
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePredicting absorption of compounds from an in vivo liver surface based on molecular weight or in vitro release using a dialysis membrane in combination with lipophilicity†
Fei Yuan ,
Ayaka Torigoe,
Nao Mitsudome,
Hirotaka Miyamoto,
Shintaro Fumoto,
Akira Toriba and
Koyo Nishida*
,
Ayaka Torigoe,
Nao Mitsudome,
Hirotaka Miyamoto,
Shintaro Fumoto,
Akira Toriba and
Koyo Nishida*
Graduate School of Biomedical Sciences, Nagasaki University, 1-7-1 Sakamoto, Nagasaki 852-8501, Japan. E-mail: koyo-n@nagasaki-u.ac.jp; Tel: +81-95-819-8566
First published on 1st May 2025
Abstract
A liver surface application (LSA) was developed to reduce the side effects of chemotherapy in liver cancer. The effects of molecular weight (MW) and lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) PC) on the absorption of hydrophilic and lipophilic compounds from the rat liver surface were examined. However, how these two factors simultaneously affect compound absorption remains unclear. The combined effects of MW and logPC on the absorption of these compounds in rats and mice were investigated. The compounds were administered to the liver surface using a cylindrical diffusion cell, and in vitro release experiments were conducted using a dialysis membrane to explore the relationship between release and absorption. The results indicate that log (PC/MW0.5) has a significant linear correlation with logPapp, absorption (Papp, apparent permeability coefficient). Similarly, a significant correlation was observed between log (PC × Papp, release) and logPapp, absorption. These two relationships observed in rats were used to predict compound absorption in mice, and the predicted values closely matched the experimental data. This implies that both combinations of MW and in vitro release with logPC can explain compound absorption from the liver surface. This study provided important information for understanding the absorption characteristics of LSA.
PC) on the absorption of hydrophilic and lipophilic compounds from the rat liver surface were examined. However, how these two factors simultaneously affect compound absorption remains unclear. The combined effects of MW and logPC on the absorption of these compounds in rats and mice were investigated. The compounds were administered to the liver surface using a cylindrical diffusion cell, and in vitro release experiments were conducted using a dialysis membrane to explore the relationship between release and absorption. The results indicate that log (PC/MW0.5) has a significant linear correlation with logPapp, absorption (Papp, apparent permeability coefficient). Similarly, a significant correlation was observed between log (PC × Papp, release) and logPapp, absorption. These two relationships observed in rats were used to predict compound absorption in mice, and the predicted values closely matched the experimental data. This implies that both combinations of MW and in vitro release with logPC can explain compound absorption from the liver surface. This study provided important information for understanding the absorption characteristics of LSA.
Introduction
The liver plays an essential role in maintaining homeostasis in the body, and many liver-related diseases are life-threatening. Primary liver cancer was ranked the sixth most prevalent type of cancer globally in 2022 and is recognized as the third leading cause of cancer-related mortality.1 Hepatocellular carcinoma (HCC) is the most common type of liver cancer and is difficult to diagnose before tumor formation, increasing the need to develop an effective treatment.2–4 In conventional chemotherapy, the therapeutic range of anticancer drugs is usually narrow and prone to severe side effects owing to their distribution throughout the body and at non-focal sites.5,6 Therefore, in recent years, surgical resection and local therapies, such as radiofrequency ablation,7,8 transcatheter arterial embolization,9 transcatheter arterial chemoembolization,10 and hepatic arterial perfusion chemotherapy,11,12 have been widely used as the main therapeutic approaches for HCC. Local therapy effectively reduces systemic toxicity by directly acting on the tumor site and is an important method of HCC treatment.13,14We are committed to developing innovative methods for drug delivery that specifically target the liver. Hence, we propose a liver surface application (LSA) method. This approach allows the delivery of drugs to the lesion site to enhance the targeted accumulation of drugs at the lesion site while reducing systemic side effects.15 We conducted experiments using water-soluble organic anions to elucidate the absorption properties and delivery mechanisms of drugs onto rat liver surfaces.16 Previous studies have demonstrated that drug absorption from the rat liver surface occurs through passive diffusion and is influenced by various factors, including the volume and surface area of the drug,17 MW,18 and lipophilicity.19 The individual effects of MW and lipophilicity on the absorption of compounds on the rat liver surface were examined. However, it remains unclear how these factors influence absorption simultaneously.
To explore the synergistic effects of MW and lipophilicity on the absorption of compounds in vitro, we selected semipermeable dialysis membranes made of regenerated cellulose for release experiments. In recent years, cell-based systems such as the Caco-2 cell line have been widely used in drug absorption and transport studies because their phenotype, morphology, and function are similar to those of cells in the small intestine.20–23 However, these cell models have certain drawbacks, such as long culture periods, complex procedures, and significant interlaboratory variability.24,25 To overcome these limitations, a simple artificial membrane system has been developed as an alternative.26,27 Berben et al. introduced an artificial membrane insert system (AMI system) made of a regenerated cellulose membrane that showed a strong correlation with the widely used Caco-2 cell model, exhibiting a Pearson correlation coefficient (R) of 0.95.27 Therefore, this system can be considered a high-throughput and cost-effective approach for analyzing the passive permeability of drugs.
In this study, different hydrophilic and lipophilic compounds were selected as research subjects to examine the combined effects of MW and lipophilicity on drug absorption from the liver. An in vitro release experiment was conducted using a dialysis membrane composed of regenerated cellulose to investigate the correlation between the in vitro release of the compounds and their absorption from the rat liver surface. We compared the differences between rats and mice in terms of the absorption characteristics of compounds from the liver surface. Additionally, we established in vitro and in vivo correlation models to predict compound absorption from the murine liver surface and subsequently verified the reliability of these models.
Experimental
Chemicals
5-Fluorouracil (5-FU), phenol sulfonphthalein (PSP), bromophenol blue (BPB), and bromosulfophthalein (BSP) were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). Doxorubicin hydrochloride (DOX) was obtained from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). Methyl p-hydroxybenzoate (methylparaben), butyl p-hydroxybenzoate (butylparaben), and antipyrine were purchased from FUJIFILM Wako Pure Chemical Inc. (Osaka, Japan). p-Hydroxybenzoic acid (PHBA), propyl p-hydroxybenzoate (propylparaben), and FITC-dextrans of different molecular weights: 4 kDa (FD-4), 10 kDa (FD-10), 40 kDa (FD-40), and 70 kDa (FD-70) were obtained from Sigma-Aldrich (St. Louis, MO, USA). All the other chemicals were of reagent grade.Measurement of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) PC
PC
The logPC of the compounds was determined using the shake-flask method.28 Briefly, n-octanol (Nacalai Tesque Inc., Kyoto, Japan) was mixed with distilled water at a 1:1 volume ratio and shaken for 4 h. This was followed by overnight phase separation to obtain water-saturated n-octanol. Next, water-saturated n-octanol was mixed with a buffer solution at a volume ratio of 1:1, and an appropriate amount of the compound to be measured was weighed, added to this mixture, shaken for 4 h, and allowed to stand overnight. Finally, the concentration of the compound in each phase was determined separately, and logPC was calculated (eqn (1)).
 | (1) |
Here, Cn-octanol is the concentration of the compound in the n-octanol phase, and Cbuffer is the concentration of the compound in the buffer solution phase.
Evaluation of the release of the lipophilic/hydrophilic compounds in vitro
The release rates of lipophilic and hydrophilic compounds were studied using a dialysis membrane. A solution of hydrophilic compounds (5-FU, PSP, BPB, BSP, FD-4, FD-10, PHBA, and DOX) and lipophilic compounds (methylparaben, butylparaben, propylparaben, and antipyrine) was sealed in a dialysis membrane and immersed in phosphate-buffered saline (PBS, pH 7.4) (300 mL) or phosphate buffer (PB, pH 7.4) at 37 °C and stirred. Tween 20 (0.2%) (Nacalai Tesque Inc.) was added to the buffer to reduce the non-specific adsorption of lipophilic compounds in the dialysis bag and beaker. The inner portion of the dialysis membrane containing the compound was placed on the donor side, whereas the outer portion was placed on the receiver side. The samples from the receiver side were collected at various time intervals. The release rate of the compounds through the dialysis membrane was calculated as a percentage of the added amount (eqn (2)).
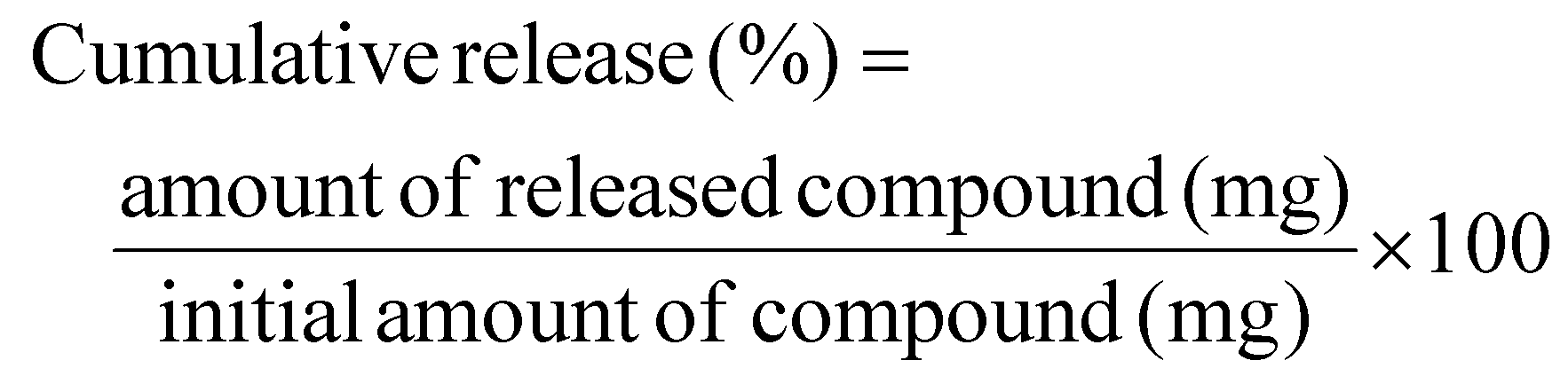 | (2) |
Animal experiment
Male Wistar rats (230–280 g) and male ICR mice (25–27 g) (Japan SLC Co., Ltd, Shizuoka, Japan) were fed standard laboratory diets. Before the experiments, the animals were housed in an air-conditioned room at ambient temperature and humidity. All animal experiments followed the Nagasaki University Animal Experimentation Guidelines.Rats (Approval Number: 2004071621) or mice (Approval Number: 2109221747) were anesthetized with a drug mix (Medetomidine hydrochloride (0.75 mg kg−1, Kyoritsu Pharmaceutical Co., Ltd, Tokyo, Japan), Midazolam (4 mg kg−1, Sandoz Pharmaceutical Co., Ltd, Tokyo, Japan), and Butorphanol tartrate (5 mg kg−1, Meiji Seika Pharma Co., Ltd, Tokyo, Japan)). During the experiments, the body temperature of the rats and mice was maintained at 37 °C using a heating lamp. A cylindrical diffusion cell was attached to the liver surface using Aron Alpha surgical adhesive (Daiichi Sankyo Co., Ltd, Tokyo, Japan). The diffusion cell used in mice had a diameter of 6 mm and an area of 0.28 cm2, while the diffusion cell used in rats had a diameter of 9 mm and an area of 0.64 cm2. Lipophilic (0.1 mL) or hydrophilic (0.05 mL) compound solutions were added dropwise directly into diffusion cells. The top of the diffusion cell was sealed with aluminum foil to prevent the evaporation of the applied solution. After a certain period, the residual solution in the diffusion cell was withdrawn for measurement.
Quantification of compounds
The concentration of the compounds released through the dialysis membrane and the concentration of the compound solution recovered in the diffusion cell were determined as follows. The concentration of 5-FU was measured spectrophotometrically using a UV-Visible spectrophotometer (UV-1600, Shimadzu Corporation, Tokyo, Japan) at a wavelength of 266 nm. The concentration of PSP was assessed spectrophotometrically at 560 nm after dilution with 1 M NaOH. The concentration of BPB was quantified spectrophotometrically at 600 nm. The concentration of BSP was determined spectrophotometrically at 580 nm after dilution with a 1 M NaOH solution. FITC-dextran concentrations were measured using a fluorescence spectrometer (RF-5300PC; Shimadzu Corp., Kyoto, Japan) at excitation and emission wavelengths of 489 and 515 nm, respectively. DOX concentrations were measured using fluorescence spectroscopy at excitation and emission wavelengths of 500 and 550 nm, respectively. The concentrations of PHBA, methylparaben, butylparaben, and propylparaben were determined spectrophotometrically at 254 nm. Finally, the antipyrine concentration was assessed using spectrophotometry at a wavelength of 242 nm.Statistical analysis
All experiments were performed in triplicate, and the means and standard deviations (SDs) were calculated. SigmaPlot (Version 14.5) (Systat Software, Inc., San Jose, CA, USA) was used for statistical analyses.Results
In vitro release of compounds from a dialysis membrane
The cumulative release of hydrophilic and lipophilic compounds with different MWs through dialysis membranes was monitored. The changes in the amount of hydrophilic compounds released to the receiver side over time are plotted (Fig. 1(a)). The release rate of the hydrophilic compounds tended to decrease with increasing MW (Table 1). The changes over time in the remaining amounts of hydrophilic compounds on the donor side at the initial stage of release were replotted on a semi-logarithmic graph (Fig. 1(b)). Under all conditions, the decreasing profiles followed a linear trend. The release of hydrophilic compounds through the dialysis membranes at the initial stage of release was considered to follow the first-order equation, as given in eqn (3).
 | (3) |
PC.
 | ||
| Fig. 1 (a) Release profiles of compounds through the dialysis membrane. (b) Semi-log plot of the remaining amounts of compounds on the donor side of the dialysis membrane release experiment (the semi-log plot of the remaining amounts of 5-FU is not displayed, as it was nearly completely released to the donor side of the dialysis membrane within 60 minutes). Each point represents the mean ± SD of three experiments. | ||
| MW | logPC |
kr (min−1 × 10−3) | |
|---|---|---|---|
| Hydrophilic compound | |||
| 5-FU | 130 | −0.87 | 45.50 |
| PHBA | 138 | −0.26 | 21.30 |
| PSP | 354 | −0.78 | 13.00 |
| DOX | 579 | −0.24 | 6.90 |
| BPB | 670 | −1.60 | 8.69 |
| BSP | 794 | −0.75 | 6.63 |
| FD-4 | 4400 | −2.87 | 2.78 |
| FD-10 | 9300 | −2.60 | 0.35 |
| Lipophilic compound | |||
| Methylparaben | 152 | 1.94 | 20.79 |
| Propylparaben | 180 | 2.77 | 16.68 |
| Antipyrine | 188 | 0.25 | 29.70 |
| Butylparaben | 194 | 3.08 | 5.11 |
Absorption properties of DOX from the rat liver surface
DOX is a widely used anthracycline anticancer drug with remarkable efficacy against a wide range of tumors, including breast cancer,29 lymphoma, leukemia,30 and liver cancer.31 In addition, its inherent fluorescent properties make it particularly suitable for studying the distribution of drugs in cells and tissues, which helps resolve the mechanism of drug action.32–34 Data on DOX absorption from the rat liver surface were incorporated into our dataset to facilitate future exploration of the spatial distribution of anticancer drugs within the liver following administration onto its surface. Consistent with findings from previous studies,16,19 the absorption process of DOX at the rat liver surface followed a first-order kinetic equation, resulting in an absorption rate constant (ka) of 0.0099 min−1 (ESI Fig. 1†).Relationship between in vitro release and in vivo absorption from the rat liver surface
To assess whether in vitro release through a dialysis membrane can predict compound absorption in LSA, we evaluated the relationship between the release of these compounds through the dialysis membrane in vitro and their absorption from the rat liver surface in vivo using the apparent permeability coefficient, Papp (eqn (4) and (5)).
 | (4) |
 | (5) |
 | ||
| Fig. 2 Correlation between Papp, absorption, and Papp and the release of compounds. The line indicates the linear regression for hydrophilic compounds. 1, 5-FU; 2, PHBA; 3, PSP; 4, BPB; 5, BSP; 6, FD-4; 7, FD-10; 8, DOX; 9, antipyrine; 10, methylparaben; 11, propylparaben; and 12, butylparaben. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. | ||
| Papp, absorption (μm min−1) | Papp, release (μm min−1) | |
|---|---|---|
| a Papp, absorption calculated based on previously reported data.18b Papp, absorption calculated based on previously reported data.19c Papp, absorption calculated based on previously reported data.35 | ||
| Hydrophilic compound | ||
| 5-FU | 25.8c | 75.83 |
| PHBA | 14.22b | 35.55 |
| PSP | 10.50a | 21.67 |
| DOX | 17.16 | 11.50 |
| BPB | 8.46a | 14.48 |
| BSP | 4.64a | 11.05 |
| FD-4 | 2.46a | 4.63 |
| FD-10 | 1.44a | 0.58 |
| Lipophilic compound | ||
| Methylparaben | 154.81b | 34.65 |
| Propylparaben | 168.58b | 27.80 |
| Antipyrine | 48.63b | 49.50 |
| Butylparaben | 157.70b | 14.10 |
A previous study indicated that when a drug is absorbed through the gastrointestinal tract (such as the stomach and small intestine) by passive diffusion, and if the drug passes through a homogeneous membrane, for instance, a pore, the following formula can be employed for description (eqn (6)).18
 | (6) |
 | ||
| Fig. 3 (a). Relationship between Papp, absorption of compounds from the rat liver surface, and 1/MW0.5. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. The line indicates the linear regression for hydrophilic compounds. (b) Relationship between the logPC and logPapp, absorption of compounds. The line indicates the linear regression for all the compounds. (c) Relationship between log(PC/MW0.5) and log Papp, absorption of compounds. The line indicated the linear regression for all compounds. 1, 5-FU; 2, PHBA; 3, PSP; 4, BPB; 5, BSP; 6, FD-4; 7, FD-10; 8, DOX; 9, antipyrine; 10, methylparaben; 11, propylparaben; and 12, butylparaben. | ||
Lipophilicity is a crucial factor influencing the absorption of compounds. Within a specific range, an increase in the logPC value correlated with improved drug absorption.19 A linear correlation was observed (R = 0.949) between logPC and logPapp, absorption of compounds on the rat liver surface (Fig. 3(b)). When examining the combined effects of the lipophilicity and MW of the compounds on absorption at the rat liver surface, we observed a strong linear correlation (R = 0.962) between these factors and compound absorption (Fig. 3(c)).
Relationship between log (PC × Papp, release) and the absorption of compounds from the rat liver surface
Based on the results shown in Fig. 2, a favorable linear relationship was observed between in vitro release and in vivo absorption of the hydrophilic compounds. In addition, the absorption of hydrophilic compounds increased to 1/MW0.5. Therefore, there may be a linear correlation between the release of hydrophilic compounds and 1/MW0.5. Inspired by Fig. 3(c), we attempted to replace 1/MW0.5 in the relationship with Papp, release. A significant linear relationship (R = 0.971) was observed between log(PC × Papp, release) and logPapp, absorption for both the hydrophilic and lipophilic compounds (Fig. 4). Furthermore, this linear correlation is better than that of 1/MW0.5 (Fig. 3(c)).
 | ||
| Fig. 4 Relationship between log(PC × Papp, release) and logPapp, absorption of hydrophilic and lipophilic compounds. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. 1, 5-FU; 2, PHBA; 3, PSP; 4, BPB; 5, BSP; 6, FD-4; 7, FD-10; 8, DOX; 9, antipyrine; 10, methylparaben; 11, propylparaben; and 12, butylparaben. | ||
Absorption characteristics of hydrophilic compounds in mice
We used mice to verify the predictability of the two relationships described above (Fig. 3(c) and 4(b)). A semi-logarithmic plot depicting the amount of hydrophilic compounds remaining in the diffusion cell following their application to the murine liver surface is shown (Fig. 5(a)). The absorption of hydrophilic compounds from the murine liver surface follows the first-order equation; the ka values of each compound were calculated from the slopes of the regression lines for each group. The remaining ratio of hydrophilic compounds 240 min post-application to the murine liver surface was investigated (Fig. 5(b)). The remaining ratio decreases with decreasing MW. The remaining ratio of FD-40, which has a MW of approximately 40 kDa, was 95.51% 240 min after administration. This suggests that although FD-40 can be absorbed by the murine liver surface, the rate of absorption is extremely slow. This indicates that the upper MW limit of the compound that can be absorbed by the murine liver surface is approximately 40 kDa. No absorption was observed for FD-70 (70 kDa) on the murine liver surface at 240 min after administration (data not shown). | ||
| Fig. 5 (a) Semi-log plot of the remaining amounts of compounds in the diffusion cell after application to the murine liver surface. (b) The remaining ratio of compounds at 240 min after application to the murine liver surface. Each point represents the mean ± SD of three experiments. | ||
The overall trend of compound absorption in rats was greater than that observed in mice (Table 3). The absorption of hydrophilic compounds in mice was significantly influenced by the MW, which was consistent with the absorption from the rat liver surface. A significant linear relationship was observed between Papp, absorption values in rats and mice (Fig. 6).
 | ||
| Fig. 6 Relationship between logPapp, absorption of hydrophilic compounds in rats and mice. 1, 5-FU; 2, PSP; 3, BPB; 4, BSP; 5, FD-4; 6, FD-10; and 7, FD-40. | ||
| Hydrophilic compound | Papp, absorption (μm min−1) | |
|---|---|---|
| Rats | Mice | |
| a Papp, absorption calculated based on previously reported data.18b Papp, absorption calculated based on previously reported data.35 | ||
| 5-FU | 25.80b | 17.38 |
| PSP | 10.50a | 4.96 |
| BPB | 8.46a | 3.79 |
| BSP | 4.64a | 3.62 |
| FD-4 | 2.46a | 2.36 |
| FD-10 | 1.44a | 1.11 |
| FD-40 | 0.23a | 0.13 |
| FD-70 | 0.10a | 0 |
Prediction of compound absorption in mice based on the absorption relationship of hydrophilic compounds in rats and mice
As shown in Fig. 6, the absorption trends of hydrophilic compounds on the liver surfaces of both rats and mice were highly consistent. Consequently, we hypothesized that a similar trend might also apply to lipophilic compounds in terms of absorption between these two species. To test this hypothesis, we predicted the absorption of the remaining compounds from the murine liver surface based on the linear relationship shown in Fig. 6.After the incorporation of logPC, the linear correlation between the absorption of hydrophilic compounds in rats and mice improved (Fig. 7(a)). We substituted the logPapp, absorption of the remaining compounds in rats for x in the linear equation presented in Fig. 7(a) to obtain the predicted value of logPapp, absorption in mice (y). The mean absolute percentage error (MAPE) was used to evaluate the predictability. MAPE is considered to have high accuracy at <10%, good accuracy at 10–20%, reasonable accuracy at 20–50%, and low accuracy at >50%. The predicted value exhibits a strong linear correlation with the experimental value, with R as high as 0.997 and MAPE as low as 1.971% (Fig. 7(b)). The results indicate that the relationship model of hydrophilic compounds in rats and mice exhibits excellent predictive performance, enabling the effective use of this model to predict the absorption of other compounds in mice. In addition, we compared the logPapp, absorption values obtained for the remaining compounds in mice with their corresponding values in rats. Consistent with our expectations, a strong linear relationship was observed between the two species, and the absorption trends were highly consistent (Fig. 8).
 | ||
| Fig. 7 (a) Relationship between log(PC × Papp, absorption) of hydrophilic compounds in rats and mice. 1, 5-FU; 2, PSP; 3, BPB; 4, BSP; 5, FD-4; and 6, FD-10. (b) Relationship between the experimental and predicted values of logPapp, absorption in mice. 7, Methylparaben; 8, propylparaben; 9, antipyrine; 10, DOX; and 11, PHBA. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. | ||
 | ||
| Fig. 8 Relationship between logPapp, absorption of compounds in rats and mice. 1, PHBA; 2, DOX; 3, antipyrine; 4, methylparaben; and 5, propylparaben. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. | ||
Comparison of predictability based on log(PC × Papp, release) or log(PC/MW0.5)
Fig. 6 and 8 show that the absorption trends of the compounds in rats and mice were highly consistent, regardless of lipophilicity. Therefore, we expected that the absorption characteristics of the murine liver surface would have a similar correlation between logPapp, absorption and log(PC × 1/MW0.5) or log(PC × Papp, release) in rats, as shown in Fig. 3(c) and 4(b). A murine model was used to verify whether these two hypothesized relationships could effectively predict the absorption of compounds on the liver surface. A linear relationship was observed between logPapp, absorption of hydrophilic compounds in mice and log(PC × Papp, release) (Fig. 9(a)). We substituted log(PC × Papp, release) of the remaining compounds for x in the linear equation shown in Fig. 9(a) to obtain the predicted value of logPapp, absorption. The results reveal a significant linear correlation between the experimental and predicted values, with an MAPE of approximately 13.054% (Fig. 9(b)). This indicates that the prediction model exhibited excellent predictive performance.
 | ||
| Fig. 9 (a) Relationship between log(PC × Papp, release) and logPapp, absorption of hydrophilic compounds in mice. 1, 5-FU; 2, PSP; 3, BPB; 4, BSP; 5, FD-4; and 6, FD-10. (b) Relationship between the experimental and predicted values of logPapp, absorption in mice. 7, Methylparaben; 8, propylparaben; 9, antipyrine; 10, DOX; and 11, PHBA. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. | ||
Similarly, we derived a linear relationship between log(PC/MW0.5) of the hydrophilic compounds and their absorption in mice (Fig. 10(a)). The log(PC/MW0.5) values of the remaining compounds were used as x in the linear equation shown in Fig. 9(a) to calculate the predicted logPapp, absorption. The results revealed a significant linear correlation between the experimental and predicted values, with an MAPE value of 10.343% (Fig. 10(b)). This demonstrates that the model exhibited excellent predictive performance.
 | ||
| Fig. 10 (a) Relationship between log(PC/MW0.5) and logPapp, absorption of hydrophilic compounds in mice. 1, 5-FU; 2, PSP; 3, BPB; 4, BSP; 5, FD-4; and 6, FD-10. (b) Relationship between the experimental and predicted values of logPapp, absorption in mice. 7, Methylparaben; 8, propylparaben; 9, antipyrine; 10, DOX; and 11, PHBA. Diamonds and circles indicate hydrophilic and lipophilic compounds, respectively. | ||
Discussion
Previously, we conducted independent investigations on the effects of MW on the absorption of hydrophilic compounds and the effects of lipophilicity on the absorption of lipophilic compounds on the rat liver surface. The results indicated that the ka of hydrophilic compounds in rat liver decreased with increasing MW.18 Within a certain range, when the logPC value increased, the ka value of the lipophilic compounds increased.19 However, the adsorption of hydrophilic and lipophilic compounds onto the liver surface was influenced by both logPC and MW. Therefore, it is essential to investigate how these two factors concurrently affect the absorption of compounds from the liver surface. For this purpose, we chose a dialysis membrane to explore the relationship between in vitro release and in vivo absorption. Furthermore, we attempted to elucidate the relationship between the physicochemical properties of the compounds (i.e., MW and logPC) and their absorption from the rat liver.
A significant linear relationship was observed between the in vitro release and in vivo absorption of hydrophilic compounds, whereas no such relationship was evident for lipophilic compounds (Fig. 2). The dialysis membrane, which is composed of regenerated cellulose, exhibits high permeability to hydrophilic compounds. Therefore, the diffusion of hydrophilic compounds was mainly affected by the MW. This phenomenon is consistent with the passive diffusion of hydrophilic compounds in a monolayer of mesothelial cells on the liver surface, which is dominated by MW.18 In contrast, there are relatively strong interactions between lipophilic compounds and dialysis membranes, which become more pronounced with increasing lipophilicity.36 This obstructs the diffusion of lipophilic compounds through dialysis membranes. However, as phospholipid bilayers constitute the cell membrane, lipophilic compounds can readily diffuse into cells through the plasma membrane. Thus, in the in vitro dialysis membrane release environment, the diffusion behavior of lipophilic compounds showed no obvious linear correlation with their absorption from the rat liver surface.
After introducing logPC, both hydrophilic and lipophilic compounds showed a strong correlation between their absorption from the rat liver surface and release in vitro through the dialysis membrane experiment, as well as their MW. Lipophilicity is a critical factor that influences drug absorption. Many previous studies have demonstrated that within a specific range, there is a significant linear relationship between the penetration of compounds on the skin surface,37 and in the oral cavity38 and logPC. The findings of this study revealed a significant linear relationship between the absorption of compounds on the rat liver surface and logPC (Fig. 3(b)). Hence, logPC can be used directly to predict drug absorption; however, its applicable scope must be ascertained. Additionally, a growing number of studies in recent years have highlighted that predicting the absorption of compounds in the gastrointestinal tract should not rely solely on logPC as a chemical descriptor.39–41 Quantitative structure–activity relationship model studies, apart from logPC, MW, hydrogen bond donors, and other chemical descriptors, have made substantial contributions to the prediction of compound absorption models.42–44 This study confirmed that the linear correlation became more pronounced upon the simultaneous introduction of MW and logPC into the model (Fig. 3(c)).
Compared with the widely used Caco-2 model for in vitro drug permeability studies, the dialysis membrane release experiment offers advantages such as good reproducibility, minimal laboratory-to-laboratory variability, and time and cost savings, allowing precise evaluation of compound permeability.24,45 However, dialysis membrane release experiments cannot simulate the active transport mechanisms of drugs or evaluate the interactions between drugs and transporter proteins. In the in vitro dialysis membrane release experiment, the compound was limited to passing through the dialysis membrane via passive diffusion alone.27,46 The surface of the liver is covered with a monolayer of mesothelial cells.47 Although there is currently no definitive evidence that mesothelial cells possess specific transport carriers, if a compound crosses or enters mesothelial cells via non-passive diffusion mechanisms (e.g., active transport), neglecting these mechanisms might lead to an underestimation of the permeability of the compound. Consequently, compound absorption from the liver surface may not exhibit a significant linear relationship with log(PC/MW0.5) or log(PC × Papp, release). Compounds with higher logPC values are likely to reach a saturation state in the liver surface cell layer, which impedes their penetration into the cell membrane and entry into the liver. This significantly limits their ability to absorb highly lipophilic compounds. Our previous studies demonstrated that the absorption of a compound from the rat liver surface is maximized when the logPC value is approximately 2.5.19 During the prediction of compound absorption from the murine liver surface, butylparaben was excluded from the prediction range because of its excessively high logPC value, which led to the predicted values significantly exceeding the experimental results. Additionally, based on our previous research, the maximum MW of compounds that can be absorbed from the rat liver surface is approximately 70 kDa.18 Therefore, when log(PC/MW0.5) or log(PC × Papp, release) is used to predict drug absorption on the liver surface, it is important to consider the specific conditions under which these linear correlations hold. These compounds cross mesothelial cells on the liver surface via passive diffusion to enter the liver. The logPC values of these compounds must meet a certain range for passage through the liver surface, and there is a MW limit for absorption at the liver surface.
After understanding the conditions under which the two linear correlations hold, we aimed to validate their predictability in mice. The use of mice for prediction can provide foundational data for characterizing pharmacokinetic features across species. In addition, mice can be used as experimental subjects to assess the effectiveness of the two relationship models in a similar physiological context, thereby verifying the universality and reliability of the models. Based on these considerations, we selected mice as the experimental subjects to validate the predictability of these two relationships.
A comparison of the absorption differences of hydrophilic compounds in rats and mice revealed that although the absorption trends in both species were similar, the absorption decreased with increasing MW (Table 3). However, the MW limit for the compounds absorbed by the murine liver surface was approximately 40 kDa (Fig. 5(b)), which was significantly different from the absorption MW limit observed in rats (70 kDa). Previous studies have demonstrated that hydrophilic macromolecular compounds can traverse various cellular barriers via paracellular pathways. For example, FITC-dextran has been shown to cross Caco-2 cell monolayers,48 epithelial barriers,49 and arterial endothelial cell monolayers50 via this route. Although there is no direct evidence that FITC-dextran crosses mesothelial cells via the paracellular pathway, the structural similarity between mesothelial tight junctions and the aforementioned barriers suggests a plausible mechanism for its passage through liver mesothelial cells. Furthermore, there may be significant differences in tight junctions and intercellular spaces of hepatic mesothelial cells between mice and rats. Murine mesothelial cells exhibit tighter junctions and smaller intercellular spaces, which could restrict the paracellular passage of hydrophilic macromolecular compounds. This structural disparity may result in a lower MW limit for absorption in mice than that in rats. However, further research is required to validate these hypotheses. Therefore, when developing a DDS (drug delivery system) for LSA, it is essential to consider species-specific differences.
Additionally, we used the linear relationship between the absorption of hydrophilic compounds from the rat liver surface and extrapolated the absorption of lipophilic compounds in mice. The results demonstrate a strong correlation between the predicted and experimental values (Fig. 7 and 8). This indicates that the absorption data from rats can be used to predict absorption in mice. Predictions in humans may be possible in the future with limited data using model compounds based on models obtained from mice or rats.
Our findings indicate that the use of the murine model validated the accuracy and reliability of log(PC × Papp, release) and log(PC/MW0.5) as predictive indicators of the absorption of compounds from the liver surface (Fig. 9 and 10). Although the MW and logPC offer initial predictions of the absorption characteristics of compounds on the liver surface, the design and application of LSA can be further refined by incorporating specific additives to modulate the absorption process. For instance, viscous additives can adjust absorption rates, thereby optimizing therapeutic outcomes. Previous studies have demonstrated that viscous additives, such as carboxymethylcellulose sodium and polyvinyl alcohol, can effectively reduce the absorption rate of the anticancer drug 5-FU on the rat liver surface, prolong its residence time in the dosing area, and thereby reduce systemic toxic reactions. Therefore, relying solely on MW and logPC may not yield accurate predictions in the presence of these additives. Therefore, log(PC × Papp, release) can be used to predict drug absorption from the liver surface in the presence of additives.
Although the absorption of compounds from the liver surface can be predicted, their spatial distribution within the liver after absorption remains unclear. Our findings demonstrate that drugs can be selectively concentrated at the site of administration in rats; however, the precise spatial distribution at the administration site, particularly at the lesion, remains unknown. Elucidation of the spatial distribution is critical for the development of future LSA formulations for clinical applications.
Conclusion
The combined effects of the MW and logPC on the absorption of compounds from the rat liver surface were investigated. A significant relationship was observed between log(PC/MW0.5) and logPapp, absorption. Additionally, a significant correlation was observed between log(PC × Papp, release) and logPapp, absorption. The absorption patterns of these compounds on the liver surfaces of rats and mice showed consistent trends. The models developed based on these two linear relationships demonstrated strong predictive capabilities for estimating the absorption of compounds from murine liver surfaces. This study provides valuable insight into the absorption characteristics of LSA.
Author contributions
Fei Yuan: writing – original draft, writing – review & editing, investigation, data curation, validation, formal analysis, and visualization; Ayaka Torigoe: investigation, data curation, and writing – review & editing; Nao Mitsudome: writing – review & editing; Hirotaka Miyamoto: writing – review & editing; Shintaro Fumoto: writing – review & editing; Akira Toriba: writing – review & editing and supervision; Koyo Nishida: conceptualization, writing – review & editing, supervision, and funding acquisition.Data availability
Original data will be available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was funded by the Grant-in-Aid for Scientific Research (C) (No. 24K09748). Fei Yuan was supported by the Otsuka Toshimi Scholarship Foundation, which contributed to the successful completion of this project.References
- N. Jokhadze, A. Das and D. S. Dizon, CA Cancer J. Clin., 2024, 74, 224–226 CrossRef PubMed.
- S. Chidambaranathan-Reghupaty, P. B. Fisher and D. Sarkar, Adv. Cancer Res., 2021, 149, 1–61 CrossRef CAS PubMed.
- J. D. Yang, P. Hainaut, G. J. Gores, A. Amadou, A. Plymoth and L. R. Roberts, Nat. Rev. Gastroenterol. Hepatol., 2019, 1–16 Search PubMed.
- S. Shetty, N. Sharma and K. Ghosh, Crit. Rev. Oncol. Hematol., 2016, 99, 129–133 CrossRef PubMed.
- A. B. El-Khoueiry, B. Sangro, T. Yau, T. S. Crocenzi, M. Kudo, C. Hsu, T.-Y. Kim, S.-P. Choo, J. Trojan, T. H. Welling, T. Meyer, Y.-K. Kang, W. Yeo, A. Chopra, J. Anderson, C. de la Cruz, L. Lang, J. Neely, H. Tang, H. B. Dastani and I. Melero, Lancet, 2017, 389, 2492–2502 CrossRef CAS PubMed.
- P. M. Lopez, A. Villanueva and J. M. Llovet, Aliment. Pharmacol. Ther., 2006, 23, 1535–1547 CrossRef CAS PubMed.
- G. Lassandro, S. G. Picchi, A. Bianco, G. Di Costanzo, A. Coppola, A. M. Ierardi and F. Lassandro, Med. Oncol., 2020, 37, 25 CrossRef CAS PubMed.
- X. M. Bai, M. Cui, W. Yang, H. Wang, S. Wang, Z. Y. Zhang, W. Wu, M. H. Chen, K. Yan and S. N. Goldberg, Radiology, 2021, 300, 458–469 CrossRef PubMed.
- C. Zhou, Q. Yao, H. Zhang, X. Guo, J. Liu, Q. Shi, S. Huang and B. Xiong, Sci. Rep., 2020, 10, 2964 CrossRef CAS PubMed.
- Y. R. Huo and G. D. Eslick, JAMA Oncol., 2015, 1, 756–765 CrossRef PubMed.
- Y. Pan, J. Mei, J. Chen, D. Zhang, J. Wang, X. Wang, M. Yi, Z. Zhou, Y. Zhang, M. Chen, R. Guo and L. Xu, Ann. Surg. Oncol., 2022, 29, 2016–2029 CrossRef PubMed.
- R. Li, W. L. Li, G. S. Yuan, H. J. Pang, Q. Li, X. Y. Hu, Y. B. Guo, J. Z. Chen and M. Y. Zang, Zhonghua Ganzangbing Zazhi, 2023, 31, 1163–1168 CAS.
- K. Kim, T. H. Kim, T. H. Kim and J. Seong, J. Hepatocell. Carcinoma, 2021, 8, 35–44 CrossRef PubMed.
- D. Kim, S. Park, W. Sohn, H. P. Hong and B. I. Kim, J. Liver Cancer, 2022, 22, 51–56 CrossRef PubMed.
- Y. Kodama, S. Fumoto, J. Nishi, M. Nakashima, H. Sasaki, J. Nakamura and K. Nishida, Biol. Pharm. Bull., 2008, 31, 1049–1052 CrossRef CAS PubMed.
- K. Nishida, N. Sato, H. Sasaki and J. Nakamura, J. Pharm. Pharmacol., 1995, 47, 227–231 CrossRef CAS PubMed.
- K. Nishida, N. Sato, Y. Nakakoga, T. Mukai, H. Sasaki and J. Nakamura, J. Pharm. Pharmacol., 1997, 49, 976–980 CrossRef CAS PubMed.
- K. Nishida, N. Sato, H. Sasaki and J. Nakamura, J. Drug Targeting, 1996, 4, 141–150 CrossRef CAS PubMed.
- K. Nishida, M. Kobayashi, H. Miyamoto, N. Yoshikawa, S. Fumoto, H. Sasaki and J. Nakamura, J. Pharm. Pharmacol., 2011, 63, 736–740 CrossRef CAS PubMed.
- X. Jin, T. L. Luong, N. Reese, H. Gaona, V. Collazo-Velez, C. Vuong, B. Potter, J. C. Sousa, R. Olmeda, Q. Li, L. Xie, J. Zhang, P. Zhang, G. Reichard, V. Melendez, S. R. Marcsisin and B. S. Pybus, J. Pharmacol. Toxicol. Methods, 2014, 70, 188–194 CrossRef CAS PubMed.
- Y. Sambuy, I. De Angelis, G. Ranaldi, M. L. Scarino, A. Stammati and F. Zucco, Cell Biol. Toxicol., 2005, 21, 1–26 CrossRef CAS PubMed.
- N. Panse and P. M. Gerk, Int. J. Pharm., 2022, 624, 122004 CrossRef CAS PubMed.
- C. L. Pires, C. Praça, P. A. T. Martins, A. L. M. Batista de Carvalho, L. Ferreira, M. P. M. Marques and M. J. Moreno, Pharmaceutics, 2021, 13, 1563 CrossRef CAS PubMed.
- P. Berben, J. Brouwers and P. Augustijns, Int. J. Pharm., 2018, 537, 22–29 CrossRef CAS PubMed.
- M. Kus, K. Gorniak, P. Czaklosz, A. Olejnik, P. Skupin-Mrugalska, I. Ibragimow and H. Piotrowska-Kempisty, Molecules, 2022, 27, 2232 CrossRef CAS PubMed.
- P. Berben, A. Bauer-Brandl, M. Brandl, B. Faller, G. E. Flaten, A.-C. Jacobsen, J. Brouwers and P. Augustijns, Eur. J. Pharm. Sci., 2018, 119, 219–233 CrossRef CAS PubMed.
- P. Berben, J. Brouwers and P. Augustijns, J. Pharm. Sci., 2018, 107, 250–256 CrossRef CAS PubMed.
- C. D. Schönsee and T. D. Bucheli, J. Chem. Eng. Data, 2020, 65, 1946–1953 CrossRef.
- R. E. Nicoletto and C. M. Ofner 3rd, Cancer Chemother. Pharmacol., 2022, 89, 285–311 CrossRef CAS PubMed.
- Q. Zhai, Y. Chen, J. Xu, Y. Huang, J. Sun, Y. Liu, X. Zhang, S. Li and S. Tang, Mol. Pharm., 2017, 14, 3888–3895 CrossRef CAS PubMed.
- R. T. P. Poon and N. Borys, Expert Opin. Pharmacother., 2009, 10, 333–343 CrossRef CAS PubMed.
- P. Mohan and N. Rapoport, Mol. Pharm., 2010, 7, 1959–1973 CrossRef CAS PubMed.
- G. Li, M. Pei and P. Liu, Colloids Surf., A, 2020, 603, 125258 CrossRef CAS.
- J. Li, Y. Yang and P. Liu, Mol. Pharm., 2023, 20, 1426–1434 CrossRef CAS PubMed.
- Y. Kodama, M. Horishita, S. Fumoto, T. Mine, H. Miyamoto, N. Yoshikawa, H. Hirata, H. Sasaki, J. Nakamura and K. Nishida, J. Pharm. Pharmacol., 2012, 64, 1438–1444 CrossRef CAS PubMed.
- L. Groth and A. Jørgensen, Anal. Chim. Acta, 1997, 355, 75–83 CrossRef CAS.
- T. Uchida, M. Yakumaru, K. Nishioka, Y. Higashi, T. Sano, H. Todo and K. Sugibayashi, Chem. Pharm. Bull., 2016, 64, 1338–1346 CrossRef CAS PubMed.
- Y. Kurosaki, N. Aya, Y. Okada, T. Nakayama and T. Kimura, J. Pharmacobio-Dyn., 1986, 9, 287–296 CrossRef CAS PubMed.
- Y. Akiyama, N. Matsumura, A. Ono, S. Hayashi, S. Funaki, N. Tamura, T. Kimoto, M. Jiko, Y. Haruna, A. Sarashina, M. Ishida, K. Nishiyama, M. Fushimi, Y. Kojima, T. Fujita and K. Sugano, Pharm. Res., 2023, 40, 359–373 CrossRef CAS PubMed.
- E. Price, J. C. Kalvass, D. DeGoey, B. Hosmane, S. Doktor and K. Desino, J. Med. Chem., 2021, 64, 9389–9403 CrossRef CAS PubMed.
- S. Sharma, C. Kogan, M. V. S. Varma and B. Prasad, Drug Metab. Pharmacokinet., 2023, 53, 100518 CrossRef CAS PubMed.
- T. J. Hou, W. Zhang, K. Xia, X. B. Qiao and X. J. Xu, J. Chem. Inf. Comput. Sci., 2004, 44, 1585–1600 CrossRef CAS PubMed.
- K. Lanevskij and R. Didziapetris, J. Pharm. Sci., 2019, 108, 78–86 CrossRef CAS PubMed.
- S. Mathew, D. Tess, W. Burchett, G. Chang, N. Woody, C. Keefer, C. Orozco, J. Lin, S. Jordan, S. Yamazaki, R. Jones and L. Di, J. Pharm. Sci., 2021, 110, 1799–1823 CrossRef CAS PubMed.
- M. di Cagno, H. A. Bibi and A. Bauer-Brandl, Eur. J. Pharm. Sci., 2015, 73, 29–34 CrossRef CAS PubMed.
- H. A. Bibi, M. di Cagno, R. Holm and A. Bauer-Brandl, Int. J. Pharm., 2015, 493, 192–197 CrossRef CAS PubMed.
- S. E. Mutsaers, Int. J. Biochem. Cell Biol., 2004, 36, 9–16 CrossRef CAS PubMed.
- M. Maruyama, Y. Nishida, H. Tanaka, T. Minami, K. I. Ogawara, M. Miyake, Y. Takamura, H. Kakuta and K. Higaki, Eur. J. Pharm. Biopharm., 2022, 180, 332–343 CrossRef CAS PubMed.
- Y. Matsukawa, V. H. Lee, E. D. Crandall and K. J. Kim, J. Pharm. Sci., 1997, 86, 305–309 CrossRef CAS PubMed.
- R. Hashida, C. Anamizu, Y. Yagyu-Mizuno, S. Ohkuma and T. Takano, Cell Struct. Funct., 1986, 11, 343–349 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5pm00054h |
| This journal is © The Royal Society of Chemistry 2025 |