
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photochemical action plots evidence UV-promoted radical ring-opening polymerisation of cyclic ketene acetals†
Till
Meissner
 abc,
Peter
Friedel
a,
Joshua A.
Carroll
c,
Christopher
Barner-Kowollik
*cd and
Jens
Gaitzsch
*a
abc,
Peter
Friedel
a,
Joshua A.
Carroll
c,
Christopher
Barner-Kowollik
*cd and
Jens
Gaitzsch
*a
aLeibniz-Institut für Polymerforschung Dresden e.V., Hohe Strasse 6, 01069 Dresden, Saxony, Germany. E-mail: gaitzsch@ipfdd.de
bFaculty of Chemistry and Food Chemistry, Organic Chemistry of Polymers, Technische Universität Dresden, Bergstrasse 66, 01059 Dresden, Saxony, Germany
cSchool of Chemistry and Physics, Centre for Materials Science, Queensland University of Technology (QUT), 2 George Street, 4000 Brisbane, QLD, Australia. E-mail: christopher.barnerkowollik@qut.edu.au
dInstitute of Nanotechnology (INT), Karlsruhe Institute of Technology (KIT), Herrmann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany
First published on 17th December 2024
Abstract
Radical ring-opening polymerisation (RROP) of cyclic ketene acetals (CKAs) is a powerful avenue for the synthesis of biodegradable polyesters with the potential to replace non-decomposable conventional polymers. The radical polymerisation of CKAs is – surprisingly – accelerated by UV light, yet to-date the cause of the acceleration is unknown. We herein demonstrate how highly wavelength-resolved photochemical action plots of the light-induced RROP of 2-methylene-1,3,6-trioxocane (MTC) provide key information for understanding the light-prompted acceleration. We showcase that two wavelengths, 275 and 350 nm, are critical for the acceleration, with the first one facilitating the ring-opening step of the key CKA-intermediate and the second one promoting free radical initiator decay in a wavelength-orthogonal fashion. In contrast to previous studies, we aimed at unravelling the photochemically-driven monomer conversion by performing RROP at room temperature. Computational studies on the MTC radical formed during RROP indicated the cause of the acceleration: a delocalisation of the radical within the ring, which are calculated to be excited by wavelengths close to those identified experimentally. Thus, remarkably, 275 nm light critically accelerates the rate-determining ring-opening step during RROP, suggesting that photons can be used as a traceless reagent in an unexpected fashion to expedite RROPs.
Introduction
The ever-growing environmental pollution with non-(bio)degradable polymers forces humanity to seek more sustainable alternatives and substitutes. As stated by Walker et al., merely close to 9% of globally produced plastic waste has ever been recycled and 12% incinerated whilst the remaining 79% have accumulated in natural ecosystems.1 As a result of the abundance and persistence of plastics in the environment, these compounds can even be detected in humans as microplastics and are likely to pose severe health hazards for all organisms.2 In order to address these issues, it is vital that research into (bio)degradable and eco-friendly polymers is accelerated. An important class of polymers potentially able to address the noted issues are polyesters, as they entail biodegradable ester moieties in every repeating unit of their main chain. Common representatives include poly(lactic acid; PLA), poly(ε-caprolactone; PCL) and poly(β-hydroxybutyrate; PHB).3 Polyesters are classically synthesised via either ring-opening polymerisation or polycondensation, whereby the majority of these approaches require either toxic catalysts, feature low tolerance of participating functional groups or afford polymers with limited structural control.4 Radical ring-opening polymerisation (RROP) of cyclic ketene acetals (CKAs) is an alternative for synthesising polyesters and has gained significant research interest in the last decades.5–10 CKAs are able to undergo radical polymerisation due to their exo-cyclic double bond resulting in a cleavable ester group upon ring-opening.5,11 RROP of CKAs entails unique features, such as branching originating from intra- and/or inter-molecular H-transfer reactions as pioneered by Jin and Gonsalves as well as the opportunity to include pH-sensitivity from amine-bearing CKAs.10,12–15As promising candidates, poly(CKA)s and structurally similar poly(cyclic allylic sulfides) and poly(thionolactones) have been assessed and investigated as biodegradable polymers.16–18 Common CKAs that have been mapped amply by several research groups are for example 2-methylene-1,3-dioxepane (MDO), 2-methylene-4-phenyl-1,3-dioxolane (MPDL), 2-methylene-1,3,6-trioxocane (MTC) and even amine-bearing CKAs, such as N-iso-propyl-2-methylene-1,3,6-dioxozocane (iPr-MAC).6,8,11,19–22
Implementing biodegradable units into vinylic polymers exhibiting C–C backbones by copolymerisation with CKAs is generally well-established. Here, the properties of the already existing vinylic part govern the properties of the final material.23–25 Several research groups have focused on the adaptation of typical polymerisation techniques applied to the copolymerisation of vinylic monomers to CKAs, paving the way for new biodegradable polymers. Although RROP of CKAs represents a viable methodology to yield polyesters, structural limitations of CKAs due to their acetal moiety are a continuing limitation. For example, the ubiquitous issue of CKA hydrolysis was addressed by Kordes et al. and Carter et al., who recently investigated the hydrolytic behaviour of MDO for a possible use in an aqueous polymerisation protocol.26,27 Nicolas and coworkers have shown that this copolymerisation approach is even applicable for the generation of nanoparticles via radical ring-opening copolymerisation-induced self-assembly (RROPISA).28–30
While the above approach is generally functioning well, the resulting polymers still suffer from incomplete degradation of the backbone following the use of vinylic comonomers.28 To enable complete disintegration, CKAs can be polymerised on their own using RROP, generating an array of fully degradable polyesters, including self-assembling and pH-sensitive ones.8,15,28,31 In doing so, a focus is placed on the reaction mechanism of RROP, including the omnipresent branching reactions. Branching within poly(2-methylene-1,3-dioxepane) (PMDO), for example, can be utilised to control the melting temperature of this PCL analogue. Kinetic studies on RROP of MTC revealed that branching was merely dependent on monomer conversion and showed an unexpected, yet intriguing accelerated reactivity under broadband ultraviolet light irradiation of 200–400 nm.10,32 Even though the RROPs of MTC were compared with two different initiators in this study, the UV-polymerisation rate as well as macroscopic properties of the obtained polymer differed in such a substantial manner from the thermal approach, that merely relying on the influence of the employed initiator seemed not sufficient to rationalise these observations. Taking this finding into account and considering previous studies, UV light exposure has thus been noted as a promising leverage point for the acceleration of RROP of the MTC system, sparking interest to combine photochemistry with RROP of CKAs.8,32 Herein, we critically advance the mechanistic understanding of RROP in monochromatic photonic fields and determine the principle behind the observed unexpected acceleration. By addressing the main disadvantage of RROP of CKAs, i.e. their slow polymerisation rate, we will critically advance the polymerisation to make it more widely applicable.
In the field of photochemistry, photochemical action plot (AP) measurements have been pioneered by the Barner-Kowollik group as a pivotal methodology assessing the overall effectiveness of photochemical processes in a highly wavelength-resolved fashion.33–35 Modern day APs broke the previous reliance on predicting the reactivity of a photochemical system solely based on its absorbance. For a broad range of photochemical systems, a strong disparity between the absorbance maximum and reactivity maximum has been observed via careful AP mapping, often presenting a strong red shift.34,36 This bathochromism of the reactivity maximum had prevented chemists from employing the most efficient wavelength for photochemical processes.37 Establishing the true reactivity maxima and minima allows for individually addressing photo-responsive moieties, giving access to controlling photochemical processes with two colours of light synergistically, orthogonally, and antagonistically.38,39 Thus, photochemical APs are an ideal tool to gain holistic wavelength-resolved insights into photochemical processes.33
Herein, we determine the mechanistic cause of the reported UV light-induced polymerisation rate acceleration of the RROP of MTC by means of a deep photochemical AP analysis (Scheme 1). Without having access to absorbance spectra of the photoactive species, we critically advance the range of applications of APs. Our herein presented monochromatic wavelength-by-wavelength analysis allows to unpack the effects different wavelengths have on the polymerisation process, carefully separating the effect of MTC's reactivity from the initiation process. In contrast to previous studies combining thermal- and light-driven CKA conversion, we herein unravel the photochemical contribution by performing all reactions at room temperature. We underpin our wavelength-resolved findings with computational studies with the key ring radical of MTC. A kinetic study subsequently allows for in-depth insights into the properties of the final polymer for two distinct wavelengths (Scheme 1). Our study thus critically expands the use of APs and advances the photochemical and mechanistic understanding of the RROP of CKAs and simultaneously shows how RROPs can be enhanced in a wavelength-orthogonal fashion. The latter will enhance their use as a viable biodegradable alternative for common commodity plastics.
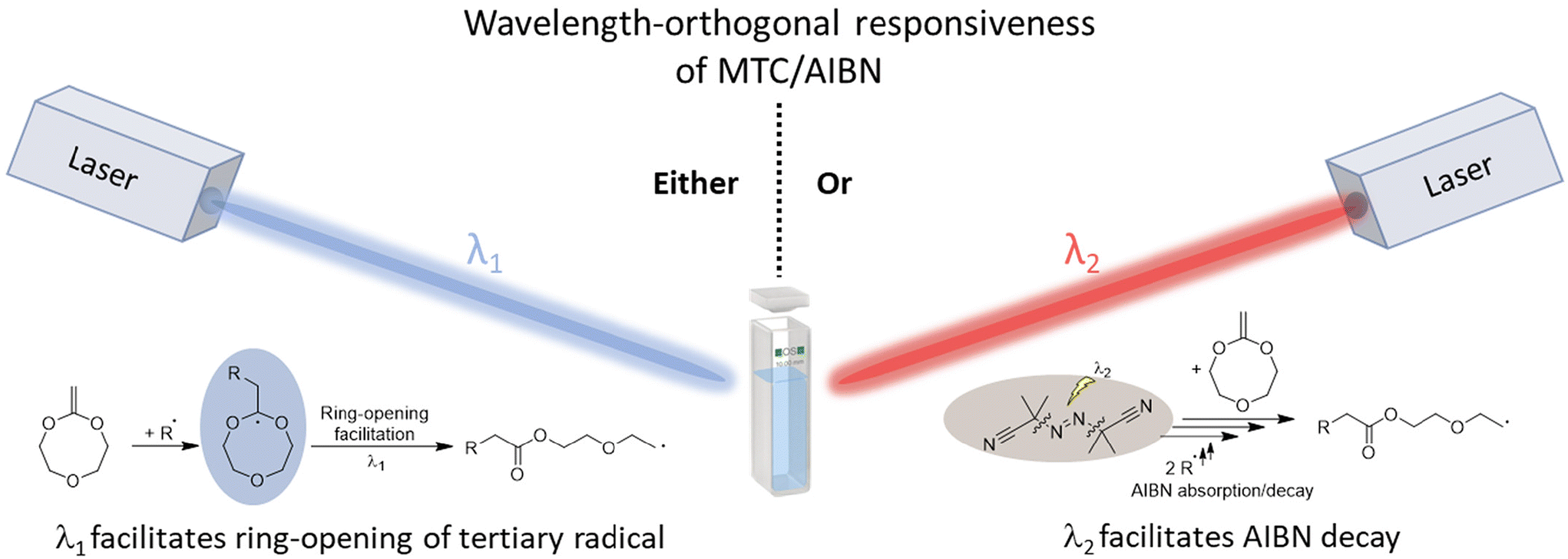 | ||
| Scheme 1 AIBN-initiated RROP of MTC exhibits a wavelength-orthogonal behaviour established by photochemical action plot measurements, i.e. either enhancement of the ring-opening propensity of the tertiary radical intermediate at λ1 = 275 nm (blue) or photolytic AIBN decay at λ2 = 350 nm (red). | ||
Results and discussion
To explore the photochemical activity of MTC, its free radical polymerisation using 2,2′-azobis(2-methylpropionitrile) (AIBN) as initiator was initially examined. The AP methodology has been described in substantial detail elsewhere (refer to Scheme S3†).33 Briefly, a monochromatic tuneable optical parametric oscillator (OPO, herein referred to as laser) was employed to map the wavelength-dependent monomer conversion of MTC via1H-NMR spectroscopy, resulting in a photochemical AP ranging from 235 to 420 nm (Scheme 1, Tables S1, S4 and Fig. S3†).The AP assessment of MTC/AIBN revealed two conversion maxima at two different wavelengths in the UV region (Fig. 1A). Whilst one maximum is located close to 275 nm, the second conversion maximum covers the spectral range between 300 and 380 nm. Interestingly, the conversion maxima are connected via an intermediate region, which is only slightly below both peaks. In order to separate the photochemically-induced radical degeneration of AIBN from the suspected self-acceleration effect of the MTC polymerisation, a control AP of an MMA/AIBN system was recorded under identical conditions with regards to photon flux and species concentration. The AP of the MMA/AIBN system displays only one high conversion plateau from 300 to 380 nm and no MMA conversion was observed close to 275 nm. While the range above 300 nm – perhaps not surprisingly – resembles the reactivity profile of the MTC/AIBN system, the now absent reactivity peak at 275 nm – critically and indeed surprisingly – strongly suggests that this wavelength region is responsible for the photochemically enhanced polymerisation rate of MTC (Fig. 1A). This finding is further underpinned by the absence of MTC conversion below 300 nm when no AIBN has been used, emphasising the need of a radical supply for commencing the FRP of MTC (Fig. S3†).
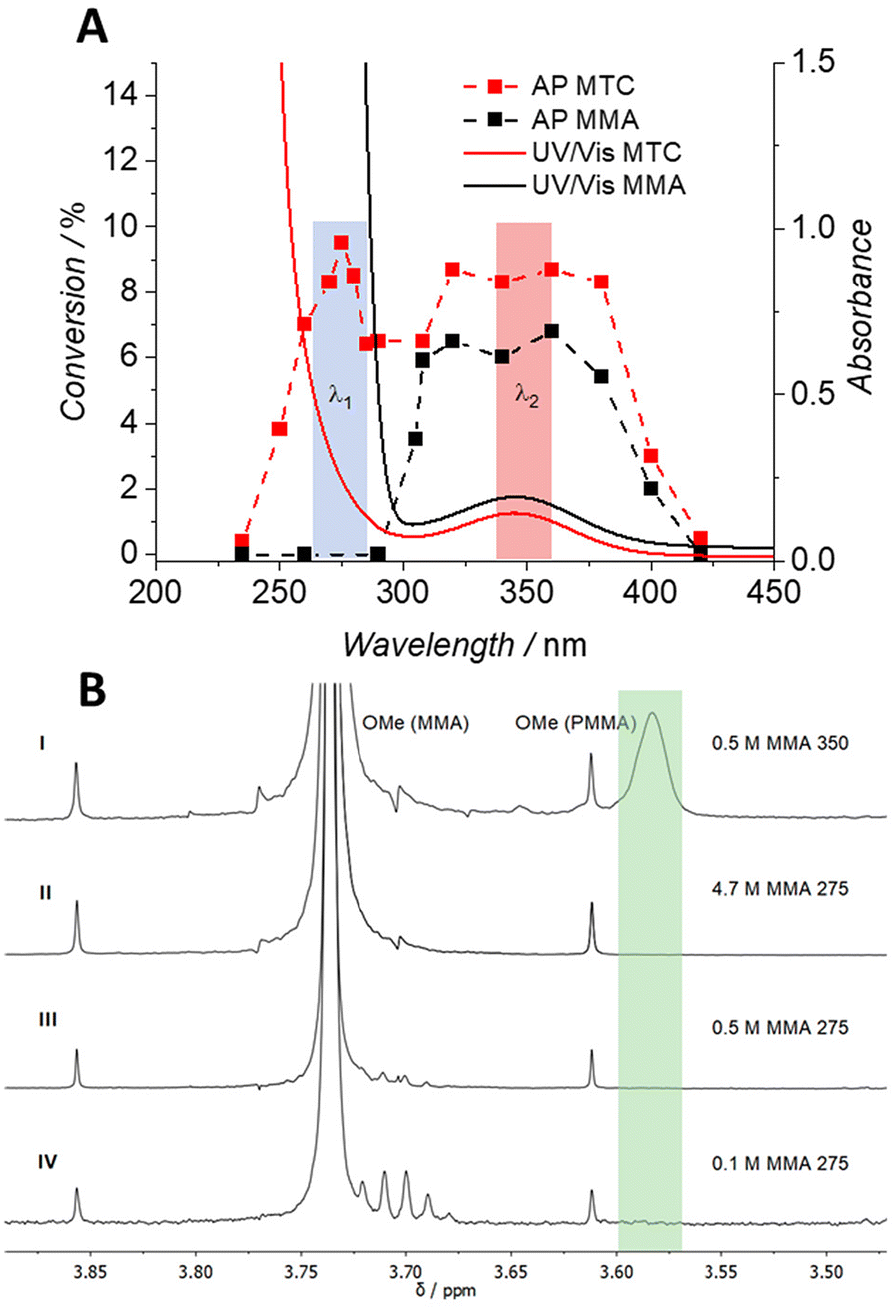 | ||
| Fig. 1 (A) Overlay of the action plots of MTC and MMA with AIBN in tBuOH at RT (dashed curves) and the corresponding normalised UV/Vis spectra (solid curves) of the stock reaction solutions emphasising wavelengths of interest, i.e. λ1 at 275 nm and λ2 at 350 nm and (B) 1H-NMR spectra of different MMA concentrations irradiated with 275 nm, showcasing the absence of PMMA formation whereas a reference experiment conducted at 350 nm indicates MMA polymerisation. | ||
Both obtained APs, i.e. of MTC/AIBN and MMA/AIBN, were overlaid with their corresponding UV/Vis spectra, both of which show a broad absorption peak at 340 nm and absorptions below 300 nm (refer to Fig. S3 and S6† for the APs for both systems). As AIBN was present in both systems, the absorption band close to 350 nm was associated with AIBN (positive control in Fig. S10†) and the congruent broad conversion maxima in both APs with photo-scission of AIBN initiating the free radical polymerisation of the respective monomers.
The critical finding from the APs is that although both polymerisation mixtures, i.e. MTC/AIBN and MMA/AIBN strongly absorb below 300 nm, only the MTC system shows polymerisation activity in that region (Fig. 1A). As the MMA system exhibits – surprisingly – no polymerisation propensity below 300 nm, we can infer that the number of radicals generated from AIBN at these wavelengths does not suffice to initiate the FRP of MMA.
In the subsequent step, it was critical to validate these findings. We assessed the monomer to polymer conversion of MMA via1H-NMR spectroscopy (Fig. 1B), while irradiating the samples with either 275 or 350 nm. The irradiation experiment with 0.5 M MMA at 350 nm clearly displays a resonance at δ = 3.57 ppm and thus radical formation that induced PMMA formation (Fig. S8A†). Reference experiments at 275 nm with variable concentrations of MMA were conducted (4.7 to 0.1 M at identical AIBN concentrations (Table S3†)), displaying no resonance at δ = 3.57 ppm, indicating the absence of PMMA formation at any of the assessed concentrations (Fig. 1B, refer to Fig. S7† for full MMA concentration series). Notably, for 0.5 M MMA, the absorption of MMA was below 0.7 at 275 nm, suggesting that MMA itself was not responsible for the observed lack of polymerisation by absorbing the majority of the incident photon flux (Fig. S9†). These observations underpin that AIBN does not decompose below 300 nm and remains photochemically silent particularly at 275 nm – which is a highly remarkable finding in itself. As a final control to evidence the absence of AIBN decay at 275 nm, the MMA analogue methyl isobutyrate was illuminated at 275 nm in the presence of AIBN and UV/Vis spectra of the mixture were recorded prior to and after irradiation. The obtained spectra clearly showed no AIBN decay at 275 nm due to an identical intensity of the absorbance band of AIBN at 350 nm after irradiation (Fig. S8B†). UV-acceleration of RROP of MTC at 275 nm was thus wavelength-orthogonal to the AIBN decay promoted at 350 nm.
In turn, we hypothesise that as radical supply for initiating the RROP of MTC at 275 nm, a slow thermal background decay of AIBN at room temperature took place rather than photo-induced cleavage at 350 nm. This limited number of generated radicals sufficed to commence the polymerisation of MTC, promoted by an intermediately formed primary radical present after ring-opening. However, the slow decay of AIBN was – compared to the MTC/AIBN system – not sufficient to initiate the free radical polymerisation of MMA, which is critically promoted by an intermediately generated tertiary radical. This radical is stabilised by a carbonyl group, kinetically slowing subsequent chain propagation. Despite the absence of PMMA formation due to an insufficient radical supply from AIBN at 275 nm and the fact that AIBN only contributes a small number of primary radical at room temperature, it can of course not be excluded that photochemical background initiation also contributes to the provision of primary radicals at 275 nm.
We note, however, that MTC also forms an intermediate tertiary radical on the acetal ring during RROP (blue ellipse in Scheme 1). Thus, a possible explanation for the UV-accelerated polymerisation of MTC is that 275 nm light excites and promotes the opening of the ring radical towards a highly reactive primary radical. Such light-responsiveness would, however, require a delocalised radical that is susceptible to UV light. Computational studies were thus conducted to closely examine the radical within the ring-opening step. Please note that a CH3 radical was chosen as an attacking radical (3D structure in Fig. 2A, schematic structure in Fig. 2B (I)). While this type of radical does not reflect the true nature of the initiator or intermediately formed tertiary radical, this simplification of the molecular structure allowed for the application of a broad range of basis sets at a reduced computational cost.
 | ||
| Fig. 2 (A) Ball-rod-model of the intermediately formed tertiary radical I with methyl substituent as R; (B) 2D-visualisation indicating delocalisation of the radical via the α-positioned oxygens (II + II′) and labelled atoms (III); (C) summarised computational values of interest determined via basis set STO-6G for radical I poised to undergo UV-promoted ring-opening at 275 nm; and (D) computationally-determined transition moment energies translated into wavelengths of radical I for increasingly complex basis sets. | ||
The intermediately formed tertiary radical I and two reference structures containing either no oxygen atoms or no radical were modelled and analysed (Fig. 2 and Fig. S12†) for bond length and bond order using Slater-type-orbitals based on Gaussian functions (STO-6G see chapter 1.3 in the ESI†). Atoms were numbered starting from the radical-bearing carbon atom (C1) and then following the ring to O2′/O2′′ and C3′/C3′′ on either side of C1. Both C–O bonds starting from C1 (C1–O2′/2′′ in Fig. 2B with 140.0 and 140.3 pm, respectively) were 3.5 to 4 pm shorter than the following C–O bonds for carbon atoms C3′/C3′′ (O2′–C3′/O2′′–C3′′ in Fig. 2B with 143.7 and 144.1 pm, respectively) To put this in perspective, C–O single bond lengths are generally defined by values of 143 pm and C–O bonds with a partial double bond character exhibit bond lengths as low as 136 pm. Consequentially, the herein depicted C1–O2′/2′′ bond lengths of close to 140 pm in radical I indeed indicate a slight double bond character and hence a delocalised electron.40 Both calculated bond orders of the C1–O2′/2′′ bonds (1.024 and 0.991, respectively) are approximately 0.05 higher than the ones of the O2′–C3′/O2′′–C3′′ bonds (0.954 and 0.950, respectively), also supporting the theory of slightly delocalised electrons within the acetal moiety. Further, an intriguing and highly surprising O2′–O2′′ bond order of 0.052 across the ring molecule I was found. Note that the equivalency of the O2′–O2′′ bond order with the amount by which the C1–O2′/2′′ bonds were stronger, underpins the presence of a slightly delocalised radical. Consequently, suitable mesomeric resonance structures of radical I were modelled (structures II and II′ in Fig. 2B). Contrary to the electronegativity values of oxygen and carbon, these suggested slightly positively charged oxygen atoms O2′/O2′′ as well as negatively charged radical-bearing C1. Our calculations confirm this notion with charges of 0.019 and 0.009 for O2′ and O2′′, respectively, and a charge of −0.140 for C1 (Fig. 2C).
In contrast to radical I, neither a saturated reference molecule without the tertiary radical at C1 (Fig. S12A†) nor a radical without adjacent oxygen atoms (Fig. S12B†) showed deviating bond lengths, deviating bond orders, positively charged oxygens O2′/O2′′ or a negatively charged carbon C1. The structural peculiarities of acetal radical I were solidified by these negative controls and may thus be associated with the observed light-responsiveness. We were thus able to infer a slight but critical delocalisation of the radical over the entire three-atom entity of the acetal moiety, capable of absorbing incident UV irradiation at 275 nm. Radical I was thus better represented by a delocalised three-way bond (structure III in Fig. 2B).
Completing the link to light-responsiveness are final computational studies to assess a possible interaction of molecule I with light of 275 nm. Thus, the transition moments (HOMO–LUMO gap) of tertiary radical I were calculated for increasingly complex basis sets (more expensive in calculation time). Starting with the basis set STO-6G, a transition moment of 473 nm could be identified.41 The more complex, and hence more accurate basis sets CCD and ACCD from the Dunning family42 as well as basis set KTZV from the Ahlrich group43,44 resulted in transition moments of 311–312 nm for all of them. This range was verified by APCseg-0 and PCseg-0 from the Jensen family with 316 and 307 nm, respectively.45 (Fig. 2D, see Table S6† for a comprehensive overview of all basis sets). Following this considerable decrease in wavelength (i.e. increase in energy gap) with increasing accuracy of basis set, it is reasonable to assume that the real transition would occur at an even lower wavelength (i.e. require more energy). Taking a likely hypsochromic shift towards the reactivity maximum at 275 nm into account, as elaborated via action plots for a broad range of chromophores, a reasonable match between the computed absorption maximum of radical I and experimental reactivity maximum of the RROP of MTC is observed.33,34,36 Being able to solely accelerate RROP at 275 nm and having identified the underlying mechanistic reason will allow for faster development of this chemistry.
To elaborate the scope of the identified photo-sensitivity of RROP of MTC, we subsequently explored possible influences of the utilised wavelength on the polymerisation kinetics. Following the argument of wavelength-orthogonality, polymerisations conducted separately at 275 nm (accelerating RROP) and 350 nm (AIBN cleavage) were thus analysed for characteristic properties, such as monomer conversion, polymerisation rate, density of branches (DB), molecular weight and dispersity (Fig. 3). Following a previously published protocol, these experiments were carried out in bulk as well as in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dilution with tert-butanol, however in the current study, we performed RROP exclusively at room temperature.32
:1 dilution with tert-butanol, however in the current study, we performed RROP exclusively at room temperature.32
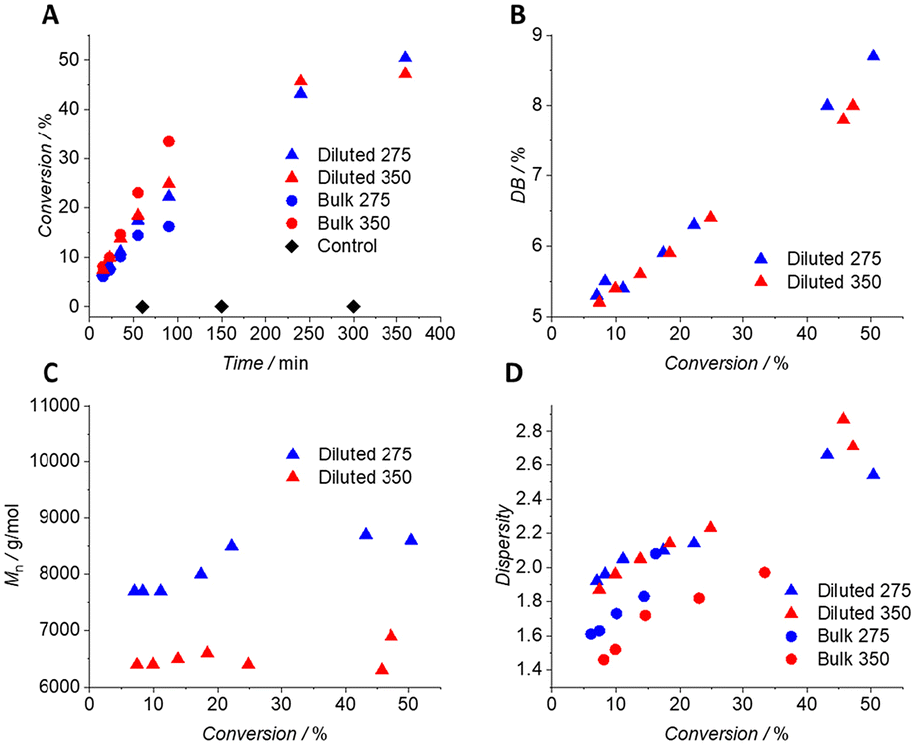 | ||
| Fig. 3 Kinetic studies of the RROP of MTC under UV light of 275 and 350 nm at room temperature entailing (A) MTC conversion vs. time development for all kinetics together with control samples kept under light exclusion; (B) DB vs. MTC conversion development for 1:1 diluted kinetics; (C) relatively determined Mnvs. MTC conversion development for 1:1 diluted kinetics; and (D) dispersity vs. MTC conversion correlation for all kinetics. | ||
In dilution, monomer conversion, DB and dispersity revealed similar absolute values under both 275 and 350 nm (Fig. 3, Fig. S4 and Table S2†). In both cases, MTC conversion increases in a logarithmic manner from 7% after 15 min to 50% after 360 min of irradiation, whereas no MTC conversion was monitored at any time under light exclusion (Fig. 3A). Using UV light of 275 nm, allowed for a 50% conversion of MTC after 360 min at room temperature. A similar value (about 55%) had been reached in the thermal polymerisation protocol of MTC at 65 °C after the same period of time. Via this comparison, the strong acceleration of RROP of MTC under irradiation with UV light of 275 nm is evident.32 The dispersity increased from close to 1.9 to approx. 2.7 in the same time frame (Fig. 3D). The DB of PMTC increased linearly from close to 5% to approximately 9% from 10% to 50% conversion, respectively, underpinning the previously suggested exclusive dependence of the DB on monomer conversion and no other external parameters (Fig. 3B).26,32,46 The DBs for the bulk kinetics were not determined due to unknown other side reactions that hindered a clear read of the CH3-signal in the 1H-NMR spectrum required for DB determination.32
Size exclusion chromatography (SEC), however, revealed a notable difference for the polymers originating from the two distinct incident wavelengths. A general trend of decreased elution times for polymers from 275 nm over polymers from 350 nm was observed (Fig. S5A†, Fig. 3C, refer to Fig. S5B† for the complete data set). Using a universal calibration of linear PMMA standards, these decreased elution times translated into increased molecular weights. While the absolute numbers of the determined molecular weight of branched PMTC polymers may thus not agree with their true values, the relative difference in molecular weight between the polymers is still valid.10,12,32 PMTC formed at 275 nm exhibits increased molecular weights by approximately 20–30% compared to 350 nm, ranging from 7700 to 8600 g mol−1 for 275 nm and from 6400 to 6900 g mol−1 for 350 nm – again from 10 to 50% conversion, respectively (Fig. S5B†). As possible rationalisation, the dependence of the molecular weight on the initiator concentration can be invoked. In a simplified manner, more initiator leads to lower molecular weights of the resulting polymers.47 Due to the UV-induced dissociation of AIBN at 350 nm, a larger number of radicals is formed, resulting in lower molecular weights of the obtained polymers.47 The slow thermal and photochemical background decay of AIBN at 275 nm leads to a smaller number of radicals to initiate RROP, thus the obtained polymers display a larger molecular weight. We note that inherent transfer reactions occurring during RROP of CKAs mitigate the influence of the AIBN radical flux during the reaction, ultimately lowering the disparity in the observed molecular weights.32 Note that the herein determined molecular weights appear to be relatively low, i.e. 8500 g mol−1 at 50% monomer conversion (for 1:1 dilution at 275 nm). It should be noted that the formed polyester exhibits a significant absorption band at 275 nm, which could absorb a certain amount of incident photons, eventually disallowing quantitative monomer conversion and, as a possible consequence, higher molecular weight (Fig. S11†). This potential limit is particularly pronounced when compared to previous studies by Mehner et al. for a thermal RROP protocol, achieving 17000 g mol−1 at similar conversions.32 However, these studies have been performed at elevated temperature as opposed to the current work. In addition, as these molecular weights from thermal polymerisation were obtained using absolute molecular weight determination, they cannot be compared to the current study using relative calibration.
Varying the initial MTC concentration from 1:1 dilution to bulk revealed a disparate polymerisation behaviour, particularly at 275 nm. While the polymerisation rate increased by a factor of 2.5 for the bulk kinetic at 350 nm, it significantly decreased at 275 nm (Fig. S4 and Table S2†). In addition, SEC measurements reveal consistently lower molecular weights at 275 compared to 350 nm, which is in contrast to previous findings in 1:1 dilution and to literature.32 These data can be rationalised when inspecting the UV/Vis spectrum of PMTC, which absorbs light at 275 nm, but not at 350 nm (Fig. S11†). When working in bulk, any formed PMTC now absorbs incident photons, which subsequently cannot perform the critical acceleration of the ring-opening step. Thus, no acceleration of ring-opening occurs at 275 nm, but rather a deceleration of the overall monomer consumption, concomitantly affecting the molecular weight of the resulting PMTC as just discussed. Intriguingly, the obtained dispersities for PMTC prepared in bulk exhibit consistently lower values compared to solution RROP, which contradicts previously published data of the homopolymerisation of MTC.32 This contradiction remains the subject of further investigations.
Conclusions
We herein employ the powerful photochemical action plot methodology to determine why the polymerisation rate of the ring-opening polymerisation of MTC is critically accelerated by UV light of specific wavelengths. All reactions were carried out at room temperature to minimise thermally-driven MTC conversion. In doing so, we determined two conversion maxima in the MTC/AIBN polymerisation system, i.e. at 275 and 350 nm. Key reference experiments utilising MMA/AIBN allowed us to allocate 350 nm to UV-promoted AIBN decay and 275 nm to accelerated RROP of MTC. The identified wavelength selectivity was corroborated by computational studies. Bond lengths, bond orders, and charge distributions of the intermediate closed-ring radical suggested a slight but notable delocalisation of the intermediate radical over the acetal unit, rendering the molecule susceptible to UV light. Applying the trend of decreasing excitation wavelengths with increasingly complex basis sets aligned well with the noted reactivity maximum at 275 nm, considering the ubiquitous bathochromic shift of photochemical reactivity for a vast array of chromophores. Subsequent kinetic measurements revealed the enhancement of monomer conversion by UV light of 275 nm in conjunction with an increase of molecular weight for polymers generated at 275 nm compared to ones formed at 350 nm. The behaviour is rationalised by the higher number of radicals formed through photo-activated AIBN scission upon incident UV light of 350 nm. Additional efforts will be focused on modulating the macroscopic PCKA properties by varying the incident photon flux. Both the reference experiments as well as theoretical considerations suggest that the MTC/AIBN system responds to UV light in a wavelength-orthogonal fashion. While radiation with 275 nm light uniquely expedites the rate of the ring-opening step of the key CKA intermediate, radiation with 350 nm only promotes photolytic AIBN decay.The herein exemplified MTC-specific interaction with UV irradiation paves the way for exploiting this mechanism for other CKAs, including copolymerisation and controlled radical polymerisation protocols. This critical acceleration could now be expanded to other CKAs and allows RROP to be employed in a broader synthetic field as their major drawback – a slow reaction rate – can now be overcome with UV light. As photochemical APs allow for allocating the acceleration wavelength without previous knowledge of the absorption maximum of the photochemically active species, our study critically broadens the application of photochemical APs in modern synthetic macromolecular chemistry.
Author contributions
All authors contributed to the writing, editing, and formatting of the manuscript. All authors have given approval to the final version of the manuscript.Data availability
Experimental data is available in the ESI.† Additional data for this article, including [NMR spectra, UV-Vis spectra, computational data] are available at Zenodo at https://doi.org/10.5281/zenodo.13141568.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B.-K. acknowledges the Australian Research Council (ARC) for a Laureate Fellowship enabling his photochemical research program as well as the QUT Centre for Materials Science for continued support (Grant number FL170100014). T. M. acknowledges the German Academic Exchange Service (DAAD) for a PROMOS scholarship as financial support. T. M. and J. G. acknowledge the German Research Foundation (DFG) for financial support as part of the DFG project GA 2051/7-1. The Central Analytical Research Facility (CARF) at QUT is gratefully acknowledged. The authors acknowledge discussions with Prof. Albena Lederer (IPF), providing valuable scientific discussions regarding the SEC data analysis.References
- T. R. Walker and L. Fequet, Current trends of unsustainable plastic production and micro(nano)plastic pollution, TrAC, Trends Anal. Chem., 2023, 160, 116984 CrossRef CAS.
- S. Kasavan, S. Yusoff, M. F. Rahmat Fakri and R. Siron, Plastic pollution in water ecosystems: A bibliometric analysis from 2000 to 2020, J. Cleaner Prod., 2021, 313, 127946 CrossRef.
- G. Z. Papageorgiou, Thinking Green: Sustainable Polymers from Renewable Resources, Polymers, 2018, 10, 952 CrossRef PubMed.
- P. M. Schäfer and S. Herres-Pawlis, Robust Guanidine Metal Catalysts for the Ring-Opening Polymerization of Lactide under Industrially Relevant Conditions, ChemPlusChem, 2020, 85, 1044–1052 CrossRef PubMed.
- W. J. Bailey, Z. Ni and S.-R. Wu, Synthesis of poly-ε-caprolactone via a free radical mechanism. Free radical ring-opening polymerization of 2-methylene-1,3-dioxepane, J. Polym. Sci., Polym. Chem. Ed., 1982, 20, 3021–3030 CrossRef CAS.
- S. Agarwal, Chemistry, chances and limitations of the radical ring-opening polymerization of cyclic ketene acetals for the synthesis of degradable polyesters, Polym. Chem., 2010, 1, 953–964 RSC.
- A. Tardy, J.-C. Honoré, D. Siri, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, A comprehensive kinetic study of the conventional free-radical polymerization of seven-membered cyclic ketene acetals, Polym. Chem., 2017, 8, 5139–5147 Search PubMed.
- J. Folini, W. Murad, F. Mehner, W. Meier and J. Gaitzsch, Updating radical ring-opening polymerisation of cyclic ketene acetals from synthesis to degradation, Eur. Polym. J., 2020, 134, 109851 CrossRef CAS.
- S. Reddy Mothe, J. S. J. Tan, L. R. Chennamaneni, F. Aidil, Y. Su, H. C. Kang, F. C. H. Lim and P. Thoniyot, A systematic investigation of the ring size effects on the free radical ring-opening polymerization (rROP) of cyclic ketene acetal (CKA) using both experimental and theoretical approach, J. Polym. Sci., 2020, 58, 1728–1738 CrossRef CAS.
- F. Mehner, M. Geisler, K. Arnhold, H. Komber and J. Gaitzsch, Structure–Property Relationships in Polyesters from UV-Initiated Radical Ring-Opening Polymerization of 2-Methylene-1,3-dioxepane (MDO), ACS Appl. Polym. Mater., 2022, 4, 7891–7902 CrossRef CAS.
- A. Tardy, J. Nicolas, D. Gigmes, C. Lefay and Y. Guillaneuf, Radical Ring-Opening Polymerization: Scope, Limitations, and Application to (Bio)Degradable Materials, Chem. Rev., 2017, 117, 1319–1406 CrossRef CAS PubMed.
- S. Jin and K. E. Gonsalves, A Study of the Mechanism of the Free-Radical Ring-Opening Polymerization of 2-Methylene-1,3-dioxepane, Macromolecules, 1997, 30, 3104–3106 Search PubMed.
- S. Agarwal, Microstructural Characterisation and Properties Evaluation of Poly (methyl methacrylate-co-ester)s, Polym. J., 2007, 39, 163–174 CrossRef CAS.
- S. Agarwal and C. Speyerer, Degradable blends of semi-crystalline and amorphous branched poly(caprolactone): Effect of microstructure on blend properties, Polymer, 2010, 51, 1024–1032 CrossRef CAS.
- Y. Deng, A. Frezel, F. Mehner, P. Friedel and J. Gaitzsch, Amine-bearing cyclic ketene acetals for pH-responsive and degradable polyesters through radical ring-opening polymerisation, Polym. Chem., 2023, 14, 4275–4281 RSC.
- P. T. Do, B. L. J. Poad and H. Frisch, Programming Photodegradability into Vinylic Polymers via Radical Ring-Opening Polymerization, Angew. Chem., Int. Ed., 2023, 62, e202213511 CrossRef CAS PubMed.
- R. Abu Bakar, K. S. Hepburn, J. L. Keddie and P. J. Roth, Degradable, Ultraviolet-Crosslinked Pressure-Sensitive Adhesives Made from Thioester-Functional Acrylate Copolymers, Angew. Chem., Int. Ed., 2023, 62, e202307009 CrossRef CAS PubMed.
- M. Lages, N. Gil, P. Galanopoulo, J. Mougin, C. Lefay, Y. Guillaneuf, M. Lansalot, F. D'Agosto and J. Nicolas, Degradable Latexes by Nitroxide-mediated Aqueous Seeded Emulsion Copolymerization Using a Thionolactone, Macromolecules, 2023, 56, 7973–7983 Search PubMed.
- Y. Deng, F. Mehner and J. Gaitzsch, Current Standing on Radical Ring-Opening Polymerizations of Cyclic Ketene Acetals as Homopolymers and Copolymers with one another, Macromol. Rapid Commun., 2023, 44, 2200941 CrossRef CAS PubMed.
- W. J. Bailey, S.-R. Wu and Z. Ni, Synthesis and free radical ring-opening polymerization of 2-methylene-4-phenyl-1,3-dioxolane, Macromol. Chem. Phys., 1982, 183, 1913–1920 CrossRef CAS.
- M. R. Hill, E. Guégain, J. Tran, C. A. Figg, A. C. Turner, J. Nicolas and B. S. Sumerlin, Radical Ring-Opening Copolymerization of Cyclic Ketene Acetals and Maleimides Affords Homogeneous Incorporation of Degradable Units, ACS Macro Lett., 2017, 6, 1071–1077 CrossRef CAS PubMed.
- G. G. Hedir, C. A. Bell, R. K. O'Reilly and A. P. Dove, Functional Degradable Polymers by Radical Ring-Opening Copolymerization of MDO and Vinyl Bromobutanoate: Synthesis, Degradability and Post-Polymerization Modification, Biomacromolecules, 2015, 16, 2049–2058 Search PubMed.
- J. Undin, P. Plikk, A. Finne-Wistrand and A.-C. Albertsson, Synthesis of amorphous aliphatic polyester-ether homo- and copolymers by radical polymerization of ketene acetals, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4965–4973 CrossRef CAS.
- P. Galanopoulo, L. Sinniger, N. Gil, D. Gigmes, C. Lefay, Y. Guillaneuf, M. Lages, J. Nicolas, F. D'Agosto and M. Lansalot, Degradable vinyl polymer particles by radical aqueous emulsion copolymerization of methyl methacrylate and 5,6-benzo-2-methylene-1,3-dioxepane, Polym. Chem., 2023, 14, 1224–1231 RSC.
- T. Pesenti, E. Gillon, S. Ishii, S. Messaoudi, Y. Guillaneuf, A. Imberty and J. Nicolas, Increasing the Hydrophilicity of Cyclic Ketene Acetals Improves the Hydrolytic Degradation of Vinyl Copolymers and the Interaction of Glycopolymer Nanoparticles with Lectins, Biomacromolecules, 2023, 24, 991–1002 CrossRef CAS.
- B. R. Kordes, L. Ascherl, C. Rüdinger, T. Melchin and S. Agarwal, Competition between Hydrolysis and Radical Ring-Opening Polymerization of MDO in Water. Who Makes the Race?, Macromolecules, 2023, 56, 1033–1044 CrossRef CAS.
- M. C. D. Carter, A. Hejl, M. Janco, J. DeFelippis, P. Yang, M. Gallagher and Y. Liang, Emulsion Polymerization of 2-Methylene-1,3-Dioxepane and Vinyl Acetate: Process Analysis and Characterization, Macromolecules, 2023, 56, 5718–5729 CrossRef CAS.
- E. Guégain, C. Zhu, E. Giovanardi and J. Nicolas, Radical Ring-Opening Copolymerization-Induced Self-Assembly (rROPISA), Macromolecules, 2019, 52, 3612–3624 CrossRef.
- M. Lages, T. Pesenti, C. Zhu, D. Le, J. Mougin, Y. Guillaneuf and J. Nicolas, Degradable polyisoprene by radical ring-opening polymerization and application to polymer prodrug nanoparticles, Chem. Sci., 2023, 14, 3311–3325 Search PubMed.
- G. G. Hedir, A. Pitto-Barry, A. P. Dove and R. K. O'Reilly, Amphiphilic block copolymer self-assemblies of poly(NVP)-b-poly(MDO-co-vinyl esters): Tunable dimensions and functionalities, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 2699–2710 CrossRef CAS.
- Y. Deng, S. Schäfer, D. Kronstein, A. Atabay, M. Susewind, E. Krieg, S. Seiffert and J. Gaitzsch, Amphiphilic Block Copolymers PEG-b-PMTCs: Synthesis, Self-Assembly, Degradation Properties and Biocompatibility, Biomacromolecules, 2024, 25, 303–314 CrossRef CAS PubMed.
- F. Mehner, T. Meissner, A. Seifert, A. Lederer and J. Gaitzsch, Kinetic studies on the radical ring-opening polymerization of 2-methylene-1,3,6-trioxocane, J. Polym. Sci., 2023, 61, 1882–1892 CrossRef CAS.
- I. M. Irshadeen, S. L. Walden, M. Wegener, V. X. Truong, H. Frisch, J. P. Blinco and C. Barner-Kowollik, Action Plots in Action: In-Depth Insights into Photochemical Reactivity, J. Am. Chem. Soc., 2021, 143, 21113–21126 CrossRef CAS PubMed.
- S. L. Walden, J. A. Carroll, A.-N. Unterreiner and C. Barner-Kowollik, Photochemical Action Plots Reveal the Fundamental Mismatch Between Absorptivity and Photochemical Reactivity, Adv. Sci., 2024, 11, 2306014 CrossRef CAS PubMed.
- P. W. Kamm, J. P. Blinco, A.-N. Unterreiner and C. Barner-Kowollik, Green-light induced cycloadditions, Chem. Commun., 2021, 57, 3991–3994 Search PubMed.
- I. Mohamed Irshadeen, V. X. Truong, H. Frisch and C. Barner-Kowollik, Simultaneously recorded photochemical action plots reveal orthogonal reactivity, Chem. Commun., 2023, 59, 11959–11962 RSC.
- D. E. Fast, A. Lauer, J. P. Menzel, A.-M. Kelterer, G. Gescheidt and C. Barner-Kowollik, Wavelength-Dependent Photochemistry of Oxime Ester Photoinitiators, Macromolecules, 2017, 50, 1815–1823 Search PubMed.
- H. Frisch, F. R. Bloesser and C. Barner-Kowollik, Controlling Chain Coupling and Single-Chain Ligation by Two Colours of Visible Light, Angew. Chem., Int. Ed., 2019, 58, 3604–3609 CrossRef CAS.
- C. G. Bochet, Two Decades of Chromatic Orthogonality, Isr. J. Chem., 2021, 61, 486–495 CrossRef CAS.
- N. N. Greenwood and A. Earnshaw, Chemistry of the Elements, Elsevier, 2012 Search PubMed.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- T. H. Dunning, P. J. Hay and H. F. Schaefer, Methods of electronic structure theory, Mod. Theor. Chem., 1977, 3, 1–28 CAS.
- A. Schäfer, H. Horn and R. Ahlrichs, Fully optimized contracted Gaussian basis sets for atoms Li to Kr, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- F. Jensen, Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets, J. Chem. Theory Comput., 2014, 10, 1074–1085 CrossRef CAS.
- M. Mousa, M. Jonsson, O. Wilson, R. Geerts, H. Bergenudd, C. Bengtsson, A. Larsson Kron and E. Malmström, Branched polyesters from radical ring-opening polymerization of cyclic ketene acetals: synthesis, chemical hydrolysis and biodegradation, Polym. Chem., 2023, 14, 5154–5165 RSC.
- H.-G. Elias, Makromoleküle: Chemische Struktur und Synthesen, Wiley-VCH, 6th edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data and analyses. See DOI: https://doi.org/10.1039/d4py00847b |
| This journal is © The Royal Society of Chemistry 2025 |