or 130 °C), the reaction is scalable (1.3 mmol) and has a short duration. Furthermore, this methodology is applicable to a range of substrates utilising different O- and N-protecting groups which is useful for late-stage functionalisation. Notably here, β-selectivity is achieved in preference, whereby methods (a), (c) and (d) in Table 4 generate the α-anomers as the major product. Method (b) is β-selective but is lower yielding (33%) and incompatible with glycosyl substrates prone to elimination under basic conditions.
| Method | Donor (equiv.) | Coumarin (equiv.) | Promotor (equiv.) | Time (h) | Temp. (°C) | Scale (mmol) | Yield | α:β ratio |
|---|---|---|---|---|---|---|---|---|
| N.B. promotors for the described methods are as follows: this work.a BF3·Et2O.b NaOH.c Cu(OTf)2.d PPh3. | ||||||||
| This work | 1.0 | 1.2 | 1.0 | 2.0 | −25 | 1.3 | 76% | 2:1–0:1 |
| Ref. 13a |
2.0 | 1.0 | 15 | 5.0 | 60 | 2.0 | 54% | α major |
| Ref. 17b |
1.0 | 2.0 | 2.0 | 3.0 | RT | 0.6 | 33% | 0:1 |
| Ref. 19c |
1.0 | 4.0 | 0.15 | 0.5 | 130 | 1.0 | 53% | α major |
| Ref. 20d |
1.0 | 8.0 | 1.3 | 10.0 | 130 | 0.05 | 80% | 9:1 |
Conclusion
We have developed an O-glycosylation protocol for installing the commonly used aglycon coumarin onto D-GalN monosaccharides and disaccharides (chondroitin sulfate precursors). First optimising a methodology towards simple D-Glc donors, we selected imidates as the leaving group and BF3·Et2O as Lewis acid activator. Moving to D-GalN, the protecting group used on nitrogen proved optimal as N-Troc at monosaccharide level and either N-Phth or N-Troc for disaccharides containing a D-GlcA-(1,3)-β-D-GalN motif. Here the diastereoselectivity outcome was also dependant on the coumarin acceptor used, with higher β-selectivity observed for 4-MU versus a difluorinated analogue. Overall, the methodology is accessible and applicable, harnessing mild reaction conditions, short reaction times and delivering good yields (64%−76%) across a panel of fourteen glycosyl donors screened. Furthermore, access to orthogonally protected disaccharides used in late-stage oligosaccharide synthesis is shown and serves to demonstrate a broader application potential for glycosidation of fluorogenic motifs to study carbohydrate active enzymes.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
UK Research and Innovation are thanked for project grant funding to G. J. M. (Future Leaders Fellowship MR/T019522/1 & MR/Z000025/1) and the Engineering and Physical Sciences Research Council (EPSRC, EP/T007397/1). We also thank Keele University for PhD funding to H. S. W., the HRMS Facility within the Faculty of Natural Sciences at Keele University and the UK National Mass Spectrometry Facility (NMSF) at Swansea University.References
- M. K. Hussain, S. Khatoon, M. F. Khan, M. S. Akhtar, S. Ahamad and M. Saquib, Coumarins as Versatile Therapeutic Phytomolecules: A Systematic Review, Phytomedicine, 2024, 134, 155972, DOI:10.1016/j.phymed.2024.155972.
- W. A. El-Sayed, F. M. Alminderej, M. M. Mounier, E. S. Nossier, S. M. Saleh and A. F. Kassem, New 1,2,3-Triazole-Coumarin-Glycoside Hybrids and Their 1,2,4-Triazolyl Thioglycoside Analogs Targeting Mitochondria Apoptotic Pathway: Synthesis, Anticancer Activity and Docking Simulation, Molecules, 2022, 27(17), 5688, DOI:10.3390/molecules27175688.
- S. Ranđelović and R. Bipat, A Review of Coumarins and Coumarin-Related Compounds for Their Potential Antidiabetic Effect, Clin. Med. Insights: Endocrinol. Diabetes, 2021, 14, 1–9, DOI:10.1177/11795514211042023.
- R. K. Arora, N. Kaur, Y. Bansal and G. Bansal, Novel Coumarin–Benzimidazole Derivatives as Antioxidants and Safer Anti-Inflammatory Agents, Acta. Pharm. Sin. B, 2014, 4(5), 368–375, DOI:10.1016/j.apsb.2014.07.001.
- S. N. Mangasuli, K. M. Hosamani, H. C. Devarajegowda, M. M. Kurjogi and S. D. Joshi, Synthesis of Coumarin-Theophylline Hybrids as a New Class of Anti-Tubercular and Anti-Microbial Agents, Eur. J. Med. Chem., 2018, 146, 747–756, DOI:10.1016/j.ejmech.2018.01.025.
- M. Z. Hassan, H. Osman, M. A. Ali and M. J. Ahsan, Therapeutic Potential of Coumarins as Antiviral Agents, Eur. J. Med. Chem., 2016, 123, 236–255, DOI:10.1016/j.ejmech.2016.07.056.
- B. L. Wilkinson, L. F. Bornaghi, T. A. Houston, A. Innocente, C. T. Supuran and S. A. Poulsen, A Novel Class of Carbonic Anhydrase Inhibitors: Glycoconjugate Benzene Sulfonamides Prepared by “Click-Tailing”, J. Med. Chem., 2006, 49(22), 6539–6548, DOI:10.1021/jm060967z.
- D. Iacopini, M. Santi, M. C. Santangelo, G. Sardelli, L. Piazza, R. Mosca, L. M. Comparini, C. Granchi, M. Pineschi, S. D. Pietro, G. Signore and V. D. Bussolo, Glycoconjugate Coumarins Exploiting Metabolism-Enhanced Fluorescence and Preferential Uptake: New Optical Tools for Tumor Cell Staining, Bioorg. Chem., 2024, 153, 107836–107836, DOI:10.1016/j.bioorg.2024.107836.
- T. Cai and B. Cai, Pharmacological Activities of Esculin and Esculetin: A Review, Medicine, 2023, 102(40), e35306, DOI:10.1097/md.0000000000035306.
- T. Kizhner, Y. Azulay, M. Hainrichson, Y. Tekoah, G. Arvatz, A. Shulman, I. Ruderfer, D. Aviezer and Y. Shaaltiel, Characterization of a Chemically Modified Plant Cell Culture Expressed Human α-Galactosidase-A Enzyme for Treatment of Fabry Disease, Mol. Genet. Metab., 2015, 114(2), 259–267, DOI:10.1016/j.ymgme.2014.08.002.
- S. Dwivedi, S. Dey and A. Sau, Sugar Functionalized Coumarin Motifs: Synthesis and Applications, Carbohydr. Res., 2024, 544, 109244, DOI:10.1016/j.carres.2024.109244.
- S. Kumar, A. Arora, R. Kumar, N. N. Senapati and B. K. Singh, Recent Advances in Synthesis of Sugar and Nucleoside Coumarin Conjugates and Their Biological Impact, Carbohydr. Res., 2023, 530, 108857, DOI:10.1016/j.carres.2023.108857.
- X. Wei, Y. Ma, Q. Wu, J. Zhang, Z. Cai and M. Lu, An Improved Helferich Method for the α/β-Stereoselective Synthesis of 4-Methylumbelliferyl Glycosides for the Detection of Microorganisms, Molecules, 2015, 20(12), 21681–21699, DOI:10.3390/molecules201219789.
- K. F. Chilvers, J. D. Perry, A. L. James and R. H. Reed, Synthesis and Evaluation of Novel Fluorogenic Substrates for the Detection of Bacterial β-Galactosidase, J. Appl. Microbiol., 2001, 91(6), 1118–1130, DOI:10.1046/j.1365-2672.2001.01484.x.
- J. Liu, K. A. Schleyer, T. L. Bryan, C. Xie, G. Seabra, Y. Xu, A. Kafle, C. Cui, Y. Wang, K. Yin, B. Fetrow, P. K. P. Henderson, P. Z. Fatland, J. Liu, C. Li, H. Guo and L. Cui, Ultrasensitive Small Molecule Fluorogenic Probe for Human Heparanase, Chem. Sci., 2020, 12(1), 239–246, 10.1039/d0sc04872k.
- C. Malet, J. L. Viladot, A. Ochoa, B. Gallego, C. Brosa and A. Planas, Synthesis of 4-Methylumbelliferyl-D-Glucan Oligosaccharides as Specific Chromophoric Substrates of(1-3),(1-4)-D-Glucan 4-Glucanohydrolases, Carbohydr. Res., 1995, 285–301 CrossRef CAS PubMed.
- S. Park and I. Shin, Profiling of Glycosidase Activities Using Coumarin-Conjugated Glycoside Cocktails, Org. Lett., 2007, 9(4), 619–622, DOI:10.1021/ol062889f.
- A. V. Stachulski and G. V. Jenkins, The Synthesis of O -Glucuronides, Nat. Prod. Rep., 1998, 15(2), 173–186, 10.1039/a815173y.
- Q. Zhang, Z. Li, T. Chernova, V. Saikam, R. Cummings, X. Song, T. Ju, D. F. Smith and P. G. Wang, Synthesis and Characterization of Versatile O-Glycan Precursors for Cellular O-Glycomics, ACS Synth. Biol., 2019, 8(11), 2507–2513, DOI:10.1021/acssynbio.9b00168.
- A. Imamura, H. Ando, H. Ishida and M. Kiso, Di-Tert-Butylsilylene-Directed α-Selective Synthesis of 4-Methylumbelliferyl T-Antigen, Org. Lett., 2005, 7(20), 4415–4418, DOI:10.1021/ol051592z.
- H. S. Wootton, S. S. Berry, E. L. Ferguson, C. S. Mahon and G. J. Miller, Adaptable Synthesis of Chondroitin Sulfate Disaccharide Subtypes Preprogrammed for Regiospecific O–Sulfation, Eur. J. Org. Chem., 2024, 27(40), e202400587, DOI:10.1002/ejoc.202400587.
- A. N. Cheallaigh, S. E. Guimond, S. Oscarson and G. J. Miller, Chemical Synthesis of a Sulfated D-Glucosamine Library and Evaluation of Cell Proliferation Capabilities, Carbohydr. Res., 2020, 495, 108085, DOI:10.1016/j.carres.2020.108085.
- I. Pongener, C. O'Shea, H. Wootton, M. Watkinson and G. J. Miller, Developments in the Chemical Synthesis of Heparin and Heparan Sulfate, Chem. Rec., 2021, 21(11), 3238–3255, DOI:10.1002/tcr.202100173.
- I. Pongener and G. J. Miller, D-Glucuronate and D-Glucuronate, Glycal Acceptors for the Scalable Synthesis of D-GlcN-1,4-D-GlcA Disaccharides and Modular Assembly of Heparan Sulfate, J. Org. Chem., 2023, 88(15), 11130–11139, DOI:10.1021/acs.joc.3c01108.
- I. Pongener, E. T. Sletten, J. Danglad-Flores, P. H. Seeberger and G. J. Miller, Synthesis of a Heparan Sulfate Tetrasaccharide Using Automated Glycan Assembly, Org. Biomol. Chem., 2024, 22(7), 1395–1399, 10.1039/d3ob01909h.
- C. O'Shea and G. J. Miller, Synthesis of S-Glycoside Building Blocks as Mimetics of the Repeating d-GlcN-α-1,4-d-GlcA Heparan Sulfate Disaccharide, Molecules, 2024, 29(23), 5809, DOI:10.3390/molecules29235809.
- M. C. Z. Meneghetti, L. Naughton, C. O'Shea, D. S.-E. K. Teki, V. Chagnault, H. B. Nader, T. R. Rudd, E. A. Yates, J. Kovensky, G. J. Miller and M. A. Lima, Using NMR to Dissect the Chemical Space and O-Sulfation Effects within the O- and S-Glycoside Analogues of Heparan Sulfate, ACS Omega, 2022, 7(28), 24461–24467, DOI:10.1021/acsomega.2c02070.
- L. Wu, N. Wimmer, G. J. Davies and V. Ferro, Structural Insights into Heparanase Activity Using a Fluorogenic Heparan Sulfate Disaccharide, Chem. Commun., 2020, 56(89), 13780–13783, 10.1039/d0cc05932c.
- Y. Singh and A. V. Demchenko, Koenigs–Knorr Glycosylation Reaction Catalyzed by Trimethylsilyl Trifluoromethanesulfonate, Chem. – Eur. J., 2019, 25(6), 1461–1465, DOI:10.1002/chem.201805527.
- R. Enugala, L. C. R. Carvalho, M. J. D. Pires and M. M. B. Marques, Stereoselective Glycosylation of Glucosamine: The Role of the N–Protecting Group, Chem. – Asian J., 2012, 7(11), 2482–2501, DOI:10.1002/asia.201200338.
- S. van der Vorm, T. Hansen, J. M. A. van Hengst, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Acceptor Reactivity in Glycosylation Reactions, Chem. Soc. Rev., 2019, 48(17), 4688–4706, 10.1039/c8cs00369f.
- S. van der Vorm, J. M. A. van Hengst, M. Bakker, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Mapping the Relationship between Glycosyl Acceptor Reactivity and Glycosylation Stereoselectivity, Angew. Chem., Int. Ed., 2018, 57(27), 8240–8244, DOI:10.1002/anie.201802899.
- B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira and P. H. Seeberger, Nucleophile–Directed Stereocontrol Over Glycosylations Using Geminal–Difluorinated Nucleophiles, Angew. Chem., Int. Ed., 2016, 55(46), 14431–14434, DOI:10.1002/anie.201606774.
- J. M. A. van Hengst, R. J. C. Hellemons, W. A. Remmerswaal, K. N. A. van de Vrande, T. Hansen, S. van der Vorm, H. S. Overkleeft, G. A. van der Marel and J. D. C. Codée, Mapping the Effect of Configuration and Protecting Group Pattern on Glycosyl Acceptor Reactivity, Chem. Sci., 2023, 14(6), 1532–1542, 10.1039/d2sc06139b.
- M. Marianski, E. Mucha, K. Greis, S. Moon, A. Pardo, C. Kirschbaum, D. A. Thomas, G. Meijer, G. von Helden, K. Gilmore, P. H. Seeberger and K. Pagel, Remote Participation during Glycosylation Reactions of Galactose Building Blocks: Direct Evidence from Cryogenic Vibrational Spectroscopy, Angew. Chem., Int. Ed., 2020, 59(15), 6166–6171, DOI:10.1002/anie.201916245.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ob00746a |
| This journal is © The Royal Society of Chemistry 2025 |