
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSugar-decorated cholesterol-core nanoparticles as potential targeting nanomedicines for the delivery of lipophilic drugs†
Laís Rossetto Ferraz
de Barros
a,
Carlos Eduardo
de Castro
a,
Anabella Patricia
Rosso
a,
Rodrigo
da Costa Duarte
b,
Alexandre Gonçalves
Dal-Bó
b,
Wendel Andrade
Alves
 a and
Fernando Carlos
Giacomelli
*a
a and
Fernando Carlos
Giacomelli
*a
aCentro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-580, Santo André, Brazil. E-mail: fernando.giacomelli@ufabc.edu.br
bUniversidade do Extremo Sul Catarinense – UNESC, 88806-000, Criciúma, SC, Brazil
First published on 23rd January 2025
Abstract
Targeted drug delivery is a precise and effective strategy in oncotherapy and can be achieved through sugar-decorated assemblies since glucose receptors are overexpressed on cancer cell membranes to compensate for their increased glucose demands. In this study, core–shell nanoparticles (NPs) were synthesized using amphiphilic macromolecules comprising hydrophobic cholesterol (Chol) segments conjugated to hydrophilic polyethylene oxide containing azide group (Chol-PEO22-N3) or substituted with the carbohydrate N-acetyl-D-glucosamine (Chol-PEO22-GlcNAc) via a click chemistry reaction. These self-assemblies, which are smaller than 100 nm and suitable for cancer treatment, demonstrated efficient loading efficiency (exceeding 70%) with ursolic acid (UA), a hydrophobic drug, serving as a proof-of-concept for targeted therapy using natural compounds against non-small cell lung cancer. The incorporation of sugar molecules modified the structural characteristics of the nanocarriers, resulting in larger and presumably less dense particles. This modification influenced the UA release mechanism, leading to a faster and nearly complete release over a week, whereas approximately 60% of the encapsulated UA remained entrapped in the Chol-PEO22-N3 NPs. Enhanced cell cytotoxicity was achieved with UA-loaded NPs with in vitro cell viability assays indicating at least two-fold increase in the inhibitory effect of the drug-loaded nanocarriers. The targeted delivery was also demonstrated as UA-loaded Chol-PEO22-GlcNAc NPs showed greater internalization by cancer cells than their healthy counterparts.
1. Introduction
Cancer is a significant public health concern worldwide, causing approximately 1700 deaths per day in the United States1 and more than 600 deaths per day in Brazil.2 Among the different types of cancer, lung cancer usually remains the first or second leading cause of cancer death in both men and women every year,1 and non-small cell lung cancer (NSCLC), which constitutes 80–85% of lung cancer cases, was selected as the model for this investigation. Chemotherapy is the primary approach for treating tumors and to mitigate its adverse effect such as cytotoxicity and limited selectivity, the development of nanotherapeutic platforms for cancer drug delivery has been proposed, aiming to enhance specificity and improve therapeutic efficacy.3,4The enhanced permeability and retention (EPR) effect5 allows nanoparticles (NPs) sized from 50 to 100 nm to accumulate more in tumor sites because of the imperfect architecture and irregular morphology of blood vessels in such locations. Furthermore, previous investigations have indicated that drug-loaded nanocarriers can be deposited and evade mucociliary clearance and alveolar macrophages in the lung-lining liquid, and clearance by alveolar macrophages is slower for relatively small particles (<80 nm).6,7 Additionally, the cellular uptake process is crucial, as treatment efficiency often relies on the internalization of therapeutic agents. The surface chemistry of drug-loaded nanocarriers significantly influences nanoparticle–cell membrane interactions.8 In this framework, polymer chains such as polyethylene oxide (PEO) can be used for biocompatibility and further functionalization, allowing the manufacturing of nanocarriers with targeting attributes and increased accumulation at a desired location. Small molecules can be conjugated to PEO chains through click reactions using organic azide, ensuring high-efficiency coupling in aqueous environments.9,10 In this context, short-chain carbohydrates (sugars) are being employed in the synthesis of targeted cargo delivery systems.11,12 Glycidic moieties, for instance, have a high affinity for cell membrane components such as lecithins, thus promoting increased cellular uptake.13 Sugars are particularly attractive as cancer-targeting ligands due to the augmented glycolysis in proliferating cancer cells, which leads to higher glucose uptake.14,15 Since glucose receptors are overexpressed on cancer cell membranes, galactose, mannose, and glucose monosaccharide derivatives such as N-acetyl glucosamine serve as promising targeting ligands.11,16
Taking these considerations into account, we have evaluated two amphiphilic macromolecules: one with a hydrophobic fraction of cholesterol (to allow hydrophobic drug loading) conjugated to PEO terminated with an azide group (Chol-PEO22-N3) and the other with a PEO substituted with the carbohydrate (targeting ligand) N-acetyl glucosamine (Chol-PEO22-GlcNAc). N-Acetylglucosamine (GlcNAc), which is an amide derivative of glucose, has been selected as the targeting ligand due to the enhanced metabolism, high glucose demand of cancer cells and amplified expression of glucose transporters in tumor sites.17 These amphiphilic macromolecules were self-assembled into core–shell NPs that have been evaluated as potential nanocarriers for ursolic acid (UA), a model hydrophobic chemotherapeutic drug. UA is a pharmacologically active, natural pentacyclic triterpene derived from fruits, herbs, and plants,18 and it can regulate the proliferation, metastasis, angiogenesis, and apoptosis of tumor cells by acting on a variety of cytokines.19 UA has a significant therapeutic effect on breast, lung, colorectal, liver, and prostate cancer, for instance.20 Despite its promising properties,18 the poor water-solubility, low intestinal mucosal absorption, and low bioavailability restrict its clinical application and in vivo administration. These drawbacks can be circumvented by using nanotechnology-based platforms to enable its pharmacological administration. Accordingly, herein UA-loaded cholesterol-core sugar-decorated NPs were evaluated as a proof-of-concept to improve drug solubility and bioavailability to treat non-small cell lung cancer. A549 cells were used as a model for in vitro biological assays. This investigation therefore relies on the manufacturing of UA-loaded nanocarriers and subsequent evaluation of their structural features, colloidal stability, drug release profile, cell cytotoxicity, and cellular uptake.
2. Materials and methods
2.1. Amphiphilic macromolecules and other chemicals
The amphiphile azide-poly(ethylene glycol)-cholesterol conjugate Chol-PEO22-N3 and the glycosurfactant N-acetyl-β-D-glucosaminyl-poly(ethylene glycol)-cholesterol conjugate Chol-PEO22-GlcNAc were synthesized according to previously reported procedures13,21–23 with the methodology described in the ESI† along with respective product characterization by FTIR and 13C NMR. All solvents, coumarin-6, and UA were of analytical grade purchased from Sigma-Aldrich. The water used in sample preparation was pretreated with Milli-Q® Plus System (Millipore Corporation).2.2. Preparation of the UA-loaded and coumarin-6-loaded NPs
The NPs produced using Chol-PEO22-N3 and Chol-PEO22-GlcNAc were prepared by nanoprecipitation. In this process, 5.0 mg of amphiphilic macromolecules were dissolved in 2.0 mL of organic solvent mixture (THF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acetone 25:75 v/v). The formation of the NPs was subsequently induced by injecting the organic phase (1 mL min−1) into 5.0 mL of water. The organic solvent was further evaporated at 30 °C for 48 h, and the final volume was adjusted to 5.0 mL. Negligible amounts of organic solvent at the final samples can be expected by using this protocol.24 To produce drug-loaded NPs, 0.25 mg of UA (5% w/w) was added during amphiphile dissolution in the organic phase. Probe-loaded NPs were similarly produced using 0.01 mg of coumarin-6 (0.2% w/w).
:acetone 25:75 v/v). The formation of the NPs was subsequently induced by injecting the organic phase (1 mL min−1) into 5.0 mL of water. The organic solvent was further evaporated at 30 °C for 48 h, and the final volume was adjusted to 5.0 mL. Negligible amounts of organic solvent at the final samples can be expected by using this protocol.24 To produce drug-loaded NPs, 0.25 mg of UA (5% w/w) was added during amphiphile dissolution in the organic phase. Probe-loaded NPs were similarly produced using 0.01 mg of coumarin-6 (0.2% w/w).
2.3. Structural characterization of the NPs
Dynamic light scattering (DLS) data were acquired using an ALV/CGS-3 compact goniometer. The autocorrelation functions were collected in triplicate (30 s counting time), further averaged, and analyzed using the Cumulant method to determine the values of the mean relaxation time (τ), and the values of hydrodynamic radius (RH) were further determined using the straightforward Stokes–Einstein relation. The Cumulant method also allows to probe the polydispersity index (PDI) of the samples (μ2/Γ2).The electrophoretic mobility (UE) of the colloidal materials was measured using a Zetasizer Nano-ZS ZEN 3600 instrument (Malvern Instruments). Each sample was measured five times with three sub runs of 30 s counting time. The values were further converted to ζ-potential (mV) using the Henry's equation and Smoluchowski approximation.
Cryo-TEM images were acquired at the Brazilian Nanotechnology National Laboratory (LNNano) using a Talos F200C (Thermo Fisher Scientific) high-resolution microscope at an accelerating voltage of 200 kV. Two microliters of each sample were loaded into 01895 – Lacey Carbon, 300 mesh copper electron microscopy grids (Electron Microscopy Science). Excess sample was removed by blotting for 3 s, and the grids were immediately plunged into liquid ethane. The specimens were stored in liquid nitrogen and transferred, under controlled conditions, to an autoloader cassette and then to a microscopic column for imaging.
2.4. Determination of UA loading content (LC) and loading efficiency (LE)
The UA loading efficiency was determined by phase separation using Amicon® Ultra-15 centrifuge tubes (Merck Millipore, 10000 MWCO). One milliliter of the drug-loaded NPs was placed in the reservoirs and centrifuged at 20 °C for 5 min and 5000 rpm. The volume of the filtrate was then set to 1.0 mL using a 8:2 v/v mixture of MeOH:water. This solvent mixture was selected in order to disassemble the core–shell nanoparticles. The UV-Vis absorbance of the filtrates was subsequently measured in triplicate (averaged values are provided) at λ = 200 nm (the maximum absorbance of UA) using a Varian Cary 50 Bio UV-Vis spectrophotometer. To determine the concentrations of UA, an analytical curve was generated with linear response (R2 = 0.99) in the range 0.005–0.25 mg mL−1 in the same medium. The values of loading content (LC) and loading efficiency (LE) were calculated by using the following equations: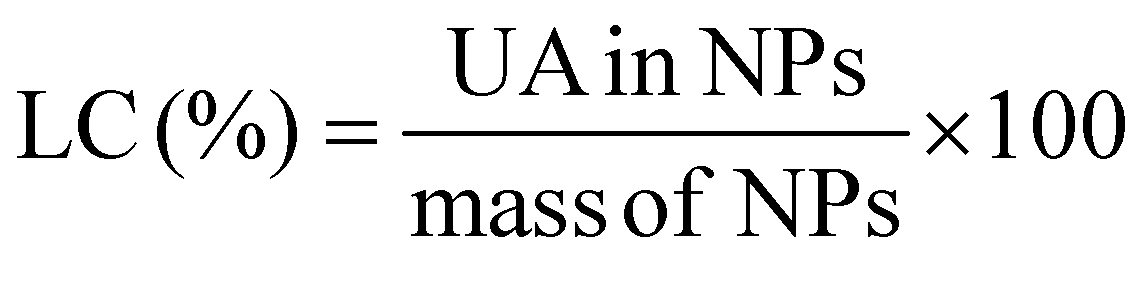 | (1) |
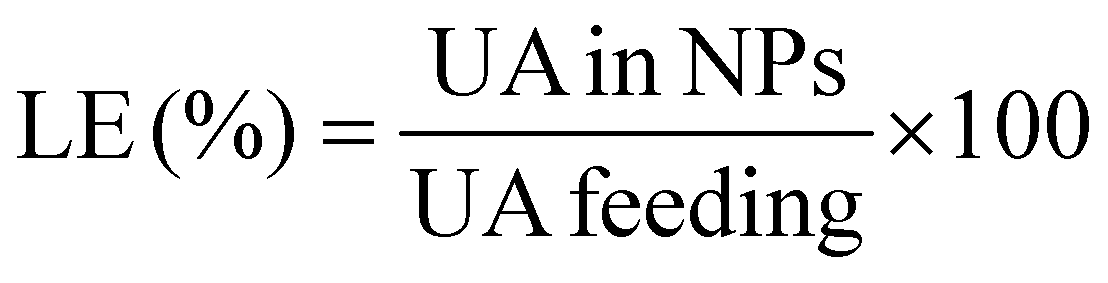 | (2) |
2.5. Evaluation of UA release kinetics
UA release kinetics were evaluated using cellulose dialysis membranes (MWCO 8–10 kDa, Spectra-Por® Float-A-Lyzer® G2) using well-established protocols regularly used by us25–27 as well as by other research groups.28,29 The tubes were filled with 5.0 mL of the UA-loaded NPs and immersed in 2 L of phosphate-buffered saline (PBS) solution at pH 7.4 and 37 °C with gentle stirring. At predetermined times, 80 μL of the samples were removed from the dialysis tube and diluted with 320 μL of MeOH to determine the quantity of UA remaining in the samples. An equal volume of nanoparticle suspension was immediately replaced. The remaining amount of UA was determined by measuring the UV-Vis absorbance acquired in triplicate (averaged values are provided) at λ = 200 nm (the maximum absorbance of UA) using a Varian Cary 50 Bio UV-Vis spectrophotometer. Concentrations were calculated based on the calibration curve described in the previous section. The UA release kinetics were evaluated using different kinetic models: zero-order, first-order, Higuchi, and Korsmeyer-Peppas, as discussed in the Results and discussion sections.2.6. Biological assays
000 U mL−1 penicillin, and 10000 μg mL−1 streptomycin at 37 °C under CO2 atmosphere.
The cellular uptake of the NPs was further quantified by flow cytometry analysis using essentially the same procedures described above, except that 1 × 104 cells per well were plated, incubated for 1 h, washed three times with PBS, harvested by trypsinization using trypsin–EDTA solution, fixed with 4% paraformaldehyde for 10 min, and finally resuspended. Measurements were performed using a BD FACS Canto II flow cytometer (using a FITC filter for coumarin-6) and analyzed using the FlowJo V.6 software. Flow cytometry analyses were performed in triplicate (10000 events were collected for each experiment), and the data are presented as the mean ± standard deviation. Statistical analyses were performed using a one-way ANOVA followed by Tukey's test. A p-value of 0.05 or greater was considered statistically significant.
3. Results and discussion
3.1. Synthesis of the amphiphilic macromolecules
The synthetic strategy used to prepare the amphiphile azide-poly(ethylene oxide)-cholesterol conjugate Chol-PEO22-N3 (2) from the initial reactant cholesteryl hemisuccinate (1) and the glycosurfactant N-acetyl-β-D-glucosaminyl-poly(ethylene oxide)-cholesterol conjugate Chol-PEO22-GlcNAc (3) is illustrated in Fig. 1. | ||
| Fig. 1 Synthetic strategy used in the preparation of (2) azide-poly(ethylene oxide)-cholesterol conjugate Chol-PEO22-N3 and (3) glycosurfactant N-acetyl-β-D-glucosaminyl-poly(ethylene glycol)-cholesterol conjugate Chol-PEO22-GlcNAc: (a) N,N′-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), mono-azide-PEO (PEO22-N3), CH2Cl2, 72%; (b) copper sulfate (CuSO4), sodium ascorbate (C6H7O6Na), propargyl-2-N-acetamido-2-deoxy-β-D-glucopyranoside (C7H17NO6, H2O/THF, 70%). | ||
The synthesis of the amphiphilic macromolecules has been detailed in previous publications.13,21–23 Briefly, the hemisuccinate cholesteryl group was conjugated to the PEO chains via an esterification reaction using dicyclohexylcarbodiimide (DCC), resulting in a 72% yield of “ready-to-click” Chol-PEO22-N3. Propargyl β glycosides of N-acetyl-glucosamine were further introduced at the polar head of the surfactant via copper-catalyzed azide–alkyne Huisgen cycloaddition. The reaction was carried out at 40 °C with copper/ascorbate as a catalyst in a water/THF mixture to ensure good solubility of the amphiphile. The glycosylated conjugate Chol-PEO22-GlcNAc was isolated in 70% yield after purification by silica gel chromatography. Characterization of the amphiphiles was conducted using FTIR and 13C NMR analyses, where the characteristic signals of the aliphatic chains, ethylene oxide, and the carbohydrate unit, as well as the signals of the triazolyl ring, were identified. These experimental data are provided in the ESI (Fig. S1–S4†) along with further details of the employed synthetic approach.
3.2. Manufacturing and structural characterization of the nanocarriers
Dynamic light scattering (DLS) autocorrelation functions and respective size distributions for the produced amphiphilic NPs are shown in Fig. 2, highlighting sub-100 nm self-assemblies with sizes ranging from 36 to 79 nm.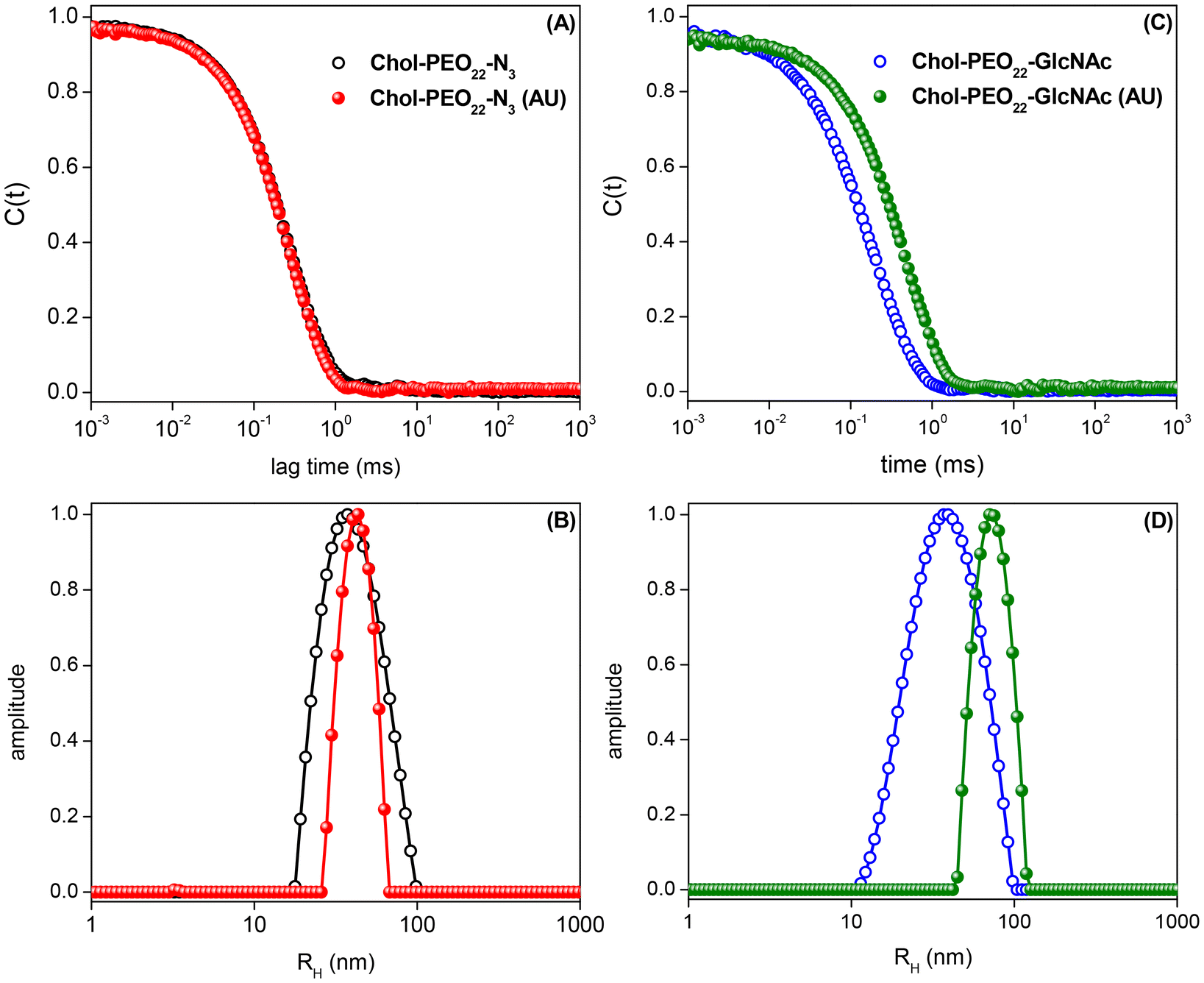 | ||
| Fig. 2 Dynamic light scattering data acquired for drug-free and 5% w/w UA-loaded Chol-PEO22-N3 and Chol-PEO22-GlcNAc NPs: (A and C) autocorrelation functions and (B and D) respective distributions of sizes. | ||
Table 1 lists the quantitative data. The sizes of the nanocarriers are suitable for cancer treatment as they are larger than the threshold for fast renal clearance (DH = 2RH > 10 nm)30,31 and smaller than 100 nm, which is the average pore size of tumor sites, enabling enhanced accumulation via the EPR effect.5,32 The particle size increases with UA (5% w/w) loading, reflecting changes in the internal structure of the nanocarriers,33 and the surface charge is possibly the results of ionized groups (probably hydroxide ions) at the interface between water and the organic shells.34 The UA loading does not significantly affect the surface charge of the NPs.
| Nanoparticle | R H (nm) | PDI | ζ (mV) |
|---|---|---|---|
| Chol-PEO22-N3 | 40 | 0.37 | −25.1 ± 0.8 |
| Chol-PEO22-N3 (UA) | 48 | 0.20 | −22.5 ± 0.7 |
| Chol-PEO22-GlcNAc | 36 | 0.47 | −19.2 ± 0.7 |
| Chol-PEO22-GlcNAc (UA) | 79 | 0.22 | −22 ± 1 |
The negative zeta potential of the assemblies is expected to provide enhanced colloidal stability. Furthermore, positively charged NPs may indeed induce fast nanoparticle clearance due to protein adsorption whereas negative surfaces can impart protein-repelling features since main protein components at the bloodstream are negatively charge at physiological pH, thus enabling longer blood circulation time. The surface charge, size and size distribution of the manufactured soft colloids have been frequently checked over 4 months and no signs of nanoparticle aggregation when stored at 4 °C have been detected. Due to the presence of fairly negative values of ζ-potential, the colloidal stability is supposed to come at least partially from electrostatic effects since repulsive Coulomb forces acting between the charged colloidal particles are certainly present.
Morphological observations through cryo-TEM (Fig. 3) showed homogenous and spherical structures in reasonable agreement with the size range obtained by DLS. The images confirm also the formation of NPs in the presence of UA.
 | ||
| Fig. 3 Cryo-TEM images for (A) Chol-PEO22-N3 and (B) Chol-PEO22-N3 NPs loaded with 5% w/w UA, (C) Chol-PEO22-GlcNAc and (D) Chol-PEO22-GlcNAc NPs loaded with 5% w/w UA. The value of 5% w/w refers to the UA feeding. | ||
Differences regarding in the cryo-TEM images may be the result of disparate electron density contrast in the presence and in the absence of UA nevertheless, core–shell structures are more likely to be produced taking into account the weight fraction of the hydrophobic segment (roughly 30%). Additionally, discrepancies when comparing cryo-TEM and DLS data are to some extent acceptable since light scattering intensity is heavily weighted by the particle size implying that DLS is particularly sensitive to larger compared to smaller scattering objects.
3.3. Evaluation of UA loading content (LC) and loading efficiency (LE)
The determined values of UA loading content (LC) and loading efficiency (LE) are listed in Table 2. The experimental data show that UA was encapsulated to a slightly greater extent in Chol-PEO22-GlcNAc NPs, possibly reflecting the larger volume of these nanocarriers. However, the size of the UA-loaded Chol-PEO22-GlcNAc (Table 1) was probably balanced by a lower particle density; therefore, the LE and LC values were not significantly different.| Nanoparticle | LE (%) | LC (%) |
|---|---|---|
| Chol-PEO22-N3 | 71 ± 1 | 3.6 ± 0.1 |
| Chol-PEO22-GlcNAc | 81 ± 2 | 4.1 ± 0.1 |
The LE is approximately 70–80%, which is higher than values reported for UA loaded into other assemblies, such as dendrimer-like NPs (UA-G4K)33 or poly(lactic-co-glycolic acid)-b-polyethylene glycol diblock copolymer NPs.35 The LE for Chol-PEO22-GlcNAc NPs is similar to that reported for chitosan NPs cross-linked with sodium tripolyphosphate.36 High LE values are hardly achieved during UA loading,19 making this an outstanding feature of the investigated systems, possibly due to the high degree of core hydrophobicity and therefore, favorable core–drug intermolecular interactions. The UA water solubility is equal to 0.005 mg mL−1.33–35 Considering the values of LC and LE reported in Table 2, the UA solubility in the formulated samples is approximately 40 times higher than as free drug. Because of its high degree of hydrophobicity, which contributes to its affinity for nonpolar organic solvents, UA migrates towards the cholesteric hydrophobic cores of the assemblies during nanoprecipitation. Thus, this method has proven efficient in developing cargo delivery systems aimed at increasing the solubility of lipophilic drugs for systemic administration.18
3.4. In vitro release of UA
Subsequently, the UA release profiles were examined. Drug release from soft NPs may occur via dissolution, diffusion, partitioning, osmosis, swelling, or erosion.37 The release mechanism can be understood using mathematical models to fit the experimental data. The cumulative release of UA was evaluated in PBS (pH 7.4), and the experimental data are shown in Fig. 4.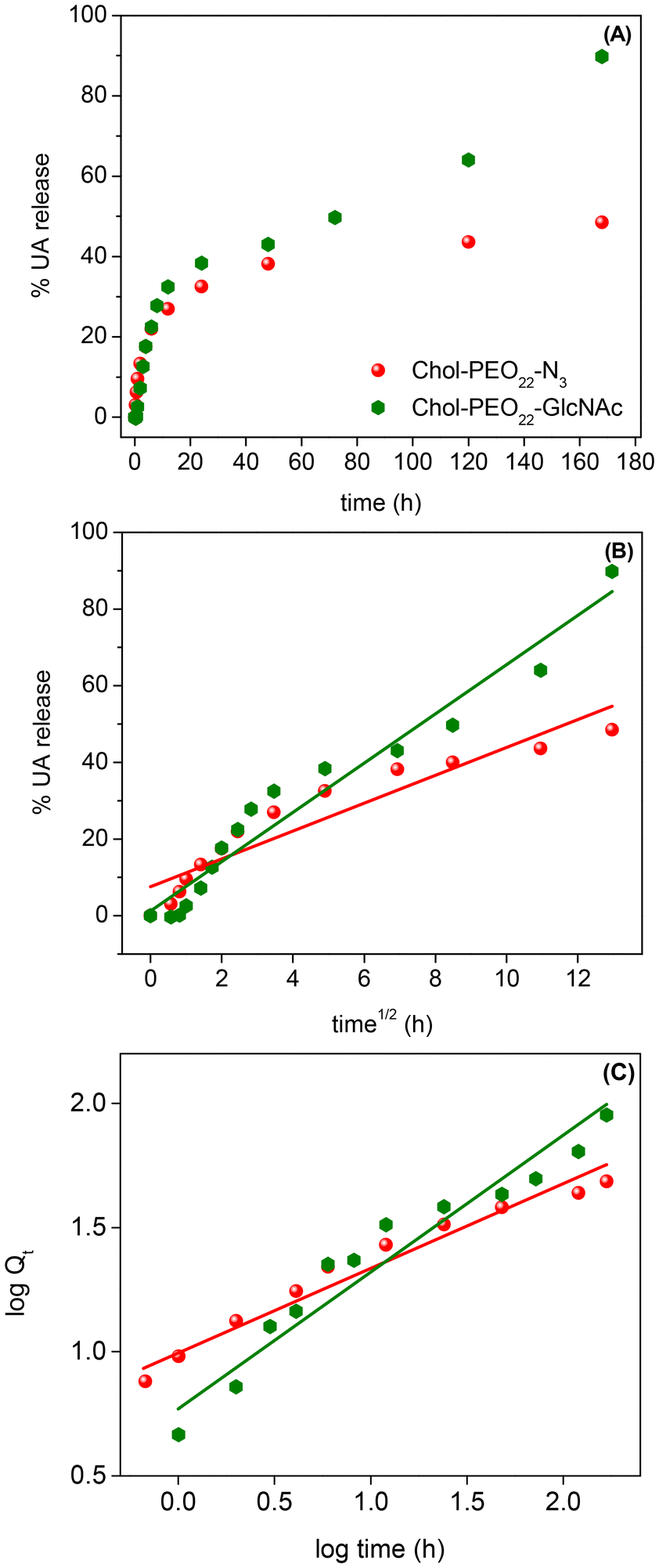 | ||
| Fig. 4 (A) UA release profiles and UA release model fitting using the Higuchi (B) and the Korsmeyer-Peppas (C) approaches from Chol-PEO22-N3 and Chol-PEO22-GlcNAc NPs. | ||
The UA release showed a biphasic pattern, with an initial rapid release from Chol-PEO22-N3 (up to 12 h), followed by slower release, achieving a maximum release of approximately 40%. In contrast, the drug release from Chol-PEO22-GlcNAc NPs was initially rapid (up to 12 h) and then slower. However, it was faster than that for Chol-PEO22-N3 with nearly quantitative elution. This feature is remarkable because the complete release of UA was achieved from Chol-PEO22-GlcNAc, whereas approximately 60% of UA remained encapsulated in the Chol-PEO22-N3 counterparts after a week. The larger size of UA-loaded Chol-PEO22-GlcNAc NPs (Table 1) and the presence of the sugar moieties likely lead to greater swelling in the assemblies and lower particle density, facilitating the complete UA release. This sustained release over a week is desirable for targeted delivery at specific locations. To quantitatively evaluate the kinetics of UA release, the profiles in Fig. 4A were analyzed using zero-order, first-order, Higuchi, and Korsmeyer-Peppas models. Briefly, the zero-order model describes the drug release from NPs at a constant rate, independent of concentration. The first-order model indicates a release rate dependent on the remaining drug concentration. The Higuchi model addresses drug release from an insoluble matrix as the square root of a time-dependent process based on Fickian diffusion, whereas the Korsmeyer-Peppas model describes drug release from various systems, including Fickian and non-Fickian diffusion. This model is useful when the release mechanism is unknown, or when more than one type of drug release mechanism is involved.37 The models are mathematically described by the following equations:
| Qt = Q0 − K0t | (3) |
| lnQt = lnQ0 − K1t | (4) |
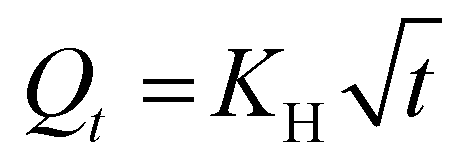 | (5) |
| logQt = logKKP + nlogt | (6) |
Q t is the quantity of drug released at time t, and Q0 is the initial quantity at t = 0 h. The zero-order, first-order, Higuchi, and Korsmeyer-Peppas constants are K0, K1, KH, and KKP, respectively. The suggested release mechanism is discussed based on the kinetic models and the values of R2 reported in Table 3. These values suggest that the Korsmeyer-Peppas model provided the best fit for Chol-PEO22-N3 NPs, whereas the Higuchi model was most suitable for Chol-PEO22-GlcNAc NPs. However, the Korsmeyer-Peppas model showed relatively high R2 values. The Higuchi and Korsmeyer-Peppas fittings are shown in Fig. 4B and C, and the others have been omitted for brevity.
| Parameter | Chol-PEO22-N3 | Chol-PEO22-GlcNAc |
|---|---|---|
| Zero-order | ||
| r 2 | 0.65 | 0.85 |
| K 0 (h−1) | 0.24 ± 0.05 | 0.48 ± 0.05 |
| First-order | ||
| r 2 | 0.37 | 0.24 |
| K 1 (h−1) | 0.010 ± 0.004 | 0.018 ± 0.008 |
| Higuchi | ||
| r 2 | 0.87 | 0.95 |
| K H (h−1/2) | 3.6 ± 0.4 | 6.4 ± 0.4 |
| Korsmeyer-Peppas | ||
| r 2 | 0.96 | 0.93 |
| N | 0.33 | 0.51 |
| K KP (h−n) | 10.4 | 6.6 |
In the Korsmeyer-Peppas plot, n (eqn (6)) indicates the release exponent, and this value indicates the mechanism of drug release. For spherical particles, n ≤ 0.43 suggests drug release via Fickian diffusion, and values of 0.43 < n < 0.85 indicate anomalous transport that usually occurs due to a combination of diffusion and erosion processes; for n ≥ 0.85, the erosion mechanism is dominant.37 The value of n = 0.33 for Chol-PEO22-N3 (Table 3) suggests UA release dominated by a Fickian diffusion, while UA release from Chol-PEO22-GlcNAc fits better with the Higuchi model, indicating drug release occurring through the porosity of the matrix,37 resulting in negligible matrix dissolution and constant drug diffusion. The differing release mechanisms likely result from the size of the NPs, with larger Chol-PEO22-GlcNAc NPs having a lower particle density and a higher surface-to-volume ratio compared to their counterparts.
3.5. Biological assays
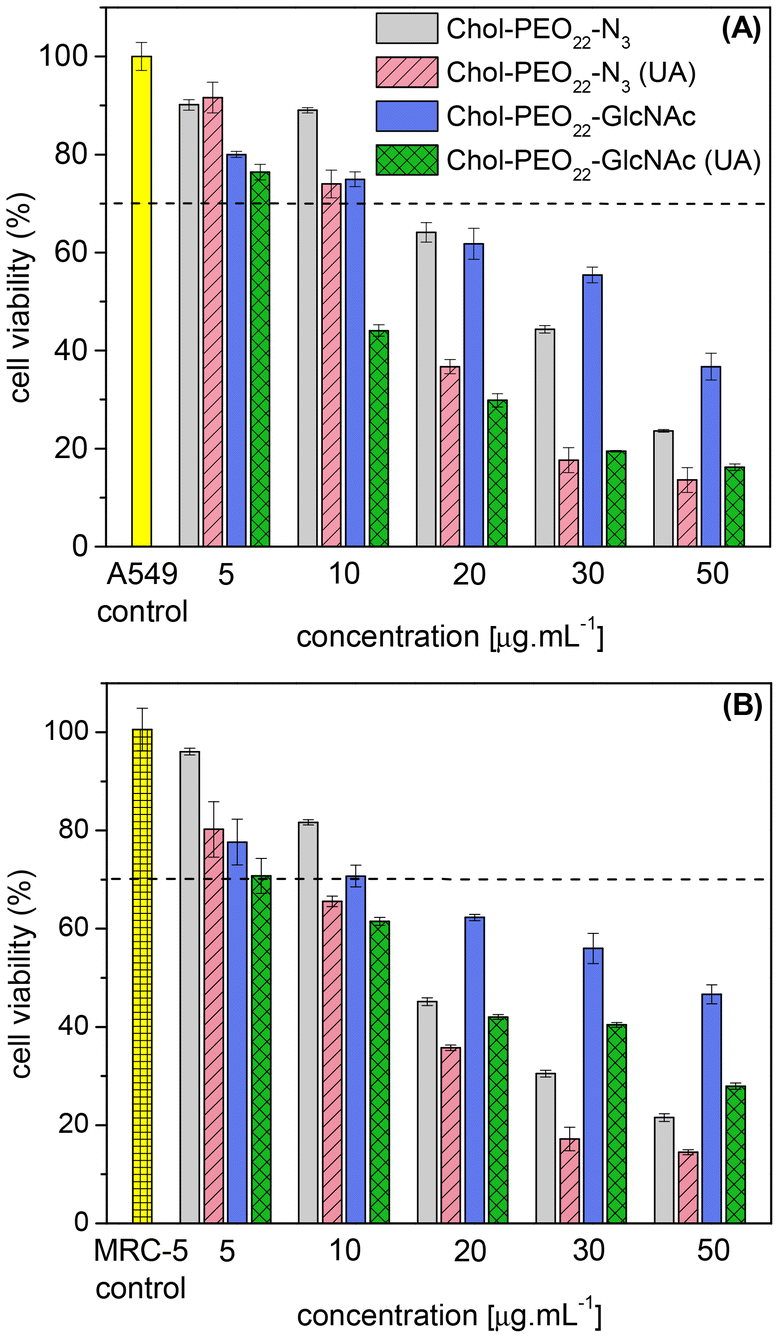 | ||
| Fig. 5 Viability of (A) A549 and (B) MRC-5 cells after 24 h exposure to various concentrations of NPs. The results were normalized to 100% viability from control cells (n ≥ 3). | ||
When the ISO 10993 5:2009 standard is considered, cytotoxic effects are considered when the cell viability is reduced by more than 30% compared to the control (untreated cells). In this regard, drug-free NPs are non-toxic at concentrations of 5 μg mL−1 and 10 μg mL−1, while UA-loaded NPs are not cytotoxic only if administered at a maximum concentration of 5 μg mL−1. The experimental data indeed clearly confirms that the UA loaded into the NPs promotes an additive cytotoxic effect.
Furthermore, when the different phenotypes are considered, there is a notably enhanced reduction in cell viability particularly for UA-lading Chol-PEO22-GlcNAc in contact with A549 cells. This can be seen, for instance, by comparing the experimental data at 30 μg mL−1. Accordingly, the presence of sugar-decorated particles reduces the toxicity of the NPs and, simultaneously, it promotes a higher cytotoxicity selectively for lung tumor A549 cells in the presence of the natural active agent.
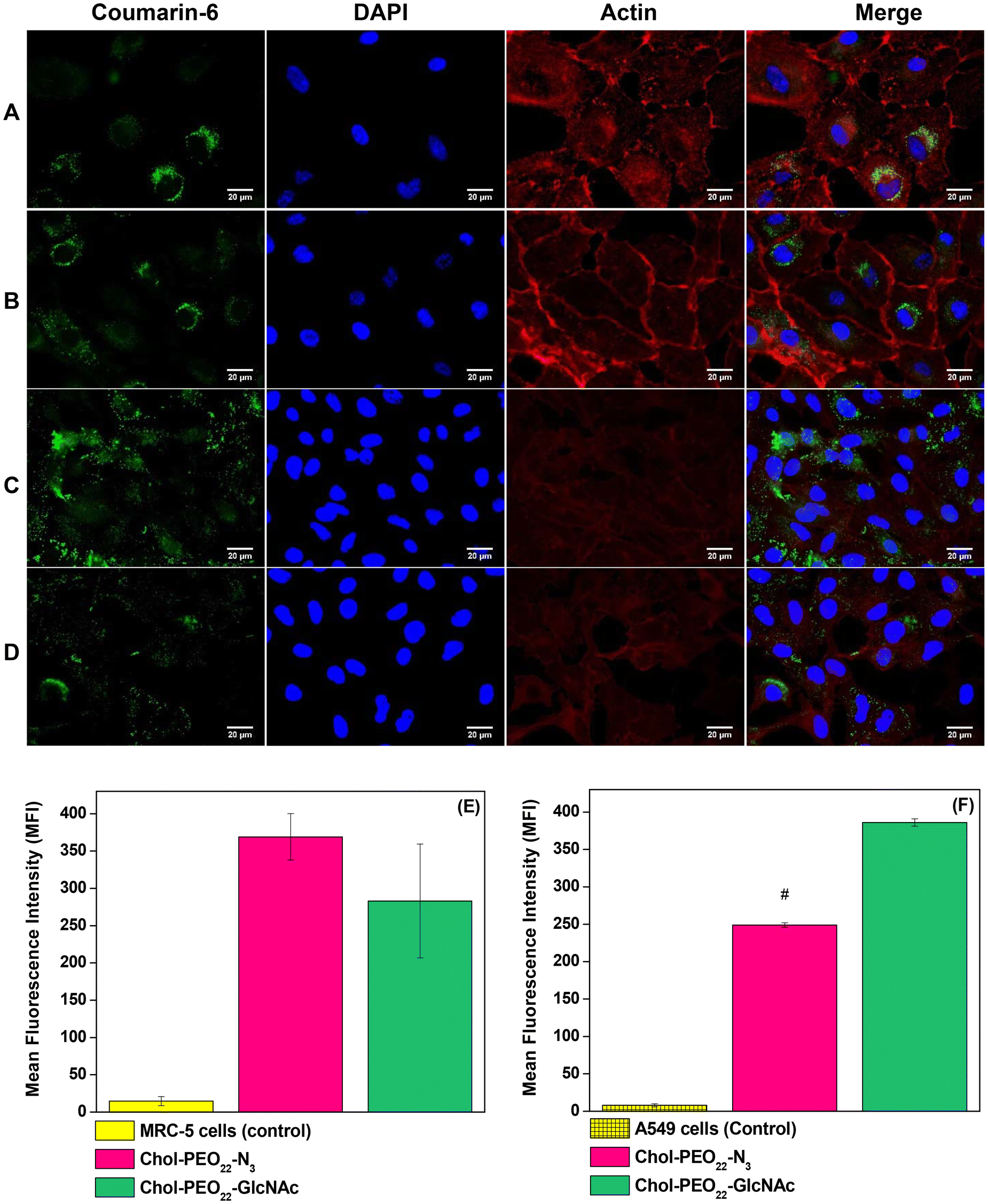 | ||
| Fig. 6 (Top) Fluorescence microscopy images of MRC-5 cells incubated for 1 h with coumarin-6-loaded (A) Chol-PEO22-GlcNAc and (B) Chol-PEO22-N3 NPs; A549 cells incubated for 1 h with coumarin-6-loaded (C) Chol-PEO22-GlcNAc and (D) Chol-PEO22-N3 NPs. (Bottom) Flow cytometry data (mean fluorescence intensity – MFI) for (E) MRC-5 and (F) A549 cells incubated for 1 h with coumarin-6-loaded NPs. | ||
The mean fluorescence intensity (MFI) of flow cytometry data (Fig. 6E and F) revealed no significant difference in uptake between Chol-PEO22-N3 and Chol-PEO22-GlcNAc NPs by MRC-5 cells. However, the MFI value for Chol-PEO22-GlcNAc NPs was significantly higher than that for Chol-PEO22-N3 NPs in A549 cells. Because glucose transporters and lectin-like receptors are overexpressed in almost all tumor cells in lung tissues11 these data suggest an increased affinity of sugar-decorated nanostructures to A549 cells. This characteristic is particularly significant for cancer therapy because tumor cells can internalize a higher quantity of sugar-decorated NPs, thereby minimizing off-target effects and enhancing therapeutic efficacy.
4. Conclusions
Herein, the manufacture of UA-loaded, sugar-decorated cholesterol-core NPs with less than 100 nm in size that are suitable for cancer treatment is reported. The nanocarriers demonstrated a high LE for UA (>70%) while maintaining colloidal stability. The in vitro cell viability assays indicated at least a two-fold increase in the inhibitory effect of the drug-loaded nanocarriers compared with their unloaded counterparts. Chol-PEO22-GlcNAc NPs are larger and presumably less dense than their sugar-free assemblies, and such features enable a more rapid and complete release of the loaded UA within approximately one week. In contrast, approximately 60% of the loaded amount was still entrapped at the core of the sugar-free self-assemblies after 7 days. Furthermore, the Chol-PEO22-GlcNAc NPs demonstrated targeted in vitro delivery, as they were internalized to a greater extent by A549 cancer cells compared to non-decorated NPs. These findings suggest significant potential for the development of advanced nanoplatforms for tumor therapy. The cholesterol core's ability to encapsulate highly hydrophobic drugs aligns with the properties of several chemotherapeutics, while the sugar molecules serve as effective ligands for targeting receptors overexpressed in cancer cells. This research article opens avenues for future studies to explore the therapeutic potential of sugar-decorated cholesterol-core nanoparticles across a broader range of cancer types and to investigate their in vivo efficacy and safety profiles. Additionally, further research could focus on optimizing the nanoparticle design to enhance targeted drug delivery and improve clinical outcomes in cancer treatment.Data availability
The relevant data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
This work was supported by FAPESP (grants 2021/12071-6, 2022/14753-0 and 2023/00558-3), CNPq (grant no. 303268/2020-4 and 305574/2023-0), and the National Institute of Science and Technology in Bioanalytics-INCTBio (FAPESP grant 2014/50867-3 and CNPq grant 465389/2014-7). We thank CEM at UFABC for granting access to its light scattering facilities, and the LNNano (CNPEM, Brazil) is acknowledged for granting access to the cryo-TEM facilities (BAG proposal 20233074). A. P. R. acknowledges the scholarship granted by FAPESP (grant 2022/14668-2), and A. G. D. acknowledges financial support from FAPESC (grant 2023TR000872) and CNPq (grant 303985/2021-6).References
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed.
- Estatísticas de Câncer no Brasil, https://www.inca.gov.br/numeros-de-cancer.
- P. Kumari, B. Ghosh and S. Biswas, J. Drug Targeting, 2016, 24, 179–191 CrossRef CAS PubMed.
- H. Zhang, X. Li, J. Ding, H. Xu, X. Dai, Z. Hou, K. Zhang, K. Sun and W. Sun, Int. J. Pharm., 2013, 441, 261–268 CrossRef CAS PubMed.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- I. M. El-Sherbiny, N. M. El-Baz and M. H. Yacoub, Glob. Cardiol. Sci. Pract., 2015, 2015, 2 CrossRef PubMed.
- E. Lepeltier, C. Bourgaux and P. Couvreur, Adv. Drug Delivery Rev., 2014, 71, 86–97 CrossRef CAS PubMed.
- C. E. de Castro, C. A. S. Ribeiro, A. C. Alavarse, L. J. C. Albuquerque, M. C. C. da Silva, E. Jäger, F. Surman, V. Schmidt, C. Giacomelli and F. C. Giacomelli, Langmuir, 2018, 34, 2180–2188 CrossRef CAS PubMed.
- S. Zhang, K. H. Chan, R. K. Prud'Homme and A. J. Link, Mol. Pharm., 2012, 9, 2228–2236 CrossRef CAS PubMed.
- H. Koo, S. Lee, J. H. Na, S. H. Kim, S. K. Hahn, K. Choi, I. C. Kwon, S. Y. Jeong and K. Kim, Angew. Chem., Int. Ed., 2012, 51, 11836–11840 CrossRef CAS PubMed.
- D. Pooja, T. Srinivasa Reddy, H. Kulhari, A. Kadari, D. J. Adams, V. Bansal and R. Sistla, Eur. J. Pharm. Biopharm., 2020, 154, 377–386 CrossRef CAS PubMed.
- H. Freichels, R. Jérôme and C. Jérôme, Carbohydr. Polym., 2011, 86, 1093–1106 CrossRef CAS.
- A. G. Dal Bó, V. Soldi, F. C. Giacomelli, B. Jean, I. Pignot-Paintrand, R. Borsali and S. Fort, Soft Matter, 2011, 7, 3453–3461 RSC.
- S.-G. Sampathkumar, M. B. Jones, M. A. Meledeo, C. T. Campbell, S. S. Choi, K. Hida, P. Gomutputra, A. Sheh, T. Gilmartin, S. R. Head and K. J. Yarema, Chem. Biol., 2006, 13, 1265–1275 CrossRef CAS PubMed.
- M. G. V. Heiden, L. C. Cantley and C. B. Thompson, Science, 2009, 324, 1029–1033 CrossRef PubMed.
- L. Lavín de Juan, V. García Recio, P. Jiménez López, T. Girbés Juan, M. Cordoba-Diaz and D. Cordoba-Diaz, J. Drug Delivery Sci. Technol., 2017, 42, 126–133 CrossRef.
- S. Yan, J. Na, X. Liu and P. Wu, Pharmaceutics, 2024, 16, 1–19 Search PubMed.
- L. Wang, Q. Yin, C. Liu, Y. Tang, C. Sun and J. Zhuang, Front. Pharmacol., 2021, 12, 706121 CrossRef CAS PubMed.
- A. Miatmoko, E. A. Mianing, R. Sari and E. Hendradi, Front. Pharmacol., 2021, 12, 1–19 Search PubMed.
- S. Zafar, K. Khan, A. Hafeez, M. Irfan, M. Armaghan, A. ur Rahman, E. S. Gürer, J. Sharifi-Rad, M. Butnariu, I. C. Bagiu and R. V. Bagiu, Cancer Cell Int., 2022, 22, 1–15 CrossRef PubMed.
- Y. M. S. Micheletto, N. P. da Silveira, D. M. Barboza, M. C. dos Santos, V. R. de Lima, F. C. Giacomelli, J. C. V. Martinez, T. E. A. Frizon and A. G. D. Bó, Colloids Surf., A, 2015, 467, 166–172 CrossRef CAS.
- A. G. Dal Bó, V. Soldi, F. C. Giacomelli, C. Travelet, R. Borsali and S. Fort, Carbohydr. Res., 2014, 397, 31–36 CrossRef PubMed.
- M. Cardoso dos Santos, B. Silva de Farias, D. da Costa Cabrera, T. Roberto, St’. A. Cadaval Junior, L. Antonio de Almeida Pinto, A. Gonçalves Dal-Bó and V. R. de Lima, Chem. Phys. Lipids, 2021, 235, 105027 CrossRef CAS PubMed.
- F. A. de Oliveira, L. J. C. Albuquerque, K. A. Riske, E. Jäger and F. C. Giacomelli, J. Colloid Interface Sci., 2020, 574, 260–271 CrossRef CAS PubMed.
- A. P. Rosso, F. A. de Oliveira, P. Guégan, E. Jager and F. C. Giacomelli, J. Colloid Interface Sci., 2024, 671, 88–99 CrossRef CAS PubMed.
- L. J. C. Albuquerque, V. Sincari, A. Jäger, J. Kucka, J. Humajova, J. Pankrac, P. Paral, T. Heizer, O. Janouškova, I. Davidovich, Y. Talmon, P. Pouckova, P. Štěpánek, L. Sefc, M. Hruby, F. C. Giacomelli and E. Jäger, J. Controlled Release, 2021, 332, 529–538 CrossRef CAS PubMed.
- E. Jäger, P. Černoch, M. Vragovic, L. J. C. Albuquerque, V. Sincari, T. Heizer, A. Jäger, J. Kučka, O. Š. Janoušková, E. Pavlova, L. Šefc and F. C. Giacomelli, Biomacromolecules, 2024, 25, 4192–4202 CrossRef PubMed.
- C. Sanson, C. Schatz, J. F. Le Meins, A. Soum, J. Thévenot, E. Garanger and S. S. Lecommandoux, J. Controlled Release, 2010, 147, 428–435 CrossRef CAS PubMed.
- J. S. Katz, K. A. Eisenbrown, E. D. Johnston, N. P. Kamat, J. Rawson, M. J. Therien, J. A. Burdick and D. A. Hammer, Soft Matter, 2012, 8, 10853–10862 RSC.
- A. Sánchez, S. P. Mejía and J. Orozco, Molecules, 2020, 25, 1–45 Search PubMed.
- N. D. Donahue, H. Acar and S. Wilhelm, Adv. Drug Delivery Rev., 2019, 143, 68–96 CrossRef CAS PubMed.
- Y.-R. Zhang, R. Lin, H.-J. Li, W.-L. He, J.-Z. Du and J. Wang, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2019, 11, e1519 Search PubMed.
- S. Alfei, A. M. Schito and G. Zuccari, Nanomaterials, 2021, 11, 2196 CrossRef CAS PubMed.
- L. S. McCarty and G. M. Whitesides, Angew. Chem., Int. Ed., 2008, 47, 2188–2207 CrossRef CAS PubMed.
- A. Markowski, A. Jaromin, P. Migdał, E. Olczak, A. Zygmunt, M. Zaremba-Czogalla, K. Pawlik and J. Gubernator, Int. J. Mol. Sci., 2022, 23, 5536 CrossRef CAS PubMed.
- L. Zhao, X. Duan, W. Cao, X. Ren, G. Ren, P. Liu and J. Chen, Foods, 2021, 10, 2470 CrossRef CAS PubMed.
- M. L. B. T.-S. to M. the D. R. from P. S. Bruschi, Ed., Woodhead Publishing, 2015, pp. 63–86.
- B. Arechabala, C. Coiffard, P. Rivalland, L. J. Coiffard and Y. de Roeck-Holtzhauer, J. Appl. Toxicol., 1999, 19, 163–165 CrossRef CAS PubMed.
- C. A. S. Ribeiro, L. J. C. Albuquerque, C. E. de Castro, B. L. Batista, A. L. M. de Souza, B. L. Albuquerque, M. S. Zilse, I. C. Bellettini and F. C. Giacomelli, Colloids Surf., A, 2019, 580, 123690 CrossRef CAS.
- X. Zhang, A. Ji, Z. Wang, H. Lou, J. Li, L. Zheng, Y. Zhou, C. Qu, X. Liu, H. Chen and Z. Cheng, J. Med. Chem., 2021, 64, 11543–11553 CrossRef CAS PubMed.
- C. E. De Castro, C. A. S. Ribeiro, A. C. Alavarse, L. J. C. Albuquerque, M. C. C. Da Silva, E. Jäger, F. Surman, V. Schmidt, C. Giacomelli and F. C. Giacomelli, Langmuir, 2018, 34, 2180–2188 CrossRef CAS PubMed.
- C. E. De Castro, C. A. S. Ribeiro, M. C. C. Da Silva, A. Goncąlves Dal-Bó and F. C. Giacomelli, Langmuir, 2019, 35, 8060–8067 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00317a |
| This journal is © The Royal Society of Chemistry 2025 |