
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNon-invasive quantitative chemical measurements of liposomal formulations using Raman spectroscopy†
Elizabeth J.
Legge
 a,
Ryan T.
Coones
a,
William A.
Lee
a,
Yiwen
Pei
a,
Natalie A.
Belsey
ab and
Caterina
Minelli
*a
a,
Ryan T.
Coones
a,
William A.
Lee
a,
Yiwen
Pei
a,
Natalie A.
Belsey
ab and
Caterina
Minelli
*a
aNational Physical Laboratory, Teddington TW11 0LW, UK. E-mail: caterina.minelli@npl.co.uk
bSchool of Chemistry & Chemical Engineering, University of Surrey, Guildford GU2 7XH, UK
First published on 3rd January 2025
Abstract
With a growing interest towards low batch-volume personalised medicines and continuous manufacturing of pharmaceuticals, the need for robust non-invasive quality control analytical methods is becoming increasingly important. Current methods for the quantification of total and encapsulated drug in a liposomal formulation include reversed-phase high-performance liquid chromatography with ultraviolet or fluorescence spectroscopy, which requires sample consumption after procedures such as ultrafiltration to separate the free drug from the encapsulated drug. We have developed and tested a method to perform non-invasive Raman spectroscopy measurements on liposomal doxorubicin. Raman spectroscopy provides chemically specific, potentially quantitative information, with measurements able to be performed on the contents of a sealed glass vial. We developed and validated the method by using a model system of polystyrene (PS) nanospheres and produced calibration curves for the concentration of PS at sizes of 40 nm, 125 nm and 200 nm. We then applied the same method to a liposomal doxorubicin formulation to measure the concentration of lipidic and drug components, and differences in the percentage of encapsulated drug. Our results show that by this method we can measure differences in doxorubicin concentration of 0.25 mg ml−1 and distinguish between free and encapsulated doxorubicin down to a minimal relative concentration of 2.3%.
Introduction
The approval of the first liposomal drug formulation in 1995 provided momentum for a large area of nanomedicine to develop centred around lipid-based particle delivery methods for therapeutics, with uses ranging from the original Doxil® liposome1 used in the treatment of cancer to the recent COVID-19 vaccine.2 Innovation is also occurring in the mode of manufacturing, with new trends including continuous manufacturing3,4 and personalised medicines.5 With these new developments, the need for suitable robust analytical methods and related standards to support product development, manufacturing and regulatory qualification is becoming increasingly important. Rapid and non-invasive techniques that can be applied on-line have the potential to enable real-time quality assessment and decision making, with minimal or no sample consumption.The therapeutic index, which compares the drug toxicity to the drug efficacy, is an important factor in the success of a medicine. Complex medicines, where the active ingredient is encapsulated in a carrier, offer a strategy to protect the cargo and target its delivery.6 The quantity and form of encapsulated versus free drug is one of the characteristics that impacts the potential drug toxicity7 and therefore is one of the attributes commonly assessed in liposomal drug formulations. The most common method for the quantification of drug in a liposomal formulation is reversed-phase high-performance liquid chromatography (RP-HPLC) with ultra-violet (UV) or fluorescence spectroscopy detectors. This requires the release of encapsulated drug from the liposomes, achieved by dispersion in surfactants or solvents.8,9 Centrifugal filtration is typically required during the sample preparation process when measuring the free drug content, in order to separate the free drug from the liposomes and encapsulated drug.10 RP-HPLC provides accurate results, reported to be within 5% of theoretical concentrations,11 however, quantification can be time consuming and the measurement is destructive to the sample, therefore it is not suitable for on-line analysis. Asymmetric-flow field-flow fractionation (AF4) is another, newer separation technique used to measure the encapsulated and free drug, with a promising outlook for achieving accurate results for purposes such as quality control.10,12 Both RP-HPLC and AF4 require the use of sample material that cannot be totally recovered afterwards, which limits quality control in areas such as personalised medicines where only small volumes are produced. The use of sample preparation methods and separation processes also introduces uncertainties, for example due to the loss of analytes in filters, membranes or separation columns.
Raman spectroscopy is chemically specific,13 non-destructive,13 and linear with concentration.14 Raman spectroscopy therefore can be used for measuring the concentration of molecules in liquids.15 Fourier transform (FT)-Raman spectroscopy has previously been shown as an alternative technique to HPLC, where FT-Raman spectroscopy is faster15 and more accurate,14 mainly because the sample preparation method for HPLC is longer and can introduce systematic errors. However, it should be noted that the detection limit of HPLC is typically 2–3 orders of magnitude better than Raman spectroscopy,15 therefore the method choice depends on the nature of the sample of interest and the objective of the analysis. Raman spectroscopy and optical trapping methods have also been used to measure differences between single particles16 and to detect glutamate encapsulated in liposomes,17 demonstrating promising applications of Raman spectroscopy for liposomes. Raman spectroscopy has also been shown to be suitable to measure changes to phospholipids in giant liposomes,18 and changes to doxorubicin when bound to squalene.19 However, few studies have investigated the use of Raman spectroscopy for the measurement of particle concentration20 and less so in the case of particle-based drug delivery systems, or how this relates to the amount of encapsulated drug.
In this work, we developed a method with minimal sample preparation to chemically identify and non-invasively measure the relative concentration of nanoparticles in a solution inside a sealed glass vial, by Raman spectroscopy. Firstly, we used polystyrene (PS) nanoparticles of different sizes as a model system, to develop a robust measurement method. PS was chosen because it is typically used as a reference material for testing the Raman shift for Raman spectrometers,21 and PS nanoparticles are well monodispersed particle systems which are typically used as a size standard to calibrate particle sizing and counting instruments.22 We then applied this method to study liposomes and measure the concentration of different components including the lipid carriers and the active ingredient (doxorubicin hydrochloride). We show that Raman spectroscopy is suitable to distinguish between different forms of doxorubicin and therefore useful towards the quantification of changes in encapsulated (and crystalline) versus free drug in the liquid formulation.
Materials & methods
Materials
Polystyrene nanoparticles (3000 Series Nanosphere™ Size Standards) with a diameter of 40 nm, 125 nm and 200 nm and reported concentration of 1% w/v solids, were purchased from Fisher Scientific (UK). Dilutions were performed volumetrically in ultrapure water (resistivity = 18.2 MΩ cm). Samples were sonicated in an ultrasonic bath for 5 minutes before measurements, and very gently shaken immediately before measurements to reduce any sedimentation.Doxoves® liposomal doxorubicin hydrochloride (HCl) and plain control liposomes containing ammonium sulphate, both constituted of hydrogenated soy L-α-phosphatidylcholine (HSPC), cholesterol (Chol) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (mPEG2000-DSPE), with a lipid composition of HSPC/Chol/mPEG2000-DSPE (56.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38.4:5.3 mol%) were purchased from Stratech (manufacturer FormuMax, CA). The concentrations declared by the manufacturer were (29.7 ± 0.3) mg ml−1 lipid in both the Doxoves® and control liposomes samples, and (4.0 ± 0.2) mg ml−1 doxorubicin HCl in the Doxoves® sample. The doxorubicin encapsulation efficiency declared by the manufacturer was >97%, which has been confirmed in studies within the literature on fluorescence lifetime microscopy23 and capillary electrophoresis with UV-Vis detection.24 Doxoves® itself has been characterised with cryo-TEM in previous studies.25 All gravimetric dilutions were performed using a calibrated Sartorius CPA 224 S Balance (220 g × 0.1 mg).
:38.4:5.3 mol%) were purchased from Stratech (manufacturer FormuMax, CA). The concentrations declared by the manufacturer were (29.7 ± 0.3) mg ml−1 lipid in both the Doxoves® and control liposomes samples, and (4.0 ± 0.2) mg ml−1 doxorubicin HCl in the Doxoves® sample. The doxorubicin encapsulation efficiency declared by the manufacturer was >97%, which has been confirmed in studies within the literature on fluorescence lifetime microscopy23 and capillary electrophoresis with UV-Vis detection.24 Doxoves® itself has been characterised with cryo-TEM in previous studies.25 All gravimetric dilutions were performed using a calibrated Sartorius CPA 224 S Balance (220 g × 0.1 mg).
Doxorubicin hydrochloride was purchased from Stratech (manufacturer Biorbyt, UK). Doxorubicin hydrochloride has the hazard codes H302, H340, H350 and H360FD, therefore sample preparation was performed in a ducted fume hood with appropriate personal protective equipment (PPE), and sample access was controlled. A buffer solution of 10% w/v sucrose, 10 mM histidine, pH 6.5, according to manufacturer instructions, was purchased from Stratech (manufacturer FormuMax, CA).
Particle tracking analysis (PTA)
Particle tracking analysis (PTA) measurements were carried out with a Horiba ViewSizer3000 (Horiba Scientific, Irvine, CA). The cell volume was illuminated using three lasers at 450 nm (blue), 532 nm (green) and 635 nm (red). Sample dilutions from the stock samples were performed gravimetrically in a buffer solution of 10% w/v sucrose, 10 mM histidine, pH 6.5. All samples were vortexed for 30 seconds to ensure the homogeneity of the suspension. For each sample, five sequential replicate videos of 60 seconds were recorded using a camera recording at 30 frames per second. The data were analysed using the VU-3000 Measure software (GUI software version 2.13, Processing library version 1.0.9, WeekBuild 2920).Multi angle DLS (MADLS)
Multi angle DLS (MADLS) measurements were performed on a Zetasizer Ultra (Malvern Panalytical Ltd, UK) using a 10 mW He–Ne laser at 633 nm. Samples were contained in disposable DTS0012 cuvettes (Malvern Panalytical, UK). MADLS measurements combined three scattering angles of 173°, 90°, and 17° to give a single particle size distribution for any given MADLS measurement. Liposomes were measured in water where the viscosity of water was set, in the analysis software, to 0.8872 mPa s with a refractive index value of 1.33. The refractive index of the liposomes was set at 1.45 in the analysis software. The working concentration range was determined through a series of measurements at different dilutions with analysis according to ISO 22412:2017.26UV-visible and fluorescence spectroscopy
UV-visible extinction and fluorescence spectra were acquired using matched quartz cuvettes (Hellma, semi-micro quartz Suprasil™) with an optical path length of 10 mm. UV-visible spectra were acquired using a Cary 60 UV-visible spectrophotometer (Agilent, Santa Clara, CA). Samples were diluted gravimetrically. Fluorescence measurements were performed using a Perkin Elmer SL 55 Luminescence Spectrometer. Fluorescence emission spectra were acquired at an excitation wavelength of 470 nm. In both cases spectra were baseline subtracted using the spectrum of ultrapure water (resistivity = 18.2 MΩ cm).Raman spectroscopy
Raman spectroscopy measurements were performed with an inVia Qontor spectrometer (Renishaw plc, UK) with a 50×, 0.5 NA objective lens (Leica Microsystems Ltd, Germany). A backscattered configuration was used with a grating of 1200 lines per mm and a charge coupled device (CCD) detector (Renishaw Centrus 0JPN74–1040 × 256). The Raman shift was calibrated each day using the peak at 520.5 cm−1 of an internal silicon sample. Raman spectra were acquired with a 830 nm laser (line focus), a power at the sample of (92.0 ± 1.6) mW and a theoretical diffraction limited line profile spot size of ∼2 μm in x-axis, ∼7 μm in y-axis, and >10 μm in z-axis. The 830 nm laser excitation was chosen over other wavelengths due to the high fluorescence of doxorubicin.For Raman spectroscopy, dilutions of the Doxoves® and control liposomes samples were performed gravimetrically in ultrapure water for quantification of doxorubicin HCl and liposomes in the solution, to concentrations of 1.0–1.5 mg ml−1 (doxorubicin HCl) and 1.5–11.1 mg ml−1 (total lipids). Doxorubicin hydrochloride was diluted to a concentration of 4.0 mg ml−1 in a solution of 10% w/v sucrose, 10 mM histidine, pH 6.5 and is referred to as 100% free doxorubicin as there are no liposomes in this sample. Doxorubicin hydrochloride was also dispersed in an aqueous solution of 250 mM ammonium sulphate at a concentration of 1.5 mg ml−1. Samples containing different concentrations of free and liposomal doxorubicin were prepared by adding different volumes of a solution containing doxorubicin HCl (10.3 mg ml−1) in buffer (free), to a solution of Doxoves® (diluted to 2 mg ml−1 doxorubicin HCl) in buffer (liposomal doxorubicin). The 100% free sample contained only doxorubicin HCl in buffer, whereas the 100% encapsulated sample contained only Doxoves®.
Each PS, liposome and free drug Raman spectrum was acquired with 2 accumulations of 60 s (with 3 measurements acquired at different positions along the length of the vial on the XY plane, subsequently averaged). The acquisition time for 3 samples with an expected encapsulation efficiency of >90% was increased to 4 accumulations of 90 s in order to detect small changes. For the doxorubicin HCl in ammonium sulphate solution, the measurement was performed on an aggregate. Depth profile measurements were performed on a water and a PS suspension inside a glass vial, where a spectrum was acquired at different z-axis intervals, starting from above the vial and moving down into the suspension. For the depth profile measurements, at each position the spectrum was acquired with 2 accumulations of 30 s (PS depth profiles), and 2 accumulations of 20 s (water depth profile). In-depth sample optimisation was required to achieve repeatable results, and the methods for this optimisation are described within the results of this manuscript.
Data analysis was performed in WiRE 5.3 for component subtraction, baseline subtraction and peak fitting. For quantitative analysis, a linear baseline was fitted between two points on the spectra, where the two points were either side of the peak of interest. The regions of the linear baseline were 970 cm−1–1055 cm−1 (PS peak) and 1485 cm−1–1860 cm−1 (water peak) in Fig. 2 and 3, 1165 cm−1–1365 cm−1 (lipid and doxorubicin peaks) and 1515 cm−1–1850 cm−1 (water peak) for the spectra analysed to build Fig. 4c, and 1165 cm−1–1505 cm−1 (lipid and doxorubicin peaks) in Fig. 4d and 5. It should be noted that in the case of the spectra analysed to build Fig. 2c, due to the overlap of the vial and water peaks at the vial–liquid interface (Fig. S1†), an identical position for the limits of the linear baseline was not possible, therefore a linear function with floating limits was fitted to the data between 1300 cm−1 and 2000 cm−1 using the ‘intelligent fitting’ function in WiRE 5.3. Furthermore, for qualitative analysis and visualisation of the spectra (Fig. 4a and b), a polynomial curve of order 10 was fitted to the full spectra with the ‘intelligent fitting’ function in WiRE 5.3, and subtracted as the baseline.
Component subtraction of the buffer was performed in some cases (Fig. 4c and d for the lipid and doxorubicin peaks and Fig. 5), to obtain information from the particles alone. For this, the contribution of the buffer component was ascertained through spectral fitting within a region containing peaks only attributed to the buffer. The relevant contribution of the buffer component was then subtracted from the spectrum of interest.
For the peak fitting used for Fig. 2 and 3, the PS and water peaks were fitted separately in each region of the spectrum, shown in Fig. 2a and b. However, for the peak fitting of the spectra used to build Fig. 2c, the two peaks of interest overlapped and were therefore fitted together, centred at 1794 cm−1 (vial peak) and 1638 cm−1 (water peak). In Fig. 4c, 6 peak functions were fitted to the region containing the water peak, as described in Fig. S2.† In Fig. 5, a different approach was taken, due to the multiple overlapping peaks and the need to separate out different component contributions, therefore component fitting was performed, as shown in Fig. S3 and S4.†
For the purposes of visualisation, spectra were normalised in Fig. 4a, b, d and 5a, to the peak stated in the text.
For the depth profile measurements in Fig. 2 and Fig. S5,† graphs containing values for the area of the peak at 1794 cm−1 (vial) were differentiated, then a Voigt peak function was fitted to the derivative, and the peak centre (position of greatest rate of change) was taken to be 0 μm. In Fig. 3 and Fig. S6–S8,† the depth position was estimated through focussing on the outside glass vial interface and moving to the expected z-axis offset from there.
Linear component fits were performed in Microsoft Excel (version 2308) and graphs were produced in Origin 2023. For the PS suspension where individual peak fitting was possible, the intensities are reported as areas, however in liposome samples with multiple overlapping peaks the height intensities were used. More detailed information on the analysis is provided within the results. The analytical approach chosen for this work served the purpose to monitor and quantify changes between spectra. Alternative approaches to data analysis are currently being investigated and will be subject of future work.
Results
This study focussed on demonstrating non-invasive Raman spectroscopy measurements of the bulk material of a sample contained within a sealed 2 ml glass vial, therefore requiring minimal sample preparation and with no loss of material. Measurements were acquired through the side wall of the glass vial, at a point in which the laser beam is perpendicular Fig. 1. The sample filled the whole vial, so that there was no air gap between the glass and the sample to minimise any distortion to the laser beam. Samples were gently shaken and inverted immediately before placing inside the spectrometer, to ensure a homogenous distribution of particles in the solution and minimise sedimentation.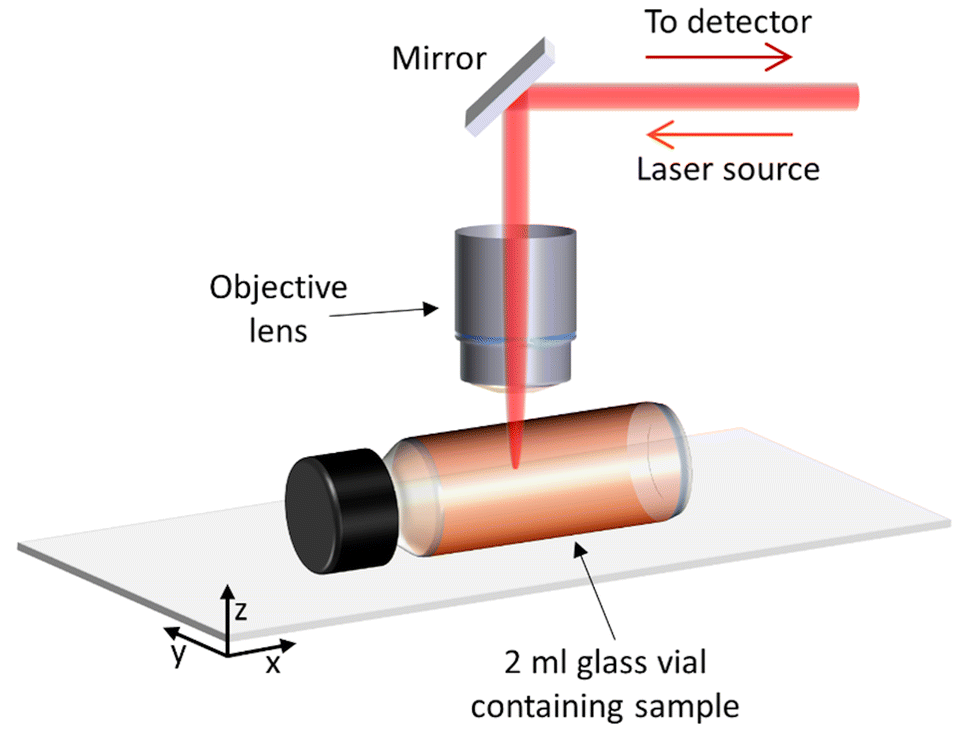 | ||
| Fig. 1 Set-up and position of the sample in relation to the laser when performing Raman spectroscopy measurements. | ||
PS nanospheres were used as a model system, to test the suitability of confocal Raman spectroscopy for measuring the relative mass concentration of particles while simultaneously obtaining chemical information. The Raman spectrum for PS exhibits a sharp strong band at ∼1002 cm−1 attributed to the carbon aromatic ring breathing mode27,28 and some less intense bands at 1032 cm−1 (C–H in-plane deformation), 1583 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretch) and 1602 cm−1 (ring-skeletal stretch),29 while the Raman spectrum for water contains a broad peak around 1638 cm−1 attributed to the O–H vibrational bending mode.20 The PS peak at ∼1002 cm−1 (Fig. 2a) is well separated from the characteristic water peak at 1638 cm−1, and it was therefore selected to monitor the concentration of PS in water. The water peak at 1638 cm−1 overlaps with some low intensity PS bands, however the contributions of the individual peaks can be easily resolved by peak-fitting (Fig. 2b). Raman spectra of the individual components can be found in the ESI (Fig. S9†).
C stretch) and 1602 cm−1 (ring-skeletal stretch),29 while the Raman spectrum for water contains a broad peak around 1638 cm−1 attributed to the O–H vibrational bending mode.20 The PS peak at ∼1002 cm−1 (Fig. 2a) is well separated from the characteristic water peak at 1638 cm−1, and it was therefore selected to monitor the concentration of PS in water. The water peak at 1638 cm−1 overlaps with some low intensity PS bands, however the contributions of the individual peaks can be easily resolved by peak-fitting (Fig. 2b). Raman spectra of the individual components can be found in the ESI (Fig. S9†).
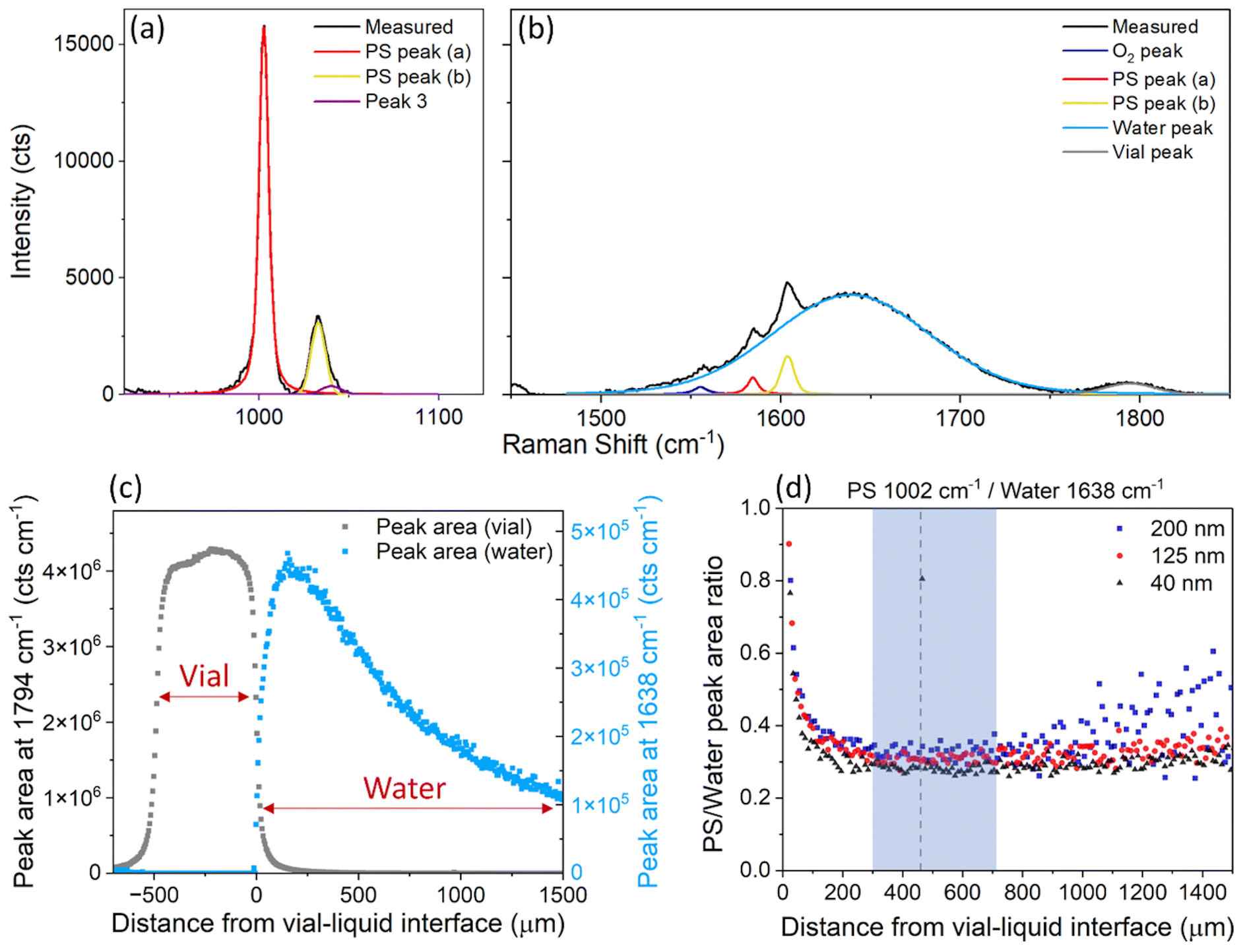 | ||
| Fig. 2 (a) and (b) Average Raman spectrum from 3 measurements of 40 nm PS in water, at a concentration of 4 mg ml−1, after baseline subtraction and peak fitting of (a) the PS peak at 1002 cm−1 and (b) the water peak at 1638 cm−1 and vial peak at 1794 cm−1. (c) Area of the peak centred at 1794 cm−1 (grey, vial) and 1638 cm−1 (blue, water), measured at different depths inside the glass vial. (d) Ratio of the area of the PS peak at 1002 cm−1 and the water peak at 1638 cm−1, for PS nanosphere sizes of 40 nm, 125 nm and 200 nm dispersed in ultrapure water at a concentration of 4 mg ml−1, at different depths inside the sample. The shaded blue area shows the depths where the measurements were most reproducible. The dotted line at 460 μm is the depth at which all other point measurements were acquired. An example of the Raman spectra at different points of the depth scan, and the equivalent graph for the individual PS and water peak areas can be found in the ESI (Fig. S5†). | ||
Raman spectra of the water sample and the ‘PS particles in water’ samples were acquired at different depths into the vial, to determine the optimal position for measurement, i.e. where the most repeatable results could be obtained. The depth scan started from above the glass vial (z = −690 μm) and moved down into the glass wall (∼490 μm thick) to reach the interface between the vial and sample (z = 0 μm) and measured the sample down to 1500 μm inside the vial. Fig. 1 demonstrates the direction of the z-axis plane.
For the sample containing only water, between z = −490 μm and z = 0 μm, the main Raman peak that was observed was at 1794 cm−1 and attributed to the vial (Fig. 2c). From z = 0 μm onwards, the water peak at 1638 cm−1 became the most dominant. The water peak had the highest intensity just inside the vial interface around z = 150 μm. However, for samples containing PS, the PS peak had the highest intensity at around z = 20 μm (Fig. S5†), in a depth profile measurement with a step size of 10 μm, which suggests the z-axis spot size is close to 10–20 μm. The maximum intensity of the water peak is further inside the sample, likely due to the presence of the peak at 1794 cm−1 (vial) which interferes with the analysis of the water peak between z = 0 μm and z = 150 μm (Fig. S1†). After z = 200 μm, the water peak decreased in intensity the further the distance inside the sample (Fig. 2c). This decrease in intensity is expected, due to the increased path length of the laser and therefore increased scattering and absorption of light by the sample. It is therefore beneficial to measure samples at a distance far enough inside the sample to have minimal interference from the glass vial, but not so far that the signal is too low for rapid analysis.
PS particles with nominal diameters of 40 nm, 125 nm and 200 nm were all measured, and the ratio of the selected PS and water peak areas was monitored. For a stable sample, i.e. in the absence of particle sedimentation, this ratio is expected to remain constant irrespective of the measurement depth. For all sizes, the optimal region for measurement was identified to be between z = 300 μm and z = 700 μm inside the sample (Fig. 2d). At less than 300 μm depth, the glass vial interfered with the measurement and analysis, but more than 700 μm inside the vial, the spectral intensity dropped too low for analysis of acquisition times of 1 minute, particularly for the larger PS particles (200 nm). These measurements informed all further point measurements for both the PS particles in water and liposome samples, which were taken at a depth of 460 μm from the vial–liquid interface.
In the depth interval between 300 μm and 700 μm not only is the ratio between the selected polystyrene and water peak areas constant, but it is similar for all three polystyrene samples. This similarity was expected, because all samples were measured at a nominal concentration of 4 mg ml−1 and the Raman signal intensity of a chemical component is proportional to its volume fraction.
With increasing concentration of particles in a sample, the intensity of laser light reaching the point of focus decreases, and backscattered signals decrease, due to increased light absorption and scattering by the particles. Therefore, the Raman signal intensity is expected to decrease with increasing concentration of particles, and this attenuation is expected to be more significant for larger particles because they scatter more light than small particles.30–32 UV-vis absorption spectra (Fig. S10†) show that more scattering occurs for larger PS particle diameters at the same nominal mass concentration. The UV-vis absorption, scaled to the same nominal mass concentration, at a wavelength of 800 nm was 0.1 for the 40 nm PS sample, 57.5 for the 125 nm PS sample, and 182.4 for the 200 nm PS sample. The increase in absorption between the 40 nm PS sample and 200 nm sample, is a factor of ∼3.2 times larger than between the 40 nm PS sample and the 125 nm PS sample.
Fig. 3a shows the PS peak areas as a function of the particle concentration and shows that the 200 nm particles at higher concentrations have a smaller peak area than smaller particles with the same concentration. The overall change in intensity of the Raman spectrum is confirmed by data in Fig. 3b, which shows the water peak areas as a function of particle concentration and diameter. For the 40 nm sample, changes in concentration of PS have little effect on the water concentration within the focal volume and the intensity of the water peak is very similar at all mass concentrations. For the 125 nm and 200 nm PS samples, the increased scattering of light at higher concentrations and particles sizes, despite little difference in water concentration within the focal volume (Tables S1–4†), results in a reduction of the water peak area (Fig. 3b). The reduction in the Raman spectroscopy (830 nm excitation wavelength) water peak area compared to the 40 nm PS sample, is a factor of ∼3.5 times larger for the 200 nm sample than the 125 nm sample at a concentration of 4 mg ml−1, which is similar to the increased absorption factor (∼3.2) from the UV-vis measurements However, these effects are cancelled when the ratio of the peak areas is considered instead of the single peak areas, as shown in Fig. 3c.
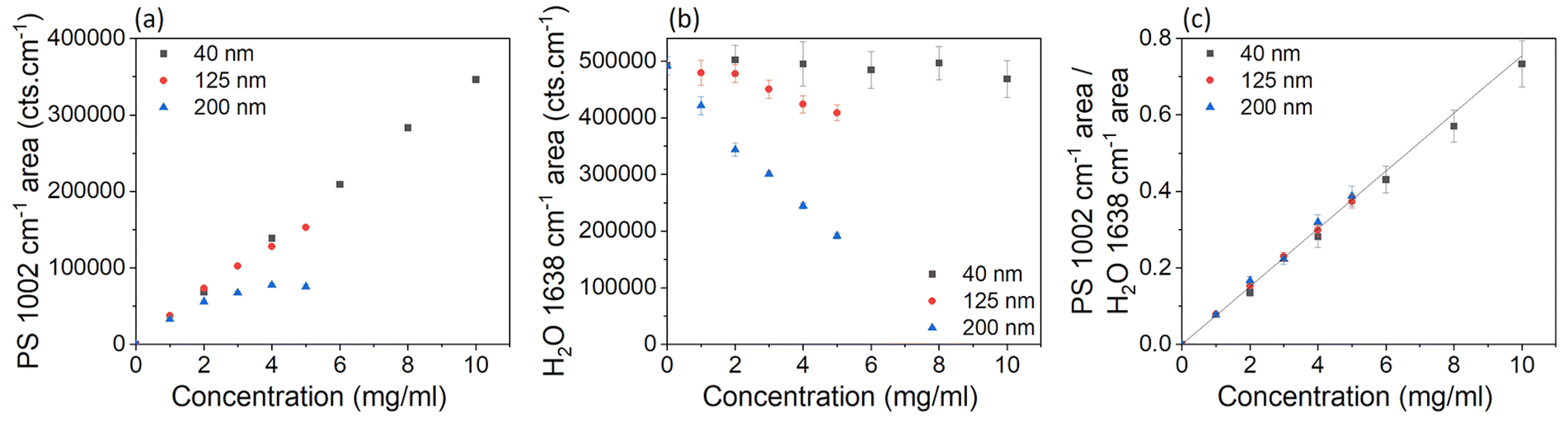 | ||
| Fig. 3 (a) Peak area of the fitted peak in the Raman spectra for PS (1002 cm−1), where the error bars are smaller than the data points, for samples containing different sizes and concentrations of PS. (b) Peak area of the ultrapure water (1638 cm−1) peak. (c) Ratio of the area of the fitted peaks in the Raman spectra for PS at 1002 cm−1 and water at 1638 cm−1, for samples containing different sizes and concentrations of PS. Each point on the graph is an average of 3 spectra, acquired at different places (but the same depth of 460 μm) through the vial. Three different methods of baseline subtraction were investigated for each peak (Fig. S6 and S7†), with the most consistent method chosen to calculate the peak area. The error bars represent the standard deviation of the fitted areas from each baseline subtraction method, which was found to be the largest source of error. A linear curve was fitted to all the data in (c), with the intercept set to 0, and represented as a black line. Corresponding data on peak heights are in the ESI (Fig. S8†). | ||
Fig. 3c shows the ratios of the PS (1002 cm−1) and water (1638 cm−1) peak areas as a function of the concentration of the particle dispersions in water. Within error, the ratios showed a linear (R2 = 0.82) increase with the concentration of PS, for all three particle sizes. The fit to the data provides us with a calibration curve for dilute PS solutions, to convert the ratio of the Raman peak areas of polystyrene and water to a particle mass concentration. We note that for this conversion we used the value of the mass concentration of the as-purchased solution declared by the manufacturer.
The Raman method we developed with PS particles was applied to the study of the liposomal doxorubicin formulation Doxoves® which, according to the manufacturer, has a doxorubicin concentration of 4 mg ml−1. Measurements were first performed with UV-vis spectroscopy, fluorescence spectroscopy, PTA, and MADLS to characterise the liposomes in a dispersion. Both MADLS and PTA show monomodal particle size distributions centred at a diameter of ≈93 nm (Fig. S11†), which is within the range of sizes initially investigated using PS. These results indicate the liposomes are well dispersed in the buffer at a range of concentrations with no apparent agglomeration. UV-vis absorption spectra (Fig. S12a†) show that the spectrum for a stock solution of doxorubicin hydrochloride has a maximum absorbance at 490 nm, which is shifted to higher wavelengths and has a reduced intensity at 490 nm in the spectrum for Doxoves® at the same concentration in water. This effect has been previously reported23,33 and is attributed to the aggregation of doxorubicin within the liposomes, therefore indicating the effective encapsulation of the drug. Similarly, we observed a significant reduction in intensity in the fluorescence spectra of Doxoves® when compared to a doxorubicin hydrochloride stock solution of equivalent doxorubicin concentration (Fig. S12b†), which is consistent with the literature.23,34 The reduced intensity of the encapsulated doxorubicin signal for both absorption at 490 nm and fluorescence spectra with respect to that of doxorubicin dispersed in water makes it challenging to produce a meaningful calibration curve using doxorubicin in water for the measurement of encapsulated doxorubicin concentration, and is likely to result in an underestimation of the doxorubicin concentration of the Doxoves®. We note that doxorubicin within the liposome is in a crystal form, but it is not possible to build a calibration curve using the dispersion in ammonium sulphate due to doxorubicin precipitation. In contrast, we show that our Raman method is able to differentiate between the two forms, and therefore we further investigate the suitability of Raman spectroscopy for measurements of doxorubicin concentrations.
Fig. 4a shows the Raman spectra of liposomal doxorubicin, along with the spectra of the empty liposomes and that of the sucrose and histidine buffer used for the liposome dispersions. Each spectrum is an average of 3 spectra acquired from each sample, which were also monitored to ensure there were no destructive changes to the samples (Fig. S13†). The Raman spectrum of liposomal doxorubicin showed characteristic peaks around 1210 cm−1 and 1240 cm−1 which were not observed in the spectra of empty liposomes or the buffer and are consistent with the Raman spectrum and tentative peak assignments (O–H⋯O bending, CH2 out of plane, C–OH bending, N–H2 and ring vibrations for 1210 cm−1 and CH2, C–H, O–H and C–O–C vibrations for 1240 cm−1) of doxorubicin HCl in the literature.19,35 We therefore attribute these peaks to the doxorubicin HCl. The peaks around 430–470 cm−1 (O–H⋯O wagging and C–H wagging vibrations tentatively assigned to 444 cm−1 and phenyl ring bending, C–H wagging and CO bending vibrations tentatively assigned to 466 cm−1)35 were also attributed to doxorubicin HCl, however they overlapped with a small peak in the buffer spectrum. Additionally, we observed a peak at 1297 cm−1 (attributed to the CH2 twisting vibration typically found in lipids36,37) in the spectra of both the empty and loaded liposomes that was not present in the spectrum of the buffer solution (Fig. 4a). The 1297 cm−1 peak was therefore attributed to the lipidic part of the liposomes. The peaks at 1210 cm−1 and 1297 cm−1 were selected to measure respectively the relative concentration of liposomal doxorubicin and lipids in the samples. The peak at 524 cm−1 was observed to have a similar intensity in all samples, this peak was tentatively attributed to the buffer solution and in some instances selected to measure the relative concentration of different components.
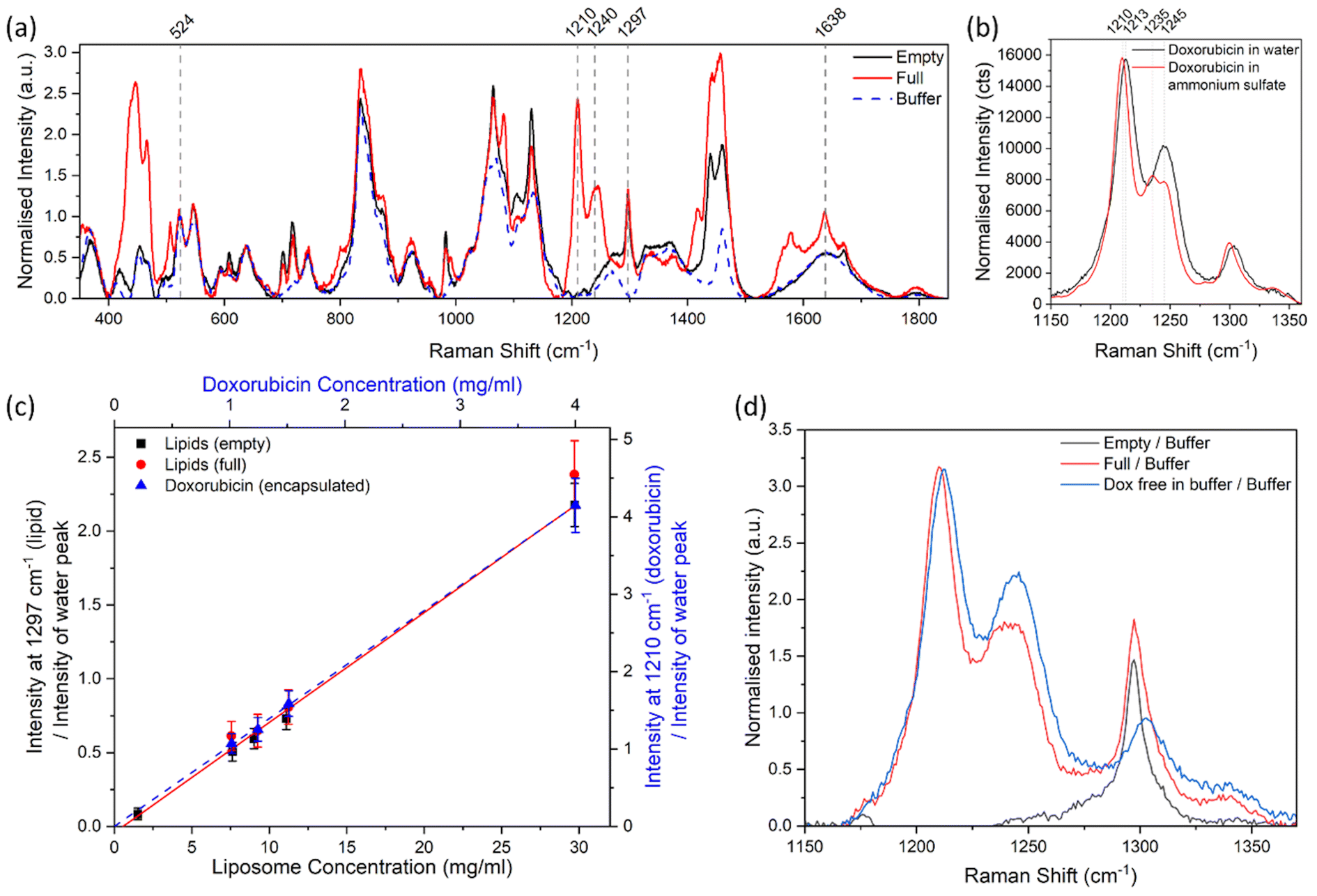 | ||
| Fig. 4 (a) Raman spectrum of a buffer solution of 10% sucrose 10 mM histidine, without liposomes (buffer), with Doxoves® (full), and with plain control liposomes (empty), after baseline subtraction and normalisation to the buffer peak at 524 cm−1. (b) Raman spectrum of doxorubicin HCl dispersed in ultrapure water and an ammonium sulphate solution, after baseline subtraction and normalisation to the peak at 1210–1213 cm−1. (c) Analysis of Doxoves® and plaincontrol liposomes, where the peak height intensity ratio of the 1297 cm−1 peak (for lipids) or the 1210 cm−1 peak (for doxorubicin) to the water peak is compared to the concentration calculated from dilution factors and the manufacturers’ reported concentration for the stock solution. Uncertainties were calculated from the peak–peak noise level as a percentage of the signal. A linear curve (lipids shown in red and doxorubicin shown in blue dashes) was fitted to each data set in (c), where the intercept is set to 0. (d) Raman spectrum of doxorubicin HCl, Doxoves®, and plain control liposomes, dispersed in a buffer solution of 10% sucrose 10 mM histidine, after subtraction of the buffer component and subsequent baseline subtraction. | ||
The doxorubicin molecules inside the liposomes are initially dispersed in ammonium sulphate, which promotes the formation of a crystalline structure.38–40 On the other hand, the free doxorubicin remains dispersed in the water-based buffer.38,39 The different forms of doxorubicin are reflected in different Raman spectral characteristics. The Raman spectrum of doxorubicin HCl in ultrapure water (Fig. 4b) exhibited a peak at 1213 cm−1, whereas the same peak in a Raman spectrum of doxorubicin HCl in ammonium sulphate is shifted to 1210 cm−1. More significantly, the relative intensity and shape of the peaks at 1235–1245 cm−1 appear to change depending on the form and environment of the doxorubicin. This may be due to the conversion of the NH2 group of the doxorubicin molecule to a NH3+ group in the presence of ammonium sulphate39 and consequent complexation with sulphate and other protonated doxorubicin molecules, which in turn alters the vibrations of the connected aromatic ring. The latter are likely to form the main peaks between 1235–1245 cm−1 (C–H2, C–H, O–H and C–O–C vibrations).35 It is also possible that the expected longitudinal stacking of doxorubicin within the liposomes changes the Raman spectra.39 These features in the spectra can potentially be exploited to identify and quantify the different forms of the active ingredient in the product. However, there is a need to establish the sensitivity of the approach, the impact of spectral resolution, and the limits of detections.
Similarly to the case of the polystyrene samples, the ratio of the lipid peak and the water peak produced a linear response with an increase in concentration of liposomes, for both the loaded and empty liposome samples (Fig. 4c, red circles and black squares respectively). The stock solutions appeared to contain similar levels of lipid content in both the empty and loaded samples (Fig. 4c). The intensity ratio of the 1210 cm−1 (doxorubicin HCl) and water peaks in the full liposomes also had a linear response with the nominal concentration of doxorubicin (Fig. 4c, blue triangles). These curves can be used to calibrate the Raman spectroscopy method for the measurement of mass concentration of active and lipidic components in a sample of liposomal doxorubicin with an unknown concentration.
The method used in Fig. 4c to calculate the ratio of the intensity of doxorubicin or lipidic peaks to water peaks, while similar to the PS analysis, proved challenging; the water peak in the Raman spectrum overlaps with some doxorubicin and liposome peaks, which increases the resulting uncertainty on the fitting (Fig. S2†). To reduce the measurement uncertainty, the buffer solution was preferred to water as a dispersant of the liposomes and its Raman peaks were used for the analysis in place of the water peaks.
To compare the spectra from samples with similar concentrations, the stock solutions of Doxoves® (4 mg ml−1 Doxorubicin HCl, 29.7 mg ml−1 lipids), control liposomes containing ammonium sulphate only (29.7 mg ml−1 lipids), and doxorubicin HCl (4 mg ml−1) dispersed in a buffer solution were analysed by confocal Raman and results compared (Fig. 4d). The peak at 524 cm−1, which is likely attributed to the sucrose in the buffer solution,41 was used to calculate the contribution of the buffer to the sample spectra. This was done by scaling the intensity of the buffer spectrum to match the intensity of the spectrum of interest between 516–572 cm−1, containing peaks at 527 cm−1, and 552 cm−1 attributed to sucrose41 and a peak at 544 cm−1 attributed to histidine.42 The scaled buffer component was subsequently subtracted from the Raman spectrum of the sample (Fig. S14a and b†), after which a linear baseline was subtracted between 1165–1505 cm−1 (Fig. S14c and d†). The resulting spectra are attributed to the liposome and doxorubicin HCl materials only. To compare the concentrations of the liposome and doxorubicin components, the spectra were normalised to the intensity of the previously scaled buffer peak (Fig. S14c and d†), so that the peak intensities related to components which have the same concentration in each sample should be equal. Fig. 4d shows that after this operation, the intensity of the 1210/1213 cm−1 peak is the same for both encapsulated doxorubicin and free doxorubicin of the same nominal concentration, while the peaks around 1240 cm−1 are different. The shape of these peaks, compared with the spectra shown in Fig. 4b indicates the presence of doxorubicin in a crystal form within the liposomes. The high chemical specificity of Raman spectroscopy enables these observations over techniques such as UV-vis or fluorescence spectroscopy.
Quantification of lipids in this sample is challenging due to the lack of lipidic peaks that are isolated and have high intensity. The lipidic peak at 1297 cm−1 is the most prominent lipidic peak and was therefore used to estimate the lipidic contribution (Fig. 4c). The lipidic peak at 1297 cm−1 overlaps a low intensity doxorubicin peak at ∼1302 cm−1 and has an increased background or broad peak, which is difficult to resolve. Interestingly, the peak at 1297 cm−1 appears to have similar intensities for the loaded and empty liposomes (Fig. 4d), confirming similar lipid concentrations, and confirming the suitability of this peak for monitoring lipidic contributions.
We assessed the usefulness of Raman spectroscopy for measuring the encapsulation efficiency of doxorubicin within the liposomes. Samples containing different concentrations of free and liposomal doxorubicin were prepared, after which Raman spectra were acquired (Fig. 5a). While there is a repeatable shift in the 1210 cm−1 peak to 1213 cm−1 from crystalline to dispersed doxorubicin, it should be noted that the spectral resolution of our system is ∼3 cm−1 and therefore the peak intensity ratios of the fitted components are a preferable measurand. As before, the buffer component was scaled between 516–572 cm−1 and subtracted from each Raman spectrum (Fig. S3a and b†), followed by a linear baseline subtraction between 1165–1505 cm−1. The Raman spectra of the liposomal doxorubicin sample (after subtraction of the Raman spectrum of the plain control liposomes sample to remove the lipid components), free doxorubicin (100% free), and plain control liposomes samples were fitted to the spectrum of the combined samples (Fig. S3c and d†) to evaluate the relative concentrations of these components. The height intensities of the liposomal doxorubicin Raman spectrum at 1211 cm−1 and the 100% free doxorubicin Raman spectrum at 1213 cm−1, both resulting from the fit, were taken to represent the “encapsulated” drug and “free” drug contributions respectively. The measured encapsulated drug percentage was calculated from the intensity of the encapsulated drug component as a percentage of the total intensity of both the encapsulated and free drug components. The liposomal doxorubicin sample and free doxorubicin sample were assumed to be 100% and 0% encapsulated, respectively. It should be noted that this is an approximation, because there may be some amount of free drug in the original liposomal doxorubicin sample, which is below 3% according to the manufacturer. The percentage of encapsulated doxorubicin measured in this way by Raman spectroscopy was compared to the expected percentage value. This is shown in Fig. 5b and resulted in a linear correlation (R2 = 0.9991). The measured percentages are equal within uncertainties to the expected values, with one of the largest contributions to measurement variability being the fit of the baseline. The measurement uncertainties in Fig. 5b result from the standard deviation from the data obtained from 3 independent linear baseline fits, where each fit is between different x-axis boundaries (1165 cm−1–1505 cm−1, 1135 cm−1–1505 cm−1 and 1035 cm−1–1505 cm−1). These fit boundaries were chosen to only include the peaks of interest to minimise the uncertainty. Many different baselines could be used, such as a polynomial fitted to the whole spectrum, but this may significantly increase the measurement variability.
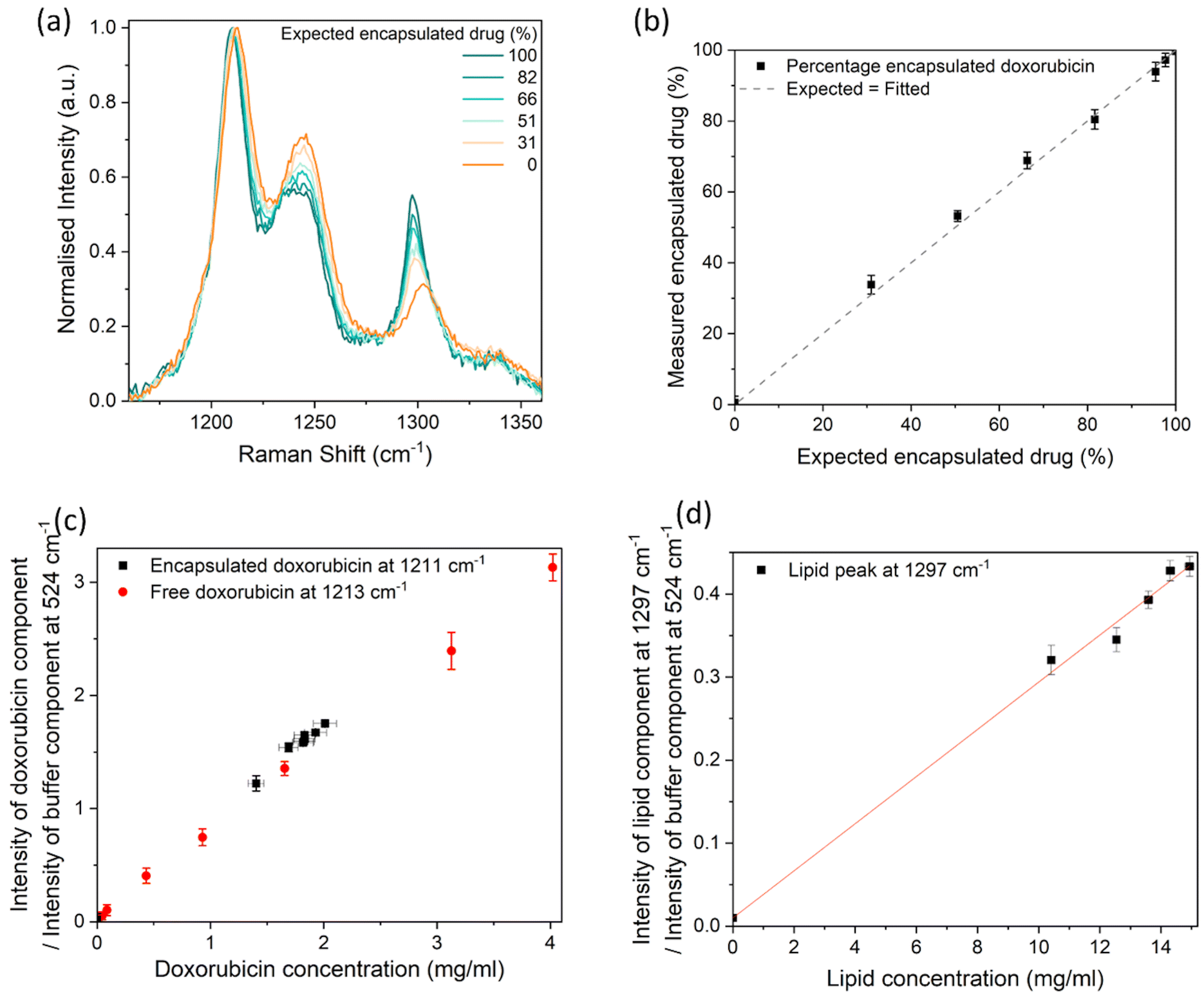 | ||
| Fig. 5 (a) Raman spectrum of samples containing a combination of free and encapsulated doxorubicin, after subtraction of the buffer component (Fig. S3†), baseline subtraction, and normalisation between 0 and 1. The Raman spectrum for samples with smaller encapsulated doxorubicin increments (95, 98 and 100%) can be found in the ESI (Fig. S15†). (b) The measured percentage of encapsulated doxorubicin, compared to the expected percentage of encapsulated doxorubicin, with an assumption that the stock solution contained 100% encapsulated doxorubicin, and an assumption that the 100% free doxorubicin contained 0% encapsulated doxorubicin. The dashed line shows the identity relationship. (c) Height intensity ratio of the fitted doxorubicin component measured at 1211–1213 cm−1 and the buffer component measured at 524 cm−1. (d) Height intensity ratio of the liposome component measured at 1297 cm−1 and the buffer component measured at 524 cm−1. A linear curve was fitted to the data set in (d). The data points on all graphs are from the optimal baseline fit (linear fit between 1165 cm−1–1505 cm−1), however the y-axis error bars are the standard deviation of 3 different baseline fits, to demonstrate one of the main sources of uncertainty. Each point is the average value from 3 analysed spectra, where the standard deviation was smaller than the uncertainty due to baseline fits. The x-axis error bars are the uncertainties due to the manufacturer's specifications, some of which are smaller than the data points. | ||
The ratio of the intensity of the 1210–1213 cm−1 (doxorubicin) peak (from the fitted liposomal doxorubicin and free doxorubicin components) and the 524 cm−1 peak (from the fitted buffer component) was used to measure the concentration of the free and encapsulated doxorubicin components. Fig. 5c shows that these measured ratios both have a linear relationship with the expected concentration (R2 = 0.9985 for free drug, R2 = 0.9925 for encapsulated drug). Similarly to Fig. 4d, the peak intensity ratios of the doxorubicin peak and the buffer peak result in similar values for free and encapsulated doxorubicin of a similar nominal concentration. The peak intensity ratio of the lipid peak resulting from the fit and the buffer peak from the fit, also exhibited a linear response (R2 = 0.9987) with expected concentration (Fig. 5d), confirming that the lipid concentration in a buffer could be monitored with Raman spectroscopy.
These results suggest Raman spectroscopy could be a useful method for monitoring changes in encapsulation efficiency. We have detected differences as small as 2.3% for encapsulation efficiencies above 90%, where expected encapsulation efficiencies of 95.4 ± 0.2% and 97.7 ± 0.1% were measured as 93.9 ± 2.6% and 97.2 ± 1.9%. The uncertainties were calculated from the standard deviation of datasets resulting from fits performed with linear baselines of different end points. Different baseline fits typically result in an intensity shift of the whole data set, therefore provided the baseline fit is kept consistent it is repeatedly possible to distinguish between the 95.4% sample and 97.7% sample.
Discussion
We have developed non-invasive Raman spectroscopy measurements of nanoparticle-based formulations and demonstrated its application to liposomal doxorubicin. The measurements can be performed on a closed vial and do not require sample preparation or the consumption of any product material. Each spectrum can be acquired in as little as 2 minutes. As shown in Fig. 1, measurements were performed in a reflection configuration, while transmission Raman spectroscopy could also be implemented to analyse larger sample volumes. This flexibility in the type of configuration means that from an application point of view the Raman instrument can be integrated, for example, within other analytical instrumentation and/or manufacturing lines.To enable quantitative chemical measurements, we developed a method to identify the optimal depth within the sample from which to perform the measurement, such that the confocal volume was fully contained within the sample. Polystyrene particles were suitable as a model system for this purpose. To investigate the impact of light absorption and scattering on the measured Raman signal, we utilised spherical particles with diameters ranging from 40 nm to 200 nm. We also investigated larger particles with diameters up to a micron, but we found them prone to significant sedimentation within the duration of the measurements, which affected their concentration within the measurement region. For this reason, they were excluded from the analysis. Fig. 3 shows that the larger the particles and the higher the concentration, the more the detected Raman spectra is attenuated for both the polystyrene particles and the water. However, the intensity ratios between the polystyrene and the water peaks are linear with the nominal mass concentration of polystyrene for all particles, irrespective of the size. We note that in this concentration range the volume occupied by the particles is <1% (Tables S1–4†), with respect to the volume of water, so the mass of the water can be considered constant for all samples and the ratio is linear with the volume fraction of the polystyrene.
The linear relationship in Fig. 3c offers a strategy to calibrate the Raman method for concentration measurements. Fig. 3c is effectively a calibration curve that converts the Raman ratio discussed above into a polystyrene mass concentration. With knowledge of the particle shape and size, or by using a particle-by-particle counting method for number concentration, it is also possible to compare the ratio with a number concentration. Nevertheless, for therapeutics and complex medicines, it is useful to refer to the dose of the active ingredients in terms of their mass concentration and thus this is the approach taken in this work.
The method developed using PS particles was applied to the study of liposomal doxorubicin formulations. The use of UV-Vis absorption spectroscopy to confirm the nominal doxorubicin concentration in liposomal formulations declared by the manufacturer presents a number of challenges. One problem presented here is that while doxorubicin HCl disperses well in water, it forms crystals and precipitates when dispersed in ammonium sulphate, where the latter is representative of the environment doxorubicin is experiencing when first encapsulated inside liposomes. As a result of the doxorubicin aggregates in the liposomal formulation, the maximum absorbance of its peak around 490 nm in the UV-vis spectrum shifts to higher wavelengths and has a lower absorbance compared to the free dispersed drug at the same concentration. The UV-vis method (Fig. S12a†) therefore appears to underestimate the encapsulated doxorubicin concentration.33 A similar conclusion is reached when considering fluorescence measurements.34 The uncertainties on the absorption behaviour of doxorubicin in different environments supported the need for an alternative and quantitative spectroscopical analysis approach, such as Raman spectroscopy.
As far as the quantification of overall doxorubicin in the sample is concerned, Fig. 4c, 5c and d show that the Raman spectroscopy approach we developed with the PS model systems can be replicated to measure not only the concentration of the doxorubicin, but also that of the lipid in the formulations, by selecting peaks at 1210/1213 cm−1 and 1297 cm−1 respectively. Two approaches were demonstrated for measuring the component concentrations. The first approach replicated the PS analysis with the use of the water peak at 1638 cm−1 (Fig. 4c), whereas the second approach utilised the buffer peaks around 516–572 cm−1 (Fig. 5). Both approaches produced a linear response with the nominal concentration, however the second approach reduced the uncertainty of the results. The first approach required baseline subtraction and peak fitting of multiple overlapping components to calculate the water contribution, introducing fitting uncertainties for both the baseline and peaks. Whereas the second approach utilised a region containing purely the buffer component and fitted the component before any baseline subtraction. Advanced data analysis methods such as spectral unmixing and multivariate analysis, have the potential to further enhance the quantification in future work.
We then investigated the possibility to quantify the relative amount of free versus encapsulated doxorubicin. We note that the similarities in the UV-Vis absorption spectra prevent this endeavour by UV-Vis spectroscopy without applying a pre-analytical separation method to separate the two forms of the active ingredient.33 In Raman spectroscopy, however, we can take advantage of the difference in the spectra observed for doxorubicin dispersed in water and ammonium sulphate. From the fitting of the Raman spectrum, we calculated the percentage contribution of the encapsulated doxorubicin component out of the total doxorubicin components (free and encapsulated, as shown in Fig. 5b). This approach showed that we could identify differences in concentrations of free doxorubicin hydrochloride down to ∼2.3%. The empty liposome sample with a total lipid concentration of 1.5 mg ml−1 exhibited a lipid peak with a very low intensity (Fig. 4c), resulting in >50% uncertainty, suggesting these samples at this concentration and lower are below the limit of detection for these particular acquisition settings. The total lipid concentration in Doxil® is ∼16 mg ml−1,43 which is above the limit of detection. Additional repeats, longer acquisition times, higher laser powers, or optimised spectrometer setups have the potential to reduce measurement uncertainties and increase the confidence level in the detection of lower concentrations of free doxorubicin. However, the main source of uncertainty we encountered was the baseline fitting, which can be improved using further data analysis techniques such as spectral unmixing. The limit of detection for HPLC is much better than Raman spectroscopy,15 and future work will compare these techniques in more detail.
The approach we described is useful to monitor changes in the encapsulation efficiency of doxorubicin and to study the release behaviour. It is not useful, however, to measure the amount of non-encapsulated doxorubicin present in the initial sample, because the spectrum of the initial sample has been used in the analysis to represent the 100% condition. We investigated the possibility to extend the approach to the measurement of non-encapsulated doxorubicin in the initial sample. To this aim, we fitted the Raman spectra of the mixtures of liposomal and free doxorubicin using the spectrum of doxorubicin in ammonium sulphate in place of that of the stock liposomal doxorubicin (Fig. S4†). The free drug in the stock solution resulting from this fit is ∼19%, which is higher than the non-encapsulated value declared by the manufacturer (<3%). One reason for this discrepancy is that the drug molecules associated with the Doxoves® liposomes can have different forms. Specifically, along with the characteristic doxorubicin crystal, isolated or small clusters of doxorubicin molecules are also found within and associated with the liposomal membrane.21 These forms of doxorubicin are expected to have different Raman spectra, but the fit we performed can only discriminate between crystalline and isolated doxorubicin molecules, with no insight on whether the signal originated from within or outside a liposome. The approach is therefore likely to overestimate the non-encapsulated drug. However, the expected amount of isolated doxorubicin molecules within the liposome membrane (0.66%23) is not such to justify the observed discrepancy. A different reason could relate to the spectrum of doxorubicin in ammonium sulphate not being representative of the doxorubicin crystals found inside the liposomes. When doxorubicin is dispersed in this solvent, it precipitates forming large crystals. On the other hand, the doxorubicin crystals within the liposomes are smaller in size, with a significant number of edge molecules that may not be fully crystalised. It is possible then that the Raman spectrum associated with the encapsulated doxorubicin contains contributions from both the doxorubicin crystal and other components due to the edge molecules and molecules within the liposomal membrane. The use of methods enabling the measurement of Raman spectra of single liposomes such as tip-enhanced Raman spectroscopy and SPARTA44 could help address this question. In this study, we have taken the decision to focus on quantifying the changes to the nominally >97% encapsulated sample in the presence of free drug, for purposes of monitoring changes in the stock sample.
The major advantage of the use of the Raman spectroscopy method described in this work with respect to other types of spectroscopies and analytical techniques is the ability to measure doxorubicin and lipid concentrations rapidly and non-invasively. This can be of advantage during scale up, manufacturing and storage of the product. Due to the significant cost and footprint of state-of-the-art Raman spectrometers such as the one used in this work, portable Raman spectrometer probes are growing in use and represent a more suited implementation route in manufacturing. A study of the impact of the spectrometer resolution and signal noise on the measurement of concentration would be useful to further explore this approach. Because typical concentrations of free doxorubicin in commercial samples is around 2% and below, HPLC would remain the method of choice for the measurement of free doxorubicin for qualification purposes. In this respect it appears that this Raman method is best suited for rapid screening, monitoring changes in the sample quality during storage, or for measuring the concentration of a specific drug form non-invasively and in situ, for example during release experiments.
Conclusion
We demonstrated the use of Raman spectroscopy for the identification and quantification of chemical components in nanoparticle formulations. We measured a linear response from particles (PS and liposomal) with increasing concentration of the constituent ingredients, which inform a strategy for the calibration of this spectroscopic method for relative and absolute concentration measurements. Measurements were performed on samples within closed vials, indicating this is a technique suitable for in-line measurements. This Raman spectroscopy method allows for rapid and non-invasive measurements without the need for sample preparation, whereas methods such as HPLC require longer sample preparation times and are destructive to the sample. However, the detection limit of HPLC is better than Raman spectroscopy, therefore the method of choice depends on the nature of the sample of interest and the objective of the analysis. We suggest Raman spectroscopy as a future validation technique for measuring concentrations of liposomes and other chemicals and propose it as a tool to support manufacturing.Author contributions
Elizabeth J. Legge: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft preparation. Ryan T. Coones: investigation, formal analysis, writing – reviewing and editing. William Lee: investigation, formal analysis. Yiwen Pei: supervision, writing – reviewing and editing. Natalie A. Belsey: supervision, writing – reviewing and editing. Caterina Minelli: funding acquisition, conceptualization, methodology, supervision, writing – reviewing and editing.Data availability
The data supporting this article has been included either in the main text or as part of the ESI.† Any other relevant data is available on request via the authors at caterina.minelli@npl.co.uk and elizabeth.legge@npl.co.uk.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge funding from the National Measurement System programme of the UK Department for Science, Innovation and Technology. We thank Keith Paton and Alex Shard at the National Physical Laboratory for insightful discussions.References
- A. Gabizon, T. Peretz, A. Sulkes, S. Amselem, R. Ben-Yosef, N. Ben-Baruch, R. Catane, S. Biran and Y. Barenholz, Eur. J. Cancer Clin. Oncol., 1989, 25, 1795–1803 CrossRef.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, J. L. Perez, G. Pérez Marc, E. D. Moreira, C. Zerbini, R. Bailey, K. A. Swanson, S. Roychoudhury, K. Koury, P. Li, W. V. Kalina, D. Cooper, R. W. Frenck, L. L. Hammitt, Ö. Türeci, H. Nell, A. Schaefer, S. Ünal, D. B. Tresnan, S. Mather, P. R. Dormitzer, U. Şahin, K. U. Jansen and W. C. Gruber, N. Engl. J. Med., 2020, 383, 2603–2615 CrossRef.
- E. Kastner, R. Kaur, D. Lowry, B. Moghaddam, A. Wilkinson and Y. Perrie, Int. J. Pharm., 2014, 477, 361–368 CrossRef.
- M. Sheybanifard, L. P. B. Guerzoni, A. Omidinia-Anarkoli, L. De Laporte, J. Buyel, R. Besseling, M. Damen, A. Gerich, T. Lammers and J. M. Metselaar, Lab Chip, 2023, 23, 182–194 RSC.
- H. J. Han, C. Ekweremadu and N. Patel, J. Drug Delivery Sci. Technol., 2019, 52, 1051–1060 CrossRef.
- D. Papahadjopoulos, T. M. Allen, A. Gabizon, E. Mayhew, K. Matthay, S. K. Huang, K. D. Lee, M. C. Woodle, D. D. Lasic, C. Redemann and F. J. Martin, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 11460–11464 CrossRef PubMed.
- S. G. M. Ong, L. C. Ming, K. S. Lee and K. H. Yuen, Pharmaceutics, 2016, 8(3), 25 CrossRef PubMed.
- M. Kato, Appl. Spectrosc. Rev., 2018, 53, 279–289 CrossRef.
- J. K. Lang, J. Chromatogr. A, 1990, 507, 157–163 CrossRef.
- Y. Hu, R. M. Crist and J. D. Clogston, Anal. Bioanal. Chem., 2020, 412, 425–438 CrossRef PubMed.
- M. Oswald, M. Platscher, S. Geissler and A. Goepferich, Int. J. Pharm., 2016, 497, 293–300 CrossRef.
- F. Caputo, D. Mehn, J. D. Clogston, M. Rösslein, A. Prina-Mello, S. E. Borgos, S. Gioria and L. Calzolai, J. Chromatogr. A, 2021, 1635, 461767 CrossRef CAS.
- R. S. Das and Y. K. Agrawal, Vib. Spectrosc., 2011, 57, 163–176 CrossRef.
- T. R. M. De Beer, G. J. Vergote, W. R. G. Baeyens, J. P. Remon, C. Vervaet and F. Verpoort, Eur. J. Pharm. Sci., 2004, 23, 355–362 CrossRef PubMed.
- M. G. Orkoula, C. G. Kontoyannis, C. K. Markopoulou and J. E. Koundourellis, Talanta, 2007, 73, 258–261 CrossRef PubMed.
- J. Penders, I. J. Pence, C. C. Horgan, M. S. Bergholt, C. S. Wood, A. Najer, U. Kauscher, A. Nagelkerke and M. M. Stevens, Nat. Commun., 2018, 9, 1–11 CrossRef PubMed.
- K. Masui, Y. Nawa, S. Tokumitsu, T. Nagano, M. Kawarai, H. Tanaka, T. Hamamoto, W. Minoshima, T. Tani, S. Fujita, H. Ishitobi, C. Hosokawa and Y. Inouye, ACS Omega, 2022, 7, 9701–9709 CrossRef.
- H. Noothalapati, K. Iwasaki, C. Yoshimoto, K. Yoshikiyo, T. Nishikawa, M. Ando, H. O. Hamaguchi and T. Yamamoto, Spectrochim. Acta, Part A, 2017, 187, 186–190 CrossRef PubMed.
- H. Rammal, A. Al Assaad, F. Dosio, B. Stella, A. Maksimenko, S. Mura, L. Van Gulick, M. Callewaert, D. Desmaële, P. Couvreur, H. Morjani and A. Beljebbar, Nanomedicine, 2021, 35, 102404 CrossRef PubMed.
- M. R. Pollard, K. Sparnacci, L. J. Wacker and H. Kerdoncuff, Chemosensors, 2020, 8(1), 21 CrossRef.
- A. Ntziouni, J. Thomson, I. Xiarchos, X. Li, M. A. Bañares, C. Charitidis, R. Portela and E. Lozano Diz, Appl. Spectrosc., 2022, 76, 747–772 CrossRef PubMed.
- G. W. Mulholland and B. J. Bauer, J. Nanopart. Res., 2000, 2, 5–15 CrossRef.
- P. Tentori, G. Signore, A. Camposeo, A. Carretta, G. Ferri, P. Pingue, S. Luin, D. Pozzi, E. Gratton, F. Beltram, G. Caracciolo and F. Cardarelli, Nanoscale, 2022, 14, 8901–8905 Search PubMed.
- S. M. Ansar, W. Jiang and T. Mudalige, Int. J. Pharm., 2018, 549, 109–114 CrossRef.
- S. Zhu, L. Ma, S. Wang, C. Chen, W. Zhang, L. Yang, W. Hang, J. P. Nolan, L. Wu and X. Yan, ACS Nano, 2014, 8, 10998–11006 CrossRef.
- International Organization for Standardization, ISO 22412:2017: Particle size analysis—Dynamic light scattering (DLS), ISO, Switzerland, 2017 Search PubMed.
- T. E. Bridges, M. P. Houlne and J. M. Harris, Anal. Chem., 2004, 76, 576–584 CrossRef PubMed.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, 1991 Search PubMed.
- M. Mazilu, A. C. De Luca, A. Riches, C. S. Herrington and K. Dholakia, Opt. Express, 2010, 18, 11382 CrossRef.
- Y. Guo, A. Jarabo and S. Zhao, ACM Trans. Graph., 2021, 40(6), 1–12 Search PubMed.
- P. G. Cummins and E. J. Staples, Langmuir, 1987, 3, 1109–1113 CrossRef.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, John Wiley & Sons, 1983 Search PubMed.
- D. Mehn, P. Iavicoli, N. Cabaleiro, S. E. Borgos, F. Caputo, O. Geiss, L. Calzolai, F. Rossi and D. Gilliland, Int. J. Pharm., 2017, 523, 320–326 CrossRef PubMed.
- P. Changenet-Barret, T. Gustavsson, D. Markovitsi, I. Manet and S. Monti, Phys. Chem. Chem. Phys., 2013, 15, 2937–2944 RSC.
- G. Das, A. Nicastri, M. L. Coluccio, F. Gentile, P. Candeloro, G. Cojoc, C. Liberale, F. De Angelis and E. Di Fabrizio, Microsc. Res. Tech., 2010, 73, 991–995 CrossRef PubMed.
- F. Rodà, S. Picciolini, V. Mangolini, A. Gualerzi, P. Seneci, A. Renda, S. Sesana, F. Re and M. Bedoni, Nanomaterials, 2023, 13(4), 699 CrossRef.
- K. Czamara, K. Majzner, M. Z. Pacia, K. Kochan, A. Kaczor and M. Baranska, J. Raman Spectrosc., 2015, 46, 4–20 CrossRef.
- X. Wei, D. Shamrakov, S. Nudelman, S. Peretz-Damari, E. Nativ-Roth, O. Regev and Y. Barenholz, ACS Omega, 2018, 3, 2508–2517 CrossRef.
- S. A. Abraham, D. N. Waterhouse, L. D. Mayer, P. R. Cullis, T. D. Madden and M. B. Bally, Methods Enzymol., 2005, 391, 71–97 Search PubMed.
- X. Li, D. J. Hirsh, D. Cabral-Lilly, A. Zirkel, S. M. Gruner, A. S. Janoff and W. R. Perkins, Biochim. Biophys. Acta, Biomembr., 1998, 1415, 23–40 CrossRef PubMed.
- E. Wiercigroch, E. Szafraniec, K. Czamara, M. Z. Pacia, K. Majzner, K. Kochan, A. Kaczor, M. Baranska and K. Malek, Spectrochim. Acta, Part A, 2017, 185, 317–335 CrossRef PubMed.
- G. P. De Sousa, P. T. C. Freire, J. M. Filho, F. E. A. Melo and C. L. Lima, Braz. J. Phys., 2013, 43, 137–144 CrossRef.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 1–33 CrossRef PubMed.
- C. Saunders, J. E. J. Foote, J. P. Wojciechowski, A. Cammack, S. V. Pedersen, J. J. Doutch, H. M. G. Barriga, M. N. Holme, J. Penders, M. Chami, A. Najer and M. M. Stevens, ACS Nano, 2023, 17, 11713–11728 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00238e |
| This journal is © The Royal Society of Chemistry 2025 |