
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comprehensive physicochemical, biophysical, and in vitro characterization of lung surfactant SP-A peptidomimetics
David Encinas-Basurtoab,
Priya Muralidharana,
M. D. Saiful Islama,
Ernest L. Vallorza,
Stephen M. Blackcd,
Monica Kraftcefg,
Julie G. Ledford*cef and
Heidi M. Mansour *acdf
*acdf
aThe University of Arizona College of Pharmacy, Tucson, AZ, USA
bUniversity of Sonora, Mexico
cThe University of Arizona College of Medicine, Tucson, AZ, USA. E-mail: hmansour@fiu.edu; jledford@arizona.edu
dFlorida International University, Center for Translational Science, Port St. Lucie, FL, USA
eThe University of Arizona Health Sciences Asthma and Airway Disease Research Center, Tucson, AZ, USA
fThe University of Arizona, the BIO5 Research Institute, Tucson, AZ, USA
gDepartment of Medicine, Icahn School of Medicine at Mount Sinai, Mount Sinai Health System, New York, NY, USA
First published on 6th May 2025
Abstract
Surfactant protein-A (SP-A) is an endogenous and essential lung surfactant-specific protein that is integral to pulmonary immunity, including inhibition of asthma exacerbations. This study aims to comprehensively characterize two peptides (10-AA and 20-AA) of SP-A which confer activity similar to the full-length oligomeric SP-A protein. Spectroscopic and chromatographic analyses revealed that the phosphate (PS) and acetate (AC) salts exhibited distinct solubility and log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P partitioning behavior, impacting their physicochemical properties. MD simulations and circular dichroism showed that SP-A 10-AA initially adopts an α-helical structure but loses helicity over time, while SP-A 20-AA remains disordered. Differential scanning calorimetry confirmed variations in thermal stability between salt forms and zeta potential measurements showed that PS salts had a more negative surface charge, potentially influencing membrane interactions. In vitro studies showed high cell viability (>90%) and stable TEER values at the air–liquid interface, confirming biocompatibility and potential epithelial permeability. These findings provide crucial insights into the structural and functional properties of SP-A peptides, supporting their potential as therapeutic agents for pulmonary diseases.
P partitioning behavior, impacting their physicochemical properties. MD simulations and circular dichroism showed that SP-A 10-AA initially adopts an α-helical structure but loses helicity over time, while SP-A 20-AA remains disordered. Differential scanning calorimetry confirmed variations in thermal stability between salt forms and zeta potential measurements showed that PS salts had a more negative surface charge, potentially influencing membrane interactions. In vitro studies showed high cell viability (>90%) and stable TEER values at the air–liquid interface, confirming biocompatibility and potential epithelial permeability. These findings provide crucial insights into the structural and functional properties of SP-A peptides, supporting their potential as therapeutic agents for pulmonary diseases.
Introduction
Surfactant Protein A (SP-A), one of the major protein constituents of pulmonary surfactant, is an important mediator of pulmonary host defense. SP-A is encoded by two genes, SP-A1 and SP-A2, and forms a highly oligomeric octodecamer structure. Specific to asthma, it was previously shown that SP-A function is defective, which could arise from genetic variation.1–3 Hence, a 10-amino acid (AA) active region of SP-A2 has been identified that significantly reduces phenotypes associated with asthma in human cells and in murine models.4 This 10-AA (1.25 kDa) size SP-A peptide is an ideal candidate to package into an inhaler for airway delivery.Inhaled medicine has become the preferred route of administration for targeted drug delivery directly to the lung tissue by a noninvasive route for local and systemic action.5 Inhaled medicines as marketed products are the first-line therapies used clinically in the treatment and preventative management of several pulmonary diseases of global health importance (i.e. asthma, COPD, cystic fibrosis, pulmonary fibrosis, and pulmonary infections) and noninvasive systemic therapy through the pulmonary route (i.e. Afrezza® inhaled insulin dry powder inhaler and Adasuve® inhaled antipsychotic marketed pharmaceutical products).
Peptides and proteins are complex macromolecules. Consequently, the first step in development of a protein or peptide-based drug product involves comprehensive characterization to understand their chemical structure and physicochemical properties that pertains to its biological activity and stability. The advancement in analytical chemistry field provides a plethora of techniques for evaluation of these biopharmaceuticals in the different stages of product development. Infrared spectroscopy, Raman spectroscopy, circular dichroism, solid state nuclear magnetic resonance are powerful techniques for structure elucidation. Thermal analytical techniques like differential scanning calorimetry, thermogravimetry and X-ray diffraction is used to probe thermal behavior, molecular mobility, kinetics of solid state stability as well as extent of crystallinity of peptide.6,7 It is imperative to identify these properties of peptide since it can affect the processability, chemical stability and eventually in vivo efficacy of these biomolecules. Therefore, a combination of these techniques is utilized to thoroughly characterize the peptide before and after development of the final formulation or drug product.
The work presented in this manuscript is the first report to the Authors’ knowledge on the comprehensive physical and biophysical characterization, in vitro cellular properties, in silico molecular modeling, and in vitro human cell properties of the innovative SP-A 10-AA and SP-A 20-AA peptides as two different salt forms (phosphate (PS) and acetate (AC)). The results presented in this study will serve as a reference for the physicochemical properties of the two SP-A peptides, which can be compared with the final drug formulation intended for lung inhalation.
Materials and methods
Materials
SP-A 10-AA has an amino acid sequence of Lys-Glu-Gln-Cys-Val-Glu-Met-Tyr-Thr-Asp (KEQCVEMYTD) and SP-A 20-AA sequence is Pro-Ala-Gly-Arg-Gly-Lys-Glu-Gln-Cys-Val-Glu-Met-Tyr-Thr-Asp-Gly-Gln-Trp-Asn-Asp (PAGRGKEQCVEMYTDGQWND). The PS and AC 10 AA, PS and AC 20 AA peptides with molecular mass of 1245.38 g mol−1 and 2284.45 g mol−1 respectively, were synthesized by GenScript (Piscataway, NJ) with >98% purity. Hydranal®-Coulomat AD, trifluoroacetic acid (TFA) and 1-Octanol were obtained from Sigma-Aldrich (St Louis, MO). Phosphate buffered saline (PBS) pH 7.4 (1×) was obtained from Life Technologies (Carlsbad, CA) and Emprove (absolute ethanol) USP grade was obtained from VWR (Radnor, PA). Methanol and Acetonitrile (ACN) HPLC grade were obtained from Thermo Fisher Scientific (Waltham, MA). Resazurin sodium salt was from Thermo Fisher Scientific (Waltham, MA). The peptides were stored in sealed glass desiccators over indicating Drierite/Drierite™ desiccant at −20 °C under ambient pressure. Other chemicals were stored under room conditions.Methods
:50 v/v octanol:water ratio in a glass vial and allowed to shake in temperature-controlled water bath at 37 °C. After 24 hours, the shaker was stopped and the vial was undisturbed for 24 hours to allow the solvents to separate. An aliquot (20 μL) of water phase and octanol phase was injected into HPLC to quantify the peptide concentration in each phase.The assay was conducted using similar previously reported conditions.16 Briefly, in Nunc™ MicroWell™ 96-well optical-bottom plates with polymer base (ThermoScientific Waltham, MA) each cell line was seeded at a concentration of 5000 cells in 100 μL per well (six wells, n = 6) and allowed to attach for 48 hours. The cells were treated with 100 μL volume of different concentrations (0.1–1000 μM) of the peptides dissolved in the respective media. After 72 hours, the plate was treated with 20 μL of 10 μM resazurin solution. After 3–4 hours, the fluorescence intensity was read at 514 nm (excitation) and 590 nm (emission) wavelengths using the Synergy H1 Multi-Mode Reader (BioTek Instruments, Inc., Winooski, Vermont). The corresponding growth media was used as control for each cell line. Relative viability was calculated as a ratio of fluorescence intensity of the sample to the control.
 000 cells per well in proliferation medium (0.5 ml in the apical and 1.5 ml in the basolateral chambers). The seeding day was defined as Day 0 and the cells were allowed to attach. The basal media was changed every other day until the formation of a monolayer. After the cells reached confluence, the polarization medium was removed from the apical compartment, leaving the apical surface of the cells exposed to air i.e. air–liquid (ALI) culture condition. The polarization medium was made up of base medium RPMI 1640 containing 4% FBS, 1% penicillin–streptomycin, 1% GlutaMAX, 1% insulin-transferrin-selenium (ITS, Thermo Fisher, Auckland, New Zealand), and 200 nM dexamethasone (Sigma, Auckland, New Zealand). Medium was changed every two days. The maximum ALI TEER value expected was around 250 Ω cm2. Once they reached that value, cells were dosed with 100 μM of each peptide. TEER values were read before the treatment and 3 hours after treatment and every day to monitor the behavior of the monolayer.
000 cells per well in proliferation medium (0.5 ml in the apical and 1.5 ml in the basolateral chambers). The seeding day was defined as Day 0 and the cells were allowed to attach. The basal media was changed every other day until the formation of a monolayer. After the cells reached confluence, the polarization medium was removed from the apical compartment, leaving the apical surface of the cells exposed to air i.e. air–liquid (ALI) culture condition. The polarization medium was made up of base medium RPMI 1640 containing 4% FBS, 1% penicillin–streptomycin, 1% GlutaMAX, 1% insulin-transferrin-selenium (ITS, Thermo Fisher, Auckland, New Zealand), and 200 nM dexamethasone (Sigma, Auckland, New Zealand). Medium was changed every two days. The maximum ALI TEER value expected was around 250 Ω cm2. Once they reached that value, cells were dosed with 100 μM of each peptide. TEER values were read before the treatment and 3 hours after treatment and every day to monitor the behavior of the monolayer.Results
Scanning electron microscopy/energy dispersive X-ray spectroscopy (SEM/EDX)
SEM is a tool to study the microstructural properties of biomolecules. It is important to study the microstructure of the peptides to understand the influence of different conditions that will be encountered in drug product development. The surface structure and morphology of the peptide/s could change with varying techniques of treatment. The microscopic image of both counter-ion SP-A peptides 10-AA and 20-AA can be observed in Fig. 1. SP-A peptides show a network which is representative of the complex folding of the peptides. | ||
| Fig. 1 Scanning electron microscopy (SEM) images of SP-A peptides highlighting morphology variations between acetate (AC) and phosphate (PS) salts. (A) SP-A_10-AA PS shows dispersed aggregates, while (B) SP-A_10-AA AC forms fibrillar structures. (C) SP-A_20-AA PS presents loosely packed sheets, whereas (D) SP-A_20-AA AC forms compact clusters. Scale bars: 100 μm. | ||
EDX is used to determine the molecular composition of the peptide that provides useful information about the atomic percent and stoichiometric ratios. The EDX analysis of the peptides showed peaks corresponding to carbon, oxygen, nitrogen, and sulfur for both peptides as seen from Fig. 2. Additionally, peaks corresponding to carbon (C) and gold (Au) were seen at 0.28 keV and 2.1 keV respectively which is an artifact from the sample preparation for SEM imaging. It can be observed phosphorus confirming the counter-ion used in Fig. 2A and C.
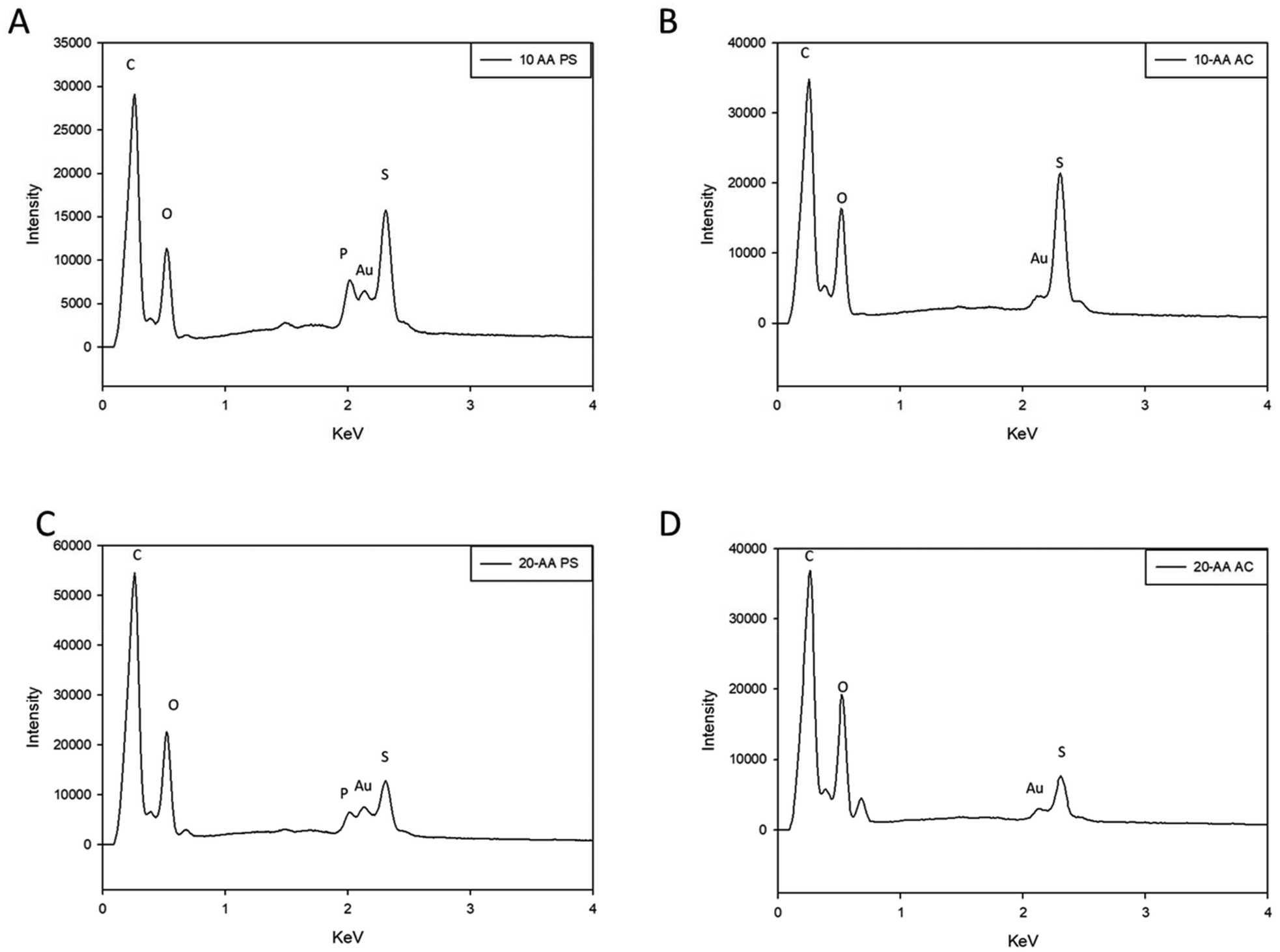 | ||
| Fig. 2 Energy dispersive X-ray (EDX) spectra of SP-A peptides: (A) SP-A 10-AA PS; (B) SP-A 10-AA AC; (C) SP-A 20-AA PS; and (D) SP-A 20-AA AC. | ||
Circular dichroism spectroscopy for secondary structure analysis of SP-A peptides
The Circular Dichroism (CD) spectra for SP-A -10 AA and SP-A -20 AA peptides at different concentrations are shown in Fig. 3. The spectra confirm that SP-A 10-AA and SP-A 20-AA peptides predominantly assume a disordered conformation, consistent with their short sequence length, which limits the formation of stable secondary structures.18 No significant α-helical or β-sheet content was detected in either peptide, across all conditions tested (temperature and counterion variation). Although a negative peak around 202 nm is observed in SP-A 10, particularly in PS conditions, this does not indicate a well-defined secondary structure, but rather a typical signal of disordered peptides with minor transient conformations. Moreover, the absence of a clear 222 nm minimum and the lack of structural persistence across conditions confirm that SP-A 10-AA does not form a stable α-helix or β-sheet structure. This is further supported by molecular simulations, which demonstrate that the peptide remains predominantly disordered in solutions, although the longer sequence might be expected to stabilize some secondary structure, its CD spectra does not exhibit well-defined α-helical or β-sheet structures. Instead, its CD spectra features remain consistent with a random coil conformation, similar to SP-A 10-AA, across both 25 °C and 37 °C. | ||
| Fig. 3 Circular dichroism (CD) of SP-A peptides at 2 different temperatures: (A) SP-A 10-AA and SP-A 20-AA at 25 °C (B) SP-A 10-AA and SP-A 20-AA at 37 °C. (N = 3). | ||
Additionally, temperature does not significantly alter peptide conformation, nor does the choice of counterion lead to major structural stabilization. This suggests that ionic interactions alone are not sufficient to induce secondary structure formation in these short peptides. These findings are consistent with studies on disordered peptides, where local sequence propensity and environmental conditions can influence transient conformations but do not necessarily stabilize well-defined structures.10
Differential scanning calorimetry for thermal stability and phase transition analysis
The DSC thermograms of SP-A 10-AA and SP-A -20 AA peptides in PS and AC salt forms exhibit unique thermal responses, indicating structural and stability variations (Fig. 4). The 10-AA AC peptide exhibits a broad endothermic transition about 150 °C, indicating a phase transition or unfolding event, but the 10-AA PS peptide has a sharper endothermic peak around 200 °C, indicating a more defined thermal transition. The AC and PS forms of the 20-AA peptides exhibit several endothermic transitions, with the AC form showing a prominent peak around 200 °C and the PS form transitioning at both 150 °C and 200 °C. The DSC thermogram of each peptide shows a characteristic endothermic peak at 168 °C, 171 °C, 158 °C and 166 °C for SP-A 10-AA AC, PS and 20 AC, PS, respectively, which is suggestive of their respective melting temperatures (Tm). The enthalpy value (ΔH) was measured to be 1.96 J g−1, 3.08 J g−1, 1.08 J g−1, and 1.63 J g−1 for SP-A 10-AA -mer AC, SP-A 10-AA PS, SP-A 20-AA AC, and SP-A PS, respectively. This thermodynamic parameter indicates the heat required to break the molecular bonds that will lead to melting of the compound eventually.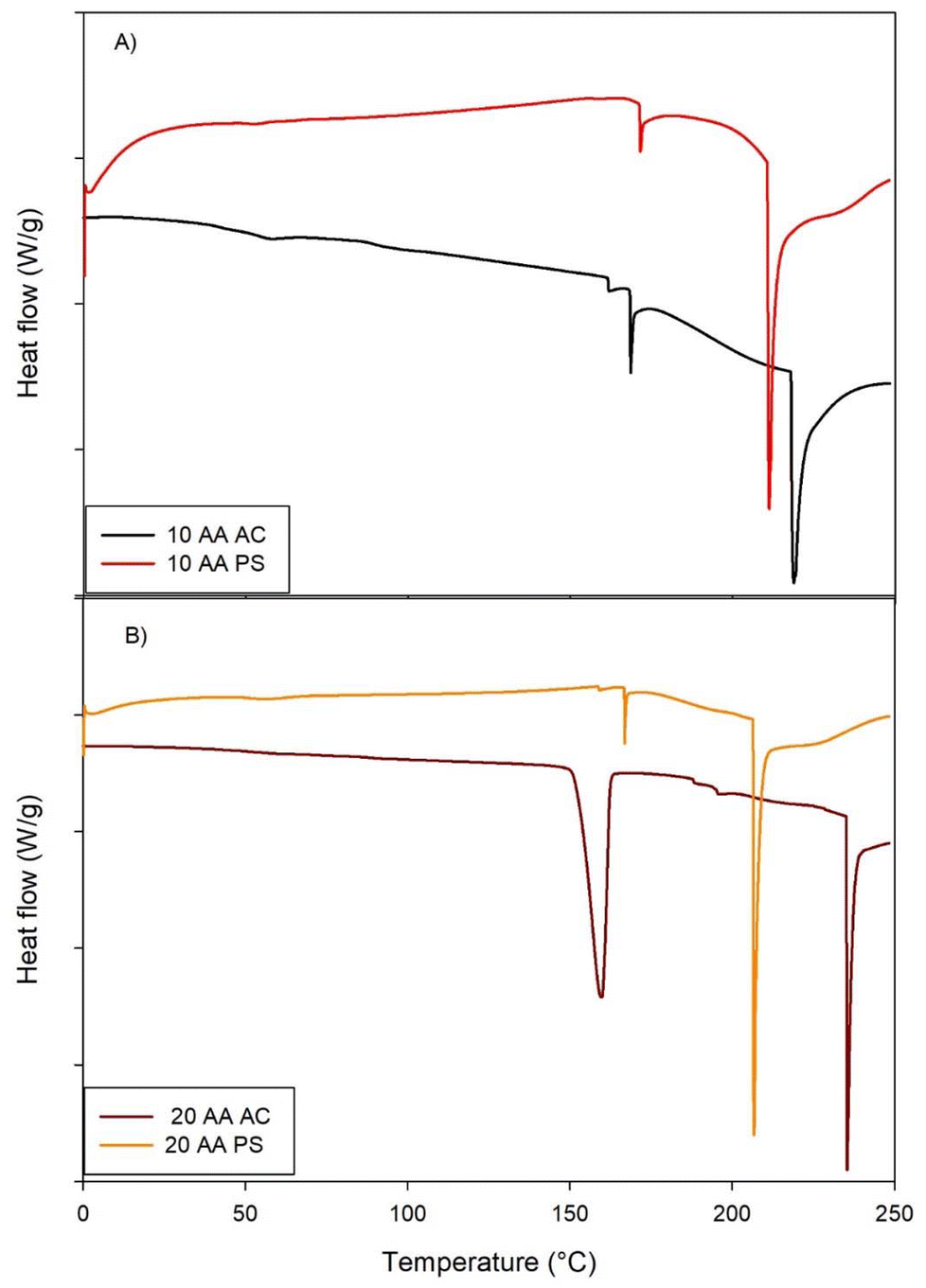 | ||
| Fig. 4 Differential scanning calorimetry (DSC) thermograms of SP-A: (A) 10-AA and SP-A (B) 20-AA Peptides. (Exothermic peak is up. Endothermic peak is down). (N = 3). | ||
X-ray powder diffraction for crystallinity and structural analysis
The structural properties of the two SP-A peptides salts were studied using XRPD. The main diffraction peaks observed for both the peptides are shown in Fig. 5. In solid state, both peptides exhibited a peak around 19.5° 2θ at room temperature. Hence, the peak seen at 19.5° 2θ indicates that the peptides can have a degree of order, similar to other peptides reported.11 The 20-AA AC peptide showed the sharp diffraction peak pattern of a highly crystalline powders.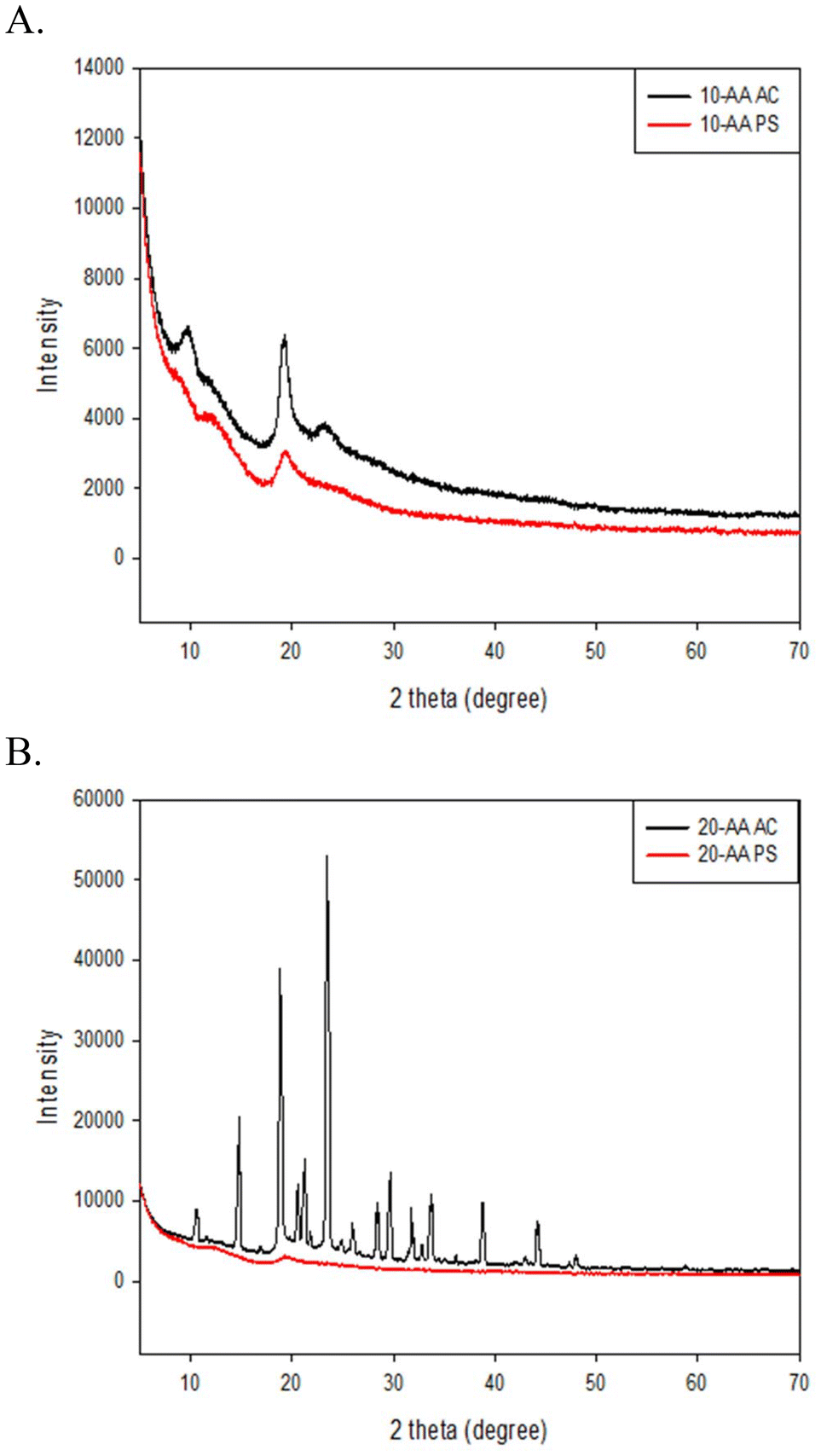 | ||
| Fig. 5 X-ray powder diffraction (XRPD) of SP-A peptides: (A) SP-A 10-AA AC and SP-A 10-AA PS; and (B) SP-A 20-AA AC and SP-A 20-AA PS. | ||
Attenuated total reflectance-Fourier transform infrared spectroscopy (ATR-FTIR) for structural and chemical analysis
IR spectrum is used to detect and identify the chemical functional groups present in the compound which can be used as molecular fingerprint. The several molecular vibrations in a macromolecule leads to absorption peaks, hence it is difficult to assign every single peak to its corresponding bond. However, there are some characteristic peaks that are used to identify and study the secondary structure of proteins and peptides. Amide linkage in peptide/protein molecules are very active in IR mode which enables the characterization of biomolecules including a strong band at 1,600–1690 cm−1 (amide I), 1560–1540 cm−1 for amide II and 1370–1350 cm−1 for C–N.The ATR-FTIR spectra of SP-A 10 and SP-A 20-AA peptides in their AC and PS salt forms were analyzed in the amide I region (1600–1700 cm−1) to determine their secondary structure (Fig. 6). All spectras exhibit a predominant band centered at 1624–1627 cm−1, whose intensity and width suggest an extended or disordered structural conformation. Spectral deconvolution indicates that this signal accounts for 47% to 86% of the total structural content, with the highest proportion observed in SP-A 10 AC (86.13%), suggesting a strong tendency toward aggregation. Although the 1624–1627 cm−1 region is dominant, its broad profile suggests that these signals may arise from not only β-sheet structures but also aggregated disordered conformations.19 The higher proportion of signal in the 1650–1661 cm−1 region in SP-A 20-AA PS and SP-A 20-AA AC indicates increased structural flexibility, possibly influenced by the phosphate counterion.20
 | ||
| Fig. 6 ATR-FTIR spectra of SP-A peptides: (A) SP-A 10-AA; and (B) SP-A 20-AA Peptides. (N = 3). | ||
Raman spectroscopy for molecular fingerprinting and structural characterization
The Raman spectra of SP-A -10 AA and SP-A -20 AA peptides show unique vibrational characteristics, indicating structural changes (Fig. 7). The peaks in the 2800–3000 cm−1 area, which indicate C–H stretching vibrations, are stronger for SP-A 10-AA. This indicates a higher concentration or a different environment for aliphatic chains in this peptide. The peaks in the 1600–1700 cm−1 area belong to amide I vibrations, linked to the peptide backbone's secondary structure, with significant intensity changes between the peptides. SP-A -20 AA exhibits distinct peaks in the 800–1200 cm−1 range, representing C–C, C–N, and N–H bending vibrations, and in the 1200–1400 cm−1 area, representing CH2 and CH3 bending modes. These spectrum discrepancies imply variations in backbone conformation and side-chain interactions, which may reflect differences in secondary structures like alpha-helices, beta-sheets, or random coils.21 | ||
| Fig. 7 Confocal Raman spectra of SP-A 10-AA and SP-A 20-AA peptides. | ||
Zeta potential measurement for peptides charge
The zeta potential (ζ) results are shown in Table 1. Both the SP-A 10-AA and 20-AA peptides show net negative surface charge under these conditions, as expected from their amino acid sequence. The zeta potential values more positive than −20 mV in <30 mM diluent indicating a relatively low anionic surface charge.22 The 10-AA become less net anionic with increasing salt concentration for both salts. The SP-A 20-AA PS peptide became more net anionic with increasing salt concentrations, ζ of −20 mV at 0.5× PBS and NS. SP-A 20-AA AC peptide showed the same trend as the SP-A 10-AA peptide. This cutoff has been shown to correlate with aggregation, with lower absolute zeta being more likely to aggregate when charge is the predominant driver of aggregation.22,23| Sample | ζ (mV) | |||||
|---|---|---|---|---|---|---|
| Phosphate buffered saline | Normal saline | |||||
| 0.1× | 0.2× | 0.5× | 0.1× | 0.2× | 0.5× | |
| SP-A 10-AA PS | −7.7 ± 0.25 | −9.6 ± 2.09 | −2.7 ± 0.84 | −6.5 ± 0.81 | −7.4 ± 1.66 | −3.5 ± 1.34 |
| SP-A 20-AA PS | −5.5 ± 0.56 | −11.1 ± 0.38 | −21.4 ± 1.42 | −3.1 ± 1.03 | −1.6 ± 1.6 | −18.4 ± 4.76 |
| SP-A 10-AA AC | −7.9 ± 1.5 | −3.9 ± 1.9 | −3.7 ± 1.06 | −5.6 ± 1.2 | −5.36 ± 0.25 | −4.9 ± 0.48 |
| SP-A 20-AA AC | −9.0 ± 0.16 | −2.8 ± 1.52 | −5.2 ± 1.01 | −7.8 ± 0.62 | −4.91 ± 0.63 | −3.0 ± 0.75 |
Coulometric Karl Fischer titration for precise moisture content analysis
Karl Fisher titration is performed to measure the residual water content in the powder. Residual water content (Table 2) of the two peptides were measured to be 6.31 ± 1.30% and 3.12 ± 0.36% w/w for SP-A 10-AA PS and 20-AA PS peptides, respectively. On the other hand, SP-A 10-AA AC and 20-AA AC peptide powders show residual water contents of 4.6 ± 1.12% w/w and 1.9 ± 0.21% w/w, respectively.| SP-A peptide | Residual water content (% w/w) |
|---|---|
| 10-AA PS | 6.31 ± 1.30 |
| 10-AA AC | 4.6 ± 1.12 |
| 20-AA PS | 3.12 ± 0.36 |
| 20-AA AC | 1.9 ± 0.21 |
In vitro solubility assessment for peptide formulation and bioavailability
The in vitro solubility results are presented in Table 3. The solubility of the SP-A 10-AA PS peptide was measured to be 12.2 ± 0.05 mg mL−1 in MQ water, 1.81 ± 0.02 mg mL−1 in PBS, 2.06 ± 0.03 mg mL−1 in EtOH, and 4.25 ± 0.01 mg mL−1 in methanol. The SP-A 10-AA AC showed less solubility in aqueous solutions i.e. 0.42 ± 0.040 mg mL−1 in MQ water, 1.16 ± 0.01 mg mL−1 in PBS, 0.04 ± 0.01 mg mL−1 in EtOH, and 0.01 ± 0.0003 mg mL−1 in methanol.| Solvent | 10-AA PS solubility (mg mL−1) | 10-AA AC solubility (mg mL−1) | 20-AA PS solubility (mg mL−1) | 20-AA AC solubility (mg mL−1) |
|---|---|---|---|---|
| MilliQ water | 12.2 ± 0.05 | 0.42 ± 0.040 | 10.36 ± 0.034 | 0.08 ± 0.029 |
| PBS (pH 7.4) | 1.81 ± 0.02 | 1.16 ± 0.01 | 0.47 ± 0.004 | 0.30 ± 0.01 |
| Absolute ethanol | 2.06 ± 0.03 | 0.04 ± 0.01 | 0.11 ± 0.05 | 0.02 ± 0.02 |
| Methanol | 4.25 ± 0.01 | 0.01 ± 0.0003 | 1.45 ± 0.02 | 0.02 ± 0.005 |
For the solubility between SP-A 20-AA PS and AC different salts, same trend was observed. SP-A 20-AA PS solubility was measured to be 10.36 ± 0.034 mg mL−1 in MQ water, 0.47 ± 0.004 mg mL−1 in PBS, and 0.11 ± 0.05 mg mL−1 in EtOH 1.45 ± 0.02 mg mL−1 in methanol. The AC 20-AA SP-A salt shows very different solubility properties, namely, 0.08 ± 0.029 mg mL−1 in MQ water, 0.30 ± 0.01 mg mL−1 in PBS, 0.02 ± 0.02 mg mL−1 in EtOH, and 0.02 ± 0.005 mg mL−1 in methanol. The solubility properties demonstrate the effect of the counterion type on the solubility value independent of the same length-peptide.
Octanol/water partition coefficient for lipophilicity and drug permeability assessment
The octanol/water partition coefficient (logP) studies of the SP-A 10-AA and SP-A 20-AA peptides in PS and AC salt forms indicate unique hydrophilic and hydrophobic qualities that are critical for their biological activity. As shown in Table 4, the 10-AA peptide in both salt forms is more soluble in water than in octanol, with logP values of −1.79 PS and −1.07 AC, indicating a hydrophilic character. The 20-AA peptide has hydrophilic properties, with logP values of −0.81 PS and −0.91 AC, yet it is somewhat more hydrophobic than the 10-AA peptide.
P of SP-A 10-AA and SP-A 20-AA peptide at T = 37 °C (N = 3, mean ± standard deviation)
| Solvent | 10-AA PS | 10-AA AC | 20-AA PS | 20-AA AC |
|---|---|---|---|---|
| MilliQ water | 1.12 ± 0.5 mg mL−1 | 2.06 ± 0.03 mg mL−1 | 1.49 ± 0.015 mg mL−1 | 0.91 ± 0.01 mg mL−1 |
| Octanol | 0.47 ± 0.004 mg mL−1 | 0.17 ± 0.02 mg mL−1 | 0.22 ± 0.009 mg mL−1 | 0.11 ± 0.01 mg mL−1 |
| LogP |
−1.79 ± 0.4 | −1.07 ± 0.2 | −0.81 ± 0.3 | −0.91 ± 0.2 |
High performance liquid chromatography
The HPLC chromatogram of peptides 10-AA and 20-AA are shown in Fig. 8. The retention time of 10-AA peptide was around 7.6 minutes and 20-AA peptide was around 7.9 minutes. The peak seen around 22 minutes is probably from the change in polarity of the mobile phase, since this peak was also seen in blank solvent injection. | ||
| Fig. 8 High performance liquid chromatography of SP-A Peptides: (A) SP-A 10-AA; and (B) SP-A 20-AA. | ||
In vitro cell viability assessment for biocompatibility and cytotoxicity evaluation
The in vitro cell viability experiments performed with different pulmonary cell lines exhibits the cytotoxic effect of the four peptides in Fig. 9. The A549 and H441 cell lines showed no cytotoxicity effect but the 20-AA AC by affecting the relative cell viability for SP-A concentration higher than 100 μM. For H38 cell line, the only peptide that had a cell viability effect was the 20-AA PS at its highest concentration. The results indicate that A549 and H441 cells maintained high viability across all peptide treatments, with no statistically significant reductions, except for 20-AA AC, which significantly reduced cell viability at concentrations above 100 μM (p < 0.05). In contrast, the H358 cell line exhibited a statistically significant decrease in viability only with 20-AA PS at 1000 μM (p < 0.05), suggesting a selective cytotoxic response in this cell line. Most peptide formulations exhibit no significant cytotoxic effects, supporting their biocompatibility for potential pulmonary applications and that is a concentration-dependent cytotoxicity in 20-AA AC and 20-AA PS highlights the importance of formulation selection for optimizing peptide-based inhalation therapies. | ||
| Fig. 9 In vitro cell viability of human lung cells at various doses of SP-A 10-AA and SP-A 20-AA peptides: (A) H441; (B) A549; and (C) H358. (N = 6). Significant differences are indicated by different letters (p < 0.05). | ||
In vitro transepithelial electrical resistance (TEER) measurement for barrier integrity and permeability assessment
The barrier properties of the H-441 cell line were evaluated at the air–liquid interface (ALI). The TEER values of H-441 cell lines treated with both salts of 10-AA and 20-AA peptides showed similar patterns in Fig. 10. The electrical resistance of the cells treated with peptides were similar to the control cells which was treated with the RPMI media. | ||
| Fig. 10 Transepithelial electrical resistance (TEER) following dosing of SP-A 10-AA and SP-A 20-AA peptides on H441 human pulmonary cells at the air–liquid interface (ALI). (N = 3). | ||
Molecular dynamics simulation for structural stability and conformational analysis of SP-A peptides
The Fig. 12 presents the structural evolution and secondary structure stability of SP-A 10 and SP-A 20-AA peptides over a 10 ns MD simulation. To further understand secondary structure retention and dynamic flexibility, we assessed the conformational stability of the 10-AA and 20-AA peptides throughout a 10 ns simulation. Fig. 12 presents the structural evolution and secondary structure stability of SP-A 10-AA and SP-A 20-AA peptides over a 10 ns MD simulation. Fig. 12A–D show the initial (0 ns) and final (10 ns) conformations of each peptide, highlighting structural changes, in SP-A 10-AA, a progressive loss of α-helical content is observed after 5 ns, indicating structural flexibility and instability, in contrast, SP-A 20-AA does not exhibit any α-helical secondary structure, maintaining a predominantly disordered conformation throughout the simulation. The Fig. 12E and F shows the evolution of α-helical content over simulation time, with SP-A 10-AA gradually decreasing while SP-A 20-AA remains as random coil structure. MD simulations provided insights into the structural stability and conformational evolution of the SP-A 10-AA and SP-A 20-AA peptides over a 10 ns simulation. The results revealed significant differences in their ability to maintain secondary structure.Discussion
The objective of this study was to characterize the physicochemical properties and in vitro cellular effects of SP-A peptides, particularly their biocompatibility and interaction with lung epithelial cells. The in vitro results demonstrated that SP-A peptides maintain high cell viability, do not significantly alter epithelial barrier integrity, and do not increase membrane permeability. These findings indicate that the peptides are well tolerated in lung models, reinforcing their potential as safe and effective candidates for inhaled therapeutic applications. It is has been identified that the 10-AA derived from carbohydrate recognition domain of SP-A2 has activity similar to the full length oligomeric protein in treating inflammatory aspects of asthma.4,24 The use of small SP-A-derived peptides offers several advantages over full-length SP-A for therapeutic applications, including enhanced stability, greater bioavailability, and improved formulation feasibility, such as drug delivery. Furthermore, XRD examination shows that the 20-AA peptide has higher crystallinity, indicating a more organized structure equivalent to that found in full-length SP-A. The DSC study also confirms this by revealing increased heat stability, which is critical for retaining structural integrity during inhalation administration.Additionally, SP-A peptides presenting the CRD domain have been demonstrated to retain functional activity, as seen in previous studies by Kraft and Ledford.25 showing that SP-A peptides covering the CRD domain maintain bioactivity while successfully reducing airway hyperresponsiveness and inflammation in preclinical asthma models. Our peptides’ structural stability, as validated by XRD, CD, and DSC studies, strengthens their capacity to modulate surfactant activity and immunological responses in the same way that full-length SP-A work. Furthermore, given the association between SP-A deficiency and increased airway inflammation, these peptides may be potential candidates for inhalation therapy to restore SP-A-like function in respiratory disorders such as asthma.
Hence, the goal is to develop this peptide into a novel therapy to treat asthma that will be delivered locally to the respiratory tract. Currently, inhaled drugs targeted directly to the lungs has become the preferred route of drug delivery used clinically as successful pharmaceutical products in many respiratory diseases. Aerosol therapeutic drug development is a complex process where the particles or droplets need to be of sufficient size distribution for targeted delivery.26 Consequently, several novel technologies are involved in development and optimization of these inhaled aerosols that can expose the drug compound to different conditions with respect to temperature, pressure, pH etc. Therefore, it is imperative to characterize the therapeutic peptides to understand the important biophysical and chemical properties such as phase transition, thermal behavior, and secondary structure that contribute to its development.
Microscopic imaging enables us to see the secondary structure, the three-dimensional structure of the peptide, and the self-assembly or aggregation of the peptide, if any. Hence, this observation in unprocessed peptides will help understand the changes that the peptide may undergo during formulation development. Similarly, techniques such as DSC, XRPD, ATR-FTIR, Raman spectroscopy are used to characterize the peptide before and after processing to study the physical or chemical changes that might occur to the peptide. This information is key for the development of therapeutic peptides since it may affect the stability and biological activity of the peptide. The elemental characterization technique, EDX, determined that the peptides were mainly composed of carbon, oxygen, and nitrogen, but elements such as sulfur were found in both 10-AA and 20-AA peptides. The source of sulfur atoms will be the cysteine and methionine amino acids that are part of the primary structure of both peptides.
Circular Dichroism is the commonly used light absorption technique to detect and determine the secondary structure of peptides and proteins. The CD spectra of the two SP-A peptides in solution are shown in Fig. 3. The length of the amino acid chain in peptides has a significant impact on their secondary structure and stability, as shown for SP-A -10 AA and SP-A 20-AA peptides. The CD spectra confirm that the SP-A 10-AA and SP-A 20-AA peptides predominantly adopt a disordered conformation, as expected due to their short length, which prevents the formation of stable secondary structures.27 Our analysis shows that SP-A 10-AA at both temperatures (25 °C and the 37 °C) exhibits a characteristic profile of disordered peptides, with minimal to no α-helical or β-sheet content. SP-A 10-AA exhibits a modest negative ellipticity about 202 nm, indicating that a small portion of its population may briefly adopt an α-helical shape. One possible explanation for this behavior is electrostatic interactions between the counterion and the peptide, which may temporarily reduce backbone solvation, allowing short-range hydrogen bonding to persist.28 While this helicity is marginal and does not constitute a stable secondary structure, it aligns with prior studies in short peptides, where local sequence propensity and ionic interactions can induce partial helicity in otherwise disordered peptides.18
Trifluoroethanol (TFE) has been shown to reduce backbone solvation and promote intramolecular hydrogen bonding in disordered peptides, leading to α-helical structures.29 Given that SP-A 10-AA and 20-AA were derived from a portion of the domain of SP-A carbohydrate recognition, it was expected that they would exhibit some structural and physical characteristics comparable to those of the natural protein. This finding is consistent with previous research on peptides generated from proteins that perform comparable biological roles.30 CD analysis is an important tool in the creation and evaluation of therapeutic peptides because it reveals information about their secondary structure in solution and their capacity to preserve structural integrity under physiological conditions.
ATR-FTIR spectroscopy was used to analyze the secondary structure of SP-A peptides, focusing on the Amide I region (1600–1700 cm−1). Deconvolution of the Amide I region in the ATR-FTIR spectra revealed a dominant band around 1624–1627 cm−1, which is best attributed to aggregated random coil conformations. This assignment is strongly supported by CD spectra, which indicate a predominant random coil signature, and MD simulations, which show a progressive loss of secondary structure, with the peptides transitioning into fully disordered states over time. The broad nature of this FTIR band suggests the presence of strong inter-chain hydrogen bonding within disordered aggregates, a feature commonly observed in peptides that lack stable secondary structures but tend to self-associate. The combination of these findings—CD spectra showing a random coil signature, MD simulations revealing structural destabilization, and ATR-FTIR indicating aggregated disordered conformations.31–33
ATR-FTIR provided greater sensitivity to structural changes than CD spectroscopy, but the combined data indicate that the observed secondary structure should be interpreted as aggregated disordered structures rather than a defined secondary structure, which has important implications for formulation stability and peptide interactions.34,35
Using energy minimization in molecular modeling software programs (ChemSite Pro® and Molecular Modeling Pro®), Fig. 11 shows that both the α-helix and β-sheet secondary structure conformations are thermodynamically stable. Calculations from Fig. 11 for 10-AA peptide, the α-helix structure (Fig. 11A) reveals that the distance, d, from lysine 1 to Asp 10 is 18.53 Å, whereas β-sheet (Fig. 11B) calculations shows that the d is 26.72 Å i.e. predictive longer length for the β-sheet conformation. For the 20-AA peptide, α-helix and β-sheet structures showed an intramolecular distance from Pro 1 to Aps 20 of 31.39 Å and 66.16 Å, respectively. After energy minimization for all peptide structures, the predicted total free energy was −130.24 kcal mol−1, −178.4 kcal mol−1, −65.05 kcal mol−1, and −7.4 kcal mol−1 for 10-AA α-helix, 20-AA α-helix, 10-AA β-sheet, and 20-AA β-sheet structures, respectively, suggesting that the α-helix conformation is more stable in these minimized structures. However, CD spectra indicate that the peptides predominantly adopt a random coil conformation rather than a well-defined α-helix in the solid state, suggesting that these secondary structures are not maintained in experimental conditions. These energy minimization results provide a static representation of potential conformations but do not account for structural fluctuations in solution, which are better captured through MD simulations.
 | ||
| Fig. 11 Energy minimized 3-D models of SPA peptides by molecular modeling (Molecular Modeling Pro® Software) for: (A) SPA 10-AA; (B) SPA 10-AA energy minimized; (C) SPA 20-AA; and (D) SPA 20-AA energy minimized. | ||
Hence, this suggests that the α-helix structure is favorable in the peptide, which agrees with the CD spectra, which show the presence of a marginally α-helix secondary structure in a solid state. These energy minimization results provide a static representation of the peptides’ initial conformations. However, they do not account for structural fluctuations in solution, which are better captured through MD simulations.
MD simulations provide important information about the structural behavior and stability of peptides under physiological settings. In agreement with the small negative ellipticity observed at 202 nm in the CD spectra, MD reveals that SP-A 10-AA exhibits transient α-helical structure, though it remains unstable and does not persist throughout the simulation (Fig. 12). This aligns with prior studies suggesting that short peptides can form momentary helices due to sequence propensity and ionic interactions, but these do not constitute stable secondary structures. Notably, SP-A 20-AA does not exhibit significant helicity in either CD or MD, reinforcing its inherently disordered nature. The MD data also reveals the presence of transient β-sheet formation in SP-A 20-AA, which was not evident in the CD spectra, likely due to its low population and instability over time.
 | ||
| Fig. 12 Structural evolution and α-helical stability of SP-A 10-AA and SP-A 20-AA peptides from MD simulations. MD simulation results of SP-A 10-AA and SP-A 20-AA. (A–D) Structural snapshots at 0 ns and 10 ns. (E and F) Secondary structure evolution over time. | ||
The anionic 10-AA PS and 20-AA PS salt peptides showed more hydrophilic behavior than their AC salt form with good solubility in various aqueous solvents as evident from Table 3. This is consistent with the known hydrophobic property of the AC salt. Both peptide salts had limited solubility in common alcohols. The octanol/water partition study also suggests that the peptides have higher affinity to the aqueous phase than the organic phase. The predicted logP of 10-AA PS and 20-AA PS peptides were −1.79 ± 0.4 and −0.81 ± 0.2, respectively. LogP values for the AC salts were 1.07 ± 0.3 and −0.91 ± 0.2 for 10-AA AC and 20-AA AC, respectively. The negative logP value agrees with the very low solubility shown in less polar solvents as ethanol and methanol in Table 4. The AC versions of both peptides have better water solubility and smaller negative logP values than their PS counterparts, implying that the AC salt promotes hydrophilicity. These changes in solubility and partitioning qualities affect the peptides’ bioavailability and cellular uptake, both of which are crucial for their efficacy in pulmonary applications, emphasizing the need of optimizing salt forms for therapeutic usage. The AC versions of both peptides had a higher solubility in water and consequently lower negative logP values, demonstrating that AC salts increase the peptides’ hydrophilicity. As a result, the salt form used can have a direct impact on peptide bioavailability, cellular uptake, and overall therapeutic efficacy.
A peptides solubility is regulated by a variety of physicochemical characteristics, including its net charge at physiological pH, hydrophilic–hydrophobic balance, and the presence of particular counterions. We investigated the solubility of SP-A peptides in relation to their estimated pI and experimentally measured logP values. The estimated pI values for SP-A 10 and SP-A 20-AA peptides were 3.69 and 3.93, respectively, revealing their charge distribution in aqueous environments and implying that their solubility is significantly based on the solution pH relative to their pI. Peptides have lower solubility near their pI due to reduced electrostatic repulsions, which promote aggregation and precipitation, but they are more soluble when carrying a net charge at pH values above or below their pI.35
Our experimental solubility findings align with theoretical predictions, demonstrating that peptide solubility is significantly higher in conditions where they carry a net charge, confirming the electrostatic contribution to solubility. The presence of PS and AC counterions influenced hydration shells and electrostatic interactions, leading to ion-specific effects on solubility.34 These ion-specific solubility effects are particularly relevant for peptide formulation, as salt selection plays a critical role in preventing aggregation and enhancing shelf-life stability. Understanding these counterion-dependent solubility trends provides valuable insights for optimizing peptide formulations, particularly for pharmaceutical and biotechnological applications.
The logP parameter describes how peptides partition between aqueous and lipidic environments, affecting their interaction with biological membranes and potential for drug administration.36 A balanced hydrophilic-lipophilic profile is essential for successful dissolving in aqueous settings while retaining the ability to interact with cell membranes, especially in inhalable formulations that need diffusion across the lung epithelium. Our findings show that SP-A 10-AA and SP-A 20-AA peptides have distinct hydrophilic and hydrophobic properties, with logP values indicating that they can preserve solubility while retaining membrane affinity, which is critical for pulmonary delivery and therapeutic efficacy.37
The formulation and bioavailability of therapeutic peptides depend largely on their solubility and partitioning characteristics.38 According to our research, the peptide AC salt is more soluble in water than its PS version due probably to AC ions having weaker ionic interactions and less kosmotropic effects.39 The PS salt, on the other hand, has somewhat reduced solubility, which could be explained by more robust electrostatic interactions that encourage aggregation. Furthermore, logP research indicates that PS may promote interactions with lipid membranes, which could affect absorption and bioactivity, whereas AC prefer hydrophilic environments, improving systemic bioavailability in aqueous formulations.20 These results highlight how crucial counter-ion selection is when refining peptide formulations for certain therapeutic applications.
The ΔH value of 20-AA is higher than the 10-AA due to the increased aa length that requires more energy to break the intermolecular bonds before melting. No other transitions were observed before the melting transition indicating that these peptides are thermodynamically stable at room and biological temperatures.40 The longer chain of the 20-AA peptide results in more complex thermal behavior, which could be attributed to higher intramolecular interaction and structural stability. AC forms had larger and lower temperature transitions than PS forms, indicating that the salts influence peptide stability and interactions differently.
The high melting temperatures observed in the DSC thermograms suggest that intermolecular interactions, rather than a traditional folding–unfolding event, are the primary cause of the thermal changes of the SP-A 10-AA and SP-A 20-AA peptides. Strong peptide-peptide interactions and potential dehydration effects are indicated by the observed transitions, although it is not likely that these peptides would form highly stable secondary structures due to their short length. Further evidence that counterion-specific electrostatic interactions support thermal stability is provided by the differences between phosphate and acetate salts, with phosphate perhaps promoting aggregation or intermolecular hydrogen bonding. These results concur with earlier studies that found counterions affect the stability and self-assembly of peptides at higher temperatures.41,42 These findings are critical for understanding the peptides’ stability and applicability for therapeutic uses, as heat stability might affect storage, handling, and biological activity.
A peptide salt form can affect its solubility, thermal stability, and intermolecular interactions—all of which can be measured with DSC.43 The AC and PS salt versions of the peptides showed significant variations in heat stability, according to DSC themograms. The multivalent character of PS ions could mitigate charge repulsion and strengthen intra- and intermolecular contacts, as seen by the PS form's greater Tm, which suggests improved electrostatic stability. The AC form, on the other hand, showed a somewhat lower Tm, suggesting a less stable structure, possibly as a result of weaker ionic connections.20
XRPD is used to determine degree of crystallinity/non-crystallinity, polymorphs, and peptide secondary structures in the solid-state, as our group has reported.11,14,44For example, insulin is a protein macromolecule has been extensively characterized using crystallography and its several polymorphs of insulin were discovered using XRPD.45
Spectroscopic techniques such as FTIR and Raman can be used to characterize peptides and proteins due to the presence of the amide (peptide) bonds which is characteristic of the peptide/protein folding. The IR peaks seen in (Fig. 6) amide I (1700–1600 cm−1) region is primarily due to carbonyl groups in peptide (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), while amide II (1500–1600 cm−1) and amide III (1220–1320 cm−1) regions consists of C–N stretching and N–H bending.31 These three amide regions are generally used to identify conformational changes of proteins and peptides. Complimenting this technique, Raman spectra (Fig. 7) also exhibits absorption bands in the three amide regions. The strongest peak seen in Raman, for both peptides, around 2900–3200 cm−1 is the C–H vibration in the peptides. Also, seen in the region of 2500–2600 cm−1 is the stretching vibration of sulfhydryl group.46 When using IR spectroscopy, a combination of the three amide regions are generally used to deduce the structure of the peptide/protein due to multiple overlapping from other bands. Among these, amide II band is the less sensitive and less frequently used for structural analysis,47 possibly due to its conformational sensitivity.31,48 Goormaghtigh et al. have reported that only three wavenumbers are useful in deducing the secondary structure of protein. The best wavenumbers identified to predict secondary structure are 1545 cm−1 for α-helix, 1656 cm−1 for β-sheet, 1677 cm−1 for β-turn and 1544 cm−1 for random structure. Absence of any of these bands in the spectra observed in this study suggests the complexity of characterizing peptide secondary structure in solid state. However, these spectroscopic data will serve as a qualitative tool to compare any changes that might occur to the peptides during formulation development or in the final drug product.7 In general, Raman and IR spectral data is processed using advanced chemometric analysis to determine the composition and secondary structure of protein/peptide and it is also common to correlate the structural determination obtained from vibrational spectroscopy with CD.
O stretching), while amide II (1500–1600 cm−1) and amide III (1220–1320 cm−1) regions consists of C–N stretching and N–H bending.31 These three amide regions are generally used to identify conformational changes of proteins and peptides. Complimenting this technique, Raman spectra (Fig. 7) also exhibits absorption bands in the three amide regions. The strongest peak seen in Raman, for both peptides, around 2900–3200 cm−1 is the C–H vibration in the peptides. Also, seen in the region of 2500–2600 cm−1 is the stretching vibration of sulfhydryl group.46 When using IR spectroscopy, a combination of the three amide regions are generally used to deduce the structure of the peptide/protein due to multiple overlapping from other bands. Among these, amide II band is the less sensitive and less frequently used for structural analysis,47 possibly due to its conformational sensitivity.31,48 Goormaghtigh et al. have reported that only three wavenumbers are useful in deducing the secondary structure of protein. The best wavenumbers identified to predict secondary structure are 1545 cm−1 for α-helix, 1656 cm−1 for β-sheet, 1677 cm−1 for β-turn and 1544 cm−1 for random structure. Absence of any of these bands in the spectra observed in this study suggests the complexity of characterizing peptide secondary structure in solid state. However, these spectroscopic data will serve as a qualitative tool to compare any changes that might occur to the peptides during formulation development or in the final drug product.7 In general, Raman and IR spectral data is processed using advanced chemometric analysis to determine the composition and secondary structure of protein/peptide and it is also common to correlate the structural determination obtained from vibrational spectroscopy with CD.
A dose–response cell viability study was performed on different pulmonary cell lines using the two SP-A peptides salts at different concentrations. The relative viability of the A549 cells remained high for the peptides at all tested concentrations. The 20-AA PS showed decreased viability at higher concentrations in the H-358 cell line and the 20-AA AC showed a reduction in viability at higher concentrations in H-441 cell lines. This in vitro cellular demonstrates that SP-A peptides are safe for pulmonary cell lines up to 100 μM. Fig. 9 shows cell viability values of cells treated with the peptides relative to control cells treated with cell growth media. Generally, a cell viability of 80% or higher is considered safe for in vitro lung studies. The TEER values observed in H441 human lung cells were similar to a previous study where the TEER value observed was a plateau.17 The TEER value observed in that study was that of naïve cells. A similar value seen in this study, where the H-441 cells were treated with the SP-A peptides demonstrate that the peptides didn't adversely affect the tight membrane interactions.
Their smaller size facilitates diffusion and deposition in the lung epithelium, making them ideal candidates for inhalation therapy. Additionally, peptide-based approaches are more cost-effective, scalable, and reduce immunogenicity concerns compared to recombinant full-length SP-A. These findings reinforce the relevance of SP-A-derived peptides in pulmonary drug development and highlight their advantages as therapeutic agents for respiratory diseases.
The high aqueous solubility of the PS peptides suggests that an aqueous-based liquid aerosol (solution) formulation will be a straightforward approach for the early phase testing of the safety and efficacy of the peptides in vivo models. However, highly hydrophilic compounds pose the risk of poor membrane permeability. For AC peptides a formulation using dry powder inhalers will be the more suitable pathway to produce a SP-A drug. Another key factor to consider is the chemical and conformational instability of proteins leading to denaturation and degradation in solutions.
Therefore, a liquid aerosol formulation of the SP-A peptides must consider excipients that will enhance the stability and permeation of the peptides. Alternately, the SP-A peptides AC can be developed in solid state as dry power aerosol which is less likely to affect chemical stability. In solid-state formulation chemical and conformational stability can be achieved by the right choice and concentration of excipients that can improve the physicochemical property. The most common approach is through glass stabilization, where the excipient and drug form a glassy amorphous structure (with limited molecular mobility) whose glass transition is higher than the peptide, rendering it stable over the usable temperature range. Other possible mechanism is through hydrogen bonding between the peptide chain and the excipient thus stabilizing the structure49 for longer period. The high temperature and drying cycles commonly encountered during the micronization process can affect the stability, in which case, cryoprotectants like sugar, amino acid,50 or organic salt can prevent the crystallization of protein that may occur during processing. Each physical state of the peptide has its own advantages and challenges. Hence, it is important to consider the possible options for the development of SP-A peptides formulation that can locally deliver to the lungs.
The 20-AA peptide's structural stability, as shown by CD spectra, is consistent with techniques employed in commercial therapeutic peptides like GLP-1 receptor agonists. The engineering of exenatide and liraglutide demonstrates the significance of secondary structure preservation in peptide drug design to preserve α-helical integrity in order to improve receptor affinity and bioactivity.51 As with these formulations, maintaining our peptides’ stability may be essential to their possible therapeutic use, impacting formulation techniques to increase bioavailability and preserve functional activity. Additionally, understanding the thermal properties of therapeutic peptides is essential for formulation development. The transition of liraglutide from a crystalline to an amorphous state during encapsulation highlights the impact of formulation processes on peptide stability. Similarly, the thermal stability of SP-A derived peptides will inform regarding storage conditions, delivery mechanisms, and potential excipient selection to maintain peptide integrity and therapeutic efficacy.
The observed transient helicity in SP-A 10-AA aligns with previous studies on short peptides, where local sequence composition and electrostatic interactions can induce partial helicity but fail to stabilize it over extended timescales. For example, studies on antimicrobial peptides, such as LL-37,52 have shown that helical stability is often context-dependent, requiring interactions with lipid membranes or other biomolecules to maintain secondary structure. The behavior of SP-A 10-AA may follow a similar paradigm, where the peptide exhibits α-helical character only in specific microenvironments, such as when bound to a receptor or embedded in a lipid bilayer.
Conclusion
This study provides a comprehensive characterization of SP-A-derived peptides, integrating experimental and computational approaches to better understand their structural behavior, solubility, and biocompatibility, which are key factors for future formulation development. The findings reveal that SP-A 10-AA exhibits transient α-helicity (∼80–90% at 0 ns, decreasing over time), while SP-A 20-AA remains largely disordered throughout the simulation, suggesting that secondary structure stability is sequence-dependent and may influence formulation performance. Differences in solubility and partitioning behavior were also observed, with phosphate salts exhibiting higher aqueous solubility (logP ∼−2.5) compared to acetate salts (logP ∼−1.8), which could impact their dissolution rate and bioavailability in pulmonary delivery systems.
These insights emphasize the importance of physicochemical stability, secondary structure, and solubility in the design of peptide-based therapeutics. The observed differences in secondary structure and solubility among formulations suggest that selecting the appropriate salt form and excipients could enhance peptide stability, aerosolization properties, and bioavailability in inhalable formulations. Future studies focusing on membrane interactions, functional assays, and in vivo evaluations will be essential to further optimize these formulations and advance their potential as peptide-based treatments for pulmonary diseases.
Ethical approval
This article does not contain any studies with human participants nor animals performed by any of the authors.Data availability
Data available upon request.Conflicts of interest
Dr Encinas-Basurto, Dr Muralidharan, Dr Islam, Dr Vallorz, Professor Black, and Professor Mansour declare no competing interests. Dr Ledford and Dr Kraft are co-founders of RaeSedo Inc., a startup company out of the University of Arizona aimed at the development of SP-A peptide-based therapeutics for the treatment of asthma.Acknowledgements
The authors gratefully acknowledge the financial support from NIH NIAD R21 AI135935 (awarded to J. G. L., H. M. M., M. K., and S. M. B.) and the CONACyT (National Council of Science and Technology of Mexico) postdoctoral fellowship (awarded to D. E. B. and mentor H. M. M.). SEM images and data were collected in the W.M. Keck Center for Nano-Scale Imaging in the Department of Chemistry and Biochemistry at the University of Arizona with funding from the W.M. Keck Foundation Grant. Raman data was collected in the Imaging Cores – Materials Imaging and Characterization Facility supported by the office of Research, Discovery and Innovation at the University of Arizona. This material is based upon work supported by the National Science Foundation under Grant Number #0619599 and Arizona Proposition 301: Technology and Research Initiative Fund (A.R.S.§15–1648). X-ray diffraction data were collected in the X-Ray Diffraction Facility of the Department of Chemistry and Biochemistry, The University of Arizona.References
- J. Floros, R. Fan, S. Diangelo, X. Guo, J. Wert and J. Luo, Surfactant protein (SP) B associations and interactions with SP-A in white and black subjects with respiratory distress syndrome, Pediatr. Int., 2001, 43(6), 567–576 CrossRef CAS PubMed.
- M. M. Pettigrew, J. F. Gent, Y. Zhu, E. W. Triche, K. D. Belanger and T. R. Holford, et al., Respiratory symptoms among infants at risk for asthma: association with surfactant protein A haplotypes, BMC Med. Genet., 2007, 8, 15 CrossRef PubMed.
- Y. Wang, D. R. Voelker, N. L. Lugogo, G. Wang, J. Floros and J. L. Ingram, et al., Surfactant protein A is defective in abrogating inflammation in asthma, Am. J. Physiol.: Lung Cell. Mol. Physiol., 2011, 301(4), L598–L606 CrossRef CAS PubMed.
- D. Francisco, Y. Wang, C. Marshall, M. Conway, K. J. Addison and D. Billheimer, et al., Small Peptide Derivatives Within the Carbohydrate Recognition Domain of SP-A2 Modulate Asthma Outcomes in Mouse Models and Human Cells, Front. Immunol., 2022, 13, 900022 CrossRef CAS PubMed.
- H. M. Mansour and A. J. Hickey, Delivery of drugs by the pulmonary route, in Modern pharmaceutics Volume 2: applications and advances Drugs and pharmaceutical sciences series. 2, ed. A.T Florence and J. Siepmann, Taylor & Francis, Inc., New York, 2009, ch. 5, pp. 191–219 Search PubMed.
- E. F. Haney, K. Nazmi, J. G. Bolscher and H. J. Vogel, Structural and biophysical characterization of an antimicrobial peptide chimera comprised of lactoferricin and lactoferrampin, Biochim. Biophys. Acta, 2012, 1818(3), 762–775 CrossRef CAS PubMed.
- W. P. Kelley, S. Chen, P. D. Floyd, P. Hu, S. G. Kapsi and A. S. Kord, et al., Analytical characterization of an orally-delivered peptide pharmaceutical product, Anal. Chem., 2012, 84(10), 4357–4372 CrossRef CAS PubMed.
- X. Li, F. G. Vogt and D. Hayes Jr., Mansour HM. Physicochemical characterization and aerosol dispersion performance of organic solution advanced spray-dried microparticulate/nanoparticulate antibiotic dry powders of tobramycin and azithromycin for pulmonary inhalation aerosol delivery, Eur. J. Pharm. Sci., 2014, 52, 191–205 CrossRef CAS PubMed.
- P. Muralidharan, D. Hayes Jr., S. M. Black and H. M. Mansour, Microparticulate/nanoparticulate powders of a novel Nrf2 activator and an aerosol performance enhancer for pulmonary delivery targeting the lung Nrf2/Keap-1 pathway, Mol. Syst. Des. Eng., 2016, 1(1), 48–65 RSC.
- S. M. Kelly, T. J. Jess and N. C. Price, How to study proteins by circular dichroism, Biochim. Biophys. Acta, 2005, 1751(2), 119–139 CrossRef CAS PubMed.
- H. M. Mansour, S. Damodaran and G. Zografi, Characterization of the in situ structural and interfacial properties of the cationic hydrophobic heteropolypeptide, KL4, in lung surfactant bilayer and monolayer models at the air-water interface: implications for pulmonary surfactant delivery, Mol. Pharm., 2008, 5(5), 681–695 CrossRef CAS PubMed.
- J. C. Corbett, M. T. Connah and K. Mattison, Advances in the measurement of protein mobility using laser Doppler electrophoresis - the diffusion barrier technique, Electrophoresis, 2011, 32(14), 1787–1794 CrossRef CAS PubMed.
- A. Aodah, A. Pavlik, K. Karlage and P. B. Myrdal, Preformulation Studies on Piperlongumine, PLoS One, 2016, 11(3), e0151707 CrossRef PubMed.
- W. Alabsi, M. F. Acosta, F. A. Al-Obeidi, M. Hay, R. Polt and H. M. Mansour, Synthesis, Physicochemical Characterization, In Vitro 2D/3D Human Cell Culture, and In Vitro Aerosol Dispersion Performance of Advanced Spray Dried and Co-Spray Dried Angiotensin (1–7) Peptide and PNA5 with Trehalose as Microparticles/Nanoparticles for Targeted Respiratory Delivery as Dry Powder Inhalers, Pharmaceutics, 2021, 13(8), 1278 CrossRef CAS PubMed.
- E. Gasteiger, C. Hoogland, A. Gattiker, S. E. Duvaud, M. R. Wilkins and R. D. Appel, et al., Protein identification and analysis tools on the ExPASy server, The proteomics protocols handbook, 2005, pp. 571–607 Search PubMed.
- S. A. Meenach, K. W. Anderson, J. Z. Hilt, R. C. McGarry and H. M. Mansour, High-performing dry powder inhalers of paclitaxel DPPC/DPPG lung surfactant-mimic multifunctional particles in lung cancer: physicochemical characterization, in vitro aerosol dispersion, and cellular studies, AAPS PharmSciTech, 2014, 15(6), 1574–1587 CrossRef CAS PubMed.
- J. J. Salomon, V. E. Muchitsch, J. C. Gausterer, E. Schwagerus, H. Huwer and N. Daum, et al., The cell line NCl-H441 is a useful in vitro model for transport studies of human distal lung epithelial barrier, Mol. Pharm., 2014, 11(3), 995–1006 CrossRef CAS PubMed.
- A. E. García and K. Y. Sanbonmatsu, α-Helical stabilization by side chain shielding of backbone hydrogen bonds, Proc. Natl. Acad. Sci. U. S. A., 2002, 99(5), 2782–2787 CrossRef PubMed.
- S. Sukumaran, Protein secondary structure elucidation using FTIR spectroscopy, Thermo Fisher Scientific, 2017 Search PubMed.
- K. Sikora, M. Jaśkiewicz, D. Neubauer, D. Migoń and W. Kamysz, The role of counter-ions in peptides—an overview, Pharmaceuticals, 2020, 13(12), 442 CrossRef CAS PubMed.
- R. Tuma, Raman spectroscopy of proteins: from peptides to large assemblies, J. Raman Spectrosc., 2005, 36(4), 307–319 CrossRef CAS.
- S. Lazzari, D. Moscatelli, F. Codari, M. Salmona, M. Morbidelli and L. Diomede, Colloidal stability of polymeric nanoparticles in biological fluids, J. Nanopart. Res., 2012, 14(6), 920 CrossRef PubMed.
- S. Bhattacharjee, DLS and zeta potential - What they are and what they are not?, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- A. B. C. Dy, S. Tanyaratsrisakul, D. R. Voelker and J. G. Ledford, The emerging roles of surfactant protein-A in asthma, J. Clin. Cell. Immunol., 2018, 9(4), 553 Search PubMed.
- M. Kraft and J. Ledford, Surfactant Protein A: An Innate Immune Modulator And Therapeutic In Asthma, Trans. Am. Clin. Climatol. Assoc., 2024, 134, 94 Search PubMed.
- J. K. Lam and Q. Zhou, Advances in Pulmonary Drug Delivery Systems and Inhalation Formulations, Pharm. Res., 2023, 40(5), 1013–1014 CrossRef CAS PubMed.
- M. Wojciechowska, J. Miszkiewicz and J. Trylska, Conformational changes of Anoplin, W-MreB1–9, and (KFF) 3 K peptides near the membranes, Int. J. Mol. Sci., 2020, 21(24), 9672 CrossRef CAS PubMed.
- R. F. Sommese, S. Sivaramakrishnan, R. L. Baldwin and J. A. Spudich, Helicity of short E–R/K peptides, Protein Sci., 2010, 19(10), 2001–2005 CrossRef CAS PubMed.
- A. K. Prasad, R. Samajdar, A. S. Panwar and L. L. Martin, Origin of Secondary Structure Transitions and Peptide Self-Assembly Propensity in Trifluoroethanol–Water Mixtures, J. Phys. Chem. B, 2024, 128(32), 7736–7749 CrossRef CAS PubMed.
- F. J. Walther, R. David-Cu, C. Leung, R. Bruni, J. Hernández-Juviel and L. M. Gordon, et al., A synthetic segment of surfactant protein A: structure, in vitro surface activity, and in vivo efficacy, Pediatr. Res., 1996, 39(6), 938–946 CrossRef CAS PubMed.
- A. D. G. Adochitei, Rapid characterization of peptide secondary structure by FT-IR spectroscopy, Rev. Roum. Chim., 2011, 56, 783–791 CAS.
- J. S. Cobb, V. Zai-Rose, J. J. Correia and A. V. Janorkar, FT-IR, spectroscopic analysis of the secondary structures present during the desiccation induced aggregation of elastin-like polypeptide on silica, ACS Omega, 2020, 5(14), 8403–8413 CrossRef CAS PubMed.
- A. Jabs, Determination of secondary structure in proteins by fourier transform infrared spectroscopy (FTIR), Jena Library Biol. Macromol., 2005, Available online: http://jenalib.leibniz-fli.de/ImgLibDoc/ftir/IMAGE_FTIR.html (accessed on 6 January 2025) Search PubMed.
- S. H. Surasarang, G. Florova, A. A. Komissarov, S. Shetty, S. Idell and R. O. Williams III, Formulation for a novel inhaled peptide therapeutic for idiopathic pulmonary fibrosis, Drug Dev. Ind. Pharm., 2018, 44(2), 184–198 CrossRef PubMed.
- M. Oeller, R. J. Kang, H. L. Bolt, A. L. Gomes dos Santos, A. L. Weinmann and A. Nikitidis, et al., Sequence-based prediction of the intrinsic solubility of peptides containing non-natural amino acids, Nat. Commun., 2023, 14(1), 7475 CrossRef PubMed.
- A. Falanga, R. Bellavita, S. Braccia and S. Galdiero, Hydrophobicity: The door to drug delivery, J. Pept. Sci., 2024, 30(5), e3558 CrossRef CAS PubMed.
- J. Renukuntla, A. D. Vadlapudi, A. Patel, S. H. Boddu and A. K. Mitra, Approaches for enhancing oral bioavailability of peptides and proteins, Int. J. Pharm., 2013, 447(1–2), 75–93 CrossRef CAS PubMed.
- A. Bak, D. Leung, S. E. Barrett, S. Forster, E. C. Minnihan and A. W. Leithead, et al., Physicochemical and formulation developability assessment for therapeutic peptide delivery—a primer, AAPS J., 2015, 17, 144–155 CrossRef CAS PubMed.
- M. T. Zafarani-Moattar and S. Hamzehzadeh, Salting-out effect, preferential exclusion, and phase separation in aqueous solutions of chaotropic water-miscible ionic liquids and kosmotropic salts: Effects of temperature, anions, and cations, J. Chem. Eng. Data, 2010, 55(4), 1598–1610 CrossRef CAS.
- M. I. Abd-Elrahman, M. O. Ahmed, S. M. Ahmed, T. aboul-Fadl and A. El-Shorbagi, Kinetics of solid state stability of glycine derivatives as a model for peptides using differential scanning calorimetry, Biophys. Chem., 2002, 97(2–3), 113–120 CrossRef CAS PubMed.
- P. Luo and R. L. Baldwin, Mechanism of helix induction by trifluoroethanol: a framework for extrapolating the helix-forming properties of peptides from trifluoroethanol/water mixtures back to water, Biochemistry, 1997, 36(27), 8413–8421 CrossRef CAS PubMed.
- P. Barth, J. Schonbrun and D. Baker, Toward high-resolution prediction and design of transmembrane helical protein structures, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(40), 15682–15687 CrossRef CAS PubMed.
- A. D. Ozkan, A. B. Tekinay, M. O. Guler and E. D. Tekin, Effects of temperature, pH and counterions on the stability of peptide amphiphile nanofiber structures, RSC Adv., 2016, 6(106), 104201–104214 RSC.
- B. B. Eedara, W. Alabsi, D. Encinas-Basurto, R. Polt and H. M. Mansour, Spray-dried inhalable powder formulations of therapeutic proteins and peptides, AAPS PharmSciTech, 2021, 22(5), 185 CrossRef CAS PubMed.
- F. Karavassili, A. Valmas, S. Fili, C. D. Georgiou and I. Margiolaki, In Quest for Improved Drugs against Diabetes: The Added Value of X-ray Powder Diffraction Methods, Biomolecules, 2017, 7(3), 63 CrossRef PubMed.
- R. Tuma, Raman spectroscopy of proteins: from peptides to large assemblies, J. Raman Spectrosc., 2005, 36(4), 307–319 CrossRef CAS.
- B. R. Singh, Basic Aspects of the Technique and Applications of Infrared Spectroscopy of Peptides and Proteins, Infrared Analysis of Peptides and Proteins, ACS Symposium Series 750, American Chemical Society, 1999, pp. 2–37.
- E. Goormaghtigh, J. M. Ruysschaert and V. Raussens, Evaluation of the information content in infrared spectra for protein secondary structure determination, Biophys. J., 2006, 90(8), 2946–2957 CrossRef CAS PubMed.
- O. N. Ogain, J. Li, L. Tajber, O. I. Corrigan and A. M. Healy, Particle engineering of materials for oral inhalation by dry powder inhalers. I-Particles of sugar excipients (trehalose and raffinose) for protein delivery, Int. J. Pharm., 2011, 405(1–2), 23–35 CrossRef CAS PubMed.
- S. Claus, C. Weiler, J. Schiewe and W. Friess, Optimization of the fine particle fraction of a lyophilized lysozyme formulation for dry powder inhalation, Pharm. Res., 2013, 30(6), 1698–1713 CrossRef CAS PubMed.
- F. S. Willard and K. W. Sloop, Physiology and emerging biochemistry of the glucagon–like peptide–1 receptor, J. Diabetes Res., 2012, 2012(1), 470851 Search PubMed.
- L. Zhao, Z. Cao, Y. Bian, G. Hu, J. Wang and Y. Zhou, Molecular dynamics simulations of human antimicrobial peptide LL-37 in model POPC and POPG lipid bilayers, Int. J. Mol. Sci., 2018, 19(4), 1186 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |