
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimizing mesoporous silica synthesis procedures to enhance their potential as nanoplatforms in therapeutic applications†
Olia Alijanpourtoloutia,
Gamini Senanayakea,
Sulev Koksb and
David J. Henry *a
*a
aSchool of Mathematics, Statistics, Chemistry and Physics, College of Science, Technology, Engineering and Math, Murdoch University, WA 6150, Australia. E-mail: d.henry@murdoch.edu.au
bCentre for Molecular Medicine and Innovative Therapeutic, Murdoch University, WA 6150, Australia
First published on 15th May 2025
Abstract
PARK7 mRNA encodes the DJ-1 protein, which functions as a protective agent against oxidative stress and cell damage within brain cells. Mutations in the mRNA can lead to reduced production of DJ-1 and initiate brain diseases such as Parkinson's disease. Transport of appropriate mRNA to damaged brain cells may provide a suitable treatment. Mesoporous silica nanoparticles (MSNPs), particularly pore-expanded and dye-labeled varieties, are regarded as potential carriers for large therapeutic agents such as mRNA. This study explored the influence of alterations in reaction conditions on the structural characteristics of MSNPs to produce nanoparticles with favorable characteristics for delivering large therapeutic agents to target sites. One-stage and two-stage procedures were compared for the introduction of 3-aminopropyltriethoxysilane (APTES) and an APTES−dye adduct, in conjunction with two different surfactants, cetyltrimethylammonium bromide (CTAB) and cetyltrimethylammonium chloride (CTAC). Analysis of the MSNPs shows that the two-stage method using CTAB as a surfactant produced amine-functionalized, dye-labelled particles with smaller overall size and better uniformity than the one-stage approach. However, due to their small pore size (<10 nm), these particles were unable to encapsulate the PARK7 mRNA (926 nucleotides). The one-stage method via CTAC produced MSNPs with a large pore size (150 nm), broad pore distribution (10–20 nm), and high aggregation, limiting their suitability for brain-targeted gene delivery. In comparison, the two-stage method using CTAC yielded well-ordered MSNPs with an optimal size (80 nm) and pore diameters (15–20 nm), enabling effective encapsulation of the large PARK7 mRNA and offering strong potential for future brain gene therapy studies.
1. Introduction
Parkinson's and Alzheimer's diseases are brain disorders that slowly destroy memory and thinking skills. The role of congenital genetic mutations and abnormal regulation of gene expression in the early stages of brain disorders has been demonstrated.1 Gene therapy offers the potential to treat both genetic and acquired brain disorders.2 While gene therapy using small interfering RNA (siRNA) is showing success in various clinical trials,3 delivering larger therapeutic agents such as DNA and RNA remains challenging due to their high molecular weight and negative charge, which limit their ability to cross cellular membranes.4 Moreover, they can be easily degraded by the enzymes in the complex physiological microenvironment. The complexity of the blood–brain barrier (BBB) presents another limitation by effectively blocking most therapeutic agents from reaching brain cells. Therefore, an efficient delivery system capable of carrying and protecting these large therapeutic agents while crossing the BBB is crucial for the successful gene therapy of brain disorders.4 In the past decade, various delivery systems have been explored, including lipid nanoparticles, viruses, and ligand-conjugated RNAs.5 Among them, mesoporous silica nanoparticles (MSNPs) offer a promising solution to overcome this challenge by facilitating the delivery of therapeutic agents into damaged brain cells.6MSNPs are a type of nanomaterial characterized by a well-ordered pore structure with pore sizes typically ranging from 2 to 50 nanometers.7 The unique properties of MSNPs such as high surface area, easy control of internal and external surface properties, tuneable pore size, the ability of surface functionalization with targeting ligands, biocompatibility, and the ability to cross the BBB make them desirable nanocarriers for loading and delivering of large quantities of therapeutic agents to damaged brain cells.8,9 The capacity of MSNPs for encapsulating therapeutic agents and their release rate largely depend on the overall dimensions and the pore size of MSNPs. While small-molecule delivery with conventional MSNPs with pore sizes ranging from 2 to 5 nm has demonstrated success and notable benefits, the encapsulation of larger therapeutic biomolecules is constrained, leading to pore blockage.6 To enhance the transfection efficacy of larger bioactive molecules and to mitigate cytotoxicity and enzymatic degradation, it is imperative to optimize the pore size of MSNPs based on the cargo size and the constraints of controlled release.10,11 While a smaller pore size might result in restricted loading and release of a payload at a target site, a larger pore size can result in premature release of the payload before reaching the target site, causing unwanted side effects or toxicity.12 Numerous instances of successful MSNP-mediated delivery of small biomolecules exist incorporating various release mechanisms including pH responsiveness,13–15 magnetic responsiveness,16–18 redox responsiveness,19,20 enzyme responsiveness,21,22 and hypoxia responsiveness23,24 while large biomolecules can only be carried on the nanoparticle surface25,26 unless MSNPs with large enough pores are obtained.27–29
The pore size of hexagonal mesoporous silica can be increased to more than 300 Å by increasing the hydrophobic volume of the self-assembled aggregates. This can be achieved by changing the copolymer composition or block sizes, or by adding cosolvent organic hydrophobic molecules.30 A modified Stober's process with the incorporation of site-directing agents such as polystyrene-block-poly(ethylene oxide)-block-polystyrene,31 poly(methyl methacrylate)-block-poly(ethylene oxide)32 diblock copolymers, poly(ethylene oxide)-block-poly(methyl methacrylate)-block-polystyrene,33 and (F127, Pluronic® P123) triblock copolymers34 has been used to synthesize MSNPs with pore sizes ranging between 2 and 50 nm.12 The size of pores and specific surface area of the mesoporous silica are controlled by the morphologies of the microdomains formed in the silica matrices, which are quite dependent on the compositions, molecular weights, and architectures of the block copolymers.35 Although applying copolymers as a template resulted in the preparation of well-ordered hexagonal mesoporous silica structures, this method is an expensive process due to the high cost of surface-active polymers as well as the difficulties of removing these template molecules.36 The challenge remains to find an inexpensive and simple method to obtain uniform mesoporous silica with large pores. An alternative strategy employing a variety of hydrophobic additives has been employed for pore size expansion, including aromatic hydrocarbons,37 long-chain alkanes,38 and long-chain alkylamines39 that enlarge the micelle size of the templates and enlarge the pore size. In the biphasic stratification method, the self-assembly of silicate species with a surfactant at the interface of the immiscible phases of water and oil, results in dendrimer-like mesoporous silica nanoparticles with very uniform pore sizes.40 1,3,5-Trimethylbenzene (TMB),41,42 ethylbenzene,43 decane,38,44 hexane,45,46 heptane,47 1,3,5-triisopropylbenzene (TIPB),48,49 cyclohexane,50,51 octane,44 and N,N-dimethylhexadecylamine52 are the most commonly used pore expanders used in this method. For example, Fuertes et al. used N,N-dimethylhexadecylamine (DMHA) and TMB as swelling agents to synthesize pore-expanded MSNPs which can adsorb a large amount of protein.53 Shafiee et al. obtained well-tuned pores in MSNPs using n-heptane, which were suitable for drug delivery.54 Paris et al. prepared MSNPs with four different pore sizes using cyclohexane.51 In the Ashley group, mesoporous silica nanoparticle-supported lipid bilayers were synthesized for loading siRNAs and delivering them to Hep3B and hepatocyte cells. These nanoparticles had a diameter of 180 nm and a pore size ranging from 20–30 nm. The synthesis involved utilizing an oil phase consisting of hexadecane with 3 wt% ABIL EM 90, a non-ionic emulsifier soluble in the oil phase.55 The binding of siRNA-loaded MSNPs to cells is dependent on the presence of an appropriate targeting peptide. This binding occurs through an endocytic pathway and leads to the induction of gene expression targeted by the siRNAs carried by the nanoparticles. Fujimoto et al. reported the synthesis of 100 nm spherical MSNPs in the presence of five different hydrophobic agents (benzene, toluene, TMB, hexane, and cyclohexane) through a biphasic stratification approach, and compared the effect of hydrophobic agents on pore size distribution. Unlike benzene and toluene, which were almost ineffective in pore expansion, effective pore expansions on the MSNPs synthesized in the presence of TMB, hexane, and cyclohexane were observed. The largest pore size was obtained for the MSNPs synthesized with cyclohexane, followed by hexane, TMB, toluene, and benzene. In addition, excluding hexane and cyclohexane, the particle size was increased significantly when other hydrophobic agents were used.56 An important advantage of the biphasic approach is that it enables the pore size and particle morphology to be tuned with minimal modifications of synthetic conditions and also allows facile incorporation of dyes and imaging agents and modification of particle and pore surface chemistry, depending on the intended application, without shape or uniformity alteration.40,57
In addition to the pore size, the overall size of nanoparticles is another important factor for the efficient performance of nanocarriers. Torrano and co-workers demonstrated that nanoparticles in the 50–100 nm range can escape the usual endocytic pathway, moving directly into the cytosol and, hence, have high potential to become essential platforms for intracellular-targeted drug and gene delivery. In comparison, particles larger than 150 nm are internalized by conventional endocytosis.58 One study investigated the biodistribution and excretion of polyethylene glycol (PEG)-MSNPs with different particle sizes (80, 120, 200, and 360 nm) in an animal model through tail-vein injection. This study revealed that small particles could easily escape from the liver and spleen and had slower biodegradation and a much lower excretion rate of biodegradation products.59 Chen et al. compared the efficiency of a series of uniform MSNP structures with various surface charges and two different sizes (50 and 200 nm), on passing the blood–brain barrier (BBB). Zebrafish were used as a model for screening in vivo BBB permeability and toxicity tests. The results indicated that only negatively charged MSNPs could successfully penetrate the BBB while the detection of MSNPs in the brain of zebrafish decreased for the large (200 nm) MSNPs. These results suggested that the transport of MSNPs across the BBB is charge- and size-dependent.60 The Betzer group61 also reported that only NPs smaller than 100 nm can overcome the BBB. Based on the findings outlined above, MSNPs with a large pore size (>15 nm) and an overall size smaller than 100 nm should be suitable for drug and gene delivery vectors for the treatment of brain diseases.62
An important benefit of MSNPs is the facility to selectively functionalize the external and internal surfaces with organic and inorganic groups.63 In clinical applications, the external surface is commonly conjugated with targeting molecules to achieve a selective recognition of a specific target cell. In comparison, internal functionalization is often performed to control therapeutic agents’ loading and release kinetics.64 For efficient therapeutic nucleic acid loading and delivery, positive charges are needed on the MSNP's internal surface, capable of interacting with negatively charged nucleic acids to form a delivery complex.65 The methods of introducing positive charge into MSNPs typically involve surface grafting with positively charged functional groups.11 Suitable functional groups include short-chain amines66 incorporated using APTES67 and APTMS,68 and cationic polymers such as PEI69 and PDEAEMA70 that form strong covalent linkages with silanol groups. These functional groups are also highly desirable for further coupling of various bioactive molecules.71 Amino-functionalized MSNPs can be prepared by two methods, co-condensation and post-synthesis grafting of amines. Co-condensation is the functionalization of the inorganic support by incorporating the organic group directly during the synthesis of the particles in a single-stage “one-pot” process. Grafting is a post-synthesis treatment, where the organic functionality is covalently attached to the inorganic network through silanol groups. However, the co-condensation method has been proven to ensure a more homogeneous distribution of amino groups on both the inner and outer surfaces of MSNPs,72 while maintaining the overall spherical morphology.73
Imaging plays a key role in the preclinical evaluation of drug delivery systems, and it has provided important insights into their mechanism of action and therapeutic effects.74 Fluorescent dye molecules incorporated inside mesoporous silica show favourable optical characteristics due to their controlled host–guest interactions which make them suitable for bioimaging and bioscience.75 Dye molecules can also be entrapped into the pores of the growing silica matrix by electrostatic interactions with surfactant molecules. After the removal of the surfactant, dye molecules reside inside the mesopores due to the interaction of the positively charged dye with negatively charged silanol groups present on the surface of the particles.76 Wei and co-workers synthesized fluorescent MSNPs via a one-pot surfactant templated method using derivatives of 9,10-distyrylanthracene with alkoxyl end groups as fluorogenic agents and a cationic surfactant as a structure-directing template through a non-covalent interaction for both cell imaging and cancer therapy applications.77
In most cases, covalent attachment is preferred because it can reduce the dye leakage to the surrounding environment and hence increase the photostability of the dye molecule.78 The first report of covalent attachment of dye molecules into the matrix of silica NPs was by van Blaaderen and Vrij.79 This involved the coupling of fluorescein isothiocyanate (FITC) to an oxysilane coupling agent (3-aminopropyl) triethoxysilane (APTES), and the controllable incorporation of the reaction product into the silica matrix.79 Other dye−amino silane adducts have been reported for dyes with highly reactive groups. For example, the carboxyl group of rhodamine B (RhB) can react with the amino groups of APTES through an amidation reaction to make a dye−APTES adduct.80,81
As mentioned above, methods for synthesizing large-pore MSNPs are well-established. However, modifications of MSNPs through dye labeling and amine functionalization can affect the physical and chemical properties of MSNPs,82 including accessible pore space and overall size, which can limit their application, especially for encapsulating and delivering large biomolecules across the BBB.83,84 The aim of this study was to develop a suitable synthesis procedure for amine-functionalized dye-labeled MSNPs with large pores to potentially accommodate large biomolecules while preserving overall dimensions <100 nm and examine the ability of these nanoparticles for encapsulation of PARK7 mRNA, which acts as a protective agent against oxidative stress and cell damage within brain cells.
2. Materials and methods
2.1. Chemicals and reagents
Tetraethyl orthosilicate (TEOS) (Sigma-Aldrich, 78-10-4, 98%), CTAB (Sigma-Aldrich, 57-09-0, ≥99%), CTAC (25% in water, Sigma-Aldrich, 112-02-7), APTES (Sigma-Aldrich, 919-30-2, ≥98%), rhodamine B (RhB) (Sigma-Aldrich, 81-88-9, ≥95%), triethanolamine (Sigma-Aldrich, 102-71-06, 99%), cyclohexane (Chem-supply, CA019, ≥99%), ammonium hydroxide (NH4OH) (Chem-supply, UN2672, 28–30% w/v), and heparin (Sigma-Aldrich, H3393-10KU, grade A) were used as received. Absolute ethanol (Ajax Fine Chem, 99.5%) and Millipore-filtered deionized water (D.H2O) were used throughout the experimental work. HPLC and LC/MS grade acetonitrile, water, acetonitrile with formic acid (0.1%), TURBO DNA-free™ Kit (catalog number AM1907), and RNaseOUT™ (catalog number 10777019) were procured from Fisher Chemical. PARK7 (mRNA) was prepared by the Queensland University (mRNA ID 2022-50_M6). Monarch® RNase A (lot number 10137958) was supplied from New England Biolabs (NEB). The SH-SY5Y cell line was obtained from American Type Culture Collection (ATCC) and cultured in Opti-MEM media supplemented with 10% fetal bovine serum, 1 mM sodium pyruvate, MEM nonessential amino acids (Gibco), and 1% penicillin/streptomycin at 37 °C under a humidified atmosphere of 5% CO2 for 24 h. Cells were plated at a density of 1 × 104 cells per well onto 12-well plates and incubated for another 24 h to be ready for the cell viability test.2.2. Synthesis of MSNPs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.39 CTAC:0.16 TEA:0.058 APTES:0.0016 Rh-B:21.14 cyclohexane:263.4 deionized water:1.12 ethanol.:0.15 CTAB:0.15 NH4OH:370.7 H2O:3.82 EtOH:2.45 cyclohexane. Then, the nanoparticles were washed twice with deionized water (D·H2O) and once with absolute ethanol. Next, the surfactant removal process involved refluxing the nanoparticles in acidic ethanol (20 mL of 37% HCl in 100 mL of absolute ethanol) at 60 °C for 12 h under stirring at 700 rpm. The resulting precipitate was collected via centrifugation and subjected to three washes with absolute ethanol. The final step in the nanoparticle synthesis involved drying the sample using a freeze-dryer.:0.15 CTAB:0.15 NH4OH:0.094 APTES:5.58 × 10−4 rhodamine B:370.7 H2O:3.82 EtOH:2.45 cyclohexane.:0.058 APTES. The product was named ADP-MSNPs-CTAB3.
:2.39 CTAC:0.16 TEA:0.058 APTES:0.0016 Rh-B:21.14 cyclohexane:263.4 deionized water:1.12 ethanol.:0.15 CTAB:0.15 NH4OH:370.7 H2O:3.82 EtOH:2.45 cyclohexane. Then, the nanoparticles were washed twice with deionized water (D·H2O) and once with absolute ethanol. Next, the surfactant removal process involved refluxing the nanoparticles in acidic ethanol (20 mL of 37% HCl in 100 mL of absolute ethanol) at 60 °C for 12 h under stirring at 700 rpm. The resulting precipitate was collected via centrifugation and subjected to three washes with absolute ethanol. The final step in the nanoparticle synthesis involved drying the sample using a freeze-dryer.:0.15 CTAB:0.15 NH4OH:0.094 APTES:5.58 × 10−4 rhodamine B:370.7 H2O:3.82 EtOH:2.45 cyclohexane.:0.058 APTES. The product was named ADP-MSNPs-CTAB3.The general procedure for LC-MS. The RhB standard solutions and ADP-MSNP samples were prepared and passed through a 0.2 μm syringe filter. The separation was performed using the isocratic flow of equal contributions of ammonium acetate (4 mM) and acetonitrile (with 0.1% formic acid) as the mobile phases, while MS studies were conducted in positive ionization mode, optimizing various MS conditions. The flow rate was set at 0.4 mL min−1. The injection volume was 1 μL. The interface temperature was set 300 °C while the desolvation line and heat block temperatures were set at 250 and 400 °C, respectively. The interface voltage was maintained at 3 kV. Nitrogen was used as the nebulizing gas at a flow of 3 L min−1, while drying gas and heating gas flows (also nitrogen) were maintained at 10 L min−1. Argon was used as the collision gas at 230 kPa.
The general procedure for HPLC. RhB standard solutions and supernatants from ADP-MSNP suspension samples were filtered using a 0.45 μm syringe filter prior to HPLC-UV analysis. RP-HPLC was performed with acetonitrile/water/phosphoric acid (30
:70:0.1) as mobile phase A and acetonitrile as mobile phase B, implemented under a gradient program that decreased polarity. The elution gradient began at 0% phase B, where it was held for 2 min, before ramping to reach 90% phase B at 15 min, maintaining this for 5 min. Between samples, the post-run equilibration time was 2 min. The analysis parameters included a UV wavelength of 553 nm, a flow rate of 0.4 mL min−1, and an injection volume of 10 μL.
 | (1) |
The measurements were carried out in triplicate, and data are shown as mean ± SEM.
PARK7 mRNA encapsulation ability of MSNPs. To prepare each complex, 5 μl of naked PARK7 mRNA (0.1 μg μL−1) was incubated with 5 μL of ADP-MSNPs at varying concentrations (1, 2, 4, 8, and 12 mg mL−1) at 4 °C in PBS. After 1 h, 5 μL of loading dye (15% Ficoll, 0.5% bromophenol blue, and xylene cyanol FF 0.25%) was added to 5 μL of the mixtures; then 4 μL of these were loaded onto a 2% agarose gel. Electrophoresis was performed at 100 V for 1 h in TAE buffer (1×). Following 10 minute staining with 0.004% Redsafe in TAE and 1 h destaining in deionized water, the gel was ready for visualization using a UV transilluminator.
Optimizing the time effect of RNase A on PARK7 mRNA. Gel electrophoresis was performed for three different RNase A
:mRNA ratios (1:20, 1:30, and 1:50) with a fixed mRNA amount of 0.5 μg after 5 and 30 min of incubation. The results were compared to evaluate the time-dependent effect of RNase A on mRNA degradation.
Optimization of the PARK7 mRNA/RNase A ratio. Solutions with varying RNase A
:mRNA ratios (1:20, 1:30, 1:50, 1:100, 1:1000, 1:2000, and 1:3000) were prepared, each containing a fixed mRNA amount of 0.5 μg, and incubated for 5 min at 37 °C. A gel electrophoresis assay (2% agarose gel, 120 mV, 1 h) was conducted to assess mRNA degradation by RNase A.
Inactivation of RNase A with RNaseOUT™ (HPRI). Two experiments were conducted to confirm the inactivation of RNase A by HPRI:
(A) Two sets of solutions with varying RNase A:PARK7 mRNA ratios (1:1000, 1:2000, and 1:3000) were incubated at 37 °C for 5 min. One of them was then further treated with 40 units of HPRI and incubated for an additional 30 min at 37 °C. Gel electrophoresis was performed to compare the band patterns of both sets and analyze the results.
(B) RNase A (0.0005 μg) was incubated with two different amounts of HPRI (10 and 40 units) for 30 min to test its inactivation. Subsequently, mRNA (0.5 μg) was added to the solutions. To further confirm the ineffectiveness of heparin—a reagent required for releasing encapsulated mRNA from the MSNP structure—similar experiments were performed by 30 min additional incubation with heparin. Gel electrophoresis was performed for all samples at 120 mV for 1 h.
RNase A protection assay. PARK7 mRNA/MSNP complexes were prepared by mixing 60 μg of MSNPs (ADP-MSNPs-CTAB2 & ADP-MSNPs-CTAC2) with 0.5 μg of mRNA in a final volume of 10 μL in PBS, and stored at 4 °C for 1 h. Naked mRNA (0.5 μg) was dissolved in 10 μL of PBS. To generate complex solution of mRNA/RNase A, 0.5 μg of mRNA in 5 μL of PBS was incubated with 5 μL of RNase I (0.0005 μg) in a buffer containing 10 mM Tris-HCl, 2.5 mM MgCl2, and 0.5 mM CaCl2 (pH 7.6) at 37 °C for 5 min. Then, RNase A was inactivated by adding 1 μL of HPRI (40 unit) and incubating for 30 min. The mRNA was then released from mRNA/MSNP complexes by treatment with 5 μL of heparin (10 mg mL−1) at 37 °C for 30 min. Finally, all samples were loaded onto a 2% agarose gel, and after electrophoresis at 120 mV for 1 h, the bands in the gel were compared.
DNase I protection assay. PARK7 mRNA/MSNP complexes were prepared by mixing 60 μg of ADP-MSNPs with 0.5 μg of mRNA in a final volume of 10 μL in PBS and stored at 4 °C for 1 h. Naked mRNA (0.5 μg) was dissolved in 10 μL of PBS. The turbo DNA-free kit was used as a DNase I reagent. To prepare the mRNA/DNase I complex solution, 0.5 μg of mRNA was incubated with 5 μL of DNase I (0.25 U μL−1, diluted in a buffer containing 10 mM Tris-HCl, 2.5 mM MgCl2, and 0.5 mM CaCl2, pH 7.6) at 37 °C for 30 min. Then, DNase I was inactivated by adding 5 μL of 25 mM EDTA. The mRNA was then released from mRNA/MSNP complexes by treatment with 5 μL of heparin (10 mg mL−1) at 37 °C for 30 min. Finally, all samples were loaded onto a 2% agarose gel, and after electrophoresis at 120 mV for 1 h, the bands in the gel were compared.
2.3. Characterization techniques
Bright field transmission electron microscopy (TEM) images were acquired with a Gatan OneView camera on a JEOL F200-HR(CF) TEM operating at 200 kV. EDS spectra were acquired on the same TEM using a JEOL SD100WL 100 mm2 SDD EDS detector run through Oxford Instruments’ AZtec software. To analyze the porosity of the synthesized mesopore structures, nitrogen adsorption–desorption isotherms were generated at 77 K using the Micromeritics Tristar II instrument, following degassing of the samples at 90 °C using the Micromeritics vacuum degasser system 061 VacPrep. The specific surface area and pore size distribution were calculated using the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. Elemental composition was determined through X-ray photoelectron spectroscopy (XPS) using a Kratos Axis Ultra-XPS with a hybrid lens and a 50 W monochromatic Al Kα (1486.6 eV) radiation source at 15 kV (10 mA); resolution levels of 160 and 40 nm were chosen for full (wide) spectra and high-resolution spectra, respectively. The raw data were analyzed using CasaXPS, with which the XPS spectra results were mathematically corrected and fitted using the linear background and calibrated to the C–C/C–H peak (284.8 eV) from adventitious carbon contamination. X-ray diffraction (XRD) measurements were carried out with an EMMA instrument (enhanced multi materials analyser, 25 mA, 10 kV) operating under Cu-Kα (λ = 1.5418 Å) radiation at a scanning rate of 2° min−1 from 2θ, 15°–45°. The amino group modifications of mesoporous silica nanoparticles were characterized using a PerkinElmer attenuated total reflectance Fourier transform infrared (ATR-FTIR) spectrometer. The spectra were collected over the range of 400–4000 cm−1 at a resolution of 4 cm−1 using dried powdered samples. Ultraviolet–visible (UV-Vis) spectra were recorded on a Cary 3500 UV-vis spectrophotometer. Dye leakage was studied using a Shimadzu liquid chromatograph mass spectrometer (LCMS-8045), featuring a CBM-20A communication bus module, a DGU-20A5R degassing unit, LC-30AD pumps, an SIL-30AC autosampler, and a CTO-20AC column oven. The chromatographic separation utilized a Thermo Scientific Accucore C18 column (50 × 3 mm, 2.6 μm), kept at a consistent oven temperature of 40 °C. RP-HPLC analysis was conducted with a Shimadzu Prominence series composed of an LC-20AT, an SIL-20AC autosampler, an SPD-M20A photodiode array (PDA) detector with Labsolutions software, a CBM-20A system controller, a DGU-20A3 degasser, and a CTO-20A column oven (Shimadzu Corp., Kyoto, Japan). The analytical column (Alltech prevail C18 column, 100 × 2.1 mm, particle size 3 μm) was kept at 40 °C. Dynamic light scattering (DLS) and zeta potential (ZP) measurements were carried out with Litesizer DLS 500 (Anton Paar). Data are presented as mean ± standard deviation. An unpaired two-tailed t-test was used to compare the two groups in the CCK-8 assay. A p-value of <0.05 was considered statistically significant.3. Results and discussion
3.1. Physiochemical characterization of MSNPs synthesized using CTAC and CTAB as surfactants
The formation of MSNPs was confirmed by analyzing the XRD, DLS, and FTIR patterns of the powdered samples, along with BET analysis and TEM images. The broad XRD pattern of the peak at 2θ around 22–24° (Fig. S1†) confirmed the amorphous nature of the silica nanoparticles based on previously reported data.89 Energy dispersive spectroscopy (EDS) of the mesoporous silica products revealed only the anticipated peaks for Si, O, C, and N, highlighting the purity (Fig. S1†).The N2 adsorption–desorption isotherm curves of the various types of MSNPs synthesised in this study are displayed in Fig. 1. Except for ADP-MSNPs-CTAC1, all MSNPs exhibit a type IV isotherm curve according to the IUPAC classification, featuring an H1-type hysteresis loop in the latter half (at P/P0 of 0.8–1.0). This suggests a typical mesoporous structure with a narrow range of uniform cylindrical pores.90,91 For ADP-MSNPs-CTAC1, a type IV isotherm curve with a H4 hysteresis loop from P/P0 = 0.45 to 1.0 indicates the presence of narrow slit-like pores, particles with irregular shape and broad size distribution.92 Additionally, a distinctive tail at P/P0 ∼ 1.0 also implies the presence of macropores among the particles.93 The presence of an H4-type hysteresis loop is also noted in materials with aggregated mesoporous and micro–mesoporous characteristics.94
 | ||
| Fig. 1 N2 adsorption–desorption isotherm study of pore-expanded and amine-functionalized dye-labeled MSNP samples synthesized using CTAB and CTAC surfactants. | ||
The TEM results illustrate that MSNPs created using the CTAC (Fig. 2) surfactant exhibited a larger average particle size and pore size compared to those synthesized with CTAB (Fig. 3). This size difference can be attributed to the presence of different halide ions in their chemical structures (bromide vs. chloride), which can influence their interactions with the aqueous environment.95 However, the exact size difference may depend on the specific synthesis conditions.96,97 While both types show potential for drug delivery, MSNPs-CTACs with their larger pore size and overall size below 100 nm present an opportunity for delivering larger therapeutic agents, which remains a current challenge.6 Additionally, it is worth noting that ADP-MSNPs created using the one-stage method (Fig. 2b and 3c) demonstrate larger sizes and increased aggregation when compared with those produced through the two-stage method (Fig. 2c and 3b), potentially diminishing their effectiveness in drug delivery applications. This finding is also supported by BET and DLS data. Table 1 provides an overview of the BET surface analysis data, TEM results, and DLS measurements. Based on the DLS data, the average diameters of MSNPs produced via the one-stage method are 4 to 10 times larger than those of the corresponding particles obtained via the two-stage method. This is partly due to larger individual particle sizes but mainly due to aggregation among particles synthesized via the one-stage method, as shown in the TEM images.
 | ||
| Fig. 2 TEM images of MSNPs-CTAC. (a) P-MSNPs-CTAC: pore-expanded MSNPs using the CTAC surfactant, (b) ADP-MSNPs-CTAC1: amine-functionalized dye-labelled pore-expanded MSNPs synthesized via one-stage synthesis, (c) ADP-MSNPs-CTAC2: amine-functionalized dye-labelled pore-expanded MSNPs synthesized via two-stage synthesis. | ||
 | ||
| Fig. 3 TEM images of MSNPs-CTAB. (a) P-MSNPs-CTAB: pore-expanded MSNPs synthesized via the CTAB surfactant, (b) ADP-MSNPs-CTAB2: amine-functionalized dye-labelled pore-expanded MSNPs synthesized via two-stage synthesis, (c) ADP-MSNPs-CTAB1: amine-functionalized dye-labelled pore-expanded MSNPs synthesized via one-stage synthesis, and (d) ADP-MSNPs-CTAB3: one-stage amine-functionalized dye-labelled pore-expanded MSNPs synthesised using half the amount of APTES (amine) compared to the ADP-MSNPs-CTAB1 preparation method. | ||
| Type of sample | SBETa (m2 g−1) | BJH avg pore diameterb (nm) | TEM avg diameter (nm) | TEM avg pore diameter (nm) | DLS avg diameter (nm) | Zeta potential (mV) |
|---|---|---|---|---|---|---|
| a Total surface area calculated by the BET method.b BJH desorption average pore diameter (4 V A−1). | ||||||
| P-MSNPs-CTAB | 263.9 | 6.5 | 28.5 ± 1.5 | 6.5 ± 0.5 | 40.0 ± 4.5 | −30.4 ± 1.5 |
| ADP-MSNPs-CTAB1 | 97.8 | 6.8 | 51.7 ± 11.5 | 8.5 ± 2.2 | 444.6 ± 87.7 | 229.9 ± 3.3 |
| ADP-MSNPs-CTAB2 | 194.8 | 5.9 | 28.6 ± 1.3 | 6.3 ± 1.0 | 43.9 ± 3.0 | 245.0 ± 6.9 |
| ADP-MSNPs-CTAB3 | 128.1 | 6.9 | 39.7 ± 6.4 | 7.6 ± 2.0 | 279.0 ± 83.5 | 30.9 ± 3.7 |
| P-MSNPs-CTAC | 587.2 | 22.2 | 76.7 ± 3.8 | 17.9 ± 3.1 | 75.1 ± 10.4 | −26.8 ± 0.6 |
| ADP-MSNPs-CTAC1 | 35.1 | 4.2 | 149.9 ± 11.2 | 11.2 ± 1.3 | 335.2 ± 63.1 | 37.2 ± 0.7 |
| ADP-MSNPs-CTAC2 | 363.8 | 17.4 | 78.1 ± 1.6 | 13.8 ± 1.8 | 88.5 ± 11.3 | 33.8 ± 0.4 |
The incorporation of amine functionalization and dye labelling into the MSNP synthesis via the two-stage method had a negligible effect on the overall particle size (Table 1) but resulted in a slight decrease in pore size, likely due to the embedding of the dye and APTES within the pore structure.98 Following dye labelling and amine functionalization, the zeta potential of MSNPs changed from negative to positive, affirming the successful bonding of dye and amine groups. Additionally, ADP-MSNPs-CTAB3 displayed a reduced positive charge compared to ADP-MSNPs-CTAB1 and ADP-MSNPs-CTAB2, which have a higher APTES/TEOS ratio.
Another intriguing finding is related to ADP-MSNPs-CTAC2 particles that were synthesized using a two-stage method with varying addition speeds of the TEOS/cyclohexane solution (2, 2.5, and 3 mL min−1). This analysis indicates that a higher addition speed leads to smaller particle and pore sizes, as illustrated in Fig. 4.
 | ||
| Fig. 4 TEM images of ADP-MSNPs-CTAC2 synthesized at different rates of TEOS/cyclohexane solution addition. | ||
FTIR and XPS analysis were employed to investigate the attachment of amine groups on the surface of MSNPs. The FTIR spectra of MSNPs (Fig. 5) displayed absorption bands at 3400 cm−1, 1628 cm−1, and 1059 cm−1, corresponding to silanol (Si-OH) symmetric stretching, bending vibrations, and Si–O–Si bending, respectively. Notably, the intense peaks at 950 cm−1 and 800 cm−1 signify the asymmetric and symmetric stretching vibrations of Si–O–Si, respectively.99 Additionally, the peak at 450 cm−1 is associated with the deformation mode of O–Si–O. After APTES treatment, the band observed at 1580 cm−1 was attributed to the bending vibration of NH2.99 Additionally, a weak-to-medium absorption band appearing in the 1470–1540 cm−1 range could be associated with the symmetric or asymmetric N–H bending vibrations.99,100 However, the N–H band of primary amines is shifted to a lower range compared to those in aqueous solutions (1500–1600 cm−1). This shift is attributed to hydrogen bonding interactions between amines, which act as hydrogen acceptors, and silanol groups. These interactions can lead to the formation of various hydrogen-bonded surface conformations.99,101 The FT-IR spectra of MSNPs also have a weak broadband at 2800–3500 cm−1, which covers a wide range of vibrations from 2800 to 3000 cm−1 (–C–CH3 stretching vibration), 2916, 2837 cm−1 (–C–CH2–C– stretching vibration), and a broad signal at 3000–3500 cm−1, which can arise from the stretching and bending vibrations of aliphatic amine (N–H) and hydroxyl (O–H) groups within these samples.102,103
 | ||
| Fig. 5 FTIR spectra of MSNPs. | ||
XPS measurements (Fig. S2 and S3†) were carried out to better elucidate if APTES and RhB were successfully grafted on ADP-MSNPs. Five peaks were observed at binding energies around 153 eV, 102 eV, 532 eV, 400 eV, and 284 eV, corresponding to Si 2s, Si 2p, O 1s, N 1s, and C 1s, respectively. As detailed in Table S1,† the O 1s spectrum was deconvoluted into four peaks with binding energies attributed to Si-OH, Si–O–Si, (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–N (amide), and O–Si–C/C–O–C (aromatic).99,104,105 The N 1s spectrum was categorized into three distinct components, corresponding to the binding energies of primary amine,106,107 quaternary/tertiary amine,108 and (CO)–N.109 Meanwhile, the Si 2p spectrum showed two peaks linked to the binding energies of SiO4 and O3–Si–C.110,111 The O–Si–C, O3–Si–C, and primary amine peaks obtained from the deconvolution of the O 1s, Si 2p, and N 1s spectra, respectively, confirm the successful grafting of APTES into MSNPs. Additionally, the presence of (CO)–N in the O 1s and N 1s spectra indicates an amide reaction between RhB and APTES within the MSNP framework. As anticipated, the ADP-MSNPs-CTAB synthesized using the two-stage method show a higher percentage of primary amine in the deconvolution of the N 1s peak compared to the one-stage method. This confirms that more APTES was present on the surface of MSNPs produced by the two-stage method compared to the one-stage method. Furthermore, the reduction in APTES concentration during the synthesis of ADP-MSNPs-CTAB3, compared to ADP-MSNPs-CTAB1, led to a decrease in the percentage of detected primary amine in the deconvolution of the N 1s peak. For ADP-MSNPs-CTAC2, the deconvolution of the N 1s peak from MSNPs produced via the two-stage method reveals all three types of amines available in the MSNP structure: primary amine of APTES, quaternary amine, and amide-related to rhodamine B. In contrast, the MSNPs synthesized using the one-stage method only show primary amines, likely due to substantial aggregation of the ADP-MSNPs-CTAC1 particles that limit access of XPS to all surface areas of NPs. Although XPS spectra provide valuable insights, the quantitative comparison of peaks for MSNPs is challenging because of substantial aggregation in certain samples. Furthermore, the porous structure of the MSNPs allows X-ray photons to access both internal and external surfaces, as supported by the deconvoluted XPS data. However, they cannot access the entire internal surface, as the penetration depth of X-ray photons is approximately 10–100 Å.112 These limitations can hinder the quantitative analysis of the XPS peaks.
O)–N (amide), and O–Si–C/C–O–C (aromatic).99,104,105 The N 1s spectrum was categorized into three distinct components, corresponding to the binding energies of primary amine,106,107 quaternary/tertiary amine,108 and (CO)–N.109 Meanwhile, the Si 2p spectrum showed two peaks linked to the binding energies of SiO4 and O3–Si–C.110,111 The O–Si–C, O3–Si–C, and primary amine peaks obtained from the deconvolution of the O 1s, Si 2p, and N 1s spectra, respectively, confirm the successful grafting of APTES into MSNPs. Additionally, the presence of (CO)–N in the O 1s and N 1s spectra indicates an amide reaction between RhB and APTES within the MSNP framework. As anticipated, the ADP-MSNPs-CTAB synthesized using the two-stage method show a higher percentage of primary amine in the deconvolution of the N 1s peak compared to the one-stage method. This confirms that more APTES was present on the surface of MSNPs produced by the two-stage method compared to the one-stage method. Furthermore, the reduction in APTES concentration during the synthesis of ADP-MSNPs-CTAB3, compared to ADP-MSNPs-CTAB1, led to a decrease in the percentage of detected primary amine in the deconvolution of the N 1s peak. For ADP-MSNPs-CTAC2, the deconvolution of the N 1s peak from MSNPs produced via the two-stage method reveals all three types of amines available in the MSNP structure: primary amine of APTES, quaternary amine, and amide-related to rhodamine B. In contrast, the MSNPs synthesized using the one-stage method only show primary amines, likely due to substantial aggregation of the ADP-MSNPs-CTAC1 particles that limit access of XPS to all surface areas of NPs. Although XPS spectra provide valuable insights, the quantitative comparison of peaks for MSNPs is challenging because of substantial aggregation in certain samples. Furthermore, the porous structure of the MSNPs allows X-ray photons to access both internal and external surfaces, as supported by the deconvoluted XPS data. However, they cannot access the entire internal surface, as the penetration depth of X-ray photons is approximately 10–100 Å.112 These limitations can hinder the quantitative analysis of the XPS peaks.
Ninhydrin reagent was utilized to assess the extent of amine grafting on ADP-MSNPs, following the method outlined by Lu et al.86 The primary/secondary amine groups on the surface of the ADP-MSNPs reacted with the ninhydrin, producing a purple solution that confirmed the presence of primary/secondary amines. The quantity of APTES grafted onto the ADP-MSNPs surface, and the surface density of amine groups are calculated using eqn (2) and (3), respectively, with the concentration of reacted ninhydrin derived from the standard curve (Fig. S4†). The result is summarized in Table 2.
| Molar quantity of grafted amine = M/Ws [mol g−1] | (2) |
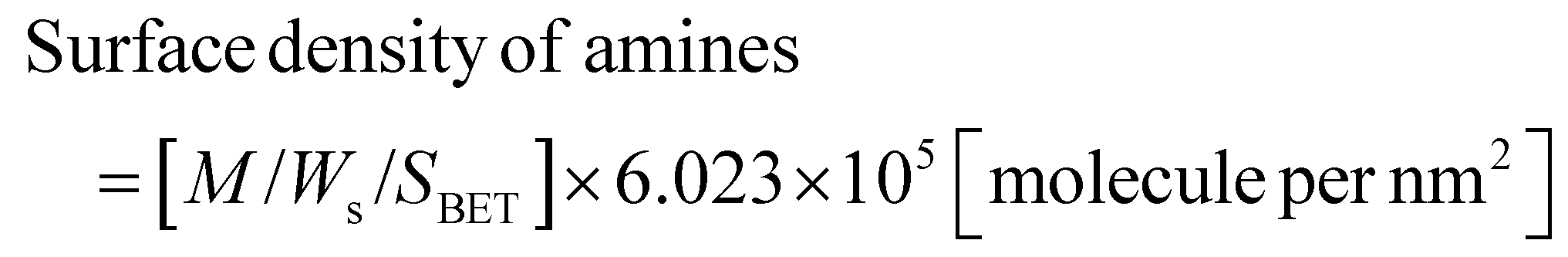 | (3) |
| Type of sample | Amine grafting on ADP-MMSNs [mmol APTES per g of MSNPs] | Surface density of amine groups [APTES molecule per nm2] |
|---|---|---|
| ADP-MSNPs-CTAB1 | 1.40 ± 0.06 | 8.65 |
| ADP-MSNPs-CTAB2 | 1.71 ± 0.07 | 5.28 |
| ADP-MSNPs-CTAB3 | 1.29 ± 0.03 | 6.06 |
| ADP-MSNPs-CTAC1 | 0.55 ± 0.11 | 9.43 |
| ADP-MSNPs-CTAC2 | 0.92 ± 0.10 | 1.52 |
A comparison of the amine grafting data for ADP-MSNPs reveals that the two-stage method is more efficient for amine grafting than the one-stage method. Although the surface density of amine groups on particles from the one-stage method appears higher than for the particles from the two-stage method; this is largely due to the reduced surface area (SBET values) (Table 1) arising from aggregation of the particles.
The successful encapsulation of RhB dye within the mesoporous silica matrix was validated using the absorption spectra of ADP-MSNPs (Fig. 6A). The RhB solution displays a prominent absorption peak at 553 nm, indicative of the π–π* electronic transitions of the CN and CO groups in the RhB monomer, while a shoulder peak at 515 nm corresponds to its dimer.113 Additionally, the RhB-doped nanoparticles exhibited vibrant fluorescence when illuminated with UV light (Fig. 6B).
 | ||
| Fig. 6 (A) UV-vis spectrum and (B) bright fluorescence under UV light illumination of ADP-MSNPs-CTAC2, respectively. | ||
Dye loading and encapsulation efficiencies were studied using UV-vis spectroscopy. Dye loading efficiency refers to the amount of dye per mg of sample, determined quantitatively by measuring the absorbance of the solution containing ADP-MSNPs against that of the free dye solution and comparing the results with the standard curve of dye at 553 nm (Fig. S5†). Encapsulation efficiency [EE%] is the percentage of dye entrapped within the silica matrix, calculated using the equation below.

The results for loading efficiency and encapsulation efficiency are summarized in Table 3. The stability of dye labeling in ADP-MSNPs was evaluated by measuring dye release from the nanoparticles at a specific time point using HPLC and LC/MS (Table 3). The data indicate that ADP-MSNPs synthesized via the two-stage method demonstrate higher encapsulation efficiency and lower dye leakage compared to those produced using the one-stage method.
| Type of sample | Loading efficiency (μg of dye per mg of MSNPs) | Encapsulation efficiency (%) | Dye leakage (%) (HPLC)a | Dye leakage (%) (LC/MS) |
|---|---|---|---|---|
| a LOD = 0.012 μg mL−1, LOQ = 0.374 μg mL−1. | ||||
| ADP-MSNPs-CTAB1 | 4.31 ± 0.040 | 35.92 | Below the limit of detection | 0.17 ± 0.005 |
| ADP-MSNPs-CTAB2 | 7.41 ± 0.313 | 61.77 | Below the limit of detection | 0.10 ± 0.02 |
| ADP-MSNPs-CTAB3 | 3.86 ± 0.070 | 32.15 | Below the limit of detection | 0.25 ± 0.03 |
| ADP-MSNPs-CTAC1 | 5.48 ± 0.184 | 45.64 | 0.88 ± 0.14 | 0.55 ± 0.03 |
| ADP-MSNPs-CTAC2 | 7.87 ± 0.327 | 65.58 | Below the limit of detection | 0.049 ± 0.005 |
3.2. Cytotoxicity of ADP-MSNPs-CTAC2 and ADP-MSNPs-CTAB2
Cell viability and cytotoxicity assays are important tools for evaluating the toxic effects of different chemicals on cells.114 For gene therapy, gene carriers should ideally have low cytotoxicity and high biocompatibility. CCK-8 allows sensitive colorimetric assays for the determination of the number of viable cells in the proliferation and cytotoxicity assays.115,116 The assay uses a water-soluble tetrazolium salt that is reduced by living cells to produce an orange-colored formazan dye. The amount of formazan dye produced, which is directly proportional to the number of living cells, is measured by recording changes in absorbance at 570 nm using a plate reading spectrophotometer.117,118 As aggregation can impair the stability and efficacy of nanocarriers,119,120 MSNPs-CTAC2 and MSNPs-CTAB2 which exhibited the lowest aggregation were selected for the biological study. To assess the toxicity of the synthesized MSNPs by the CCK-8 assay, SH-SY5Y cells were treated with various concentrations of MSNPs-CTAC2 and MSNPs-CTAB2, and cell viability was determined as mentioned in section 2.2.12.As illustrated in Fig. 7, ADP-MSNPs at a concentration of 1000 μg mL−1 reduced cell viability to below 80%. At other concentrations, cell viability ranged from 80% to 100%, and neither MSNP exhibited significant toxicity. MSNPs-CTAC2 appeared to be more toxic than MSNPs-CTAB2. This can be attributed to the larger size of MSNPs-CTAC2 compared to MSNPs-CTAB2, which may result in lower clearance rates, longer retention periods, and greater damage to cells.6 However, the statistical comparison (t-test) of the data for MSNPs-CTAC2 and MSNPs-CTAB2 at different concentrations revealed no significant differences, as all P-values were greater than 0.05.
 | ||
| Fig. 7 CCK-8 assay of SH-SY5Y cell viability after treatment with different concentrations of MSNPs-CTAC2 and MSNPs-CTAB2 for 24 h. The error bar indicates the standard deviation (±SD) for three measurements. | ||
3.3. mRNA nanocarrier potential of MSNPs
:1 weight ratio of ADP-MSNPs-CTAC2 to mRNA, the mRNA was fully loaded into the MSNPs, leading to the complete disappearance of the mRNA band on the gel, indicating a loading capacity of approximately 1.25 wt% for ADP-MSNPs-CTAC2. In contrast, ADP-MSNPs-CTAB2 exhibits a lower mRNA loading capacity, likely due to its smaller pore size and the blockage of its pores by the large mRNA molecules.
 | ||
| Fig. 8 (A) Gel retardation assay demonstrating mRNA nanoparticle complexation with increasing amounts of MSNPs, (B) RNase A protection assay, and (C) DNase I protection assay. | ||
:mRNA ratios. Comparison of the electrophoresis data (Fig. S6†) for RNase A:mRNA solutions incubated at two different times confirms that 5 minutes of incubation was sufficient to degrade all available PARK7. Additionally, the results shown in Fig. S7† indicate that an RNase A:mRNA ratio of 1:1000 completely degraded the mRNA, leaving no intact mRNA bands visible in the gel electrophoresis.
Comparing electrophoresis bands of RNase A. mRNA for different ratios in the presence and absence of HPRI (Fig. S8†) showed that ratios of 1
:2000 and 1:3000 (RNase A:mRNA) were partially preserved following the addition of HPRI (40 units). Fig. S9† provides further confidence regarding the inactivation of RNase A by HPRI (40 units), but not by HPRI (10 units). Specifically, the electrophoresis band of mRNA added to the solution after RNase A inactivation with HPRI (40 units) appears like naked mRNA, whereas it disappears completely when HPRI (10 units) is used for RNase A inactivation. Additionally, the data confirm that heparin does not have any effect on the results.The efficacy of the two selected types of MSNPs (ADP-MSNPs-CTAC2 and ADP-MSNPs-CTAB2) for encapsulating mRNA and protection against RNase A degradation was also investigated. The data (Fig. 8(B)) show that ADP-MSNPs-CTAC2 effectively encapsulates mRNA, as demonstrated by the disappearance of the mRNA band in the mRNA/ADP-MSNPs-CTAC2 complex and the reappearance of the band after using heparin. Heparin is a negatively charged competing biomolecule that can displace siRNA from cationic gene carriers via ion exchange.122,123 In contrast, ADP-MSNPs-CTAB2 appears to adsorb mRNA only on its surface, as no changes were observed in the mRNA band after mixing heparin with them.
RNase A, an enzyme secreted by the human body into fluids such as tears, saliva, mucus, and perspiration as a defense mechanism against invading microorganisms, was selected to assess the ability of MSNPs to protect mRNA.115 Neither type of nanoparticle effectively protected mRNA from degradation by RNase A, as no mRNA band appeared for the mRNA/MSNP complex after the following steps: adding RNase A, inhibiting RNase A activity with HPRI, and releasing the mRNA from the MSNP structure using heparin.
3.4 Discussion
Previous studies have shown that the transport of MSNPs across the BBB is size-dependent and that only NPs smaller than 100 nm can overcome the BBB.60,61 The two-stage synthesis method reported in this study using CTAB as a surfactant produced amine-functionalized, dye-labelled particles (ADP-MSNPs-CTAB2) with smaller overall size and better uniformity (28.6 ± 1.3 nm) than the one-stage approach (51.7 ± 11.5 nm) based on TEM analysis. Both of these types of particles appear to be less than 100 nm and therefore suitable for crossing the BBB. However, DLS analysis suggests that there may be aggregation of the particles from the one stage method (ADP-MSNPs-CTAB1) with the aggregate dimensions being much larger than 100 nm. The DLS measurements of ADP-MSNPs-CTAB2 particles indicate little or no aggregation making them suitable candidates to cross the BBB. However, due to their small pore size (<10 nm), these particles were unable to encapsulate the PARK7 mRNA (926 nucleotides), which would make them unsuitable as nanocarriers for large biomolecules to treat brain disorders.The one-stage synthesis method via CTAC produced MSNPs with a large size (150 nm), broad pore distribution (10–20 nm), and high aggregation, which would also limit their suitability for brain-targeted gene delivery. In comparison, the two-stage method using CTAC yielded well-ordered MSNPs with an optimal size (80 nm) and pore diameters (15–20 nm), enabling effective encapsulation of the large PARK7 mRNA and offering strong potential for future brain gene therapy studies. However, these particles could not protect the mRNA from RNase A degradation, highlighting the need for further optimization to enhance mRNA stability in biological environments. Despite this limitation, ADP-MSNPs-CTAC2 can be considered a promising substrate to develop carriers for gene delivery to brain cells, thanks to their ability to encapsulate large biomolecules and their particle size of under 100 nm after amine functionalization and dye labelling.
4. Conclusion
Findings indicate that ADP-MSNPs produced via the two-stage method exhibit a narrow size distribution, reduced aggregation, and superior structure compared to those created using the 1-stage method. Furthermore, the two-stage approach enables greater control over the functional groups on both the pores and the external surfaces of the MSNPs. This will allow the synthesis of MSNPs with more accessible pores and specific attachments for encapsulating various therapeutic agents in the initial stage. During the second stage, the surface of the MSNPs can be easily modified with different functional groups, allowing for a range of applications, including targeted drug delivery and specific conjugations for specialized purposes. When comparing ADP-MSNPs synthesized with CTAB and CTAC as surfactants, each type offers MSNPs tailored for specific applications. Using CTAB as a surfactant, we synthesized MSNPs smaller than 50 nm with approximately 5 nm pores, making them ideal for delivering small therapeutic agents. In contrast, using CTAC, we produced MSNPs having size under 100 nm and pores with a diameter of 20 nm, which we hope have the potential to overcome the complexity of the BBB and serve as a valuable vehicle for delivering large therapeutics to brain cells, as we successfully encapsulated PAR7 (mRNA, 926 nt) into the pores of two-stage MSNPs synthesized using CTAC. However, further research is needed to protect encapsulated mRNA from degradation by biological enzymes.Data availability
The relevant data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the TEM facility at the Centre for Microscopy, Characterization, and Analysis, The University of Western Australia [CMCA UWA], a facility funded by the University, State and Commonwealth Governments, and Associate Professor Martin Saunders at CMCA UWA for his assistance during TEM imaging. We would like to thank the Faculty of Science and Engineering of Curtin University for undertaking the XPS analyses. We also thank Murdoch University for providing a PhD scholarship to Olia Alijanpourtolouti.References
- J. Andrade-Guerrero, A. Santiago-Balmaseda, P. Jeronimo-Aguilar, I. Vargas-Rodríguez, A. R. Cadena-Suárez, C. Sánchez-Garibay, G. Pozo-Molina, C. F. Méndez-Catalá, M.-C. Cardenas-Aguayo, S. Diaz-Cintra, M. Pacheco-Herrero, J. Luna-Muñoz and L. O. Soto-Rojas, Int. J. Mater. Sci., 2023, 24, 3754 CAS.
- J. K. W. Lam, M. Y. T. Chow, Y. Zhang and S. W. S. Leung, Mol. Ther. Nucleic Acids, 2015, 4, e252 CrossRef CAS PubMed.
- J. Zhang, B. Chen, C. Gan, H. Sun, J. Zhang and L. Feng, Int. J. Nanomed., 2023, 18, 7605–7635 Search PubMed.
- Q. Bao, X. Liu, Y. Li, T. Yang, H. Yue, M. Yang and C. Mao, Mater. Adv., 2024, 5, 1626–1630 RSC.
- D. Wu, Q. Chen, X. Chen, F. Han, Z. Chen and Y. Wang, Signal Transduction Targeted Ther., 2023, 8, 217 CrossRef PubMed.
- U. Niroumand, N. Firouzabadi, G. Goshtasbi, B. Hassani, P. Ghasemiyeh and S. Mohammadi-Samani, Front. Mater., 2023, 10, 1189463 CrossRef.
- R. Narayan, U. Y. Nayak, A. M. Raichur and S. Garg, Pharmaceutics, 2018, 10, 118 CrossRef CAS PubMed.
- F. Ahmadi, A. Sodagar-Taleghani, P. Ebrahimnejad, S. Pouya Hadipour Moghaddam, F. Ebrahimnejad, K. Asare-Addo and A. Nokhodchi, Int. J. Pharm., 2022, 625, 122099 CrossRef CAS PubMed.
- T. I. Janjua, Y. Cao, F. Kleitz, M. Linden, C. Yu and A. Popat, Adv. Drug Delivery Rev., 2023, 203, 115115 CrossRef CAS PubMed.
- S. B. Hartono, M. Yu, W. Gu, J. Yang, E. Strounina, X. Wang, S. Qiao and C. Yu, Nanotechnology, 2014, 25, 055701 CrossRef CAS PubMed.
- Y. Li, M. Hei, Y. Xu, X. Qian and W. Zhu, Int. J. Pharm., 2016, 511, 689–702 CrossRef CAS PubMed.
- V. Selvarajan, S. Obuobi and P. L. R. Ee, Front. Chem., 2020, 8, 602 Search PubMed.
- J. Du, L. A. Lane and S. Nie, J. Controlled Release, 2015, 219, 205–214 CrossRef CAS PubMed.
- A. F. Moreira, V. M. Gaspar, E. C. Costa, D. De Melo-Diogo, P. Machado, C. M. Paquete and I. J. Correia, Eur. J. Pharm. Biopharm., 2014, 88, 1012–1025 CrossRef CAS PubMed.
- L.-L. Hu, J. Meng, D.-D. Zhang, M.-L. Chen, Y. Shu and J.-H. Wang, Talanta, 2018, 177, 203–211 CrossRef CAS PubMed.
- S. Giri, B. G. Trewyn, M. P. Stellmaker and V. S.-Y. Lin, Angew. Chem., Int. Ed., 2005, 44, 5038–5044 CrossRef CAS PubMed.
- P.-J. Chen, S.-H. Hu, C.-S. Hsiao, Y.-Y. Chen, D.-M. Liu and S.-Y. Chen, J. Mater. Chem., 2011, 21, 2535 RSC.
- S. Mukherjee, L. Liang and O. Veiseh, Pharmaceutics, 2020, 12, 147 CrossRef CAS PubMed.
- M. Chen, J. Hu, L. Wang, Y. Li, C. Zhu, C. Chen, M. Shi, Z. Ju, X. Cao and Z. Zhang, Sci. Rep., 2020, 10, 14447 CrossRef CAS PubMed.
- L. Sun, D. Wang, Y. Chen, L. Wang, P. Huang, Y. Li, Z. Liu, H. Yao and J. Shi, Biomaterials, 2017, 133, 219–228 CrossRef CAS PubMed.
- S. Naz, M. Wang, Y. Han, B. Hu, L. Teng, J. Zhou, H. Zhang and J. Chen, Int. J. Nanomed., 2019, 14, 2533–2542 CrossRef CAS PubMed.
- W. Luo, X. Xu, B. Zhou, P. He, Y. Li and C. Liu, Mater. Sci. Eng., C, 2019, 100, 855–861 CrossRef CAS PubMed.
- E. H. Jang, G. L. Kim, M. G. Park, M. K. Shim and J.-H. Kim, J. Drug Delivery Sci. Technol., 2020, 56, 101543 Search PubMed.
- S. Khatoon, H. Han, J. Jeon, N. Rao, D.-W. Jeong, M. Ikram, T. Yasin, G.-R. Yi and J. Park, Polymers, 2018, 10, 390 Search PubMed.
- D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216–13217 Search PubMed.
- H. H. P. Yiu, S. C. McBain, A. J. El Haj and J. Dobson, Nanotechnology, 2007, 18, 435601 Search PubMed.
- S. B. Hartono, W. Gu, F. Kleitz, J. Liu, L. He, A. P. J. Middelberg, C. Yu, G. Q. (Max) Lu and S. Z. Qiao, ACS Nano, 2012, 6, 2104–2117 CrossRef CAS PubMed.
- B. G. Cha, J. H. Jeong and J. Kim, ACS Cent. Sci., 2018, 4, 484–492 CrossRef CAS PubMed.
- L. Chen, G. Zhu, D. Zhang, H. Zhao, M. Guo, W. Shi and S. Qiu, J. Mater. Chem., 2009, 19, 2013 Search PubMed.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- E. Bloch, T. Phan, D. Bertin, P. Llewellyn and V. Hornebecq, Microporous Mesoporous Mater., 2008, 112, 612–620 CrossRef CAS.
- J. Wei, Y. Deng, J. Zhang, Z. Sun, B. Tu and D. Zhao, Solid State Sci., 2011, 13, 784–792 CrossRef CAS.
- J. Zhang, Y. Deng, J. Wei, Z. Sun, D. Gu, H. Bongard, C. Liu, H. Wu, B. Tu, F. Schüth and D. Zhao, Chem. Mater., 2009, 21, 3996–4005 CrossRef CAS.
- L. Yang, H. Wu, J. Jia, B. Ma and J. Li, Microporous Mesoporous Mater., 2017, 253, 151–159 CrossRef CAS.
- M. Huang, L. Li and S. Zheng, Microporous Mesoporous Mater., 2016, 225, 9–25 CrossRef CAS.
- M. Ulfa, K. S. Aristia and D. Prasetyoko, 3rd International Seminar on Chemistry: Green Chemistry and Its Role for Sustainability, ed. F. Kurniawan, Y. Kusumawati, S. Fatmawati, H. Juwono and A. S. Purnomo, 2018. (AIP Conference Proceedings) Search PubMed.
- S. K. Jana, A. Mochizuki and S. Namba, Catal. Surv. Asia, 2000, 8, 1–13 CrossRef.
- J. L. Blin, C. Otjacques, G. Herrier and B.-L. Su, Langmuir, 2000, 16, 4229–4236 CrossRef CAS.
- A. Sayari, Angew. Chem., Int. Ed., 2000, 39, 2920–2922 CrossRef CAS PubMed.
- A. Noureddine, A. Maestas-Olguin, L. Tang, J. I. Corman-Hijar, M. Olewine, J. A. Krawchuck, J. Tsala Ebode, C. Edeh, C. Dang, O. A. Negrete, J. Watt, T. Howard, E. N. Coker, J. Guo and C. J. Brinker, ACS Nano, 2023, 17, 16308–16325 CrossRef CAS PubMed.
- J. Ma, Q. Liu, D. Chen, S. Wen and T. Wang, Micro Nano Lett., 2015, 10, 140–144 CrossRef.
- C. Xin, N. Zhao, H. Zhan, F. Xiao, W. Wei and Y. Sun, J. Colloid Interface Sci., 2014, 433, 176–182 Search PubMed.
- L. Huang and M. Kruk, J. Colloid Interface Sci., 2012, 365, 137–142 Search PubMed.
- K.-C. Kao and C.-Y. Mou, Microporous Mesoporous Mater., 2013, 169, 7–15 CrossRef CAS.
- S. Song, X. Zhou, A. Duan, Z. Zhao, K. Chi, M. Zhang, G. Jiang, J. Liu, J. Li and X. Wang, Microporous Mesoporous Mater., 2016, 226, 510–521 Search PubMed.
- M. G. Colmenares, U. Simon, F. Schmidt, S. Dey, J. Schmidt, A. Thomas and A. Gurlo, Microporous Mesoporous Mater., 2018, 267, 142–149 Search PubMed.
- E. M. Johansson, J. M. Córdoba and M. Odén, Microporous Mesoporous Mater., 2010, 133, 66–74 Search PubMed.
- H. Yamada, H. Ujiie, C. Urata, E. Yamamoto, Y. Yamauchi and K. Kuroda, Nanoscale, 2015, 7, 19557–19567 Search PubMed.
- Y. Awoke, Y. Chebude and I. Díaz, Molecules, 2020, 25, 4909 Search PubMed.
- X. Hong, X. Zhong, G. Du, Y. Hou, Y. Zhang, Z. Zhang, T. Gong, L. Zhang and X. Sun, Sci. Adv., 2020, 6, eaaz4462 CrossRef CAS PubMed.
- J. L. Paris, C. Monío, A. M. Pérez-Moreno, R. Jurado-Escobar, G. Bogas, T. D. Fernández, M. I. Montañez, C. Mayorga and M. J. Torres, Adv. Healthcare Mater., 2023, 12, 2203321 CrossRef CAS PubMed.
- A. Sayari, M. Kruk, M. Jaroniec and I. L. Moudrakovski, Adv. Mater., 1998, 10, 1376–1379 CrossRef CAS.
- A. B. Fuertes, P. Valle-Vigón and M. Sevilla, J. Colloid Interface Sci., 2010, 349, 173–180 CrossRef CAS PubMed.
- M. Shafiee, S. Abolmaali, M. Abedanzadeh, M. Abedi and A. Tamaddon, Iran. J. Med. Sci., 2021, 46, 475–486, DOI:10.30476/ijms.2020.86173.1595.
- C. E. Ashley, E. C. Carnes, K. E. Epler, D. P. Padilla, G. K. Phillips, R. E. Castillo, D. C. Wilkinson, B. S. Wilkinson, C. A. Burgard, R. M. Kalinich, J. L. Townson, B. Chackerian, C. L. Willman, D. S. Peabody, W. Wharton and C. J. Brinker, ACS Nano, 2012, 6, 2174–2188 CrossRef CAS PubMed.
- K. Fujimoto, K. Watanabe, S. Ishikawa, H. Ishii, K. Suga and D. Nagao, Colloids Surf., A, 2021, 609, 125647 CrossRef CAS.
- A. Noureddine, E. A. Hjelvik, J. G. Croissant, P. N. Durfee, J. O. Agola and C. J. Brinker, J. Sol-Gel Sci. Technol., 2019, 89, 78–90 CrossRef CAS.
- A. A. Torrano, R. Herrmann, C. Strobel, M. Rennhak, H. Engelke, A. Reller, I. Hilger, A. Wixforth and C. Bräuchle, Nanoscale, 2016, 8, 13352–13367 RSC.
- Q. He, Z. Zhang, F. Gao, Y. Li and J. Shi, Small, 2011, 7, 271–280 CrossRef CAS PubMed.
- Y.-P. Chen, C.-M. Chou, T.-Y. Chang, H. Ting, J. Dembélé, Y.-T. Chu, T.-P. Liu, C. A. Changou, C.-W. Liu and C.-T. Chen, Front. Chem., 2022, 10, 931584 CrossRef CAS PubMed.
- O. Betzer, M. Shilo, R. Opochinsky, E. Barnoy, M. Motiei, E. Okun, G. Yadid and R. R. Popovtzer, Nanomed., 2017, 12, 1533–1546 Search PubMed.
- S. Mukherjee, V. S. Madamsetty, D. Bhattacharya, S. R. Chowdhury, M. K. Paul and A. Mukherjee, Adv. Funct. Mater., 2020, 30, 2003054 CrossRef CAS.
- B. G. Trewyn, I. I. Slowing, S. Giri, H.-T. Chen and V. S.-Y. Lin, Acc. Chem. Res., 2007, 40, 846–853 CrossRef CAS PubMed.
- Z. Tao, RSC Adv., 2014, 4, 18961 RSC.
- J. L. Paris and M. Vallet-Regí, Pharmaceutics, 2020, 12, 526 CrossRef CAS PubMed.
- J. L. Steinbacher and C. C. Landry, Langmuir, 2014, 30, 4396–4405 CrossRef CAS PubMed.
- K. Zheng, H. Yang, L. Wang, S. Jing, H. Huang, J. Xu and P. Zhang, J. Porous Mater., 2013, 20, 1003–1008 CrossRef CAS.
- A. M. Carvalho, R. A. Cordeiro and H. Faneca, Pharmaceutics, 2020, 12, 649 CrossRef CAS PubMed.
- X. Li, Y. Chen, M. Wang, Y. Ma, W. Xia and H. Gu, Biomaterials, 2013, 34, 1391–1401 CrossRef CAS PubMed.
- J.-T. Sun, C.-Y. Hong and C.-Y. Pan, J. Phys. Chem. C, 2010, 114, 12481–12486 CrossRef CAS.
- M. V. Cabañas, D. Lozano, A. Torres-Pardo, C. Sobrino, J. González-Calbet, D. Arcos and M. Vallet-Regí, Mater. Chem. Phys., 2018, 220, 260–269 CrossRef.
- B. M. Estevão, I. Miletto, N. Hioka, L. Marchese and E. Gianotti, ChemistryOpen, 2021, 10, 1251–1259 CrossRef PubMed.
- H. Zha, T. Zhou, F. Gan, B. Wang, Z. Dai and X. Jiang, Catalysts, 2022, 12, 620 CrossRef CAS.
- F. Man, T. Lammers and R. T. M. De Rosales, Mol. Imaging Biol., 2018, 20, 683–695 CrossRef PubMed.
- I. Slowing, J. Viveroescoto, C. Wu and V. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278–1288 CrossRef CAS PubMed.
- L. Xu, X. Jiang, K. Liang, M. Gao and B. Kong, Aggregate, 2022, 3, e121 CrossRef CAS.
- X. Zhang, X. Zhang, S. Wang, M. Liu, Y. Zhang, L. Tao and Y. Wei, ACS Appl. Mater. Interfaces, 2013, 5, 1943–1947 CrossRef CAS PubMed.
- V. Gubala, G. Giovannini, F. Kunc, M. P. Monopoli and C. J. Moore, Cancer Nanotechnol., 2020, 11, 1 CrossRef CAS.
- A. Van Blaaderen and A. Vrij, Langmuir, 1992, 8, 2921–2931 CrossRef CAS.
- X. Gao, J. He, L. Deng and H. Cao, Opt. Mater., 2009, 31, 1715–1719 CrossRef CAS.
- M. Wang, Y. Li, L. Chen, W. Xu, Z. Huang, L. Gao, W. Zhang and L. Gao, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2016, 46, 1432–1435 CrossRef CAS.
- H. A. Ajiz, W. Widiyastuti, H. Setyawan and T. Nurtono, Heliyon, 2024, 10, e26691 CrossRef PubMed.
- X. Pan, H. Veroniaina, N. Su, K. Sha, F. Jiang, Z. Wu and X. Qi, Asian J. Pharm. Sci., 2021, 16, 687–703 Search PubMed.
- J. D. Torres-Vanegas, J. C. Cruz and L. H. Reyes, Pharmaceutics, 2021, 13, 428 CrossRef CAS PubMed.
- D. Shen, J. Yang, X. Li, L. Zhou, R. Zhang, W. Li, L. Chen, R. Wang, F. Zhang and D. Zhao, Nano Lett., 2014, 14, 923–932 CrossRef CAS PubMed.
- H. T. Lu, Colloid J., 2013, 75, 311–318 Search PubMed.
- Z. B. Kaya, V. Santiago-Padilla, M. Lim, S. L. Boschen, P. Atilla and P. J. McLean, Sci. Rep., 2024, 14, 4775 CrossRef CAS PubMed.
- W. Zhang, Y. Chen, X. Pei, Y. Zang and S. Han, Saudi J. Biol. Sci., 2018, 25, 242–247 Search PubMed.
- P. B. Sarawade, J.-K. Kim, A. Hilonga and H. T. Kim, J. Hazard. Mater., 2010, 173, 576–580 CrossRef CAS PubMed.
- G. P. Androutsopoulos and C. E. Salmas, Ind. Eng. Chem. Res., 2000, 39, 3747–3763 CrossRef CAS.
- A. Ojuva, Unpublished, 2009, preprint, DOI:10.13140/RG.2.1.5023.2807.
- K. Yao, J. Anthony, R. Maghirang, D. Hagstrum, K. Zhu and S. Bhadriraju, Grain Oil Sci. Technol., 2020, 3, 127–137 CrossRef.
- X. Liu, C. Culhane, W. Li and S. Zou, ACS Omega, 2020, 5, 24367–24378 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CrossRef CAS.
- D. V. Wellia, A. Syafawi, Y. E. Putri and Muldarisnur, RSC Adv., 2023, 13, 29645–29656 Search PubMed.
- A. Stolaś, I. Darmadi, F. A. A. Nugroho, K. Moth-Poulsen and C. Langhammer, ACS Appl. Nano Mater., 2020, 3, 2647–2653 CrossRef.
- Z. Singh and I. Singh, Sci. Rep., 2019, 9, 5880 Search PubMed.
- H. Abdul Ajiz, W. Widiyastuti, H. Setyawan and T. Nurtono, Heliyon, 2024, 10, e26691 Search PubMed.
- N. Bouazizi, J. Vieillard, B. Samir and F. Le Derf, Polymers, 2022, 14, 378 Search PubMed.
- H. He, Y. Hu, S. Chen, L. Zhuang, B. Ma and Q. Wu, Sci. Rep., 2017, 7, 3913 CrossRef PubMed.
- C.-H. Chiang, H. Ishida and J. L. Koenig, J. Colloid Interface Sci., 1980, 74, 396–404 Search PubMed.
- V. Nairi, S. Medda, M. Piludu, M. F. Casula, M. Vallet-Regì, M. Monduzzi and A. Salis, Chem. Eng. J., 2018, 340, 42–50 Search PubMed.
- S. Zhou, L. Kong, C. Yan, Y. Zhou, X. Qiu and C. Liu, RSC Adv., 2020, 10, 26813–26823 RSC.
- M. S. Chandrasekar and N. R. Srinivasan, Ceram. Int., 2016, 42, 8900–8908 CrossRef CAS.
- K. Choi, S. Lee, J. O. Park, J.-A. Park, S.-H. Cho, S. Y. Lee, J. H. Lee and J.-W. Choi, Sci. Rep., 2018, 8, 1438 CrossRef PubMed.
- Z. Mo, H. Liu, R. Hu, H. Gou, Z. Li and R. Guo, Ionics, 2018, 24, 1505–1513 Search PubMed.
- C. M. Goodwin, Z. E. Voras, X. Tong and T. P. Beebe, Coatings, 2020, 10, 967 CrossRef CAS.
- Q. Zhou, W. Chen, X. Jiang, H. Liu, S. Ma and B. Wang, Sci. Rep., 2020, 10, 1353 CrossRef CAS PubMed.
- M. Kehrer, J. Duchoslav, A. Hinterreiter, M. Cobet, A. Mehic, T. Stehrer and D. Stifter, Plasma Processes Polym., 2019, 16, 1800160 CrossRef.
- P. Post, L. Wurlitzer, W. Maus-Friedrichs and A. P. Weber, Nanomaterials, 2018, 8, 530 CrossRef PubMed.
- W. Lu, A. T. Tarekegne, Y. Ou, S. Kamiyama and H. Ou, Sci. Rep., 2019, 9, 16333 CrossRef PubMed.
- F. A. Stevie and C. L. Donley, J. Vac. Sci. Technol., A, 2020, 38, 063204 CrossRef CAS.
- K. N. Nono, A. Vahl and H. Terraschke, Nanomaterials, 2024, 14, 849 CrossRef PubMed.
- M. Wu, Y. Wang, H. Liu, F. Chen, Y. Zhang, P. Wu, Z. Deng and L. Cai, Mater. Des., 2023, 227, 111705 CrossRef CAS.
- X. Hu, W. Xiong, X. Liu, J. Wang, S. Wang, Q. Chen, K. Gao, C. Li and Y. Li, Mater. Des., 2024, 241, 112899 CrossRef CAS.
- A. Ahmadi, M. Sokunbi, T. Patel, M.-W. Chang, Z. Ahmad and N. Singh, Nanomaterials, 2022, 12, 2016 CrossRef CAS PubMed.
- T. L. Riss, R. A. Moravec, A. L. Niles, H. A. Benink, T. J. Worzella and L. Minor, Cell Viability Assays, in Assay Guidance Manual. Bethesda (MD): Eli Lilly & Company and the National Center for Advancing Translational Sciences, 2004 Search PubMed.
- L. Cai, X. Qin, Z. Xu, Y. Song, H. Jiang, Y. Wu, H. Ruan and J. Chen, ACS Omega, 2019, 4, 12036–12042 CrossRef CAS PubMed.
- S. Z. Alshawwa, A. A. Kassem, R. M. Farid, S. K. Mostafa and G. S. Labib, Pharmaceutics, 2022, 14, 883 CrossRef CAS PubMed.
- R. Singh and J. W. Lillard, Exp. Mol. Pathol., 2009, 86, 215–223 Search PubMed.
- A. R. Chandrasekaran, Nat. Rev. Chem., 2021, 5, 225–239 CrossRef CAS PubMed.
- C. Wang, Z. Li, P. Xu, L. Xu, S. Han and Y. Sun, J. Nanobiotechnol., 2022, 20, 476 CrossRef CAS PubMed.
- E. Oner, M. Kotmakci, A.-M. Baird, S. G. Gray, B. D. Butuner, E. Bozkurt, A. G. Kantarci and S. P. Finn, J. Nanobiotechnol., 2021, 19, 71 CrossRef CAS PubMed.
- J. M. Gallup, K. Kawashima, G. Lucero and M. R. Ackermann, Biol. Proced. Online, 2005, 7, 70–92 CrossRef CAS PubMed.
- J. A. Santos, G. A. Luz, K. P. Oliveira, L. F. Oliveira, A. S. Andrade Júnior, S. E. S. Valente and P. S. C. Lima, Genet. Mol. Res., 2016, 15 DOI:10.4238/gmr15049073.
- M.-H. Kim, H.-K. Na, Y.-K. Kim, S.-R. Ryoo, H. S. Cho, K. E. Lee, H. Jeon, R. Ryoo and D.-H. Min, ACS Nano, 2011, 5, 3568–3576 CrossRef CAS PubMed.
- R. Heidari, P. Khosravian, S. A. Mirzaei and F. Elahian, Sci. Rep., 2021, 11, 20531 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5pm00066a |
| This journal is © The Royal Society of Chemistry 2025 |