
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe rise of mRNA therapeutic vaccines†
Jinlu Du *ad,
Ziling Fanab,
Jiangming Huangab,
Zhengyuan Lic,
Hongguo Hua and
Yanxia Liab
*ad,
Ziling Fanab,
Jiangming Huangab,
Zhengyuan Lic,
Hongguo Hua and
Yanxia Liab
aYaoPharma Co, Ltd, No. 100, Xingguang Ave, Chongqing, 401121, China
bJisikai (Suzhou) Pharmaceutical Co. Ltd, No. 40 Suhong West Road, Suzhou Industrial Park, Jiangsu Province, China
cSchool of Chemical and Bioprocess Engineering, University College Dublin Belfield, Dublin 4, Ireland
dJnovapharma Co. Ltd, No. 5, Yongchang Middle Road, Daxing District Economic and Technology Development Zone Beijing, Beijing, 100176, China
First published on 23rd January 2025
Abstract
During the COVID-19 pandemic, messenger ribonucleic acid (mRNA) vaccines were developed and approved to curb the spread of coronavirus. After over 16 billion doses of the Pfizer-BioNTech-Fosun and Moderna mRNA vaccines were administered, the immune protection and clinical value of the lipid nanoparticle (LNP) platform were fully demonstrated. Herein, we provide a detailed overview of the mRNA–LNP structure and immunogenicity function and provide mechanistic insights into the ability of the LNP to elicit an immune response to combat diseases. The challenges and solutions to address these are discussed. Finally, by learning from the fast-growing and most recent advances in mRNA therapeutic vaccines, from both pre-clinical and clinical aspects, we can further expand the mRNA platform to develop a new generation of mRNA therapeutic vaccines, satisfying unmet medical needs beyond COVID-19.
Introduction
It is well known that mRNA-based vaccines are evolving rapidly. In 1990, mRNA-mediated gene transfer was first validated in vivo with functional reporter–protein expression after direct injection into mouse muscle.1 In 1992, an mRNA-mediated therapeutic product was developed by intrahypothalamic injection of vasopressin mRNA, and temporary reversal of diabetes insipidus was observed.2 In 1993, the influenza vaccine was developed by formulating mRNA encoding the influenza virus nucleoprotein with liposomes, and anti-influenza cytotoxic T lymphocytes were successfully induced.3 Intramuscular injection of antigen mRNA (human carcinoembryonic antigen) or DC vaccine, sensitized OVA mRNA, can protect mice challenged with antigen-expressing tumor cells.4,5 However, appropriate mRNA delivery technology was required for better delivery of the antigen and more efficient vaccination.In 2018, the FDA approved the first lipid nanoparticle (LNP) product, Onpattro™, for treating hereditary transthyretin amyloidosis.6,7 The LNP formulation used in Onpattro™ can deliver short interfering RNA (siRNA) to liver cells. This suggests that the clinical development of nucleic acid-based therapeutics is possible with suitable LNP delivery technology. During the COVID-19 pandemic, mRNA–LNP vaccines (Comirnaty™ and Spikevax™) were successfully developed at an unprecedented pace. These mRNA vaccines received emergency use authorization (EUA) in 2020 and full approval of a biologics license application (BLA) in 2021.8 The success of the mRNA vaccine against COVID-19 could not be accomplished without decades of research on mRNA–LNP, virology, and immunology.9,10 The mRNA vaccine emerged as technology to combat infectious diseases and cancer. Previous mRNA-related reviews focused on prophylactic vaccines, and therapeutic vaccine reviews had limited content on mRNA–LNP technology and its underlying mechanisms. So, in this review article, we describe the structure, function, and immune-stimulating properties of LNPs, and then summarize recent progress in mRNA therapeutic vaccine development.
Development of mRNA–LNP vaccines
LNPs: structure and function
Since canonical linear mRNA easily suffers from poor stability and a high innate immune response, currently approved linear mRNA vaccines utilize nucleotide modification like pseudo-uridine or N-1-methyuride in their products (modRNA).14 Some studies showed that the modified nucleotide increased the risk of ribosomal frameshifting and increased the mRNA therapeutics’ safety risks.15 Several new forms of mRNA have been designed to tackle mRNA stability and efficiency problems. For example, Son et al. reported an Additional Chimeric Element incorporated mRNA (ACE mRNA).16 A well-designed sequence was elongated to a traditional mRNA 3′ end poly A structure for hybridization with complementary DNA sequences. In several cell lines, this new form of unmodified mRNA showed similar expression levels to those of pseudo-uridine-modified mRNA for RFP, firefly luciferase, and Cas9 protein. Similarly, Tockary et al. developed a comb structure by hybridizing immunostimulatory double-stranded RNA (dsRNA) teeth with an mRNA sequence encoding target drug proteins. The therapeutic potential of comb-structured mRNA was evaluated in subcutaneous lymphoma mice expressing the OVA model antigen and subcutaneous B16F0 melanoma mice, and the tooth-structured mRNA reduced the tumor volume.17 Chen et al. also reported a new form of mRNA with chemical incorporation of branched poly A tails.18 The multi-poly A mRNA showed increased stability in cell lines with ∼4.7–19.5-fold higher luminescence signals than the control mRNA from 24 to 72 h post-transfection, and in vivo with a signal that was detectable for 14 d longer than that of traditional mRNA (less than 7 d). The modified poly(A) tail structure achieved efficient multiplexed genome editing of the clinically relevant genes Pcsk9 and Angptl3 in mouse liver with a minimal mRNA dosage. These new mRNA structures increased the diversity of mRNA therapeutics and showed promising results for cancer immunotherapy. However, given the complexity of the human immune system, more proof-of-concept experiments (especially in large animal models), safety-related evaluations, and CMC scaling-up and comparability studies data are still needed for these innovative mRNA structures as candidates for human cancer therapeutics.
Overview of mRNA–LNP drug product. As described, lipid nanoparticles (LNPs) have emerged as a highly effective platform for the delivery of mRNA therapeutics due to their stability, low toxicity, and relatively straightforward manufacturing process.19 Unlike traditional therapies, mRNA–LNP therapy utilizes the body's own cellular mechanisms to directly encode and produce target proteins, leading to therapeutic effects. This method provides a flexible and efficient way to treat various diseases, with particular potential for treating emerging diseases and in personalized cancer medicine. LNPs function as the principal delivery platform for mRNA, encapsulating and protecting mRNA from enzymatic degradation, and facilitating its delivery into target cells. The incorporation of four distinct lipids ensures the stability and efficacy of the drug product, facilitating its delivery and distribution. The ionizable lipids within the LNP play a pivotal role in the formation of complexes with negatively charged mRNA, ensuring both stability in circulation and efficient release in the environment of endosomes, where the mRNA is delivered into the cytoplasm.20 The structural integrity and membrane fusion capabilities of LNPs are enhanced by the contribution of cholesterol and phospholipids, collectively referred to as “helper lipids”. These lipids facilitate the interaction of LNPs with cell membranes and promote mRNA take up.21 Polyethylene glycol (PEG) lipids enhance the stability of nanoparticles and prolong the circulation time by preventing aggregation. However, they can also reduce cellular take up by limiting interactions with target tissues.22
The precise composition and structure of LNPs facilitate the maximization of mRNA drug product efficacy, ensuring the safe delivery of mRNA to target cells and the induction of therapeutic protein expression.
LNP structure and its impact on drug product. LNPs, as a vehicle for mRNA delivery, typically comprise four principal lipid components: ionizable lipids, cholesterol, phospholipids, and polyethylene glycol (PEG) lipids. Each of these plays a specific role in maintaining the structure of nanoparticles and facilitating mRNA delivery.23,24
1. Ionizable lipids
The term “liposome” was defined in 1965, when the self-assembling property of amphiphilic phospholipids in the aqueous phase was discovered.25 Cationic lipid DOTMA and DOTAP were used as the key components of the liposomes, known as RNA lipid particle aggregates (LPAs) or RNA-lipoplexes (RNA-LPX), in cancer mRNA vaccine development. In human trials, these RNA-carrying liposomes elicited strong and rapid immune responses in glioblastoma and pancreatic cancer.26–28 These cationic lipids contain a headgroup with permanent positive charges, while ionizable lipids have one or several ionizable headgroups with the property of pH-dependent ionization.29 In this section, we focus on ionizable lipids, LNPs and their functions in therapeutic vaccine development.
Ionizable lipids are of significant importance in mRNA–LNPs. The proportion of ionizable lipids in the total lipid content is 30–50%.29 The differing electronegativity of the various types of RNA will result in disparate electrostatic adsorption. Therefore, the ratio of this lipid must be adjusted to achieve optimal release of RNA. Ionizable lipids are electrically neutral under physiological conditions and can be rapidly cleared from the bloodstream to avoid causing an immune response. Ionizable lipids contains three fundamental structures: head group, linker, and hydrophobic tail.30 The head group is typically composed of simple tertiary amines and branched or cyclic compounds of polyamines including piperazine, diketopiperazine or benzene, which can be readily protonated under acidic conditions.31 A recent study showed that the amine headgroups of the ionizable lipids were linked to both innate and adaptive immune responses, via Toll-like receptor 4 and CD1d and facilitated lipid-raft formation.32 The linker substantially determines the biodegradability of the ionizable lipid; certain linkers, containing ester bonds, can be hydrolyzed to facilitate elimination.33 Furthermore, the tails of the organic chains are typically unsaturated, which can facilitate mRNA delivery. In an acidic environment, ionizable lipids become positively charged due to protonation, enabling them to interact with negatively charged mRNA through electrostatic forces. Following the replacement of the buffer by dialysis or tangential flow filtration, the mRNA–LNP also becomes electrically neutral. Upon injection of the mRNA–LNP, the formation of an acidic environment within the endosomes results in the protonation of the ionizable liposomes, which then bind to the negatively charged phospholipids present in the cell membrane, thereby destroying its structure and releasing the mRNA into the cytoplasm.34 Several ionizable lipids were successfully developed in the last two decades. The lipid MC3 was created based on prior research on DODMA and D-Lin-DMA.35 The MC3-based LNPs were developed to mediate potent hepatic gene silencing for RNAi therapeutics in 2012;35 MC3-based LNPs encapsulating siRNA, Onpattro™, were approved by the FDA and the EMA in 2018.35,36 Nevertheless, MC3-based LNPs can be used for therapeutic vaccine development in many preclinical studies.37–40 The ground-breaking work of MC3 promoted both ionizable lipid research and the development of LNP-based RNA therapeutics. In 2013, L319, a novel biodegradable lipid, outperformed the previous generation of ionizable MC3 lipid with substantially improved tolerability and potency in vivo. The biodegradability of the ionizable lipid was improved by the ester bonds in both the linker and lipidic tails.33 More biodegradable lipids, such as Moderna's lipid H(SM102) and Acuitas’ ALC0315, were screened and reported in 2019.41,42 And SM102 and ALC0315 were used in the mRNA–LNP vaccines Spikevax™ and Comirnaty™, respectively.8 In addition to biodegradability, another important factor of the ionizable lipid is pKa: pKa values in the range of 6.6–6.9 are ideal for IM, immunogenicity in mice;41 a pKa between 6.2 and 6.5 is suitable for siRNA delivery.35 For instance, the pKa of lipid 16(MC3) is 6.44,35 the apparent pKa values of SM-102 and ALC-0315 are determined to be 6.68, and 6.2, respectively.30 To improve the immunogenicity and delivery efficiency, the optimization and fine-tuning of the ionizable lipid pKa should be based on the indication of the therapeutic vaccine, mRNA coding the antigen, and targeted cells or tissues.43
Rational design, screening and validation of ionizable lipids are required for LNP development, because the ionizable lipid has a huge impact on mRNA–LNP immunogenicity.41 Novel ionizable lipid discovery and development with improved performance, such as higher transfection efficiency and immunogenicity, can be accelerated by combinatorial chemistry techniques and machine learning technology.44,45 Li et al. presented an approach for identifying effective ionizable lipids for mRNA delivery by integrating machine learning with advanced combinatorial chemistry. Using a simple four-component reaction platform, a foundational dataset was experimentally generated for machine learning model training. The transfection efficiency of a virtual library of 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 lipids was computationally predicted and the top 16 predicted candidates were experimentally validated. Finally, lipid 119-23 was identified, which outperformed established benchmarks in transfecting muscle and immune cells across multiple tissues.46 Similarly, researchers took advantage of machine learning for LNP lipid formulation optimization. Maharjan et al. assembled the XGBoost/Bayesian model to optimize the process and predict the lipid mixture ratio through which important critical product qualities were improved, including particle size, polydispersity index (PDI) and encapsulation efficiency.47 Recently, Witten et al. introduced a method utilizing a deep learning model, a directed message-passing neural network specifically, for the delivery efficacy prediction of lipids with diverse structures and two promising lipids with improved delivery efficiency were reported in this work.48
000 lipids was computationally predicted and the top 16 predicted candidates were experimentally validated. Finally, lipid 119-23 was identified, which outperformed established benchmarks in transfecting muscle and immune cells across multiple tissues.46 Similarly, researchers took advantage of machine learning for LNP lipid formulation optimization. Maharjan et al. assembled the XGBoost/Bayesian model to optimize the process and predict the lipid mixture ratio through which important critical product qualities were improved, including particle size, polydispersity index (PDI) and encapsulation efficiency.47 Recently, Witten et al. introduced a method utilizing a deep learning model, a directed message-passing neural network specifically, for the delivery efficacy prediction of lipids with diverse structures and two promising lipids with improved delivery efficiency were reported in this work.48
2. Phospholipids
Phospholipids, though only 10–20% of the total lipids in LNPs, are crucial for their function.29 They enhance phase-transition temperatures and form lipid bilayers.49 Typically, they have quaternary amine headgroups, which boost the proton sponge effect, and unsaturated tails, which can improve membrane fluidity, aiding endosomal escape. DSPC, a saturated phospholipid with a cylindrical shape, stabilizes lamellar phases but limits endosomal escape.31,49 Phospholipids play an essential role in enhancing the transport efficiency of LNPs. Moreover, there is scope for further optimization with regard to unsaturation and chain length.
3. PEGylated lipids
PEGylated lipids typically constitute 0.5% to 2.5% of the total lipid content, primarily enhancing particle stability and extending the circulation time in vivo.50 The PEG chains create a hydrophilic barrier that minimizes protein adsorption on the nanoparticle surface, thereby reducing recognition and clearance by the immune system, which prolongs the particle's half-life in the bloodstream.50,51 Furthermore, PEG facilitates the prevention of particle aggregation, thereby ensuring good dispersibility and uniformity. However, while PEG improves stability, excess amounts may impede membrane fusion and reduce the drug release efficiency. Modifying the PEG content enables control of the LNP particle size, with higher PEG concentrations generally producing smaller LNPs.51 Notably, LNPs with a particle size of approximately 65 nm exhibit superior absorption in vivo.52 Therefore, it is essential to balance stability and delivery efficiency during the formulation development process.
4. Cholesterol
Cholesterol typically comprises 20% to 50% of the total lipid content in LNP formulations.29,31 The incorporation of cholesterol into the lipid bilayer of nanoparticles enhances their stability during circulation in vivo.50 It was demonstrated that cholesterol concentration directly influenced the membrane's curvature and the phase transition of LNPs, thereby modulating their behavior within endosomes. This property facilitates the more efficient release of the drug within the target cells.53 Moreover, an elevated cholesterol concentration extends the half-life of LNPs in circulation and enhances the delivery efficiency by stimulating endocytosis and membrane fusion.54
Given the intrinsic liver-targeting properties of cholesterol, the identification of alternative lipids that enable targeted delivery to other tissues represents a promising strategy in the field of LNP research.
The structure of LNPs is primarily dependent on the lipid formulation. During the formulation process, lipids with varying compositions give rise to an internal structure that is disordered and inverted hexagonal, with a characteristic distance of approximately 6 nm. This structure is not observed in empty LNPs that lack mRNA. Typically, the core of an LNP is composed of ionizable liposomes, cholesterol, mRNA, and water, with a diameter of approximately 54 nm. The core is surrounded by a monolayer composed of lipids ∼2.4 nm thick. A second outer layer, ∼4 nm thick, corresponds to a PEG lipid layer in a mushroom-like configuration.52,55 Furthermore, alterations in the size and surface composition of LNPs are shown to influence protein expression significantly. For instance, Yanez Arteta et al. demonstrated that in adipocytes, protein expression levels could vary by up to 50-fold between adipose and liver cells when using 130 nm LNPs.52
Formulation and stability of drug product. The lipid composition of LNPs varies, influencing their performance, as seen in the two COVID-19 vaccines from Pfizer and Moderna, each with slightly different formulations. Pfizer's LNPs contain 46.3% ALC-0315 (ionizable lipid), 9.4% DSPC (phospholipid), 42.7% cholesterol, and 1.6% ALC-0159 (PEG-lipid). In contrast, Moderna's LNPs use 50% SM-102 (ionizable lipid), 10% DSPC, 38.5% cholesterol, and 1.5% PEG2000-DMG (PEG-lipid).56 Despite the small differences in PEG-lipid amounts, both vaccines achieve similar encapsulation efficiencies and immune responses.8 These differences contribute to the vaccines’ unique stability, delivery efficiency, and immune response characteristics.
The term “stability” in the context of LNPs is typically used to describe their physicochemical characteristics, such as size, charge, and encapsulation efficiency. LNPs interact with one another through a combination of hydrophobic and hydrophilic forces and electrostatic interactions, forming self-assembled structures. Although this process results in a tightly packed and robust formulation, the thermodynamically unfavorable state of these structures renders them inevitably prone to instability and eventual disintegration over time.57,58 Generally, two types of mechanisms have been identified for referring to LNP instability. The first is mechanical stress, which is shown to cause damage to the membrane of liposomes. The second mechanism involves a chemical transformation, which can alter the in vivo behavior of LNPs by impacting their loading efficiency and release kinetics.59 In addition, several common factors can lead to the leakage of LNPs, such as the hydrolysis and oxidation of phospholipids, the aggregation of LNPs, and the progressive permeabilization of their membranes. These issues undermine the structural integrity of LNPs, leading to their destabilization.19,60
Therefore, stabilizers are commonly added to enhance and prolong the stability and shelf life of LNPs. These include ethylene glycol and cryoprotectants such as sucrose, trehalose, and mannitol.61
LNP adjuvant effect and strategies to boost immunogenicity
LNPs are immunostimulatory and can be used as an adjuvant component for either mRNA vaccines or protein-based vaccines, with elevated Tfh cell and humoral responses. The empty LNP adjuvant outperformed the Addavax adjuvant in influenza HA protein-based vaccine development. It was found that only the empty LNP with an ionizable lipid possessed adjuvant properties, while empty DOTAP–LNP had no adjuvant effect. These data highlighted the importance of ionizable lipids in therapeutic mRNA vaccines.62 After intramuscular injection, only LNP-formulated mRNA can induce secondary lymphoid organ expression, which is recognized as the main driver for an adaptive immune response. Only mice treated with LNP-formulated mRNA, encoding the spike protein antigen, had strong cellular and humoral immune responses. In contrast, free spike-protein-encoding mRNA with/without co-administered adjuvanted LNPs had little immune response.37
Katalin and Weissman found that the TLR3, TLR7, and TLR8 signaling pathways could be triggered by a foreign RNA structure, and the incorporation of modified nucleosides, such as m5C, m6A, m5U, s2U, and pseudo-uridine, could minimize such an immune response. Nucleoside modifications suppress the potential RNA-DC activation by mimicking the endogenous mammalian total RNA, which is highly modified in nucleosides. The innate immune system detects foreign RNA via the nucleoside modification status. Specifically, the mRNA, probably through its secondary structure, proved to be an endogenous ligand and activator for TLR3, and the DC's response to mRNA maturation was inhibited by an antagonistic TLR3-specific antibody.64,65 The dsRNA impurity has a huge impact on immunogenicity; as previously discussed, the mRNA utilized in COVID-19 vaccines is deliberately engineered with various modifications to diminish its immunogenicity. Nucleoside-modified mRNA–LNP vaccines outperformed adjuvanted protein and inactivated virus vaccines and pathogen infection, with higher levels of Tfh and GC B cells, more robust polyfunctional antigen-specific CD4+ cell response, and neutralizing antibody production. The incorporation of noninflammatory, modified nucleotides is vital in mRNA therapeutic vaccine development.66
Antigen structures. Immunogenicity can be altered by adding certain gene-coding polypeptides with specific functions. Some signal peptides, such as endoplasmic reticulum translocation, secretion, transmembrane peptide, or polymerization signals, were introduced into the ORF.67 In previous protein-based respiratory syncytial virus vaccine development, the T4 fibritin-derived foldon trimerization domain was used in the structure-based design of a fusion glycoprotein antigen, and the stability and immunogenicity of antigen siteØ were improved.68,69 During the COVID-19 pandemic, the T4 fibrin-derived folded trimerization domain was also applied to promote immunogenicity and display of the spike RBD antigen in the BNT162b1 mRNA vaccine. A robust RBD-specific humoral immune response, CD4+ and CD8+ T cell responses, and favorable cytokine responses were induced by the BNT162b1 mRNA vaccine, according to the phase I/II data.10,67,70 Al-Wassiti's team found that the transmembrane domain and cytoplasmic tail of the spike protein were vital for immunogenicity and reactogenicity, and they proposed that antigens expressed in a membrane-anchored manner could be a critical determinant of the mRNA vaccine product development, especially for SARS-CoV2 and related mutant viruses. Regarding Omicron BA.1 and BA.5, mRNA vaccines developed via such an RBD-TM domain platform showed 12 and 22-fold higher neutralizing activity against the RBD targets than the corresponding whole-spike variants.71 Furthermore, Hendricks and colleagues computationally designed and optimized the RBD mRNA vaccine. They engineered the spike RBD antigen onto the exterior surface of the 60-subunit, icosahedral symmetric protein complex by fusing the secreting signal, RBD mutant gene Rpk9, and I3-01NS (nanoparticle gene) into one mRNA construct (Rpk9-I3-01NS). It was observed that such mRNA-launched protein nanoparticles were successfully translated, self-assembled, efficiently secreted, and possessed by the immune system. Delivering protein nanoparticle immunogens with mRNA vaccines has dual advantages: 5- to 28-fold higher levels of neutralizing antibodies were generated when compared with the membrane-anchored spike mRNA vaccine; a more robust CD8 T response was elicited when compared with an adjuvanted protein nanoparticle with the same immunogen.72
Particle size. Using in vivo imaging technology, two different LNP fates after intramuscular administration were compared and studied: DOG-IM4-LNP with a particle size of 184 nm and MC3 LNP with a particle size of 114 nm. It was found that mRNA from the DOG-IM4 LNPs persisted at the injection region, whereas mRNA from the MC3 LNPs quickly moved to the draining lymph nodes. Furthermore, MC3 LNPs induced the fastest increase in blood neutrophil counts after injection and more significant inflammation, as shown by IL-1RA, IL-15, CCL-1, and IL-6 concentrations in nonhuman primate sera. These observations highlight the influence of the nature of the LNP on mRNA vaccine distribution and early immune responses. The particle size of the LNP contributed to the different performances in LNP immunogenicity.73 The vaccine size does matter in antigen take up and processing. As a result of evolution, APCs can capture and process any antigen with a geometric structure (20–100 nm) similar to that of virus pathogens. So, manufacturing vaccines with proper sizes may enhance the antigens’ take up, processing, and presentation by APCs.74 Moderna did tremendous research on the relationship between LNP particle size and immunogenicity. The mRNA LNP particle size can be controlled by the manufacturing process rather than formulation compositions, as determined by many quality control methods, such as dynamic light scattering (DLS), nanoparticle tracking analysis (NTA), cryo-electron microscopy (cryo-EM), and small-angle X-ray scattering (SAXS). LNPs with a diameter of around 100 nm were most effective at eliciting immune responses in mice. Smaller diameter LNPs (64 nm) were less immunogenic in mice. Still, all particle sizes (60–150 nm) tested yielded robust immune responses in non-human primates, suggesting that the best LNP size for immunogenicity might differ between species.75 LNP particle size partially correlates with in vitro potency under certain conditions. The particle size was measured by DLS, and HepG2 cells were used for the RSV LNP vaccine potency assay. It was reported that when the LNP size increased, the cell-based potency decreased; 50% potency is lost if the LNP size is 130 nm.76 The vaccine containing mRNA–LNPs with a size range of 80–100 nm demonstrated the best stability and protection under storage conditions of both 4 °C and −20 °C. Smaller particles (60–80 nm) showed poorer stability, with increased particle size and an increase in polydispersity index (PDI) after storage, indicating potential aggregation or precipitation. The immunogenicity of mRNA–LNPs in mice showed that vaccines stored for up to 6 months maintained their bioactivity, with the 80–100 nm mRNA–LNPs showing the highest antibody titers and cytokine levels. This provides evidence that the size of LNPs affects the stability and immunogenicity of mRNA vaccines, emphasizing the importance of LNP size in vaccine design. The findings suggest that optimizing LNP size could improve the quality and effectiveness of mRNA vaccines, which could influence future vaccine development. Freezing the vaccine at −20 °C is more appropriate for maintaining long-term stability than storage at 4 °C. It is demonstrated that the vaccine containing 80–100 nm mRNA–LNPs showed the best stability and protection at 4 °C and −20 °C. Freezing the vaccine at −20 °C is more appropriate for maintaining stability in the long term. These efforts are poised to provide a scientific basis for improving the quality of ongoing mRNA vaccine endeavors and providing information for developing novel products.77 The particle size of ALC0315-formulated LNPs can be fine-tuned by adjusting the aqueous to lipid phase ratios. Furthermore, LNPs with different size ranges showed different in vitro and in vivo expression efficacies. LNPs, prepared with the lowest phase ratio, show increased particle size (70 nm to 140 nm) and reduced expression levels. LNP particle size affects the cargo expressed in vivo and thus impacts the immunogenicity.78
Injection routes. Injection routes are another important factor affecting immunogenicity since different administration routes cause different immunogenic outcomes. The in vivo fate of LNPs in mice was reported in 2016. LNP-encapsulated mRNA can induce protein production at the injection site for up to 10 days via subcutaneous, intramuscular, and intradermal injection routes.79 For therapeutic mRNA vaccine development, the LPX vaccine was optimized to be injected intravenously (IV),26,80 while LNP vaccines, such as mRNA-4157, were injected intramuscularly.22 In developing H10N8 and H7N9 mRNA, intramuscular and intradermal injection were compared. In the low dosage group (25 μg), the ID (intradermal) injection route induced HAI titers >1
:40 in 64.7% of participants compared to 34.5% of participants of the IM injection routine. In contrast, in the high dosage group (100 μg), the IM route led to seroconversion rates of 100% in the induced HAI titers ≥1:40. The ID injection route was reported to be highly related to solicited adverse events.81 In the mouse model, it was found that a subcutaneous (s.c.) injection routine could reduce the pro-inflammatory response rate and related systemic AEs, compared to an intramuscular injection routine. Changing the administration routine is possible to improve post-vaccination fatigue and other systemic adverse events. Since IM is widely accepted and easy to administer, IM injection is the most preferred administration route for the LNP vaccine.82
IL12 is a frequently studied cytokine used in mRNA therapeutic vaccines. The delivery of long-lasting IL-2 mRNA can restore immune cell infiltration, IFNγ induction, and transition of the highly proinflammatory TME signature, thus overcoming the immune desertification and therapeutic resistance caused by MHC class I loss of tumor cells. When IL-2 mRNA treatment was performed, antigen presentation proficiency and other M1-phenotype-associated features were changed so that both TME resident macrophages and correlated neoantigens targeting CD8+ T cells were facilitated to improve the effectiveness of the treatment.83 It was observed that co-delivering IL-12 and OVA mRNA could elevate OVA specific CD8+ T proliferation and the effector function and promote the expansion of the memory CD8+ T population. The improved CD8+ T cell-mediated protective response via IL-12 mRNA was proved in Listeria monocyte-OVA and B16 F0-OVA melanoma models.84 Brook et al. evaluated the adjuvant LNP encapsulating interleukin-12p70 mRNA with a multiorgan protection (MOP) sequence. Admixing IL-12-MOP (CTX-1796) with the BNT162b2 SARS-CoV-2 vaccine can amplify both humoral and cellular immunity, prolong vaccination protection (over 1 year in mice), restore immunity in aged mice, and enhance the vaccine-derived DC and GC responses.85 Another new lipid nanoparticle (DMT7-IL12 LNP), encapsulating IL12 mRNA, was evaluated with a STING agonist MSA2. It was observed that T cell function was restored, and the exhausted T cell phenotype was reversed. More importantly, the preclinical efficacy and safety were proved in melanoma and lung metastasis models.86
In addition, many cytokines have been shown to be effective vaccine adjuvants. A natural adjuvant derived from the C3 complement protein was developed by fusing the C3 gene with an antigen. It was found that the fusion of the C3d mRNA sequence with the antigen sequence into a single transcript improved the mRNA vaccine by generating a higher level of antibodies in mice at a relatively low dosage.39 To promote the efficacy of the mRNA vaccine in prostate cancer, ImmunER (immune-enhancing adjuvant) was co-delivered with an mRNA TAA vaccine called Tetra. ImmunER, consisting of 4-1BBL, OX40L, and CCR7 coding mRNA, can promote dendritic cell maturation and migration (as indicated by upregulated CD80 CD86 and MHCII) and improve antigen presentation at both the cellular and animal levels (as increased CD8+ T cell infiltration and activation in RM-1-PSMA tumor tissues). It was found that a combination of TAA mRNA vaccine and ImmunER could enhance the specific T cell cytotoxicity required for tumor elimination.87 Zhivaki et al. reported that the immunostimulatory effect of mRNA-encoded antigens targeted to DCs could be further elevated via cGASΔN LNPs. cGASΔN-LNP, as an adjuvant, can produce cyclic dinucleotide cGAMP, which binds the protein STING and activates the innate immune response. The enzyme can facilitate the antibody response to antigen-LNP biased toward the type I isotype. As a catalytic adjuvant, the active mutant of the enzyme cGAS, cGASΔN, can induce durable antigen-specific IFNγ-producing T cells and Th1-biased antibody isotypes.88 In developing the HPV mHTV-02 mRNA vaccine, Flt3LI (extracellular domain of Fms-like tyrosine kinase-3 ligand) was fused with the tumor antigen ORF. The induction of Flt3L can facilitate both antigen presentation and intratumoral DC infiltration.89 Similarly, in the development of the mRNA vaccine for malaria, macrophage inflammatory protein 3 alpha (MIP3α) was fused with the circumsporozoite protein (CSP) malaria antigen. Such fusion mRNA was engineered to enhance PfCSP antigen presentation via immature dendritic cells (iDC) and elicited a robust CD4+ T cell response. Another advantage of the CSP-MIP3α fusion vaccine is that it provides better protection against liver infection when challenged with P. berghei PfCSP transgenic sporozoites, considering that the liver is an organ with specific immune tolerance properties. Such a MIP3α adjuvant fusion mRNA vaccine generates significantly greater TNF, IL2, 426, and IFN responses in CD4+ T cells.90,91 Researchers from Moderna reported that a constitutively active mutation (V155M) of the stimulator of interferon (IFN) genes (STING), as a genetic adjuvant, could effectively induce CD8+ T cell responses at specific antigen/adjuvant mass ratio. The adjuvant LNP encapsulating STINGV1155M mRNA enhanced the type I IFN responses via the nuclear factor κB (NF-κB) and IFN-stimulated response element (ISRE) pathways, resulting in efficient antigen-specific T cell responses. More importantly, the adjuvant effect of mRNA-encoded STINGV155M LNPs was proved in the in vivo assay with the HPV E6 E7 mRNA vaccine.92
Researchers from the Mitchell lab developed an innovative approach for enhancing the immune response of mRNA vaccines. The research demonstrates that the incorporation of an adjuvant lipidoid into lipid nanoparticles (LNPs) used in mRNA vaccines significantly boosts the immune response. This adjuvant lipidoid enhances the delivery of mRNA and imparts Toll-like receptor 7/8 (TLR7/8) agonistic activity to the LNPs, leading to increased innate immunity in mice. The optimized vaccine formulation with adjuvant lipidoid substitution elicited potent neutralizing antibodies against multiple SARS-CoV-2 pseudovirus variants. It also stimulated strong Th1-biased cellular immunity, robust B cell responses, and long-lived plasma cell responses. The researchers developed a novel component for LNPs. This adjuvant lipidoid improves mRNA delivery and serves as a functional moiety to increase the adjuvanticity of LNPs. TLR7/8 agonistic activity: the adjuvant lipidoid was designed to have TLR7/8 agonistic properties, which are known to activate dendritic cells and promote a robust adaptive immune response. The substitution of a standard lipidoid with an adjuvant lipidoid in LNPs enhanced mRNA delivery and expression in lymph nodes and other immune-relevant organs, which are critical for initiating an effective immune response. The study suggests that the adjuvant lipidoid may enhance the endosomal escape of LNPs, facilitating the release of mRNA into the cytoplasm and subsequent translation into the antigen. The adjuvant effect of the lipidoid is synergistic with the use of nucleoside-modified mRNA, which, while reducing innate immune responses, is essential for increased translational capacity and biological stability of the mRNA. The study's findings significantly contribute to mRNA vaccine development by offering a strategy to enhance the potency and effectiveness of vaccines against infectious diseases like COVID-19.38 After screening a library of 480 biodegradable ionizable lipids, researchers discovered lipid 331 with a dose-dependent immunostimulatory effect. In mice, intramuscular or intranasal administration of LNPs with lipid 331 and C3 adjuvant with the antigen protein (either the spike protein or the receptor-binding domain of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)) can increase the titers of antibodies against SARS-CoV-2 tenfold.39
Gu's team reported that the incorporation of Pam2Cys, a Toll-like receptor 2/6 (TLR2/6) agonist, into mRNA vaccines significantly enhanced their efficacy against both cancer and infectious diseases. Pam2Cys was identified as an effective adjuvant due to its ability to signal through the TLR2/6 pathway, which triggers both humoral and cellular adaptive immune responses. Its lipophilic nature enables it to be easily incorporated into lipid nanoparticles (LNPs) used in mRNA vaccines without additional chemical modifications. The inclusion of Pam2Cys in mRNA–LNPs leads to a robust immune response in the draining lymph nodes, as characterized by the induction of cytokines such as IL-12 and IL-17, which are critical for Th1 and Th17 immune responses, respectively. Preliminary safety profiles of these new vaccines in murine models show no significant adverse effects, suggesting their potential for clinical application. The addition of Pam2Cys to mRNA vaccines, LNP formation specifically, can significantly boost their effectiveness against diseases by enhancing immune responses and this has the potential to improve the outcomes of cancer and infectious disease treatments.93 mRNA vaccines are more complicated and less understood than protein vaccines. For instance, TLR4 activation can inhibit mRNA translation without affecting LNP take up; in contrast, TLR4 inhibition or downregulation of the TLR4 downstream effector PKR can improve LNP delivery. The TLR4 pathway has a discrepant impact on LNP take up and translation, as elevated immune activation and cytokine over-production can prevent efficient mRNA translation. It is important to consider the effect of the adjuvant and potential adjuvant–LNP interactions.94
A new delivery vehicle called charge-altering releasable transporters (CARTs) was developed with inherently nonimmunogenic characteristics. Further improvement of the vaccine immunogenicity was achieved with co-formulated adjuvants, such as oligodeoxynucleotides with CpG motifs (CpG-ODN). The mice vaccination data proved that CpG-CART generated more RBD-neutralizing antibodies in the circulation and lung bronchial fluids. Furthermore, CpG-CART vaccination elicited strong and long-lasting RBD-specificTH1 T cell responses, including CD4+ and CD8+ T cell memory.95 The efficient activation of antigen-presenting cells—such as dendritic cells (DCs)—in tumors and lymph nodes is critical for the design of next-generation cancer vaccines and may be able to provide anti-tumor effects by itself through immune stimulation. The challenge is to stimulate these cells without causing excessive toxicity. It is hypothesized that a multi-pronged combinatorial approach to DC stimulation would enable dose reductions of innate immune receptor-stimulating TLR3 agonists while enhancing drug efficacy. Here, a hybrid lipid nanoparticle (LNP) platform is developed and tested for double-stranded RNA (polyinosinic:polycytidylic acid for TLR3 agonism) and immune modulator (L-CANDI) delivery. This study shows that the ≈120 nm hybrid nanoparticles-in-nanoparticles effectively eradicate tumors and generate long-lasting, durable anti-tumor immunity in mouse models.96 Although various mRNA-based vaccines have been explored, the optimal conditions for the induction of both humoral and cellular immunity remain unknown. This study evaluated mRNA vaccines of nucleoside-modified mRNA in lipoplexes (LPXs) or lipid nanoparticles (LNPs) after administration in mice through different routes, assessing mRNA delivery, tolerability, and immunogenicity. In addition, they investigated whether mRNA vaccines could benefit from incorporating the adjuvant alpha-galactosylceramide (αGC), an invariant natural killer T (iNKT) cell ligand. Intramuscular (IM) vaccination with ovalbumin (OVA)-encoding mRNA encapsulated in LNPs adjuvanted with αGC showed the highest antibody- and CD8+ T cell responses. Furthermore, they observed that signal peptides and endocytic sorting signals of either LAMP1 or HLAB7 in the OVA-encoding mRNA sequence further enhanced CD8+ T cell activation, reducing the induction of IgG antibody responses. Moreover, mRNA LNPs with the ionizable lipidoid C12–200 exhibited higher pro-inflammatory- and reactogenic activity than mRNA LNPs with SM-102, correlating with increased T cell activation and antitumor potential. It was also observed that αGC could further enhance the cellular immunity of clinically relevant mRNA LNP vaccines, promoting therapeutic antitumor potential. Finally, a Listeria monocytogenes mRNA LNP vaccine supplemented with αGC showed synergistic protective effects against listeriosis, highlighting a key advantage of co-activating iNKT cells in antibacterial mRNA vaccines. Taken together, those studies offered multiple insights for optimizing the design of mRNA vaccines for disease applications, such as cancer and intracellular bacterial infections (Table 1).97
| Indication | COVID | COVID & tumor | COVID & tumor | Tumor | Tumor & Listeria monocytogenes |
|---|---|---|---|---|---|
| Antigen | SARS-CoV-2 mRNA | SARS-CoV-2 mRNA OVA | OVA; CT26 neoantigen EGFP; COVID spike | NA | OVA Lmon0149 |
| Antigen fusion domain | NA | C3 | HLA-A, HLA-B T&CD | NA | Signal peptide\MITD\LAMP1 |
| Ionizable lipid | SM-102, MC3 | ALC0315 MC3 lipid331 | ALC0315 | C12-200 | C12–200, SM102 |
| Formulation | LNP | LNP | LNP | LNP | LNP |
| Adjuvant | C12-TLRa | Lipid 331 | Pam2Cys | Poly I:C | αGC |
| Ref. | 38 | 39 | 93 | 96 | 97 |
Machine learning related mRNA product development
The mRNA sequence can be divided into UTR regions, including 5′UTR and 3′UTR, and the ORF (open reading frame) region.98 UTR regions are located upstream and downstream of the ORF and control mRNA translation rates. The ORF region determines the sequence of translated protein, and this region is the main body of a mRNA molecule, thus greatly influencing the structure of the mRNA sequence. Due to the enormous design space, computational methods have been exploited in mRNA sequence design and optimization.Computational UTR design to improve vaccine properties is one of the active fields in vaccine development. In mRNA-based therapies, a commonly adopted approach has been the use of naturally occurring UTRs with high expression capability derived from human beings or other eukaryotic organisms.99 There has recently been a shift toward designing novel UTR elements with improved characteristics compared to their natural sequences. With the rapid development of high-throughput experimental reporter techniques and next-generation sequencing (NGS), researchers can rapidly generate enough data for machine learning model training and explore the massive design space.100,101 This model can simulate UTR behavior with extra wet experiments and predict their effects on mRNA translation, improving the efficacy of mRNA-based therapies to the next level. These innovations have increased the potency of mRNA therapies and helped identify the most effective UTR components from alternative sources. For example, Castillo-Hair trained a convolutional neural network (CNN) model with a natural UTR sequence with several modifications for ribosome loading quantification, which further represented translation efficiency.102 Gong et al. took it one step further when they also considered the ORF sequence when a neural network model was trained for UTR optimization.103 Applying genetic algorithms and diverse prognostic models inspired by natural selection has further advanced the design of 5′UTRs. Sample et al. combined the deep learning model with a genetic algorithm, and this method could be used to engineer new 5′UTR directing specified levels of ribosome loading. This precise control of translation efficiency enabled the tuning of sequences for optimal protein expression.104 Moreover, recent studies used deep generative models to create novel and improved regulatory sequence elements with similar patterns to those of existing sequences. In one study, researchers trained a GAN on natural genomic and transcriptomic data.105 The training sequence traversed the entire DNA regulatory region, including UTRs, promoters, and terminators, and produced de novo functional regulatory DNA that outperformed highly expressed natural controls in Saccharomyces cerevisiae. This comprehensive approach highlights the importance of considering interactions between regulatory elements and coding sequences, which is also relevant for mRNA-based therapies where synthetic UTRs must be designed by coding sequences and other regulatory mRNA components like the poly(A) tail.
The ORF region determines the downstream translated protein sequence and largely influences an mRNA molecule's secondary structure, further affecting drug efficacy.106 The codon degeneracy phenomenon and different abundancy of tRNA in different organisms necessitate codon optimization for desired biological properties, including translation elongation, efficiency, fidelity and mRNA stability.107–109 Various computational strategies have been developed to enhance these properties, primarily focusing on substituting synonymous codons in the open reading frame sequence. One commonly used methodology involves replacing rare codons in the sequence with synonymous codons with higher abundance, which does not change the amino acid sequence. For example, the codon adaptation index (CAI), has been proposed to compute codon optimality or translational fitness. The concept of the CAI was proposed by Sharp et al. and it measured codon optimality based on the geometric mean of relative codon frequencies calculated from highly expressed genes in a specific organism.110 Codon bias observed in these genes informs this measure, and maximum CAI values can be achieved by selecting the most frequent codons for each amino acid. Approved mRNA vaccines, BNT-162b2 from BioNTech and mRNA-1273 from Moderna, are found to focus on optimizing their mRNA sequences based on the CAI and exhibit significantly higher CAI values (>0.9) compared to wild-type antigen sequences.111 Beyond classic measures of codon usage, research has revealed that codon composition impacts mRNA stability, with studies demonstrating that the codon content is a general determinant of mRNA decay rates across species. For example, Presnyak et al. proposed the codon stabilization coefficient (CSC), which correlated codon frequency with mRNA half-life data to predict stability.109 Additionally, the increased usage of the GC replacement of AT and decreased uridine usage are always preferred in mRNA sequence design to enhance mRNA stability and decrease immunogenicity.88,109
RNA structure is another critical factor influencing molecular stability and its degradation in living organisms. RNA sequences translating the same protein sequence can exhibit vastly different secondary structures due to differences in nucleic acid sequence affecting molecular in vivo stability. Metrics like minimum free energy (MFE) are commonly used to evaluate mRNA stability, and several computational methods have been proposed to design RNA sequences of optimal secondary structures with minimal MFE. For instance, Terai et al. developed the CDSfold program to find the RNA sequence with the most stable structure using a dynamic programming algorithm in only cubic time of the sequence.112 CDSfold optimized the computing time by applying amino acid constraints in the sequence for comparison with the Zuker algorithm through a similar methodology.113 Another stability metric, average unpaired probability (AUP), measures the likelihood of unpaired nucleotides in a sequence, and algorithms like RiboTree have been developed to minimize AUP.114
The challenge of optimizing mRNA sequences for vaccines is compounded when considering multiple objectives, such as codon usage, structural stability, and GC enrichment. Multi-objective optimization algorithms have proved to be effective at designing synthetic RNA sequences with enhanced functionality. Deterministic algorithms like LinearDesign, which employ finite-state automata, offer a promising alternative by simultaneously optimizing multiple objectives.115 Moreover, more complete neural network models, more suitable for solving a muti-module problem compared to the abovementioned simple model, have also been investigated for the problem of optimizing more than one objective.116,117
To maintain the molecule's integrity and deliver it the target organ, mRNA molecules must be properly encapsulated as the final drug product. In vaccine development and optimization, aside from the mRNA sequence, LNP formulation and the structure of the ionizable lipids are two primary aspects researchers focus on to improve mRNA vaccine efficacy. Current research attempts to apply machine learning to facilitate LNP formulation optimization. Wang et al. collected more than 300 samples with both formulation composition and animal in vivo IgG data and trained a predictive model through the lightGBM method. The evaluation results showed that this method was valuable for accelerating the formulation optimization process.118 Ionizable lipids play a vital role in targeting the LNP drug and cellular uptake efficiency.20 Xu et al. developed AGILE combining combinatorial chemistry and deep learning. Ionizable lipids selected by AGILE showed expected cell-specific preferences that addressed the complex needs of mRNA delivery in clinical practice.45
CMC consideration of mRNA therapeutic vaccine development
Manufacturing
The production of mRNA vaccines is significantly more streamlined than traditional vaccines, as it bypasses the need for culturing cells or viruses. Their production relies on in vitro synthesis technology, eliminating the time-consuming steps required for conventional vaccine manufacturing and accelerating production and scalability.119Key steps in conventional linear mRNA vaccine production include (a) preparation of the DNA template of the antigen sequence for in vitro transcription (IVT); (b) IVT reaction for mRNA synthesis and mRNA purification; (c) LNP formulation and encapsulation; and (d) sterile filtration and filling.119 The process begins with the design of the target antigen sequence, which is optimized and inserted into a plasmid backbone designed for suitable copy number DNA replication and compatible for the sequential IVT reaction for mRNA production. The plasmid is then propagated in bacteria to produce a certain amount of plasmid for sequential mRNA production. Plasmid DNA is extracted and purified as the template for IVT, and restriction enzymes are usually used in the linearization step. This DNA serves as a template for the IVT process and mRNA molecules are synthesized from the T7 promoter. In IVT, the DNA template and necessary components like NTPs, ribonuclease inhibitors, and other cofactors produce the desired mRNA molecules. The reaction lasts 3–5 h, yielding target quantities of mRNA based on the amount of original DNA template and the abovementioned reaction components.
The final mRNA product is often 5′ end capped, and this capping is essential for evading immune detection and facilitating protein translation.120 The Cap 1 structure is an advancement on the Cap 0 structure, where further methylation at the 2′-O position improves translation efficiency and helps avoid innate immune responses.121 Two primary methods are used for capping mRNA in vitro: co-transcriptional capping and enzymatic capping. In co-transcriptional capping, the cap structure is added during transcription, as seen with technologies like CleanCap, which offers the advantage of being a one-step process with a capping efficiency of around 94%.120 However, this method requires expensive cap analogs and incomplete capping may occur, necessitating additional processing of uncapped RNA with alkaline phosphatase to prevent immune detection. Moreover, the DNA template may need modifications for optimal performance. On the other hand, enzymatic capping is a two-step process where the vaccinia capping enzyme first forms the Cap 0 structure, which is then converted into Cap 1 by 2′-O-methyltransferase.120 While this method ensures a high capping efficiency, it is more complex and time-consuming, requiring multiple buffer exchanges. Additionally, secondary structures at the RNA's 5′ end can hinder the efficiency of the enzymatic process.
After transcription and capping, mRNA undergoes a purification process to eliminate impurities such as buffers, proteins, DNA templates, dsRNA, and short RNA fragments.122 This purification involves several steps, including DNase I digestion to break down DNA templates, oligo dT affinity chromatography to capture full-length mRNA molecules with poly-A tails and eliminate undesired impurities, such as dsRNA, which is a side product of the IVT reaction, tangential flow filtration (TFF) to filter out small molecules, and sterile filtration to eliminate particulate contaminants. More purification steps may be added to improve purity, such as cellulose chromatography, anion exchange chromatography, and hydrogen bond chromatography.123
Naked mRNA molecules are degraded easily once introduced into the body. Therefore, a specifically designed delivery system is required for drug administration.124 The production of lipid nanoparticles (LNPs) for mRNA delivery begins by preparing a mixture of four essential lipids: ionizable lipid, DSPC, cholesterol, and PEG-lipid, each dissolved in ethanol at specific concentrations. Simultaneously, mRNA is diluted in a buffer solution at a pH of approximately 4 to form the aqueous phase. This pH environment maintains a desirable static charge for mRNA molecules to interact with lipid molecules and maximize encapsulation efficiency.125 The lipid solution and mRNA are then mixed in a microfluidic or T-junction channel, where the ionizable lipid, which is positively charged in the acidic environment (pH 5.5), binds electrostatically to the negatively charged mRNA. This interaction promotes the formation of lipid vesicles, which encapsulate the mRNA.125 Recently, advanced stainless-steel crossflow membrane micromixing technology was developed for LNP preparation with comparable performance to that of microfluidic and jet mixers. Jet mixers and membrane-based production systems provide better scalability and reusability.126 Following this, dilution and ultrafiltration are carried out to remove ethanol and replace the buffer, raising the pH and causing the ionizable lipid to become hydrophobic and uncharged.127 The resulting stable mRNA–LNP spherical complex is filtered and prepared for final formulation through sterile filtration and filling.
Analytical methods
Despite the inspiring performance of mRNA drugs, the analytical and quality control strategies for mRNA as a therapeutic are still under development. The high molecular weight of the mRNA sequence, high heterogenicity, poor stability, and lack of industrial standards hinder the further development of mRNA therapeutics. Meanwhile, innate immunity also requires consideration for mRNA product-related and process-related impurities.From an industrial manufacturing point of view, the production of mRNA drug substances and drug product formulation are complicated compared with traditional protein drugs. Thus, a robust and effective analytical strategy is vital to ensure better safety and quality of the final mRNA drug products. The United States Pharmacopeia (USP) has released three quality draft editions on analytical procedures for the quality of mRNA vaccines and therapeutics (https://www.usp.org/mrna-quality). The drafts aim to share understanding and provide comprehensive lists of analytical methods and procedures for mRNA drug quality evaluation from the pharmaceutical points of view. Typical pharmaceutical quality attributes include the identities of the drug substance and drug product, drug content determination, molecular integrity, purity, structure, bioactivity, safety (such as bioburden, endotoxin, and sterility), and so on. Compared with the two previous editions, the latest version incorporated more impurity control considerations such as residual T7 polymerase activity by ELISA, free nucleotide determination by LC-MS/MS, and aggregates analysis by SEC-HPLC. It can be concluded that the future direction of mRNA therapeutics will focus on completing mRNA-based drug impurity analysis.
mRNA therapeutics remain a broad research area and are being developed rapidly. This review mainly focuses on progress made for several critical quality attributes related to identity, purity or integrity, and impurities.
In addition to traditional DNA/mRNA sequencing, several other works have been published on LC/MSMS-based mRNA drug sequencing.129,130 By dedicated experimental controls, batch-to-batch consistency analysis can be achieved for large mRNA molecules (more than 1000 nucleotides). The reported quantitation ability of LC-MS/MS techniques can be lower as 1% for mRNA sequence variants such as single nucleotide polymorphisms (SNPs), if the oligonucleotide containing the SNP is unique. This method also shows potential for the rapid screening of possible modifications of mRNA molecules. Partial RNase digestions using immobilized RNase T1 were used for mRNA digestion followed by high-resolution LC-MS/MS. However, large mRNA therapeutics showed more duplicate subsequences compared with proteins, making oligonucleotide mapping more challenging than peptide mapping in method reproducibility. Since Sanger sequencing or NGS testing are usually outsourced due to high instrument costs, quality control for such outsourced analytical data is more challenging than in-house analysis. From this respect, the LC-MS/MS-based method shows some advantages for pharmaceutical companies once the sequencing method is stable and the workflow is more simplified.
In addition to mRNA drug substances, the formulation process and control of co-formulated components are essential for maintaining good therapeutic performance, as these might modify and lead to the malfunction of mRNA molecules under improper storage conditions. Packer et al. reported that reversed-phase ion-pair high-performance liquid chromatography (RP-IP HPLC) and mass spectrometry were used to identify a class of impurity formed through lipid–mRNA reactions; such reactions are typically undetectable by traditional analytical techniques for determining mRNA purity. The impurities include the product of the oxidation and subsequent hydrolysis of the tertiary amine.124
Current status of mRNA therapeutic vaccine development
Virus-related therapeutic vaccines
000 CHB-related deaths reported, according to the 2019 World Health Organization (WHO) report.134 In most high-risk HCC areas, such as China and South Korea, the key determinants include chronic HBV infection.135 In addition to an HBV prophylactic vaccine,136 currently licensed therapies against HBV are nucleoside analogs (NAs) and pegylated interferons (Peg-IFNs) to suppress viral replication. Unfortunately, neither of these two widely used therapies has a significant effect on the covalently closed circular DNA (cccDNA) of the viral genome responsible for viral persistence, and thus, life-long treatment is required.137The integration of HBV DNA, previously recognized as a byproduct of the HBV life cycle with an unclear function, is now recognized as a pivotal element in the pathogenesis of HBV. The integrated DNA can sustain HBsAg levels even without HBV virus replication, and the secreted HBsAg further promotes HBV pathogenesis by disturbing the host's immune response as a result of complicated virus–host interactions during evolution. Meanwhile, the escalated DNA integration frequency can activate oncogenes and growth-promoting genes, which can cause tumor progression.138 Current clinically available agents cannot eradicate HBV from the CHB patients, and it was reported that the HBV genome might be persistently retained in infected liver cells among those patients who recovered from previous acute or chronic HBV infections.139 LNP technology can be applied to cure HBV. One strategy is to deliver mRNA coding therapeutic products, such as IL2, IL21, or anti-HBsAg antibody140–142 or editing genes (CAS9, ARCUS nuclease, CAS13b, CBE) to the liver cells for HBV treatment.140,143–146 In previous reports, such strategies proved to be potential cures for CHB infection in pre-clinical stages, by triggering strong HBV-specific CD4+ and CD8+ T cell responses,141 a reduction in the serum HBsAg level,142,143,147 or even a decreased cccDNA level.140,143,148 To achieve therapeutic goals safely, the LNPs’ immunogenicity must be well controlled. Such LNPs can be developed in a re-dosing, relatively high dosage, target-specific-cell, and transient expression manner.144
One alternative strategy is to boost the CHB patients’ immune response against HBV via a therapeutic mRNA vaccine, as explained in the following section. mRNA COVID vaccine can induce neutralizing antibodies and a poly-specific T cell response in humans;14 the HBV mRNA vaccine may induce similar responses against HBV in CHB patients with proper medical management. In 2016, Merck-proprietary empty LNPs, in combination with IMO-2125, were used as adjuvants to enhance immune responses to the hepatitis B virus surface antigen. Such LNPs were recognized as a potential sub-unit vaccine adjuvant playing an important role in boosting both B-cell and T-cell responses to the therapeutic HBV vaccine.149 In 2017, researchers from South Africa developed an mRNA therapeutic vaccine for HBV prevention and treatment. They prepared LHB and SHB mRNA and tested them in transfected cells. The in vitro data showed that LHB and SHB proteins were produced; the former was retained intracellularly while the SHBs were secreted.150
The breakthrough COVID-19 vaccines ignited the development of mRNA therapeutic vaccines, especially the HBV vaccine. In 2024, researchers from the China Pharmaceutical University reported a mRNA vaccination for CHB treatment. Their mRNA vaccine demonstrated potent therapeutic efficacy in two different mouse models of chronic hepatitis B (CHB), showing efficient and persistent virologic suppression. The vaccine induced a robust immune response, including strong innate immune activation, high-level virus-specific antibodies, memory B cells, and T cells. The vaccine provided full protection against subsequent viral re-exposure and maintained virologic suppression for an extended period, suggesting the potential for long-term immunity. The vaccine showed limited cytotoxicity and no hepatotoxicity or liver injury, indicating its safety for use. It was the first attempt to use an mRNA platform as a therapeutic vaccine against CHB; this approach is innovative, as mRNA vaccines have shown superior immunogenicity compared to other types of vaccines. The vaccine utilizes an artificial intelligence-based algorithm to design mRNA with optimal folding stability and codon usage, contributing to a high translation efficiency. The study suggests that the mRNA vaccine may function through a non-cytotoxic mechanism to eliminate the virus, which significantly advances vaccine development to avoid liver damage.151 Based on their previous extensive experience with HBV therapeutic vaccine research, researchers from Johnson & Johnson and the University of Antwerp developed an HBV mRNA therapeutic vaccine. According to the AAV HBV mouse model data, the mRNA vaccine, formulated in lipid nanoparticles (LNPs), demonstrated immunogenicity by inducing strong antigen-specific polyfunctional T cell responses in mouse models, along with the production of anti-HBs and anti-HBe antibodies. After three immunizations, the vaccine induced a significant reduction in systemic HBV surface antigen (HBsAg) levels, achieving up to 1.7log10 IU mL−1 reduction in 50% of AAV-HBV-transduced mice without additional adjuvants or HBsAg reducing agents. A transient drop in systemic HBeAg levels was observed, but this effect was not sustained in the long term. Despite the reduction in HBsAg, no treatment-related effect on viremia was observed in the liver, indicating that the vaccine's impact on infected hepatocytes was limited. The results suggest that the mRNA vaccine could be a candidate for inclusion in a multimodal therapeutic regimen for treating chronic HBV infection, potentially in combination with other treatments to reduce immune tolerance. The vaccine includes three different HBV antigens (core, polymerase, and surface antigens), which are believed to be essential for creating HBsAg-specific T cells and antibodies to target infected hepatocytes. This study reports an mRNA therapeutic vaccine with the potential to induce a strong immune response and reduce HBsAg levels in an animal model of chronic HBV infection, offering a promising direction for new treatment strategies for CHB.152 These preclinical data indicated that the mRNA vaccine could be developed for CHB treatment. However, it is worth noting that the HBV–host interaction is far more complicated, and the immune response of CHB patients is continuously attenuated. Yosuke's team reported preliminary clinical data in 2023. They found a significant increase in the number of CHB patients who experienced a rapid reduction in HBsAg levels after the initiation of the mRNA COVID-19 vaccination program compared to the period before the program started. A greater proportion of patients showed a reduction in HBsAg levels by more than 50% per year following mRNA COVID-19 vaccination, suggesting a possible association between the mRNA vaccine and the reduction of HBsAg. Although the study failed to provide statistically significant proof that COVID-19 vaccines were involved in HBsAg reduction, the trend observed was suggestive of a potential link. It was hypothesized that the immunological response induced by mRNA vaccines, including the production of type 1 interferons and activation of immune cells, might reduce HBsAg levels.153 In Rhesus Macaques, Liang and his colleagues reported that intramuscularly administered LNP/mRNA could induce rapid and local infiltration of neutrophils, monocytes, and dendritic cells (DCs) to the site of administration and the draining lymph nodes (LNs). They observed up-regulation of the type I IFN inducible gene and innate immune activation. They demonstrated that mRNA-based vaccines induced type-I IFN-polarized innate immunity and, when combined with antigen production in APCs, led to the generation of potent vaccine-specific responses.154 Silva's team described the case of a 6-month-old female infant who received the equivalent of 6 adult doses of the COVID-19 Pfizer vaccine due to an immunization error. The child showed no severe adverse effects associated with the vaccine overdose; on the other hand, immune response evaluation showed a strong expression of cytokines related to the Th2 profile and a well-controlled inflammatory state on the tenth day. Forty-three days after vaccine administration, the inflammation status remained, with a predominance of the cellular immune response; IFN-γ expression was increased compared to the previous evaluation and a robust antiviral state was in place. A strong expression of type I interferons and cytokines, including IL6, IL10, and IL4, was observed in the infant. These data suggested that the COVID-19 mRNA vaccine showed a strong antiviral response, which might contribute to the HBV response in a systematic manner.153,155 In addition, West China Hospital initiated the first human trial of the HBV mRNA vaccine (NCT05738447) in 2023. They optimized the design of the mRNA sequence and delivery vehicle. Their preclinical research was completed, and the vaccine safety and efficacy data were verified. The clinical study is ongoing; no AEs severer than grade 3 were observed. The study included patients with advanced hepatocellular carcinoma who failed second-line standard treatment or could not receive standard treatment.156 These reports highlighted that the HBV mRNA therapeutic vaccine might be important in CHB treatment. The importance of mRNA therapeutic vaccines in CHB management should not be underestimated. LNP technology remains a promising vaccination route for optimizing the T cell response in CHB patients. T cell function is vital for CHB treatment, and a proportion of CHB patients can develop long-term immunological control of the virus, as indicated by the reduced HBsAg level. The mRNA therapeutic vaccination has the advantage of evoking and restoring an HBV-specific immune response mediated by T cells. By using the mRNA CHB therapeutic vaccine in combination with immunotherapies or sequential treatment, the ambitious goal of the “Elimination of Hepatitis by 2030” may be achieved.157
HPV therapeutic vaccine, mHTV-02 vaccine, is formulated with lipid nanoparticles (LNPs) and induces a robust antigen-specific cellular immune response and memory T-cell immunity in mice. This response was associated with significant CD8+ T-cell infiltration and cytotoxicity in tumors expressing HPV E6/E7, leading to tumor regression and prolonged survival. The design of the vaccine to include specific signal peptides and fusion with Flt3L, a factor involved in dendritic cell development and activation, is an innovative strategy to enhance antigen presentation and immune response. The finding that different routes of administration can significantly affect the vaccine's therapeutic efficacy highlights the importance of delivery optimization for mRNA vaccines. The study found that intramuscular or intratumoral injection of mHTV-02 displayed significant therapeutic effects, while intravenous delivery showed minimal benefits in reducing tumor size or improving survival. Mice that experienced complete tumor regression following mHTV-02 treatment exhibited long-term immunological memory against tumor re-challenge, suggesting the vaccine's potential to induce lasting protection. They reported a promising therapeutic mRNA vaccine candidate, mHTV-02, for treating malignancies caused by HPV16 or HPV18 infections, demonstrating strong immunogenicity and therapeutic potential in preclinical models.89
Wang and colleagues developed an innovative mRNA-based therapeutic vaccine, mHTV-03E2, targeting high-risk human papillomavirus (HPV)-related malignancies, precisely HPV types 16 and 18. The vaccine induced strong antigen-specific cellular immune responses, leading to significant CD8+ T cell infiltration and cytotoxicity in tumor models, which are crucial for eliminating cancer cells. mHTV-03E2 demonstrated substantial tumor regression and prolonged survival in animal models with HPV-induced tumors, highlighting its potential as an effective therapeutic agent. The vaccine elicited robust memory T cell responses against HPV16/18 E6/E7 antigens for up to 4 months post-vaccination, suggesting long-term protection against tumor recurrence. The study showed that mHTV-03E2 worked in synergy with immune checkpoint blockade to inhibit tumor growth and extend animal survival, indicating its potential for use in combination therapies for enhanced cancer treatment. The mRNA–LNP platform used for mHTV-03E2 offers many advantages in safety, efficacy, and rapid production. The E2 antigen, in addition to E6 and E7, is an innovative aspect of vaccine design. E2 is highly expressed during early infection and is critical for HPV's transcriptional regulation, making it a promising target for therapeutic vaccines. The vaccine's ability to activate antibody-dependent cell-mediated cytotoxicity (ADCC) through the E2 antigen represents a novel mechanism of action in the fight against HPV-associated cancers. mHTV-03E2 is a promising candidate for a therapeutic mRNA vaccine for treating malignancies caused by HPV16 or HPV18 infections.161 Researchers from Brazil presented a groundbreaking study on the development and evaluation of a novel mRNA therapeutic vaccine for human papillomavirus (HPV)-associated tumors. They designed and tested many mRNA antigen combinations and found that the fusion protein of HPV-16 E7 oncoprotein and herpes simplex virus type 1 glycoprotein D (gDE7) strongly enhanced the immune response. The direct comparison of different mRNA vaccine modalities (self-amplifying and non-replicating) and their comparison with DNA and protein-based vaccines provided valuable insights into the relative performance of these platforms. In summary, the study presents a promising mRNA therapeutic vaccine candidate for HPV-related tumors, with innovative design elements that enhance its immunogenicity and therapeutic potential. The findings support the further evaluation of these mRNA vaccines in clinical trials, offering a new avenue for managing HPV-associated cancers.162
Cancer therapeutic vaccines
ESI Table S1† summarizes current mRNA-based therapeutics in the clinical trial registry information provided by the Trialtrove database164 on CiteLine (https://clinicalintelligence.citeline.com/trials/results), which excludes prophylactic vaccines and neoantigen/individualized mRNA therapeutics in clinical trials. A total of 74 clinical trial records were found after being manually refined by a date of October 14, 2024. It can be seen that oncology remains the most studied therapeutic area, with 50 trials out of 74 entries registered (Fig. 1a). Metabolic diseases rank second in the non-virus therapeutic area, with 11 related studies focusing on methylmalonic acidemia, propionic acidemia, phenylalanine hydroxylase deficiency, etc. Other new therapeutic areas are being investigated, such as spot-RNA01 (NCT06567119), which was registered most recently by SIPO Biotech and focuses on skin aging. Collagen 1 alpha 1 (COL1A1) was loaded on extracellular vehicles to induce collagen protein grafts in dermal tissue, aiming at supplementing collagen in collagen-depleted skin. Among oncology diseases, melanoma ranks top, followed by a broad targeting of solid tumors (Fig. 1b). Besides, early studies mostly used DCs loaded with mRNA encoding target drug proteins165 or combined them with other therapies (peptide, chemotherapy, etc.), while in the past five years, lipid nanoparticle, lipoplex, or extracellular vehicle trials have increased.
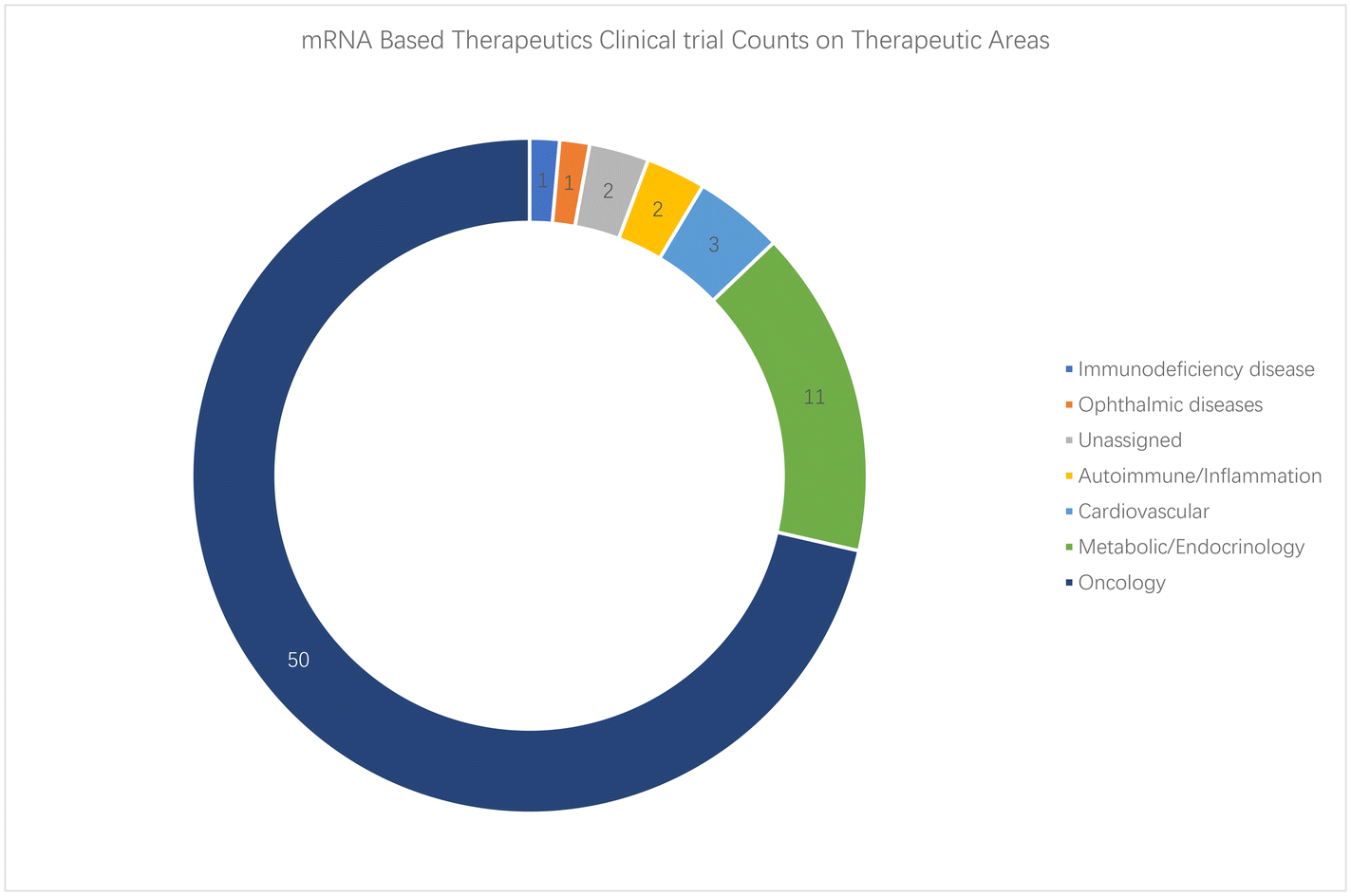 | ||
| Fig. 1 Therapeutic areas of clinical trials on mRNA based therapeutics. | ||
In addition to the registered studies, academic research using newly developed mRNA technology has also been reported. Zai et al. developed an mRNA encoding IL-2 combined with RNAi therapy for treating hepatitis B virus infection. HO-PEG2000-DMG was used to replace mPEG2000-DMG for LNP formation.141 As a result, mRNA therapeutics targeting tumor-associated antigens showed great potential in therapeutic areas and are growing rapidly post-COVID-19 (Fig. 2).
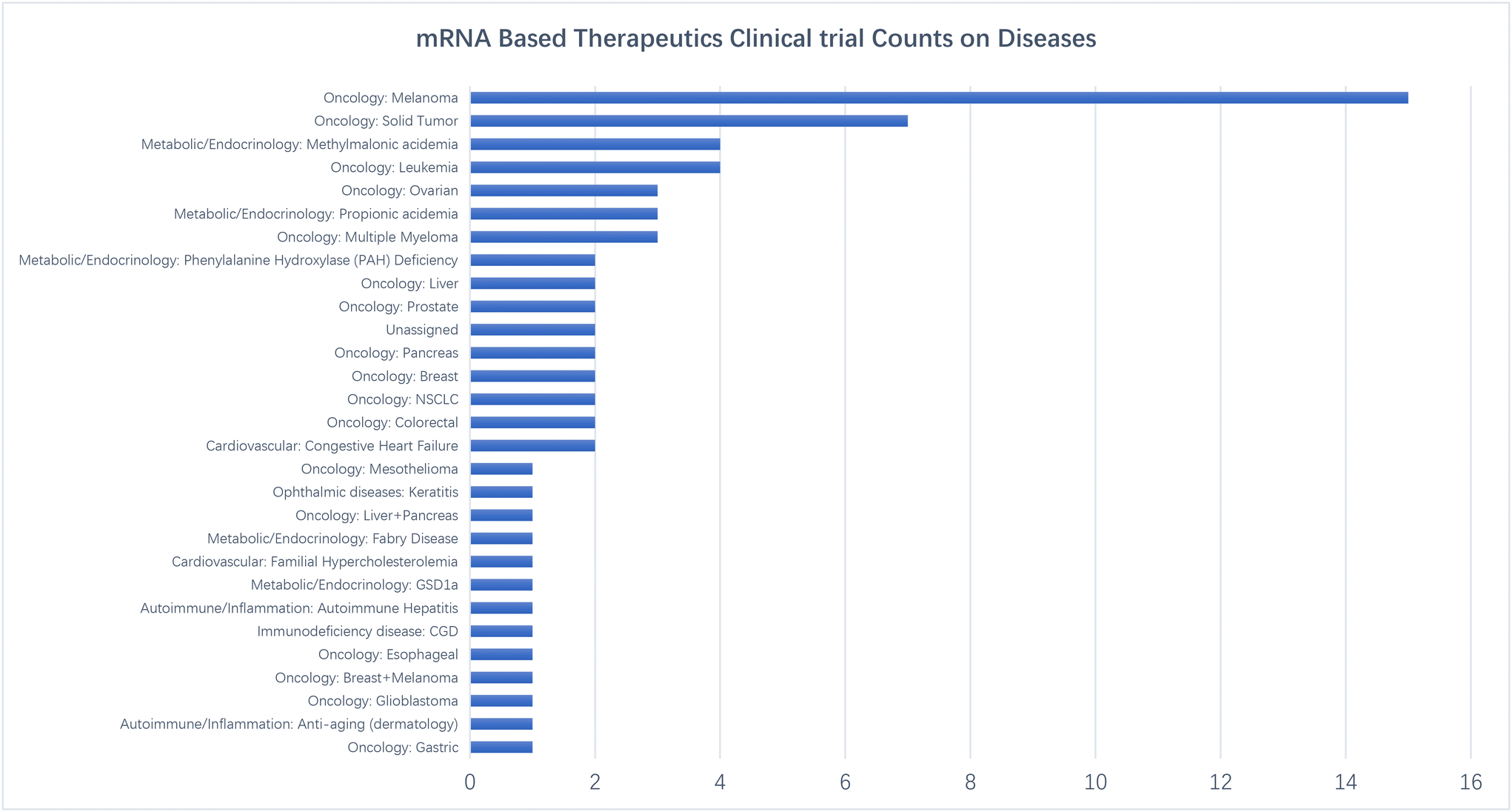 | ||
| Fig. 2 Disease types targeted in clinical trials on mRNA based therapeutics. | ||
The workflow for creating an mRNA-based personalized cancer vaccine (PCV) begins with obtaining a tumor biopsy from the patient, which is then subjected to genomic sequencing to identify mutations present in the tumor DNA.166 Bioinformatics tools analyze the sequences to identify neoantigens—unique peptides resulting from the tumor-specific mutations that are crucial for eliciting an immune response. Selected neoantigens are then used to design an mRNA sequence, which is engineered to encode these neoantigens, ensuring effective stimulation of the immune response. The designed mRNA is synthesized in the laboratory using IVT techniques from a plasmid DNA template, followed by formulation with lipid nanoparticles or other delivery systems. Rigorous quality control checks are performed to ensure the mRNA is correctly synthesized, free of contaminants, and stable. Once prepared, the personalized mRNA vaccine is usually administered via i.v. or s.c. injection. Moderna has launched two phase 3 trials for mRNA-4157, a PCV pipeline, against melanoma and non-small cell lung cancer, combined with pembrolizumab (Keytruda), an anti-PD1 monoclonal antibody as an immune checkpoint inhibitor from Merck, as combination therapy for high-risk patients who have undergone surgery.1 Similarly, BNT-122, developed by BioNTech in collaboration with Genentech is currently in a Phase II clinical trial aimed at treating resected pancreatic ductal adenocarcinoma (PDAC).28 The trial is testing the vaccine in combination with atezolizumab, an immunotherapy drug, and chemotherapy. This clinical trial aims to assess the vaccine's efficacy and safety compared to standard chemotherapy alone.
The selection of neoantigens, which possess strong immunogenicity from hundreds of mutations screened from sequencing data, plays a vital role in determining the efficacy of a PCV treatment. Currently, the immunogenicity prediction is always accomplished through the prediction of affinity between major histocompatibility complex (MHC), human leukocyte antigens (HLA) in humans, and mutated peptides identified by gene sequencing. Higher binding affinity to HLA indicates a higher likelihood of being presented on the cell surface and inducing antigen-specific killing of cancer cells by activating CD8+ cytotoxic T cells and CD4+ helper T cells. Racle et al. developed a motif deconvolution algorithm, similar to a convolutional neural network, to first identify MHC II-binding motifs from a protein sequence, and the deconvoluted proteomic datasets were then used to train a model called MixMHC2pred for affinity prediction.168 Reynisson et al. developed NetMHCpan and NetMHCIIpan utilizing the NNAlign model trained on integrated binding affinity or mass spectrometry-eluted ligand values and achieved boosted predictive performance.169 This toolset is widely recognized by pharmaceutical companies developing PCV pipelines integrated into their neoantigen prediction workflow.170,171 In practice, after identifying candidate neoantigens, further experimental analyses are conducted to evaluate their immunogenicity by ELISpot assay.
PCV can be delivered in mRNA-LNP as well as other drug modalities, such as peptide-based and dendritic cells. The diversity in strategies reflects a growing understanding of effectively engaging and manipulating the immune system for cancer treatment. mRNA-based personalized cancer vaccines offer several significant advantages that enhance their potential as a treatment option. Firstly, they allow for precision targeting by encoding specific neoantigens derived from an individual's tumor mutations, enabling a highly personalized approach focusing on cancer cells while sparing healthy tissue. Then, mRNA–LNP vaccines can induce robust immune responses, stimulating CD8+ cytotoxic T cells and CD4+ helper T cells, which are crucial for effectively recognizing and attacking cancer cells. Additionally, the time-efficient development and production of these vaccines is another benefit. It has been reported that it only takes around 5–6 weeks to complete the entire process, from sample collection to delivery of the lipid-nanoparticle-encapsulated mRNA. This time can potentially be further shortened to within a month.172 Furthermore, mRNA vaccines have a favorable safety profile, as they do not rely on live pathogens or viral vectors, reducing the risk of infections and complications, and the body quickly degrades mRNA after translation, minimizing long-term side effects.170,171 These vaccines can be effectively combined with other immunotherapies, such as checkpoint inhibitors, to enhance overall treatment efficacy and help overcome resistance mechanisms. Finally, mRNA vaccines can be easily modified to include new neoantigens if a patient's tumor evolves, allowing for adaptive treatment strategies. Together, these advantages position mRNA-based personalized cancer vaccines as a promising tool in the evolving landscape of cancer immunotherapy, offering hope for improved patient outcomes.
Summary and future perspectives
In the past 3 years, the real-world value of the mRNA vaccine has been fully proved after its administration to millions of people; the LNP platform has been validated and accepted by regulatory authorities. Beyond the COVID-19 vaccine, mRNA–LNP technology is vital in therapeutic vaccine development. Through appropriate design, the mRNA therapeutic vaccine offers many advantages: high immunogenicity, ease of scale-up and manufacture, low cost, flexibility for tailored medicine, and increasing acceptance by regulatory agencies. The future perspectives for such a new modality of vaccine are encouraging.Data availability
Data availability is not applicable to this review article as no new data were created or analyzed in this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support received from the YaoPharma Co., Ltd. The authors also appreciate the valuable suggestions provided by the reviewers.References
- J. A. Wolff, R. W. Malone, P. Williams, W. Chong, G. Acsadi, A. Jani and P. L. Felgner, Science, 1990, 247, 1465–1468 CrossRef CAS PubMed.
- G. F. Jirikowski, P. P. Sanna, D. Maciejewski-Lenoir and F. E. Bloom, Science, 1992, 255, 996–998 CrossRef CAS PubMed.
- F. Martinon, S. Krishnan, G. Lenzen, R. Magné, E. Gomard, J. Guillet, J. Lévy and P. Meulien, Eur. J. Immunol., 1993, 23, 1719–1722 CrossRef CAS PubMed.
- D. Boczkowski, S. K. Nair, D. Snyder and E. Gilboa, J. Exp. Med., 1996, 184, 465–472 CrossRef CAS PubMed.
- R. M. Conry, A. F. LoBuglio, M. Wright, L. Sumerel, M. J. Pike, F. Johanning, R. Benjamin, D. Lu and D. T. Curiel, Cancer Res., 1995, 55, 1397–1400 CAS.
- A. V. Kristen, S. Ajroud-Driss, I. Conceição, P. Gorevic, T. Kyriakides and L. Obici, Neurodegener. Dis. Manage., 2019, 9, 5–23 CrossRef PubMed.
- I. Fyfe, Nat. Rev. Neurol., 2018, 14, 509–509 CrossRef PubMed.
- L. Zhang, K. R. More, A. Ojha, C. B. Jackson, B. D. Quinlan, H. Li, W. He, M. Farzan, N. Pardi and H. Choe, npj Vaccines, 2023, 8, 156 CrossRef CAS PubMed.
- L. A. Jackson, E. J. Anderson, N. G. Rouphael, P. C. Roberts, M. Makhene, R. N. Coler, M. P. McCullough, J. D. Chappell, M. R. Denison, L. J. Stevens, A. J. Pruijssers, A. McDermott, B. Flach, N. A. Doria-Rose, K. S. Corbett, K. M. Morabito, S. O'Dell, S. D. Schmidt, P. A. Swanson, M. Padilla, J. R. Mascola, K. M. Neuzil, H. Bennett, W. Sun, E. Peters, M. Makowski, J. Albert, K. Cross, W. Buchanan, R. Pikaart-Tautges, J. E. Ledgerwood, B. S. Graham and J. H. Beigel, N. Engl. J. Med., 2020, 383, 1920–1931 CrossRef CAS PubMed.
- M. J. Mulligan, K. E. Lyke, N. Kitchin, J. Absalon, A. Gurtman, S. Lockhart, K. Neuzil, V. Raabe, R. Bailey, K. A. Swanson, P. Li, K. Koury, W. Kalina, D. Cooper, C. Fontes-Garfias, P. Y. Shi, Ö. Türeci, K. R. Tompkins, E. E. Walsh, R. Frenck, A. R. Falsey, P. R. Dormitzer, W. C. Gruber, U. Şahin and K. U. Jansen, Nature, 2020, 586, 589–593 CrossRef CAS PubMed.
- A. D. Perenkov, A. D. Sergeeva, M. V. Vedunova and D. V. Krysko, Vaccines, 2023, 11, 1600 CrossRef CAS PubMed.
- K. Bloom, F. Van Den Berg and P. Arbuthnot, Gene Ther., 2021, 28, 117–129 CrossRef CAS PubMed.
- A. T. He, J. Liu, F. Li and B. B. Yang, Signal Transduction Targeted Ther., 2021, 6, 185 CrossRef CAS PubMed.
- U. Sahin, A. Muik, I. Vogler, E. Derhovanessian, L. M. Kranz, M. Vormehr, J. Quandt, N. Bidmon, A. Ulges, A. Baum, K. E. Pascal, D. Maurus, S. Brachtendorf, V. Lörks, J. Sikorski, P. Koch, R. Hilker, D. Becker, A. K. Eller, J. Grützner, M. Tonigold, C. Boesler, C. Rosenbaum, L. Heesen, M. C. Kühnle, A. Poran, J. Z. Dong, U. Luxemburger, A. Kemmer-Brück, D. Langer, M. Bexon, S. Bolte, T. Palanche, A. Schultz, S. Baumann, A. J. Mahiny, G. Boros, J. Reinholz, G. T. Szabó, K. Karikó, P. Y. Shi, C. Fontes-Garfias, J. L. Perez, M. Cutler, D. Cooper, C. A. Kyratsous, P. R. Dormitzer, K. U. Jansen and Ö. Türeci, Nature, 2021, 595, 572–577 CrossRef CAS PubMed.
- T. E. Mulroney, T. Pöyry, J. C. Yam-Puc, M. Rust, R. F. Harvey, L. Kalmar, E. Horner, L. Booth, A. P. Ferreira, M. Stoneley, R. Sawarkar, A. J. Mentzer, K. S. Lilley, C. M. Smales, T. Von Der Haar, L. Turtle, S. Dunachie, P. Klenerman, J. E. D. Thaventhiran and A. E. Willis, Nature, 2024, 625, 189–194 CrossRef CAS PubMed.
- S. Son, M. Park, J. Kim and K. Lee, Adv. Sci., 2024, 11, 2307541 CrossRef CAS PubMed.
- T. A. Tockary, S. Abbasi, M. Matsui-Masai, A. Hayashi, N. Yoshinaga, E. Boonstra, Z. Wang, S. Fukushima, K. Kataoka and S. Uchida, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2214320120 CrossRef CAS PubMed.
- H. Chen, D. Liu, J. Guo, A. Aditham, Y. Zhou, J. Tian, S. Luo, J. Ren, A. Hsu, J. Huang, F. Kostas, M. Wu, D. R. Liu and X. Wang, Nat. Biotechnol., 2024 DOI:10.1038/s41587-024-02174-7.
- M. Kamiya, M. Matsumoto, K. Yamashita, T. Izumi, M. Kawaguchi, S. Mizukami, M. Tsurumaru, H. Mukai and S. Kawakami, Pharmaceutics, 2022, 14, 2357 CrossRef CAS PubMed.
- L. Xu, X. Wang, Y. Liu, G. Yang, R. J. Falconer and C.-X. Zhao, Adv. NanoBiomed Res., 2022, 2, 2100109 CrossRef CAS.
- J. A. Kulkarni, M. M. Darjuan, J. E. Mercer, S. Chen, R. van der Meel, J. L. Thewalt, Y. Y. C. Tam and P. R. Cullis, ACS Nano, 2018, 12, 4787–4795 CrossRef CAS PubMed.
- X. Hou, T. Zaks, R. Langer and Y. Dong, Nat. Rev. Mater., 2021, 6, 1078–1094 CrossRef CAS PubMed.
- S. Wu, L. Lin, L. Shi and S. Liu, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2024, 16, e1978 CAS.
- J. Wang, Y. Ding, K. Chong, M. Cui, Z. Cao, C. Tang, Z. Tian, Y. Hu, Y. Zhao and S. Jiang, Vaccines, 2024, 12, 1148 CrossRef CAS PubMed.
- A. D. Bangham, M. W. Hill and N. G. A. Miller, in Methods in Membrane Biology, ed. E. D. Korn, Springer US, Boston, MA, 1974, vol. 1, pp. 1–68 Search PubMed.
- H. R. Mendez-Gomez, A. DeVries, P. Castillo, B. D. Stover, S. Qdaisat, C. Von Roemeling, E. Ogando-Rivas, F. Weidert, J. McGuiness, D. Zhang, M. C. Chung, D. Li, C. Zhang, C. Marconi, Y. Campaneria, J. Chardon-Robles, A. Grippin, A. Karachi, N. Thomas, J. Huang, R. Milner, B. Sahay, W. G. Sawyer, J. A. Ligon, N. Silver, E. Simon, B. Cleaver, K. Wynne, M. Hodik, A. Molinaro, J. Guan, P. Kellish, A. Doty, J.-H. Lee, S. Carrera-Justiz, M. Rahman, S. Gatica, S. Mueller, M. Prados, A. Ghiaseddin, D. A. Mitchell and E. J. Sayour, 2023, preprint, DOI:10.1101/2023.03.12.23287108.
- L. M. Kranz, M. Diken, H. Haas, S. Kreiter, C. Loquai, K. C. Reuter, M. Meng, D. Fritz, F. Vascotto, H. Hefesha, C. Grunwitz, M. Vormehr, Y. Hüsemann, A. Selmi, A. N. Kuhn, J. Buck, E. Derhovanessian, R. Rae, S. Attig, J. Diekmann, R. A. Jabulowsky, S. Heesch, J. Hassel, P. Langguth, S. Grabbe, C. Huber, Ö. Türeci and U. Sahin, Nature, 2016, 534, 396–401 CrossRef PubMed.
- L. A. Rojas, Z. Sethna, K. C. Soares, C. Olcese, N. Pang, E. Patterson, J. Lihm, N. Ceglia, P. Guasp, A. Chu, R. Yu, A. K. Chandra, T. Waters, J. Ruan, M. Amisaki, A. Zebboudj, Z. Odgerel, G. Payne, E. Derhovanessian, F. Müller, I. Rhee, M. Yadav, A. Dobrin, M. Sadelain, M. Łuksza, N. Cohen, L. Tang, O. Basturk, M. Gönen, S. Katz, R. K. Do, A. S. Epstein, P. Momtaz, W. Park, R. Sugarman, A. M. Varghese, E. Won, A. Desai, A. C. Wei, M. I. D'Angelica, T. P. Kingham, I. Mellman, T. Merghoub, J. D. Wolchok, U. Sahin, Ö. Türeci, B. D. Greenbaum, W. R. Jarnagin, J. Drebin, E. M. O'Reilly and V. P. Balachandran, Nature, 2023, 618, 144–150 CrossRef CAS PubMed.
- Y. Eygeris, M. Gupta, J. Kim and G. Sahay, Acc. Chem. Res., 2022, 55, 2–12 CrossRef CAS PubMed.
- P. Sharma, D. Hoorn, A. Aitha, D. Breier and D. Peer, Adv. Drug Delivery Rev., 2024, 205, 115175 CrossRef CAS PubMed.
- X. Han, H. Zhang, K. Butowska, K. L. Swingle, M.-G. Alameh, D. Weissman and M. J. Mitchell, Nat. Commun., 2021, 12, 7233 CrossRef CAS PubMed.
- N. Chaudhary, L. N. Kasiewicz, A. N. Newby, M. L. Arral, S. S. Yerneni, J. R. Melamed, S. T. LoPresti, K. C. Fein, D. M. Strelkova Petersen, S. Kumar, R. Purwar and K. A. Whitehead, Nat. Biomed. Eng., 2024, 8, 1483–1498 CrossRef CAS PubMed.
- M. A. Maier, M. Jayaraman, S. Matsuda, J. Liu, S. Barros, W. Querbes, Y. K. Tam, S. M. Ansell, V. Kumar, J. Qin, X. Zhang, Q. Wang, S. Panesar, R. Hutabarat, M. Carioto, J. Hettinger, P. Kandasamy, D. Butler, K. G. Rajeev, B. Pang, K. Charisse, K. Fitzgerald, B. L. Mui, X. Du, P. Cullis, T. D. Madden, M. J. Hope, M. Manoharan and A. Akinc, Mol. Ther., 2013, 21, 1570–1578 CrossRef CAS PubMed.
- I. Hafez, N. Maurer and P. Cullis, Gene Ther., 2001, 8, 1188–1196 CrossRef CAS PubMed.
- M. Jayaraman, S. M. Ansell, B. L. Mui, Y. K. Tam, J. Chen, X. Du, D. Butler, L. Eltepu, S. Matsuda, J. K. Narayanannair, K. G. Rajeev, I. M. Hafez, A. Akinc, M. A. Maier, M. A. Tracy, P. R. Cullis, T. D. Madden, M. Manoharan and M. J. Hope, Angew. Chem., Int. Ed., 2012, 51, 8529–8533 CrossRef CAS PubMed.
- A. Akinc, M. A. Maier, M. Manoharan, K. Fitzgerald, M. Jayaraman, S. Barros, S. Ansell, X. Du, M. J. Hope, T. D. Madden, B. L. Mui, S. C. Semple, Y. K. Tam, M. Ciufolini, D. Witzigmann, J. A. Kulkarni, R. van der Meel and P. R. Cullis, Nat. Nanotechnol., 2019, 14, 1084–1087 CrossRef CAS PubMed.
- A. Takanashi, C. W. Pouton and H. Al-Wassiti, Mol. Pharmaceutics, 2023, 20, 3876–3885 CrossRef CAS PubMed.
- X. Han, M.-G. Alameh, K. Butowska, J. J. Knox, K. Lundgreen, M. Ghattas, N. Gong, L. Xue, Y. Xu, M. Lavertu, P. Bates, J. Xu, G. Nie, Y. Zhong, D. Weissman and M. J. Mitchell, Nat. Nanotechnol., 2023, 18, 1105–1114 CrossRef CAS PubMed.
- B. Li, A. Y. Jiang, I. Raji, C. Atyeo, T. M. Raimondo, A. G. R. Gordon, L. H. Rhym, T. Samad, C. MacIsaac, J. Witten, H. Mughal, T. M. Chicz, Y. Xu, R. P. McNamara, S. Bhatia, G. Alter, R. Langer and D. G. Anderson, Nat. Biomed. Eng., 2023 DOI:10.1038/s41551-023-01082-6.
- M. Zhang, J. Sun, M. Li and X. Jin, Mol. Ther.–Methods Clin. Dev., 2020, 18, 702–712 CrossRef CAS PubMed.
- K. J. Hassett, K. E. Benenato, E. Jacquinet, A. Lee, A. Woods, O. Yuzhakov, S. Himansu, J. Deterling, B. M. Geilich, T. Ketova, C. Mihai, A. Lynn, I. McFadyen, M. J. Moore, J. J. Senn, M. G. Stanton, Ö. Almarsson, G. Ciaramella and L. A. Brito, Mol. Ther.–Nucleic Acids, 2019, 15, 1–11 CrossRef CAS PubMed.
- S. M. Ansell and X. Du, Lipids and lipid nanoparticle formulations for delivery of nucleic acids, US Pat., US10166298B2, 2019.
- P. Patel, N. M. Ibrahim and K. Cheng, Trends Pharmacol. Sci., 2021, 42, 448–460 CrossRef CAS PubMed.
- Y. Xu, A. Golubovic, S. Xu, A. Pan and B. Li, J. Mater. Chem. B, 2023, 11, 6527–6539 RSC.
- Y. Xu, S. Ma, H. Cui, J. Chen, S. Xu, F. Gong, A. Golubovic, M. Zhou, K. C. Wang, A. Varley, R. X. Z. Lu, B. Wang and B. Li, Nat. Commun., 2024, 15, 6305 CrossRef CAS PubMed.
- B. Li, I. O. Raji, A. G. R. Gordon, L. Sun, T. M. Raimondo, F. A. Oladimeji, A. Y. Jiang, A. Varley, R. S. Langer and D. G. Anderson, Nat. Mater., 2024, 23, 1002–1008 CrossRef CAS PubMed.
- R. Maharjan, K. H. Kim, K. Lee, H.-K. Han and S. H. Jeong, J. Pharm. Anal., 2024, 14, 100996 CrossRef PubMed.
- J. Witten, I. Raji, R. S. Manan, E. Beyer, S. Bartlett, Y. Tang, M. Ebadi, J. Lei, D. Nguyen, F. Oladimeji, A. Y. Jiang, E. MacDonald, Y. Hu, H. Mughal, A. Self, E. Collins, Z. Yan, J. F. Engelhardt, R. Langer and D. G. Anderson, Nat. Biotechnol., 2024 DOI:10.1038/s41587-024-02490-y.
- M. L. Guevara, F. Persano and S. Persano, Front. Chem., 2020, 8, 589959 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- D. Chen, S. Ganesh, W. Wang and M. Amiji, Nanoscale, 2019, 11, 8760–8775 RSC.
- M. Yanez Arteta, T. Kjellman, S. Bartesaghi, S. Wallin, X. Wu, A. J. Kvist, A. Dabkowska, N. Székely, A. Radulescu, J. Bergenholtz and L. Lindfors, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E3351–E3360 CrossRef PubMed.
- M. Ramezanpour and D. P. Tieleman, Langmuir, 2022, 38, 7462–7471 CrossRef CAS PubMed.
- H. Abumanhal-Masarweh, D. da Silva, M. Poley, A. Zinger, E. Goldman, N. Krinsky, R. Kleiner, G. Shenbach, J. E. Schroeder and J. Shklover, J. Controlled Release, 2019, 307, 331–341 CrossRef CAS PubMed.
- D. Marsh, R. Bartucci and L. Sportelli, Biochim. Biophys. Acta, Biomembr., 2003, 1615, 33–59 CrossRef CAS PubMed.
- L. Schoenmaker, D. Witzigmann, J. A. Kulkarni, R. Verbeke, G. Kersten, W. Jiskoot and D. J. A. Crommelin, Int. J. Pharm., 2021, 601, 120586 CrossRef CAS PubMed.
- H. T. Phan and A. J. Haes, J. Phys. Chem. C, 2019, 123, 16495–16507 CrossRef CAS PubMed.
- R. Ball, P. Bajaj and K. Whitehead, Int. J. Nanomed., 2016, 12, 305–315 CrossRef PubMed.
- M. Guimarães Sá Correia, M. L. Briuglia, F. Niosi and D. A. Lamprou, Int. J. Pharm., 2017, 516, 91–99 CrossRef PubMed.
- N. M. Payton, M. F. Wempe, Y. Xu and T. J. Anchordoquy, J. Pharm. Sci., 2014, 103, 3869–3878 CrossRef CAS PubMed.
- B. Kim, R. R. Hosn, T. Remba, D. Yun, N. Li, W. Abraham, M. B. Melo, M. Cortes, B. Li, Y. Zhang, Y. Dong and D. J. Irvine, J. Controlled Release, 2023, 353, 241–253 CrossRef CAS PubMed.
- M.-G. Alameh, I. Tombácz, E. Bettini, K. Lederer, S. Ndeupen, C. Sittplangkoon, J. R. Wilmore, B. T. Gaudette, O. Y. Soliman, M. Pine, P. Hicks, T. B. Manzoni, J. J. Knox, J. L. Johnson, D. Laczkó, H. Muramatsu, B. Davis, W. Meng, A. M. Rosenfeld, S. Strohmeier, P. J. C. Lin, B. L. Mui, Y. K. Tam, K. Karikó, A. Jacquet, F. Krammer, P. Bates, M. P. Cancro, D. Weissman, E. T. Luning Prak, D. Allman, B. Z. Igyártó, M. Locci and N. Pardi, Immunity, 2021, 54, 2877–2892 CrossRef CAS PubMed.
- C. Xie, R. Yao and X. Xia, npj Vaccines, 2023, 8, 162 CrossRef PubMed.
- K. Karikó, H. Ni, J. Capodici, M. Lamphier and D. Weissman, J. Biol. Chem., 2004, 279, 12542–12550 CrossRef PubMed.
- K. Karikó, M. Buckstein, H. Ni and D. Weissman, Immunity, 2005, 23, 165–175 CrossRef PubMed.
- N. Pardi, M. J. Hogan, M. S. Naradikian, K. Parkhouse, D. W. Cain, L. Jones, M. A. Moody, H. P. Verkerke, A. Myles, E. Willis, C. C. LaBranche, D. C. Montefiori, J. L. Lobby, K. O. Saunders, H.-X. Liao, B. T. Korber, L. L. Sutherland, R. M. Scearce, P. T. Hraber, I. Tombácz, H. Muramatsu, H. Ni, D. A. Balikov, C. Li, B. L. Mui, Y. K. Tam, F. Krammer, K. Karikó, P. Polacino, L. C. Eisenlohr, T. D. Madden, M. J. Hope, M. G. Lewis, K. K. Lee, S.-L. Hu, S. E. Hensley, M. P. Cancro, B. F. Haynes and D. Weissman, J. Exp. Med., 2018, 215, 1571–1588 CrossRef CAS PubMed.
- A. B. Vogel, I. Kanevsky, Y. Che, K. A. Swanson, A. Muik, M. Vormehr, L. M. Kranz, K. C. Walzer, S. Hein, A. Güler, J. Loschko, M. S. Maddur, A. Ota-Setlik, K. Tompkins, J. Cole, B. G. Lui, T. Ziegenhals, A. Plaschke, D. Eisel, S. C. Dany, S. Fesser, S. Erbar, F. Bates, D. Schneider, B. Jesionek, B. Sänger, A. K. Wallisch, Y. Feuchter, H. Junginger, S. A. Krumm, A. P. Heinen, P. Adams-Quack, J. Schlereth, S. Schille, C. Kröner, R. de la Caridad Güimil Garcia, T. Hiller, L. Fischer, R. S. Sellers, S. Choudhary, O. Gonzalez, F. Vascotto, M. R. Gutman, J. A. Fontenot, S. Hall-Ursone, K. Brasky, M. C. Griffor, S. Han, A. A. H. Su, J. A. Lees, N. L. Nedoma, E. H. Mashalidis, P. V. Sahasrabudhe, C. Y. Tan, D. Pavliakova, G. Singh, C. Fontes-Garfias, M. Pride, I. L. Scully, T. Ciolino, J. Obregon, M. Gazi, R. Carrion, K. J. Alfson, W. V. Kalina, D. Kaushal, P. Y. Shi, T. Klamp, C. Rosenbaum, A. N. Kuhn, Ö. Türeci, P. R. Dormitzer, K. U. Jansen and U. Sahin, Nature, 2021, 592, 283–289 CrossRef CAS PubMed.
- J. S. McLellan, Science, 2013, 342, 931 CrossRef CAS PubMed.
- A. Krarup, D. Truan, P. Furmanova-Hollenstein, L. Bogaert, P. Bouchier, I. J. M. Bisschop, M. N. Widjojoatmodjo, R. Zahn, H. Schuitemaker, J. S. McLellan and J. P. M. Langedijk, Nat. Commun., 2015, 6, 8143 CrossRef PubMed.
- U. Sahin, A. Muik, E. Derhovanessian, I. Vogler, L. M. Kranz, M. Vormehr, A. Baum, K. Pascal, J. Quandt, D. Maurus, S. Brachtendorf, V. Lörks, J. Sikorski, R. Hilker, D. Becker, A. K. Eller, J. Grützner, C. Boesler, C. Rosenbaum, M. C. Kühnle, U. Luxemburger, A. Kemmer-Brück, D. Langer, M. Bexon, S. Bolte, K. Karikó, T. Palanche, B. Fischer, A. Schultz, P. Y. Shi, C. Fontes-Garfias, J. L. Perez, K. A. Swanson, J. Loschko, I. L. Scully, M. Cutler, W. Kalina, C. A. Kyratsous, D. Cooper, P. R. Dormitzer, K. U. Jansen and Ö. Türeci, Nature, 2020, 586, 594–599 CrossRef CAS PubMed.
- H. A. Al-Wassiti, S. A. Fabb, S. L. Grimley, R. Kochappan, J. K. Ho, C. Y. Wong, C. W. Tan, T. J. Payne, A. Takanashi, C. L. Lee, R. S. Mugan, H. Sicilia, S. L. Y. Teo, J. McAuley, P. Ellenberg, J. P. Cooney, K. C. Davidson, R. Bowen, M. Pellegrini, S. Rockman, D. I. Godfrey, T. M. Nolan, L.-F. Wang, G. Deliyannis, D. F. J. Purcell and C. W. Pouton, Mol. Ther. -Methods Clin. Dev., 2024, 32(4), 101380 CrossRef CAS PubMed.
- G. G. Hendricks, L. Grigoryan, M. J. Navarro, N. J. Catanzaro, M. L. Hubbard, J. M. Powers, M. Mattocks, C. Treichel, A. C. Walls, J. Lee, D. Ellis, J. Y. (John) Wang, S. Cheng, M. C. Miranda, A. Valdez, C. W. Chao, S. Chan, C. Men, M. R. Johnson, H. Hui, S.-Y. Wu, V. Lujan, H. Muramatsu, P. J. C. Lin, M. M. H. Sung, Y. K. Tam, E. M. Leaf, N. Pardi, R. S. Baric, B. Pulendran, D. Veesler, A. Schäfer and N. P. King, 2024, preprint, DOI:10.1101/2024.07.22.604655.
- K. Lemdani, R. Marlin, C. Mayet, V. Perkov, Q. Pascal, M. Ripoll, F. Relouzat, N. Dhooge, L. Bossevot, N. Dereuddre-Bosquet, G. Dargazanli, K. Thibaut-Duprey, J. Haensler, C. Chapon, C. Prost and R. Le Grand, npj Vaccines, 2024, 9, 113 CrossRef CAS PubMed.
- M. F. Bachmann and G. T. Jennings, Nat. Rev. Immunol., 2010, 10, 787–796 CrossRef CAS PubMed.
- K. J. Hassett, J. Higgins, A. Woods, B. Levy, Y. Xia, C. J. Hsiao, E. Acosta, Ö. Almarsson, M. J. Moore and L. A. Brito, J. Controlled Release, 2021, 335, 237–246 CrossRef CAS PubMed.
- X. Tong, J. Raffaele, K. Feller, G. Dornadula, J. Devlin, D. Boyd, J. W. Loughney, J. Shanter and R. R. Rustandi, Vaccines, 2024, 12, 169 CrossRef CAS PubMed.
- R. Shi, X. Liu, Y. Wang, M. Pan, S. Wang, L. Shi and B. Ni, Hum. Vaccines Immunother., 2024, 20(1), 2342592 CrossRef PubMed.
- C. McMillan, A. Druschitz, S. Rumbelow, A. Borah, B. Binici, Z. Rattray and Y. Perrie, RSC Pharm., 2024, 1, 841–853 RSC.
- N. Pardi, S. Tuyishime, H. Muramatsu, K. Kariko, B. L. Mui, Y. K. Tam, T. D. Madden, M. J. Hope and D. Weissman, J. Controlled Release, 2015, 217, 345–351 CrossRef CAS PubMed.
- C. Grunwitz, N. Salomon, F. Vascotto, A. Selmi, T. Bukur, M. Diken, S. Kreiter, Ö. Türeci and U. Sahin, OncoImmunology, 2019, 8, 1–11 CrossRef PubMed.
- R. A. Feldman, R. Fuhr, I. Smolenov, A. (Mick) Ribeiro, L. Panther, M. Watson, J. J. Senn, M. Smith, Ö. Almarsson, H. S. Pujar, M. E. Laska, J. Thompson, T. Zaks and G. Ciaramella, Vaccine, 2019, 37, 3326–3334 CrossRef CAS PubMed.
- A. Syenina, E. S. Gan, J. Z. N. Toh, R. de Alwis, L. Z. Lin, C. Y. L. Tham, J. X. Yee, Y. S. Leong, H. Sam, C. Cheong, Y. E. Teh, I. L. E. Wee, D. H. L. Ng, K. R. Chan, J. X. Y. Sim, S. Kalimuddin, E. Z. Ong, J. G. Low and E. E. Ooi, PLoS Biol., 2022, 20, e3001643 CrossRef CAS PubMed.
- J. D. Beck, M. Diken, M. Suchan, M. Streuber, E. Diken, L. Kolb, L. Allnoch, F. Vascotto, D. Peters, T. Beißert, Ö. Akilli-Öztürk, Ö. Türeci, S. Kreiter, M. Vormehr and U. Sahin, Cancer Cell, 2024, 42, 1467–1470 CrossRef CAS PubMed.
- E. A. Aunins, A. T. Phan, M.-G. Alameh, E. Cruz-Morales, D. A. Christian, M. E. Bunkofske, G. Dwivedi, R. Kedl, D. Weissman and C. A. Hunter, bioRxiv, DOI:10.1101/2024.07.29.605626.
- B. Brook, V. Duval, S. Barman, L. Speciner, C. Sweitzer, A. Khanmohammed, M. Menon, K. Foster, P. Ghosh, K. Abedi, J. Koster, E. Nanishi, L. R. Baden, O. Levy, T. VanCott, R. Micol and D. J. Dowling, Sci. Transl. Med., 2024, 16, eadm8451 CrossRef CAS PubMed.
- B. Wang, M. Tang, Q. Chen, W. Ho, Y. Teng, X. Xiong, Z. Jia, X. Li, X. Xu and X.-Q. Zhang, ACS Nano, 2024, 18, 15499–15516 CrossRef CAS PubMed.
- Z. Xu, Z.-X. Xiao, J. Wang, H.-W. Qiu, F. Cao, S.-Q. Zhang, Y.-D. Xu, H.-Q. Lei, H. Xia, Y.-R. He, G.-F. Zha and J. Pang, OncoImmunology, 2024, 13, 2373526 CrossRef PubMed.
- D. Zhivaki, E. A. Gosselin, D. Sengupta, H. Concepcion, C. Arinze, J. Chow, A. Nikiforov, V. Komoroski, C. MacFarlane, C. Sullivan and J. C. Kagan, mBio, 2023, 14(6), e0250623 CrossRef PubMed.
- J. Wang, L. Ma, Y. Chen, R. Zhou, Q. Wang, T. Zhang, D. Yi, Q. Liu, Y. Zhang, W. Zhang, Y. Dong and S. Cen, Life Sci. Alliance, 2024, 7, e202302448 CrossRef CAS PubMed.
- S. Yanik, V. Venkatesh, J. T. Gordy, M. Gabriel-Alameh, J. Meza, Y. Li, E. Glass, Y. Flores-Garcia, Y. Tam, N. Chaiyawong, D. Sarkar, D. Weissman, R. Markham and P. Srinivasan, Res. Sq., 2024, rs.3.rs-4656309 Search PubMed.
- P. Kubes and C. Jenne, Annu. Rev. Immunol., 2018, 36, 247–277 Search PubMed.
- S.-W. Tse, K. McKinney, W. Walker, M. Nguyen, J. Iacovelli, C. Small, K. Hopson, T. Zaks and E. Huang, Mol. Ther., 2021, 29, 2227–2238 CrossRef CAS PubMed.
- Y. Gu, J. Yang, C. He, T. Zhao, R. Lu, J. Liu, X. Mo, F. Wen and H. Shi, Signal Transduction Targeted Ther., 2023, 8, 273 Search PubMed.
- M. P. Lokugamage, Z. Gan, C. Zurla, J. Levin, F. Z. Islam, S. Kalathoor, M. Sato, C. D. Sago, P. J. Santangelo and J. E. Dahlman, Adv. Mater., 2020, 32, 1904905 CrossRef CAS PubMed.
- O. A. W. Haabeth, J. J. K. Lohmeyer, A. Sallets, T. R. Blake, I. Sagiv-Barfi, D. K. Czerwinski, B. McCarthy, A. E. Powell, P. A. Wender, R. M. Waymouth and R. Levy, ACS Cent. Sci., 2021, 7, 1191–1204 CrossRef CAS PubMed.
- R. Das, E. A. Halabi, I. R. Fredrich, J. Oh, H. M. Peterson, X. Ge, E. Scott, R. H. Kohler, C. S. Garris and R. Weissleder, Adv. Sci., 2023, 10, 2303576 CrossRef CAS PubMed.
- S. Meulewaeter, I. Aernout, J. Deprez, Y. Engelen, M. De Velder, L. Franceschini, K. Breckpot, S. Van Calenbergh, C. Asselman, K. Boucher, F. Impens, S. C. De Smedt, R. Verbeke and I. Lentacker, J. Controlled Release, 2024, 370, 379–391 CrossRef CAS PubMed.
- H. K. Patel, K. Zhang, R. Utegg, E. Stephens, S. Salem, H. Welch, S. Grobe, J. Schlereth, A. N. Kuhn, J. Ryczek, D. J. Cirelli and T. F. Lerch, J. Pharm. Sci., 2023, 112, 1364–1371 CrossRef CAS PubMed.
- A. G. Orlandini von Niessen, M. A. Poleganov, C. Rechner, A. Plaschke, L. M. Kranz, S. Fesser, M. Diken, M. Löwer, B. Vallazza, T. Beissert, V. Bukur, A. N. Kuhn, Ö. Türeci and U. Sahin, Mol. Ther., 2019, 27, 824–836 CrossRef CAS PubMed.
- Y. Zheng and N. J. VanDusen, J. Cardiovasc. Dev. Dis., 2023, 10, 144 Search PubMed.
- F. Lu, A. Sossin, N. Abell, S. B. Montgomery and Z. He, Nucleic Acids Res., 2022, 50, 11442–11454 CrossRef CAS PubMed.
- S. M. Castillo-Hair and G. Seelig, Acc. Chem. Res., 2022, 55, 24–34 Search PubMed.
- H. Gong, J. Wen, R. Luo, Y. Feng, J. Guo, H. Fu and X. Zhou, Briefings Bioinf., 2023, 24, bbad001 Search PubMed.
- P. J. Sample, B. Wang, D. W. Reid, V. Presnyak, I. J. McFadyen, D. R. Morris and G. Seelig, Nat. Biotechnol., 2019, 37, 803–809 CrossRef CAS PubMed.
- J. Zrimec, X. Fu, A. S. Muhammad, C. Skrekas, V. Jauniskis, N. K. Speicher, C. S. Börlin, V. Verendel, M. H. Chehreghani, D. Dubhashi, V. Siewers, F. David, J. Nielsen and A. Zelezniak, Nat. Commun., 2022, 13, 5099 CrossRef CAS PubMed.
- P. Gaspar, G. Moura, M. A. S. Santos and J. L. Oliveira, Nucleic Acids Res., 2013, 41, e73–e73 CrossRef CAS PubMed.
- Y. Liu, Q. Yang and F. Zhao, Annu. Rev. Biochem., 2021, 90, 375–401 Search PubMed.
- G. Hanson and J. Coller, Nat. Rev. Mol. Cell Biol., 2018, 19, 20–30 CrossRef CAS PubMed.
- V. Presnyak, N. Alhusaini, Y.-H. Chen, S. Martin, N. Morris, N. Kline, S. Olson, D. Weinberg, K. E. Baker, B. R. Graveley and J. Coller, Cell, 2015, 160(6), 1111–1124 Search PubMed.
- P. M. Sharp and W.-H. Li, Nucleic Acids Res., 1987, 15, 1281–1295 CrossRef CAS PubMed.
- X. Xia, Vaccines, 2021, 9, 734 Search PubMed.
- G. Terai, S. Kamegai and K. Asai, Bioinformatics, 2016, 32, 828–834 CrossRef CAS PubMed.
- M. Zuker and P. Stiegler, Nucleic Acids Res., 1981, 9, 133–148 Search PubMed.
- H. K. Wayment-Steele, D. S. Kim, C. A. Choe, J. J. Nicol, R. Wellington-Oguri, A. M. Watkins, R. A. Parra Sperberg, P.-S. Huang, E. Participants and R. Das, Nucleic Acids Res., 2021, 49, 10604–10617 CrossRef CAS PubMed.
- H. Zhang, L. Zhang, A. Lin, C. Xu, Z. Li, K. Liu, B. Liu, X. Ma, F. Zhao, H. Jiang, C. Chen, H. Shen, H. Li, D. H. Mathews, Y. Zhang and L. Huang, Nature, 2023, 621, 396–403 CrossRef CAS PubMed.
- J. Linder, N. Bogard, A. B. Rosenberg and G. Seelig, Genomics, 2019, preprint, DOI:10.1101/864363.
- J. Im, B. Park and K. Han, BMC Genomics, 2019, 20, 967 Search PubMed.
- W. Wang, S. Feng, Z. Ye, H. Gao, J. Lin and D. Ouyang, Acta Pharm. Sin. B, 2022, 12, 2950–2962 Search PubMed.
- D. D. Kang, H. Li and Y. Dong, Adv. Drug Delivery Rev., 2023, 199, 114961 Search PubMed.
- Y. Li, Q. Wang, Y. Xu and Z. Li, Nat. Commun., 2024, 15, 4622 Search PubMed.
- A. Ramanathan, G. B. Robb and S. H. Chan, Nucleic Acids Res., 2016, 44, 7511–7526 CrossRef PubMed.
- J. Zhang, Y. Liu, C. Li, Q. Xiao, D. Zhang, Y. Chen, J. Rosenecker, X. Ding and S. Guan, Pharmaceutics, 2023, 15, 2182 CrossRef CAS PubMed.
- X. Yuan, Z. Wu, J. Guo, D. Luo, T. Li, Q. Cao, X. Ren, H. Fang, D. Xu and Y. Cao, Adv. Mater., 2024, 36, 2303321 CrossRef CAS PubMed.
- M. Packer, D. Gyawali, R. Yerabolu, J. Schariter and P. White, Nat. Commun., 2021, 12, 6777 CrossRef CAS PubMed.
- M. M. Castellanos, H. Gressard, X. Li, C. Magagnoli, A. Moriconi, D. Stranges, L. Strodiot, M. Tello Soto, M. Zwierzyna and C. Campa, Vaccines, 2023, 11, 1153 CrossRef PubMed.
- M. Hussain, B. Binici, L. O'Connor and Y. Perrie, J. Pharm. Pharmacol., 2024, rgae122 Search PubMed.
- C. Geng, K. Zhou, Y. Yan, C. Li, B. Ni, J. Liu, Y. Wang, X. Zhang, D. Wang, L. Lv, Y. Zhou, A. Feng, Y. Wang and C. Li, J. Controlled Release, 2023, 364, 632–643 CrossRef CAS PubMed.
- H. M. Gunter, S. Idrisoglu, S. Singh, D. J. Han, E. Ariens, J. R. Peters, T. Wong, S. W. Cheetham, J. Xu, S. K. Rai, R. Feldman, A. Herbert, E. Marcellin, R. Tropee, T. Munro and T. R. Mercer, Nat. Commun., 2023, 14, 5663 CrossRef CAS PubMed.
- C. J. Vanhinsbergh, A. Criscuolo, J. N. Sutton, K. Murphy, A. J. K. Williamson, K. Cook and M. J. Dickman, Anal. Chem., 2022, 94, 7339–7349 CrossRef CAS PubMed.
- B. C. Gau, A. W. Dawdy, H. L. Wang, B. Bare, C. H. Castaneda, O. V. Friese, M. S. Thompson, T. F. Lerch, D. J. Cirelli and J. C. Rouse, Sci. Rep., 2023, 13, 9038 CrossRef CAS PubMed.
- J. Raffaele, J. W. Loughney and R. R. Rustandi, Electrophoresis, 2022, 43, 1101–1106 CrossRef CAS PubMed.
- S. Li, Y. Hu, A. Li, J. Lin, K. Hsieh, Z. Schneiderman, P. Zhang, Y. Zhu, C. Qiu, E. Kokkoli, T.-H. Wang and H.-Q. Mao, Nat. Commun., 2022, 13, 5561 CrossRef CAS PubMed.
- A.-G. Reinhart, A. Osterwald, P. Ringler, Y. Leiser, M. E. Lauer, R. E. Martin, C. Ullmer, F. Schumacher, C. Korn and M. Keller, Mol. Pharmaceutics, 2023, 20, 6492–6503 Search PubMed.
- M. Ogunnaike, S. Das, S. S. Raut, A. Sultana, M. U. Nayan, M. Ganesan, B. J. Edagwa, N. A. Osna and L. Y. Poluektova, Biomolecules, 2023, 13, 1208 Search PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, CA-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- T. Vesikari, J. M. Langley, V. Popovic and F. Diaz-Mitoma, Expert Rev. Vaccines, 2023, 22, 1041–1054 Search PubMed.
- J. Li, J. Yuan, H. Li, J. Guo, M. Li, T. Zhang, X.-J. Liang, H. Fan and X. Liu, Adv. NanoBiomed Res., 2024, 4, 2300132 CrossRef CAS.
- F. Zoulim, P.-J. Chen, M. Dandri, P. Kennedy and C. Seeger, J. Hepatol., 2024, S0168-8278(24)02343-2 Search PubMed.
- Y. Shi and M. Zheng, Br. Med. J., 2020, 370, m2200 CrossRef PubMed.
- J. Yi, X. Lei, F. Guo, Q. Chen, X. Chen, K. Zhao, C. Zhu, X. Cheng, J. Lin, H. Yin and Y. Xia, Antiviral Res., 2023, 215, 105618 CrossRef CAS PubMed.
- W. Zai, M. Yang, K. Jiang, J. Guan, H. Wang, K. Hu, C. Huang, J. Chen, W. Fu, C. Zhan and Z. Yuan, Signal Transduction Targeted Ther., 2024, 9, 150 CrossRef CAS PubMed.
- B. Chen, Y. Chen, J. Li, C. Wang, W. Song, Y. Wen, J. Lin, Y. Wu and T. Ying, mBio, 2022, 13, e01612–e01622 Search PubMed.
- C. L. Gorsuch, P. Nemec, M. Yu, S. Xu, D. Han, J. Smith, J. Lape, N. Van Buuren, R. Ramirez, R. C. Muench, M. M. Holdorf, B. Feierbach, G. Falls, J. Holt, W. Shoop, E. Sevigny, F. Karriker, R. V. Brown, A. Joshi, T. Goodwin, Y. K. Tam, P. J. C. Lin, S. C. Semple, N. Leatherbury, W. E. Delaney Iv, D. Jantz and A. Rhoden Smith, Mol. Ther., 2022, 30, 2909–2922 Search PubMed.
- M. G. Martinez, E. Smekalova, E. Combe, F. Gregoire, F. Zoulim and B. Testoni, Viruses, 2022, 14, 2654 CrossRef CAS PubMed.
- C. Seeger and J. A. Sohn, Mol. Ther.–Nucleic Acids, 2014, 3, e216 CrossRef CAS PubMed.
- D. Kostyushev, S. Brezgin, A. Kostyusheva, D. Zarifyan, I. Goptar and V. Chulanov, Cell. Mol. Life Sci., 2019, 76, 1779–1794 Search PubMed.
- L. C. McCoullough, M. Fareh, W. Hu, V. Sozzi, C. Makhlouf, Y. Droungas, C. L. Lee, M. Takawy, S. A. Fabb, T. J. Payne, C. W. Pouton, H. J. Netter, S. R. Lewin, D. F. Purcell, J. A. Holmes, J. A. Trapani, M. Littlejohn and P. A. Revill, J. Hepatol., 2024, S016882782400360X Search PubMed.
- E. M. Smekalova, M. G. Martinez, E. Combe, A. Kumar, S. Dejene, D. Leboeuf, C.-Y. Chen, J. R. Dorkin, L. S. Shuang, S. Kieft, L. Young, L. A. Barrera, M. S. Packer, G. Ciaramella, B. Testoni, F. Gregoire and F. Zoulim, Mol. Ther.–Nucleic Acids, 2024, 35, 102112 CrossRef CAS PubMed.
- G. Swaminathan, E. A. Thoryk, K. S. Cox, S. Meschino, S. A. Dubey, K. A. Vora, R. Celano, M. Gindy, D. R. Casimiro and A. J. Bett, Vaccine, 2016, 34, 110–119 CrossRef CAS PubMed.
- C. Lamb and P. Arbuthnot, Master of Science (MSc), University of the Witwatersrand, Johannesburg, South Africa, 2017.
- H. Zhao, X. Shao, Y. Yu, L. Huang, N. P. Amor, K. Guo, C. Weng, W. Zhao, A. Yang, J. Hu, H. Yang, Z. Liu, Q. Han, L. Shi, S. Sun, J. Zhang, A. Lin and Y. Yang, npj Vaccines, 2024, 9, 22 CrossRef CAS PubMed.
- D. De Pooter, W. Pierson, S. Pourshahian, K. Dockx, B. De Clerck, I. Najera, H. Davis, E. Van Gulck and D. Boden, Vaccines, 2024, 12, 237 CrossRef CAS PubMed.
- Y. Osawa, T. Ohtake, D. Suto, T. Akita, H. Yamada, Y. Kohgo and K. Murata, Intern. Med., 2023, 62, 51–57 CrossRef PubMed.
- F. Liang, G. Lindgren, A. Lin, E. A. Thompson, S. Ols, J. Röhss, S. John, K. Hassett, O. Yuzhakov, K. Bahl, L. A. Brito, H. Salter, G. Ciaramella and K. Loré, Mol. Ther., 2017, 25, 2635–2647 CrossRef CAS PubMed.
- J. M. E. Silva, J. B. Guimarães, S. R. Abu Jamra, B. S. Mirante, C. F. Capato, D. M. De Melo Jorge and B. A. L. Da Fonseca, Vaccine: X, 2023, 15, 100395 Search PubMed.
- Y.-S. Wang, M. Kumari, G.-H. Chen, M.-H. Hong, J. P.-Y. Yuan, J.-L. Tsai and H.-C. Wu, J. Biomed. Sci., 2023, 30, 84 CrossRef CAS PubMed.
- S. Akbar, M. Mahtab, S. Khan, O. Yoshida and Y. Hiasa, Vaccines, 2022, 10, 1644 CrossRef CAS PubMed.
- A. Chabeda, R. J. R. Yanez, R. Lamprecht, A. E. Meyers, E. P. Rybicki and I. I. Hitzeroth, Papillomavirus Res., 2018, 5, 46–58 Search PubMed.
- F. S. Alhamlan, M. B. Alfageeh, M. A. Al Mushait, I. A. Al-Badawi and M. N. Al-Ahdal, Adv. Exp. Med. Biol., 2021, 1313, 1–14 CrossRef CAS PubMed.
- Study Details | A Clinical Trial Investigating the Safety, Tolerability, and Therapeutic Effects of BNT113 in Combination With Pembrolizumab Versus Pembrolizumab Alone for Patients With a Form of Head and Neck Cancer Positive for Human Papilloma Virus 16 and Expressing the Protein PD-L1 | ClinicalTrials.gov, https://clinicaltrials.gov/study/NCT04534205?cond=HPV%20mRNA&&rank=8&&tab=results#study-overview, (accessed September 30, 2024).
- J. Wang, Q. Wang, L. Ma, K. Lv, L. Han, Y. Chen, R. Zhou, H. Zhou, H. Chen, Y. Wang, T. Zhang, D. Yi, Q. Liu, Y. Zhang, X. Li, T. Cheng, J. Zhang, C. Huang, Y. Dong, W. Zhang and S. Cen, Mol. Ther., 2024, 32, 2340–2356 Search PubMed.
- J. Ramos Da Silva, K. Bitencourt Rodrigues, G. Formoso Pelegrin, N. Silva Sales, H. Muramatsu, M. De Oliveira Silva, B. F. M. M. Porchia, A. C. R. Moreno, L. R. M. M. Aps, A. A. Venceslau-Carvalho, I. Tombácz, W. L. Fotoran, K. Karikó, P. J. C. Lin, Y. K. Tam, M. De Oliveira Diniz, N. Pardi and L. C. De Souza Ferreira, Sci. Transl. Med., 2023, 15, eabn3464 CrossRef CAS PubMed.
- B. Wang, J. Pei, S. Xu, J. Liu and J. Yu, Front. Immunol., 2023, 14, 1246682 CrossRef CAS PubMed.
- S. Stergiopoulos, K. A. Getz and C. Blazynski, Ther. Innov. Regul. Sci., 2019, 53, 307–317 Search PubMed.
- P. E. Saw and E. Song, Cell Rep. Med., 2024, 5, 101555 Search PubMed.
- N. Xie, G. Shen, W. Gao, Z. Huang, C. Huang and L. Fu, Signal Transduction Targeted Ther., 2023, 8, 9 Search PubMed.
- G. Fotakis, Z. Trajanoski and D. Rieder, Immunooncol. Technol, 2021, 12, 100052 CrossRef CAS PubMed.
- J. Racle, J. Michaux, G. A. Rockinger, M. Arnaud, S. Bobisse, C. Chong, P. Guillaume, G. Coukos, A. Harari, C. Jandus, M. Bassani-Sternberg and D. Gfeller, Nat. Biotechnol., 2019, 37, 1283–1286 Search PubMed.
- B. Reynisson, B. Alvarez, S. Paul, B. Peters and M. Nielsen, Nucleic Acids Res., 2020, 48, W449–W454 Search PubMed.
- J. S. Weber, M. S. Carlino, A. Khattak, T. Meniawy, G. Ansstas, M. H. Taylor, K. B. Kim, M. McKean, G. V. Long, R. J. Sullivan, M. Faries, T. T. Tran, C. L. Cowey, A. Pecora, M. Shaheen, J. Segar, T. Medina, V. Atkinson, G. T. Gibney, J. J. Luke, S. Thomas, E. I. Buchbinder, J. A. Healy, M. Huang, M. Morrissey, I. Feldman, V. Sehgal, C. Robert-Tissot, P. Hou, L. Zhu, M. Brown, P. Aanur, R. S. Meehan and T. Zaks, Lancet, 2024, 403, 632–644 Search PubMed.
- L. A. Rojas, Z. Sethna, K. C. Soares, C. Olcese, N. Pang, E. Patterson, J. Lihm, N. Ceglia, P. Guasp, A. Chu, R. Yu, A. K. Chandra, T. Waters, J. Ruan, M. Amisaki, A. Zebboudj, Z. Odgerel, G. Payne, E. Derhovanessian, F. Müller, I. Rhee, M. Yadav, A. Dobrin, M. Sadelain, M. Łuksza, N. Cohen, L. Tang, O. Basturk, M. Gönen, S. Katz, R. K. Do, A. S. Epstein, P. Momtaz, W. Park, R. Sugarman, A. M. Varghese, E. Won, A. Desai, A. C. Wei, M. I. D'Angelica, T. P. Kingham, I. Mellman, T. Merghoub, J. D. Wolchok, U. Sahin, Ö. Türeci, B. D. Greenbaum, W. R. Jarnagin, J. Drebin, E. M. O'Reilly and V. P. Balachandran, Nature, 2023, 618, 144–150 CrossRef CAS PubMed.
- T. Carvalho, Nat. Med., 2023, 29, 2379–2380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00309h |
| This journal is © The Royal Society of Chemistry 2025 |